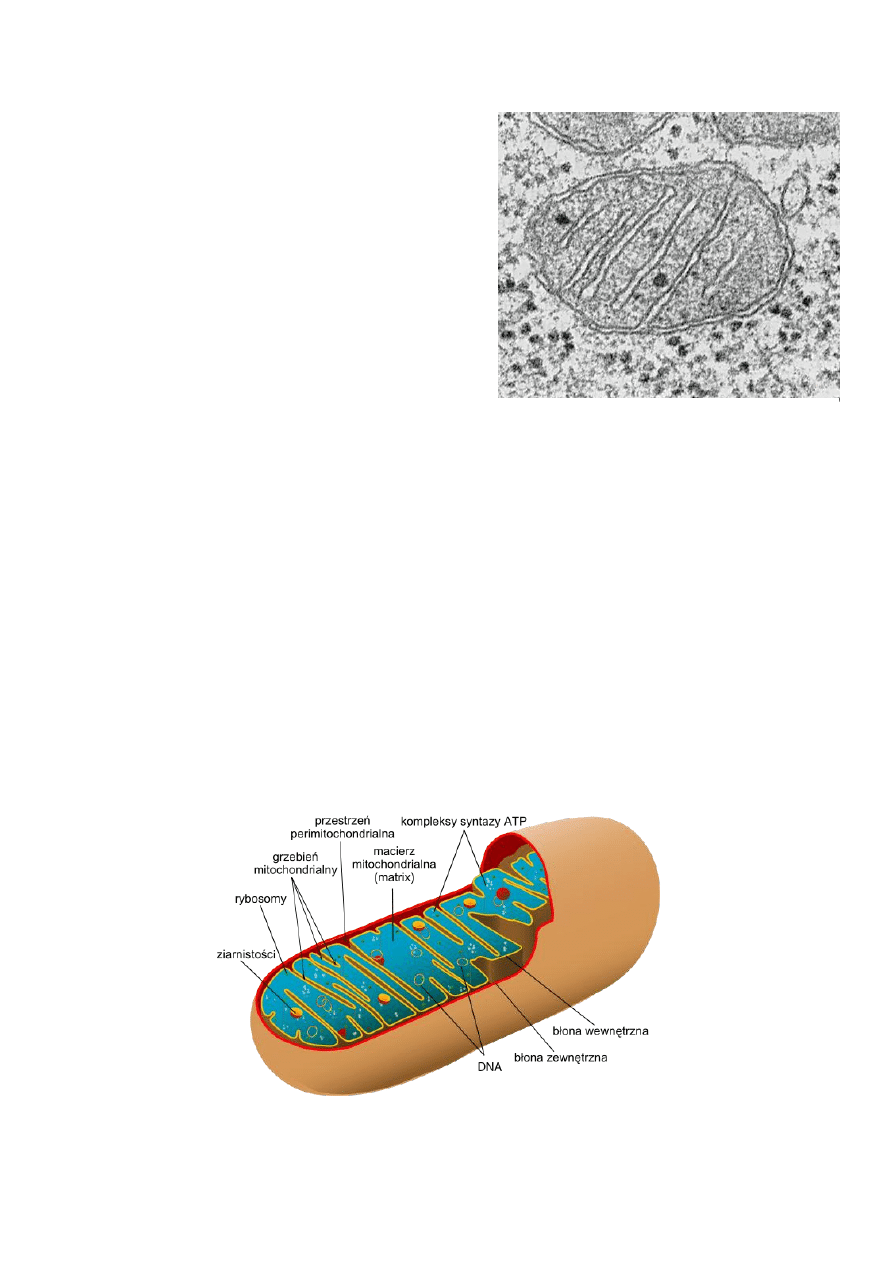
Mitochondrium - budowa i funkcje
Mitochondria to organelle komórkowe, w których
odbywa się proces oddychania tlenowego. Ich zadanie polega
na przechwytywaniu energii uwalnianej stopniowo podczas
rozpadu cząsteczek pokarmowych i wykorzystywaniu jej do
syntezy ATP (adenozynotrifosforanu), uniwersalnego nośnika
energii użytecznej dla komórki. Mitochondria są dużych
rozmiarów (przeważnie od 2 do 8 μm) i można je
zaobserwowad nawet pod mikroskopem świetlnym. Znajdują
się prawie we wszystkich komórkach eukariotycznych - oprócz
erytrocytów. W różnych komórkach mitochondria mogą
przybierad odmienne kształty: okrągły, owalny lub rzadziej
niciowaty i rozgałęziony. Ich liczba w komórce wynosi od
1
u
organizmów
jednokomórkowych
do
1000-2000
w hepatocytach (komórkach o dużej aktywności metabolicznej).
Lokalizacja mitochondriów w komórce nie jest stała. W wyniku ruchów cytoplazmy lub dzięki związaniu się
z elementami cytoszkieletu, mitochondria mają zdolnośd do przemieszczania się w kierunku miejsca o zwiększonym
zapotrzebowaniu na energię. Mitochondria mogą też przejściowo magazynowad wapo, co stanowi częśd procesów
odpowiedzialnych za zachowanie równowagi wapniowej w komórce.
Mitochondria są otoczone dwiema błonami, zbudowanymi z dwuwarstwy lipidowej. Wewnętrzna błona
mitochondrialna, w przeciwieostwie do zewnętrznej jest silnie pofałdowana w liczne grzebienie mitochondrialne
skierowane ku wnętrzu organelli. Obie błony dzielą mitochondrium na dwa przedziały: dużą wewnętrzną przestrzeo
nazwaną matriks (lub macierzą mitochondrialną) i wąską przestrzeo międzybłonową. Matriks zawiera koliste
cząsteczki mitochondrialnego DNA (mtDNA), zestaw specyficznych tRNA, rybosomy (70S - w przeciwieostwie do
rybosomów cytoplazmatycznych 80S) i różne enzymy. Dzięki obecności kanałów białkowych zewnętrzna błona
mitochondrialna jest przepuszczalna dla większości małych cząsteczek i jonów, natomiast błona wewnętrzna nie
przepuszcza prawie żadnych jonów i obdarzonych ładunkiem cząstek. W wewnętrznej błonie mitochondrialnej
znajduje się znacznie więcej białek niż w błonie zewnętrznej - są to białka odpowiedzialne za oddychanie komórkowe
oraz nośniki transportujące metabolity i jony.
Ryc. 1 Mitochondrium
Ryc. 2 Schemat budowy mitochondriom
Genom mitochondrialny
Mitochondria to tzw. organelle
półautonomiczne. Pochodzą one od
bakterii tlenowych (najprawdopodobniej
proteobakterii), które setki milionów lat
temu zostały wchłonięte na drodze
fagocytozy przez prymitywne komórki
i nie doszło do ich strawienia. Teoria
endosymbiozy (zaproponowana po raz
pierwszy w 1909 roku przez Konstantina
Mereszkowskiego) tłumaczy obecnośd w
mitochondriach: DNA, rybosomów 70S
(takich jak u bakterii), dwóch błon
mitochondrialnych (wewnętrzna błona
jest
własną
błoną
Proteobacteria,
a zewnętrzna błoną fagocytarną) oraz
aparatu biosyntezy białek, podobnego jak
u prokariontów. Nowe mitochondria powstają przez wzrost i podział już istniejących, które rozdzielają się losowo
między komórki potomne.
Genom komórki tworzą wszystkie cząsteczki DNA. Genom eukariontów składa się z kilku części. Większośd
informacji genetycznej jest zapisana w DNA przechowywanym w jądrze komórkowym (genom jądrowy), jednak jej
częśd znajduje się w organellach komórkowych: mitochondriach i chloroplastach (w przypadku roślin). Genomy
mitochondrialne ssaków są bardo małe, natomiast grzybów i roślin większe. Wielkośd mitochondrialnego genomu
u roślin wacha się w granicach 200-2400 kpz, natomiast u zwierząt w granicach 10-16 kpz.
Ryc. 3 Proteobacteria
Ryc. 4 Schemat genomu mitochondrialnego człowieka
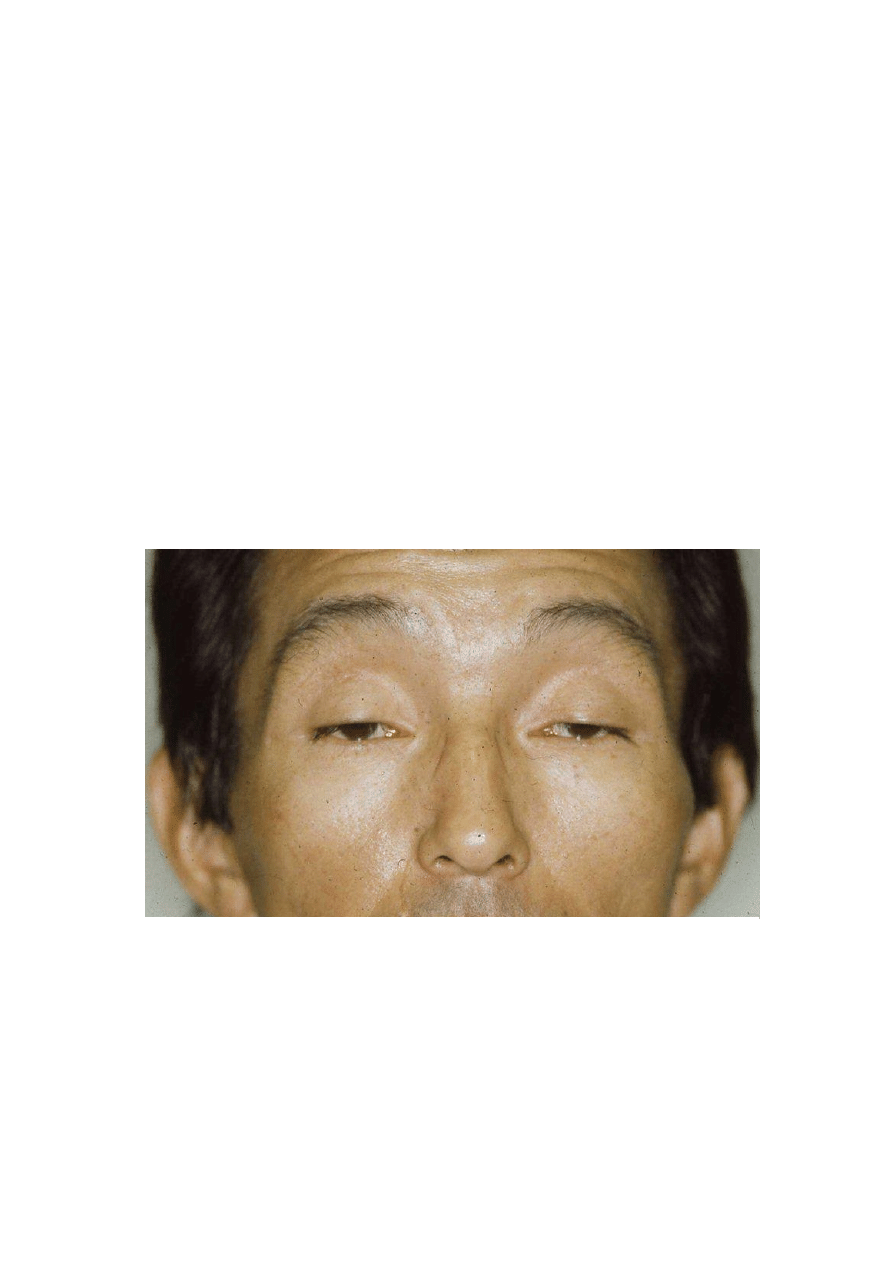
W organizmie człowieka, każde mitochondrium zawiera 4-10 cząsteczek kolistego DNA o długości około
16 kpz. Znajduje się ono w matriks i koduje 37 genów: 13 odpowiedzialnych za polipeptydy, 22 za tRNA i 2 za rRNA.
Białka kodowane przez mtDNA to częśd mitochondrialnych białek łaocucha oddechowego, jednak większośd białek
wchodzących w jego skład jest kodowana przez genom jądrowy. tRNA i rRNA biorą udział w biosyntezie białka
odbywającej się w mitochondriach. Geny mitochondrialne ludzi nie zawierają intronów. Nici pojedynczej cząsteczki
mtDNA są oznaczane jako H (ang. heavy - ciężka) i L (ang. light - lekka). Kod genetyczny mitochondriów różni się od
kodu genetycznego w genomie jądrowym. UGA (jądrowy kodon STOP) w mitochondriach oznacza tryptofan, AUA
(izoleucyna) - metioninę, a AGA i AGG (arginina) są mitochondrialnymi kodonami STOP. W sumie, w każdej komórce
występują setki kopii mtDNA (poliplazmia) i w prawidłowych komórkach są one identyczne (homoplazmia), gdyż
pochodzą tylko od jednego z rodziców - są przekazywane niemal wyłącznie w linii żeoskiej. Heteroplazmia, będąca
wynikiem mutacji to obecnośd w komórce więcej niż jednego rodzaju mtDNA. W mitochondrialnym DNA znajduje się
tzw. obszar hiperzmienny. Jest to niekodujący fragment genomu mitochondrialnego, który bardzo różni się między
ludźmi i jest wykorzystywany do badao genetyki populacyjnej i w medycynie sądowej do ustalania tożsamości.
Mutacje w genach mitochondrialnych powodują choroby mitochondrialne. Są to choroby wielosystemowe
i trudne do zdiagnozowania, a ich leczenie jest objawowe. Dotyczą głównie tkanek o największym zapotrzebowaniu
energetycznym - tkanki mięśniowej i nerwowej. Towarzyszą im zaburzenia fosforylacji oksydacyjnej i kompleksów
łaocucha oddechowego, brak aktywności niektórych enzymów oraz zmiana ultrastruktury mitochondriów. Choroby
mitochondrialne można podzielid na: rearanżacje w mtDNA (zespół szpikowo-trzustkowy Pearsona, zespół Kearnsa-
Sayre’a, zespół przewlekłej postępującej zewnętrznej oftalmoplegii - CPEO) i mutacje punktowe w mtDNA (zespół
MELAS, zespół MERF, zespół NARP, zespół Leigha).
Genomy mitochondrialne roślin są znacznie większe niż genomy mitochondrialne zwierząt. Wielkośd genomu
mitochondrialnego roślin uległa szybkim zmianom w trakcie ewolucji. Za różnice w wielkości są odpowiedzialne
wewnętrzne duplikacje oraz integracja z genomem mitochondrialnym sekwencji pochodzących z genomu jądrowego
i plastydowego. Znacznej dynamice podlega również organizacja roślinnych genomów mitochondrialnych: układ
genów w tych genomach jest bardzo różny, chod zasadniczo utrzymane są te same sekwencje kodujące.
Rozpatruje się cztery modele struktury mtDNA roślin:
genom mitochondrialny składa się z wielu kolistych cząsteczek różnej wielkości; każda z nich zawiera pełną
informacje genetyczną, ale inaczej zorganizowaną
każda z kolistych cząsteczek zawiera tylko częśd informacji genetycznej, dzięki czemu upodabniają się one do
chromosomów genomu jądrowego
Ryc. 5 Chory na CPEO (jednym z objawów jest obustronne opadanie powiek)
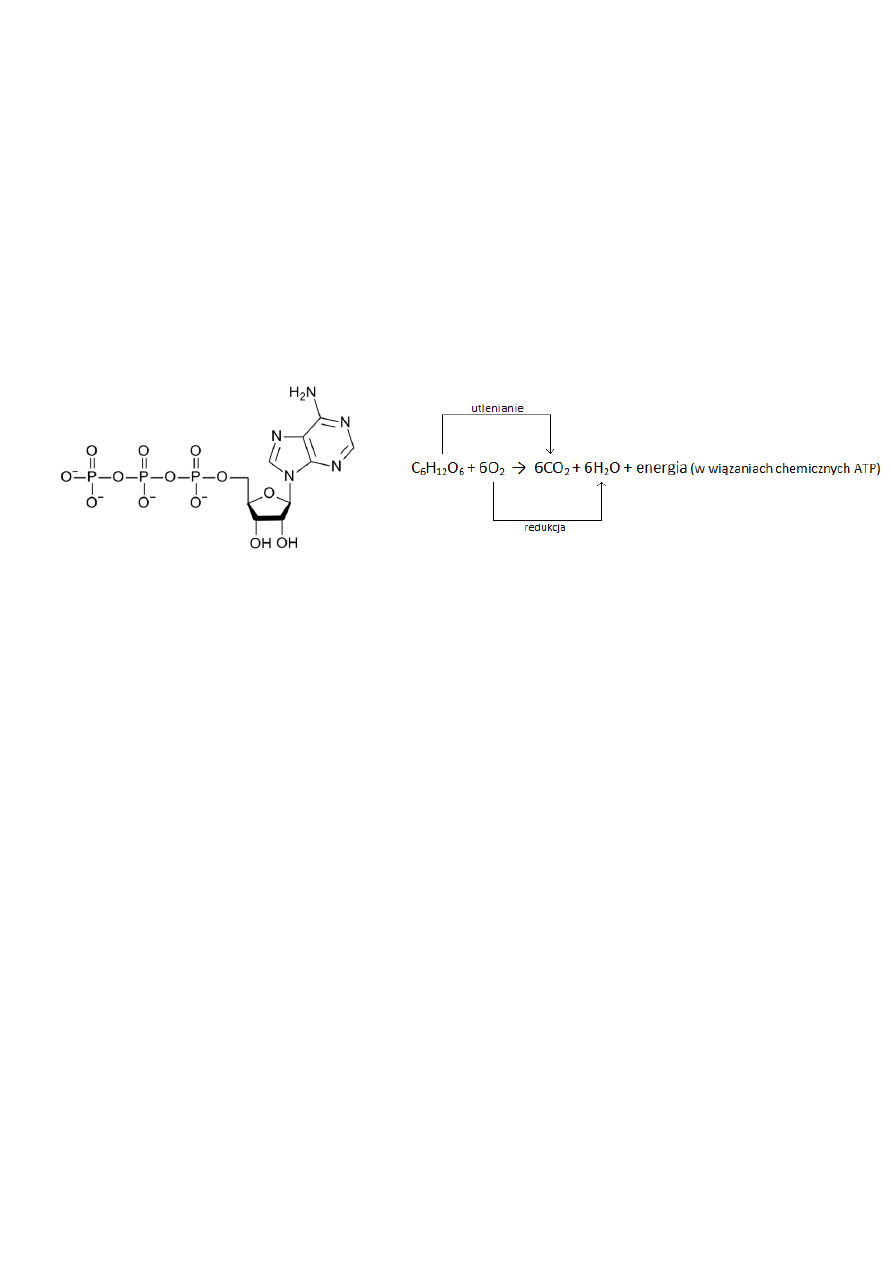
genom mitochondrialny może występowad zarówno w postaci jednej dużej kolistej cząsteczki (pojedynczego
chromosomu), jak i wielu mniejszych kolistych cząsteczek zawierających (przynajmniej w części) takie same
sekwencje
mtDNA ma postad jednej dużej, kolistej cząsteczki, tzw. chromosomu głównego (ang. master chromosome)
Oddychanie tlenowe
Oddychanie tlenowe to forma oddychania komórkowego zależna od tlenu. Podczas tego procesu składniki
pokarmu rozkładają się do dwutlenku węgla i wody. Większośd komórek wykorzystuje oddychanie tlenowe do
uzyskania energii z glukozy. Na oddychanie tlenowe składa się ciąg reakcji redoks, w których elektrony atomów
wodoru z glukozy przenoszą się na tlen, przebywając wiele etapów pośrednich. Uwolniona przy tym energia
swobodna elektronów zostaje wykorzystana do syntezy ATP.
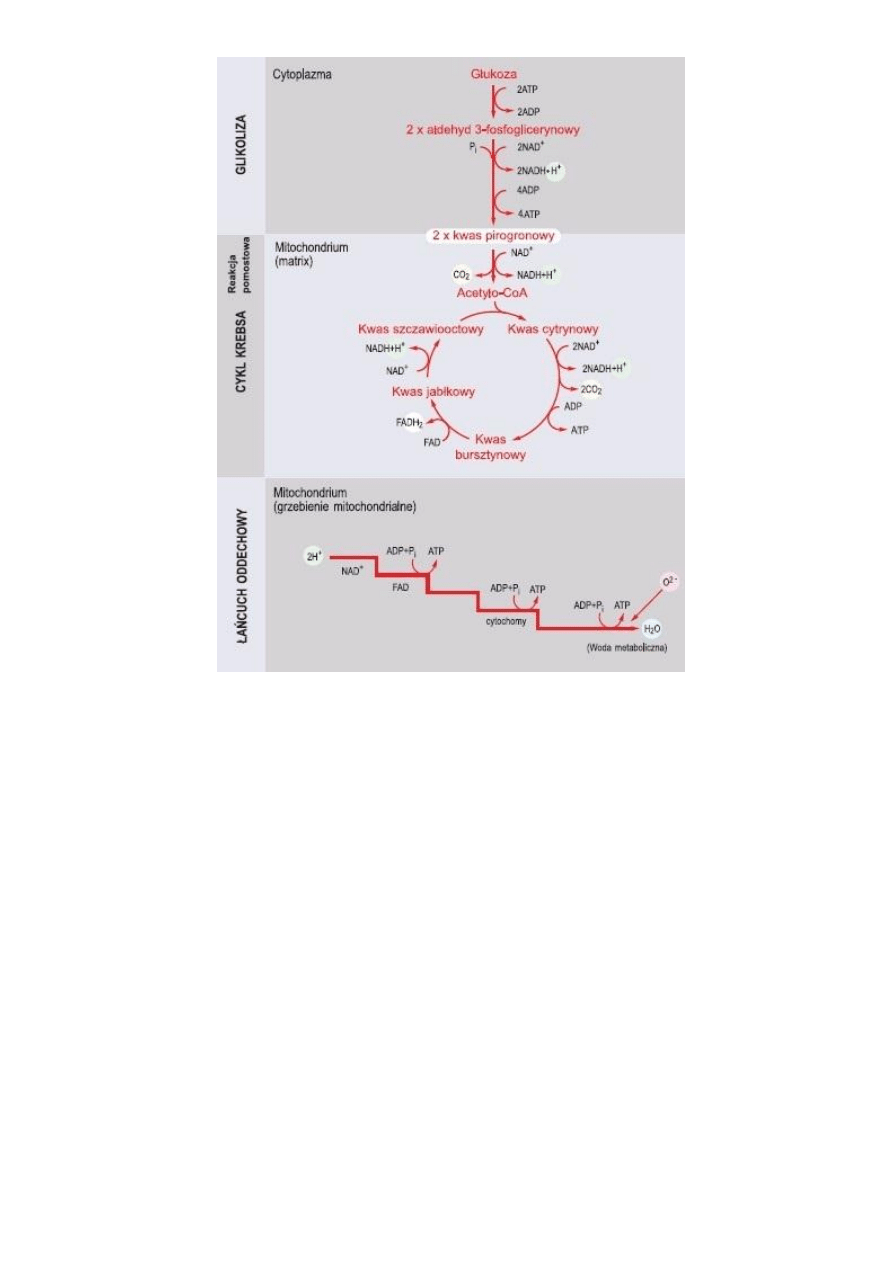
W rozkładnie glukozy podczas oddychania tlenowego wyróżnia się cztery grupy reakcji chemicznych. Są to
glikoliza, powstawanie acetylo-koenzymu A, cykl kwasu cytrynowego oraz transport elektronów i chemiosmoza. W
komórkach eukariotycznych pierwszy etap zachodzi w cytosolu, a pozostałe trzy w mitochondriach.
1. GLIKOLIZA
Glikoliza to rozpad glukozy na 2 cząsteczki pirogronianu. Może zachodzid zarówno w warunkach tlenowych, jak
i beztlenowych. Przebiega ona w cytosolu, gdzie znajdują się niezbędne reagenty, takie jak ADP
(adenozynodifosforan), NAD
+
(dinukleotyd nikotynoamidoadeninowy) i nieorganiczny fosforan. Szlak glikolityczny
składa się z serii reakcji, z których każdą katalizuje specyficzny enzym. Glikolizę dzieli się umownie na dwie fazy:
Faza inwestowania energii
W dwóch oddzielnych reakcjach fosforylacji grupa fosforanowa przenosi się z ATP na cukier. Ufosforylowany
cukier (fruktozo-1,6-bifosforan) jest mniej trwały i z udziałem odpowiedniego enzymu (aldolazy) rozpada się na
dwie 3-węglowe cząsteczki: fosfodihydroksyaceton i aldehyd 3-fosfoglicerynowy (G3P). Fosfodihydroksyaceton
pod wpływem enzymu izomerazy ulega przemianie w G3P.
glukoza + 2 ATP → 2 G3P + 2 ADP
Faza zdobywania energii
G3P utlenia się, oddając 2 elektrony (łącznie z protonami), bezpośrednio przechodzące na NAD
+
. Jako, że dwie
cząsteczki G3P powstają z każdej cząsteczki glukozy, dwie cząsteczki NAD
+
redukują się do NADH. W dwóch
reakcjach prowadzących do powstawania pirogronianu grupa fosforanowa przechodzi z ufosforylowanych
metabolitów pośrednich na ADP i powstaje ATP (fosforylacja substratowa).
2 G3P + 2 NAD
+
+ 4 ADP → 2 pirogronian + 2 NADH + 4 ATP
Ryc. 6 ATP (adenozynotrifosforan)
Ryc. 7 Sumaryczne równanie oddychania tlenowego

2. POWSTAWANIE ACETYLO-KOENZYMU A (reakcja pomostowa)
W komórkach eukariontów cząsteczki pirogronianu powstałe podczas glikolizy dostają się do mitochondriów, gdzie
tworzą acetylo-koenzym A (acetylo-CoA). W komórkach tlenowych prokariontów reakcja ta zachodzi w cotosolu.
W tej serii reakcji pirogronian podlega dekarboksylacji oksydacyjnej. Najpierw od grupy karboksylowej odłącza się
dwutlenek węgla, który dyfunduje poza komórkę, a pozostały fragment dwuwęglowy utlenia się w obecności NAD
+
.
Powstaje grupa acetylowa, która łączy się z koenzymem A dając acetylo-CoA.
2 pirogronian + 2 NAD
+
+ 2 CoA → 2 acetylo-CoA + 2 NADH + 2 CO
2
3. CYKL KWASU CYTRYNOWEGO
Cykl kwasu cytrynowego nazywany jest również cyklem Krebsa lub cyklem kwasów trójkarboksylowych (TCA).
Pierwsza reakcja cyklu jest związana z przeniesieniem dwuwęglowej grupy acetylowej z acetylo-CoA na
czterowęglowy akceptor - szczawiooctan. W wyniku tego powstaje sześciowęglowy cytrynian.
szczawiooctan (4C) + acetylo-CoA (2C) → cytrynian (6C) + CoA
Cytrynian przechodzi ciąg reakcji, w których kosztem dwóch grup karboksylowych uwalniają się 2 CO
2
. Większośd
energii wyzwolonej w reakcjach utleniania zostaje przeniesiona, wraz z elektronami na NAD
+
, dając NADH. Ponadto
elektrony służą do redukcji FAD (dinukleotyd flawinoadeninowy) do FADH
2
. W koocu każdego cyklu odtwarza się
szczawiooctan, po czym cykl powtarza się.
W cyklu Krebsa powstają 4 CO
2
, 6 NADH, 2 FADH
2
i 2 ATP na każdą cząsteczkę glukozy (Ryc. 11).
4. TRANSPORT ELEKTRONÓW I CHEMIOSMOZA
Zredukowane nukleotydy stanowią początek łaocucha transportu elektronów, w którym wysokoenergetyczne
elektrony z atomów wodoru przenoszą się na kolejne akceptory w serii reakcji redoks. Przeliczając na jedną
cząsteczkę glukozy, wcześniej powstały: 2 NADH podczas glikolizy, 2 NADH przy tworzeniu acetylo-koenzymu A oraz
6 NADH i 2 FADH
2
w trakcie cyklu Krebsa. Łaocuch transportu elektronów zawiera serie przenośników wbudowanych
w wewnętrzną błonę mitochondrialną komórek eukariotycznych. Każdy przenośnik może występowad w formie
utlenionej lub zredukowanej. Elektrony przechodzą na coraz niższe poziomy energetyczne w miarę transportu przez
cztery kompleksy łaocucha. Elektrony przekazywane w tym łaocuchu są dośd bogate w energię, którą jednak
stopniowo tracą w miarę transportu przez kolejne przenośniki. Koocowym akceptorem jest tlen i jest on niezbędny
dla wszystkich organizmów bezwzględnie aerobowych. W przypadku braku tlenu, ostatni cytochrom w łaocuchu
(Ryc. 8) zostanie nasycony elektronami (zredukowany), wskutek czego każdy poprzedni przenośnik również
pozostanie nimi nasycony, co zablokuje cały łaocuch począwszy od NADH.
Ryc. 8 Ogólny schemat łaocucha transportu elektronów
Synteza ATP (reakcja endoergiczna), polegająca na fosforylacji ADP, jest
sprzężona z egzoergicznymi reakcjami redoks w łaocuchu transportu
elektronów. Z tego względu nazywana jest fosforylacją oksydacyjną.
Sprzężenie syntezy ATP z transportem elektronów w oddychaniu
tlenowym tłumaczy model chemiosmotyczny, zaproponowany przez
Peter’a Mitchell’a w 1961 roku. Transport elektronów sprawia, że
powstaje gradient stężenia protonów - częśd energii uwalnianej podczas
tego transportu służy do przenoszenia protonów (H
+
) przez wewnętrzną
błonę mitochondrialną do przestrzeni między błonami, co sprawia że
wewnętrzna błona mitochondrialna oddziela przestrzeo o wysokim
stężeniu protonów (przestrzeo międzybłonowa) od przestrzeni o niskim
stężeniu protonów (matriks). W transporcie protonów przez
wewnętrzną błonę mitochondrialną uczestniczą trzy spośród czterech
kompleksów białkowych: I, III i IV. Gradient protonów jest formą energii
potencjalnej, która może posłużyd do syntezy ATP.
Dyfuzja protonów w odwrotnym kierunku (z przestrzeni międzybłonowej
do matriks) ogranicza się do specyficznych kanałów utworzonych przez
piąty kompleks enzymatyczny syntazę ATP. Wystaje ona częściowo nad
powietrznię wewnętrznej błony mitochondrialnej od strony matriks.
Przebiegająca zgodnie z gradientem stężenia dyfuzja protonów przez
syntazę ATP jest egzoergiczna. Proces ten dostarcza energii do
fosforylacji ADP do ATP. Syntaza ATP pracuje jak wysoce wydajny
molekularny motor. Podczas syntezy ATP z ADP i nieorganicznego
fosforanu obraca się centralna częśd syntazy ATP, przypuszczalnie na
skutek ruchu protonów przez ten enzym. Rotacja ta tak zmienia
konformację podjednostek katalitycznych, by umożliwid syntezę ATP.
Utlenienie jednej cząsteczki NADH w łaocuchu transportu elektronów wiąże się z powstaniem do 3
cząsteczek ATP. W obliczeniach trzeba wziąd pod uwagę to, że NADH powstałe w glikolizie może wygenerowad 2 lub
3 cząsteczki ATP. Wynika to stąd, że komórki eukariotyczne wydatkują częśd energii na transport powstałego
w glikolizie NADH z cytosolu do macierzy mitochondrialnej przez błonę mitochondriów. Komórki prokariotyczne nie
zawierają mitochondriów (u większości komórek
bakterii i archeabakterii wszystkie reakcje
zachodzą w cytosolu i błonie komórkowej), więc
nie muszą zużywad energii na przenoszenie
cząsteczek NADH do innego przedziału. Utlenienie
każdej
cząsteczki
FADH
2
jest
związane
z powstaniem 2 cząsteczek ATP.
Ryc. 10 przedstawia wszystkie najważniejsze
obliczenia wydajności energetycznej całkowitego
utleniania glukozy w oddychaniu tlenowym,
natomiast Ryc. 11 krótko podsumowuje przebieg
kolejnych etapów oddychania tlenowego.
Całkowite utlenienie jednej cząsteczki glukozy
prowadzi do powstania najwyżej 36-38 cząsteczek
ATP (2 w glikolizie, 2 w cyklu kwasu Krebsa
i 32-34 w łaocuchu transportu elektronów
i chemiosmozie).
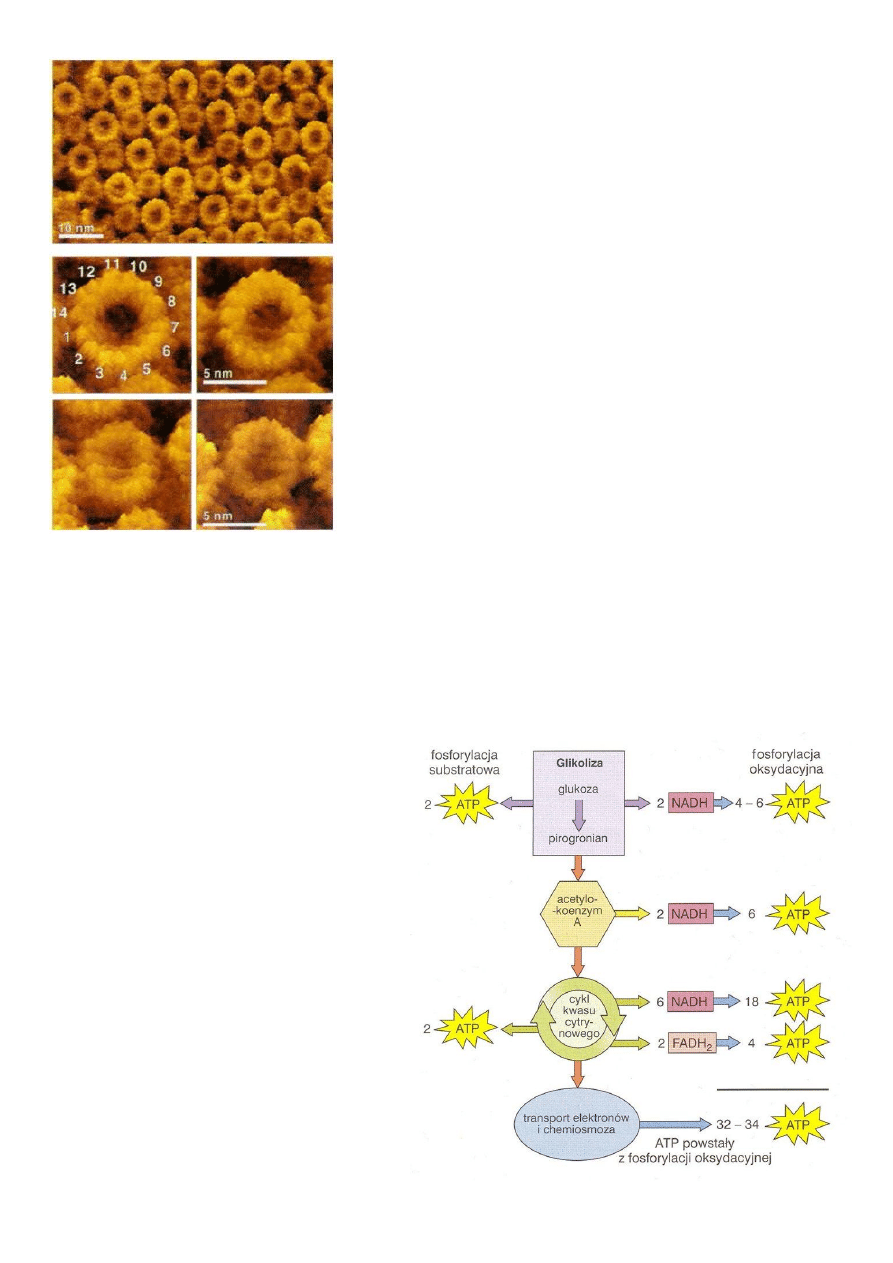
Ryc. 9 Syntaza ATP - zdjęcie zrobione za
pomocą mikroskopu AFM
Ryc. 10 Wydajnośd energetyczna całkowitego utlenienia glukozy w
oddychaniu tlenowym
Wiele istot żywych, łącznie z człowiekiem, jest uzależnionych od energii zgromadzonej w innych niż glukoza
składnikach pokarmowych. Aminokwasy ulegają deaminacji (tracą grupę aminową), a ich szkielety węglowe ulegają
przemianie w metabolity pośrednie oddychania tlenowego. Tłuszcze są bogate w energię, gdyż są w dużym stopniu
zredukowane. Całkowite utlenienie sześciowęglowego kwasu tłuszczowego daje do 44 cząsteczek ATP. Oba składniki
tłuszczu (glicerol i kwasy tłuszczowe) są wykorzystywane w oddychaniu tlenowym. Do glicerolu dołącza się fosforan,
a powstały związek ulega przemianie w G3P albo inny związek chemiczny włączający się do glikolizy. Kwasy
tłuszczowe ulegają enzymatycznemu utlenianiu do dwuwęglowych grup acetylowych związanych z koenzymem A.
Proces ten zachodzi w matriks i jest nazywany β-oksydacją. Powstałe w ten sposób acetylo-CoA mogą włączyd się
w cykl Krebsa.
Henryk Skrętny
Ryc. 11 Główne etapy oddychania tlenowego

PIŚMIENNICTWO
1. Solomon Eldra P., Berg Linda R., Martin Diana W., 2009, Biologia Villee 2009, Multico, Warszawa
2. Jerzy Duszyoski, Krystyna Grykiel, Lilla Hryniewiecka, Artur Jarmołowski, 2003, Biologia tom 1, PWN,
Warszawa
3. Grzegorek Janina, Jerzmanowski Andrzej, Staroo Krzysztof, 2003, Biologia częśd 1 tom 1, WSiP, Warszawa
4. Holak E., Lewioski W., Łaszczyca M., Skirmuntt G., Walkiewicz J., 2009, Biologia 1, Operon, Warszawa
5. Witold Mizerski, Beata Bednarczuk, Roman Mizerski, Iwona Mizerska, 2008, Tablice biologiczne, Adamantan,
Warszawa
Wyszukiwarka
Podobne podstrony:
mitochondrium, Cytologia, Fizjologia i Genetyka
6 Mitochondrium
W4 Mitochondria i chloroplasty
cytologia 2
Budowa komorki eukariotycznej czesc VI mitochondrium i jadro komorkowe
Mitochondria, biologia- studia, Biologia
Cytologia
mitochondria
MITOCHONDRIA, biologia komórki
Cytologia, Fizjoterapia, Biologia medyczna
199905 smierc mitochondriom ple
Miopatie mitochondrialne, VI rok, Genetyka, Genetyka, Egzamin
PATOMORFOLOGIA I CYTOLOGIA KOMÓRKI, Zagadnienia
Cytologia
Genom mitochondrialny
cytologia
genom mitochondrialny
więcej podobnych podstron