1
Związki aromatyczne
4 stopnie nienasycenia. Brak reakcji A
E
Źródła: - piroliza węgla
→
smoła pogazowa;
- reforming ropy naftowej
Nazewnictwo
przedrostek
– podstawnik
CH
3
Br
Br
Cl
NO
2
Br
CH
3
metylobenzen
(toluen)
1,2-dibromobenzen
o-dibromobenzen
1-chloro-3-nitrobenzen
(m-chloronitrobenzen)
1-bromo-4-metylobenzen
(p-bromo
toluen
)
OH
NH
2
CHO
OH
Br
Br
Br
benzenol
(fenol)
benzenoamina
(anilina)
benzaldehyd
2,4,6-tribromofenol
Areny
– alkilobenzeny
Ph
, Φ – C
6
H
5
-
Bn - -CH
2
C
6
H
5
(
„krewny” allilu)
Budowa
Sekstet zdelokalizowanych elektr. π; hybrydyzacja sp
2
at. C
Niskie ciepło wodorowania – miara stabilności; E
rez.
= ok. 30 kcal/mol
Energia rezonansu
– stabilizacja aromatyczna
PAHs
– karcynogeny
naftalen
antracen
2
nietrwałe, b.reaktywne
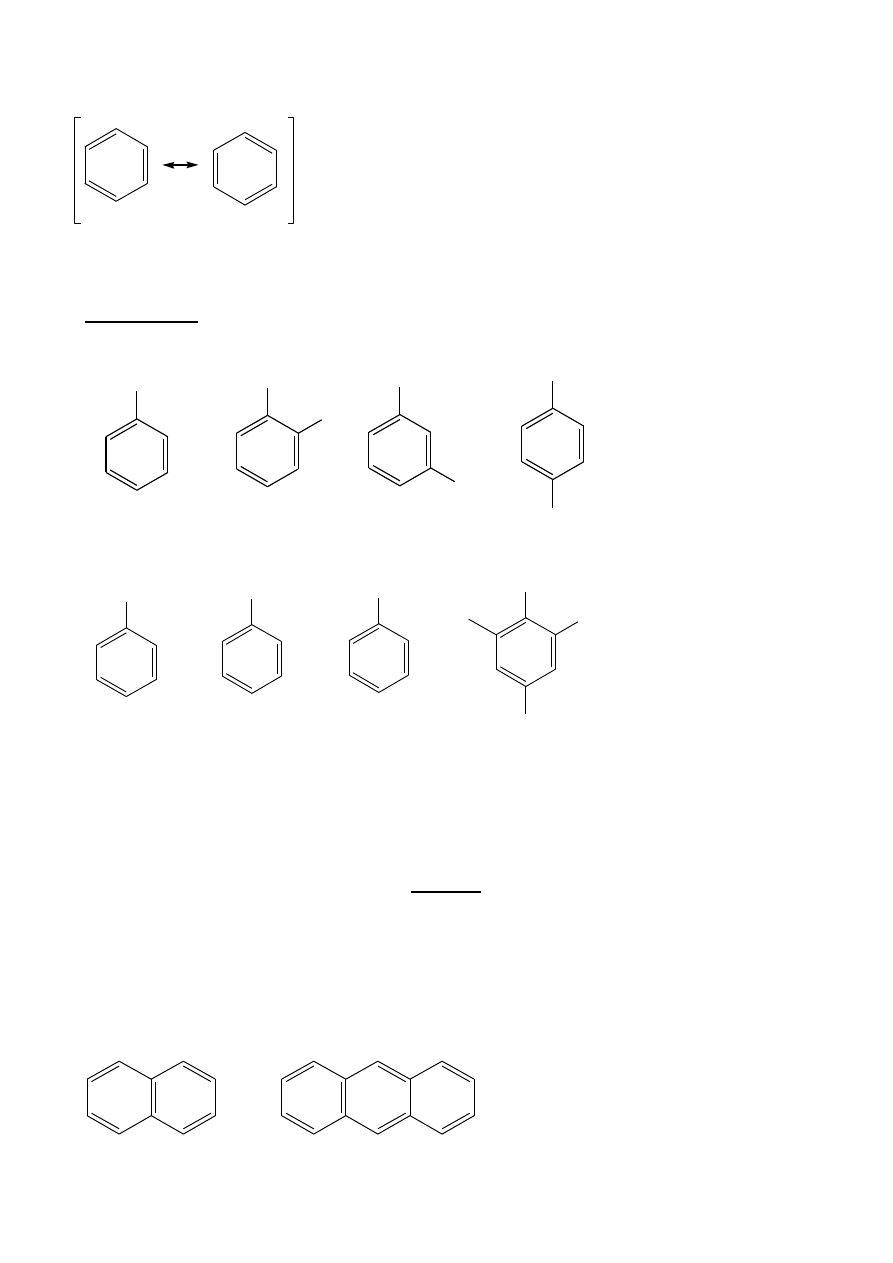
Kryteria aromatyczności:
-
układ cykliczny;
-
sprzężony układ
-
elektronowy (π = 4n + 2; Hűckel, 1931 r.);
-
wszystkie atomy układu muszą mieć niezhybrydyzowany orbital p.
Cykliczne polieny:
A.
antyaromatyczne (destabilizowane przez sprzężenie); π = 4 n
B.
niearomatyczne, niepłaskie
C.
aromatyczne;
π = 4n + 2
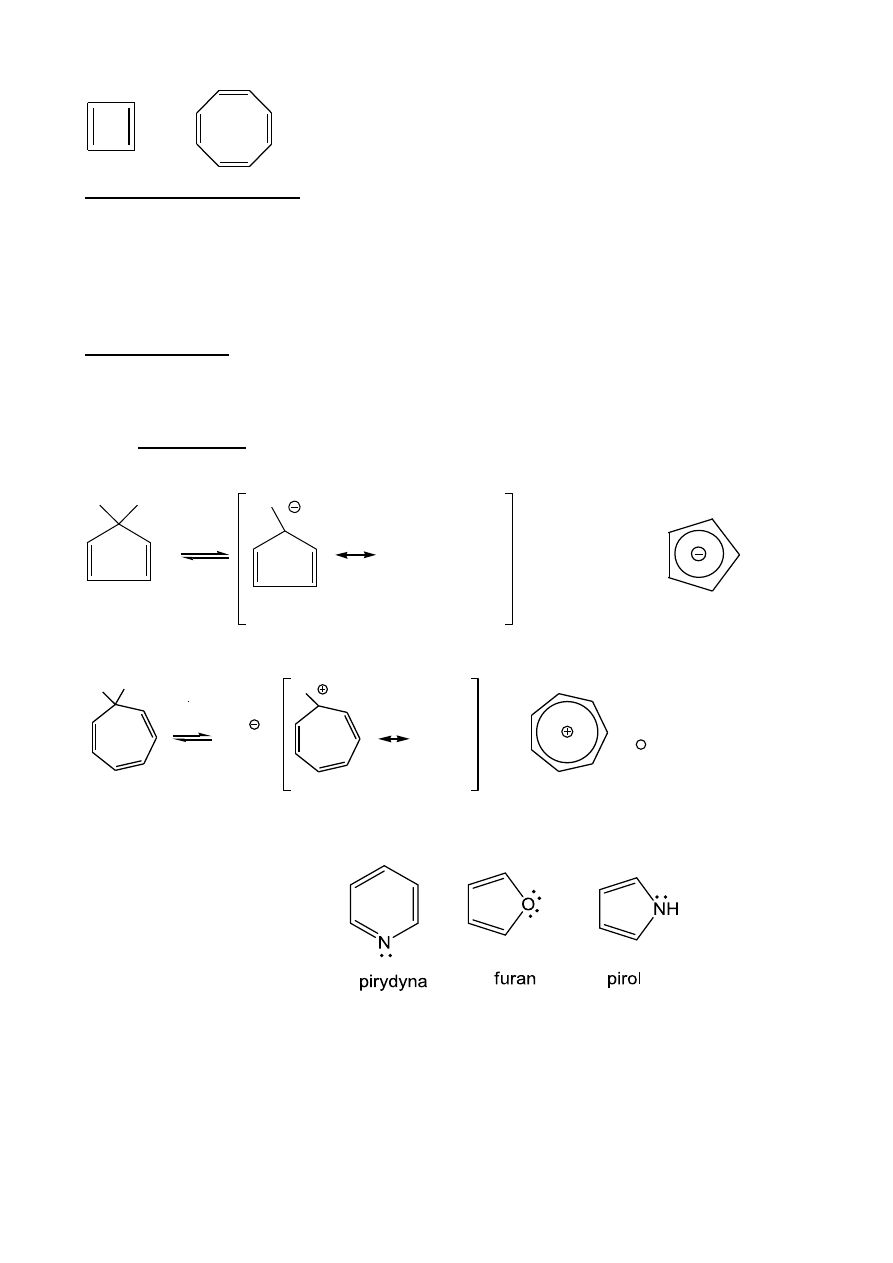
H
H
H
...
pK
a
= 16
H
H
H
Br
+
...
Br
2,
T
1891r, nieznana trwala substancja
Br
Inne aromaty:
wolna para elektronowa: orbital: ...
3
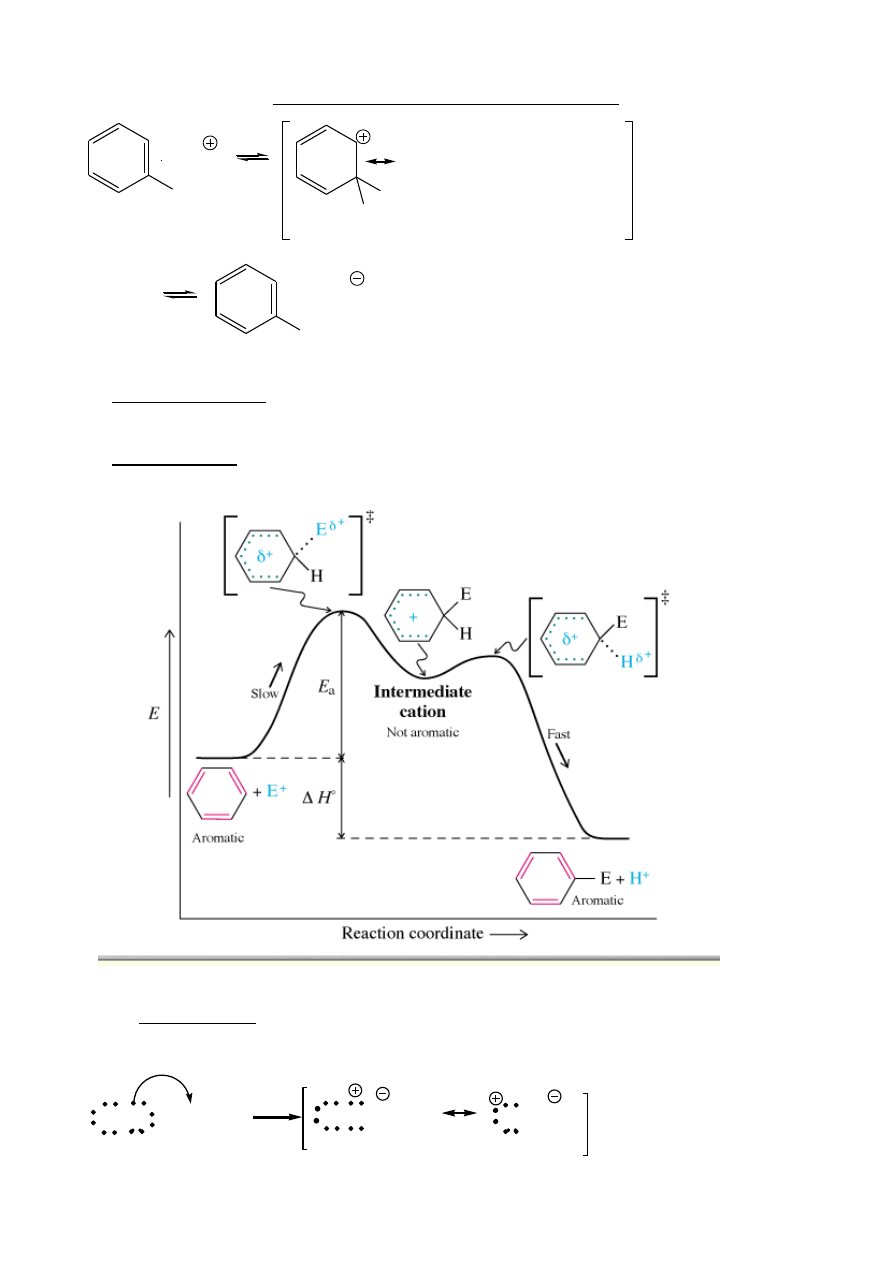
Elektrofilowe podstawienie aromatyczne
H
+ E
H
E
E
+ H
Etapy:
1. Atak elektrofilowy
– termodynamicznie niekorzystny, ładunek rozproszony, ale utrata
aromatyczności
2. Utrata protonu
– bardziej korzystny niż atak Nu (odzysk aromatyczności)
Mechanizm podstawienia elektrofilowego:
1. Halogenowanie
Katalizator: FeX
3
, AlX
3
Br-Br
+ FeBr
3
Br-Br-FeBr
3
Br FeBr
4
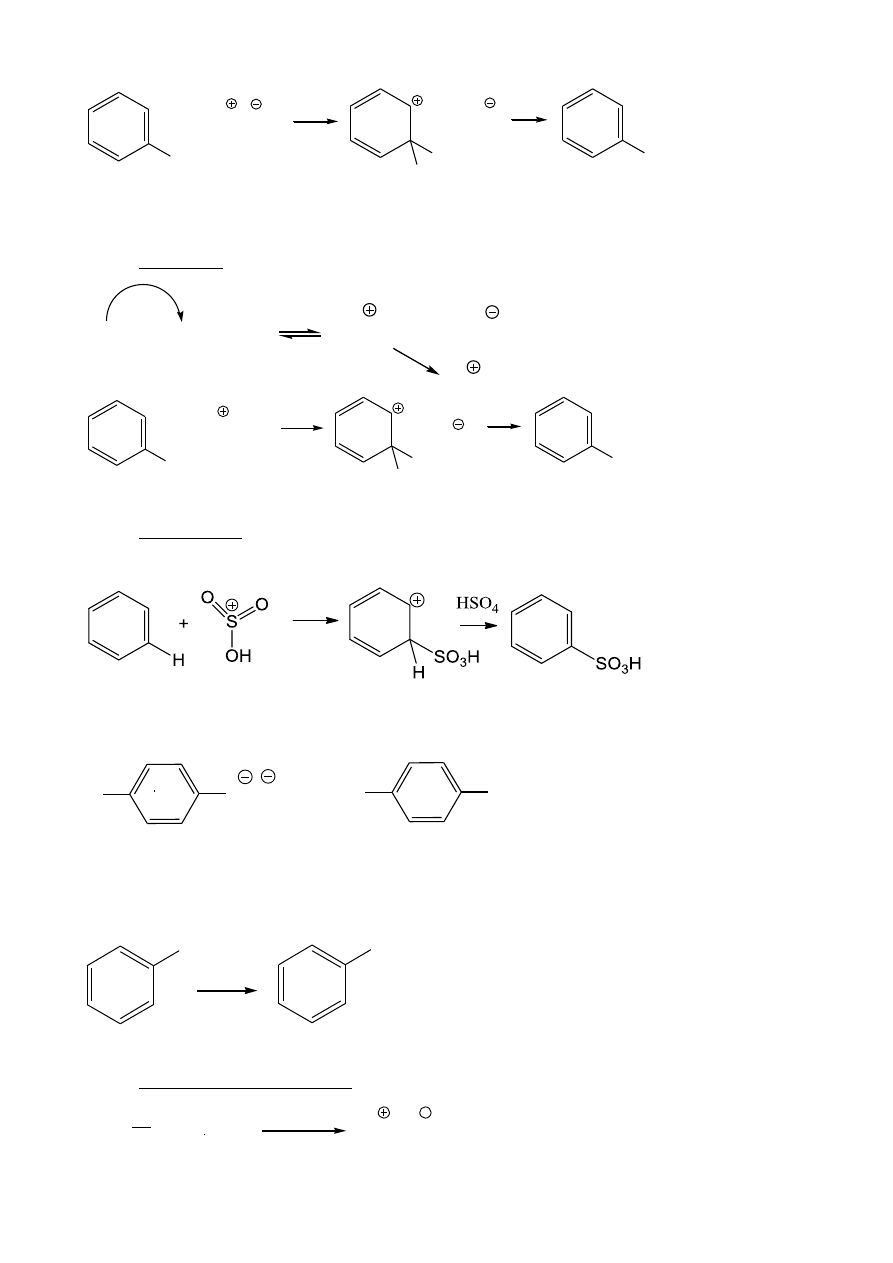
4
H
+ Br-Br-FeBr3
H
Br
+ FeBr4
Br
+ HBr + FeBr3
I -endotermiczne – nie zachodzi, F – wybuchowe
2. Nitrowanie
HO-NO
2
+ H-OSO
3
H
H
2
O-NO
2
+ HSO
4
NO
2
+ H
2
O
H
+
H
NO
2
NO
2
+ H
2
SO
4
+
HSO
4
O=N=O
3. Sulfonowanie
SO
3
– silny –I trzech O → elektrofilowy at. S
ogrzewanie z wodą → benzen
grupa sulfonowa – odwracalną gr. kierującą
R
HNR
SO
3
Na
SO
2
NHR'
detergenty (niebiodegradacyjne)
sulfonamidy
pochodne kw. benzenosulfonowego – barwniki
chlorek benzenosulfonowy – synteza (przekształcanie OH w dobrą L)
SO
3
Na
SO
2
Cl
PCl
5
+ POCl
3
+ NaCl
4. Alkilowanie Friedla – Craftsa
R
Cl + AlCl
3
R AlCl
4
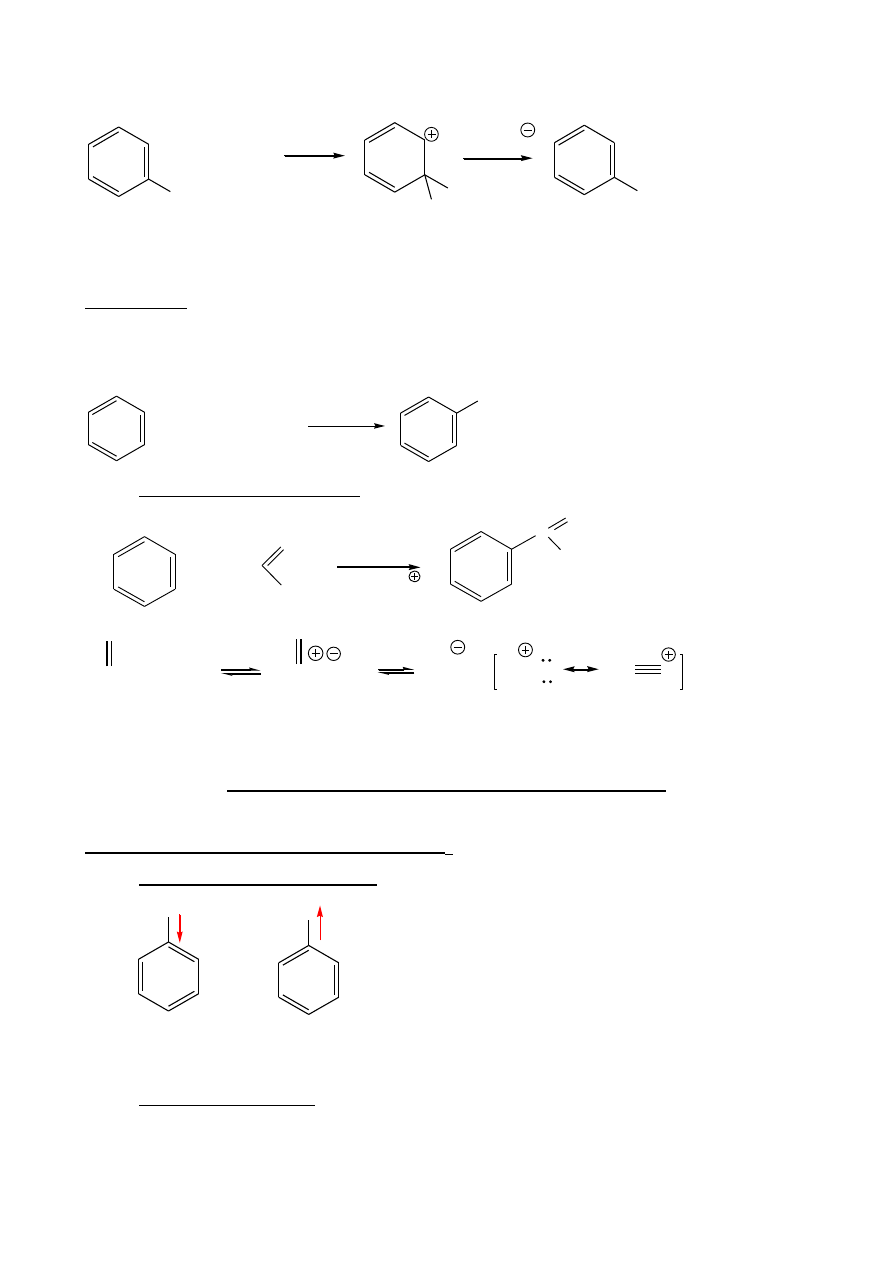
5
H
+ R-CH
2
-Cl
AlCl
3
H
CH
2
R
AlCl
4
CH
2
R
+ HCl + AlCl
3
2
, 3
R-Cl
Ograniczenia:
- polialkilowanie;
- przegrupowanie karbokationów;
- brak reakcji w układach zdezaktywowanych.
+
CH(CH
3
)
2
CH
3
CH
2
CH
2
Br
AlCl
3
+ HBr
5. Acylowanie Friedela – Craftsa
+
R-C
O
Cl
1. AlCl
3
2. H
2
O, H
C
O
R
R-C -X
O
+ AlCl
3
O
AlXCl
3
+
R-C
O
R-C-X-AlCl
3
R-C=O
Kompleks kw. Lewisa z fenyloketonami → konieczność > 1 eq. AlCl
3
, przeróbka wodna
Podstawienie elektrofilowe w pochodnych benzenu
Aktywacja i dezaktywacja pierścienia na S
E
1.
Wpływ indukcyjny ( przez
)
D
A
D - donor (alkil, aryl)
A - akceptor (-CF
3,
-NR
3,
-OR, -X, -COR, -CN, -NO
2,
-SO
3
H)
2.
Wpływ rezonansowy
Rezonansowe
„dawanie” elektronów:
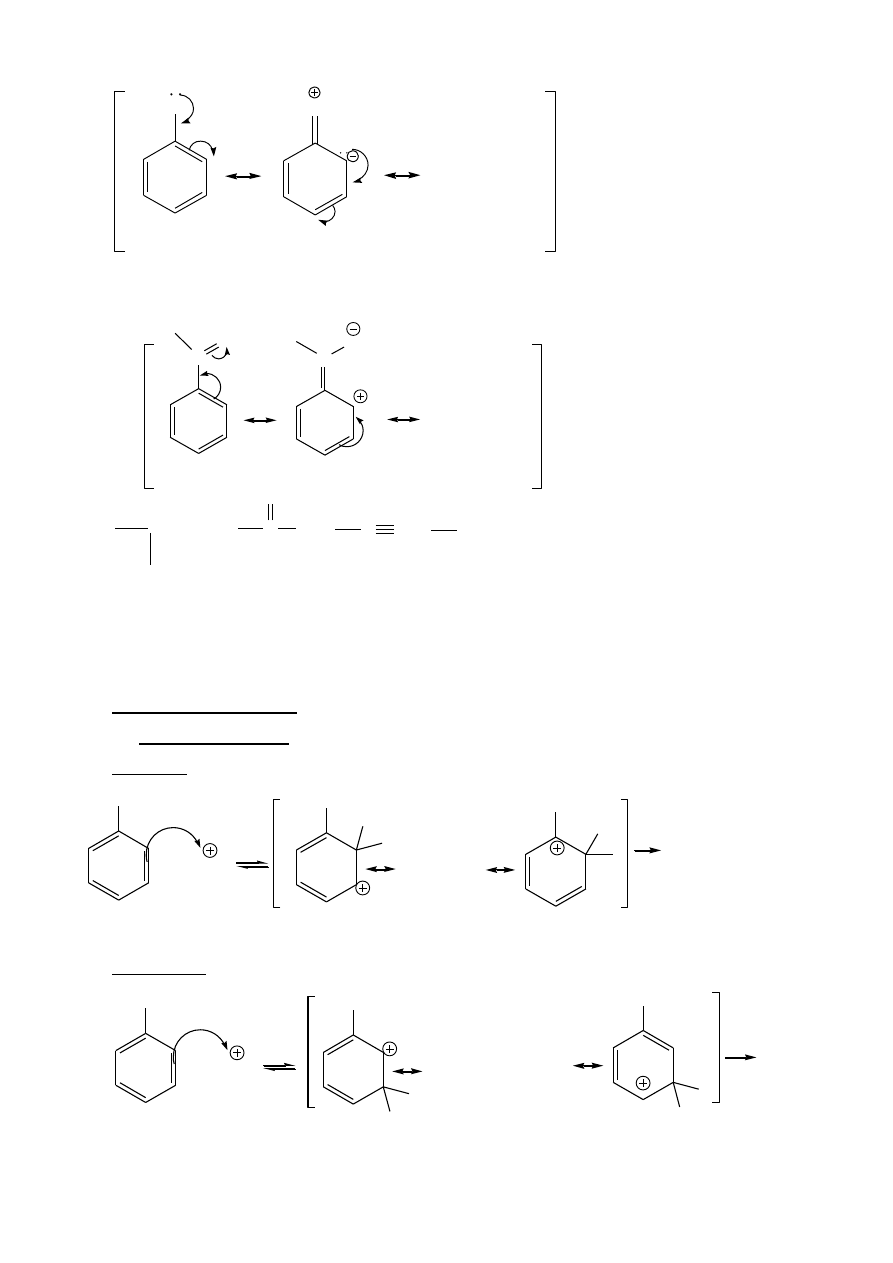
6
D
D
...
D = -NR
2
, -OR, -X
Rezonansowe „wyciąganie” elektronów:
B
...
A
B
A
B=A
=
C
O
R,
C
N,
NO
2,
-SO
3
H
Nitrowanie C
6
H
5
R (v
rel
):
R =
OH
CH
3
H
Cl CF
3
NO
2
v =
1000 25 1 0.03
3∙10
-5
6 ∙10
-8
EFEKTY KIERUJĄCE:
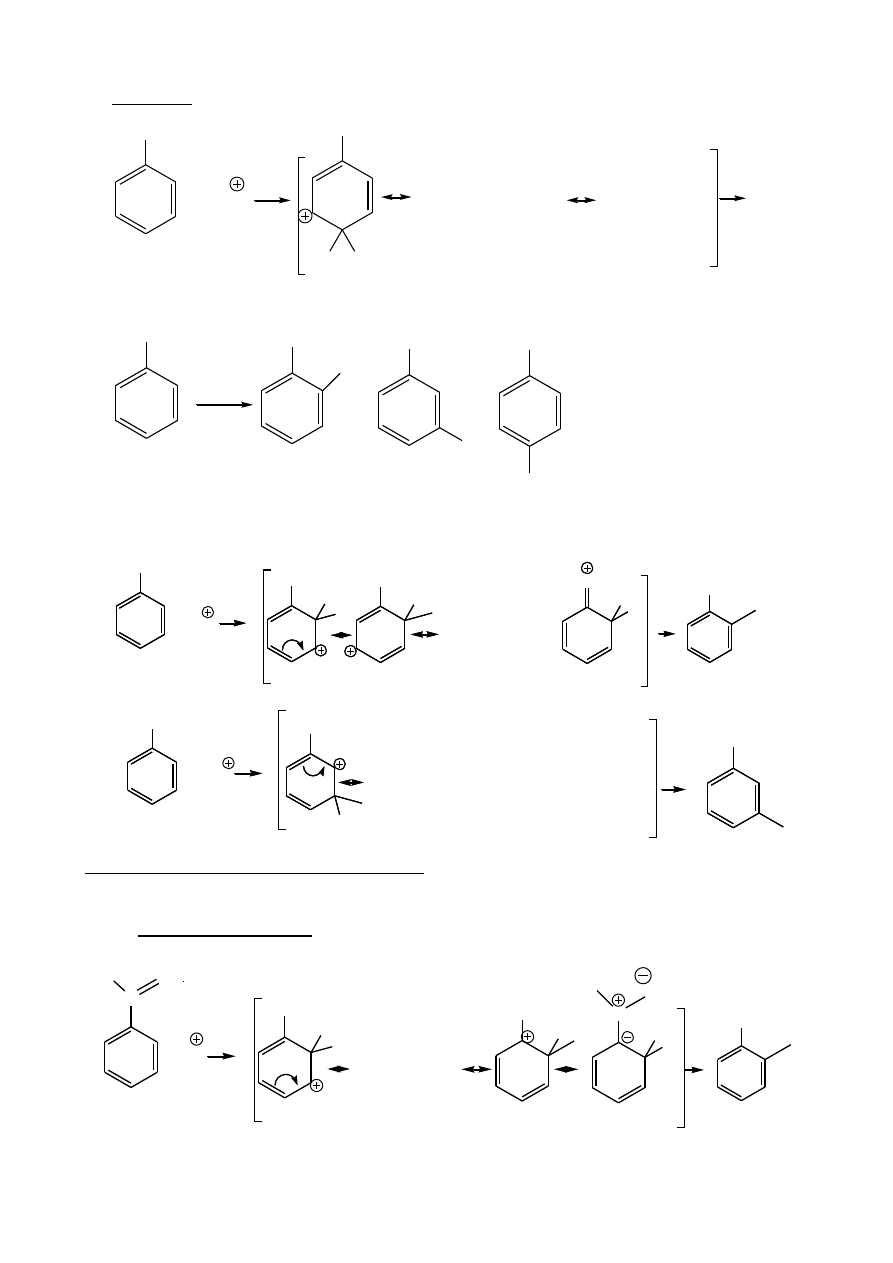
A. Grupy donorowe:
atak orto:
+ E
CH
3
H
E
CH
3
CH
3
H
E
główny kontrybutor
atak
–meta:
+ E
CH
3
CH
3
H
E
CH
3
H
E
mniej stabilny karbokation
7
atak para:
+ E
CH
3
CH
3
H
E
stabilny kation cykloheksadienylowy
CH
3
CH
3
CH
3
CH
3
Br
2,
FeBr
3
Br
Br
Br
+
+
40%
< 1%
60%
Donory „rezonansowe”:
NH
2
NH
2
+ E
+ E
NH
2
NH
2
NH
2
NH
2
H
E
H
H
E
E
NH
2
NH
2
E
E
NH
2
NH
2
+ E
+ E
atak orto
atak meta
NH
2
NH
2
NH
2
NH
2
H
E
H
H
E
E
NH
2
NH
2
E
E
Grupy donorowe kierują w poz. orto i para.
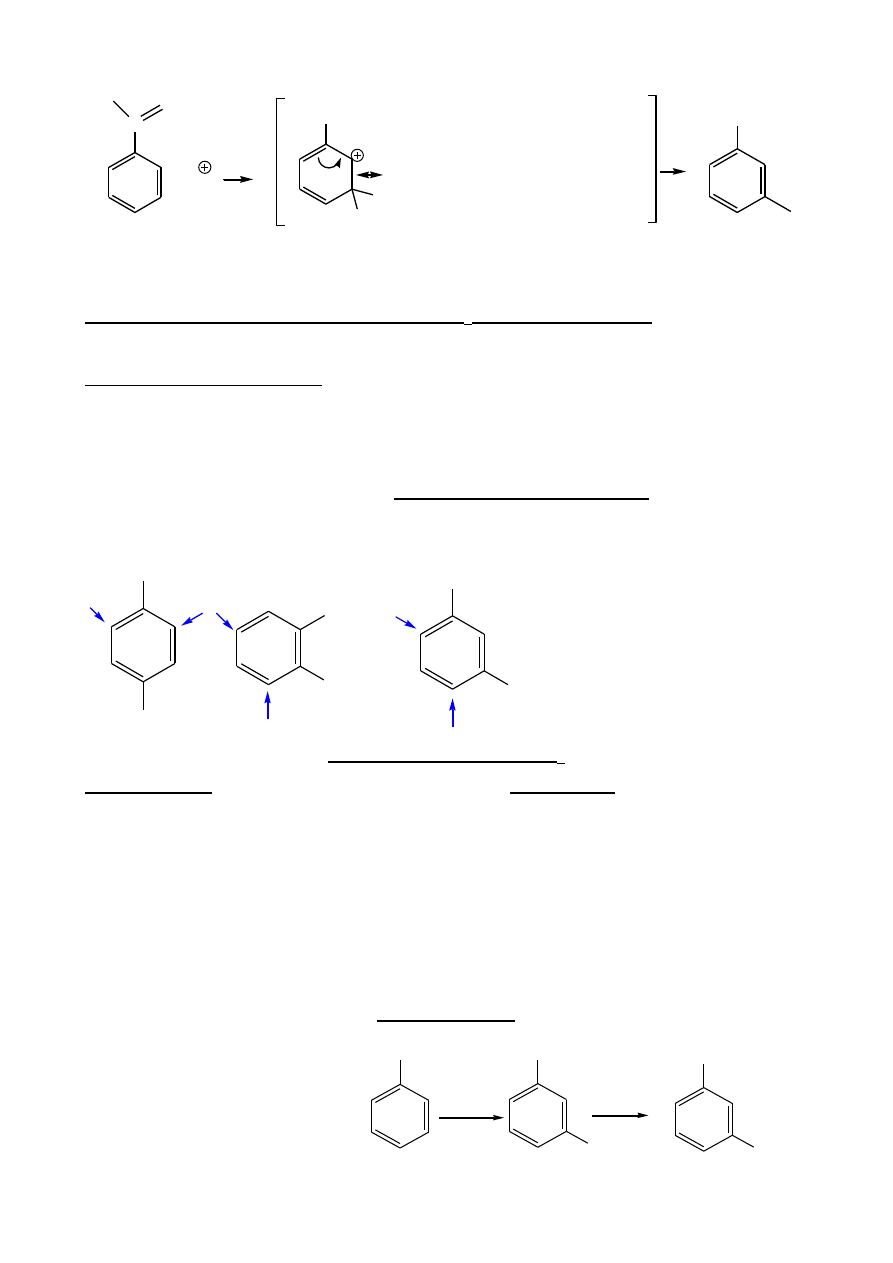
B.
Grupy akceptorowe:
C
+ E
atak orto
COOH
COOH
C
H
E
H E
H
E
COOH
E
O
HO
O
HO
8
C
+ E
atak meta
COOH
H
E
COOH
E
O
HO
kation mniej destabilizowany
atak para
– jak orto
Grupy akceptorowe dezaktywują pierścień na S
E
i kierują w poz. meta-
C. Podstawniki halogenowe
Silne indukcyjne wyciąganie elektronów – dezaktywacja
Rezonansowa stabilizacja kationu przy postawieniu orto i para. - kierowanie orto i para
S
E
w dipodstawionych benzenach
– najsilniejszy aktywator decyduje:
-NR
2
, -OR > -X, -
R > kierujące meta
OH
CH
3
NH
2
COOH
OCH
3
Br
Wpływ podstawników na S
E
kierujące o-, p-
kierujące m-
1. silne aktywatory:
1. silne dezaktywatory:
-NR
2
, -NHCOR, -OR
-NO
2
, -CF
3
, -NR
3
, -COOR, -COR,
-SO
3
H, -CN
2. słabe aktywatory:
alkil, fenyl
3. słabe dezaktywatory:
-X
Strategia syntez
3-bromobenzamina
(3-bromoanilina)
NO
2
Br
NH
2
Br
NO
2
Br
2,
FeBr
3
Fe,HCl
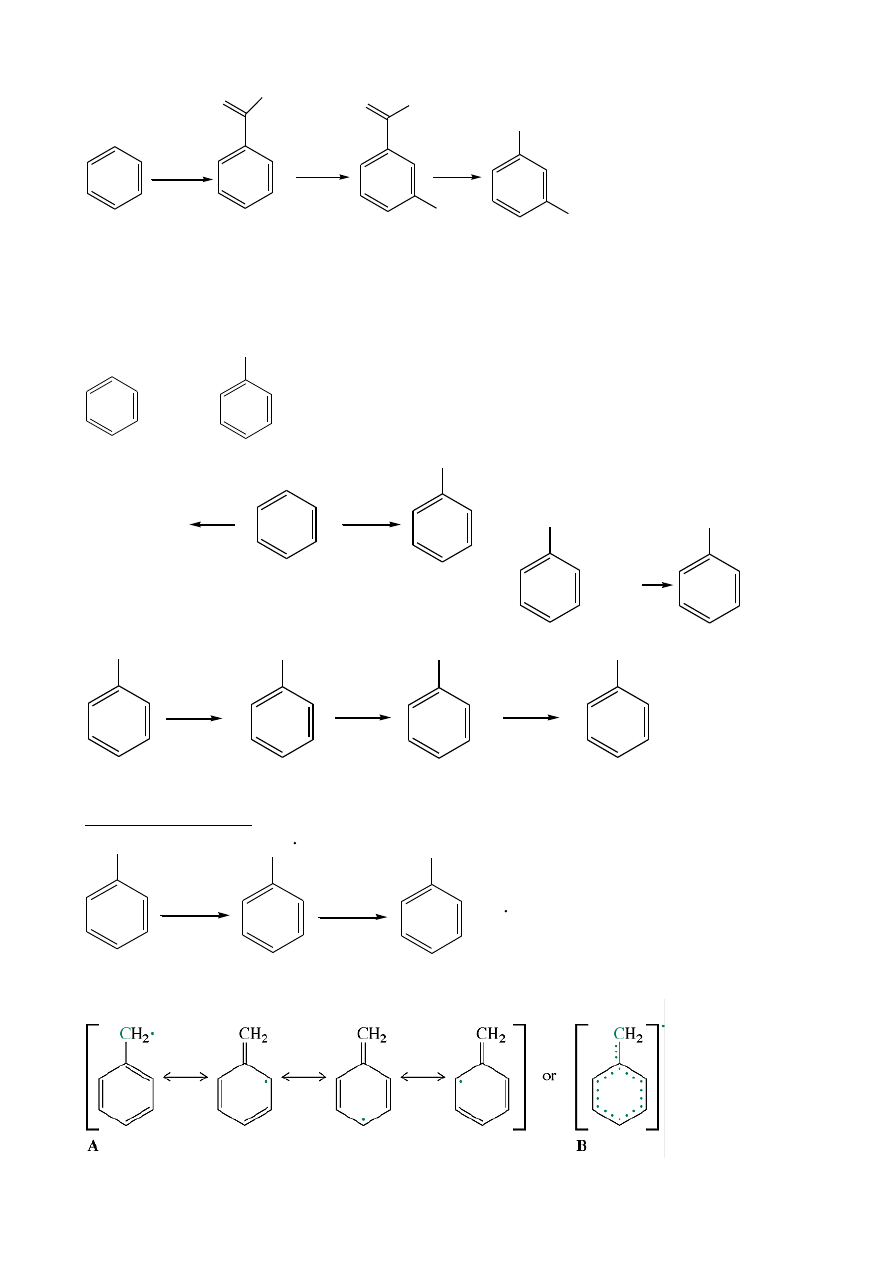
9
Cl
Cl
2,
FeCl
3
CH
2
CH
3
Cl
CH
3
COCl
O
CH
3
CH
3
O
Zn(Hg)
HCl
redukcja Clemmensena
Odwracalne sulfonowanie jako metoda blokowania lub kierowania.
Niezwykła reaktywność atomu węgla fenylometylowego (benzylowego)
CH
3
Br
brak reakcji
Br
2
Br
2,
FeBr
3
CH
2
H
CH
2
Cl
Cl
2,
hv
Cl
2,
hv
Cl
2,
hv
CHCl
2
CCl
3
trichlorometylobenzen
Mechanizm rodnikowy – jak alkany czy allilowe halogenowanie alkenów.
CH
3
CH
2
CH
2
X
+ X
X
2
-HX
CH
2
H
+ Br
2
T
CH
2
Br
+ HBr
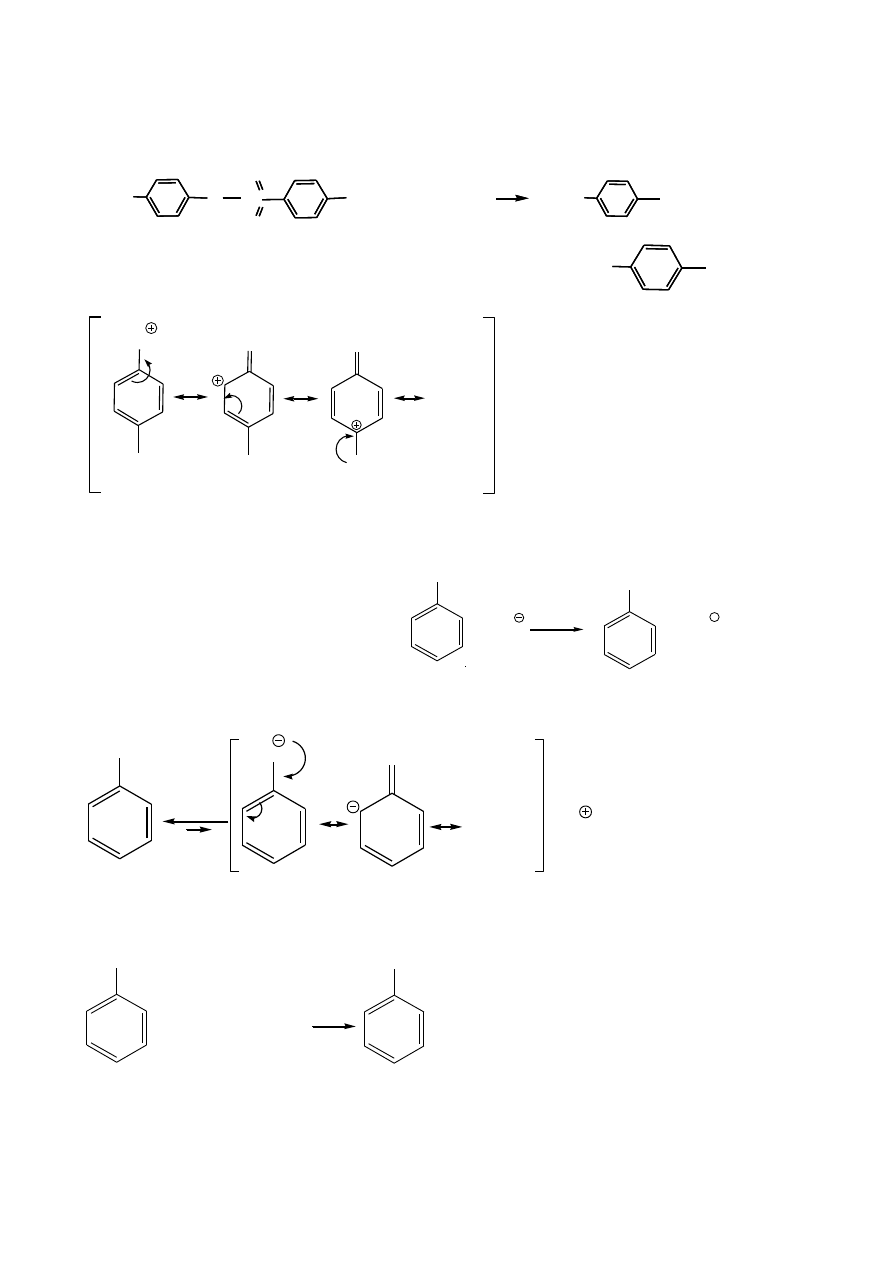
10
Stabilizacja rodnika benzylowego
C-H słabsze, bardziej reaktywne
Rezonans benzylowy silnie wpływa także na reaktywność halogenków i sulfonianów benzylowych:
H
3
CO
H
3
CO
HO
3
S
C
H
2
OS
O
O
CH
3
+ CH
3
CH
2
OH
CH
2
OC
H
2
CH
3
+
C
H
3
H
3
CO
H
3
CO
HO
3
S
C
H
2
OS
O
O
CH
3
+ CH
3
CH
2
OH
CH
2
OC
H
2
CH
3
+
C
H
3
S
N
1
CH
2
OCH
3
CH
2
OCH
3
CH
2
OCH
3
Także szybkie S
N
2 – elektrony π nakładają się z orbitalami w stanie przejściowym
ν - ok. 100 x większa niż szybkość
podstawienia w RCH
2
X
Stabilizacja rezonansowa anionu benzylowego
zwiększona kwasowość
CH
3
CH
2
+ H
CH
2
...
pK
a
= 41
CH
3
CH
2
Li
+ CH
3
CH
2
CH
2
CH
3
+ CH
3
CH
2
CH
2
CH
2
Li
CH
2
Br
+ CN
S
N
2
CH
2
CN
+ Br
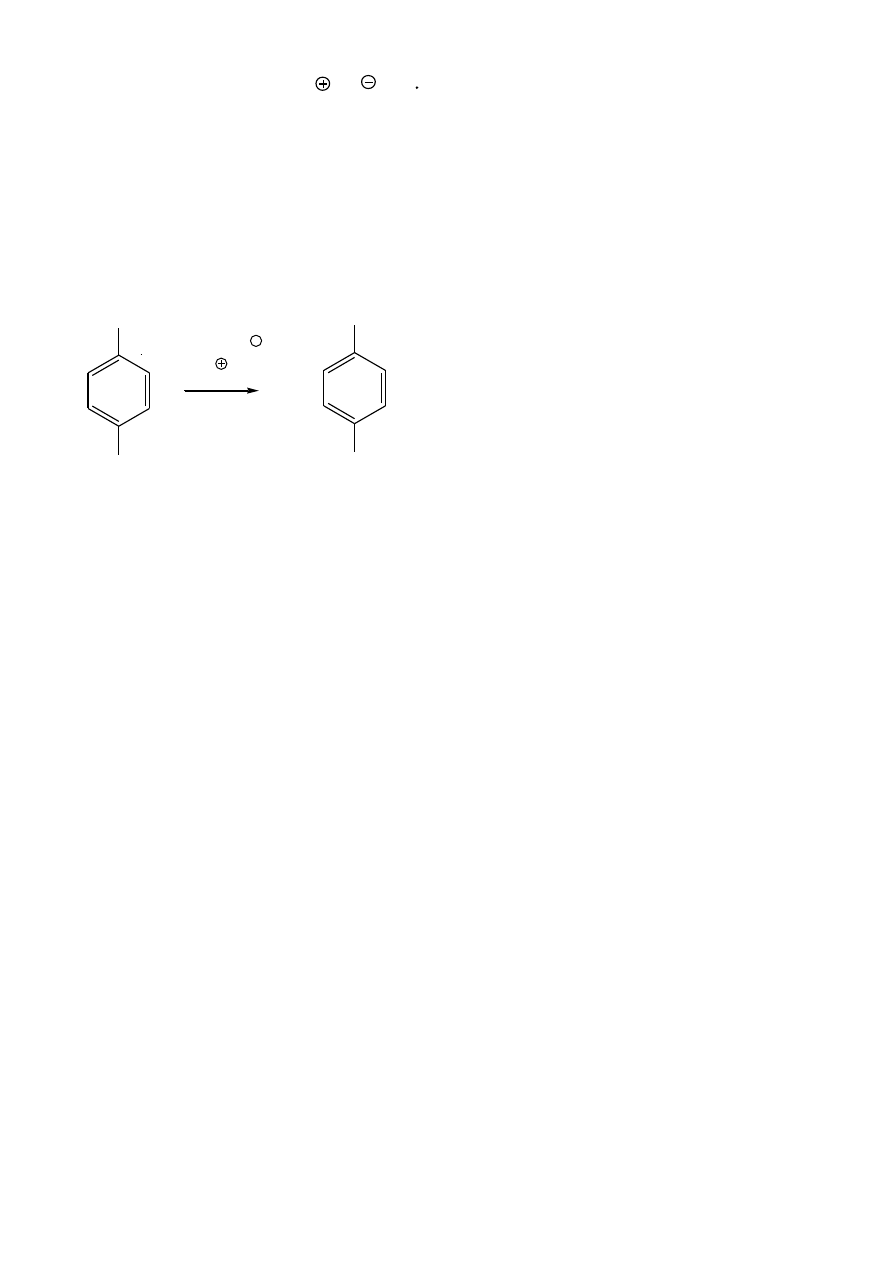
11
Stabilizacja rezonansowa
-
łatwe halogenowanie;
-
łatwe S
N
2, S
N
1;
-
zwiększona kwasowość.
Zw. aromatyczne – mało reaktywne (prócz S
E
), trudne do utlenienia, ale –
utlenianie benzylowe:
CH
3
CH
2
CH
2
CH
3
COOH
COOH
1. KMnO
4,
OH, T
2. H , H
2
O
Bn , Bn , Bn
Wyszukiwarka
Podobne podstrony:
cw 13 id 121763 Nieznany
36 13 id 36113 Nieznany (2)
7 13 id 44730 Nieznany (2)
piae wyklad3 12 13 id 356381 Nieznany
Alkohole 13 id 58087 Nieznany (2)
IMG 13 id 210986 Nieznany
G2 PB 02 B Rys 3 13 id 185405 Nieznany
13 id 189372 Nieznany (2)
(13)id 841 Nieznany
Lab 13 id 257441 Nieznany
Cwiczenie 13 id 125163 Nieznany
INS LAB PEWN 4 12 13 id 214856 Nieznany
B 13 id 74810 Nieznany (2)
INS LAB PEWN 1 12 13 id 214853 Nieznany
28 13 id 31840 Nieznany
7izostazja 2012 13 id 46496 Nieznany
2czas geologiczny 2012 13 id 32 Nieznany
więcej podobnych podstron