
Farmakologia
I kolokwium - opracowanie zagadnień
1. Lek, substancja czynna, substancja lecznicza, środek leczniczy
Lek albo produkt leczniczy (wg Prawa Farmaceutycznego) to substancja lub mieszanina substancji,
przedstawiana jako posiadająca właściwości zapobiegania lub leczenia chorób występujących u ludzi
lub zwierząt lub podawana w celu postawienia diagnozy lub w celu przywrócenia, poprawienia
lub modyfikacji fizjologicznych funkcji organizmu poprzez działanie farmakologiczne, immunologiczne
lub metaboliczne. Substancja czynna to ta część leku, która oddziałuje leczniczo na organizm zgodnie
z powyższą definicją.
Leki mogą być referencyjne, czyli innowacyjne - dopuszczone do obrotu na podstawie pełnej
dokumentacji. Zawierają one substancję czynną lub ich mieszaninę niedopuszczoną wcześniej do obrotu,
są chronione patentem.
Lek generyczny zawiera taką samą substancję czynną co lek innowacyjny, ale został wyprodukowany
po zakończeniu okresu ochrony patentowej, na podstawie formuły w zgłoszeniu patentowym.
Leki dzieli się także na:
• oficynalne, których sposób przygotowania podany jest w Farmacopei;
• magistralne - przepisywane przez lekarza na recepcie i na tej podstawie przygotowywane.
Leki magistralne przepisuje się, gdy nie mamy dostępnego danego leku gotowego lub gdy nie mamy
potrzebnej dawki w leku gotowym.
2. Rodzaje dawek leków
Działanie leków zależy od ich stężenia we krwi i w tkankach, które jest na ogół proporcjonalne
do ilości leku wprowadzonego do organizmu. W miarę zwiększania dawki leku i jego stężenia we krwi
następuje nasilenie reakcji na lek ze strony poszczególnych narządów i tkanek. Nie przebiega ono
proporcjonalnie, w pewnym zakresie dawek działanie staje się proporcjonalne do logarytmu stężenia leku,
czyli w celu uzyskania większego działania dawkę należy zwiększyć wielokrotnie, aby uzyskać odpowiednie
stężenie leku we krwi. Wielkość dawki leku zawsze musi wiązać się z ilościowym działaniem leku, często
duża dawka może wywoływać jakościowo inne działanie w porównaniu z dawką mniejszą.
Leki stosuje się:
• w dawkach jednorazowych (pro dosi);
• w dawkach dobowych (pro die) – ilość leku zastosowana w pojedynczych dawkach w ciągu doby;
jest to ważne w przypadku leków o długotrwałym działaniu, łatwo kumulujących się dla których pożądane
jest utrzymanie stałego stężenia aktywnego we krwi.
Dawka progowa lub minimalna (dosis minima) – określa najmniejszą ilość środka, wywołującą już
pewne reakcje ze strony organizmu, dawka minimalna nazywa jest też dawką subterapeutyczną.
Dawka lecznicza (dosis therapeutica) – wywołuje wyraźne zmiany w czynności narządów,
charakteryzujące się pobudzeniem lub zahamowaniem ich czynności w granicach fizjologicznych;
największa dawka lecznicza nazywa się dawką maksymalną.
Dawka uderzeniowa – stosowania np. w leczeniu antybiotykami, sulfonamidami, czy witaminą D;
początkowe dawki są znacznie większe, zazwyczaj 2-krotnie niż dawki następne podtrzymujące; stosuje
się je w celu szybszego uzyskania aktywnego stężenia leku we krwi, a tym samym skuteczniejszego
leczenia pewnych chorób.
Dawka toksyczna (dosis toxica) - zaburzenia czynności organizmu w granicach patologicznych
(działanie trujące).
Dawka śmiertelna (dosis letalis) – najmniejsza ilość leku, która poraża ważne dla życia czynności
organizmu (oddychanie, czynność serca) powodując śmierć.
1
3. Indeks terapeutyczny
Z badań wynika, że najbardziej miarodajna dla leku jest dawka wywołująca swoisty odczyn biologiczny
u połowy badanych zwierząt; wyróżnia się więc:
• dawki lecznicze dla 50% badanych zwierząt albo badanej populacji pacjentów(DE50);
• dawki toksyczne dla 50% badanych zwierząt (DT50);
• dawki śmiertelne dla 50% badanych zwierząt (DL50);
Wskaźnik (indeks) terapeutyczny – stosunek między dawką bezpieczną działającą skutecznie
a dawką mogącą wywołać niepożądane objawy toksyczne
Wskaźnik terapeutyczny = Max. dawka tolerowana/Min, dawka terapeutyczna
Wskaźnik terapeutyczny =DL50/DE50 lub DL0,01/DE0,99
Maksymalna dawka jest określana na ogół w odniesieniu do osób dojrzałych płci męskiej o masie
ciała 70 kg.
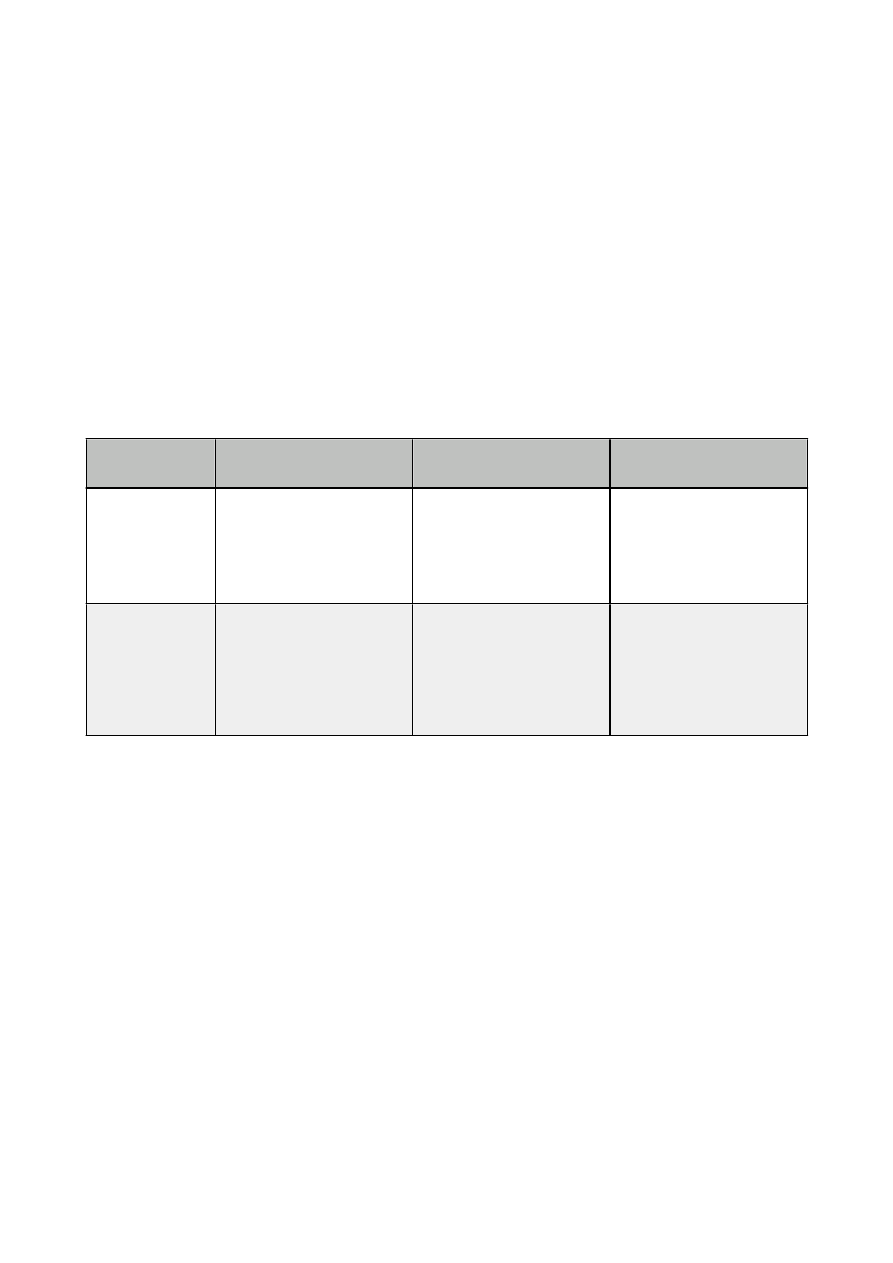
4. Drogi podania leków (tabelka)
drogi podania/
efekt działania
enteralne
parenteralne(pozajelitowe)
„powierzchniowe”
ogólnoustrojowy
podjęzykowa
dopoliczkowa
tabletki, kapsułki, leki płynne
do połykania
czopki, tabletki, kapsułki,
doodbytnicze
dożylna
dotętnicza
dosercowa
domięśniowa
podskórna
dootrzewnowa
przezskórna
inhalacje
donosowa
miejscowy
do ssania
do zębodołu
tabletki, kapsułki, leki płynne
do połykania
czopki doodbytnicze
śródskórna
dostawowa
intratekalna
doszklistkowa
do ciał jamistych
naskórna
inhalacje
postaci do oka, nosa, ucha
do pęcherza moczowego
pręciki docewkowe
tabletki, kapsułki, czopki
dopochwowe
Efekt ogólnoustrojowy - po podaniu leku mającego na celu dotarcie do krążenia ogólnego
(lek wywiera działanie w miejscu odległym od miejsca podania).
Efekt miejscowy - po podaniu leku mającego na celu działanie w obrębie tkanek pozostających
w bezpośrednim sąsiedztwie miejsca podania.
Podanie enteralne - na powierzchnię błony śluzowej przewodu pokarmowego.
Podanie parenteralne - naruszające ciągłość tkanek.
Podanie „powierzchniowe” - na powierzchnię skóry lub błony śluzowej oprócz przewodu
pokarmowego.
5. Losy leku w organizmie
Od momentu podania leku do organizmu dochodzi do licznych następujących po sobie i związanych
z sobą procesów, które obejmują dzieje leku w organiźmie. Całość toczących się procesów określamy
skrótem LADME.
L – uwalnianie
A – wchłanianie
D – dystrybucja
M – metabolizm
E – eliminacja
2

Warunkiem działania leku jest jego uwolnienie z danej postaci, ponieważ obecność leku w formie
rozpuszczonej jest niezbędnym warunkiem wchłaniania leku. Ilość uwolnionego z preparatu
i przechodzącego do roztworu leku określa się jako oddawalność, co determinuje jego dostępność
biologiczną. Etap ten zależy przede wszystkim od postaci leku (np. kapsułki o opóźnionym uwalnianiu).
Wchłanianie to przemieszczenie leku z jego miejsca podania do kompartmentu centralnego
oraz szybkość, z jaką to zachodzi. Klinicznie większe znaczenie ma biodostępność, czyli ta część dawki
leku, która trafia do miejsca działania. Absoprcja w dużej mierze zależy od drogi podania, a przy każdej
z nich pojawiają się kolejne czynniki.
Przy podaniu doustnym wchłanianie zależy od: powierzchni, przepływu krwi przez obszar
wchłaniania, fizycznej postaci leku, jego rozpuszczalności w wodzie i stężenia leku w obszarze wchłaniania.
Ze względu na przewagę absorpcji drogą dyfuzji biernej przewagę mają leki lipofilne i niezjonizowane.
Wchłanianie jest zdecydowanie szybsze w jelicie niż w żołądku ze względu na dużo większą powierzchnię.
Wynika z tego, że wchłanianie w przewodzie pokarmowym zależeć będzie także od szybkości opróżniania
żołądka.
Podanie podjęzykowe gwarantuje, że nie będzie miał miejsce efekt pierwszego przejścia,
gdyż unaczynienie z jamy ustnej prowadzi do żyły głównej.
Wchłanianie przez skórę leku zależy od powierzchni, na którą został naniesiony i od jego
rozpuszczalności w lipidach (dlatego by uzyskać szybszy efekt lek jest zawieszany w fazie olejowej).
Uszkodzenie naskórka (otarcie, oparzenie etc.) także będzie miało wpływ, gdyż skóra właściwa
jest zdecydowanie bardziej przepuszczalna dla substancji rozpuszczonych.
Podanie doodbytnicze pozwala na zmniejszenie efektu pierwszego przejścia, gdyż jedynie ok.50%
leku wchłanianego trafi do wątroby.
Podanie przeztkankowe:
• przy podaniu dożylnym proces wchłaniania jest pomijany;
• szybkość wchłaniania po podaniu podskórnym jest równomierna i powolna w czasie, więc pozwala
podtrzymać efekt działania, a okres wchłaniania można modyfikować wielkością cząsteczki,
kompleksowaniem z białkami lub zmianą pH;
• wchłanianie przy podaniu domięśniowym zależy od przepływu krwi w miejscu wstrzyknięcia
roztworu wodnego, natomiast by spowolnić proces wystarczy użyć innego nośnika niż woda albo rozpuścić
w tłuszczu;
• podanie dokanałowe umożliwia wchłanianie do OUN, które za pośrednictwem krwi jest niemożliwe
ze względu na bariery krew-mózg i krew-płyn mózgowo-rdzeniowy.
W przypadku wchłaniania płucnego dostęp do krążenia ogólnego jest szybki ze względu na dużą
powierzchnię płuc i pominięcie efektu pierwszego przejścia.
Dystrybucja tkankowa leku to dotarcie leku do płynu śródtkankowego i śródmiąższowego. Zależy
od jego podziału między krew a konkretną tkankę. I tak szybkość procesu i ilość dostarczanego leku
determinować będą pojemność minutowa serca, lokalny przepływ krwi, transport kapilarny oraz objętość
tkanki. Co za tym idzie, większość leku dostaną najpierw wątroba, nerki, mózg i inne dobrze perfundowane
narządy, a następnie - mięśnie, skóra, tkanka tłuszczowa. Zrównoważenie stężenia leku między tkankami
drugiej grupy a krwią może wymagać nawet godzin. Lek może odwracalnie wiązać się z białkami, zarówno
w osoczu, jak i w tkankach. Taka forma jest zasadniczo „nieaktywna” - nie ulega dystrybucji i metabolizmowi.
Przez wiązanie leku w tkankach może on osiągać wyższe stężenie niż w osoczu i płynie
zewnątrztkankowym. Mówi się wówczas o rezerwuarze leku w tkance albo jego kumulacji, co przedłuża
działanie, ale może powodować lokalną toksyczność.
Metabolizm leków, i w ogóle ksenobiotyków, jest ważny dla ich eliminacji z ustroju (lipofilne cząstki
są w nerce resorbowane), zaniku aktywności farmakologicznej i metabolicznej, ale także przeciwnie -
do uzyskania substancji aktywnych, zarówno o działaniu korzystnym, pożądanym, jak i toksycznym.
Metabolizm ksenobiotyków można podzielić na 2 ważne fazy:
3

I. utlenianie, redukcja, hydroliza - (przez cytochrom P450 - CYP, monooksygenazy flawinowe,
hydrolazy epoksydowe);
II.sprzęganie - tworzenie koniugatów produktów fazy 1 (przez sulfotransferazy, UDP-
glukuronozylotransferazy, glutationo-S-ransferazy, N-acetylotransferazy, metylotransferazy).
W I fazie chodzi głównie o inaktywację leku (nie zawsze!), ale także o przygotowanie miejsca
przyłączenia hydrofilowych fragmentów cząstek w fazie II, po której to metabolit jest bardziej rozpuszczalny
w wodzie i ma większą masę cząsteczkową, co ułatwi jego eliminację.
Za metabolizm ksenobiotyków odpowiedzialna jest przede wszystkim wątroba i, zależnie od drogi
podania, substancje te przechodzą przez nią dwa razy, ale w przypadku pierwszego przejścia proces
rozpocznie się już w nabłonku przewodu pokarmowego lub błonie śluzowej jamy nosowej i płuc.
Dla przemian leków znaczenie będzie miała oczywiście wydolność wątroby, ale także interakcja
z innym przyjmowanymi substancjami. Problem pojawi się wówczas, gdy oba ksenobiotyki
są metabolizowane przez ten sam enzym, albo gdy jeden ze związków jest inhibitorem dla enzymu
odpowiedzialnego za przemiany drugiego. Dotyczy to głównie enzymów CYP. Leki mogą także indukować
metabolizm, własny albo innych substancji, co może spowodować ich za niskie stężenie w osoczu i brak
efektywności.
Leki są wydalane z organizmu w postaci niezmienionej lub jako metabolity. Narządy odpowiedzialne
za eliminację (poza płucami) bardziej efektywnie usuwają substancje spolaryzowane. Największą rolę w tym
procesie odgrywają nerki. Z kałem eliminowane są leki przyjęte doustnie, które nie uległy wchłonięciu,
jako metabolity z żółcią albo wydzielone do przewodu pokarmowego i niewchłonięte. Wydalanie z mlekiem
matki ma znaczenie ze względu na możliwe działanie szkodliwe dla karmionego dziecka. Usuwanie przez
płuca jest istotne szczególnie przy pozbywaniu się gazów znieczulenia ogólnego.
Wydalanie przez nerki zależy najbardziej ogólnie od ich wydolności i perfuzji. Proces zachodzi
na każdym etapie: filtracji, sekrecji kanalikowej i resorpcji. Znaczenie będzie też miało, jak duża część leku
związana jest z białkami osocza (nieusuwane), a także od jego stopnia zjonizowania i pH moczu - fakt ten
wykorzystuje się w leczeniu zatruć.
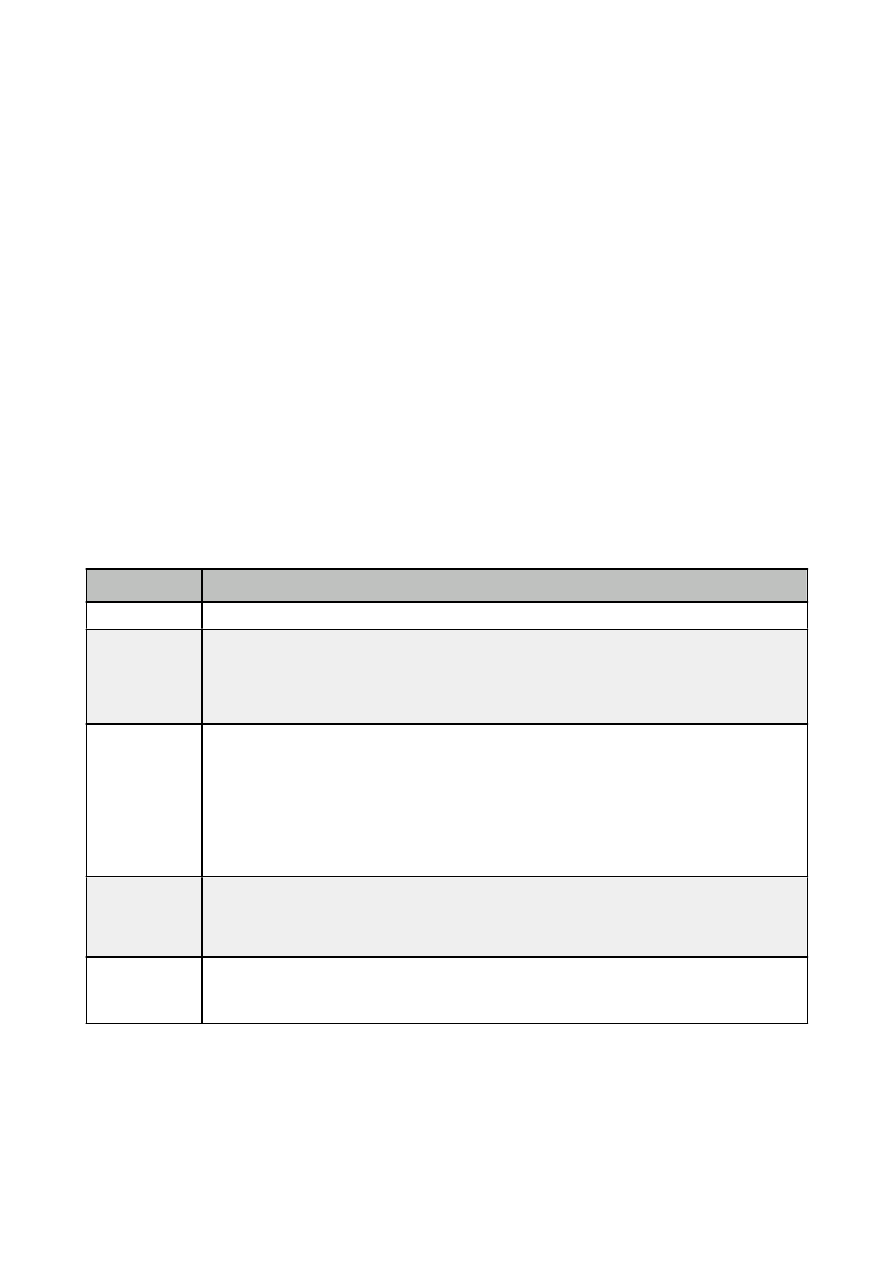
6. Induktory i inhibitory cytochromu P450
W czasach kiedy barbiturany były szeroko stosowane jako leki nasenne odkryto, że należy w czasie
leczenia zwiększać dawki leku tak, aby osiągnąć ten sam efekt nasenny. Powodem takiego stanu rzeczy
jest zwiększenie aktywności enzymów mikrosomalnych przez barbiturany, w wyniku czego stopień
metabolizmu i wydalania tych leków jest zwiększony. Ten fenomen „stymulacji” lub indukcji enzymów,
nie tylko wpływa na potrzebę zwiększenia dawki, ale jeśli inny lek, który jest metabolizowany przez tę samą
grupę enzymów jest również obecny, to także jego metabolizm enzymatyczny jest zwiększony i potrzebne są
wtedy większe dawki, aby uzyskać ten sam efekt terapeutyczny. Należy jednak zauważyć, że nie wszystkie
leki pobudzające enzymy indukują ich własny metabolizm.
Najczęściej indukowanym przez leki szlakiem metabolicznym jest I faza utleniania, w której
pośredniczą izoenzymy cytochromu P450.
CYP1A2 CYP2B6
CYP2C8 CYP2C19 CYP2C9
CYP2D6 CYP2E1
CYP3A4,5,7
tytoń
Fenobarbital
(Luminal)
N/A
Ryfampicyna
(Rifampicyna)
N/A
etanol
Karbamazepina
(Amizepin,
Tegretol)
Fenytoina
(Epanu=n
Parenteral)
Izoniazyd
(Isoniazidum)
Fenobarbital
(Luminal)
Ryfampicyna
(Rifampicyna)
Fenytoina
(Epanu=n
Parenteral)
Ryfampicyna (Rifampicyna)
Wyciągi z dziurawca
Bardziej rozpowszechniona od indukcji enzymów jest ich inhibicja. Wynikiem inhibicji enzymatycznej
jest zmniejszenie metabolizmu danego leku. Lek taki może zacząć kumulować się w organizmie, a efekt
jest zazwyczaj tak samo istotny jak w przypadku gdybyśmy zwiększyli dawkowanie. Inhibicja enzymatyczna
4

występuje bardzo szybko. Zaraz po podaniu pierwszej dawki inhibitora blokuje on metabolizm równocześnie
podanego leku. Najczęściej hamowanym szlakiem metabolizmu jest reakcja utleniania I fazy metabolizmu,
która przebiega przy udziale izoenzymów cytochromu P450.
CYP1A2
CYP2B6
CYP2C8
CYP2C19
CYP2C9
CYP2D6
CYP2E1
CYP3A4,5,7
Cymetydyna
(Cime=dine,
Altramed)
Tyklopidyna
(Aclo=n,
Ticlo)
Gemfibrozyl
(Lipozil)
Fluoksetyna
(Andepin,
Bioxe=n)
Amiodaron
(Cordarone,
Opacorden)
Amiodaron
(Cordarone,
Opacorden)
Disulfiram
(An=col)
Inhibitory
proteazy
HIV:
Indynawir
Nelfinawir
Rytonawir
Fluwoksamina
(Fevarin)
Montelukast
(Singulair)
Fluwoksamina
(Fevarin)
Flukonazol
(Flumycon)
Buproprion
(Zyban)
Amiodaron
(Cordarone,
Opacorden)
Tyklopidyna
(Aclo=n,
Ticlo)
Ketoconazol
(Ketokonazol)
Izoniazyd
(Isoniazidum)
Cymetydyna
(Cime=dine,
Altramed)
Cymetydyna
(Cime=dine,
Altramed)
Lansoprazol
(Lanzul)
Klomipramina
(Anafranil,
Hydiphen)
Klarytromycyna
(Klacid,
Fromilid)
Omeprazol
(Gasec,
Omar)
Duloksetyna
(Cymbalta)
DilDazem
(Dilzem,
Oxycardil)
Tyklopidyna
(Aclo=n,
Ticlo)
Fluoksetyna
(Andepin,
Bioxe=n)
Erytromycyna
(Erythromycinum)
Haloperydol
(Haloperidol)
Fluwoksamina
(Fevarin)
Metadon
(Methadone
Hydrochloride)
Sok z grejpfruta
Paroksetyna
(Seroxat)
Itrakonazol
(Orungal)
Chinidyna
(Chinidinum
sulfuricum)
Ketoconazol
(Ketokonazol)
Rytonawir
(Norvir)
Werapamil
(Isop=n)
7. Objętość dystrybucji
Czterema najważniejszymi parametrami decydującymi o losach leku w organizmie są:
• objętość dystrybucji - pozorna przestrzeń, w jakiej w organizmie znajduje się lek,
• okres półtrwania eliminacji - szybkość usuwania leku z organizmu,
• klirens - określa sprawność w eliminacji leku,
• biodostępność - mówi, jaka część leku, która została wchłonięta, trafi do krążenia ogólnego.
Objętość dystrybucji odnosi ilość leku w organizmie do jego stężenia we krwi. Dotyczny ona
objętości organizmu, w której zawarty byłby lek, gdyby miał występować w takim stężeniu jak we krwi.
V = ilość leku w organizmie/C
Wartość ta odzwierciedla ilość leku obecną w tkankach pozanaczyniowych oraz poza osoczem.
Zależy ona od relatywnego stopnia wiązania leku z miejscami receptorowymi i od dużym powinowactwie,
białkami osocza i tkanek, współczynnika rozmieszczenia leku w tłuszczach, a także od kumulacji w słabo
ukrwionych tkankach. Będzie różna także w zależności od wieku, płci budowy ciała pacjenta
oraz występowania choroby.
5

Zgodnie z powyższym równaniem, organizm jest traktowany jako jednorodny kompartment, ale model
ten nadaje się do stosowania w odniesieniu do większości leków.
8. Biologiczny czas półtrwania (obliczenia)
Okres półtrwania (t
1/2
) to czas, w którym stężenie osoczowe lub ilość leku podana do ustroju ulega
redukcji do 50%. Dla modelu jednokompartmentowego jest łatwy do określenia i stosowany
przy podejmowaniu decyzji o dawkowaniu.
W przybliżeniu zależność między czasem półtrwania, klirensem i objętością dystrybucji w stanie
stacjonarnym można przedstawić równaniem:
t
1/2
≅0,693·V
SS
/CL.
9. Klirens
Jest to szybkość eliminacji leku wszystkimi drogami w odniesieniu do stężenia leku w tym płynie
biologicznym, w którym możliwe jest dokonanie pomiaru. Wskazuje jaka objętość płynu biologicznego
(krew,osocze), została całkowicie oczyszczona (np. ml/min/kg).
CL=szybkość eliminacji/C
Klirens ogólnoustrojowy można obliczyć podczas stanu stacjonarnego (SS), przyjmując, że zostanie
on osiągnięty przy zrównaniu szybkości podawania i eliminacji leku, przy pełnej biodostępności:
Szybkość dawkowania = CL·C
SS
Klirens ogólnoustrojowy jest sumą klirensów dla różnych narządów.
10. Biorównoważność
Produkty lecznicze uważane są za równoważne farmaceutycznie, jeśli zawierają te same składniki
czynne i są identyczne pod względem siły działania lub stężenia, postaci leku i drogi podania.
Dwa farmaceutycznie równoważne produkty lecznicze uważane są za biorównoważne, jeśli w zakresie ilości
i szybkości biodostępność substancji czynnej w tych dwóch produktach nie różni się istotnie w odpowiednich
warunkach eksperymentalnych.
11. AUC
Powierzchnia pod krzywą przebiegu stężenia we krwi w czasie (area under the curve - AUC) może
być wykorzystana do wyliczenia klirensu dla eliminacji pierwszego rzędu:
CL=dawka/AUC.
Jest także stosowana jako miara biodostępności (100% dla leku podanego dożylnie).
12. Krążenie wątrobowo-jelitowe
Wiele leków w postaci sprzężonych metabolitów II fazy biotransformacji (np. z kwasem
glukuronowym) lub też w postaci niezmienionej jest wydalana wraz z żółcią do jelita, skąd mogą być one
wydalane z kałem lub wchłonięte do krwi. Resorpcja zwrotna niektórych metabolitów odbywa się w dalej
położonych odcinkach jelit pod wpływem enzymów hydrolitycznych bakterii flory jelitowej (np. Clostridium,
Bacteroides, bakterie grupy coli fermentujące laktozę oraz niektóre staphylokoki). Enzymy te powodują
rozszczepienie nieaktywnych metabolitów II fazy biotransformacji. Uwolniony w ten sposób środek czynny
ulega wchłonięciu i mówimy wtedy o krążeniu wątrobowo-jelitowym.
Do interakcji leków ulegających resorpcji zwrotnej w jelitach na opisanych powyżej zasadach może
dochodzić wtedy, gdy flora bakterii jelitowych zmniejszy się pod wpływem jednoczesnego podawania leków
przeciwbakteryjnych. Przykładem takich interakcji mogą być rzadkie przypadki braku skuteczności
doustnych leków antykoncepcyjnych zawierających etinyloestradiol przy jednoczesnym stosowaniu
antybiotyków takich jak ampicylina czy tetracyklina. Również w przypadku sulfasalazyny podawanie
jednocześnie antybiotyków redukujących florę jelitową (np. rymfampicyny) może zmniejszać
biotransformację sulfasalazyny przez bakterie flory jelitowej do jej aktywnego metabolitu kwasu 5-
aminosalicylowego (mesalazyny).
6
13. Krążenie jelitowo-żołądkowe
Niniejsza koncepcja opiera się na fakcie, że leki z krwi, które są słabymi zasadami (np. Diazepam)
mogą przeniknąć do światła żołądka, gdzie „ściągnie” je kwas solny, czyli dojdzie do przemieszczenia
przez błony zgodnie z gradientem pH. Następnie razem z treścią żołądkową lek wędruje do jelita, gdzie
jest ponownie wchłaniany. Zjawisko ulega nasileniu przy wzmożonym wydzielaniu kwasu solnego
do żołądka, czyli np. przy posiłku.
14. Prolek
Nieaktywne substancje lecznicze, które na drodze przemian metabolicznych uzyskują aktywność,
nazywane są prolekami.
15. Efekt pierwszego przejścia
Leki podane doustnie dostają się do krwiobiegu niemal wyłącznie przez układ krążenia wrotnego,
przez co cała wchłonięta dawka leku przechodzi najpierw przez wątrobę. W wątrobie leki poddane działaniu
enzymów ulegają biotransformacji. Jeżeli lek jest intensywnie metabolizowany, to znaczny ułamek dawki
zostaje "wyekstrahowany" z krwi i ulega przemianom, zanim dostanie się do krążenia ogólnego,
co zmniejsza ewidentnie jego dostępność biologiczną.
Czyli efekt pierwszego przejścia to zmniejszenie ilości leku dostającego się do krążenia ogólnego
po wchłonięciu, co jest związane głównie z przejściem leku przez krążenie wrotne i metabolizm w wątrobie.
16. Kategorie stosowania leków w ciąży (wg. FDA)
Amerykańska Agencja Leków i Żywności (FDA) sklasyfikowała leki pod względem bezpieczeństwa
ich stosowania w czasie ciąży.
Kategoria FDA
Opis
Kategoria A
Leki ,które były badane u kobiet w ciąży i nie wykazały szkodliwego działania na płód.
Kategoria B
Leki z którymi wykonane eksperymenty na zwierzętach nie wykazały szkodliwego działania na płód,
nie wykonano jednak badań kontrolnych u kobiet w ciąży.
lub
Leki których szkodliwe działanie na płód zostało stwierdzone na zwierzętach, lecz nie znalazło
potwierdzenia u kobiet w ciąży.
Kategoria C
Badania na zwierzętach wykazały działania niepożądane leków na płód lecz brak jest odpowiednich
i kontrolowanych badań u kobiet ciężarnych.
lub
Leki z którymi nie przeprowadzono wystarczających badań na zwierzętach i brak odpowiednich
i kontrolowanych badań u kobiet ciężarnych.
Leki te można stosować tylko w przypadkach , gdy korzyść wynikająca ze stosowania ich u matki
przewyższa ryzyko niepożądanego działania u płodu.
Kategoria D
Odpowiednie i kontrolowane badania lub obserwacje wykazały ,że leki te podawane w okresie ciąży
stanowią zagrożenie dla płodu.
Leki z tej kategorii można stosować u kobiet w ciąży w stanach zagrażających życiu matki i tylko
w przypadkach gdy leki kategorii A,B, C nie mogą być zastosowane lub są nieskuteczne.
Kategoria X
Leki o udowodnionym działaniu szkodliwym na płód.
Stosowanie tych leków jest bezwzględnie przeciwwskazane u kobiet w ciąży lub kobiet które mogą
i chcą zajść w ciąże.
17. Działania niepożądane leków – rodzaje
Typ A (Augmented effects)
Działania uboczne, które można przewidzieć – wynikają z mechanizmu działania farmakologicznego.
Ich nasilenie zależy od zastosowanej dawki. Ustępują po zmniejszeniu dawki lub odstawieniu leku.
7

Do działań niepożądanych z grupy A zalicza się także działania wynikające z interakcji leków,
które da się przewidzieć, np. efekty uboczne spowodowane zwiększeniem się stężenia jakiegoś leku
w surowicy, na skutek jego wypierania z połączeń z białkami przez zastosowany jednocześnie inny lek.
Działania tego typu stanowią ok. 70-80% wszystkich obserwowanych działań ubocznych leków.
Typ B (Bizzare effects)
Działania uboczne nieprzewidywalne, niezależne od podanej dawki leku. Działania te nie wynikają
z mechanizmu działania farmakologicznego. Często są wynikiem reakcji immunologicznej (uczulenia) na lek.
Działania tego typu stanowią ok. 20% wszystkich obserwowanych działań niepożądanych leków
i często są przyczyną wycofania leku z obrotu, lub ograniczeń w jego stosowaniu.
Typ C (Chronic effects)
Działania uboczne występujące po długotrwałym stosowaniu leku (najczęściej w terapii chorób
przewlekłych). Mechanizm ich powstawania najczęściej jest znany i wynika z właściwości farmakologicznych
leku.
Typ D (Delayed effects)
Działania uboczne pojawiające się po długim czasie od zastosowania leku, niezależnie od tego
jak długo stosowany był sam lek.
Typ E (End-of-therapy effects)
Działanie niepożądane występujące po nagłym odstawieniu leku. Zwykle jest to zaostrzenie choroby,
następujące po zaniechaniu terapii. Leki wykazujące działania niepożądane typu E powodują zwiększenie
(zaostrzenie) objawów choroby po odstawieniu leku ponad poziom na którym te objawy byłyby, gdyby leku
w ogóle nie zastosowano.
18. Mechanizmy działania leków – receptor, kanał, enzym, transporter
Receptory dla fizjologicznych cząstek regulacyjnych można zakwalifikować do kilku rodzin,
wykazujących podobieństwo zarówno pod kątem mechanizmu działania, jak i struktury cząsteczkowej:
• białkowe kinazy receptorowe i inne o aktywności enzymów,
• cyklazy guanylanowe (związane z białkami o aktywności kinaz),
• sygnalizacja receptorowa aktywowana proteazami
• kanały jonowe,
• receptory sprzężone z białkiem G (GPCR, np. cyklaza adenylanowa, fosfolipaza C, fosfodiesterazy),
• czynniki transkrypcyjne
Do rozpoczęcia konkretnych szlaków biochemicznych niektóre receptory korzystają z pośrednictwa
cząstek sygnałowych (tzw. II przekaźniki), których produkcja jest inicjowana po połączeniu ligandu
z receptorem. Tymi przekaźnikami mogą być: cAMP, cGMP, cADP, Ca
2+
, fosforan inozytolu, diacyglicerol,
NO, CO.
Punkty uchwytu dla leków mogą także stanowić transportery błonowe, np. transportery
neuroprzekaźników będące celem dla leków przeciwdepresyjnych (hamowanie wychwytu zwrotnego).
19. Agonista, antagonista, częściowy agonista
Leki, które wiążą się z receptorami fizjologicznymi i uruchamiają efekty regulacyjne endogennych
cząstek sygnalizacyjnych, nazywane są agonistami. Inne leki wiążą się z receptorami bez wywołania
efektów regulacyjnych, ale blokują wiązanie endogennych agonistów. Takie cząstki, które nie mają własnej
aktywności stymulacyjnej i mogą wywołać użyteczne efekty poprzez hamowanie działania agonisty
(np. współzawodnictwo o miejsce wiążące agonistę), określa się jako antagonistów. Czynniki, które są tylko
częściowo aktywne jak agoniści, niezależnie od ilości, w której występują, nazywane są częściowymi
agonistami, natomiast te, które stabilizują receptor w jego nieaktywnej konformacji, to odwrotni agoniści.
8

Wyróżnia się następujące rodzaje antagonizmu:
• antagonizm konkurencyjny (kompetycyjny) - 2 leki (agonista i antagonista) mające taki sam punkt
uchwytu działania konkurują o ten sam receptor, mogą się wzajemnie wypierać z wiązania z receptorem,
np. naloksanem i morfiną w obrębie receptora opioidowego;
• antagonizm funkcjonalny – 2 leki o różnym punkcie uchwytu wywołują przeciwne działanie,
np. adrenalina zwęża naczynia pobudzając w nich receptory adrenergiczne, actylocholina rozszerza
naczynia przez pobudzenie zakończeń przywspółczulnych zmniejszając pośrednio działanie presyjne
adrenaliny;
• antagonizm chemiczny – 2 leki reagują ze soba tworząc związek słabszy lub nieczynny biologicznie,
zjawisko wykorzystywane w leczeniu zatruć, np. zatrucie solami baru leczy się siarczanem sodu, wytrąca się
nietoksyczny i nierozpuszczalny w wodzie siarczan baru.
20. Up-regulation, down-regulation
Ciągła stymulacja komórki agonistą skutkuje desensytyzacją (definiowana także jako adaptacja,
refrakcja, down-regulacja) w taki sposób, że efekt, który występuje po ciągłej lub wielokrotnej ekspozycji
na to samo stężenie leku, jest zmniejszony. Gdy zjawisko to jest gwałtowne, mówi się o wystąpieniu
tachyfilaksji. Gdy przebieg adaptacji jest wolniejszy mówi się wówczas o tolerancji. Do uzyskania tej samej
reakcji staje się konieczne stopniowe zwiększanie dawki leku.
Sensytyzacja (up-regulacja), w przeciwieństwie do tolerancji, polega na stopniowym narastaniu
działania niektórych substancji. Pojawia się w wyniku wielokrotnego, lecz przerywanego, podawania.
Sensytyzacja dotyczy w szczególności aktywacji ruchowej wywoływanej przez środki psychostymulujące,
jak amfetamina, kokaina, metylfenidat, fencyklidyna, nikotyna, a także przez inne środki psychoaktywne,
jak morfina i alkohol.
21. Tachyfilaksja
Tachyfilaksja to szybkie wygaszanie działania leku, np. przy dożylnym podaniu efedryny po każdym
następnym wstrzyknięciu występuje coraz słabsze podwyższenie ciśnienia tętniczego krwi, aż w końcu
mięśnie gładkie przestają reagować na lek.
22. Tolerancja
Tolerancja jest procesem podobnym do tachyfilaksji lecz rozwijającym się wolniej, do uzyskania
tej samej reakcji konieczne staje się stopniowe zwiększanie dawki leku, np. lecznicza dawka morfiny wynosi
10-15 mg w przypadku rozwoju tolerancji może znacznie przewyższać dawkę toksyczną, ale niezbędną
do działania przeciwbólowego.
• Łatwo rozwija się w stosunku do leków nasennych (poch. kwasu barbituranowego),
przeciwbólowych, amfetaminy, lizergidu.
• Arsenofagia – zażywanie arszeniku w celu poprawy samopoczucia prowadzące do zwiększania
dawek bez objawów zatrucia.
• Związana jest z uzależnieniami lekowymi.
Mechanizmy rozwoju tolerancji mogą być różne:
• nasilenie metabolizmu leków wskutek indukowania enzymów mikrosomalnych wątroby –
barbiturany, fenytoina, NLPZ – tzw. tolerancja metaboliczna;
• zmiany wrażliwości receptorów komórkowych lub zmian biochemicznych w komórkach docelowych,
na które działają leki – leki psychotropowe;
• kilka mechanizmów łącznie – metaboliczny i komórkowy w narządach, na które działa lek,
np. tolerancja na azotany w wyniku wyczerpania się grup SH w komórkach;
• tolerancja funkcjonalna w stosunku do leków działających na OUN polegająca na powstawaniu
mechanizmów korygujących zaburzenie, np. korekcja zaburzeń koordynacji ruchowej i działania
uspokajającego etanolu i leków nasennych.
W rozwoju tolerancji istotną rolę pełni także mechanizm uczenia się.
9

23. Lekozależność
O zależności od leku (a także innych czynników nagradzających) można powiedzieć wówczas,
kiedy dochodzi do takiej zmiany w psychice, że poszukiwanie leku lub innego czynnika nagradzającego staje
się głównym motywem życia osoby uzależnionej. Nie ulega wątpliwości, że zależność rozwija się wskutek
powstawania zmian adaptacyjnych pod wpływem stosowanego przewlekle narkotyku, czyli oprócz zmian
psychicznych obecne są także fizyczne i wiąże się z nimi szereg objawów.
24. Uzależnienie fizyczne i psychiczne
Zależność fizyczna jest stanem, który rozwija się w rezultacie adaptacji, powstającej przez powstanie
nowej homeostazy spowodowane wielokrotnym przyjmowaniem narkotyku. Działa on na liczne układy,
które początkowo były w równowadze. Osoba, u której obserwuje się stan adaptacji lub zależności fizycznej,
wymaga stałego podawania narkotyków, aby móc podtrzymać normalne funkcjonowanie. Jeżeli podawanie
jakiegoś narkotyku zostanie nagle przerwane, powstają kolejne zaburzenia równowagi i układ, na który
działał narkotyk, musi przejść przez szereg procesów dostosowujących do nowej równowagi bez leków.
Zależność fizyczna jest zjawiskiem biologicznym.
Zależność psychiczna charakteryzuje się natrętnym, niekiedy niekontrolowanym dążeniem do użycia
leku (głód narkotyku), jego poszukiwaniem i dążeniem do zdobycia za wszelką cenę, oraz używaniem
mimo oczywistych poważnych zagrożeń dla zdrowia i życia.
25. Zespół abstynencyjny
Zespół abstynencji, występuje po nagłym zaprzestaniu zażywania narkotyku u uzależnionego
fizycznie. Objawy mają przynajmniej dwa źródła:
• usunięcie leku, który powodował uzależnienie
• nadmierne pobudzenie OUN, wynikające z readaptacji przy braku narkotyku.
Zespoły abstynencji są charakterystyczne dla każdej grupy substancji i zazwyczaj ich manifestacją
są efekty przeciwne niż wyjściowe efekty działania substancji, zanim pojawiła się na nią tolerancja.
Objawy tego stanu nie muszą oznaczać, że dana osoba jest uzależniona lub nadużywa substancji
psychoaktywnych. Zespół abstynencji (i zależność fizyczną) można wykazać, gdy pacjent zażywa lek
w prawidłowej dawce i zostanie on nagle odstawiony zamiast robienia tego stopniowo.
10
Wyszukiwarka
Podobne podstrony:
F Zadania do kol 1 id 167111 Nieznany
bazy danych kol 2 id 81577 Nieznany (2)
kpp arkusz 12 kol id 249459 Nieznany
farma two id 168068 Nieznany
farma gielda id 168081 Nieznany
alfabet 5 kol id 56849 Nieznany (2)
alfabet 4 kol id 56847 Nieznany (2)
alfabet 3 kol id 56845 Nieznany (2)
cecot zadania kol 1 id 109431 Nieznany
kol id 239045 Nieznany
pomocnicze dzienne 1 kol id 375 Nieznany
alfabet 2 kol id 56843 Nieznany (2)
F Zadania do kol 1 id 167111 Nieznany
bazy danych kol 2 id 81577 Nieznany (2)
Kol ME3 id 239139 Nieznany
kol kul2 id 239133 Nieznany
farma kliniczna wyklad I id 168 Nieznany
Kol PE1 10 id 239141 Nieznany
więcej podobnych podstron