
Preparaty i protokoły z chemii organicznej
1-fenyloazo-2-naftol
N
+
N
O
H
N
N
O
H
Cl
+
NaOH
W małej zlewce lub kolbie stożkowej rozpuścić 5 g (4,9 ml, 0,054 mol) aniliny w mieszaninie
16 ml stężonego kwasu solnego i 16 ml wody. Ochłodzony roztwór aminy do temp. poniżej
5
o
C (łaźnia lodowa) poddać diazowaniu dodając roztwór 4 g (0,058 mol) azotanu(III) sodu w
20 ml wody. W zlewce o pojemności 250 ml przygotować roztwór 7,8 g (0,054 mol) 2-
naftolu w 45 ml 10% roztworu NaOH i ochłodzić go do temp. 5
o
C, zanurzając zlewkę w łaźni
z lodem i jednocześnie dodając do zlewki 25g pokruszonego lodu. Do energicznie
mieszanego roztworu 2-naftolu dodawać bardzo powoli zimny roztwór soli diazoniowej;
mieszanina reakcyjna barwi się na czerwono i wydziela się czerwony osad 1-fenyloazo-2-
naftolu. Po dodaniu całego roztworu soli diazoniowej, mieszaninę pozostawić na 30 min w
łaźni lodowej, mieszając od czasu do czasu, a następnie przesączyć przez lejek Büchnera.
Osad pozostały na sączku przemyć dokładnie wodą i starannie odcisnąć płaską końcówką
szklanego korka.
Acetanilid
NH
2
+ H
3
C
O
O
O
CH
3
HN
O
CH
3
+ CH
3
COOH
W kolbie kulistej o pojemności 250 ml zaopatrzonej w chłodnicę zwrotną, umieszcza się 10
ml aniliny, 10 ml bezwodnika octowego, 10 ml lodowatego kwasu octowego i 0,1 g pyłu
cynkowego. Mieszaninę ogrzewa się łagodnie do wrzenia w ciągu 30 minut, a następnie
gorącą ciecz, mieszając wylewa cienkim strumieniem do zlewki zawierającej 250 ml wody z
lodem. Po oziębieniu w lodzie, surowy produkt odsącza się, przemywa niewielką ilością
zimnej wody i suszy się na powietrzu. Surowy produkt krystalizuje się z 250 ml wody z
dodatkiem 5 ml etanolu. T
t
= 113-114
o
C
Aspiryna (kwas acetylosalicylowy)
COOH
OH
+ (CH
3
CO)
2
O
COOH
OCOCH
3
+ CH
3
COOH
W kolbie stożkowej umieszcza się 5 g kwasu salicylowego, 7 ml bezwodnika kwasu
octowego i 3 krople stężonego kwasu siarkowego, mieszając przy tym starannie zawartość
kolby. Mieszaninę ogrzewa się na łaźni wodnej do 50-60
o
C w ciągu 15 minut. Mieszaninę
pozostawia się do ostygnięcia, wstrząsając co pewien czas dodaje się 70 ml wody, starannie
miesza i sączy pod zmniejszonym ciśnieniem. Uzyskany osad rozpuszcza się na gorąco w 20
ml etanolu. Roztwór wylewa się do 40ml gorącej wody i pozostawia do powolnego
schłodzenia. Produkt odsącza się pod zmniejszonym ciśnieniem. T
t
= 136-137
o
C
Diazoaminobenzen
N
2
Cl
+
NH
2
N
N
H
N
+ HCl
W kolbie o pojemności 100 ml umieszcza się 25 ml wody, 6,7 ml stężonego kwasu solnego i
4,6 ml aniliny. Kolbę wstrząsa się energicznie i dodaje 17g potłuczonego lodu. Następnie w
ciągu 5-10 minut, wytrząsając dodaje się roztwór wodny azotanu (III) sodu ( 1,75 g azotanu
w 4 ml wody). Mieszaninę reakcyjną pozostawia się na 15 minut, dość często wytrząsając, a
następnie w ciągu 5 minut dodaje się roztwór 7 g krystalicznego octanu sodu w 13 ml wody.
Natychmiast zaczyna wypadać żółty osad diazoaminobenzenu. Mieszaninę pozostawia się na
dalsze 45 minut, często wstrząsając i utrzymując temperaturę poniżej 20
o
C (w razie
konieczności dodaje się lodu). Żółty diazoaminobenzen odsącza się na lejku sitowym,
przemywa ok. 80 ml zimnej wody, osad odciska możliwie jak najdokładniej i suszy na bibule.
T
t
= 90-91
o
C.
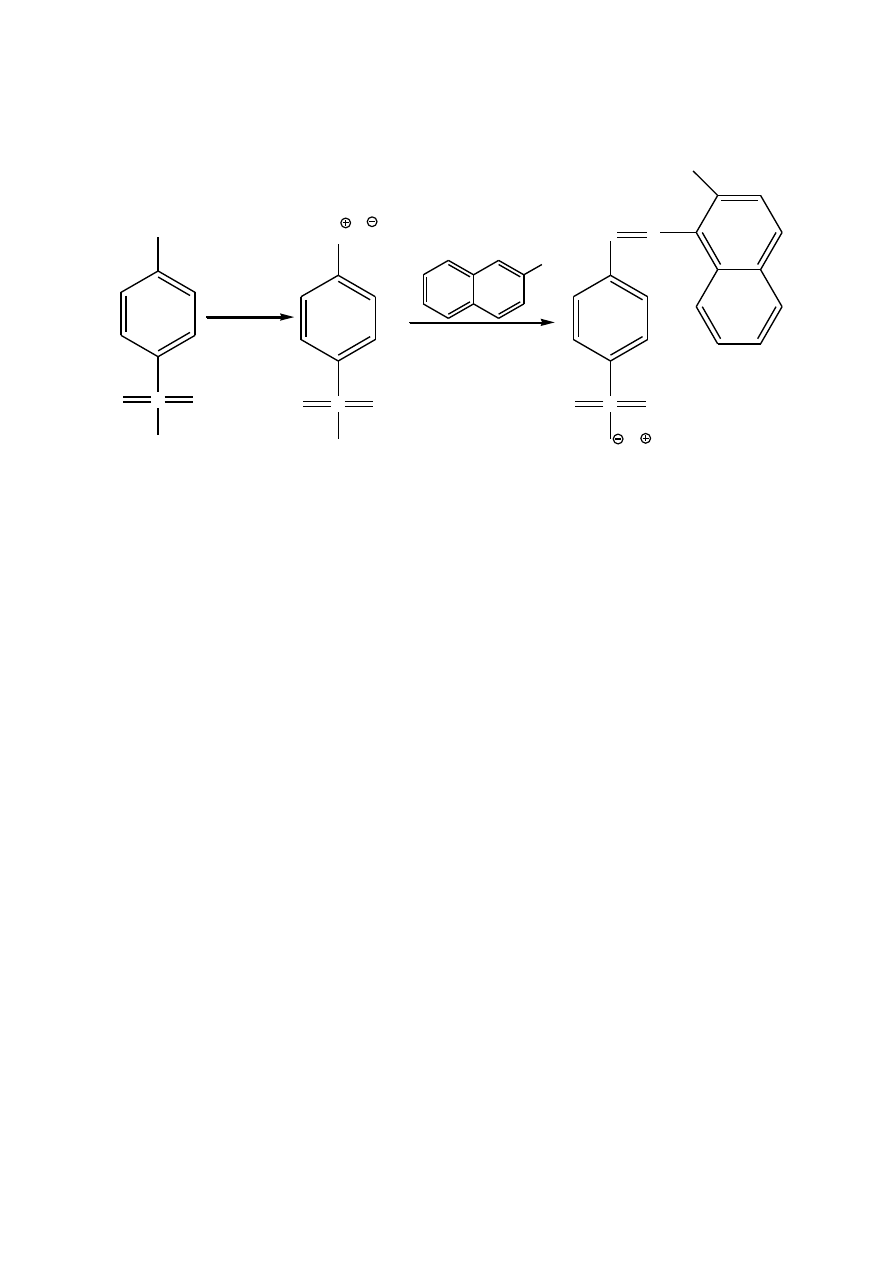
Oranż
-naftolowy
NH
2
S
OH
O
O
NaNO
2
HCl
N
2
S
OH
O
O
Cl
OH
NaOH
N
S
O Na
O
O
N
HO
2,5 g kwasu sulfanilowego rozpuszcza się w 6 ml 2 molowego roztworu NaOH, po czym
dolewa się roztwór 1g azotanu (III) sodu w 12 ml wody. Otrzymany roztwór wlewa się z
kolei, oziębiając lodem do 13 ml 4 molowego HCl. Papkowatą zawiesinę wlewa się dość
szybko, mieszając do alkalicznego roztworu
-naftolu (2 g w 25 ml 2 molowego NaOH) w
temperaturze pokojowej. Rozpoczynającą się krystalizacje barwnika (w postaci
pomarańczowych płatków) przyśpiesza się przez chłodzenie mieszaniny w łaźni lodowej.
Otrzymane kryształy odsącza się i przemywa zimną wodą.
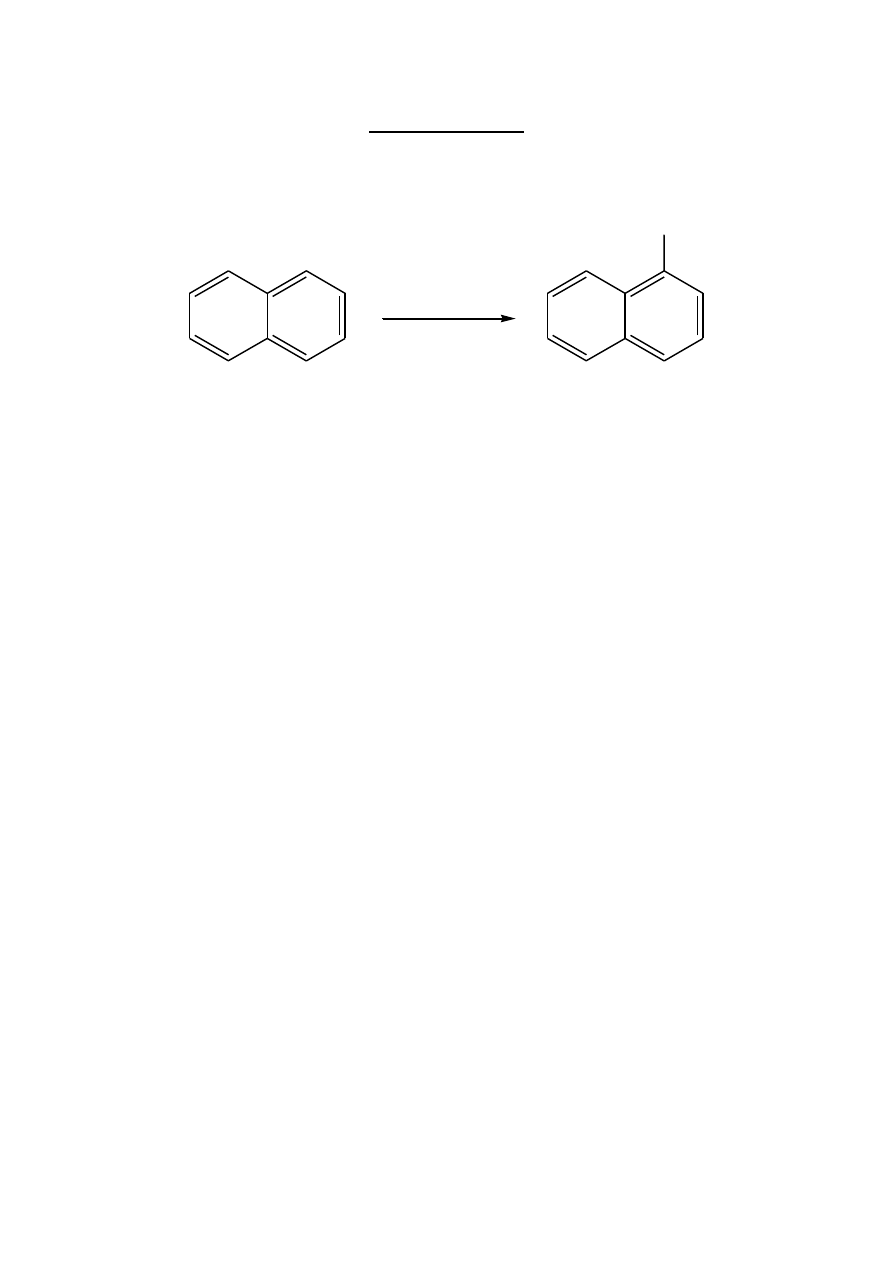
1-nitronaftalen
HNO
3
H
2
SO
4
NO
2
Do 1,85 ml stężonego HNO
3
umieszczonego w kolbie stożkowej
umieszcza się 2,85 ml stężonego H
2
SO
4
, mieszając i chłodząc zawartość kolby.
Następnie dodaje się ciągle mieszając 3,2 g sproszkowanego naftalenu, tak aby
temperatura nie przekroczyła 45
o
C. Mieszaninę ogrzewa się 1 godzinę w 60
o
C
i ciepłą wylewa do 25 ml zimnej wody. Surowy produkt gotuje się kilkakrotnie z
wodą (do zaniku odczynu kwaśnego) i stopiony nitronaftalen wlewa się cienkim
strumieniem do 50 ml wody. Po krystalizacji z etanolu otrzymuje się 1-
nitronaftalen o T
topn.
= 60-61
o
C.

Chlorek t-butylu
CH
3
C
H
3
C
CH
3
OH
+ HCl
CH
3
C
H
3
C
CH
3
Cl
+ H
2
O
W rozdzielaczu umieszcza się 25g (0,34 mola, 32 ml) alkoholu t-
butylowego, 85 ml stężonego HCl i 10 g bezwodnego CaCl
2
(zwiększa on
gęstość warstwy kwasowej, co ułatwia rozdzielenie warstw i polepsza nieco
wydajność produktu). Mieszaninę wytrząsa się co pewien czas w ciągu 20 min.
Po każdym wytrząśnięciu wyrównuje się ciśnienie w rozdzielaczu. Mieszaninę
pozostawia się na kilka minut do wyraźnego rozdzielenia się warstw, po czym
dolną warstwę kwasu spuszcza się i odrzuca. Górną warstwę chlorku t-butylu
przemywa się kolejno 20 ml 5% roztworu wodorowęglanu sodu oraz 20 ml
wody i suszy bezwodnym chlorkiem wapnia. Po odsączeniu środka suszącego
produkt destyluje się zbierając frakcję wrzącą w temp. 49-51
o
C.
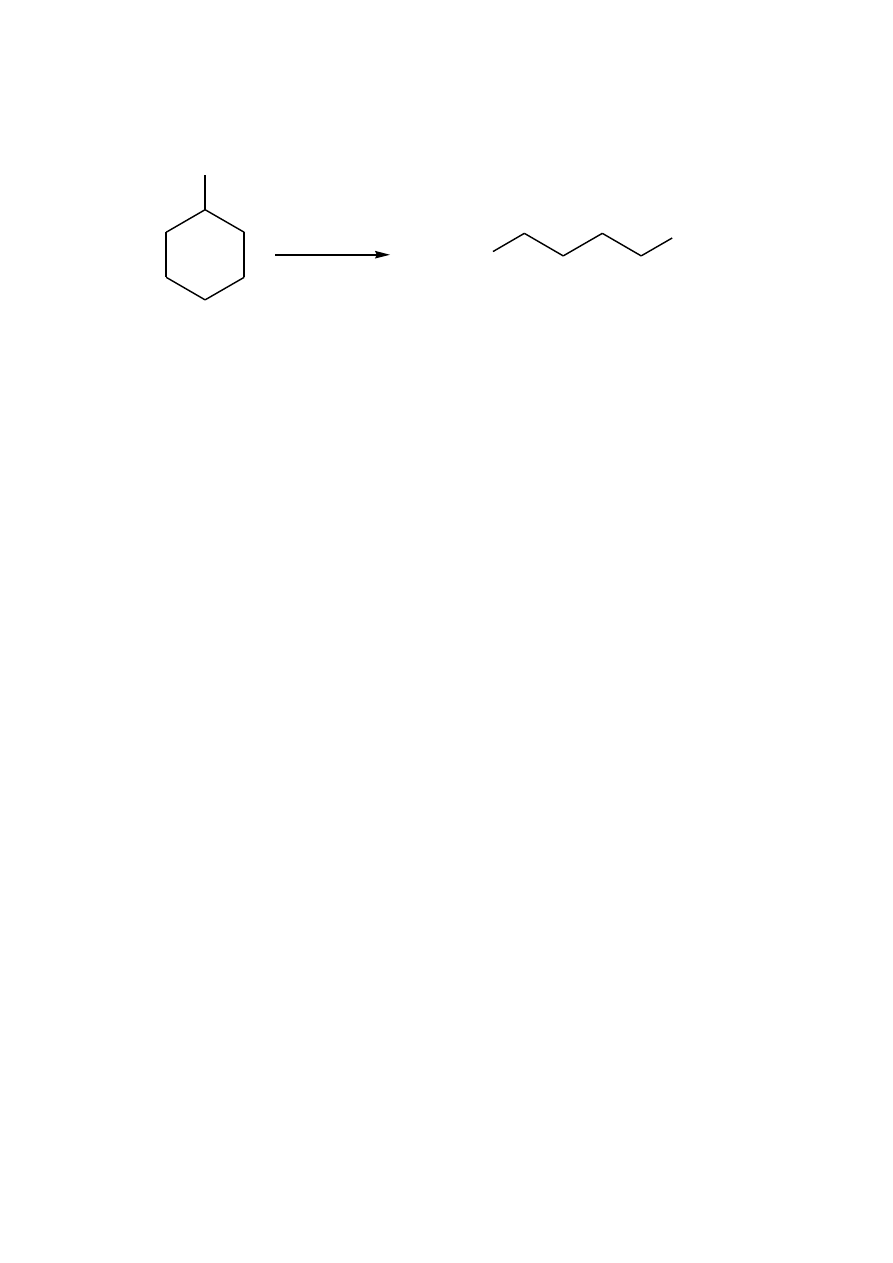
Kwas adypinowy
OH
HNO
3
HOOC
COOH
UWAGA! Reakcję prowadzi się pod wyciągiem!
W kolbie dwuszyjnej zaopatrzonej w chłodnicę i wkraplacz ogrzewa się do wrzenia 19 ml
stężonego kwasu azotowego(V). Po usunięciu płaszcza grzejnego dodaje się ostrożnie z
wkraplacza 5 g cykloheksanolu z taką szybkością, aby zawartość kolby lekko wrzała. Po
zakończeniu wkraplania mieszaninę ogrzewa się do wrzenia w ciągu 15 minut i ciepłą
przelewa do zlewki. Po oziębieniu zlewki w łaźni lodowej wykrystalizowany kwas
adypinowy sączy się przez lejek ze szkła porowatego, przemywa niewielką ilością zimnej
wody. Produkt krystalizuje się z minimalnej ilości wrzącej wody. T
t
= 151-152
o
C
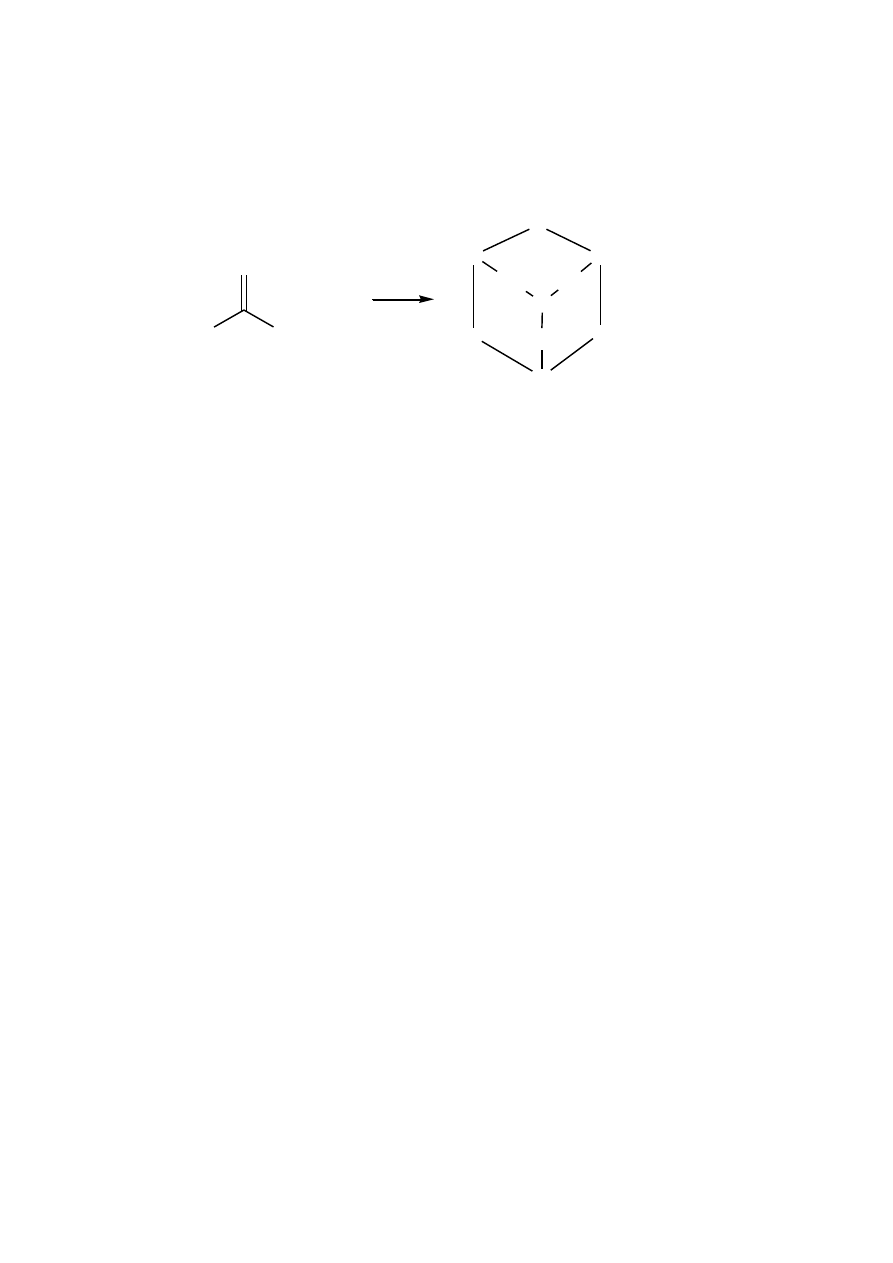
Urotropina (heksametylenotetramina)
H
H
O
+ 4 NH
3
N
H
2
C
N
CH
2
N
H
2
C
CH
2
N
H
2
C
H
2
C
+ 6 H
2
O
6
W kolbie stożkowej umieszcza się 12,5 ml 40% roztworu formaldehydu, oziębia się w wodzie
z lodem i mieszając wkrapla się 11 ml 25% amoniaku, tak regulując szybkość wkraplania,
aby temperatura mieszaniny reakcyjnej nie przekroczyła 15
o
C. Uzyskany roztwór przenosi się
do parowniczki i odparowuje na łaźni wodnej. T
t
= 260
o
C (subl.)
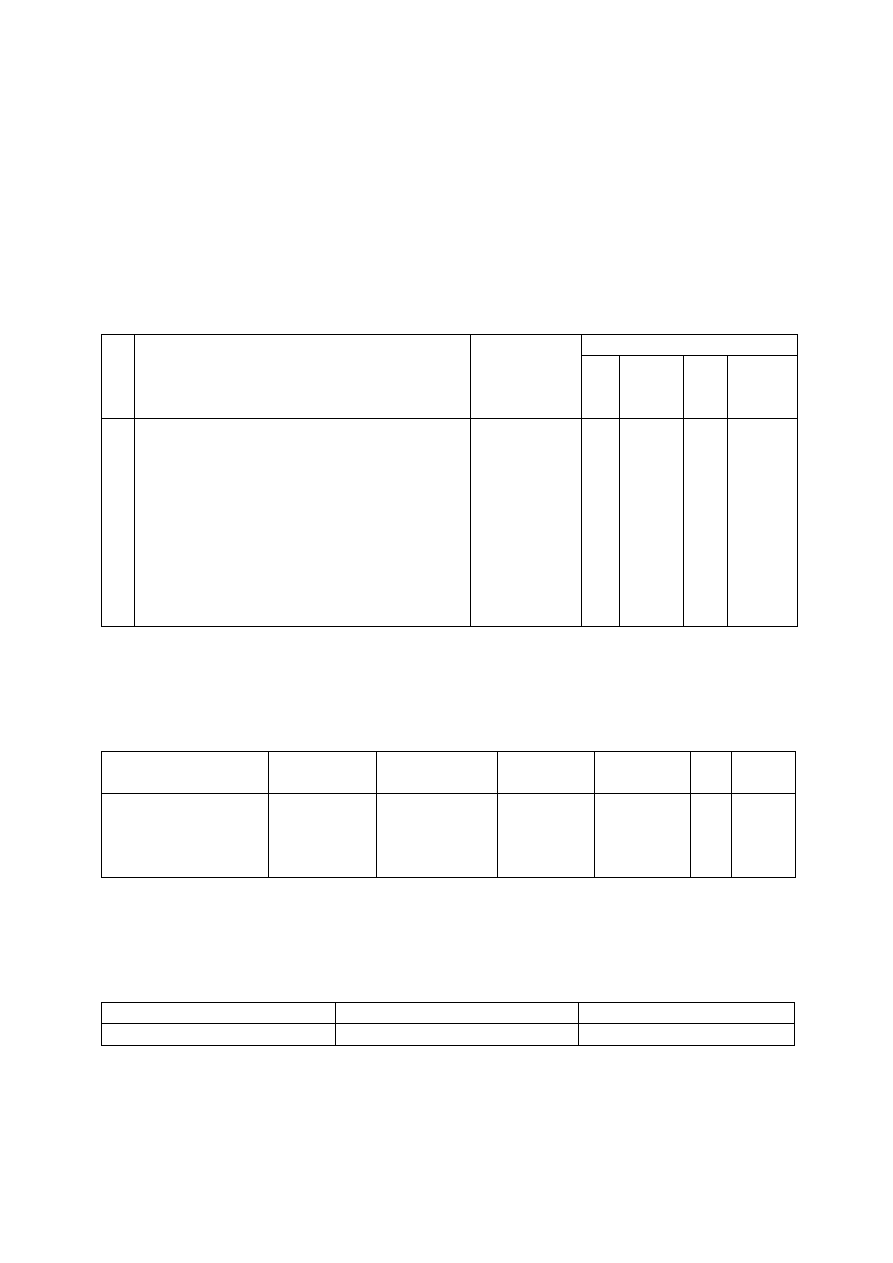
Protokół z wykonania preparatu:.........................................................
1. Schemat reakcji
2. Substraty i substancje pomocnicze
l.p.
nazwa
ciężar
cząsteczkowy
ilość
g
ciężar
wł.
[g/cm
3
]
cm
3
moli
1.
2.
.
.
3. Aparatura
4. Stałe fizykochemiczne produktu wg literatury
związek
wzór
sumaryczny
ciężar
cząsteczkowy
temperatura
topnienia
temperatura
wrzenia
n
D
20
d
[g/cm
3
]
5. Charakterystyka otrzymanego produktu
W wyniku reakcji otrzymano ...........ml cieczy / ...........g ciała stałego o następujących
parametrach fizykochemicznych:
temp. topnienia
temp. wrzenia
n
D
20
6. Obliczenie wydajności reakcji ............................................................

7. Część doświadczalna
8. Wniosek:
Otrzymałem/am ........................................... w łącznej ilości ..........ml(..........g), co
stanowi....................% wydajności teoretycznej.
Oczyszczanie substancji w wyniku sublimacji
1. Nazwa oczyszczanego związku: ...................................................................................
wzór strukturalny
masa molowa M (g/mol) ..................
2. Aparatura:
3. Opis wykonania ćwiczenia
4. Właściwości oczyszczanej substancji:
Przed oczyszczeniem Po oczyszczeniu
Według literatury
Barwa
Masa (g)
Czas sublimacji
(min.)
t.t. (
o
C)

5. Obliczenie wydajności
6. Wnioski
7. Uwagi prowadzącego ..............................................

Oczyszczanie substancji w wyniku ekstrakcji
7. Substancja użyta do ekstrakcji: ...................................................................................
8. Rodzaj ekstrakcji: a) periodyczna b) ciągła
9. Ekstrakcja: a) ciała stałego b) cieczy
10. Nazwa rozpuszczalnika do ekstrakcji ......................................................................
wzór strukturalny
masa molowa M (g/mol) ..................................................
temperatura wrzenia (
o
C)..................................................
gęstość d (g/ml).................................................................
współczynnik załamania światła n
D
..................................
11. Aparatura:
12. Opis wykonania ćwiczenia

13. Właściwości oczyszczanej substancji:
Przed oczyszczeniem Po oczyszczeniu
Według literatury
Barwa
Masa (g)
(objętość (ml))
Ilość użytego
rozpuszczalnika (ml)
14. Obliczenie wydajności
15. Wnioski
8. Uwagi prowadzącego ..............................................

Oczyszczanie substancji w wyniku krystalizacji
16. Nazwa oczyszczanego związku: ...................................................................................
wzór strukturalny
masa molowa M (g/mol) ..................
17. Nazwa rozpuszczalnika do krystalizacji ......................................................................
wzór strukturalny
masa molowa M (g/mol) ..................................................
temperatura wrzenia (
o
C)..................................................
gęstość d (g/ml).................................................................
współczynnik załamania światła n
D
..................................
18. Aparatura:
19. Opis wykonania ćwiczenia

20. Właściwości oczyszczanej substancji:
Przed oczyszczeniem Po oczyszczeniu
Według literatury
Barwa
Masa (g)
Ilość użytego
rozpuszczalnika (ml)
t.t. (
o
C)
21. Obliczenie wydajności
22. Wnioski
8. Uwagi prowadzącego ..............................................

Oczyszczanie substancji w wyniku destylacji
23. Nazwa oczyszczanego związku: ...................................................................................
wzór strukturalny
masa molowa M (g/mol) ..................
temperatura wrzenia (
o
C) ..................
gęstość d (g/ml) .................................
współczynnik załamania światła n
D
.....................
24. Aparatura:
25. Opis wykonania ćwiczenia

26. Właściwości oczyszczanej substancji:
Przed oczyszczeniem
Po oczyszczeniu
Barwa
Masa (g)
Przedgon
Fr. właściwa
Pogon
Objętość (ml)
Przedgon
Fr. właściwa
Pogon
T. wrz. (
o
C)
Przedgon
Fr. właściwa
Pogon
Gęstość d (g/ml)
Fr. właściwa
Współczynnik
załamania światła
Fr. właściwa
27. Obliczenie wydajności
Przedgon
Frakcja właściwa
Pogon
28. Wnioski
8. Uwagi prowadzącego ..............................................

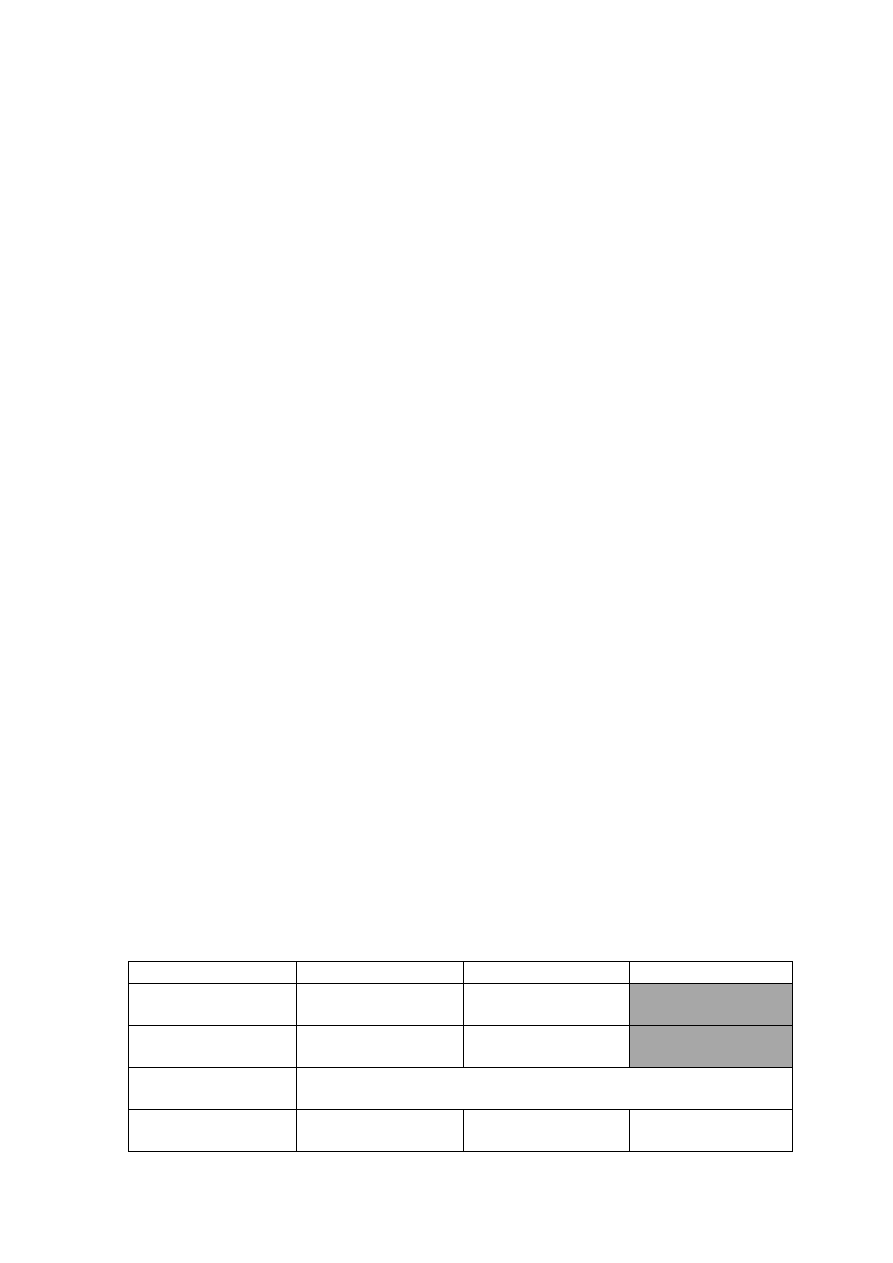
Organizacja pracowni z chemii organicznej na kierunku Biotechnologia
Pracownia 3
Pracownia 4
Pracownia 5
Pracownia 7
Pracownia 8
Pracownia 9
Pracownia 10
1.
Krystalizacja
Destylacja prosta
Ekstrakcja
Kwas sulfanilowy
Oranż
-
naftolowy
Cyklopentanon
Urotropina
2.
Destylacja prosta
Krystalizacja
Ekstrakcja
Chlorek t-butylu
Cyklopentanon
Oranż
-
naftolowy
Acetanilid
3.
Destylacja
frakcyjna
Ekstrakcja
Krystalizacja
Aspiryna
Kwas adypinowy
Acetanilid
chlorek t-
butylu
4.
Sublimacja
Krystalizacja
Destylacja prosta
1-fenyloazo-2-
naftol
Acetanilid
p-
nitroacetanilid
1-
nitronaftalen
5.
Ekstrakcja
Destylacja
frakcyjna
Krystalizacja
Cyklopentanon
kwas sulfanilowy
1-fenyloazo-2-
naftol
Aspiryna
6.
Krystalizacja
Sublimacja
Destylacja
frakcyjna
Acetanilid
p-nitroacetanilid
Urotropina
Oranż
-
naftolowy
7.
Destylacja prosta
Ekstrakcja
Krystalizacja
Diazoamino-
benzen
1-fenylo-3-
metylopirazol-
5-on
Aspiryna
Kwas
sulfanilowy
8.
Ekstrakcja
Krystalizacja
Destylacja prosta
1-fenylo-3-
metylopirazol-
5-on
Aspiryna
Chlorek t-
butylu
Diazoamino-
benzen
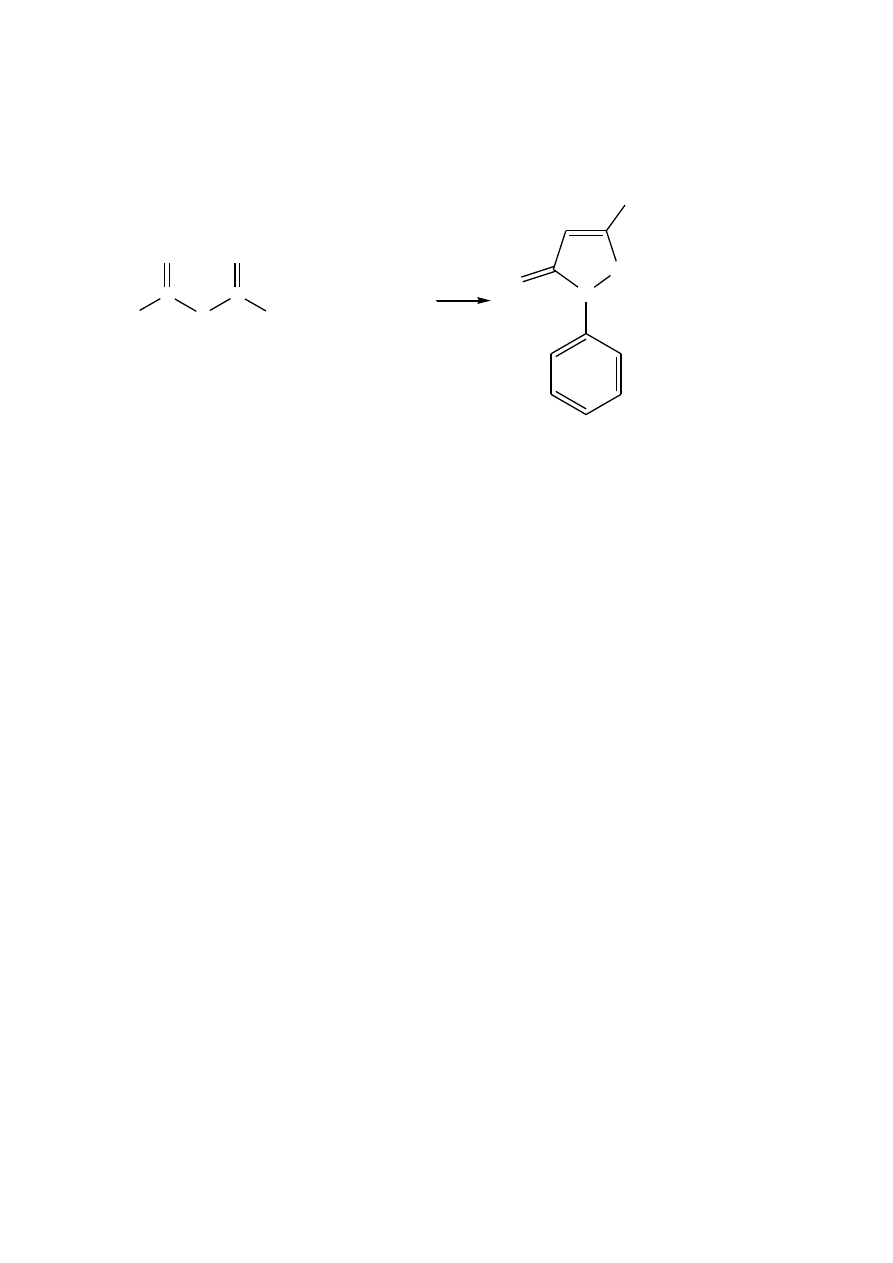
1-fenylo-3-metylopirazol-5-on
H
3
C
C
C
H
2
C
OC
2
H
5
O
O
+ NH
2
NHC
6
H
5
N
NH
CH
3
O
+ H
2
O + C
2
H
5
OH
Do kolby dwuszyjnej zaopatrzonej w chłodnicę zwrotną, wkraplacz i mieszadło magnetyczne
wprowadza się 10,8 g (9,8 ml, 0,1 mol) fenylohydrazyny. Następnie powoli mieszając,
wkrapla się 13 g (12,8 ml, 0,1 mol) acetylooctanu etylu z dodatkiem 1,3 g (1,65 ml) etanolu.
Wkraplanie należy przeprowadzić z taką szybkością, aby temperatura w kolbie nie podniosła
się wyraźnie. Po zakończeniu wkraplania zawartość kolby miesza się jeszcze 20 minut, a
następnie ogrzewa w temperaturze wrzenia przez ok. 2 godziny. Po ochłodzeniu mieszaninę
przelewa się do zlewki i pozostawia do krystalizacji. Surowy produkt odsącza się na lejku
sitowym, przemywa etanolem i krystalizuje z 50% etanolu z dodatkiem węgla aktywnego.
T
t
= 126-127
o
C.
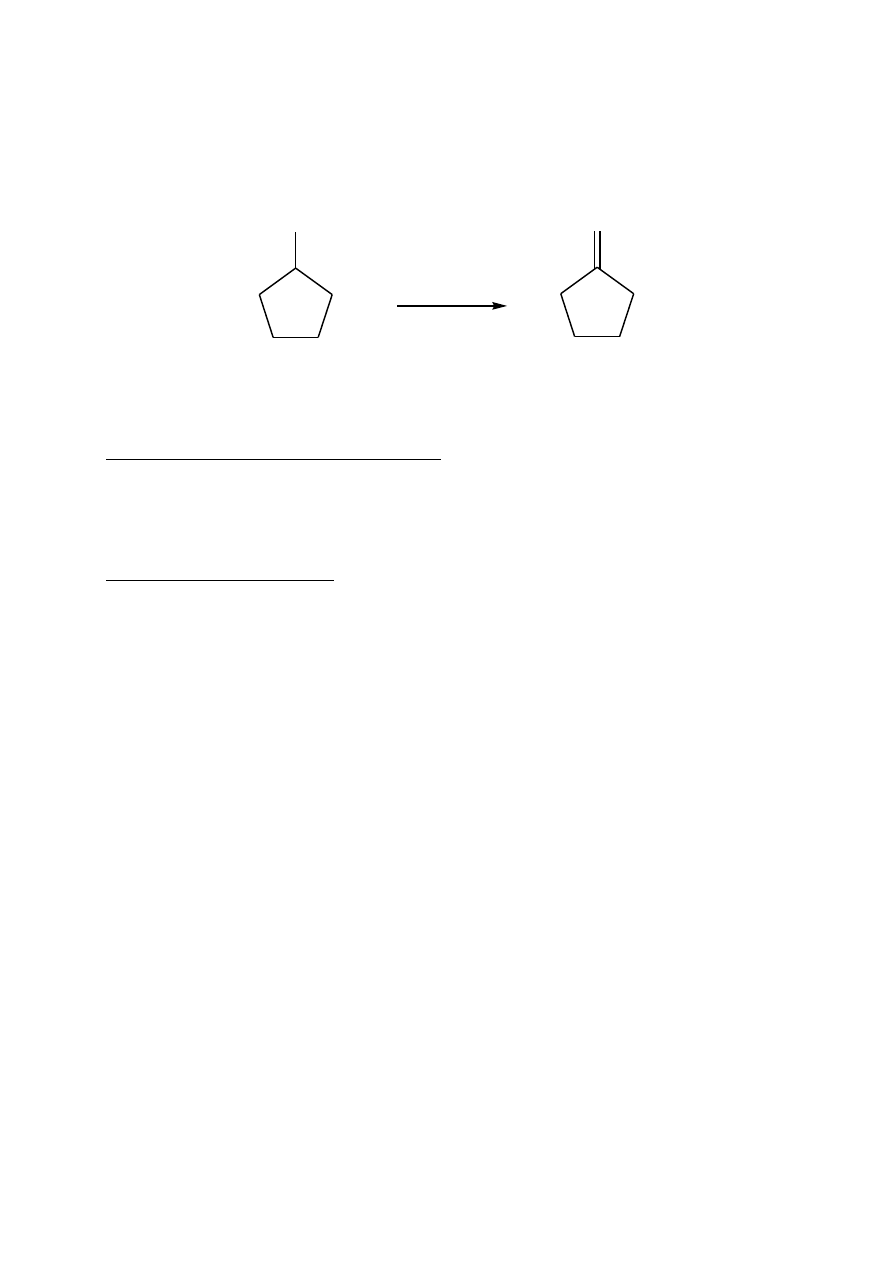
Cyklopentanon
OH
O
K
2
Cr
2
O
7
Przygotowanie mieszaniny utleniającej:
50,3g K
2
Cr
2
O
7
rozpuścić w 250 ml wody destylowanej i dodać mieszając
24 ml stężonego kwasu siarkowego (VI) a następnie ostudzić do temperatury
pokojowej.
Utlenienie cyklopentanolu:
W kolbie stożkowej o pojemności 500 ml umieścić 25 g cyklopentanolu, a następnie
dodawać małymi porcjami mieszaninę utleniającą. Temperatura mieszaniny reakcyjnej nie
powinna przekroczyć 60
o
C. Po zakończeniu wkraplania zawartość kolby mieszać przez 1
godzinę, a następnie całość rozcieńczyć 250 ml wody i destylować z kolby o poj. 1000 ml.
Oddestylować ok. 70-80 ml, wysalać NaCl, rozdzielić i wysuszyć bezwodnym MgSO
4
. Po
wysuszeniu oddestylować czysty produkt (Twrz. = 127-131
o
C, n
D
20
=1,437)
Wyszukiwarka
Podobne podstrony:
Rozkład ćwiczeń z chemii organicznej dla studentów I roku biotechnologii, chemia, organiczna
Synteza octanu n-butylu, Biotechnologia PWR, Semestr 3, Podstawy chemii organicznej - Laboratorium (
PROGRAM CWICZEN Z CHEMII ORGANICZNEJ BIOLOGIA 2010 2011
I POPRAWKA EGZAMINU Z CHEMII ORGANICZNEJ, Technologia chemiczna, Chemia organiczna, 4 semestr, organ
aminy otrzymywanie, podstawy chemii organicznej
11 Podstawy chemii organicznej Profesor Boduszek
Wprowadzenie do chemii organicznej
POPRAWKA EGZAMINU Z CHEMII ORGANICZNEJ
ROZKŁAD MATERIAŁU Z CHEMII ORGANICZNEJ
Pytanaia z chemii organicznej 13
Chamia organiczna elementy chemii organicznej
REPETYTORIUM DOTYCHCZASOWYCH WYKŁADÓW Z CHEMII ORGANICZNEJ
PODSTAWY CHEMII ORGANICZNEJ
Egzamin z chemii organicznej (2012)
więcej podobnych podstron