Immunogenetyka
cz. II
Joanna Ruszczyk
gr. 45

Celem
układu immunologicznego jest
rozpoznawanie
i
wyeliminowanie
drobnoustrojów
odpowiedzialnych
za
wywołanie infekcji.
Funkcje układu immunologicznego
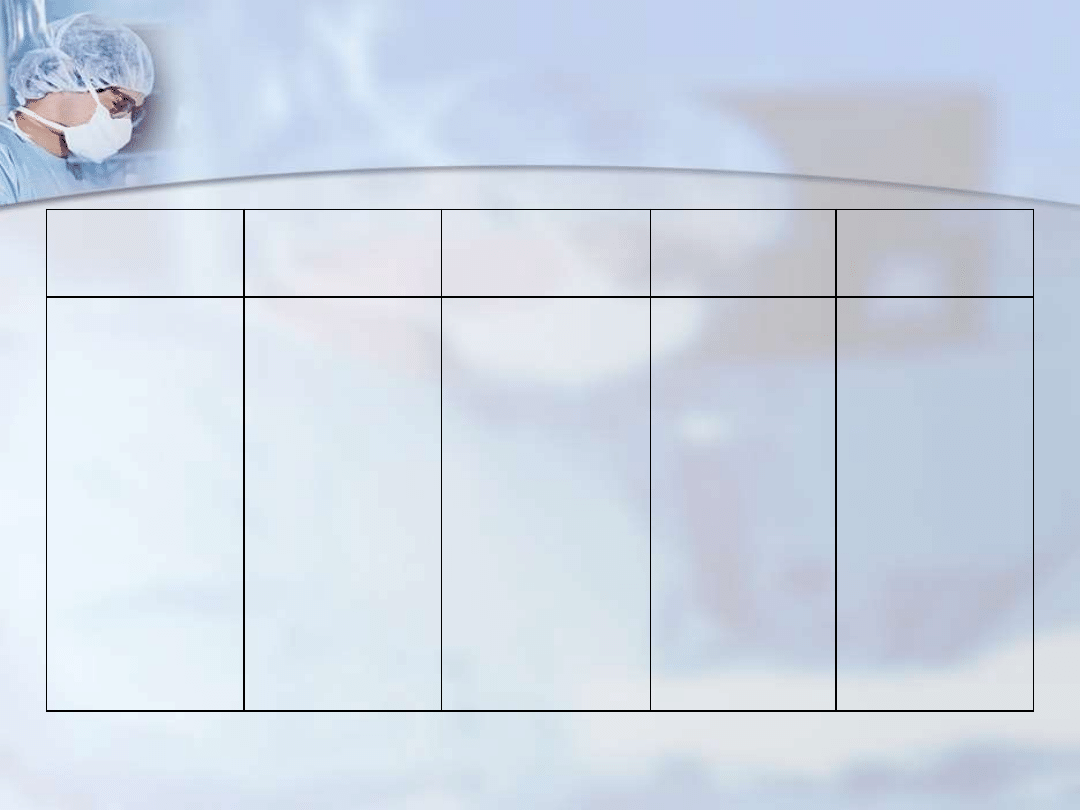
Rodzaj odporności Anygeny
Komórki
Czynniki
humoralne
Pamięć
immunologiczna
Nieswoista
(wrodzona)
Ograniczona liczba
wzorców
molekularnych
patogenów
białkowych,
lipidowych, RNA i
innych
Granulocyty
Monocyty
Makrofagi
Komórki tucze
Eozynofile
Komórki NK
Komórki NKT
Komórki
nabłonków
Komórki
środbłonków
Białka
dopełniacza
Białka ostrej fazy
Brak
Schemat odpowiedzi immunologicznej

Efekt odpowiedzi nieswoitej
1)Wytworzenie stanu zapalnego, który warunkuje aktywację
komórek i ich łatwiejsze dotarcie wraz z czynnikami humoralnymi
do miejsca infekcji
2) Niszczenie patogenów w wyniku działania czynników
cytotoksycznych wydzielanych przez komorki
Schemat odpowiedzi immunologicznej

Schemat odpowiedzi immunologicznej
Swoista
(nabyta)
Ogroma liczba
wzorców
molekularnych
białkowych,
glikoproteinwyh,
lipidowych,
glikolipidowych i
innych
Limfocyty T
Limfocyty B
Komórki
prezentujące
antygeny (APC)
Monocyty
Makrofagi
Komórki
dendrytyczn
e
Wolne
sekrecyjne
przeciwciała
Obecna pod
postacią
limfocytów T
pamięci i
limfocytów B
pamięci
Efekt odpowiedzi swoistej
Ostatecznie zniszczenie patogenów i zapewnienie skutecznej odpowiedzi
immunologicznej przy powtórnym kontakcie z tym samym antygenem w tzw. Wtórnej
odpowiedzi immunologicznej w skutek wytworzenia limfocytów T i B pamięci
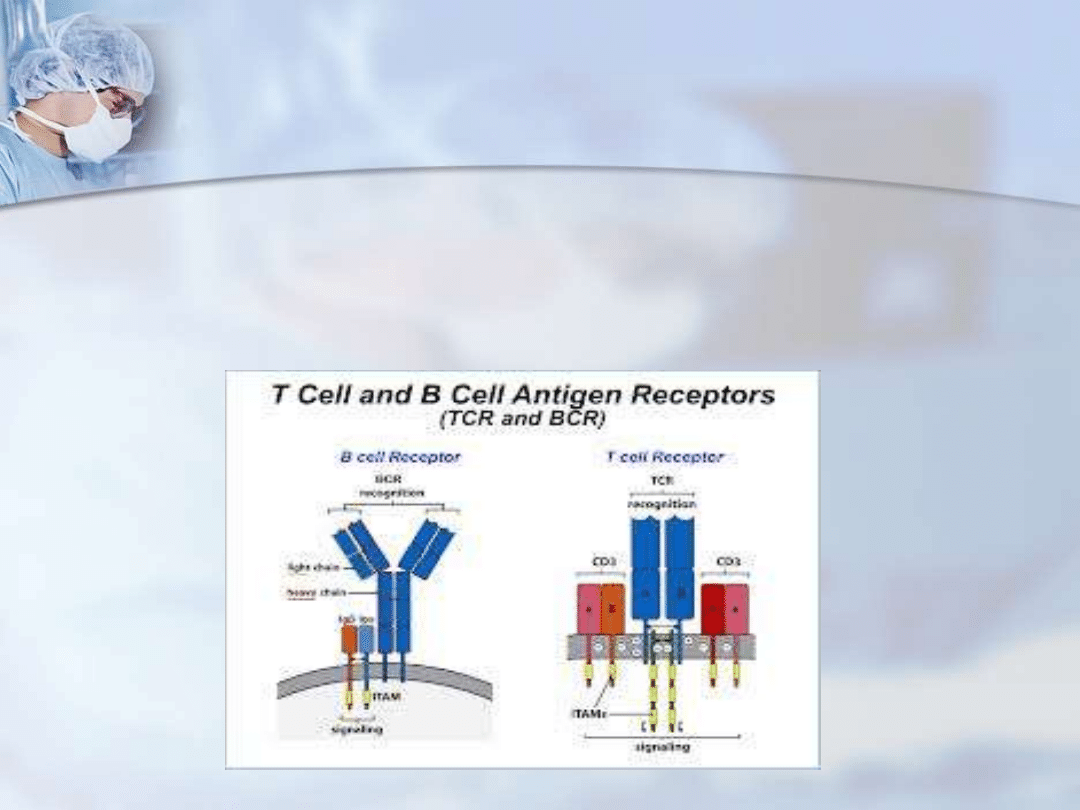
Komórki
APC
prezentują antygeny
zlokalizowane
są na
strukturach powierzchniowych
cząsteczek głównego układu
zgodności tkankowej MHC limfocytom T rozpoznającym
antygen przez receptor powierzchniowy TCR oraz limfocytom B
rozpoznającym antygen przez receptor powierzchniowy BCR.
Schemat odpowiedzi immunologicznej

Markery powierzierzchniowe limfocytów T
Antygen różnicowania komórkowego – określany w skrócie
jako CD
(ang. cluster of differentiation, gronko różnicowania)
jest standardem używanym w celu identyfikacji molekuł na
powierzchni komórek. CD mogą działać na wiele sposobów,
spełniając często funkcje receptora bądź ligandu (związku
aktywującego receptor).
Markery powierzchniowe limfocytów T i B
Zainicjowana przyłączeniem ligandu do receptora kaskada
sygnałowa zmienia zachowanie komórki. System CD jest
powszechnie wykorzystywany do oznaczania markerów
komórkowych umożliwiających rozpoznanie rodzaju komórki
na podstawie cząstek obecnych na jej powierzchni. Markery te
są często używane do przypisania komórkom właściwych im
funkcji immunologicznych.

Typ komórki
Marker CD
komórka macierzysta
CD34+,CD31-
Wszystkie grupy leukocytów
CD45+
Granulocyty
CD45+,CD15+
Monocyty
CD45+,CD14+
Limfocyty T
CD45+,CD3+
T helper
CD45+,CD3+,CD4+
komórka T cytotoksyczna
CD45+,CD3+,CD8+
Limfocyt B
CD45+, CD19+ ,CD20+
Trombocyt
CD45+,CD61+
komórka NK
CD16+,CD56+,CD3
Markery powierzchniowe limfocytów T i B
Markery powierzchniowe limfocytów T i B
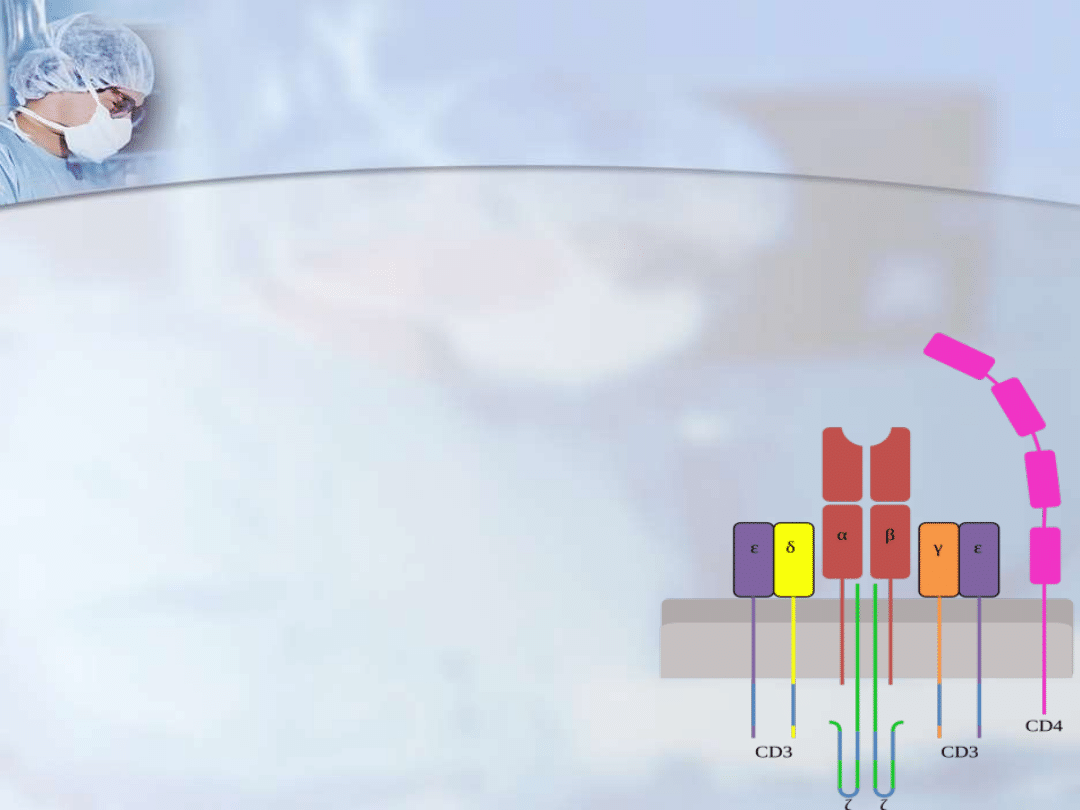
Dwie powszechnie używane cząstki CD to CD4 i CD8 - pierwsza charakteryzująca
limfocyt T pomocniczy (T helper) oraz druga dla limfocytu T cytotoksycznego.
Ze względu na ekspresję cząsteczek CD4 i CD8 wyróżniamy:
a) Limfocyty T CD4+ - rozpoznają antygeny związane z białkami MHC klasy II
b) Limfocyty T CD8+ - rozpoznają antygeny związane z białkami MHC klasy I

MHC obejmuje wiele genów odznaczających się
największym polimorfizmem z dotychczas poznanych.
Maja one podstawowe znaczenie zarówno w inicjacji jak i
w fazie efektorowej odpowiedzi immunologicznej.
Cząsteczki MHC są glikoproteinami. Istnieją cząsteczki
MHC klasy I i II różniące się pod względem budowy i
funkcji, a także cząsteczki klasy III i inne.
Główny układ zgodności tkankowej

Cząsteczki klasy I występują na powierzchni wszystkich komórek
jądrzastych, a w niewielkich ilościach również na erytrocytach.
Cząsteczki klasy II występują głównie na limfocytach B,
makrofagach, komórkach dendrytycznych, w tym na komórkach
Langerhansa, a także na komórkach nabłonkowych grasicy.
W wyniku aktywacji lub oddziaływania niektórych cytokin np.
IFN-γ, mogą się pojawić jednak na wielu innych komórkach np.
pobudzonych limfocytach T, komórkach śródbłonka, nabłonka
tarczycy,
komórkach
nabłonka
jelitowego,
fibroblastach,
keratynocytach.
U człowieka cząsteczki MHC klasy II występują konstytutywnie na
komórkach śródbłonka naczyń (w niektórych narządach np. w
sercu, nerce). Mogą być syntetyzowane selektywnie np. na 90%
monocytów znajdują się cząsteczki HLA-DR, lecz brak HLA-DQ.
Główny układ zgodności tkankowej

Główny układ zgodności tkankowej
MHC klasy III stanowią różne cząsteczki, niezwiązane z
procesem prezentacji antygenu.
O ile między klasą I i II widoczne są wybitne podobieństwa
strukturalne, o tyle MHC klasy III nie są podobne ani do
dwóch pozostałych klas, ani do siebie nawzajem.
Ludzkie MHC określane są mianem HLA (ang. human
leukocyte antigens – ludzkie antygeny leukocytarne).

U człowieka geny kodujące białka MHC znajdują się na 6
chromosomie.
Klasyczne cząsteczki MHC klasy I są kodowane przez geny
HLA-A, -B i -C, natomiast nieklasyczne – przez geny HLA-E, -
F, -G, MICA i MICB.
Nieklasyczne cząsteczki MHC klasy I mogą być także
kodowane poza regionem MHC (np. CD1).
Klasyczne cząsteczki MHC klasy II są kodowane przez geny,
leżące w regionach HLA-DP, -DQ i -DR, natomiast
nieklasyczne – HLA-DM i HLA-DO.
Pomiędzy regionem MHC klasy I, a regionem MHC klasy II,
znajduje się region kodujący MHC klasy III, choć niektóre z
tych cząsteczek są kodowane przez geny leżące w locus
MHC klasy II.
Główny układ zgodności tkankowej
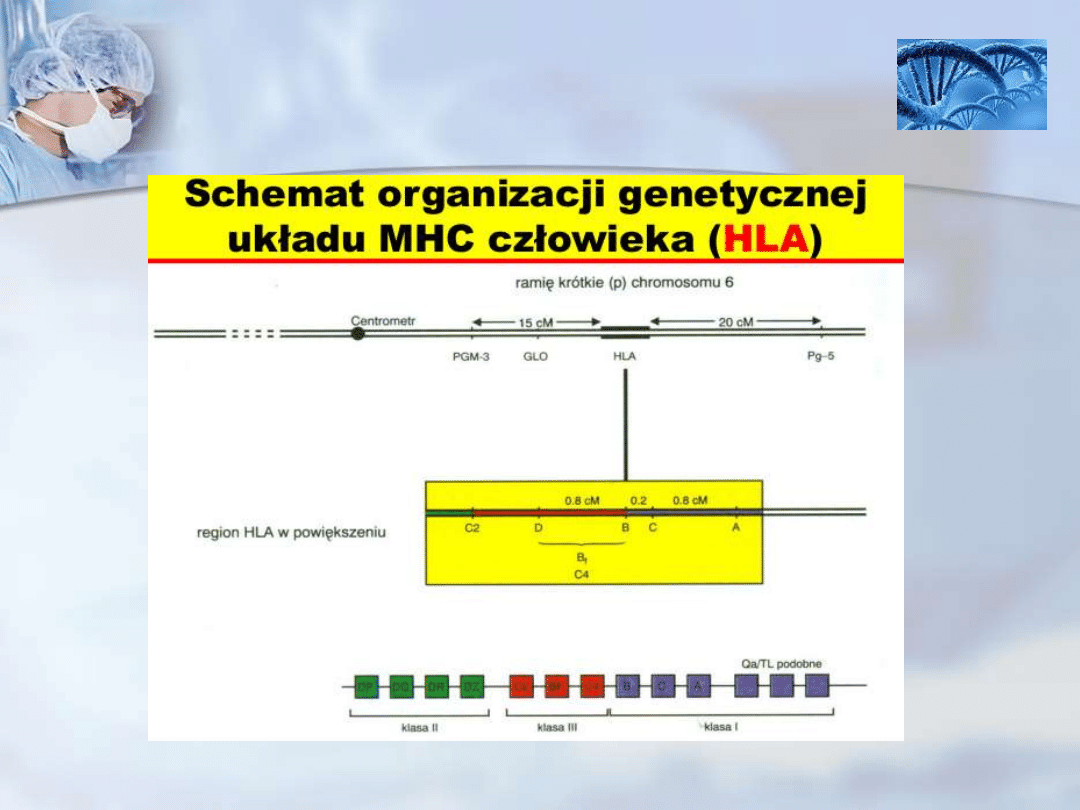
Główny układ zgodności tkankowej

Znaczenie medyczne MHC:
Znaczenie medyczne MHC wynika przede wszystkim z ich udziału
w procesach odrzucania przeszczepu. Białka te są niezwykle
silnymi, immunogennymi antygenami, w związku z tym komórki,
na których występują, są natychmiast rozpoznawane jako obce.
Stąd też dopasowanie białek MHC ma kluczowe znaczenie w
doborze dawcy i biorcy przeszczepu.
Im większa niezgodność pomiędzy allelami u dawcy i biorcy, tym
większe prawdopodobieństwo odrzucenia przeszczepu i tym
szybciej proces ten następuje
Jednak nawet wtedy, gdy MHC są idealnie dopasowane,
odrzucanie będzie zachodzić, gdyż MHC będą brały udział w
prezentacji słabych antygenów zgodności tkankowej. Jeśli będą
się one różnić (a z wyjątkiem bliźniaków monozygotycznych
będą się różnić praktycznie na pewno), to będą one
rozpoznawane jako obce i w rezultacie komórki będą niszczone
przez limfocyty Tc.
Główny układ zgodności tkankowej

Główny układ zgodności tkankowej
• Repertuar białek MHC wykazuje także powiązania z
pewnymi chorobami. W tym przypadku wystąpienie
określonych
alleli
zwiększa
prawdopodobieństwo
pojawienia
się
niektórych
chorób
(zwłaszcza
nieinfekcyjnych), np. allel HLA-B13 stwierdza się częściej u
pacjentów z łuszczycą, zaś HLA-B8 jest częstym allelem u
pacjentów z miastenią. Podłoże tego zjawiska nie jest
znane, co wiąże się z wieloczynnikową patogenezą tych
chorób oraz koniecznością analizy wielu alleli i ogromnej
liczby ich kombinacji

Oprócz cząsteczek CD swoistym markerem
limfocytów T jest receptor komórek T (TCR).
Dzięki temu receptorowi limfocyty T mają
zdolność rozpoznawania określonego
antygenu. TCR różnią się od przeciwciał w
wielu aspektach, przede wszystkim tym, że
występują jedynie w formie związanej z błoną
komórkową, natomiast przeciwciała mogą być
wydzielane przez limfocyty na zewnątrz.

Markery limfocytów B
• Limfocyty B (szpikozależne), odpowiedzialne za humoralną
odpowiedź immunologiczną (wytwarzanie przeciwciał)
charakteryzują się obecnością na powierzchni
następujących cząsteczek różnicujących:
• Kompleks receptora BCR odpowiada za aktywację limfocytów
B. BCR rozpoznaje antygen i następuje przekazanie sygnału do
wnętrza komórki. Rozpoznany już obcy antygen uruchamia
całą kaskadę reakcji, których wynikiem jest produkcja przez
limfocyt wolnych immunoglobulin. Te przeciwciała sekrecyjne
są strukturami prawie identycznymi z receptorami BCR.

Zastosowanie markerów
błonowych limfocytów
• Liczebność komórek posiadających na swojej powierzchni CD
4 i CD8 jest często używana do monitorowania progresji
infekcji wirusem
• Badanie markerów powierzchniowych limfocytów B u dzieci z
defektem biosyntezy przeciwciał (taka analiza
immunofenotypowa pozwala na wykluczenie zaburzeń
różnicowania limfocytów stadium limfopoezy i przemawia za
czynnościami zaburzeń dojrzewania limfocytów)
• Markery powierzchniowe limfocytów B to dobry cel dla
leczenia celowanego w białaczkach limfoidalnych
pochodzących z linii B-komórkowej
• Klasyfikacja chłoniaków i stanów zapalnych

Niedobory immunologiczne
PIERWOTNE, wrodzone
Nabyte, wtórne

• Wrodzone niedobory wywołane są przez defekty
genetyczne. Objawiają się one już wzmożoną
wrażliwością na infekcje już w okresie
niemowlęcym i wczesnym dzieciństwie.
• Wtórne zaś, czyli nabyte niedobory immunologiczne
rozwijają się jako konsekwencja niedożywienia,
ogólnego procesu nowotworowego, działania leków
immunosupresyjnych i chorób zaburzających
funkcję układu immunologicznego.

Agammaglobulinemia Brutona (agammaglobulinemia sprzężona z
chromosomem X) zwana również hipoimmunoglobulinemią – zespół
związany z blokadą dojrzewania limfocytów B, ujawniający się na
etapie limfocytu pre-B charakteryzujący się całkowitym brakiem
przeciwciał i śladową obecnością limfocytów B w krążeniu (poniżej 1%).
Przyczyną choroby są mutację lub delecja genu znajdującego się na
chromosomie X kodującego enzym kinazę tyrozynową (Btk).
Białko kodowane przez ten gen odgrywa rolę w dojrzewaniu
prekursorów limfocytów B i aktywacji komórek tucznych.
W szpiku kostnym wykrywa się zwiększona liczbę niedojrzałych
limfocytów pre-B, nie mających receptorów immunoglobulinowych.
Poziom limfocytów T jest prawidłowy.
Ze względu na sprzężenie z płcią agammaglobulinemia Brutona
występuje przede wszystkim u chłopców, których matki były zdrowymi
nosicielkami defektywnego genu. Częstość występowania wynosi ok. 1
na 100 tys. narodzin chłopców.
Pierwotne niedobory odporności

Pierwotne niedobory odporności
Objawy
Kliniczne objawy choroby pojawiają się około 4–6 miesiąca
życia, kiedy z krążenia zaczynają znikać przeciwciała matczyne.
Dominują
nawracające
zakażenia
bakteryjne
dróg
oddechowych. Gdy nie podejmuje się leczenia, prowadzi to do
przewlekłego zapalenia zatok i zmian rozstrzeniowych oskrzeli.
Odporność przeciwwirusowa jest sprawna, z niewiadomych
przyczyn nie dotyczy to enterowirusów.
Najczęstsza przyczyna śmierci w tym zespole jest właśnie
przewlekłe enterowirusowe zapalenia opon mózgowych i
mózgu.

Pierwotne niedobory odporności
Leczenie agammaglobulinemii Brutona polega na okresowym
substytucyjnym
uzupełnianiu
ludzkich
immunoglobulin
dożylnie (rzadziej domięśniowo lub podskórnie).
Leczenie powinno być prowadzone przez całe życie i prowadzi
do przedłużeniu długości i jakości życia.
Teoretycznie trwalsze efekty mogłaby przynieść terapia
genowa, jednak obecnie nie jest ona metodą leczniczą.

Pierwotne niedobory odporności
Ciężkie złożone niedobory odporności:
Wszystkie postacie SCID charakteryzują się albo głębokim
upośledzeniem albo całkowitym brakiem odpowiedzi humoralnej i
komórkowej.
Występują dość rzadko (2 na 75 tys. do 100 tys. urodzeń).
U dzieci dotkniętych SCID w ciągu kilku-kilkunastu tygodni od
urodzenia zaczynają pojawiać się kliniczne oznaki niedoboru
odporności: biegunki, nawracające zakażenia, do których później
dołącza się zatrzymanie wzrostu. Najczęściej spotykane są zakażenia
drożdżakami (Candida), adenowirusami, wirusami typu Herpes ( w
tym cytomegalowirusem oraz wirusem Epsteina-Barr), wirusem
paragrypy
typu
3,
a
także
patogenami
oportunistycznymi
(Pneumocystis carinii, Aspergillus sp.).
Nawet szczepionki zawierające atentowane żywe mikroorganizmy, np.
BCG, stanowią dla chorego dziecka śmiertelne zagrożenie. Dzieci z
SCID ze względu na brak funkcjonującego układu odpornościowego,
nie potrafią odrzucać przeszczepów allogenicznych.

Pierwotne niedobory odporności
Ciężki złożony niedobór odporności SCID
Wiąże się on z brakiem podstawowych receptorów komórkowych,
decydujących o odpowiedzi immunologicznej
Dochodzi do zaburzeń rozwoju limfocytów T i B na wczesnych etapach ich
różnicowania
Zespół SCID związany z upośledzeniem wytwarzania receptorów wiążących
antygeny może być wynikiem mutacji genów RAG-1 lub RAG-2
Konsekwencją tych zaburzeń jest znaczny niedobór dojrzałych limfocytów T i B
we krwi i tkankach oraz niezdolność do wytwarzania wolnych immunoglobulin

Przyczyn zespołu SCID jest jednak znacznie więcej i mogą one
wynikać z głębokich zaburzeń różnych szlaków metabolicznych
na
każdym
szczeblu
rozwoju
komórek
układu
immunologicznego
Opisano kilka przypadków SCID z mutacją genu dla łańcucha α
receptora dla IL-7. Obraz kliniczny różni się tym od mutacji γc,
że chorzy mają prawidłową liczbę komórek NK. Dojrzewanie
limfocytów T jest jednak zablokowane, co wskazuje na
niezastąpioną rolę IL-7 w różnicowaniu limfocytów T.
Mutacja genu kodującego CD45 jest również przyczyną SCID.
Cząsteczka ta jest markerem komórek hematopoetycznych. Ta
postać SCID charakteryzuje
się głęboką limfopenią z
nieznacznym spadkiem liczby komórek NK i dużą liczbą
limfocytów B.
Pierwotne niedobory odporności

Pierwotne niedobory odporności
Około 20% wszystkich przypadków SCID związanych jest z niedoborem
deaminazy adenozynowej (ADA).
Najczęstsza postać SCID spośród postaci dziedziczonych autosomalnie.
Niedobór spowodowany jest mutacją genu dla ADA, leżącym na 20
chromosomie.
ADA jest enzymem uczestniczącym w metabolizmie puryn. ADA
przekształca adenozynę oraz 2’deoksyadenozynę odpowiednio do
inozyny oraz 2’deoksyinozyny. 2’deoksyinozyna może swobodnie
dyfundować i ulegać fosforylacji do deoksyATP (dATP). Niedojrzałe
limfocyty
nie
potrafią
przekształcić
z
powrotem
dATP
w
2’deoksyinozynę i są wyjątkowo wrażliwe na jego toksyczne działanie.
dATP hamuje reduktazę rybonukleotydową, enzym niezbędny do
syntezy deoksynukleotydów, przy ich braku synteza DNA jest
niemożliwa.

Pierwotne niedobory odporności
Niedobór ADA charakteryzuje się wcześniejszymi objawami
klinicznymi w porównaniu do pozostałych postaci SCID.
Defekt dotyczy limfocytów T, B oraz komórek NK, limfopenia
jest głębsza w porównaniu do innych postaci SCID.
Oprócz klasycznych objawów w postaci zakażeń, zatrzymania
wzrostu, u połowy chorych dołączają się zaburzenia w
układzie
kostno-szkieletowym,
a
opisywane
są
też
zaburzenia neurologiczne: ślepota korowa, dystonia.
Zdarzają się przypadki niedoboru ADA o opóźnionych
objawach klinicznych ( występują nawet po kilku latach od
urodzenia). U chorych limfocytopenia rozwija się stopniowo,
nierzadko
obserwuje
się
towarzyszące
choroby
autoimmunizacyjne.

Pierwotne niedobory odporności
Ataksja teleangiektazja (zespół Louis-Bar)
Choroba
dziedziczona
autosomalnie
recesywnie,
spowodowana mutacją w genie ATM, zlokalizowanym na
chromosomie 11, którego produktem jest kinaza ATM
(
serynowo
-
treoninowa
)
Enzym ten jest regulatorem odpowiedzi komórkowej i
bierze udział w naprawie DNA
Częstość występowania 1:40 000 urodzeń
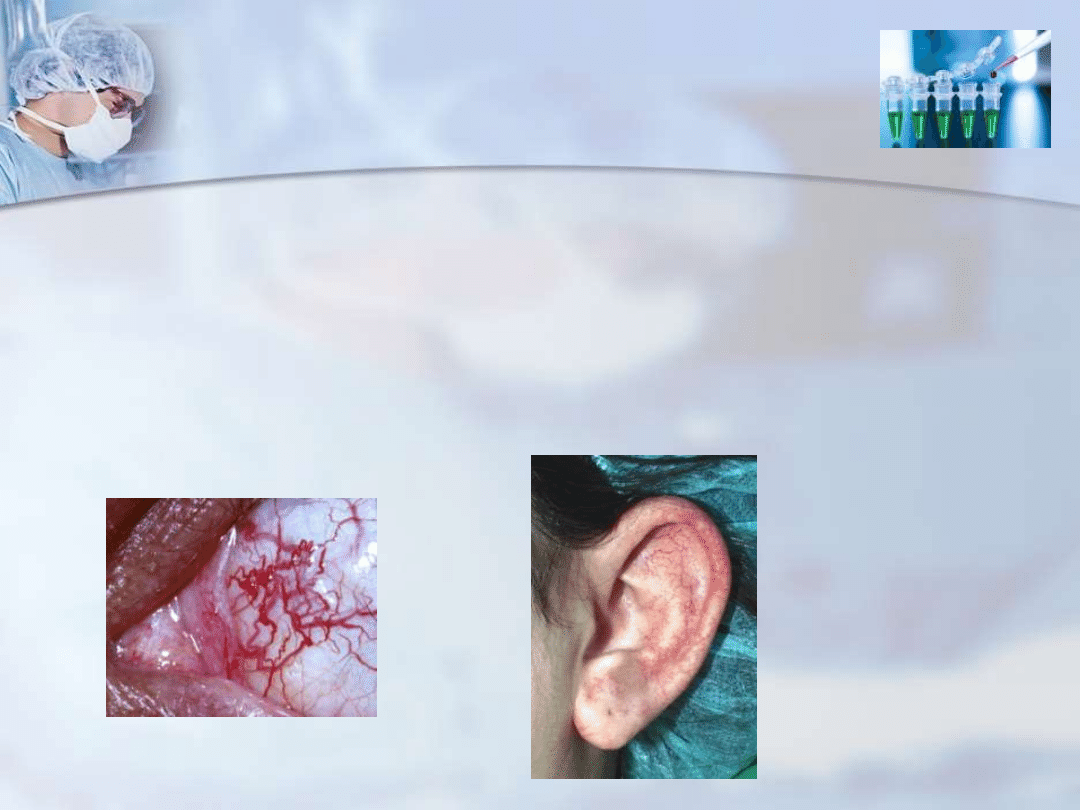
Objawy:
- zanik kory móżdżku, czego wynikiem jest postępująca
niezborność możdżkowa (ataksja)- dyslokomocja
- teleangiektazje
w obrębie skóry i gałki ocznej- rozszerzenie
naczyń włosowatych w małżowinach usznych, na spojówkach i
w innych okolicach twarzy
Pierwotne niedobory odporności

- zwiększona częstość bielactwa i plam typu cafe au lait
- spadek odporności humoralnej i komórkowej ze zwiększoną
wrażliwością na występowanie infekcji (głównie układu
oddechowego)
- wzrost zachorowania na białaczki i chłoniaki
- podwyżone stężęnie α-fetoproteiny (AFP) oraz obecność
antygenu kanceroembrionalnego (CEA)
- występowanie wielkocząsteczkowego białka należącego do
klasy IgM
- cukrzyca insulinozależna

Zespół Wiskotta-Aldricha
Zespół ten dziedziczy się z płcią.
Jego najbardziej charakterystyczną cechą jest skaza
krwotoczna.
Pierwszymi objawami są zazwyczaj krwawe biegunki i
wybroczyny skórne.
W pierwszym roku życia dołączają się zmiany skórne o
charakterze atopowego zapalenia skóry oraz infekcje
bakteryjne
(najcześciej
paciorkowce),
zakażenia
wirusowe
(przeważnie
z
grupy
Herpes)
oraz
oportunistyczne (Pneumocytis carinii).
W
późniejszych
latach
obserwuje
się
choroby
autoimmunizacyjne oraz nowotwory.
Pierwotne niedobory odporności

Pierwotne niedobory odporności
Zespół Wiskotta-Aldricha
Średni czas życia chłopców z tym zespołem nie przekracza 15
lat.
Najczęstszą przyczyną zgonów są krwawienia, zakażenia,
nowotwory, głównie chłoniaki (zwłaszcza chłoniaki Burkitta).
Obok trombocytopenii we krwi obwodowej obserwuje się
zmniejszoną liczbę limfocytów T, ale prawidłowe stężenie
przeciwciał.
Zaburzenia dotyczą zakresu antygenów rozpoznawanych
przez przeciwciała.
Molekularną przyczyną jest mutacja białka nazwanego WASP
(Wiskott-Aldricha syndrome protein), którego wybiórczą
ekspresję obserwuje się w limfocytach i megakariocytach.
Pierwotne niedobory odporności
Zespół DiGeorge’a
Zespół ten jest spowodowany wrodzoną malformacją grasicy, której
przyczyną w 90% przypadków są delecje (a częściej mikrodelecje)
fragmentu chromosomu 22.
Powoduje to selektywny brak limfocytów T przy normalnej liczbie
limfocytów B i
obniżonym stężeniu immunoglobulin w surowicy
Takie delecje
zaburzają rozwój struktur wywodzących się z 3 i 4
kieszonki
gardłowej.

Objawy:
deformacje części twarzowej czaszki
wrodzone wady serca i dużych naczyń
zaburzenia rozwoju podniebienia
trudności w uczeniu
spadek odporności pojawiający się w
pierwszych 6 miesiącach życia
Hipokalcemia spowodowana
niedorozwojem przytarczyc
Objawy infekcji w niedoborach
immunologicznych
Cechy niedoboru
Niedobór limfocytów B
Niedobór limfocytów T
Stężenie immunoglobulin
w surowicy
normalne lub
Reakcja nadwrażliwości
późnej obrazująca
odpowiedź zależną od
limfocytów T
normalna
morfologia
narządów
limfatycznych
brak lub zredukowana
wielkość
zazwyczaj normalna
wrażliwość na infekcje
wywołane przez patogeny
bakterie ropne
wirusy, grzyby

Dziękuję za uwagę
Wyszukiwarka
Podobne podstrony:
6 Immunogenetyka cz 1 Stadnik
immunoglobuliny cz II
wykłady immunologia cz 2 12 2013
6 Immunogenetyka cz 1 Stadnik
1 i 2 Podstawowe zasady dzialania ukladu immunologicznego Odpornoss nieswoista cz 1
immunologia wykłady cz.1 2012-2013, Analityka medyczna, Immunologia
Biol kom cz 1
Systemy Baz Danych (cz 1 2)
SEMINARIUM IMMUNOLOGIA Prezentacja
cukry cz 2 st
wykłady NA TRD (7) 2013 F cz`
więcej podobnych podstron