
1
Szybko reakcji chemicznej definiowana jest
jako ubytek st enia substratu lub przyrost
st enia produktu w jednostce czasu.
W definicjach szybko ci innych zjawisk wa na
jest wielko okre laj ca kinetyk w danej chwili,
tzn. wielko rzeczywista a nie rednia.
v= - dc/ dt = + dx/dt
Rzeczywista szybko
jest pochodn st enia
substratu wzgl dem czasu (ze znakiem minus,
poniewa szybko ma sens fizyczny jako
wielko dodatnia) lub pochodn st enia
produktu wzgl dem czasu
.
Szybko reakcji jest funkcja st enia reagentów. Funkcj t
zapisujemy w formie ogólnie stosowanego równania
kinetycznego.
Je eli w reakcji bior udział substraty A,B,C i reakcja jest
prosta,tzn. przebiega tylko w jednym kierunku,wówczas
v = k•[A]
a
•[B]
b
•[C]
c
Gdzie : k jest tzw. stał szybko ci reakcji, jest dla danej
reakcji wielko ci stał w danej temperaturze,zale y bardzo
silnie od temperatury.
Symbole w nawiasach kwadratowych oznaczaj st enia
poszczególnych substratów.
Współczynniki pot gowe przy st eniach poszczególnych
substratów okre laj rz d reakcji.
Rz d reakcji – odró niamy rz d reakcji sumaryczny
od rz dów reakcji wzgl dem poszczególnych
substratów.
Współczynniki pot gowe przy st eniach w
równaniach kinetycznych nie zawsze pokrywaj si
ze współczynnikami stechiometrycznymi.
Cz steczkowo reakcji – liczba cz stek
bior cych udział w elementarnej przemianie.
Zapis równania kinetycznego mówi, ze dana reakcja
jest rz du a wzgl dem substratu A, rz du b
wzgl dem B, i c wzgl dem C, a sumaryczny rz d
reakcji wynosi a+b+c.
Je eli reakcja chemiczna przebiega przez tylko jedna
elementarna przemian , wówczas wszystkie trzy wielko ci,a
wi c rz d reakcji, cz steczkowo i suma współczynników
stechiometrycznych pokrywaj si .
W reakcjach przebiegaj cych poprzez kilka stadiów przemian
elementarnych te trzy wielko ci mog by zupełnie ro ne.
Rz d reakcji stosuje si do eksperymentalnego równania
szybko ci, natomiast cz steczkowo okre la teoretyczny
mechanizm przemiany elementarnej.
Równania kinetyczne prostych reakcji.
Dla reakcji pierwszego rz du, szybko mo emy zapisa
w sposób :
-dc/dt = k•c lub dx/dt = k(a-x)
w drugim zapisie a oznacza st enie pocz tkowe
substratu, natomiast x aktualne st enie produktu.
Po scałkowaniu – dc/c = k dt + const
Otrzymujemy –lnc = kt + const. Stał całkowania
znajdziemy z warunków pocz tkowych; bior c za t=0,
const= -ln c
0
Po scałkowaniu otrzymujemy
ln a/a-x = kt
Czas połówkowy reakcji to czas, po upływie którego
st enie substratu spada do połowy pocz tkowej warto ci.
Podstawiaj c do wcze niejszego równania c=1/2c
0
otrzymujemy, e czas połówkowy I rz d = ln2/k
wynika st d, e czas ten nie zale y od st enia
pocz tkowego substratu.
W przypadku reakcji drugiego rz du równanie kinetyczne
ma posta dx/dt =k(a-x)(b-x)
Gdzie : a i b- st enia pocz tkowe substratów
a x - st enie aktualne produktu

2
W przypadku reakcji trzeciego rz du, kiedy bierze
udział w reakcji tylko jeden substrat, lub gdy st enia
substratów s jednakowe, równanie kinetyczne ma
posta - dc/dt = kc
3
po scałkowaniu przechodzi w
c
0
2
– c
2
/ 2c
2
•c
0
2
= k t
Wyznaczanie rz du reakcji
Najprostszy sposób wyznaczenia rz du reakcji, to
sprawdzanie stało ci k w równaniach kinetycznych
odpowiednich rz dów. O warto ci rz du reakcji
informuje nas tak e zachowanie si czasu
połówkowego reakcji.
Dla reakcji pierwszego rz du czas ten nie zale y od
st enia pocz tkowego. Dla innych rz dów czas ten
zale y od c
0.
ogólnie ~ 1/c
0
n-1
n-rz d reakcji
Najdokładniejsz metod wyznaczenia rz du reakcji jest
porównywanie pocz tkowej szybko ci reakcji dla
okre lonych st e pocz tkowych.
Przez zastosowanie du ego nadmiaru wszystkich
pozostałych substratów w stosunku do badanego
składnika mo na okre li rz d reakcji wzgl dem
konkretnych składników.
Badanie szybko ci reakcji w stadium pocz tkowym jest
wa ne, e eliminujemy ewentualny wpływ reakcji
odwrotnych. Trzeba podkre li ze wszystkie reakcje s
dwukierunkowe, d
do osi gni cia równowagi
termodynamicznej, w której szybko ci reakcji w obu
kierunkach si zrównuj .
Reakcje zło one
Reakcje odwrotne
S to najcz ciej spotykane reakcje zło one. Niemal wszystkie
reakcje s odwracalne i po pewnym czasie, od chwili rozpocz cia
reakcji, szybko jest ich wypadkow z reakcji w jednym i drugim
kierunku.
A
B
Tylko dla reakcji o prostym mechanizmie i niskiej całoliczbowej
rz dowo ci mo liwe jest cisłe ilo ciowe uj cie sumarycznej
kinetyki procesu.
Reakcje odwrotne
Je eli reakcje zachodz ce zarówno w prawo jak i w lewo s
pierwszego rz du, to wówczas mo na zapisa dla tej reakcji
nast puj ce równanie kinetyczne:
dx/dt = k
1
(a – x) – k
–1
x = k
1
a – (k
1
+ k
-1
)x
gdzie:
a- st enie pocz tkowe substratu k- stała szybko ci
x- aktualne st enie produktu
Reakcje odwrotne
Po scałkowaniu tego równania kinetycznego, otrzymuje si
nast puj c zale no :
ln z/z-x = (k
1
+ k
-1
)t
gdzie: z = k
1
a/(k
1
+ k
-1
)
Stosunek k
-1
/k
1
= K , gdzie K jest stał równowagi reakcji
chemicznej.

3
Reakcje równoległe
A
B
C
k
1
k
2
Je eli obie reakcje s pierwszego rz du, wówczas równanie
kinetyczne mo na przedstawi w postaci:
dx/dt = k
1
(a-x) + k
2
(a-x)= (k
1
+ k
2
) (a-x)
Gdzie: x oznacza ubytek st enia substratu (sum st e
produktów)
Reakcje równoległe
Po scałkowaniu otrzymuje si nast puj ce wyra enie:
ln a/a –x = (k
1
+ k
2
)t
Sumaryczna szybko reakcji jest zwykle uwarunkowana jedn z
dwu równoległych reakcji je eli k
1
>>
k
2
lub k
1
<<
k
2
.
Reakcje nast pcze
O reakcjach nast pczych mówimy wtedy gdy produkt
reakcji staje si substratem dla dalszej reakcji a stałe
szybko ci tych reakcji s porównywalne.
A
B
C
(a-x)
(x-y)
y
k
1
k
1
`
a-x, x-y,y oznaczaj aktualne st enia reagentów A, B, C
a- st enie pocz tkowe wyj ciowego substratu A
Reakcje nast pcze
Równania kinetyczne tych reakcji s nast puj ce:
dx/dt = k
1
(a – x)
dy/dt = k
1
`
(x – y)
Reakcje nast pcze
Rys. 1. Zmiany st e reagentów w reakcji nast pczej dla przypadku k
1
=k
1
`
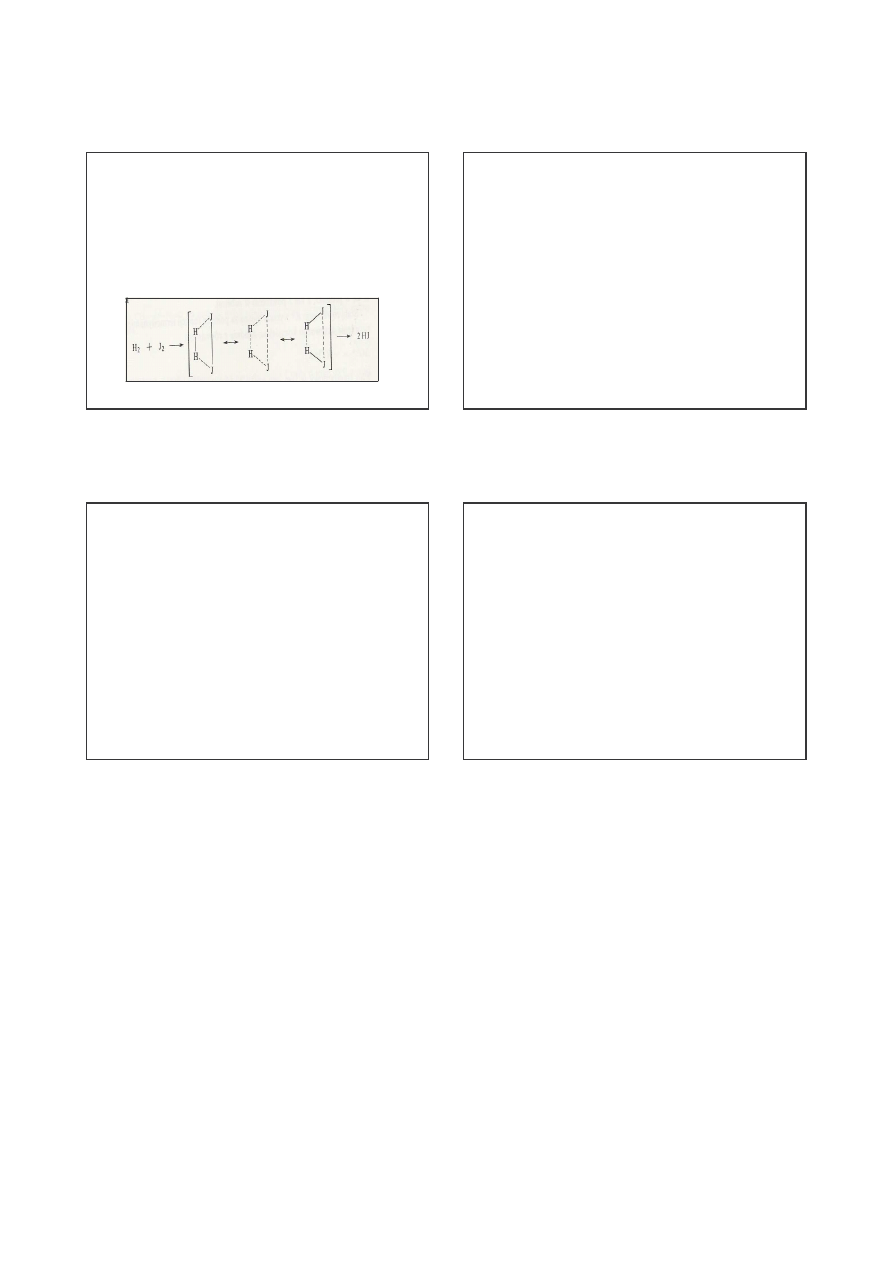
Teoria zderze aktywnych
Efektywne w sensie chemicznym s zderzenia tylko mi dzy
cz steczkami maj cymi wystarczaj cy zasób energii.
Z teorii kinetycznej gazów wiemy, e ułamek cz steczek o energii
równej lub przewy szaj cej pewn warto
a
wynosi
x = e
- a/kT
lub
x = e
-Ea/RT
je eli energi liczy na 1 mol
4
Je eli przez Z
0
oznaczymy liczb wszystkich zderze mi dzy
reaguj cymi cz steczkami, to liczba zderze aktywnych wyniesie
Z = Z
0
e
-Ea/RT
gdzie:
Z- ma wymiar szybko ci reakcji; wyra a liczb cz steczek
reaguj cych w przeci gu sekundy
Ea- energia aktywacji
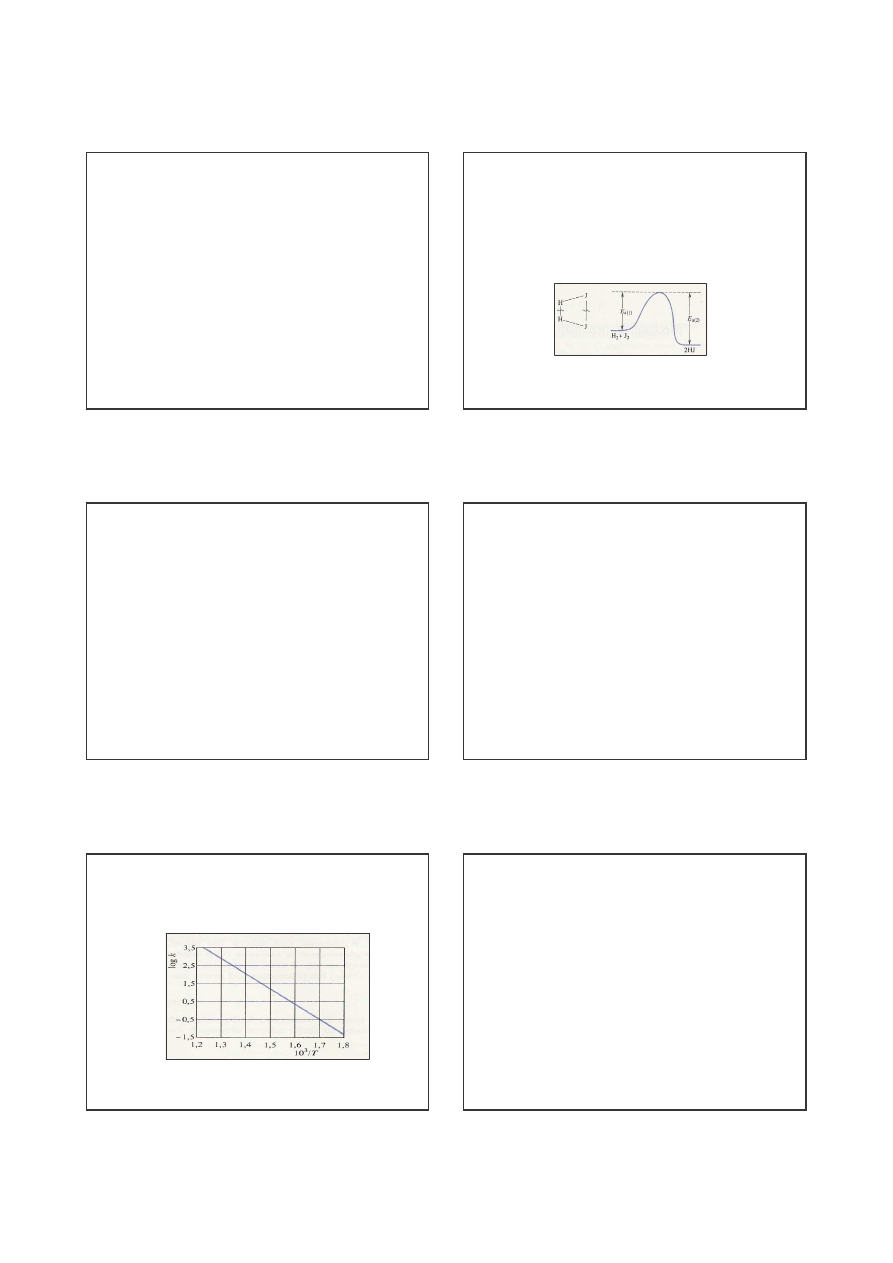
Energia aktywacji
H
2
+ J
2
= 2 HJ
Rys. 2. Bariera energetyczna reakcji chemicznej H
2
+ J
2
= 2 HJ
Równanie Arrheniusa
Równanie Z = Z
0
e
-Ea/RT
poci ga za sob fakt, e ten sam typ
zale no ci wyst pi w wyra eniu na stał szybko ci
k = Ae
-Ea/RT
gdzie:
A- czynnik cz sto ci (przedekspotencjalny), wielko stała,
niezale na od temperatury
Równanie Arrheniusa
Je eli równanie zlogarytmujemy, otrzymamy:
ln k = - E
a
/RT + ln A
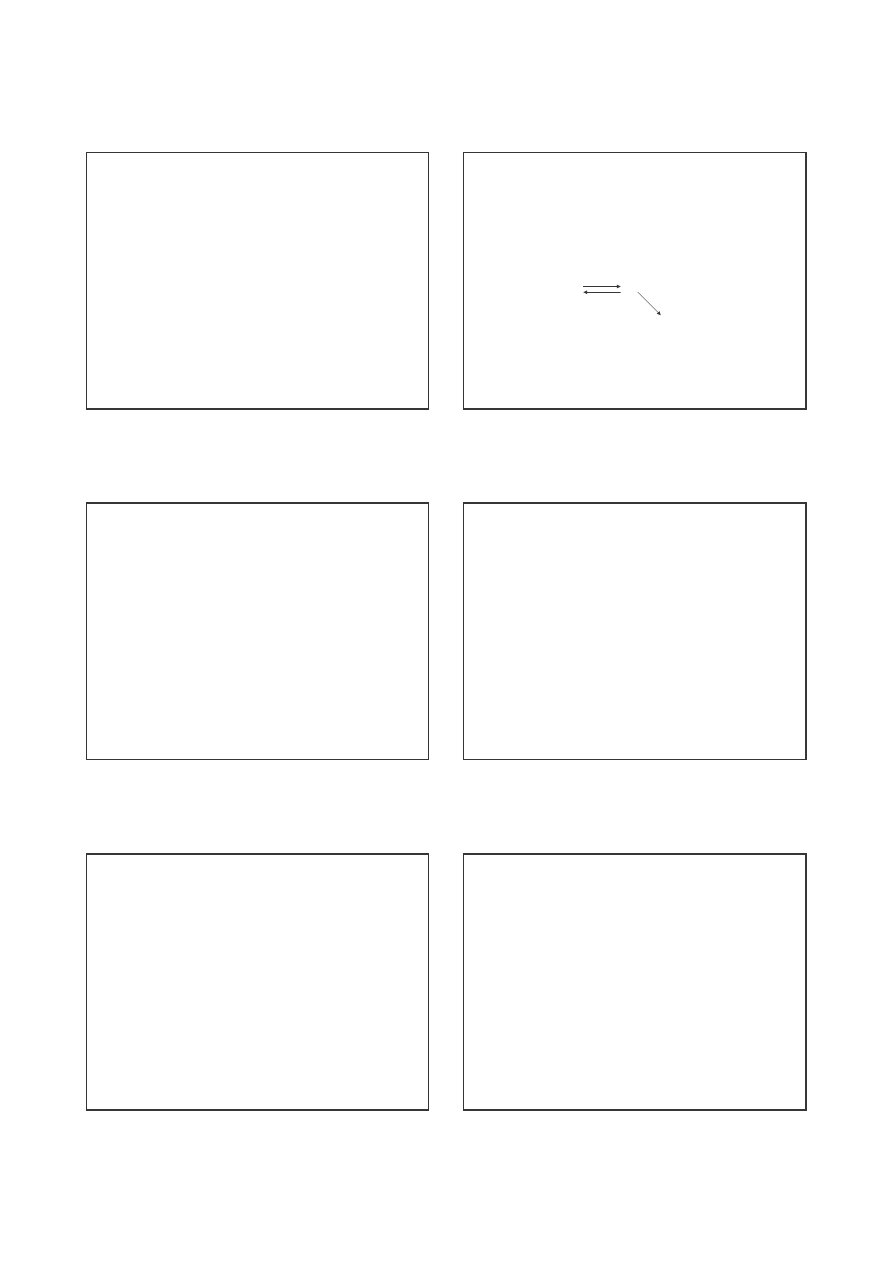
Wykre laj c zale no ln k od odwrotno ci temperatury,
otrzymujemy lini prost , której tangens k ta nachylenia okre la
warto E
a
/R
Rys. 3. Zale no logarytmu stałej szybko ci reakcji syntezy
jodowodoru od odwrotno ci temperatury.
Równanie Arrheniusa
Równanie Arrheniusa mo na ogólniej zapisa w postaci
ró niczkowej
ln k/ T = E
a
/RT
2
Po scałkowaniu w granicach T
1
od T
2
otrzymujemy przy zało eniu,
e E
a
= const jedn z najbardziej znanych postaci równania
Arrheniusa:
ln k
2
/k
1
= E
a
/R (1/T
1
– 1/T
2
)
5
Przykład
Ile razy zwi kszy si szybko reakcji chemicznej, je eli
zwi kszymy temperatur od 0 do 10°C. Energia aktywacji wynosi 40
kcal/mol.
Zgodnie z równaniem Arrheniusa:
log k
2
/k
1
= E
a
/2,303 R (1/T
1
– 1/T
2
)
log k
2
/k
1
= 4 10
4
/4,576 10/273 283 = 1,131
k
2
/k
1
= 13,5
Teoria reakcji jednocz steczkowych
W reakcji jednocz steczkowej A
→ B + C, pierwszym etapem jest
aktywacja, tj. wzbudzenie energetyczne cz steczek na drodze
zderze
A + A
A
‡
+ A
k
2
k
-2
B + C
k
1
Proces aktywacji jest procesem odwracalnym, tzn. zderzenie
cz steczki zaktywowanej ze zwykł cz steczk mo e prowadzi do
dezaktywacji.
Wymienione przemiany mo na uj za pomoc nast puj cych
równa kinetycznych
d[B]/dt = k
1
[A
‡
]
d[A
‡
]
/dt
=
k
2
[A]
2
– k
-2
[A
‡
]
[A] – k
1
[A
‡
]
Model stanu stacjonarnego
Podczas przebiegu reakcji szybko powstawania cz steczek
aktywnych równa si szybko ci ich dezaktywacji.
Stosuj c model stanu stacjonarnego do wcze niejszych rozwa a
kinetycznych mo emy stwierdzi , e [A
‡
]
jest stałe. Zatem
d[A
‡
]
/dt
= 0.
Zało enie to jest słuszne dla niedu ego przedziału
czasowego. Ponadto trzeba wzi pod uwag , e wielko [A
‡
]
jest bardzo mała, a wi c i zmiany w czasie b d małe. Mo na
wi c bez wi kszego bł du przyj pochodn równ zeru.
Poci ga to za sob :
[A
‡
] =
k
2
[A]
2
/k
-2
[A]+k
1
Szybko reakcji definiowanej jako d[B]/dt wynosi:
d[B]/dt = k
1
[A
‡
]
= k
1
k
2
[A]
2
/k
-2
[A]+k
1
Zakładaj c, e k
1
>>k
-2
[A]
2
otrzymujemy wyra enie:
d[B]/dt = k
2
[A]
2
dla zwykłej reakcji drugiego rz du.
Je eli natomiast k
-2
A>>k
1
wówczas:
d[B]/dt = k
1
k
2
/k
-2
[A] = k [A]
Otrzymujemy równanie kinetyczne reakcji pierwszego rz du.
6
Teoria stanów przej ciowych
Szczyt bariery energetycznej, która musi by pokonana podczas
przemiany, nie jest równowa ny stanowi dysocjacji. Jest to stan, w
którym działaj w równym stopniu pierwotne jak i nowe wi zania.
Stan taki nazywamy aktywnym stanem przej ciowym.
A + B
(AB)
‡
C + D
(AB)
‡
- aktywny stan przej ciowy.
Szybko tej reakcji jest proporcjonalna do st enia stanów
przej ciowych:
d[C]/dt = b[(AB)
‡
]
b- czynnik cz sto ci, okre la szybko z jak aktywny kompleks
przej ciowy przechodzi do stanu ko cowego, lub pocz tkowego
reakcji; jest odwrotno ci czasu ycia kompleksu.
b = kT/h
k- stała Boltzmana, h- stała Planca, – współczynnik transmisji
Aktywny kompleks (AB)
‡
jest w równowadze z substratami.
Stała równowagi tej reakcji wynosi:
K
‡
= [(AB)
‡
]/[A][B]
st d:
[(AB)
‡
] = K
‡
[A][B]
Szybko reakcji b dzie zatem okre lona wzorem:
d[C]/dt = bK
‡
[A][B]
Stał równowagi K
‡
wyrazi mo na za pomoc funkcji
termodynamicznych tworzenia aktywnego kompleksu, zgodnie z
równaniem:
G
0‡
= -RTlnK
c
‡
Poniewa G
0‡
= H
0‡
- T S
0‡
otrzymujemy:
k = be
S
º
/R
e
- H
º
/R
Wielko ci S
0‡
i H
0‡
nosz nazw entropii i entalpii aktywacji.
Wyszukiwarka
Podobne podstrony:
Cw 2 An kat schemat id 121639 Nieznany
An anionow cw 3 id 59349 Nieznany (2)
an widm s ok id 59366 Nieznany (2)
a an(1) id 49170 Nieznany
How to build an arbor id 206318 Nieznany
Osc an i cyfr id 293334 Nieznany
Abolicja podatkowa id 50334 Nieznany (2)
4 LIDER MENEDZER id 37733 Nieznany (2)
katechezy MB id 233498 Nieznany
metro sciaga id 296943 Nieznany
perf id 354744 Nieznany
interbase id 92028 Nieznany
Mbaku id 289860 Nieznany
Probiotyki antybiotyki id 66316 Nieznany
miedziowanie cz 2 id 113259 Nieznany
LTC1729 id 273494 Nieznany
D11B7AOver0400 id 130434 Nieznany
analiza ryzyka bio id 61320 Nieznany
więcej podobnych podstron