
Postępowanie diagnostyczno-terapeutyczne
w nowotworach tkanek miękkich u dzieci
Bernarda Kazanowska, Alicja Chybicka
Mięsaki tkanek miękkich (MTM) stanowią w Polsce około 7% wszystkich dziecięcych no−
wotworów złośliwych i po białaczkach, guzach ośrodkowego układu nerwowego, chłoniakach
i nerwiaku zarodkowym są 5. pod względem częstości grupą nowotworów występujących
u pacjentów poniżej 16. roku życia. Większość zachorowań dotyczy dzieci w wieku 2–6 lat
oraz młodzieży powyżej 12. roku życia. Wskaźnik zachorowalności jest niezależny od płci
i waha się w granicach 0,2–1,0/100 000 rocznie.
W tabeli 42 przedstawiono częstość występowania poszczególnych typów mięsaków u dzieci.
W celach terapeutycznych, uwzględniając wrażliwość na chemioterapię, MTM u dzieci
podzielono na kilka grup.
1. Grupa guzów mięsaków prążkowanokomórkowych (RMS, rhabdomysarcoma):
— z korzystnym rokowaniem:
•
typ botryoidalny zarodkowej postaci mięsaka prążkowanokomórkowego (RME, embryonal
rhabdomyosarcoma),
•
typ kolczystokomórkowy zarodkowej postaci mięsaka prążkowanokomórkowego (RME),
•
tak zwany „klasyczny” typ zarodkowej postaci mięsaka prążkowanokomórkowego
(RME);
— z niekorzystnym rokowaniem:
•
typ pęcherzykowy, łącznie z tak zwanym wariantem „litym” (RMA, alveolar rhabdomyo−
sarcoma).
Tabela 42. Częstość występowania poszczególnych typów mięsaków u dzieci
Typ mięsaka tkanek miękkich
Częstość występowania
Mięsak prążkowanokomórkowy (RMS, rhabdomyosarcoma)
69,0%
Mięsak maziówkowy (SS, synovial sarcoma)
10,2%
Włókniakomięsak (FS, fibrosarcoma)
4,0%
Pozakostny mięsak Ewinga (EES, extraskeletal Ewing sarcoma)
3,6%
Prymitywny guz neuroektodermalny (PNET, primitive neuroectodermal tumor)
6,1%
Mięsaki niezróżnicowane (UDS, undifferentiated sarcoma)
3,0%
Nerwiakowłókniakomięsak (NFS, neurofibrosarcoma)
2,2%
Mięsak gładkomórkowy (leyomyosarcoma)
1,1%
Tłuszczakomięsak (LPS, liposarcoma)
1,1%
Inne
4,3%

720
Zalecenia postępowania diagnostyczno−terapeutycznego w nowotworach złośliwych — 2009 r.
2. Grupa guzów „RMS−like” — zaliczono do niej następujące typy guzów:
— grupę pozakostnego mięsaka Ewinga (EES, extraskeletal Ewing sarcoma) obejmującą:
a) klasyczną postać mięsaka Ewinga,
b) atypową postać mięsaka Ewinga,
c) złośliwy obwodowy guz neuroektodermalny (MPNT, malignant peripheral neuroecto−
dermal tumor);
— mięsaka maziówkowego (SS, synovial sarcoma);
— mięsaki niezróżnicowane (UDS, undifferentiated sarcoma);
— mięsaki, których nie można sklasyfikować (NOS, not otherwise specified).
3. Grupa guzów nie−RMS — obejmuje następujące guzy:
— mięsak pęcherzykowaty (ASTS, alveolar soft tissue sarcoma);
— mięsak jasnokomórkowy (CCS, clear cell sarcoma) (czerniak tkanek miękkich);
— chrzęstniakomięsak poza układem szkieletowym (ECS, chondrosarcoma), w tym:
a) chrzęstniakomięsak śluzowaty (EMCS, extraskeletal myxoid chondrosarcoma),
b) chrzęstniakomięsak mezenchymalny (MCS, mesenchymal chondrosarcoma);
— włókniakomięsak (FS, fibrosarcoma), w tym:
a) wrodzona postać włókniakomięsaka (CFS, congenital fibrosarcoma);
— guzowaty włókniakomięsak skóry (DFSP, dermatofibrosarcoma protuberans);
— desmoplastyczny guz z małych owalnych komórek (DSRCT, desmoplastic small round cell
tumor);
— mięsak epitelioidalny (ES, epithelioid sarcoma);
— zapalny guz miofibroblastyczny (IMFT, inflammatory myofibroblastic tumor), w tym:
a) zapalny mięsak myofibroblastyczny (IMFS, inflammatory myofibroblastic sarcoma);
— mięśniakomięsak gładkomórkowy (LMS, leiomyosarcoma);
— tłuszczakomięsak (LPS, liposarcoma);
— mięsak histiocytarny włóknisty (MFH, fibrohistiocytoma malignum):
— mięsak mezenchymalny (MM, malignant mesenchymoma);
— złośliwy guz mięśniowopodobny (MRI, malignant rhabdoid tumor) ;
— złośliwy obwodowy guz osłonek nerwowych (MPNST); synonimy: malignant Schwannoma
oraz neurofibrosarcoma (NFS);
— barwnikowy guz neuroektodermalny wieku dziecięcego (RAT, retina anlage tumor);
— złośliwe guzy naczyniowe (VS, vascular sarcoma), w tym:
a) śródbłoniak (HE, hemangioendothelioma),
b) obłoniak (HP, hemangiopericytoma),
c) mięsak naczyniowy (AS, angiosarcoma).
Leczenie MTM u dzieci jest złożone. Strategia leczenia opiera się na wielolekowej chemio−
terapii dostosowanej do postaci histopatologicznej, immunofenotypu, stopnia zaawansowa−
nia klinicznego oraz grupy ryzyka. Zasadniczym celem chemioterapii jest eliminacja makro−
i mikroprzerzutów oraz zmniejszenie masy guza pierwotnego w celu umożliwienia lokalnej
radioterapii i radykalnej operacji ogniska pierwotnego.
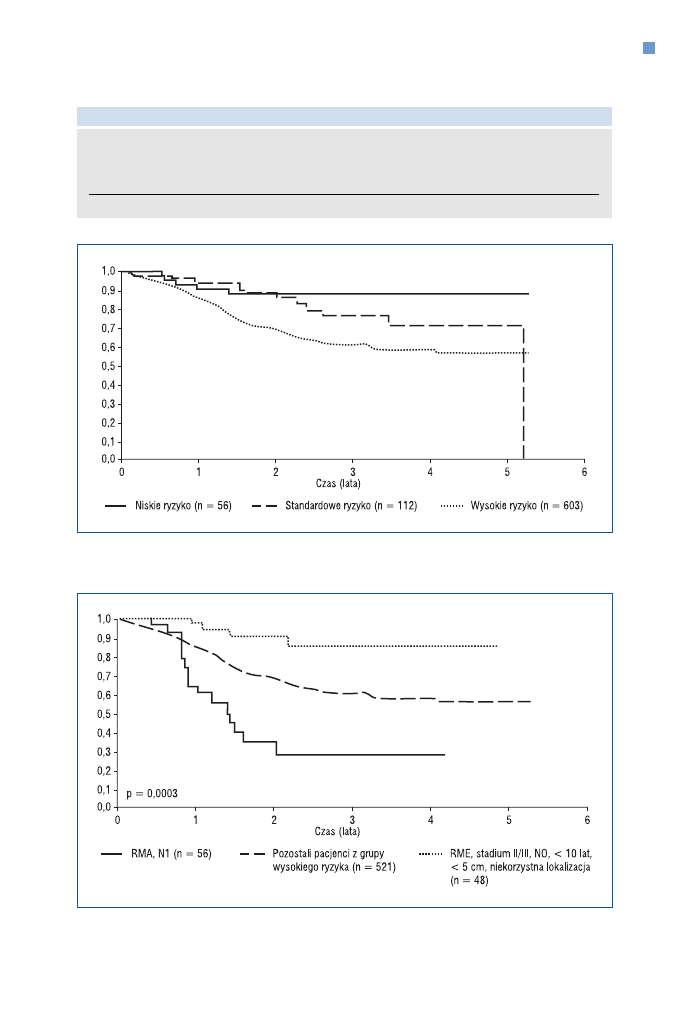
W tabeli 43 przedstawiono wyniki leczenia MTM u dzieci według protokołu CWS−96. Prze−
życie wolne od zdarzeń (EFS, events free survival) dla poszczególnych grup ryzyka w przypad−
ku guzów typu RMS według CWS−96 i ICG−96 przedstawiono na rycinie 8 (w grupie wysokiego
ryzyka RMS — ryc. 9).
Do guzów nie−RMS z dobrą prognozą, w przypadku których szansa przeżycia wynosi około
80%, zaliczają się mięsak mięśni gładkich (LMS, leiomyosarcoma), mięsak histiocytarny włók−
721
Nowotwory tkanek miękkich
Tabela 43. Guzy RMS — wyniki leczenia mięsaków tkanek miękkich u dzieci według protokołu CWS−96
Grupa ryzyka
Liczba pacjentów
3−letnie EFS
3−letnie SUR
Niskie
56
88%
97%
Standardowe
112
77%
95%
Wysokie
603
62%
78%
AS (actual survival) — przeżycie całkowite; EFS (events free survival) — przeżycie wolne od zdarzeń
Rycina 8. Przeżycie wolne od zdarzeń (EFS) w grupie guzów typu RMS według CWS−96 i ICG−96
Rycina 9. Przeżycie wolne od zdarzeń (EFS) w grupie wysokiego ryzyka RMS według programu
CWS−96/ICG−96
722
Zalecenia postępowania diagnostyczno−terapeutycznego w nowotworach złośliwych — 2009 r.
nisty (MFH, malignant fibrohistiocytoma) oraz mięsak pęcherzykowaty (AWTS, alveolar soft
tissue sarcoma). Do grupy z niekorzystnym rokowaniem należą chrzęstniakomięsak mezen−
chymalny (MesCS, mesenchymal chondrosarcoma), mięsak nabłonkowy (ES, sarcoma
epitheloides), mięsak jasnokomórkowy (CCS, sarcoma clarocellulare), złośliwy guz mięśnio−
wopodobny (MRT, malignant rhabdoid tumor) oraz drobnookrągłokomórkowy guz desmopla−
styczny (DRT, desmoplastic, small− and round cell tumor). Do grupy z pośrednim ryzykiem
zalicza się mięsaki naczyniowe i włókniakomięsaki. Na podstawie analizy multiwariacyjnej
określono najważniejsze czynniki rokownicze: typ histologiczny guza, wyjściowa wielkość guza,
radykalność zabiegu pierwotnego, status węzłów chłonnych oraz przeprowadzenie napromie−
niania lokalnego.
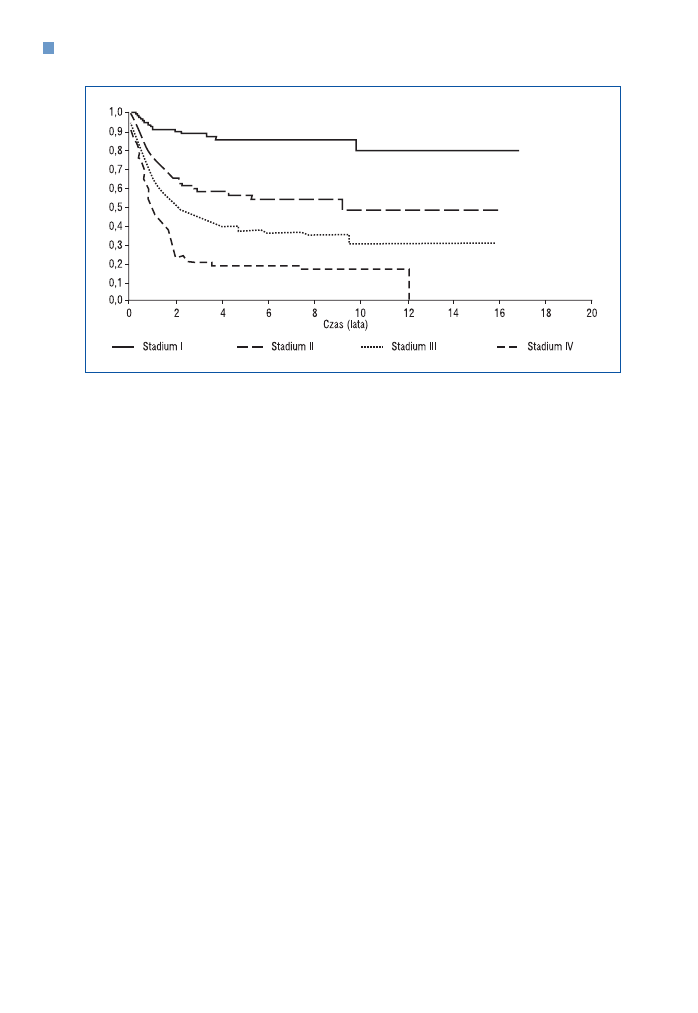
Przeżycie wolne od zdarzeń w grupie nie−RMS w zależności od stadium pooperacyjnego
przedstawiono na rycinie 10.
U około 20% pacjentów z MTM w chwili rozpoznania stwierdza się rozsianą postać choro−
by. Przeżycie 5−letnie u tych chorych jest bardzo niskie i wynosi 10–20%. W wielu opracowa−
niach wykazano, że najbardziej niekorzystnymi czynnikami rokowniczymi są: histopatologia
RMA i PNET/EES, wiek pacjenta powyżej 10 lat, zajęcie kości i/lub szpiku. Przeżycie w tej
grupie chorych wynosi około 5%. Natomiast w grupie pacjentów poniżej 10. roku życia
z zajęciem tylko jednego narządu oraz guzem o histologii RME wynosi około 50%.
Zasady klasyfikacji i istniejące systemy klasyfikacyjne
Na Międzynarodowym Spotkaniu Roboczym RMS w Jerozolimie w 1987 roku ustalono
7 lokalizacji mięsaków tkanek miękkich u dzieci:
— oczodół;
— głowa/szyja nieokołooponowo (czepiec ścięgnisty, szyja, ślinianki, jama ustna, krtań, górna
i dolna część gardła, tarczyca, policzki);
— głowa/szyja okołooponowo (jama nosowo−gardłowa, zatoki: sitowe, szczękowe, czołowe,
klinowa, jama nosowa, ucho środkowe, dół podskroniowy, dół skrzydłowo−podniebienny,
Rycina 10. Przeżycie wolne od zdarzeń (EFS) w grupie nie−RMS w zależności od stadium pooperacyjnego

723
Nowotwory tkanek miękkich
oczodół z erozją kości podstawy czaszki oraz strop oczodołu — stok, przestrzeń zaszczę−
kowa, kość klinowa, kość sitowa, podstawa nosa, erozja kości podstawy czaszki, poraże−
nie nerwów czaszkowych, obecność komórek nowotworowych w PMR;
— układ moczowo−płciowy — pęcherz i prostata;
— układ moczowo−płciowy bez pęcherza i prostaty (jądra, najądrze, okolica okołojądrowa,
prącie, srom, pochwa, jajnik, macica);
— kończyny i obręcze (w tym mięśnie pośladkowe);
— inne (miednica mniejsza, jama brzuszna, tułów, ściana klatki piersiowej, śródpiersie, skóra,
grasica).
Klasyfikacja Tumor-Nodes-Metastases (TNM) według Międzynarodowej
Unii Walki z Rakiem (UICC, International Union Against Cancer)
Guz (T)
T0: nie stwierdza się obecności guza pierwotnego
T1: guz ograniczony wyjściowo do jednego narządu lub tkanki:
T1a: największa średnica guza mniejsza lub równa 5 cm
T1b: największa średnica guza większa niż 5 cm
T2: guz wyjściowo wychodzący poza narząd lub tkankę:
T2a: największa średnica guza mniejsza lub równa 5 cm
T2b: największa średnica guza większa niż 5 cm
TX: brak adekwatnych danych o wyjściowych rozmiarach guza (oceniać jak T2).
Węzły chłonne (N)
N0: nie stwierdza się zajęcia regionalnych węzłów chłonnych
N1: zajęcie regionalnych węzłów chłonnych
NX: brak adekwatnych danych o stanie węzłów chłonnych (oceniać jak N0).
Regionalne węzły chłonne należą do okolicy lokalizacyjnie związanej z guzem pierwotnym:
— głowa/szyja — węzły chłonne szyjne i nadobojczykowe;
— brzuch/miednica — węzły chłonne podprzeponowe i biodrowo−pachwinowe;
— kończyna górna — węzły chłonne pachowe i łokciowe;
— kończyna dolna — węzły chłonne podkolanowe i pachwinowe.
Przerzuty odległe (M)
M0: nie stwierdza się przerzutów odległych ani zajęcia pozaregionalnych węzłów chłonnych
M1: przerzuty odległe lub zajęcie pozaregionalnych węzłów chłonnych
MX: brak adekwatnych danych o obecności przerzutów (oceniać jak M0).
Przerzuty odległe rozpoznaje się w przypadku zajęcia węzłów chłonnych pozaregionalnych, po
stronie guza lub zajęcia węzłów chłonnych po stronie przeciwnej, nawet jeśli odpowiadały
węzłom regionalnym.
Stadia kliniczne według Intergroup Rhabdomyosarcoma Study (IRS) zestawiono w tabeli 44.
Klasyfikacja zabiegu pierwotnego
Radykalność zabiegu pierwotnego oceniano jako:
— resekcja R0 — resekcja całkowita, margines mikroskopowo wolny od komórek nowotwo−
rowych;

724
Zalecenia postępowania diagnostyczno−terapeutycznego w nowotworach złośliwych — 2009 r.
— resekcja R1 — resekcja makroskopowo całkowita z pozostawieniem marginesu z obec−
nością komórek nowotworowych;
— resekcja R2 — resekcja z pozostawieniem makroskopowych resztek guza.
Klasyfikacja po zabiegu odroczonym
Ocena guza po operacji odroczonej (SL, second look) według patomorfologa i chirurga:
— Ips — brak komórek nowotworowych w tkance uzyskanej podczas operacji SL;
— IIps — obecność komórek nowotworowych w tkance uzyskanej podczas operacji SL;
— IIIps — obecność makroskopowych pozostałości guza.
Klasyfikację mięsaków tkanek miękkich pod względem histologii i chemiowrażliwości przed−
stawiono w tabeli 45, natomiast w tabeli 46 — system złośliwości nowotworu według Pedia−
tric Oncology Group.
Zasady rozpoznawania
Rozpoznanie powinno się opierać na anamnezie, badaniu przedmiotowym, badaniu obra−
zowym, badaniu histopatologicznym, badaniach laboratoryjnych i molekularnych. Ostateczną
diagnozę można postawić tylko na podstawie wyniku badania histopatologicznego. Diagno−
styka wstępna powinna uwzględniać przede wszystkim czynniki niezbędne do przeprowadze−
nia stratyfikacji, takie jak:
— lokalizacja ogniska pierwotnego;
— ograniczenie do narządu/tkanki wyjściowej — T−Status;
— ocena węzłów chłonnych — N−Status;
— obecność przerzutów — M−Status;
— wynik badania histopatologicznego — histologiczny podtyp guza;
— stadium po zabiegu chirurgicznym — stadium IRS I–IV;
— wiek pacjenta — poniżej lub powyżej 10. roku życia;
— wielkość guza — mniejszy lub większy niż 5 cm.
Tabela 44. Stadia kliniczne według Intergroup Rhabdomyosarcoma Study (IRS)
Stadium IRS
Określenie
Stadium pT
I
Guz usunięty doszczętnie makroskopowo i mikroskopowo,
bez zajęcia węzłów chłonnych
(IA)
— ograniczony do narządu
pT1
(IB)
— wychodzący poza narząd
pT2
II
Guz usunięty makroskopowo, lecz niedoszczętnie mikroskopowo oraz
pT3a
IIA
— regionalne węzły chłonne niezajęte
IIB
— regionalne węzły chłonne zajęte, ale usunięte
IIC
— regionalne węzły chłonne zajęte i nieusunięte
III
Niedoszczętna resekcja z pozostawieniem resztek guza
pT3b
lub tylko biopsja z rozsiewem komórek nowotworowych
do okolicznych tkanek i jam ciała
pT3c
IV
Wyjściowo przerzuty odległe lub wyjściowe zajęcie
pozaregionalnych węzłów chłonnych
pT4

725
Nowotwory tkanek miękkich
Tabela 45. Klasyfikacja mięsaków tkanek miękkich pod względem histologii i chemiowrażliwości
— objaśnienia skrótów w tekście
Grupa CWS
Grupa RMS
Grupa nie−RMS
Grupa nie−RMS
(grupa A)
(grupa B)
(grupa C)
Wrażliwość na
Dobrze odpowiadające
Średnio wrażliwe
Niewrażliwe
chemioterapię
na chemioterapię
na chemioterapię
na chemioterapię
Korzystna
Niekorzystna
histologia
histologia
Zawiera:
RME
RMA
AWTS
NFS
EES
CCS
FS
P
PNET/MPNT
ES
ECS
SS
LMS
LPS
MFH/AFH
MMM
MRT
RAT
VS (HE, HP, AS)
CFS
DRT
IMFT/IMFS
Tabela 46. System złośliwości nowotworu według Pediatric Oncology Group
Stopień 1 (G1)
Stopień 2 (G2)
Stopień 3 (G3)
Wysokozróżnicowany
Wszystkie mięsaki, nie G 1 lub G 3,
Pleomorficzny LPS
tłuszczakomięsak śluzowaty
spełniające następujące kryteria:
MesCS
(liposarcoma myxoides)
— < 15% martwicy
Kostniakomięsak
DFSP
— < 5 mitoz na 10 HPF
pozakostny (osteosarcoma)
Pozaszkieletowy MyxCS
— bez wyrażonej atypii jądrowej
Złośliwy guz Tritona
Wysokozróżnicowany LMS
— bez dużej komórkowości
(Triton−Tumor)
Wysokozróżnicowany MPNST
FS i HP malignum u dzieci poniżej
AWTS
Wysokozróżnicowany
4 lat są guzami G2 (względnie G1),
Wszystkie pozostałe mięsaki:
HP malignum
również przy dużej liczbie mitoz
— nie G 1 lub G2
Dziecięca postać FS
i proliferacji komórkowej
— > 15% martwicy
— > 5 mitoz na 10 HPF
Badania molekularne niektórych mięsaków tkanek miękkich
W niektórych mięsakach stwierdza się charakterystyczne dla danej postaci histopatolo−
gicznej patognomiczne translokacje chromosomalne. Wykazanie obecności określonej rearan−
żacji pomaga potwierdzić rozpoznanie oraz może być przydatne w monitorowaniu choroby
resztkowej (tab. 47).
RMA — translokacja t(2;13)(q35;q14) oraz t(1;13)(p36;q14)
SS — translokacja t (X;18)p11;q11)

726
Zalecenia postępowania diagnostyczno−terapeutycznego w nowotworach złośliwych — 2009 r.
EES — translokacja t(11;22)(q24;q12), translokacja t(21;22)(q22;q12), translokacja
t(7;11)(p22;q24), translokacja t(17;22)(q21;q12) oraz translokacja t(2;22)(q33q12)
W celu dokładnej oceny pierwotnego rozprzestrzeniania się guza niezbędne są następują−
ce badania:
— badanie RTG piersiowej w dwóch płaszczyznach;
— tomografia komputerowa (KT) klatki piersiowej, względnie spiralna KT, jeśli jest dostęp−
na;
— rezonans magnetyczny (MR) lub KT mózgowia przed i po podaniu kontrastu;
— badanie USG, względnie MR jamy brzusznej — badanie USG jest wystarczające, jeśli
badający ma odpowiednie doświadczenie i możliwe jest dokładne uwidocznienie narzą−
dów (pacjent bez otyłości, bez nadmiernej ilości gazów). W badaniu muszą być uwidocz−
nione i opisane wszystkie stacje węzłów chłonnych. Jeśli nie można spełnić powyższych
warunków, badanie USG należy uzupełnić o badanie MR lub KT;
— biopsja szpiku i/lub trepanobiopsja z dwóch miejsc;
— scyntygrafia układu kostnego.
Określenie czynników ryzyka
Stratyfikację w grupie RMS przedstawiono w tabeli 48. Czynniki ryzyka dla zlokalizowa−
nych guzów typu RMS zestawiono w tabeli 49. W tabeli 50 przedstawiono stratyfikację
w grupie nie−RMS, natomiast w tabeli 51 — czynniki ryzyka w tej grupie
.
Zalecane leczenie
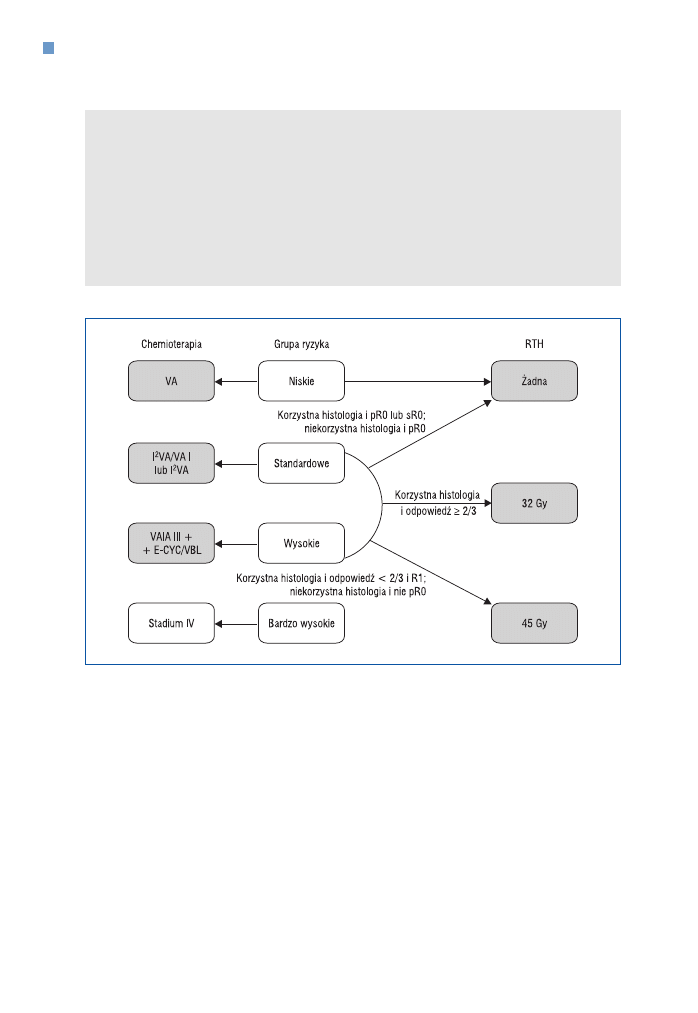
Założenia leczenia guzów typu RMS w protokole CWS−2002 przedstawiono na rycinie 11,
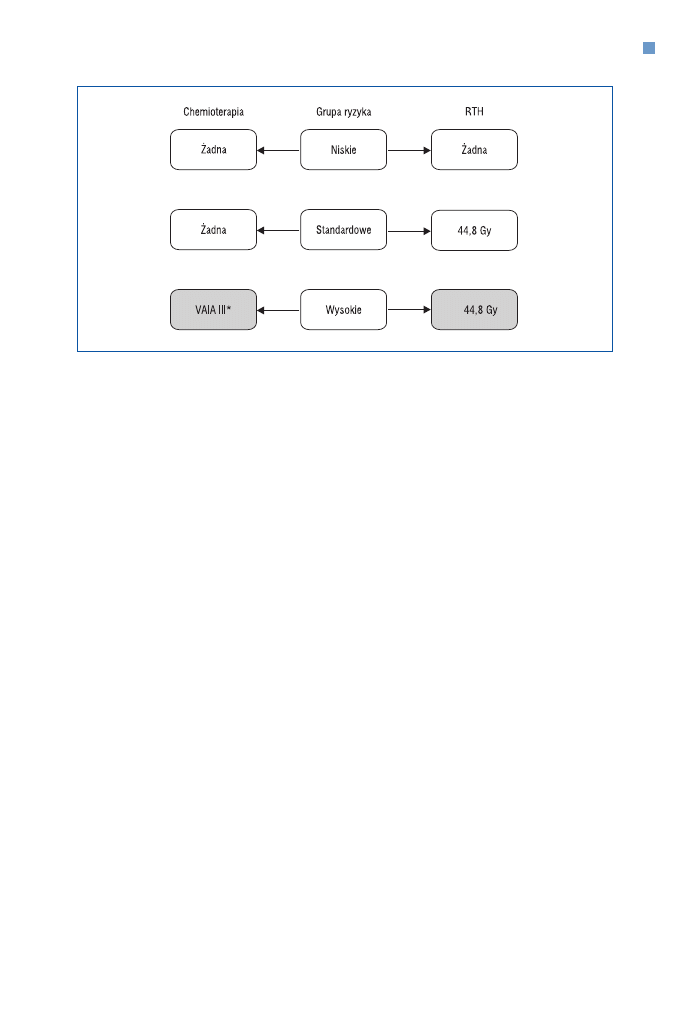
a guzów typu nie−RMS — na rycinie 12.
Winkrystyna (VCR):
1,5 mg/m
2
i.v. w bolusie 1. dnia 1., 2., 3. i 4. tygodnia bloku
Aktynomycyna D (AMD):
1,5 mg/m
2
i.v. w bolusie 1. dnia 1. i 4. tygodnia bloku
Ifosfamid (IFO):
3000 mg/m
2
i.v. w 3−godzinnym wlewie 1. i 2. dnia bloku
Adriamycyna (ADR):
40 mg/m
2
/dzień i.v. w 2 dawkach podzielonych w odstępie
12−godzinnym w 1. i 2. dniu bloku
Tabela 47. Markery immunohistochemiczne konieczne do różnicowania poszczególnych typów
mięsaków
Typ mięsaka Desmina
Wimentyna
Mioglobina
Keratyna
S−100
NSE
Aktyna MIC−2
RMS
+
+
+
–
–
–
+
–
SS fibro−
–
+
–
–
–
–
–
–
blastyczny
SS nabłon−
–
+/–
–
+
–
–
–
–
kowy
FS
–
+
–
–
–
–
–
–
LMS
+/–
–
–
–
–
–
+
–
EES
–
+
–
–
–
–
–
–
PNET
+/–
–
–
–
–/+
–/+
–
+

727
Nowotwory tkanek miękkich
Tabela 49. Czynniki ryzyka dla zlokalizowanych guzów typu RMS
Histologia
Korzystna
RME
Niekorzystna
RMA
EES
pPNET/MPNT
SS
Stadium
I
Całkowita resekcja guza
pooperacyjne
II
Mikroskopowe pozostałości guza po resekcji
(IRS)
III
Makroskopowe pozostałości guza po resekcji
Lokalizacja guza
Korzystna
Oczodół
Głowa/szyja — nieokołooponowo
Układ moczowo−płciowy bez pęcherza/prostaty
Niekorzystna
Głowa/szyja — okołooponowo
Układ moczowo−płciowy — pęcherz/prostata
Kończyny
Inne
Węzły chłonne
N0
Regionalne węzły chłonne niezajęte
N1
Regionalne węzły chłonne zajęte
Wielkość guza/
Korzystne
Wiek
£ 10 lat i wielkość guza £ 5 cm
/wiek pacjenta
Niekorzystne
Wiek > 10 lat i/lub wielkość guza > 5 cm
Tabela 50. Stratyfikacja ryzyka w grupie nie−RMS
Grupa ryzyka
Histologia
Węzły chłonne
Stadium IRS
Wyjściowa wielkość guza
Niskie
Wszystkie
N0
I
£ 5 cm
Standar−
z wyjątkiem
N0
I
> 5 cm
dowe
MRT i DRT
N0
II
Każda
N0
III
£ 5 cm
Wysokie
MRT/DRT
N0/N1
I–III
Każda
Każda
N0
III
> 5 cm
Każda
N1
I–III
Każda
Bardzo
Każda
N0/N1
IV
Każda
wysokie
Tabela 48. Stratyfikacja ryzyka w grupie RMS
Grupa ryzyka
Histologia
Stadium Lokalizacja
N−Status
Wielkość guza
IRS
i wiek pacjenta
Niskie
Korzystna
I
Wszystkie
N0
£ 5 cm i £ 10 lat
Standardowe
Korzystna
I
Wszystkie
N0
> 5 cm lub > 10 lat
Korzystna
II, III
Korzystna
N0
Wszystkie
Korzystna
II, III
Niekorzystna
N0
£ 5 cm i £ 10 lat
Wysokie
Korzystna
II, III
Korzystna
N1
£ 5 cm i £ 10 lat
Korzystna
II, III
Niekorzystna
N0/N1
> 5 cm lub > 10 lat
Niekorzystna
I, II, III
Wszystkie
N0
Wszystkie
Bardzo wysokie
Niekorzystna
I, II, III
Wszystkie
N1
Wszystkie
728
Zalecenia postępowania diagnostyczno−terapeutycznego w nowotworach złośliwych — 2009 r.
Terapię podtrzymującą (E−CYC/VBL) grupie wysokiego ryzyka kontynuuje się przez okres
6 miesięcy do 1 roku. Cyklofosfamid podaje się codziennie w dawce 2
¥ 25 mg/m
2
p.o., nato−
miast winblastynę 1 raz w tygodniu w dawce 3 mg/m
2
i.v. Po 3 tygodniach terapii następuje
1−tygodniowa przerwa, zatem w okresie 6 miesięcy pacjent otrzymuje 7 cykli, składających
się z cyklofosfamidu podawanego doustnie przez 3 tygodnie i winblastyny podawanej 3−krot−
nie na początku każdego tygodnia.
Stadium rozsiane choroby
Czynnikami niekorzystnymi prognostycznie u pacjentów z guzem pierwotnym w stadium IV są:
— wiek (niekorzystnie: > 10 lat);
Tabela 51. Czynniki ryzyka w grupie nie−RMS
Histologia
Niekorzystna
MRT, DRT
Stadium pooperacyjne
I
Całkowita resekcja guza
(z marginesem tkanek zdrowych)
II
Pozostałości mikroskopowe guza
III
Pozostałości makroskopowe guza
Węzły chłonne
N0
Regionalne węzły chłonne niezajęte
N1
Regionalne węzły chłonne zajęte
Wyjściowa wielkość guza
Korzystna
Guz
£ 5 cm
Niekorzystna
Guz > 5 cm
Rycina 11. Założenia leczenia guzów typu RMS w protokole CWS−2002
729
Nowotwory tkanek miękkich
Rycina 12. Założenia leczenia guzów typu nie−RMS w protokole CWS−2002. *Pacjenci z DRT lub MRT
otrzymują dodatkowo leczenie podtrzymujące
— histologia: typ pęcherzykowy mięsaka prążkowanokomórkowego (alveolar RMS);
— więcej niż jedno ognisko przerzutowe i/lub zajęcie kości lub szpiku kostnego.
Przebieg leczenia
Przebieg leczenia oraz strategie terapii przedstawiono w tabelach 52–59.
Wskazania i zasady leczenia chirurgicznego
oo
o
o
Przeprowadzenie pierwotnej lub wtórnej resekcji guza pierwotnego zależy od wieku chore−
go, rozległości i wielkości ogniska pierwotnego oraz jego lokalizacji.
Najlepsze wyniki w zakresie wyleczenia uzyskuje się, gdy możliwe jest przeprowadzenie
pierwotnej resekcji guza w zakresie R0. Rekomenduje się również reoperację, jeśli stwarza
ona możliwość usunięcia ewentualnych mikroskopowych pozostałości nowotworu. Jeśli wyj−
ściowo nie można usunąć guza pierwotnego bez znacznego okaleczenia pacjenta, leczenie
rozpoczyna się od chemioterapii. Stopień odpowiedzi na chemioterapię wstępną ułatwia póź−
niejsze decyzje terapeutyczne, a możliwość wykonania odroczonego, radykalnego zabiegu
chirurgicznego jest bardziej prawdopodobna.
Zabieg pierwotny
Resekcja R0 jest pierwotnym celem terapii lokalnej. W przypadku MTM nie należy jej
jednak przeprowadzać za wszelką cenę. Najczęściej można uniknąć okaleczenia pacjenta
poprzez zastosowanie terapii kombinowanej, ponieważ w przypadku guzów chemiowrażliwych,
które dobrze odpowiadają na chemio− i radioterapię, możliwe jest wyleczenie chorego także
bez resekcji R0.
W przypadku guzów nie−RMS, w których leczenie chirurgiczne jest zasadniczą metodą
leczniczą, należy jednak dążyć w miarę możliwości do wykonania pierwotnej resekcji, nawet

730
Zalecenia postępowania diagnostyczno−terapeutycznego w nowotworach złośliwych — 2009 r.
Tabela 52. Przebieg leczenia
Tydzień
Chemioterapia
1.
Topotekan/karboplatyna (TC)
2.
3.
4.
5.
6.
Remisja całkowita lub częściowa
Jeśli progresja, to zmiana na CEVAIE
7.
I VA
I VA
8.
9.
Lokalna terapia
10.
I
2
Vadr
CEV
11.
12.
13.
TC
I VE
14.
15.
16.
I VA
I VA
17.
18.
19.
I
2
Vadr
CEV
20.
21.
22.
TC
I VE
23.
24.
25.
I VA
I VA
28.–49.
Ustna terapia podtrzymująca
Ustna terapia podtrzymująca
Oceny odpowiedzi na leczenie dokonuje się w 6. tygodniu terapii
Kompletna remisja (CR)
Zanik wszystkich podstawowych zmian
Częściowa remisja (PR)
Co najmniej 30−procentowe zmniejszenie sumy maksymalnych
wymiarów wszystkich podstawowych zmian
Progresja (PD)
Co najmniej 20−procentowy wzrost sumy maksymalnych wymia−
rów; wartość referencyjna: najmniejsza osiągnięta suma mak−
symalnych wymiarów od początku terapii albo wystąpienie jed−
nej lub więcej nowych zmian
Tabela 53. Strategia terapii topotekan/karboplatyna (TC)
Dzień
Karboplatyna
Topotekan
1.
150 mg/m
2
0,75 mg/m
2
2.
150 mg/m
2
0,75 mg/m
2
3.
150 mg/m
2
0,75 mg/m
2
4.
150 mg/m
2
0,75 mg/m
2
Â
600 mg/m
2
3,0 mg/m
2

731
Nowotwory tkanek miękkich
Tabela 54. Strategia terapii ifosfamid/aktynomycyna D (I
3
VA)
Dzień
Ifosfamid
Aktynomycyna D
Winkrystyna
1.
3000 mg/m
2
+ mesna
1,5 mg/m
2
1,5 mg/m
2
2.
3000 mg/m
2
+ mesna
3.
3000 mg/m
2
+ mesna
4.
Mesna
5.
Mesna
14.
1
1,5 mg/m
2
21.
1,5 mg/m
2
Â
9000 mg/m
2
1,5 mg/m
2
4,5 mg/m
2
1
Tylko w pierwszym bloku I
2
VAdr
Tabela 55. Strategia terapii ifosfamid/adriamycyna (I
2
VAdr)
Dzień
Ifosfamid
Adriamycyna
Winkrystyna
1.
3000 mg/m
2
+ mesna
40 mg/m
2
1,5 mg/m
2
2.
3000 mg/m
2
+ mesna
40 mg/m
2
3.
Mesna
4.
Mesna
14.
2
1,5 mg/m
2
21.
1,5 mg/m
2
Â
6000 mg/m
2
80 mg/m
2
4,5 mg/m
2
2
Tylko w pierwszym bloku I
2
VAdr
Tabela 56. Strategia terapii karboplatyna/epirubicyna/winkrystyna (CEV)
Dzień
Karboplatyna
Epirubicyna
Winkrystyna
1.
500 mg/m
2
150 mg/m
2
1,5 mg/m
2
2.
Â
500 mg/m
2
150 mg/m
2
1,5 mg/m
2
Tabela 57. Strategia terapii ifosfamid/winkrystyna/etopozyd (I
3
VE)
Dzień
Ifosfamid
Etopozyd
Winkrystyna
1.
3000 mg/m
2
+ mesna
150 mg/m
2
1,5 mg/m
2
2.
3000 mg/m
2
+ mesna
150 mg/m
2
3.
3000 mg/m
2
+ mesna
150 mg/m
2
4.
Mesna
5.
Mesna
Â
9000 mg/m
2
450 mg/m
2
1,5 mg/m
2
732
Zalecenia postępowania diagnostyczno−terapeutycznego w nowotworach złośliwych — 2009 r.
Tabela 58. Strategia terapii trofosfamid/etopozyd (TE)
Dawka
skumulowana
Trofosfamid
2
¥ 75 mg/m
2
/d.
ØØ ØØ ØØ ØØ ØØ ØØ ØØ ØØ ØØ ØØ przerwa
1,5 g/m
2
VP−16
2
¥ 25 mg/m
2
/d.
ØØ ØØ ØØ ØØ ØØ ØØ ØØ ØØ ØØ ØØ przerwa
500 mg/m
2
/d.
Dzień
1.
2.
3.
4.
5.
6.
7
8.
9.
10. 11.–20.
Tabela 59. Strategia terapii trofosfamid/idarubicyna (TI)
Dawka
skumulowana
Trofosfamid
2
¥ 75 mg/m
2
/d.
ØØ ØØ ØØ ØØ ØØ ØØ ØØ ØØ ØØ ØØ przerwa
1,5 g/m
2
Idarubicyna
1
¥ 5 mg/m
2
/d.
Ø
Ø
Ø
Ø
przerwa
20 mg/m
2
/d.
Dzień
1.
2.
3.
4.
5.
6.
7.
8.
9.
10. 11.–20.
jeśli miałaby być okaleczająca, jeśli istnieją możliwości wykonania wczesnego zabiegu rekon−
strukcyjnego.
U chorych, u których resekcji R0 wyjściowo nie można przeprowadzić lub z innych powo−
dów wykonano resekcję R1, można w okresie do 4 tygodni po postawieniu diagnozy wykonać
dodatkowo resekcję odroczoną w celu uzyskania resekcji R0. Postępowanie chirurgiczne jest
szczególnie ważne w grupie pacjentów, u których stosowanie radioterapii ze względu na wiek
jest niemożliwe lub ograniczone.
Okaleczający zabieg pierwotny
Zabieg pierwotny w żadnym wypadku nie powinien być zabiegiem okaleczającym [jako
zabiegi okaleczające traktuje się: amputację kończyny, wytworzenie przetoki odbytniczej (anus
praeter naturalis), ponadpęcherzowe wyłonienie moczowodów, usunięcie macicy lub pochwy,
prostatektomię, wyłuszczenie oczodołu, jak również wszystkie inne zabiegi powodujące cięż−
kie następstwa funkcjonalne lub kosmetyczne].
W przypadku resekcji R0 guz powinien być usunięty in toto, czyli sposób cięcia i preparacji
powinno się wybrać w ten sposób, aby pomiędzy brzegiem cięcia a guzem zawsze znajdowała
się warstwa tkanki zdrowej — margines bezpieczeństwa (2–5 cm).
Biopsja węzłów chłonnych
W przypadku węzłów chłonnych wykazujących nieprawidłowości w badaniu MR, KT lub
USG należy w trakcie zabiegu pierwotnego pobrać wycinki do badania histologicznego.

733
Nowotwory tkanek miękkich
Zabieg odroczony
Po chemio− i lub radioterapii wykonuje się zabieg usunięcia ogniska pierwotnego nawet
w sposób okaleczający, jeśli umożliwia on resekcję R0. Dotyczy to szczególnie guzów nie−RMS,
które nie wykazują żadnej reakcji na chemioterapię. Guzy te powinny być, w miarę możliwości,
usuwane radykalnie (R0 i R1), czyli zawsze należy uwzględniać możliwość okaleczenia pa−
cjenta.
Wskazania i zasady radioterapii
Radioterapia jest niezbędną metodą konieczną dla miejscowej kontroli nowotworu u pa−
cjentów z mikroskopową lub makroskopową pozostałością guza po resekcji chirurgicznej lub
wstępnej chemioterapii. Wskazania do napromieniania i wybór dawki zależą od: pierwotnej
lokalizacji i wielkości guza, wieku chorego, rozpoznania histopatologicznego i rodzaju resekcji
pierwotnej.
Ogólnie rekomendowana dawka mieści się w granicach 32–54 Gy. Zaleca się unikanie
napromieniania u dzieci poniżej 3. roku życia.
Wskazania do radioterapii dla guzów typu RMS
Kwalifikacja do radioterapii odbywa się w 9. tygodniu leczenia po wykonaniu przeszacowa−
nia („re−staging”). Pacjenta przydziela się do jednej z grup:
1. Grupa chorych, u którzy nie jest konieczne napromienianie — wszyscy pacjenci z pier−
wotną resekcją R0 (typu RMS, korzystna i niekorzystna histologia) i chorzy z korzystną
histologią (RME), również przy wtórnej resekcji R0. Wyjątek: u chorych z zajętymi węzłami
i niekorzystnym wynikiem badania histologicznego należy wykonać napromienianie nawet
w przypadku resekcji R0. Guzów nie−RMS po pierwotnej lub wtórnej resekcji R0 nie na−
świetla się, chyba że wyjściowa wielkość guza wynosiła więcej niż 5 cm i/lub pacjent jest
w wieku powyżej 10 lat.
2. Grupa chorych napromienianych dawką 32 Gy — wszyscy pacjenci z korzystną histologią
(typu RMS) i dobrą albo całkowitą odpowiedzią na chemioterapię (> 2/3), niezależnie od
wyjściowego zajęcia węzłów chłonnych, jednak z dobrymi warunkami do operacji
second−look, jeśli radioterapia może jeszcze poprawić warunki zabiegu.
3. Grupa chorych, którzy muszą być napromieniani dawką 44,8 Gy — wszystkie guzy z ko−
rzystną histologią [RME (N1&N0)] i złą odpowiedzią (< 2/3 i > 1/3) i/lub z resekcją
pierwotną lub wtórną R1, jak również wszystkie histologicznie niekorzystne guzy (typu
RMS i nie−RMS), które wyjściowo nie mogły być resekowane R0 i/lub stwierdzono zajęcie
węzłów chłonnych. Ponadto do tej grupy kwalifikuje się pacjentów z korzystną histologią
po operacji second−look (R1 lub R2), bez radioterapii w przeszłości (dotyczy to również
pacjentów z niekorzystną histologią, u których niezgodnie z protokołem nie zastosowano
napromieniania przed reoperacją). Do tej grupy należą też chorzy z guzami nie−RMS więk−
szymi niż 5 cm i/lub starsi niż 10−letni, których nawet po resekcji R0 napromienia się
dawką 44,8 Gy.
U dzieci poniżej 1. roku życia radioterapię powinno się stosować tylko w wyjątkowych
wypadkach. Dawkę należy ograniczyć do maksymalnie 32 Gy. W skrajnych przypadkach nale−
ży porównać ryzyko funkcjonalnych i kosmetycznych powikłań radioterapii z następstwami
całkowitej resekcji guza (R0) i rozważyć, czy radioterapię można zastosować. Zasadniczo

734
Zalecenia postępowania diagnostyczno−terapeutycznego w nowotworach złośliwych — 2009 r.
u dzieci poniżej 1. roku życia z guzami dobrze odpowiadającymi na leczenie w miarę możliwo−
ści należy rekomendować chemioterapię. Ponownej oceny należy dokonać w 16.–18. tygo−
dniu leczenia, w celu ustalenia rodzaju terapii lokalnej (czy jest możliwa resekcja R0, czy
została osiągnięta pełna remisja?). Wiek 3 lata i mniej jest względnym przeciwwskazaniem
do radioterapii.
Monitorowanie leczenia
Do diagnostyki w trakcie leczenia należą regularne, kontrolne badania laboratoryjne
i badanie USG loży guza. Przed każdym podaniem adriamycyny powinno się przeprowadzać
badanie USG serca. Przed zastosowaniem ifosfamidu konieczne są badania wydolności ne−
rek i ocena klirensu kreatyniny.
Po zakończeniu 3. bloku terapii (9. tydzień) w przypadku guzów nieusuniętych pierwotnie
należy dokonać oceny odpowiedzi w celu określenia dalszego postępowania terapeutyczne−
go. Odpowiedź trzeba również ocenić za pomocą MR w 18. tygodniu terapii.
W zależności od wyjściowych wyników badań mogą zaistnieć wskazania do wykonania
dodatkowych badań kontrolnych, na przykład badania choroby resztkowej w szpiku u pacjen−
tów z wyjściowym rozsiewem do szpiku.
Badania kontrolne przeprowadzone na zakończenie leczenia
1. Diagnostyka obrazowa: KT/MR/USG loży guza wraz z regionalnymi węzłami chłonnymi,
RTG klatki piersiowej, KT klatki piersiowej, USG jamy brzusznej, MR mózgowia.
2. Dokładne badanie pediatryczne i neurologiczne.
3. Badania laboratoryjne: przynajmniej morfologia krwi z rozmazem, badania enzymatyczne,
klirens kreatyniny, stężenie kwasu moczowego, mocznika, badania wirusologiczne, stęże−
nie immunoglobulin, badanie ogólne moczu.
4. Badania w celu wykluczenia nefrotoksyczności indukowanej ifosfamidem:
— w surowicy: stężenie elektrolitów, w tym magnezu (Mg), nieorganicznych fosforanów, glu−
kozy, kreatyniny, gazometria, fosfataza alkaliczna, H
2
CO
3
;
— w moczu: stężenie sodu (Na), wapnia (Ca), glukozy, nieorganicznych fosforanów, kreatyni−
ny, białka całkowitego, wartość pH.
— w 24−godzinnej zbiórce moczu: klirens kreatyniny (u wszystkich dzieci powyżej 90. względ−
nie poniżej 10. percentyla masy ciała zmodyfikowanego względem wzrostu, w przeciwnym
razie wystarczające obliczenie na podstawie reguły Schwartza), 24−godzinne wydalanie
wapnia (w przypadku udowodnionej tubulopatii), frakcjonowane wydalanie fosforanów
i glukozy, maksymalne zwrotne wchłanianie fosforanów określone w stosunku do klirensu
kreatyniny.
5. Badanie USG serca i EKG.
6. Badanie EEG.
7. Badanie dna oka.
8. Audiometria.
9. Inne badania, jeśli wcześniej ich wyniki były nieprawidłowe, na przykład pozytonowa tomo−
grafia emisyjna (PET), punkcja lędźwiowa, diagnostyka układu endokrynnego.
Badania wykonane po zakończeniu leczenia zestawiono w tabeli 60.

735
Nowotwory tkanek miękkich
Terapia drugiej linii (dla grupy non-respond)
Do grupy non−respond kwalifikuje się wszystkich pacjentów, u których po 3 blokach che−
mioterapii regresja guza jest mniejsza od 50%, oraz osoby, u których mimo stosowanego
leczenia obserwuje się progresję, względnie stabilizację procesu chorobowego. Terapia dru−
giej linii składa się z naprzemiennych bloków: topotekan/karboplatyna, topotekan/cyklofos−
famid oraz karboplatyna/etopozyd (VP 16), podawanych w odstępach 3−tygodniowych.
Schematy leczenia drugiej linii przedstawiono w tabelach 61–64.
Tabela 60. Badania po zakończeniu leczenia
Okres
Badanie okolicy guza
Stadium
Badania dodatkowe
1. rok po USG (loża guza, regionalne
RTG klatki piersiowej
Ocena funkcji wątroby
leczeniu
węzły chłonne, jama
(co 3 miesiące),
i nerek (kłębuszków i cewek),
brzuszna, miednica), tomo−
scyntygrafia kości (raz/rok) echokardiografia/24−
grafia z kontrastem lub bez
[tomografia — przerzuty
−godzinne EKG,
niego (na zmianę,
(dotyczy st. IV)]
badania endokrynologiczne
co 6–12 tygodni)
(wzrost i dojrzewanie)
2. rok
Jak wyżej, tylko co
Jak wyżej, RTG klatki
Ocena zaburzeń (nietrzy−
3–6 miesiące
piersiowej co 6 miesięcy
manie moczu, uszkodzenie
wzroku i słuchu, ocena
układu ruchu)
3.–5. rok Jak wyżej, tylko co
Jak wyżej, tylko raz w roku
Badania cytogenetyczne
6–12 miesięcy
u pacjentów leczonych
preparatami G−CSF
> 5. rok
USG,
Jak wyżej, tylko co 2 lata
Inne badania (zgodnie
tomografia z kontrastem
z objawami)
(obydwa badania raz w roku)
G−CSF (granulocyte colony−stimulating factor) — czynnik pobudzający wzrost kolonii granulocytów
Tabela 61. Schemat terapii drugiej linii (grupa non−respond — SL−TECC I)
Topotekan
Topotekan
Topotekan
Karboplatyna Topotekan
Karboplatyna
Karboplatyna Karboplatyna Cyklofosfamid VP−16
Cyklofosfamid VP−16
Tydzień 1.
4.
7.
10.
13.
16.
Tabela 62. Schemat bloku topotekan/karboplatyna
Dzień
Topotekan
Karboplatyna
1
1 mg/m
2
/d. w 23−godzinnym wlewie
150 mg/m
2
/d. i.v. w 1−godzinnym wlewie
2
1 mg/m
2
/d. w 23−godzinnym wlewie
150 mg/m
2
/d. i.v. w 1−godzinnym wlewie
3
1 mg/m
2
/d. w 23−godzinnym wlewie
150 mg/m
2
/d. i.v. w 1−godzinnym wlewie
4
1 mg/m
2
/d. w 23−godzinnym wlewie
150 mg/m
2
/d. i.v. w 1−godzinnym wlewie
Dawka
4 mg/m
2
600 mg/m
2
łączna
Topotekan (Topo):
1 mg/m
2
/d. i.v. przez okres 4 dni
Karboplatyna (Carbo):
150 mg/m
2
/d. w 200 ml/m
2
, przez okres 4 dni

736
Zalecenia postępowania diagnostyczno−terapeutycznego w nowotworach złośliwych — 2009 r.
Tabela 63. Schemat bloku topotekan/cyklofosfamid
Dzień
Topotekan
Cyklofosfamid
1
1 mg/m
2
/d. w 23−godzinnym wlewie
250 mg/m
2
/d. i.v. w 1−godzinnym wlewie
+ mesna 250–300 mg/m
2
/d.
2
1 mg/m
2
/d. w 23−godzinnym wlewie
250 mg/m
2
/d. i.v. w 1−godzinnym wlewie
+ mesna 250–300 mg/m
2
/d.
3
1 mg/m
2
/d. w 23−godzinnym wlewie
250 mg/m
2
/d. i.v. w 1−godzinnym wlewie
+ mesna 250–300 mg/m
2
/d.
4
1 mg/m
2
/d. w 23−godzinnym wlewie
250 mg/m
2
/d. i.v. w 1−godzinnym wlewie
+ mesna 250–300 mg/m
2
/d
Dawka
4 mg/m
2
800 mg/m
2
łączna
Topotekan (Topo):
1 mg/m
2
/d. i.v. przez okres 4 dni
Cyklofosfamid (CYC): 250 mg/m
2
/d. i.v. w 1−godzinnym wlewie, codziennie, przez okres 4 dni
Tabela 64. Schemat bloku karboplatyna/etopozyd
Dzień
Karboplatyna
Etopozyd
1
150 mg/m
2
/d. i.v. w 1−godzinnym wlewie
150 mg/m
2
/d. i.v. w 1−godzinnym wlewie
2
150 mg/m
2
/d. i.v. w 1−godzinnym wlewie
150 mg/m
2
/d. i.v. w 1−godzinnym wlewie
3
150 mg/m
2
/d. i.v. w 1−godzinnym wlewie
150 mg/m
2
/d. i.v. w 1−godzinnym wlewie
4
150 mg/m
2
/d. i.v. w 1−godzinnym wlewie
150 mg/m
2
/d. i.v. w 1−godzinnym wlewie
Dawka 600 mg/m
2
600 mg/m
2
łączna
Etopozyd (VP−16):
150 mg/m
2
/d. i.v. przez okres 4 dni
Karboplatyna (Carbo):
150 mg/m
2
/d. przez okres 4 dni
Terapia dla wznów
Terapia wznowy zależy od rodzaju leczenia poprzedzającego. Pacjentów z grupy niskiego
ryzyka, którzy wyjściowo nie otrzymywali antracyklin ani leków alkilujących, w trakcie wznowy
powinno się leczyć zgodnie ze schematem stosowanym w grupie wysokiego ryzyka w połącze−
niu z radioterapią. U osób zakwalifikowanych wyjściowo do grupy standardowego lub wysokie−
go ryzyka stosuje się leczenie zgodne ze schematem terapii drugiej linii. U pacjentów, którzy
pierwotnie nie otrzymali napromieniania, powinno się zastosować radioterapię w trakcie le−
czenia wznowy. W przypadku pacjentów, których poprzednio napromieniano, decyzję o ewen−
tualnych naświetlaniach należy podjąć indywidualnie, po uwzględnieniu pierwotnej dawki oraz
obszaru napromieniania.
Ze względu na prognozę chorych ze wznową można podzielić na dwie grupy rokownicze:
— pacjenci z korzystną histologią (RME), wznową lokalną, z możliwością naświetlań (wyjś−
ciowo nienaświetlani lub naświetlani niższą dawką) i czasem wystąpienia wznowy powyżej
1 roku od zakończenia leczenia. Przeżycie w tej grupie chorych wynosi około 45%.
W tej grupie pacjentów należy przede wszystkim położyć nacisk na kontrolę lokalną, skła−
dającą się z zabiegu operacyjnego i radioterapii;

737
Nowotwory tkanek miękkich
— pacjenci z wysokim ryzykiem nawrotu choroby, to chorzy, u których spełniony jest przynaj−
mniej 1 z poniższych warunków: niekorzystna histologia (RMA, EES, pPNET, SS), obec−
ność przerzutów oraz brak możliwości napromieniania. Ponieważ w tej grupie pacjentów
odsetek przeżycia jest bardzo niski, należy rozważyć wykonanie radykalnego zabiegu ope−
racyjnego, nawet jeśli byłby on okaleczający. Ta opcja leczenia dotyczy również osób,
u których nie uzyskano odpowiedzi na chemoterapię wznowy.
Zalecane piśmiennictwo
Breneman J.C., Lyden E., Pappo A.S. i wsp. Prognostic factors and clinical outcomes in children
and adolescents with metastatic rhabdomyosarcoma−a report from the Intergroup Rhabdomyo−
sarcoma Study IV. J. Clin. Oncol. 2003; 1: 78–84.
Kazanowska B., Bogusławska−Jaworska J., Gorczyńska E. i wsp. Results of treatment for soft
tissue sarcoma in childhood: a report of the Polish Paediatric Solid Tumour Group (PPSTG). Med.
Pediatr. Oncol. 2001; 37: 320.
Kazanowska B., Reich A., Balcerska A. i wsp. Leczenie mięsaków tkanek miękkich grupy RMS
u dzieci w doświadczeniu Polskiej Pediatrycznej Grupy Guzów Litych (PPGGL). Nowotwory
(J. Oncol.) 2002; 53 (supl. 4): 82.
Klingebiel T., Kościelniak E., Pilz T., Treuner J. High−dose therapy versus oral maintenance: interim
results of HD CWS 96 study for treatment of patients with metastasized soft tissue sarcoma.
Monatsschr Kinderheild 1999; 147: 984.
Klingebiel T., Pertl U., Hess C.F. i wsp. Treatment of children with relapsed soft tissue sarcoma:
report of the German CESS/CWS REZ 91 trial. Med. Pediatr. Oncol. 1998; 30: 269–275.
Kościelniak E., Klingebiel T.H., Peters C. i wsp. Do patients with metastatic and recurrent rhabdo−
myosarcoma benefit from high−dose therapy with hematopoetic rescue? Report of German/
/Austrian pediatric Marrow transplantation Group. Bone Marrow Transplant. 1997; 19: 227–231.
Kościelniak E., Rosti G., Treuner J. i wsp. Prognosis in patients with primary bone and/or bone
marrow metastases of rhabdomyosarcoma (RMS). A joint report of the EBMT Solid Tumor Working
Party and the German Soft Tissue Sarcoma Study (CWS). Bone Marrow Transpl. 1998; 21: 226.
Koscielniak E., Klingebiel T., Knietig R., Morgan M., Niethammer D., Treuner J. Factors related to
prognosis in patients with primary metastatic RMS−like tumors. Analysis of the CWS−Study and
the German pediatric Stem Cell Transplantation Registry. Bone Marrow Transplant. 2001; 27: 6.
Koscielniak E., Morgan M., Treuner J. Soft tissue sarcoma in children: prognosis and manage−
ment. Pediatr. Drugs 2002; 1: 21–28.
Koscielniak E., Schmidt B., Knietig R., Mattke A., Morgan M., Treuner J. Effectivity of a 32 Gy
radiation (RT) dose in children with rhabdomyosarcoma (RMS): Report of the German Cooperati−
ve Soft Tissue Sarcoma. Med. Pediatr. Oncol. 2001; 37: 186.
Look A.T., Kirsh I.R. Molecular basis of childhood cancer. W: Pizzo P.A., Poplack D.G. (red.).
Principles and practice of pediatric oncology. JB Lippincott Co, Philadelphia 2002: 45–87.
Parham D.M. Pathologic classification of rhabdomyosarcomas and correlations with molecular
studies. Mod. Pathol. 2001; 14: 506–514.
Wyszukiwarka
Podobne podstrony:
200 t2 09 Onkologia dzieci nerczak plodowy u dzieci
200 t2 12 Onkologia dzieci Zalecenia dotycz�ce post�powania diagnostyczno leczniczego
Choroba Oparzeniowa u Dzieci Postępowanie Doraźne
onkologia dziecieca skrypt
wyklad 3 now4, Patomorfologia, Zmiany postępowe i nowotwory
Hematologia i onkologia dziecięca, klebkowe zapalenie nerek ostre rozplemowe, KZN ostre rozplemowe
Hematologia i onkologia dziecięca, ostra białaczka nielimfoblastyczna, Wzór
10 Ryzyko miarą postępu
Hematologia i onkologia dziecięca, zespol Schoenleina - Henocha, Zespół Schoenleina - Henocha
Hematologia i onkologia dziecięca, niedokrwistosc z niedoboru zelaza, niedokrwistość z niedoboru żel
Hematologia i onkologia dziecięca, niedokrwistosc z niedoboru zelaza, niedokrwistość z niedoboru żel
10 Niepełnosprawność u dzieci i młodzieży (konspekt)
Hematologia i onkologia dziecięca, limfadenopatia u dzieci, Limfadenopatia u dzieci
Zadania tekstowe 10, dla dzieci, matematyczne
więcej podobnych podstron