1
WŁASNOŚCI SPEKTRALNE NUKLEOTYDÓW PIRYDYNOWYCH
(NAD
+
, NADP
+
)
OZNACZANIE AKTYWNOŚCI TRANSAMINAZY ALANINOWEJ
WSTĘP
Nukleotydy pirydynowe (NAD
+
, NADP
+
) pełnią funkcję koenzymów dehydrogenaz przeno-
sząc jony wodorkowe (2e
-
i 1H
+
) (równoważniki redukcyjne) między utlenianym substratem, a
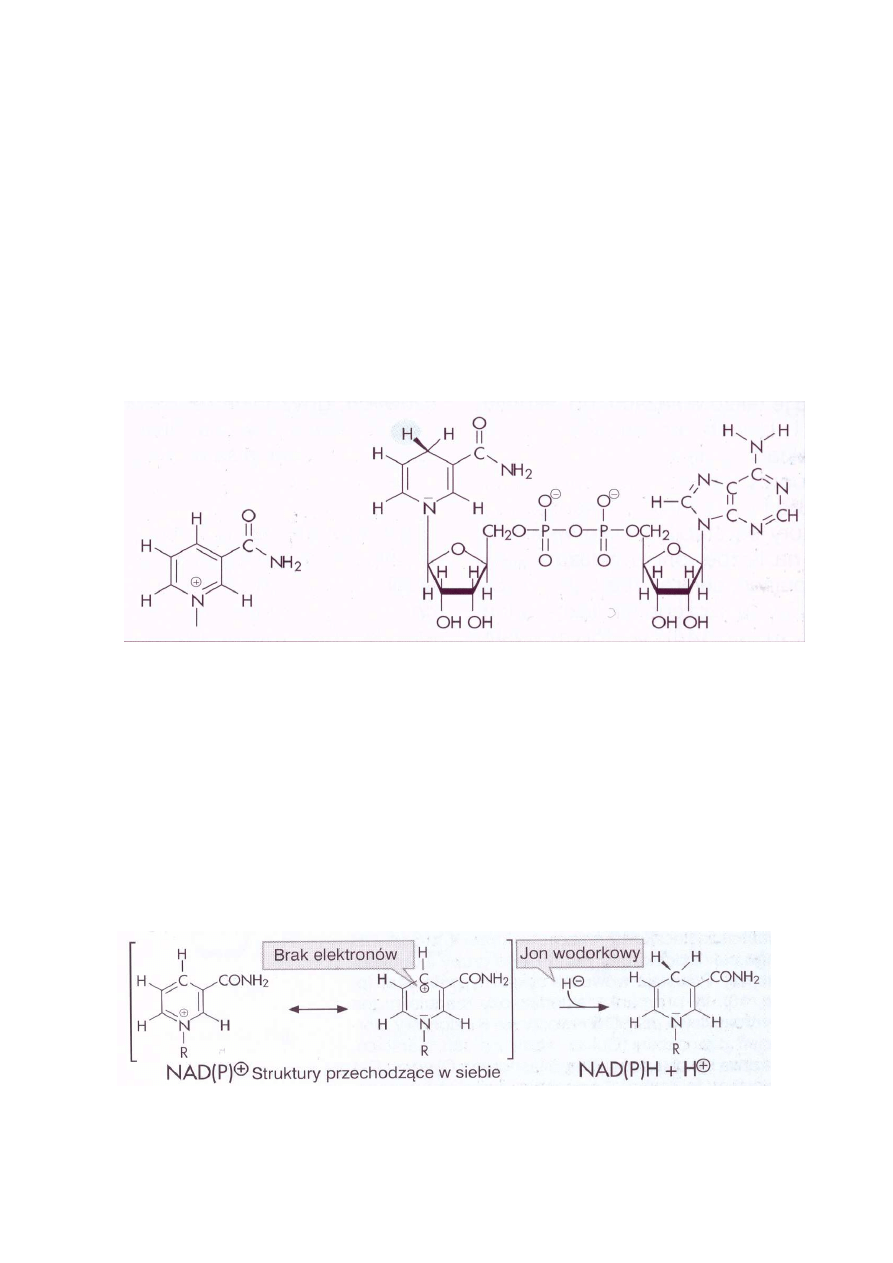
redukowanym akceptorem. Częścią aktywną koenzymów jest pierścień nikotynoamidowy (Ryc. 1)
Ryc. 1. Amid kwasu nikotynowego i dinukleotyd nikotynoamidoadeninowy, forma zredukowana
Przedstawiona na ryc. 2 forma przechodzących w siebie utlenionych struktur koenzymu NAD
+
(NADP
+
) ma w położeniu para do atomu azotu ubogi w elektrony, dodatnio naładowany atom
węgla. W to miejsce zostaje wprowadzony jon wodorkowy, tworząc zredukowane formy NADH
(NADPH). Ponieważ jednocześnie uwalniany jest proton, zredukowane koenzymy nukleotydu
pirydynowego poprawnie powinny być zapisywane jako NADH + H
+
(NADPH+ H
+
).
Ryc. 2. Formy utlenione amidu kwasu nikotynowego i przyłączenie jonu wodorkowego

2
Własności spektralne utlenionych i zredukowanych form nukleotydów pirydynowych (różnice w
widmie absorpcyjnym) są często wykorzystywane w analityce biochemicznej, np.:
1, stosując odpowiedni substrat można oznaczyć aktywność specyficznej względem niego dehy-
drogenazy na podstawie ilości zredukowanego koenzymu powstałego zgodnie z reakcją:
AH
2
+ NAD
+
⇔
A + NADH +H
+
(NADP
+
) (NADPH + H
+
)
2, stosując określoną oczyszczoną dehydrogenazę lub odpowiedni układ enzymatyczny można
oznaczać małe ilości metabolitów selektywnie utlenianych lub redukowanych przez te enzymy z
równoczesnym wytworzeniem stechiometrycznych ilości zredukowanego (utlenionego) koenzy-
mu. Przykładem takich sprzężonych reakcji jest układ używany do oznaczania ATP lub glukozy:
ATP + glukoza ⇒ ADP + glukozo-6-fosforan
glukozo-6-fosforan + NADP
+
⇒ 6-fosfoglukonolakton + NADPH +H
+
Możliwości szerokiego wykorzystania nukleotydów pirydynowych w analityce wynikają z ich
specyficznych właściwości:
-
zredukowane formy koenzymów wykazują absorbancję w 340 nm; nie wykazują jej formy
utlenione koenzymów
-
zredukowane formy są trwałe w środowisku alkalicznym i wykazują wówczas intensywną
fluorescencję „własną”. Formy utlenione ulegają w takich warunkach natychmiastowej destrukcji.
Zredukowane koenzymy są nietrwałe w środowisku kwaśnym, natomiast utlenione wykazują trwa-
łość w niskim pH. Właściwości te dają możliwość „zniszczenia”, po zakończeniu reakcji, nadmia-
ru koenzymu pozostawiając do pomiaru niezmienioną ilość trwałej w danych warunkach formy
nukleotydu – zredukowanej w środowisku alkalicznym bądź utlenionej w środowisku kwaśnym
-
zarówno zredukowana jak i utleniona postać koezymu powstałego w reakcji może być
przekształcona w silnie fluoryzującą pochodną dzięki czemu można go oznaczać w stężeniach
rzędu 10
-10
– 10
-9
M, co jest szczególnie użyteczne przy oznaczeniu stężeń metabolitów w małych
próbkach tkanek lub w pojedynczych komórkach.

3
A, WYZNACZENIE WIDMA ABSORPCYJNEGO DLA NAD
+
I NADH+H
+
Odczynniki:
•
0,05 M bufor fosforanowy pH 9,0 i pH 6,5
•
roztwór podstawowy (0,2 mM) NAD
+
w buforze fosforanowym o pH 6,5
•
roztwór podstawowy (0,2 mM) NADH + H
+
w buforze fosforanowym o pH 9,0
Wykonanie:
Dokonać pomiarów absorbancji 0,1 mM roztworu NAD
+
w zakresie widma 220-440 nm. Pomiary
wykonać w kuwetach kwarcowych względem buforu fosforanowego o pH 6,5.
Podobnych pomiarów dokonać dla 0,1 mM roztworu NADH + H
+
względem buforu o pH 9,0.
Na podstawie wykreślonego widma:
•
obliczyć teoretyczną wartość absorbancji 0,1 mM roztworu NADH + H
+
przy 340 nm wie-
dząc, że molowy współczynnik absorpcji wynosi 6220
•
określić na tej podstawie stopień czystości (%) preparatu NADH + H
+
użytego w doświad-
czeniu
B, WYKONANIE KRZYWEJ KALIBRACYJNEJ DLA NADH +H
+
METODĄ
SPEKTROFOTOMETRYCZNĄ
Odczynniki:
•
0,05 M bufor fosforanowy pH 9,0
•
roztwór podstawowy (0,2 mM) NADH + H
+
Wykonanie:
Z roztworu podstawowego (0,2 mM) NADH + H
+
, zawierającego 200 nmoli zredukowanego ko-
enzymu w 1 ml, przygotować 6 rozcieńczeń do końcowej objętości 2 ml. Rozcieńczone roztwory
powinny zawierać 20, 40, 60, 80, 100 i 150 nmoli NADH + H
+
w 1 ml roztworu. Do rozcieńczania
używać buforu fosforanowego o pH 9,0. Dokonać pomiarów absorbancji roztworów o rosnącym
stężeniu NADH + H
+
w 340 nm względem buforu używanego do rozcieńczeń.
•
Wykreślić krzywą wzorcową oraz obliczyć współczynnik kierunkowy.
4
C, OZNACZANIE AKTYWNOŚCI AMINOTRANSFERAZY ALANINOWEJ
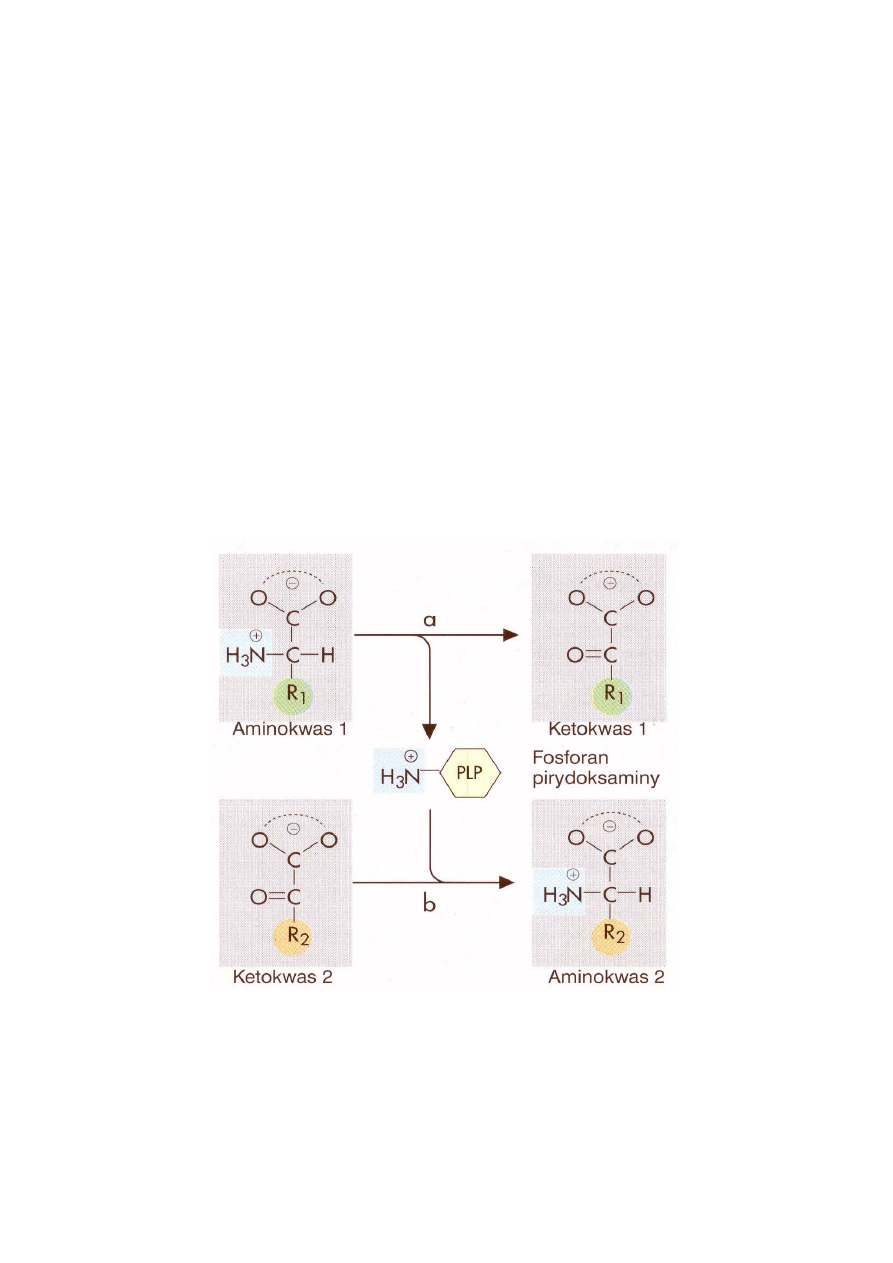
Grupa α-aminowa wielu aminokwasów jest przenoszona na α-ketoglutaran w reakcji, której me-
chanizm określany jest jako mechanizm podwójnego przeniesienia (reakcje ping-pong). Cechą
wyróżniającą ten mechanizm katalizy jest istnienie enzymu z podstawioną grupą, a więc pośred-
niej formy enzymu, który uległ czasowej modyfikacji. Tak więc, aminotransferaza alaninowa
katalizuje przeniesienie grupy aminowej z alaniny na α-ketoglutaran, a produktami reakcji są
pirogronian i glutaminian. Po związaniu alaniny do enzymu zostaje z niej usunięta grupa ami-
nową, która jest przeniesiona na fosforan pirydoksalu (PLP)(koenzym aminotransferaz i liaz), a
z aminotransferazy uwalnia się pirogronian. Drugi substrat, α-ketoglutaran, wiąże się do zmody-
fikowanego enzymu i przyjmuje od niego grupę aminową, a następnie zostaje uwolniony jako
produkt reakcji - glutaminian. Na ryc. 3 pokazany jest schemat reakcji podwójnego przeniesie-
nia, a na ryc. 4 mechanizm transaminacji.
Ryc. 3. Schemat reakcji podwójnego przeniesienia katalizowanej przez aminotransferazy
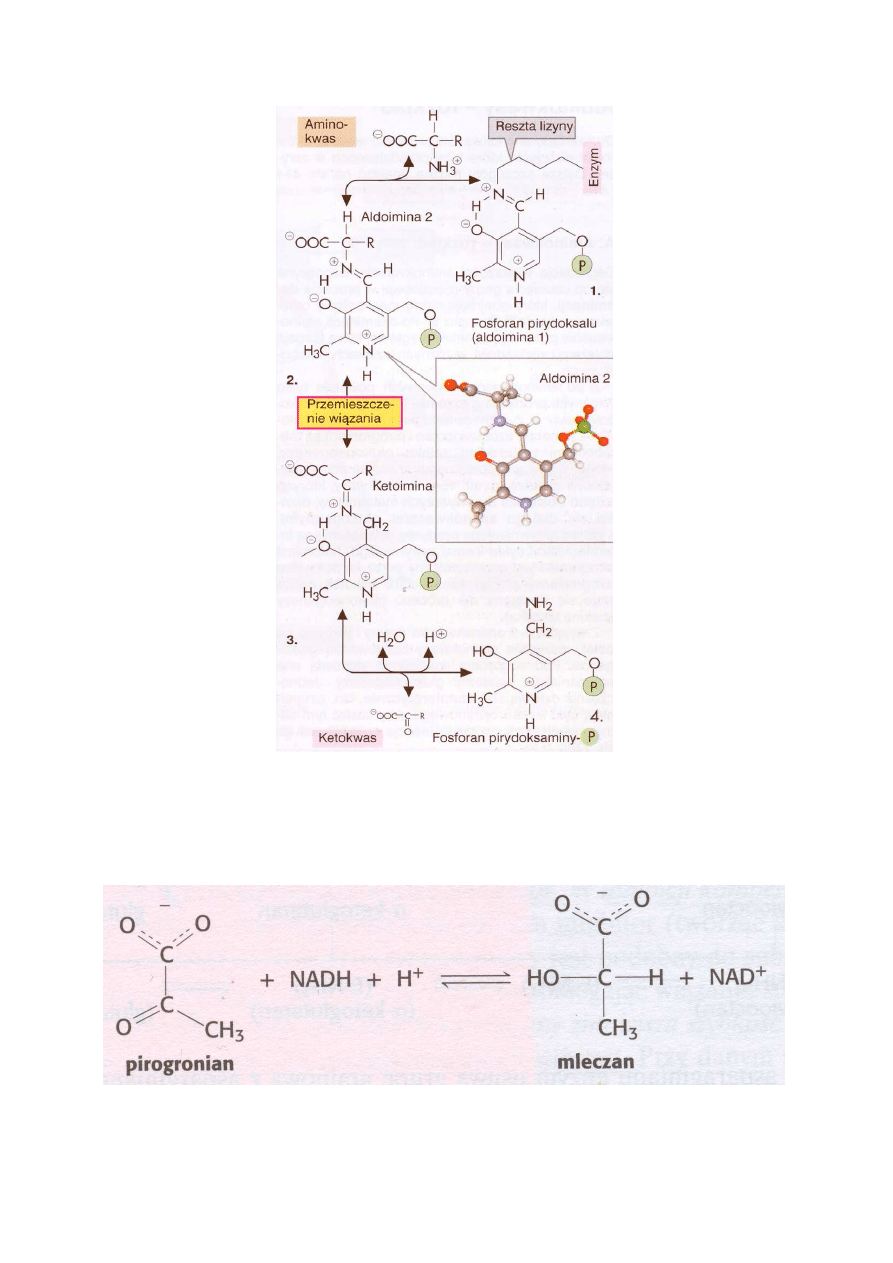
5
Ryc. 4. Mechanizm transaminacji
Powstający w reakcji pirogronian można oznaczać ilościowo zgodnie z poniższą reakcją, używając
oczyszczonej dehydrogenazy mleczanowej

6
Odczynniki:
•
100 mM roztwór buforu Tris-HCl, pH 8,0 zawierający 20 mM α-ketoglutaran
•
350 mM roztwór alaniny w 100 mM buforze Tris-HCl, pH 8,0
•
2,5 mM roztwór NADH + H
+
w 100 mM buforze Tris-HCl, pH 8,0
•
roztwór dehydrogenazy mleczanowej o aktywności około 1,2 jednostki w 100 µl (roztwór
przygotowany w 100 mM buforze Tris-HCl, pH 8,0
Materiał:
Odważyć 1 g niedojrzałych nasion grochu (zielony groszek), przenieść do małego moździerza i
rozetrzeć, najpierw „na sucho”, a potem dodając 1 ml 100 mM buforu Tris-HCl, pH 8,0. Dobrze
roztarty materiał przenieś do probówek Eppendorfa i odwirować 5 min przy 15 000 obr./min. Do
oznaczeń używać klarownego nadsączu (supernatantu) rozcieńczonego 5x buforem używanym do
homogenizacji.
Wykonanie:
Do kuwety kwarcowej napipetować 500 µl 100 mM buforu Tris-HCl, pH 8,0 zawierającego 20
mM α-ketoglutaran, następnie dodać 100 µl roztworu NADH + H
+
, 100 µl dehydrogenazy mle-
czanowej i 100 µl ekstraktu tkankowego (rozcieńczonego nadsączu). Kuwetę umieścić w spektro-
fotometrze i sprawdzić stabilność układu, a następnie reakcję transaminacji zapoczątkować doda-
jąc 200 µl 350 mM roztworu alaniny. Mierzyć spadki absorbancji przy 340 nm przez około 10
min, a następnie policzyć średnią wartość ∆A/min.
•
Obliczyć aktywność aminotransferazy alaninowej wyrażoną jako ilość nmoli pirogronianu
powstałego w reakcji enzymatycznej w ciągu 1 min.
Wyszukiwarka
Podobne podstrony:
Biol ekoIV id 87298 Nieznany
BIOL 01 id 87294 Nieznany
biol nagonasienne id 87349 Nieznany (2)
biol odp id 87936 Nieznany
Biol ekoIV id 87298 Nieznany
arch biol 20092010 sz id 67616 Nieznany
biol prob styczen 2012 id 87360 Nieznany
biol id 87286 Nieznany (2)
Kliucziewyje kompetencji ros id Nieznany
biol prob styczen 2013 id 87362 Nieznany
biol prob pp odp sty 2012 id 87 Nieznany
biol maj 2013(1) id 87306 Nieznany
biol prob pr odp sty 2012 id 87 Nieznany
biol 2 id 87289 Nieznany (2)
arch biol 20092010 sz id 67616 Nieznany
biol prob styczen 2012 id 87360 Nieznany
Abolicja podatkowa id 50334 Nieznany (2)
4 LIDER MENEDZER id 37733 Nieznany (2)
katechezy MB id 233498 Nieznany
więcej podobnych podstron