METHANESULFONIC ACID
1
Methanesulfonic Acid
MeSO
3
H
[75-75-2]
CH
4
O
3
S
(MW 96.12)
InChI = 1/CH4O3S/c1-5(2,3)4/h1H3,(H,2,3,4)/f/h2H
InChIKey = AFVFQIVMOAPDHO-QEZKKOIZCK
(cyclocondensation reagent; precursor for methanesulfonyl
chloride and anhydride; catalyst for polymerization, alkylation,
and esterification reactions)
Alternate Name:
MsOH.
Physical Data:
mp 20
◦
C; bp 167
◦
C/10 mmHg, 122
◦
C/1 mmHg;
d
1.481 g cm
−3
; n
20
D
1.3210.
Solubility:
sol water, ethanol, ether; insol hexane; very sparingly
sol benzene, toluene.
Form Supplied in:
technical quality is 95% pure containing
2% water.
Analysis of Reagent Purity:
17
O NMR.
1
Preparative Method:
by oxidation of Dimethyl Sulfide with
Dimethyl Sulfoxide
in the presence of water and a catalytic
amount of Hydrogen Bromide.
17
Purification:
stir with P
2
O
5
(20 g for 500 mL of the acid) at
100
◦
C for 0.5 h and then distill under vacuum.
Handling, Storage, and Precautions:
irritant and highly corrosive
liquid; should be stored in glass containers. Use in a fume hood.
Original Commentary
Lakshminarayanapuram R. Subramanian & Michael Hanack
Universität Tübingen, Tübingen, Germany
Antonio García Martínez
Universidad Complutense de Madrid, Madrid, Spain
Cyclization Reactions.
MsOH is a weaker acid than Trifluo-
romethanesulfonic Acid
and hence only few reports exist on the
use of pure MsOH in cyclocondensation reactions.
2
MsOH in
dichloromethane effects a symmetry allowed cyclization of the
precursor diene to afford 1,2,3,4-tetramethyl-5-(trifluoromethyl)
cyclopentadiene, which is used as a ligand in organometallic
chemistry (eq 1).
3
MeSO
3
H
CH
2
Cl
2
, rt
82%
(1)
CF
3
HO
CF
3
A 1:10 solution by weight of Phosphorus(V) Oxide in MsOH
4
is a convenient alternative to Polyphosphoric Acid for cycliza-
tion reactions. For example, the classical preparation of cyclopen-
tenones via intramolecular acylation of alkenoic acids or their
lactones (eq 2) and Beckmann rearrangement using polyphos-
phoric acid (eq 3) give comparable yields when performed with
MsOH/P
2
O
5
.
4
MeSO
3
H
P
2
O
5
92%
(2)
O
O
O
MeSO
3
H
P
2
O
5
96%
(3)
NOH
NH
O
MsOH in conjunction with P
2
O
5
has been used in the
rearrangement of 2-vinylcyclobutanones to spiro and fused
cyclopentenones (eqs 4 and 5).
5
+
O
O
O
(4)
13%
51%
MeSO
3
H
P
2
O
5
MeSO
3
H
P
2
O
5
48%
(5)
O
O
3-Unsubstituted indoles are formed regioselectively by treat-
ment of precursor hydrazones with MsOH/P
2
O
5
.
6
In the example
given in eq 6,
6
only 2–3% of the undesired 3-isomer is formed.
Because decomposition sometimes occurs, it is advisable to dilute
the reagent with a suitable polar, nonbasic solvent like sulfolane
or dichloromethane.
N
N
O
Cl
N
MeO
2
C
O
N
N
CO
2
Me
Cl
MeSO
3
H
P
2
O
5
77%
(6)
MsOH itself is a better cyclizing agent than an admixture with
P
2
O
5
for the cyclization of 3-arylpropanoic and 4-arylbutanoic
acids to 1-indanones (eq 7) and 1-tetralones (eq 8).
7
MeSO
3
H
110–115 °C
90%
(7)
CO
2
H
O
Avoid Skin Contact with All Reagents
2
METHANESULFONIC ACID
MeSO
3
H
90–95 °C
89%
(8)
HO
2
C
O
Cyclization with neat MsOH is also observed in the formation
of cyclopentenones from 2-vinylcyclobutanones (eq 9).
5
MeSO
3
H
52%
(9)
O
H
O
MsOH has also been employed successfully in cyclocondensa-
tion reactions in the field of heterocycles (eq 10).
8,18
Thus the
hexahydroimidazo[1,2-a]pyrimidine-5,7-dione shown in eq 10
gives the corresponding cyclized product in 81% yield.
8
MeO
MeO
N
N
N
O
O
MeO
MeO
N
N
N
O
MeSO
3
H
40–80 °C, 12.5 h
81%
(10)
Other Applications.
MsOH is superior to Sulfuric Acid as
the solvent and catalyst for the conversion of benzoic acid to Per-
benzoic Acid
(eq 11).
9
(11)
MeSO
3
H
70% H
2
O
2
85–90%
CO
2
H
CO
3
H
MsOH in the presence of methionine is the reagent of choice
and an excellent substitute for Boron Tribromide for O-demethy-
lation of opioid derivatives (eq 12).
10
Among the other reagents
tested, only TfOH is as effective as MsOH/methionine.
O
MeO
O
OH
N R
O
HO
O
OH
N R
(12)
MeSO
3
H, methionine
20–80 °C, 8–48 h
18–76%
R = Me, CO
2
Et
Schmidt rearrangement of optically active cyclic β-keto
esters with retention of configuration is effectively carried out
with MsOH in the presence of Sodium Azide (eq 13).
11
MeSO
3
H, NaN
3
CHCl
3
, ∆, 0.5 h
91%
O
CO
2
Et
Ph
NH
O
CO
2
Et
Ph
(13)
>95% ee
Attempted acid-induced cyclization with MsOH of an interme-
diate diazo ketone involved in the synthesis of tricyclo[5.2.1.0
4,10
]
decane-2,5,8-trione affords the corresponding methylsulfonyl-
oxy derivative via the protonated diazonium salt (eq 14).
12
CHN
2
O
O
O
H
H
CH
2
OMs
O
O
O
H
H
(14)
MeSO
3
H
71%
MsOH is a useful reagent for the condensation of 2-(hydroxy-
methylene)cyclohexanone with sulfonamides in the presence of
molecular sieves to afford products in the cis-u diastereoisomeric
forms and with >90% stereoselectivity (eq 15).
13
Ph
OH
NHSO
2
Me
OH
O
O
MeSO
2
N
Ph
H
H
O
O
MeSO
2
N
Ph
H
O
(15)
MeSO
3
H
molecular sieves 4Å
CH
2
Cl
2
, 25 °C, 24 h
22%
+
H
+
95:5
The reaction of trimethylphosphine–borane with MsOH in
dichloromethane gives the methanesulfonate derivative of the
borane (eq 16).
14
This compound can be condensed with diphenyl-
phosphine–borane in the presence of Sodium Hydride to give the
corresponding dimer. By repeating the sequences of mesylation
and condensation, a tetramer containing a linear P–B bond has
been synthesized.
14
P
Me
Me
Me
BH
3
P
Me
Me
Me
BH
2
OMs
(16)
MeSO
3
H
CH
2
Cl
2
, rt
89%
+ –
+ –
MsOH has also been used to deblock the benzyl protecting
group
15
and to carry out the acidic hydrolysis of esters.
16
First Update
Matthew M. Kreilein
University of North Carolina, Chapel Hill, NC, USA
Reagent Derivitization.
In addition to using MsOH along
with phosphorus(V) oxide to prepare Eaton’s reagent, MsOH is
also used to prepare methanesulfonic acid anhydride via dehydra-
tion with phosphorus(V) oxide followed by distillation to obtain
the desired anhydride.
19
Adsorption of MsOH onto silica gel as alumina has been docu-
mented as well. Methanesulfonic acid on silica gel actually proved
A list of General Abbreviations appears on the front Endpapers
METHANESULFONIC ACID
3
to be more acidic than MsOH alone allowing for use of an envi-
ronmentally benign heterogeneous acid that actually proved to
perform better than some of the commercially available adsorbed
acids.
20
In some transformations, MsOH/Al
2
O
3
performed worse
than MsOH and MsOH/SiO
2
and the results were unpredictable.
However, one study did show that MsOH/Al
2
O
3
was a very useful
combination for the highly selective monoesterification of diols.
21
Several amine bases can be combined with MsOH in EtOAc to
provide ammonium mesylate salts as crystalline solids.
22
In the
same publication, these mesylate salts were used to catalyze the
nucleophilic opening of α-lactams at C3 by amines (eq 17).
N
O
Ph
H
R
R
2
NH
3
+
–
OMs
R
3
NH
2
NHR
O
Ph
NR
3
H
(17)
Combining di-(t-butyl)dicarbonate (Boc
2
O) with MsOH in
the presence of 4-(N,N-dimethyl)aminopyridine (DMAP) allows
for the formation of N-Boc methanesulfonamide.
23
This reagent
has been used in Mitsunobu reactions to directly install a
Boc-protected amine functionality (eq 18).
24
C
Br
H
N
Ts
OH
C
Br
H
N
Ts
NMsBoc
(18)
n
DEAD, PPh
3
n
MsNHBoc
THF, rt
Acid-catalyzed Reactions.
Methanesulfonic acid has been
used as an acid catalyst for numerous common transformations as
it is equally as powerful as HCl, yet not as strong as TfOH. This
allows for a “strong but gentle” reactivity profile. In addition, since
MsOH can be purified and freed of H
2
O, it can be used to catalyze
reactions without hydrolysis by H
2
O if so desired.
Pummerer Rearrangement.
Certain acid-catalyzed reactions
seem to perform very well when carried out using MsOH instead
of other acids. The Pummerer rearrangement can be smoothly
catalyzed by MsOH when TfOH may be too strong for the sys-
tem in use.
25,26
A useful example of this was exhibited in efforts
toward a synthesis of apoptolidin (eq 19).
27
O
Me
S
O
Ph
O
Me
S
O
Ph
O
O
Me
PhS
(19)
–30
°C, 2 h, 90%
CH
2
Cl
2
, rt, 14 h
95%
m
-CPBA, CH
2
Cl
2
Ac
2
O, MsOH
Fries Rearrangement.
When combined with triflate Lewis
acids
28
or phosphorus oxychloride,
29
MsOH was able to smoothly
bring about the Fries rearrangement of aromatic esters. In the
reactions with POCl
3
, installation of an aromatic mesyloxy group
was possible in certain cases (eq 20).
R
1
OCOR
O C
O
MsOH
R
2
MsOH
OH
R
2
R
1
OMs
O
R
2
O
C
OMs
R
1
COR
(20)
M(OTf)
x
46–90%
M = Mg, Ca, Sc, Cu, Zn, Y, La, Nd, Dy, Yb, Bi
POCl
3
100
°C
R
1
/R
2
= H, m-Me, o-Cl, o-Me, o-Cl, m-Br, p-Cl, p-Me, etc.
+
Ritter Reactions.
The acid-catalyzed Ritter reaction proceeds
efficiently when initiated by MsOH. A large library of nitriles was
subjected to Ritter conditions with MsOH providing a wide array
of N-monosubstituted amides.
30
Several more complex examples
of Ritter reactions performed by MsOH were illustrated in the
synthesis of cis-fused indeno-pyridine ring systems (eq 21).
31
–
33
In these systems, MsOH was the only acid able to deliver the
product in very high yields (>90%). In more complex systems
of this nature, MsOH was able to catalyze the Ritter ring closure
step leaving the aryl methyl ether protecting group intact until it
was later removed using MsOH in the presence of
L
-methionine
to yield the target compounds (eq 21).
32,33
OH
OMe
O
CN
R
R
N
H
CO
2
Me
O
OH
OMe
O
CN
Ph
MeO
Ph
N
H
CO
2
Me
O
OMe
Ph
N
H
NH
2
OMe
Ph
N
H
NH
2
OH
(21)
R = H, Me, Ph, thienyl
MsOH, rt
MsOH, rt
steps
or
MsOH, PhCl, 70
°C
76%
L
-methionine
83%
MsOH
Schmidt Reaction.
Several reports also point to MsOH as
being the acid of choice in the Schmidt reaction as well.
34
–
38
One interesting application of the Schmidt reaction using MsOH
was developed using β-keto imides instead of esters, which are
Avoid Skin Contact with All Reagents
4
METHANESULFONIC ACID
typically employed in the Schmidt reaction.
39
Subjecting the start-
ing imides to the standard Schmidt condition brought about con-
version of the starting material to an oxazole product arising from
participation of the imide nitrogen lone pair in the reaction (eq 22).
X
R
1
O
O
R
2
H
O
N
R
1
R
2
X
MsOH
CHCl
3
, 0
°C to ∆
R
1
/R
2
= alkyl, aryl, H
X = N(alkyl)
2
(22)
NaN
3
Reductive Pinacol Couplings.
Methanesulfonic acid in com-
bination with zinc metal or samarium(II) iodide and Yb(OTf)
3
has been used to bring about the reductive pinacol-like coupling
of imines, oximes, and azines (eq 23).
40
–
42
It was also possible
to perform the reduction of α-iminocarbonyl compounds as well
without the pinacol coupling occurring to give rise to α-amino
carbonyl compounds (eq 23).
43
N
N
R
2
R
3
R
1
Ar
Ar
R
1
N
H
H
N
R
3
R
1
R
1
R
2
Ar
Ar
N
OH
Ar
H
2
N
Ar
Ar
NH
2
N
N
Ar
Ar
N
Ph
R
Ph
Ph
NH
2
NH
2
N
Ph
HO
CO
2
Me
NHBoc
Ph
CO
2
Me
(23)
Zn, MsOH
Zn, MsOH
or
2. (Boc)
2
O
Yb(OTf)
3
MsOH
SmI
2
1. Zn, MsOH
Regioselective Opening of Aziridines.
In the synthesis of
peptide isosteres, MsOH was used to promote the regioselective
ring opening of ethynyl aziridines and β-aziridinyl-α,β-enoates.
After activation of the aziridine functionality by MsOH, the
mesyloxy anion opened the aziridine ring. In the case of the enoate
aziridines, the trans-enoates smoothly yielded α-mesyloxyamines
while the cis-enoates gave rise to a complex mixture of products
including γ-butenolide formation (eq 24).
44
The ethynylaziri-
dines were also opened in this manner yielding an intermediate
alkynylmesyloxy amine. After treatment of this intermediate
with copper(I) bromide dimethyl sulfide complex with LiBr
in THF, a bromoallene was formed, which could be cyclized
using sodium hydride to afford cis-aziridines as the desired targets
(eq 24).
45,46
Friedel–Crafts Acylations.
Methanesulfonic acid in combi-
nation with postassium thiocyanate promotes the Friedel–Crafts
acylation of aromatic rings providing a quick synthesis of ben-
zenethioamides in good yield. A number of substituents on the
aromatic ring are stable to the reaction conditions (eq 25).
47
In another useful application of a Friedel–Crafts-type acylation,
Corey generated an iminium ion in situ, which was cyclized onto
an aromatic ring in order to synthesize the tetrahydroisoquinoline
ring of ecteinascidin 743 (eq 25).
48
N
Ph
Mts
N
CO
2
Me
Mts
OMs
NHMts
Ph
MsOH
MsOH
N
Ph
Mts
CO
2
Me
OMs
NHMts
(24)
CHCl
3
20 min, rt
from trans-enoate
CH
2
Cl
2
15 min, 0
°C
LiBr, THF
2. NaH, DMF
1. CuBr
Me
2
S
·
Ar
H
Ar
S
NH
2
HN
S CH
3
HO
CO
2
Me
MeO
BnO
N+
S
MeO
2
C
N
S CH
3
MeO
BnO
MeO
2
C
(25)
KSCN
MsOH
3 Å sieves
MsOH
30
°C
A number of examples of cyclization onto aromatic rings
using carbonyl compounds can be found throughout the litera-
ture. The cyclization onto carboxylic acids has been used to make
1-substituted fluoren-9-ones,
49
indenoisoquinolines,
50
substi-
tuted xanthones,
51
and tetralones.
52
Treatment of ketones with
MsOH and Eaton’s reagent can be followed by dehydration to form
methylchrysenes and methylbenz[a]anthracenes,
53
favelines,
54
and benzofurans.
55
Cyclization of a urethane using MsOH and
P
2
O
5
provided rapid entry into a dihydro-1(2H)-isoquinoline
(eq 26).
56
NMe
O
Me
NHCO
2
Et
Me
(26)
120
°C
72%
MsOH
P
2
O
5
Miscellaneous Acid-catalyzed Processes.
Several examples
of acid-catalyzed cyclization onto aromatic rings not involving
carbonyls can be found using MsOH. Usually, protonation of
a carbon–carbon double bond is involved and is followed by
A list of General Abbreviations appears on the front Endpapers
METHANESULFONIC ACID
5
cyclization onto the aromatic ring.
57
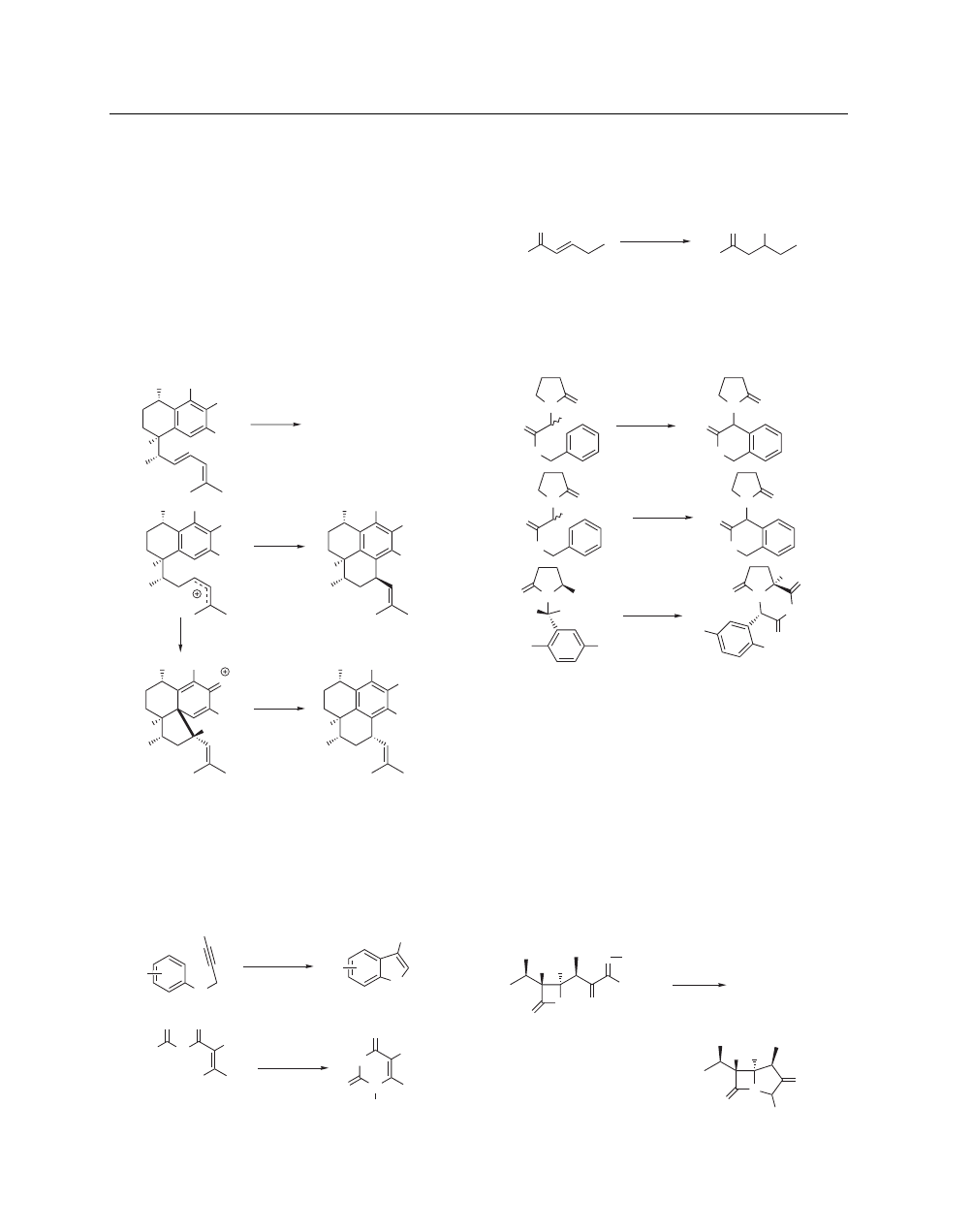
Corey utilized this strategy
in the synthesis and structural revision of the pseudopterosins,
pseudopterosin aglycon, and of helioporin E.
58,59
A tethered
diene was cyclized after protonation with MsOH. In the event,
two diastereomers of the target ring system were isolated depen-
ding on the substituent resident on the aromatic alcohol. When
the ring contained a mesyloxy substituent, smooth cyclization to
the trans-product was observed. When a TBS ether was present
on the aromatic ring, formation of the cis-product was observed
via formation of the spiro-fused five-membered ring followed by
acid-catalyzed rearrangement to the cis-six-membered ring prod-
uct (eq 27). This difference was due to the electronic effects
imparted to the system by the mesyloxy substituent in compar-
ison to the TBS ether.
OBn
OR
Me
Me
H
Me
MsOH
OBn
OR
Me
Me
H
Me
OBn
OMs
Me
Me
H
Me
OBn
OTBS
Me
Me
H
Me
H
OBn
OTBS
Me
Me
H
Me
(27)
R = Ms
R = TBS
Protonation of a TIPS-substituted triple bond was accomplished
using MsOH and was followed by cyclization and TIPS removal
in order to synthesize a library of indoles that could be used for
the synthesis of more advanced molecules (eq 28).
60
Protona-
tion of a series of di(methylthio)acryloyl ureas could also be car-
ried out with cyclization to form a series of highly functionalized
methylthio uracils (eq 28).
61
N
Ms
TIPS
R
N
Ms
Me
R
R
2
HN
O
N
H
O
R
1
MeS
SMe
HN
O
R
1
N
SMe
O
R
2
(28)
MsOH
R
1
= Me or Et
R
2
= n-Pr or Bn
HOAc
80
°C
CH
2
Cl
2
rt
MsOH
Several interesting examples of acid-catalyzed reactions can
also be carried out using MsOH. Activation of α,β-unsaturated
carbonyls in aza-Michael additions can be accomplished using
MsOH (eq 29).
62
Ph
O
Ph
O
NHCbz
(29)
cat. MsOH
CH
3
CN
H
2
NCbz
Intramolecular amidoalkylation onto aromatic rings was also
smoothly catalyzed by MsOH giving rise to isoquinoline products
via formation of an intermediate imine (eq 30).
63
HN
O
OMe
N
O
HN
O
N
O
HN
O
NHCO
2
Me
N
O
HN
O
N
O
MsOH
MsOH
MsOH
N
O
CO
2
H
H
CONH
2
R'
R
N
O
NH
H
O
O
R′
R
(30)
Methanesulfonic acid has also been shown to catalyze the
allylation of hydrates of α-keto aldehydes and glyoxylates us-
ing allyltrimethylsilane.
64
A useful transformation of dicarboxylic
acids to bisphosphonic acids was illustrated using MsOH with
phosphorus(III) chloride.
65
The MsOH allowed for the reaction
to remain liquid thereby simplifying workup and purification of
the desired bisphosphonic acids.
A useful cyclization protocol was employed for the synthesis
of bicyclic β-ketoesters of fused β-lactams. Formation of an iodo-
nium ylide was smoothly achieved and upon treatment with MsOH
in ethanol, cyclization occurred to give useful bicyclic products for
the synthesis of 1β-methylcarbapenams (eq 31).
66
Several Lewis
acids failed to catalyze the desired reaction and only sulfonic acids,
including MsOH, were able to effect this transformation.
NH
O
O
CO
2
BH
I
Ph
OH
H H
N
O
OH
H H
O
CO
2
BH
(31)
BH = benzhydryl
EtOH
15 min
MsOH
Avoid Skin Contact with All Reagents
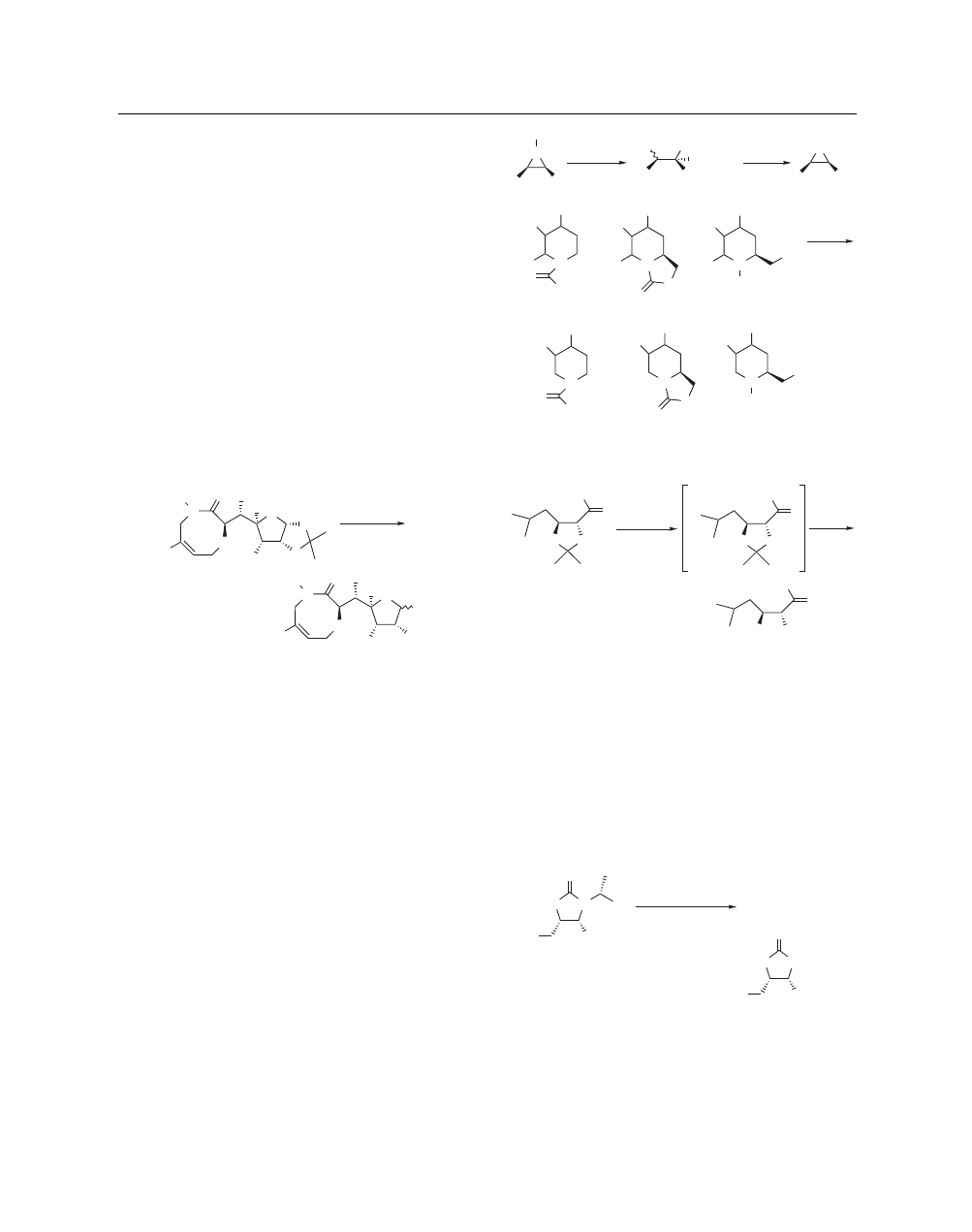
6
METHANESULFONIC ACID
As it is primarily used in a manner similar to any other
organic acid, MsOH can catalyze a great number of reactions.
It has been used to effect the following transformations: acid-
catalyzed ketalization,
67
dehydration,
68
skeletal rearrangement,
69
lactonizations,
70
–
72
cyclic amide formation,
73
amine bond for-
mation after protonation of a free hydroxyl,
74
hydrolysis of
pyrrolidin-2-ones,
75
glycosylation of nucleobases without TBS
deprotection,
76
one-step
acetal
removal
and
thiolane
installation,
77
and Wacker oxidations.
78,79
Protecting Group Removal.
Methanesulfonic acid has been
used for the cleavage of numerous acid-labile protecting groups.
While it can smoothly cleave isopropylidene acetals,
80
as well
as TBS ethers,
81
–
83
the “strong yet weak” aspect of MsOH in
comparison to TfOH makes it a very good choice for selective
deprotection. Global deprotection and chemical modification were
illustrated in the cleavage of a TBS ether with hydrolysis of an
isopropylidene acetal and concomitant glycosylation and acetyla-
tion when a sugar was treated with MsOH and acetic anhydride
(eq 32).
84
O
O
O
TBSO
H
OH
N
NCH
3
EtO
2
C
O
H
3
C
MsOH
O
OAc
AcO
H
OAc
N
NCH
3
EtO
2
C
O
H
3
C
Ac
2
O
(32)
OAc
When combined with other reagents, MsOH provides some very
useful and tunable protecting group manipulations. Smooth re-
moval of the trityl protecting group on aziridines can be achieved
by treatment of the starting material with MsOH and triethylsi-
lane upon workup with diisopropylethylamine.
85
–
89
The amine
base was necessary to complete the reaction as the intermediate in
the transformation is actually the aziridine opened to a mesylate
salt (eq 33). Another application of MsOH/Et
3
SiH was exhibited
in the selective deacylation of azasugars. Treating the systems
with MsOH/Et
3
SiH allowed for removal of the α-acetate at C2
without any deacylation or loss of the nitrogen protecting group
(eq 33).
90
Removal of several other nitrogen protecting groups has
been accomplished with MsOH as well. The cleavage of a
p
-methoxybenzyl protected-nitrogen was accomplished in good
yield using MsOH in the synthesis of naphthyridinones.
91
Removal of the Boc protecting group by MsOH is a very
commonly employed procedure that typically proceeds quite
smoothly.
Facile removal of a Boc protecting group without hydrolysis
of a t-butyl ester has been reported using MsOH with t-butyl
acetate.
92
It was also possible to remove the Boc group from
a secondary amine using MsOH without removing a primary
TBDPS ether.
93
The acidic yet water-free nature of MsOH made
it useful for stepwise Boc deprotection followed by oxazolidine
cleavage when water was added to the intermediate mesylate salt
formed in the deprotection step (eq 34).
94
N
R
R′
Tr
MsOH
O
R
AcO
AcO
OAc
N
N
O
R
AcO
OAc
N
AcO
AcO
OAc
O
O
N
AcO
OAc
O
O
R
R′
H
NH
3
+
MsO
–
OMs
AcO
AcO
OAc
CO
2
Me
OAc
N
N
AcO
OAc
CO
2
Me
OAc
i
-Pr
2
NEt
H
N
R
R′
MsOH
(33)
R = H, Ph, OMe, OBn
or
or
R = H, Ph, OMe, OBn
or
or
Et
3
SiH
0
°C
Et
3
SiH
20 min, rt
BocN
O
O
HO
HN
O
O
HO
MeSO
3
H
H
2
N
OH
O
HO
(34)
•
MsOH
H
2
O, rt
96% (one-pot)
i
-PrOH, ∆
In addition to removing the nitrogen protecting group on
oxazolidines, methanesulfonic acid in combination with anisole
has been shown to remove the 1-naphthylethyl protecting group
from the nitrogen on oxazolidinones without scrambling the stere-
ochemistry in the system and without cleavage of the oxazolidi-
none itself (eq 35).
95,96
Anisole in combination with MsOH was
also used to effect the clean removal of an Mbs-protected nitrogen
(Mbs = 4-methoxybenzensulfonyl) with concomitant hydrolysis
of a t-butyl ester.
97
In this case, a benzyl ether and several amide
linkages were left intact.
O
N
O
Ar
PMP
HO
O
NH
O
PMP
HO
(35)
MsOH, anisole
MeNO
2
, 50
°C, 6 h
Treatment of an aryl benzyl ester allowed for hydrolysis while a
t
-butyl ester present in the molecule was left intact. Only warmer
temperatures and the stronger nature of TfOH were able to bring
about hydrolysis of the t-butyl ester later in the synthesis.
98
More examples of deacylation can be seen in the one-step global
N,O
-deacylation of a NeuAc methylthioglycoside (eq 36).
99,100
A list of General Abbreviations appears on the front Endpapers
METHANESULFONIC ACID
7
O
SMe
CO
2
Me
AcO
AcHN
AcO
OAc
OAc
O
SMe
CO
2
Me
HO
H
2
N
HO
OH
OH
(36)
MsOH
MeOH
60
°C, 24 h
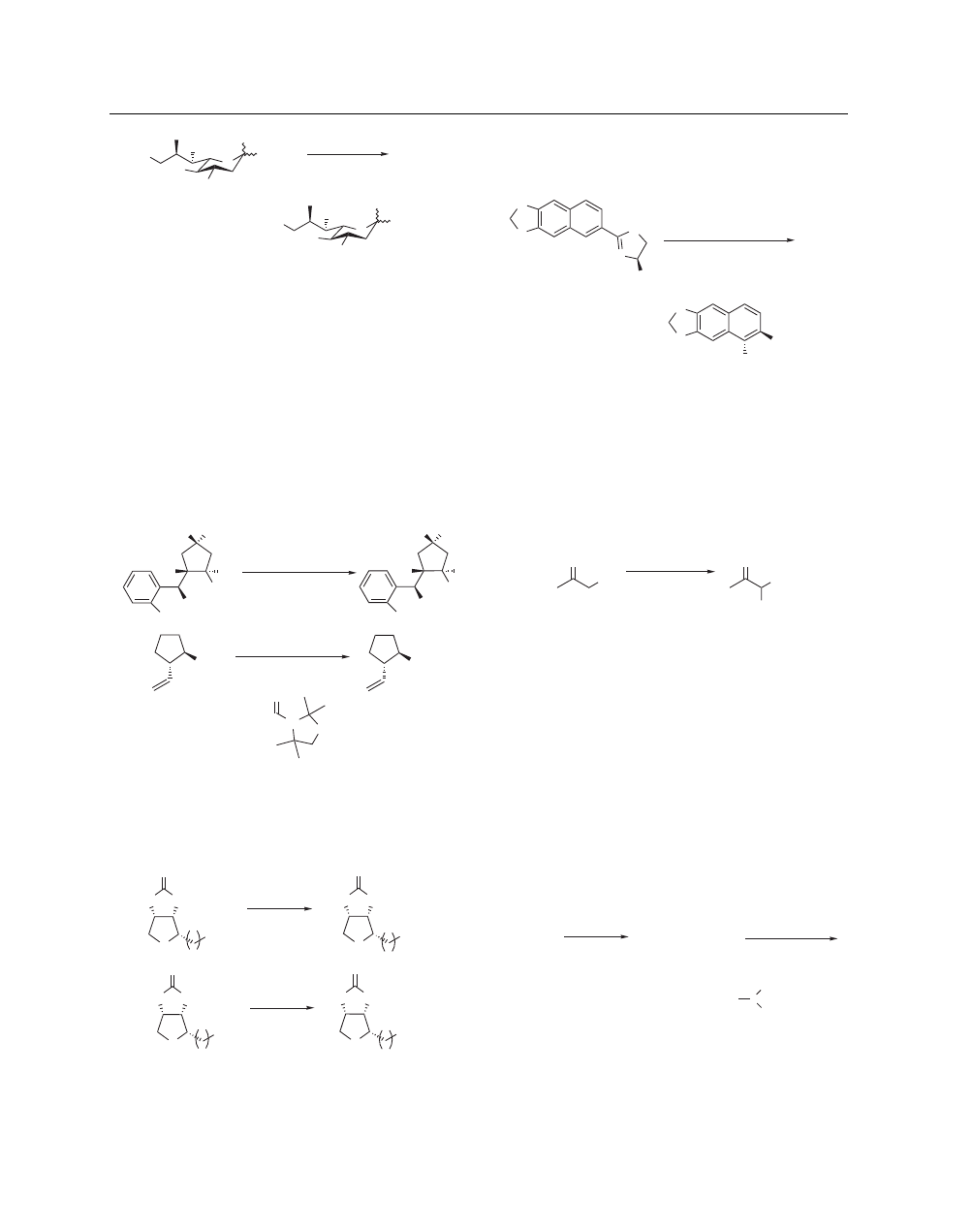
Another example of nitrogen deprotection was illustrated in
the deallylation of mono and diallyl aromatic amines when these
were treated with MsOH in refluxing EtOH. Some difficulty was
observed for diallylamines when these were positioned alpha to
a nitrogen in the aromatic ring; however, smooth removal of the
allyl groups could be effected using other means.
101
Hydroxyl groups can be protected using the Cby protecting
group (Cbyre = carbamoyloxy), which is stable to nucleophilic
attack and can be easily installed. Hydrolysis of the Cby group
can be brought about using MsOH in refluxing MeOH, with final
removal to unmask the free alcohol requiring treatment with either
barium(II) hydroxide or potassium carbonate (eq 37).
102
–
104
Removal of the Cby group can be accomplished in a manner that
is tolerant of a large number of functional groups.
R
R
H
H
OCby
OH
SiMePh
2
R
R
H
H
OH
OH
SiMePh
2
OCby
OH
O
N
O
(37)
2. Ba(OH)
2
, MeOH, ∆
2. K
2
CO
3
, MeOH, ∆
Cby =
1. MsOH, MeOH, ∆
1. MsOH, MeOH, ∆
In efforts toward a rapid synthesis of (+)-biotin, Seki and
co-workers used MsOH in mesitylene or xylene in order to
remove the benzyl protecting group resident in the cyclic urea
intermediates (eq 38).
105,106
S
NBn
BnN
O
CO
2
H
S
NH
HN
O
CO
2
H
S
NH
BnN
O
CO
2
H
S
NH
HN
O
CO
2
H
(38)
4
MsOH
4
4
MsOH
4
mesitylene
74%
xylene
84%
Hydrolysis of a carbamate has been carried out using MsOH as
well.
107
Another useful deprotection using MsOH was illustrated
with an oxazoline as both a chiral auxiliary and a masked methyl
ester.
108
After addition of an aryl Grignard reagent, the auxiliary
was hydrolyzed using MsOH in MeOH. The hydrolysis was useful
for several varieties of oxazolines (eq 39).
O
O
N
O
R
O
O
CO
2
Me
Ar
(39)
R = Me, t-Bu, Bn
Ar = 3,4,5-OMe-C
6
H
2
~
2. MsOH, MeOH, 65
°C
64–76%
1. ArBr, t-BuLi, –35
°C
Installation of Mesylates.
Installation of the methanesulfony-
loxy (mesyloxy) group can be achieved using MsOH via sev-
eral methods. Regioselective formation of the thermodynamic
enolate of ketones with subsequent mesyloxy group installation
can be achieved by treating ketones with copper(II) oxide in
refluxing acetonitrile for 14 h in the presence of MsOH. The
procedure can also be used with TsOH as well as NsOH to in-
stall tosyloxy and nosyloxy groups, respectively (eq 40).
109
O
R′
O
R′
OR
CuO, ROH
R = CH
3
SO
2
- (Ms), p-CH
3
C
6
H
4
SO
2
- (Ts), p-NO
2
C
6
H
4
SO
2
- (Ns)
(40)
CH
3
CN, ∆, 14 h
R′ = Me, Et, n-Pr, i-Pr, i-Bu
The procedure was applicable to several 2-alkanones and pro-
vided only the 3-mesyloxy-2-alkanones in 65–71% yield proving
the selective formation of the thermodynamic enolate with CuO.
Lower yields in the mesyloxy and tosyloxy compounds were
attributed to their increased water solubility in comparison to the
nosyloxy compounds.
Methanesulfonic acid can be used to synthesize 1-[hydroxy
(mesyloxy)iodo]-2,2,2-trifluoroethane, which exhibits a reacti-
vity pattern very similar to Koser’s reagent, PhI(OH)OTs.
110
–
112
Commercially available trifluoroethyl iodide is first oxidized with
trifluoroperacetic acid/trifluoroacetic acid to the corresponding
bistrifluoroacetate 2 (eq 41).
113
Treatment of 2 with MsOH in
CH
3
CN provides the target compound as a white crystalline solid
that is nonhygroscopic and stable.
CF
3
CH
2
I
CF
3
CH
2
I(O
2
CCF
3
)
2
CF
3
CH
2
I
OH
OSO
2
CH
3
(41)
CF
3
CO
2
H
0
°C, 24 h
–30
°C, 10–20 min
86%
2
3
CF
3
CO
3
H
MsOH, MeCN
Compound 3 can be reacted with silyl enol ethers to provide
mesyloxy products in the same manner that Koser’s reagent can
provide tosyloxy compounds. The stability of 3 as well as its
formation of volatile by-products (boiling point of CF
3
CH
2
I is
Avoid Skin Contact with All Reagents
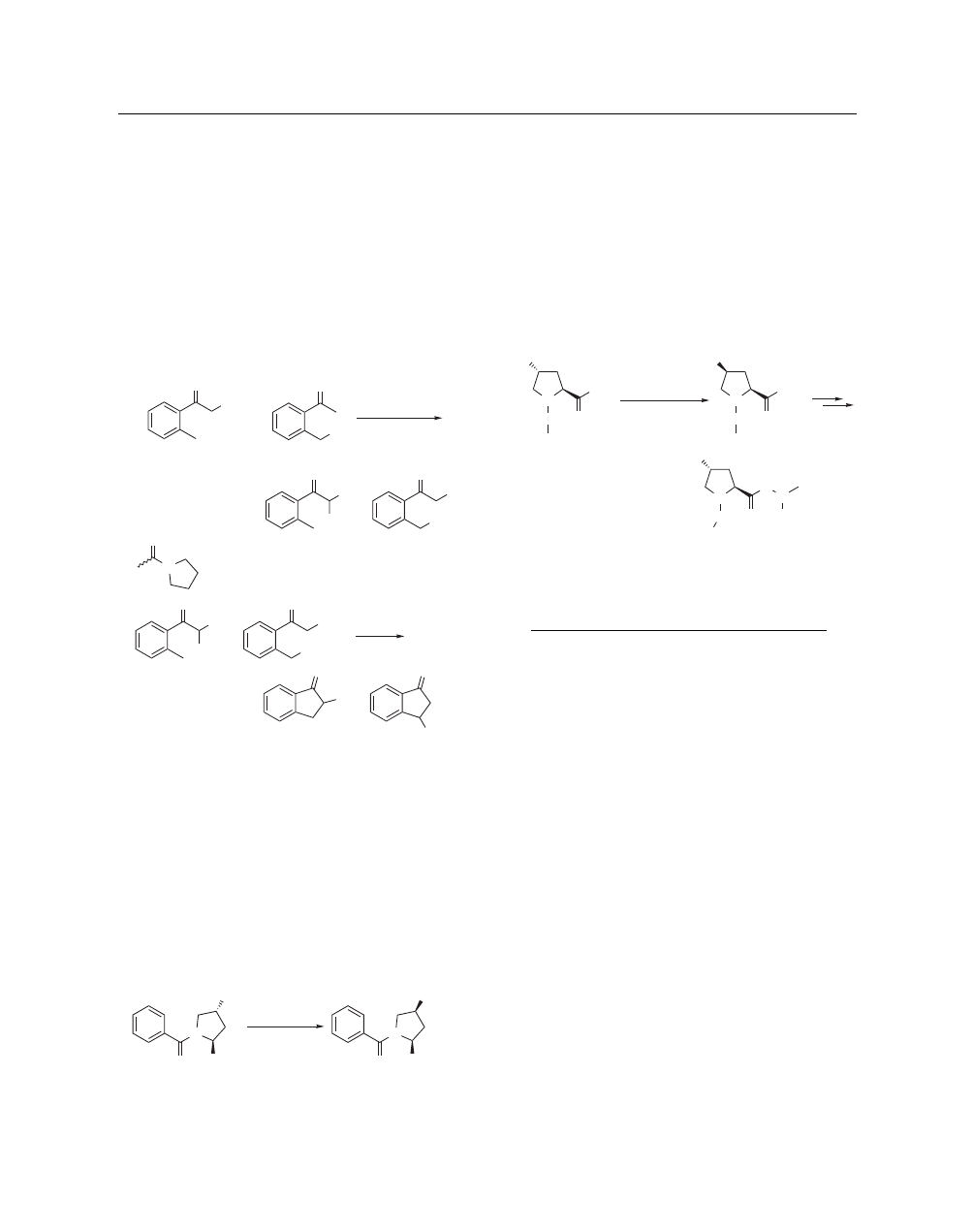
8
METHANESULFONIC ACID
55
◦
C) make it a readily available, easily prepared, and equally
reactive alternative to Koser’s reagent.
MsOH can also be used in the same manner that TsOH can
be used to prepare Koser’s reagent by treating a suspension of
iodosobenzene diacetate in CH
3
CN with the monohydrate of
methanesulfonic acid.
111,114,115
After recrystallization, hydroxy-
mesyloxyiodobenzene (PhI(OH)OMs, HMIB) can be isolated and
used in a manner similar to Koser’s reagent to install a mesyloxy
group in the alpha position of ketones. An example of this use can
be found in the synthesis of highly functionalized 1-indanones
to install a group that could be activated for radical-mediated
indanone ring closure.
116
With the mesyloxy group in place, treat-
ment with ultraviolet light brings about cyclization to the target
compounds (eq 42).
O
R
1
O
N
O
R
2
PhI(OH)OMs
O
R
1
OMs
O
R
2
OMs
O
R
1
OMs
O
R
2
OMs
O
R
1
O
R
2
(42)
R
1
= H, Me, Et, CO
2
Et,
or
R
2
= CN, CO
2
Me
or
or
hν
or
Methanesulfonic acid has also been used to install the
mesyloxy functional group with inversion of configuration via
the Mitsunobu protocol.
117,118
Using triphenylphosphine, diiso-
propyl azodicarboxylate (DIAD), triethylamine, and MsOH, the
standard Mitsunobu reaction takes place with nucleophilic attack
by the mesyloxy anion to give the mesyloxy compound with over-
all inversion of configuration. It was found that Et
3
N allowed for
smoother formation of the MsO
−
nucleophile thereby removing
the need to preform the zinc salt of methanesulfonic acid (eq 43).
Only 3% of the noninverted by-product was observed. The proto-
col could also be applied to installation of a tosyloxy group using
TsOH as well.
O
N
CO
2
Me
OH
O
N
CO
2
Me
OMs
(43)
CH
3
SO
3
H
Ph
3
P, DIAD
Et
3
N, PhCH
3
This protocol could potentially be applied to numerous sys-
tems where nucleophilic attack of an alcohol is desired but overall
retention of configuration is necessary. Performing this sequence
via double inversion with direct installation of the leaving group
in the first step allows for inversion and concern about the
hydrolysis of the intermediate ester in base sensitive substrates
is eliminated.
As with the typical Mitsunobu reaction, the sequence can
also be carried out using diethyl azodicarboxylate (DEAD) and
at lower temperatures although reaction times must be
increased.
119
–
122
In the synthesis of proline hydrazide endothelin
converting enzyme (ECE) inhibitors, Abei and Juillerat-Jeanneret
utilized this protocol to install a thiol moiety into their target com-
pounds (eq 44).
123
N
HO
O
OMe
SO
2
naphthyl
N
MsO
O
OMe
SO
2
naphthyl
N
HS
O
H
N
SO
2
naphthyl
N
SO
2
Tol
(44)
MsOH, Et
3
N
steps
PPh
3
, DEAD
toluene/THF 80
°C
Related Reagents.
Phosphorus(V) Oxide–Methanesulfonic
Acid; Polyphosphoric Acid; Trifluoroacetic Acid; Trifluorome-
thanesulfonic Acid.
1.
Ilczyszyn, M., J. Prakt. Chem. 1991, 95, 7621.
2.
Newman, M. S.; Davis, C. D., J. Org. Chem. 1967, 32, 66.
3.
Gassman, P. G.; Mickelson, J. W.; Sowa, Jr., J. R., J. Am. Chem. Soc.
1992
, 114, 6942.
4.
Eaton, P. E.; Carlson, G. R.; Lee, J. T., J. Org. Chem. 1973, 38, 4071.
5.
Matz, J. R.; Cohen, T., Tetrahedron Lett. 1981, 22, 2459.
6.
Zhao, D.; Hughes, D. L.; Bender, D. R.; De Marco, A. M.; Reider,
P. J., J. Org. Chem. 1991, 56, 3001.
7.
Premasagar, V.; Palaniswamy, V. A.; Eisenbraun, E., J. Org. Chem.
1981
, 46, 2974.
8.
Esser, F.; Pook, K.-H.; Carpy, A., Synthesis 1990, 72.
9.
(a) Silbert, L. S.; Siegel, E.; Swern, D., J. Org. Chem. 1962, 27, 1336.
(b) Silbert, L. S.; Siegel, E.; Swern, D., Org. Synth. 1963, 43, 93; Org.
Synth., Coll. Vol. 1973
, 5, 904.
10.
Andre, J.-D.; Dormoy, J.-R.; Heymes, A., Synth. Commun. 1992, 22,
2313.
11.
Georg, G. I.; Guan, X.; Kant, J., Bioorg. Med. Chem. Lett. 1991, 1, 125.
12.
Almansa, C.; Carceller, E.; Moyano, A.; Serratosa, F., Tetrahedron
1986
, 42, 3637.
13.
Hoppe, I.; Hoffmann, H.; Gärtner, I.; Krettek, T.; Hoppe, D., Synthesis
1991
, 1157.
14.
Imamoto, T.; Oshiki, T., Tetrahedron Lett. 1989, 30, 383.
15.
Loev, B.; Haas, M. A.; Dowalo, F., Chem. Ind. (London) 1968, 973.
16.
Loev, B., Chem. Ind. (London) 1964, 193.
17.
Lowe, O. G., J. Org. Chem. 1976, 41, 2061.
18.
Esser, F.; Pook, K.-H.; Carpy, A.; Leger, J. M., Synthesis 1994, 77.
19.
Kapoor, R.; Wadhawan, P.; Katyal, V.; Sood, V. R.; Kapoor, P., Can. J.
Chem. 1989
, 67, 1760.
A list of General Abbreviations appears on the front Endpapers

METHANESULFONIC ACID
9
20.
Kropp, P. J.; Breton, G. W.; Craig, S. L.; Crawford, S. D.; Durland, W.
F., Jr.; Jones, J. E.; Raleigh, J. S., J. Org. Chem. 1995, 60, 4146.
21.
Sharghi, H.; Sarvari, M. H., Tetrahedron Lett. 2003, 59, 3627.
22.
Hoffman, R. V.; Zhao, Z.; Costales, A.; Clarke, D., J. Org. Chem. 2002,
67
, 5284.
23.
Neustadt, B. R., Tetrahedron Lett. 1994, 35, 379.
24.
Ohno, H.; Hamaguchi, H.; Ohata, M.; Kosaka, S.; Tanaka, T., J. Am.
Chem. Soc. 2004
, 126, 8744.
25.
Yechezkel, T.; Ghera, E.; Ostercamp, D.; Hassner, A., J. Org. Chem.
1995
, 60, 5135.
26.
Pohmakotr, M.; Bunlaksananusorn, T.; Tuchinda, P., Tetrahedron Lett.
2000
, 41, 377.
27.
Chen, Y.; Evarts, J. B., Jr.; Torres, E.; Fuchs, P. L., Org. Lett. 2002, 4,
3571.
28.
Mouhtady, O.; Gaspard-Iloughmane, H.; Roquesb, N.; Le Rouxa, C.,
Tetrahedron Lett. 2003
, 44, 6379.
29.
Kaboudin, B., Tetrahedron Lett. 1999, 55, 12865.
30.
Lebedev, M. Y.; Erman, M. B., Tetrahedron Lett. 2002, 43, 1397.
31.
Van Emelen, K.; De Wit, T.; Hoornaert, G. J.; Compernolle, F., Org.
Lett. 2000
, 2, 3083.
32.
De Wit, T.; Van Emelen, K.; Maertens, F.; Hoornaert, G. J.;
Compernolle, F., Tetrahedron Lett. 2001, 42, 4919.
33.
Van Emelen, K.; De Wit, T.; Hoornaert, G. J.; Compernolle, F.,
Tetrahedron Lett. 2002
, 58, 4225.
34.
Ridvan, L.; Abdallah, N.; Holakovsky, R.; Tichy, M.; Závada, J.,
Tetrahedron: Asymmetry 1996
, 7, 231.
35.
Gálvez, N.; Moreno-Mañas, M.; Sebastih, R. M.; Vallribera, A.,
Tetrahedron 1996
, 52, 1609.
36.
Moreno-Mañas, M.; Trepat, E.; Sebastián, R. M.; Vallribera, A.,
Tetrahedron: Asymmetry 1999
, 10, 4211.
37.
Tanaka, M.; Oba, M.; Tamai, K.; Suemune, H., J. Org. Chem. 2001, 66,
2667.
38.
Tanaka, M.; Nishimura, S.; Oba, M.; Demizu, Y.; Kurihara, M.;
Suemune, H., Chem. Eur. J. 2003, 9, 3082.
39.
Lautens, M.; Roy, A., Org. Lett. 2000, 2, 555.
40.
Kise, N.; Oike, H.; Okazaki, E.; Yoshimoto, M.; Shonot, T., J. Org.
Chem. 1995
, 60, 3980.
41.
Kise, N.; Ueda, N., Tetrahedron Lett. 2001, 42, 2365.
42.
Annunziata, R.; Benaglia, M.; Caporale, M.; Raimondi, L.,
Tetrahedron: Asymmetry 2002
, 13, 2727.
43.
Kise, N.; Takaoka, S.; Yamauchi, M.; Ueda, N., Tetrahedron Lett. 2002,
43
, 7297.
44.
Tamamura, H.; Yamashita, M.; Nakajima, Y.; Sakano, K.; Otaka, A.;
Ohno, H.; Ibuka, T.; Fujii, N., J. Chem. Soc., Perkin Trans. 1 1999,
2983.
45.
Ohno, H.; Hamaguchi, H.; Tanaka, T., Org. Lett. 2001, 3, 2269.
46.
Ohno, H.; Ando, K.; Hamaguchi, H.; Takeoka, Y.; Tanaka, T., J. Am.
Chem. Soc. 2002
, 124, 15255.
47.
Aki, S.; Fujioka, T.; Ishigami, M.; Minamikawa, J., Bioorg. Med. Chem.
Lett. 2002
, 12, 2317.
48.
Corey, E. J.; Gin, D. Y., Tetrahedron Lett. 1996, 37, 7163.
49.
Tilly, D.; Samanta, S. S.; Faigl, F.; Mortier, J., Tetrahedron Lett. 2002,
43
, 8347.
50.
Jayaraman, M.; Fanwick, P. E.; Cushman, M., J. Org. Chem. 1998, 63,
5736.
51.
Sawyer, J. S.; Schmittling, E. A.; Palkowitz, J. A.; W. J., Smith, III, J.
Org. Chem. 1998
, 63, 6338.
52.
Ho, T.-L.; Yang, P.-F., Tetrahedron 1995, 51, 181.
53.
Kumar, S., Tetrahedron Lett. 1996, 37, 6271.
54.
Ho, T.-L.; Chen, C.-K., Tetrahedron 1995, 51, 5819.
55.
Vedejs, E.; Barda, D. A., Org. Lett. 2000, 2, 1033.
56.
Schultz, A. G.; Guzi, T. J.; Larsson, E.; Rahm, R.; Thakkar, K.; Bidlack,
J. M., J. Org. Chem. 1998, 63, 7795.
57.
Fukuyama, T.; Chen, X., J. Am. Chem. Soc. 1994, 116, 3125.
58.
Corey, E. J.; Lazerwith, S. E., J. Am. Chem. Soc. 1998, 120, 12777.
59.
Lazerwith, S. E.; Johnson, T. W.; Corey, E. J., Org. Lett. 2000, 2, 2389.
60.
Magnus, P.; Mitchell, I. S., Tetrahedron Lett. 1998, 39, 4595.
61.
Kim, D.-K.; Kim, Y.-W.; Gam, J.; Lim, J.; Kim, K. H., Tetrahedron
Lett. 1995
, 36, 6257.
62.
Wabnitz, T. C.; Spencer, J. B., Org. Lett. 2003, 5, 2141.
63.
Roth, E.; Altman, J.; Kapon, M.; Ben-Ishai, D., Tetrahedron 1995, 51,
801.
64.
Wang, M. W.; Chen, Y. J.; Wang, D., Heteroatom Chem. 2001, 12, 534.
65.
Kieczykowski, G. R.; Jobson, R. B.; Melillo, D. G.; Reinhold, D. F.;
Grenda, V. J.; Shinkai, I., J. Org. Chem. 1995, 60, 8310.
66.
Kume, M.; Ooka, H.; Ishitobi, H., Tetrahedron 1997, 53, 1635.
67.
Nicolaou, K. C.; Baran, P. S.; Zhong, Y.-L.; Choi, H.-S.; Yoon, W. H.;
He, Y.; Fong, K. C., Angew. Chem. Int. Ed. 1999, 38, 1669.
68.
Thompson, S. K.; Heathcock, C. H., J. Org. Chem. 1992, 57, 5979.
69.
Ram, V. J.; Neumeyer, J. L., J. Org. Chem. 1982, 47, 4372.
70.
Fargeas, V.; Le Ménez, P.; Berque, I.; Ardisson, J.; Pancrazi, A.,
Tetrahedron 1996
, 52, 6613.
71.
Kim, C.; Hoang, R.; Theodorakis, E. A., Org. Lett. 1999, 1, 1295.
72.
Segat-Dioury, F.; Lingibé, O.; Graffe, B.; Sacquet, M.-C.; Lhommet,
G., Tetrahedron 2000, 56, 233.
73.
Jiang, B.; Smallheer, J. M.; Amaral-Ly, C.; Wuonola, M. A., J. Org.
Chem. 1994
, 59, 6823.
74.
Orled, B. S.; Crowe, E. A., J. Chem. Soc., Perkin Trans. 1 1997, 2775.
75.
Ikemoto, T.; Nishiguchi, A.; Ito, T.; Tawada, H., Tetrahedron 2005, 61,
5043.
76.
Lim, J.; Kim, Y. H., J. Chem. Soc., Perkin Trans. 1 1999, 3239.
77.
Caballero, M.; Garcla-Valverde, M.; Pedrosa, R.; Vicente, M.,
Tetrahedron: Asymmetry 1996
, 7, 219.
78.
Kishi, A.; Higashino, T.; Sakaguchi, S.; Ishii, Y., Tetrahedron Lett.
2000
, 41, 99.
79.
Yokota, T.; Sakakura, A.; Tani, M.; Sakaguchi, S.; Ishii, Y., Tetrahedron
Lett. 2002
, 43, 8887.
80.
Li, W.; Zhang, Z.; Xiao, D.; Zhang, X., Tetrahedron Lett. 1999, 40,
6701.
81.
Martinez, M. M.; Hoppe, D., Org. Lett. 2004, 6, 3743.
82.
La Cruz, T. E.; Rychnovsky, S. D., Org. Lett. 2005, 7, 1873.
83.
Rybczynski, P. J.; Zeck, R.; Combs, E.; Turchi, D. W.; Burris, I.; Xu, T.
P.; Yangand, J. Z.; Demarest, M. K. T., Bioorg. Med. Chem. Lett. 2003,
13
, 2359.
84.
Knapp, S.; Morriello, G. J.; Doss, G. A., Org. Lett. 2002, 4, 603.
85.
Vedejs, E.; Klapars, A.; Warner, D. L.; Weiss, A. H., J. Org. Chem.
2001
, 66, 7542.
86.
Vedejs, E.; Little, J., J. Am. Chem. Soc. 2002, 124, 748.
87.
Vedejs, E.; Naidu, B. N.; Klapars, A.; Warner, D. L.; Li, V.; Na, Y.;
Kohn, H., J. Am. Chem. Soc. 2003, 125, 15796.
88.
Vedejs, E.; Little, J., J. Org. Chem. 2004, 69, 1794.
89.
Kim, M.; Vedejs, E., J. Org. Chem. 2004, 69, 7262.
90.
Furukubo, S.; Moriyama, N.; Onomura, O.; Matsumura, Y.,
Tetrahedron Lett. 2004
, 45, 8177.
91.
Valès, M.; Lokshin, V.; Pèpe, G.; Guglielmetti, R.; Samat, A.,
Tetrahedron 2002
, 58, 8543.
92.
Lin, L. S.; Lanza, T., Jr., ; de Laszlo, S. E.; Truong, Q.; Kamenecka, T.;
Hagmann, W. K., Tetrahedron Lett. 2000, 41, 7013.
93.
Kalinin, A. V.; Chauder, B. A.; Rakhitc, S.; Snieckus, V., Org. Lett.
2003
, 5, 3519.
94.
Leanna, M. R.; DeMattei, J. A.; Li, W.; Nichols, P. J.; Rasmussen, M.;
Morton, H. E., Org. Lett. 2000, 2, 3627.
Avoid Skin Contact with All Reagents

10
METHANESULFONIC ACID
95.
Park, C. S.; Kim, M. S.; Sim, T. B.; Pyun, D. K.; Lee, C. H.; Choi, D.;
Lee, W. K., J. Org. Chem. 2003, 68, 43.
96.
Sugiyama, S.; Arai, S.; Ishii, K., Tetrahedron: Asymmetry 2004, 15,
3149.
97.
Konda, Y.; Takahashi, Y.; Arima, S.; Sato, N.; Takeda, K.; Dobashi, K.;
Baba, M.; Harigaya, Y., Tetrahedron 2001, 57, 4311.
98.
Senokuchi, K.; Nakai, H.; Nakayama, Y.; Odagaki, Y.; Sakaki, K.;
Kato, M.; Maruyama, T.; Miyazaki, T.; Ito, H.; Kamiyasu, K.; Kim,
S.; Kawamura, M.; Hamanaka, N., J. Med. Chem. 1995, 38, 2521.
99.
Sugata, T.; Higuchi, R., Tetrahedron Lett. 1996, 37, 2613.
100.
Ando, H.; Koike, Y.; Ishida, H.; Kiso, M., Tetrahedron Lett. 2003, 44,
6883.
101.
Jaime-Figueroa, S.; Liu, Y.; Muchowski, J. M.; Putman, D. G.,
Tetrahedron Lett. 1998
, 39, 1313.
102.
Kleinfeld, S. H.; Wegelius, E.; Hoppe, D., Helv. Chim. Acta. 1999, 82,
2413.
103.
Papillon, J. P. N.; Taylor, R. J. K., Org. Lett. 2002, 4, 119.
104.
Christoph, G.; Hoppe, D., Org. Lett. 2002, 4, 2189.
105.
Seki, M.; Mori, Y.; Hatsuda, M.; Yamada, S., J. Org. Chem. 2002, 67,
5527.
106.
Seki, M.; Kimura, M.; Hatsuda, M.; Yoshida, S.; Shimizu, T.,
Tetrahedron Lett. 2003
, 44, 8905.
107.
Le Ménez, P.; Fargeas, V.; Berque, I.; Poisson, J.; Ardisson,
J.; Lallemand, J.-Y.; Pancrazi, A., J. Org. Chem. 1995, 60, 3592.
108.
Engelhardt, U.; Sarkar, A.; Linker, T., Angew. Chem. Int. Ed. 2003, 42,
2487.
109.
Lee, J. C.; Choi, Y., Tetrahedron Lett. 1998, 39, 3171.
110.
Koser, G. F.; Relenyi, A. G.; Kalos, A. N.; Rebrovic, L.; Wettach, R.
H., J. Org. Chem. 1982, 47, 2487.
111.
Lodaya, J. S.; Koser, G. F., J. Org. Chem. 1988, 52, 210.
112.
Moriarty, R. M.; Vaid, R. K.; Koser, G. F., Synlett 1990, 365.
113.
Zhdankin, V. V.; Kuehl, C. J.; Simonsen, A. J., Tetrahedron Lett. 1995,
36
, 2203.
114.
Rebrovic, L.; Koser, G. F., J. Org. Chem. 1984, 49, 4700.
115.
Stang, P. J.; Surber, B. W.; Chen, Z.-C.; Roberts, K. A.; Anderson, A.
G., J. Am. Chem. Soc. 1987, 109, 228.
116.
Wessig, P.; Glombitza, C.; Müller, G.; Teubner, J., J. Org. Chem. 2004,
69
, 7582.
117.
Anderson, N. G.; Lust, D. A.; Colapret, K. A.; Simpson, J. H.; Malley,
M. F.; Gougoutas, J. Z., J. Org. Chem. 1996, 61, 7955.
118.
Zhao, Y.; Zhong, Z., J. Am. Chem. Soc. 2005, 127, 17894.
119.
Davis, A. P.; Dresen, S.; Lawless, L. J., Tetrahedron Lett. 1997, 38,
4305.
120.
Ayling, A. J.; Pérez-Payán, M. N.; Davis, A. P., J. Am. Chem. Soc. 2001,
123
, 12716.
121.
Ayling, A. J.; Broderick, S.; Clare, J. P.; Davis, A. P.; Pérez-Payán, M.
N.; Lahtinen, M.; Nissinen, M. J.; Rissanen, K., Chem. Eur. J. 2002, 8,
2197.
122.
Aher, N. G.; Pore, V. S., Synlett 2005, 2155.
123.
Berger, Y.; Dehmlow, H.; Blum-Kaelin, D.; Kitas, E. A.; Löffler, B.-M.;
Aebi, J. D.; Juillerat-Jeanneret, L., J. Med. Chem. 2005, 48, 428.
A list of General Abbreviations appears on the front Endpapers
Wyszukiwarka
Podobne podstrony:
hydrobromic acid eros rh031
peracetic acid eros rp034
p toluenesulfonic acid eros rt134
glyoxylic acid eros rg009
formic acid eros rf025
hypophosphorous acid eros rh075
peroxymaleic acid eros rp041
phosphoric acid eros rp153
palladium triethylamine formic acid eros rp015
propionic acid eros rp272
zinc acetic acid eros rz002
monoperoxysulfuric acid eros rm288m
nitric acid eros rn022
boric acid eros rb242
hydrobromic acid eros rh031
peracetic acid eros rp034
p toluenesulfonic acid eros rt134
więcej podobnych podstron