
1
Biologia Komórki
Ćwiczenie I
MIKROSKOP ŚWIETLNY
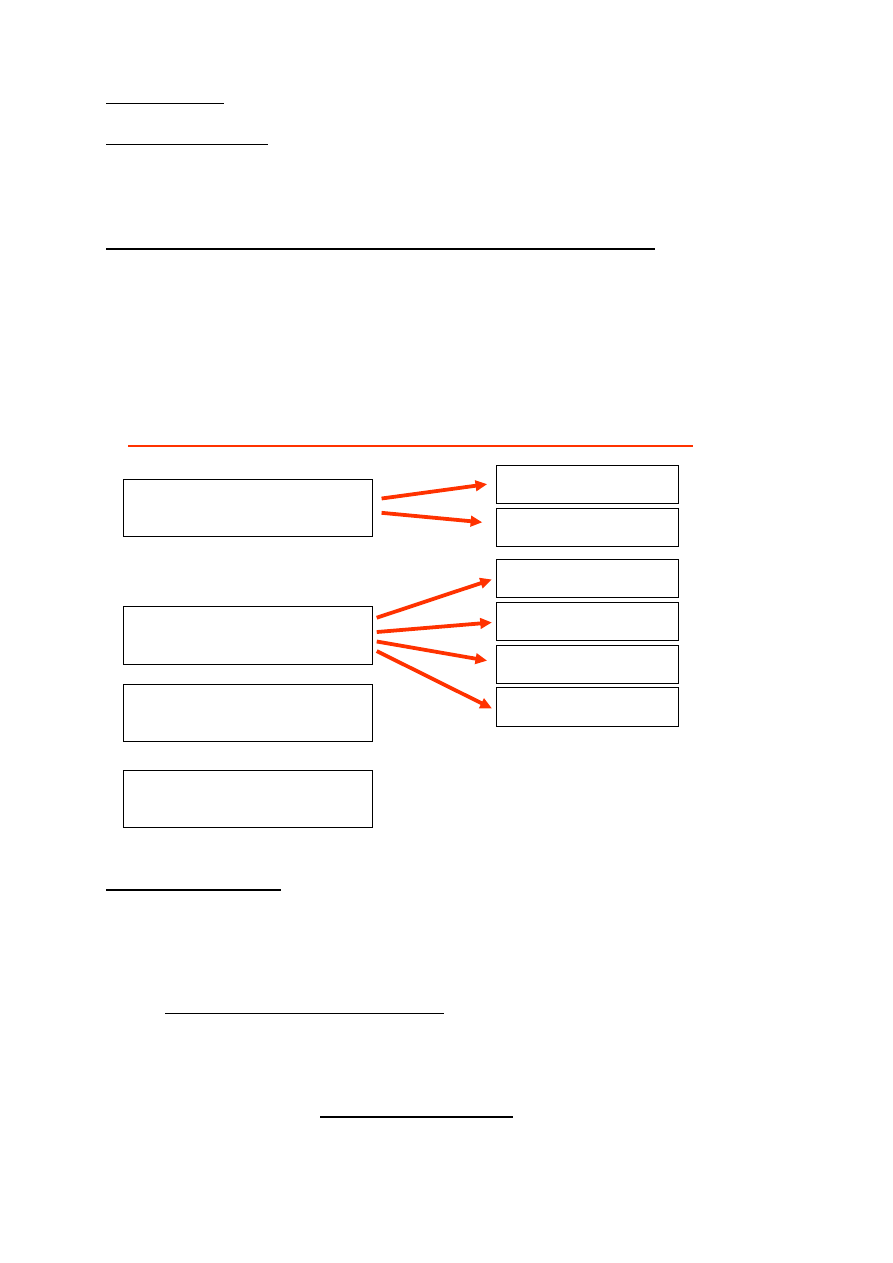
Nazwa części
Budowa
Funkcje
urz
ąd
zen
ie
ośw
iet
laj
ące
źródło
światła
żarówka halogenowa, diody
emitujące światło, żarówka
zwykła, (w starszych
mikroskopach- lusterko)
wytworzenie lub utworzenie
wiązki światła
kondensor
(aparat
Abbego)
układ soczewek
skupianie rozproszonego światła
w zbieżną wiązkę oświetlającą
pole widzenia
obiektyw
system soczewek
pierwszy z elementów tworzący
obraz powiększony
pośredni
układ
optyczny
zwierciadło lub układy
pryzmatyczne
załamanie wiązki pod
określonym kątem zgodnym z
kątem załamania tubusa (może
dzielić wiązkę na dwie części –
pierwszą skierowaną do okularu,
drugą do połączonego z
mikroskopem aparatu
fotograficznego lub kamery)
okular(y)
dwie soczewki płasko-
wypukłe
powiększanie, odwracanie i
tworzenie obrazu urojonego
Części optyczne mikroskopu świetlnego
Obraz w mikroskopie świetlnym jest pozorny (urojony), powiększony i odwrócony.
Powiększenie całkowite mikroskopu oblicza się, mnożąc powiększenie obiektywu przez
powiększenie okularu i ewentualnie przez powiększenie pośredniego układu optycznego.
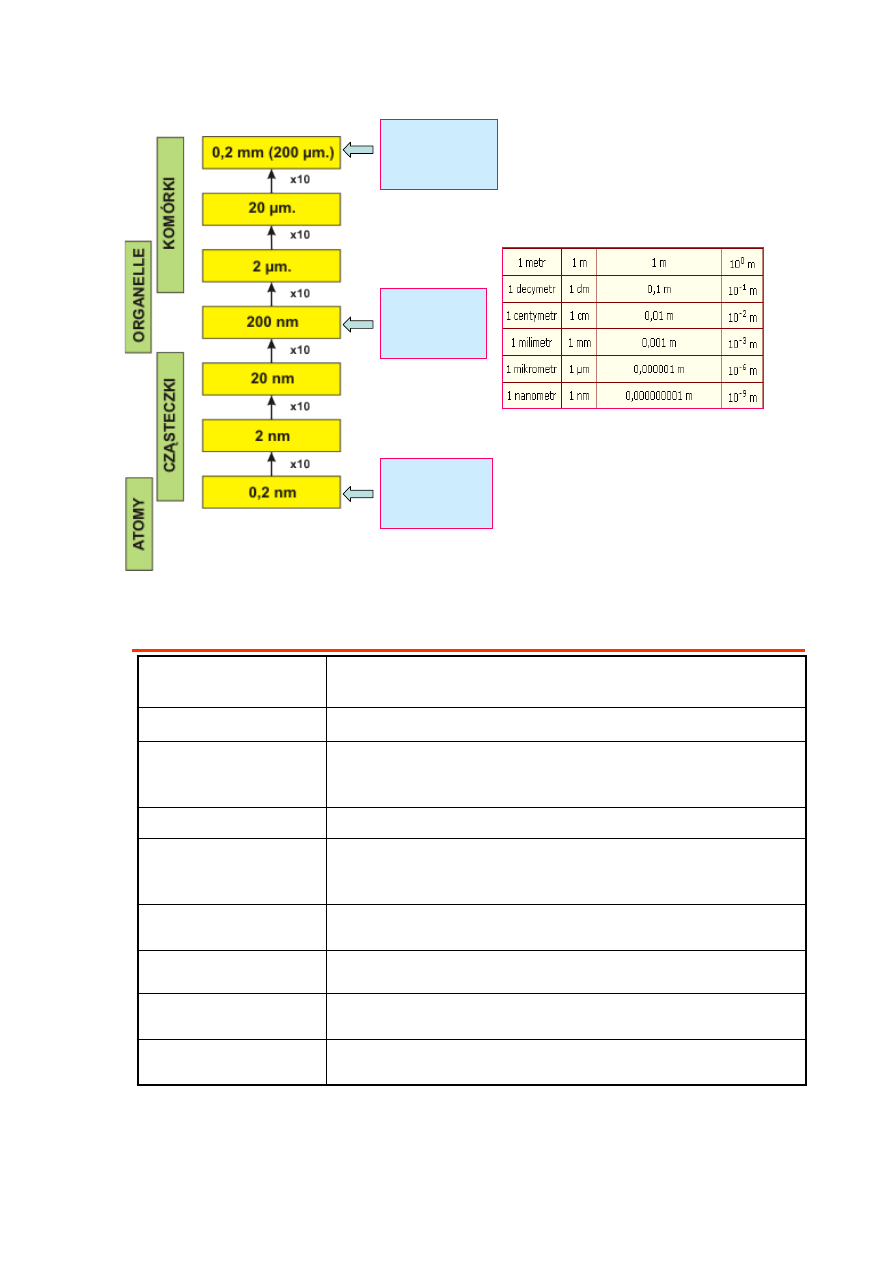
Powiększenia mikroskopów świetlnych mieszczą się w zakresie od 10-1500X.
Apertura numeryczna –
jest wielkością charakteryzującą obiektyw
An = n x sin α
n -
współczynnik załamania światła
kąt α - kąt zawarty pomiędzy osią optyczną a skrajnym promieniem trafiającym do soczewki
obiektywu
Zdolność rozdzielcza mikroskopu jest to najmniejsza odległość dzieląca dwa punkty, które w
obrazie mikroskopowym dostrzegalne są oddzielnie. Im mniejsza ta odległość tym lepsza
zdolność rozdzielcza.
E = 0,5 L / An
L -
długość fali światła (przeciętna 0,55 μm)
Obiektywy suche - An - 0,95; E -
0,3 μm
Obiektywy immersyjne wodne - An - 1,2; E -
0,24 μm
Obiektywy immersyjne olejkowe - An - 1,42; E -
0,19 μm (najlepsza zdolność rozdzielcza)
2
granica
rozdzielczości
oka
nieuzbrojonego
granica
rozdzielczości
mikroskopu
świetlnego
granica
rozdzielczości
mikroskopu
elektronowego
Rodzaje obiektywów
monochromatyczne
skorygowane na jedną określoną część widma światła
białego
achromatyczne
skorygowane na środkową część widma światła widzialnego
planachromatyczne
achromatyczne o lepszej korekcji, zwiększające ostrość
obrazu (pozwalają na korzystanie z dużego pola widzenia i
okularów o dużych powiększeniach)
apochromatyczne
prawie cały zakres fal
planapochromatyczne
apochromatyczne o lepszej korekcji, zwiększające ostrość
obrazu (pozwalają na korzystanie z dużego pola widzenia i
okularów o dużych powiększeniach)
do fluorescencji
obiektywy o bardzo dużej transmisji wiązki świetlnej,
zwłaszcza o długości fali ok. 340 nm
obiektywy suche
przy powiększeniach małych i średnich
obiektywy mokre
(immersyjne)
przy powiększeniach dużych
obiektywy o dużej
odległości roboczej
stosowane w mikroskopach odwróconych
3
Obiektyw suchy
(małych i średnich powiększeń) – część promieni świetlnych nie przechodzi
przez obiektyw, lecz odbija się na granicy szkiełka z preparatem i powietrza
Obiektyw immersyjny
(dużych powiększeń) – przestrzeń między nim a preparatem wypełnia
się olejkiem immersyjnym, którego współczynnik załamania światła jest taki sam, jak szkła –
unika się ugięcia (dyfrakcji) promieni świetlnych na granicy szkła i powietrza, co wpływa
korzystnie na jakość obrazu
Cechy charakteryzujące obiektyw (symbole umieszczone na obudowie):
Apertura numeryczna – An
Powiększenie – 10x, 20x…
Najczęściej stosowane powiększenia obiektywów to:
- obiektywy suche – 5x, 10x, 20x, 40x
- obiektywy immersyjne – 60x, 100x
Rodzaje mikroskopów świetlnych
Mikroskop świetlny klasyczny
Mikroskopy będące prostymi
modyfikacjami mikroskopu
klasycznego
Mikroskop fluoroscencyjny
Mikroskop konfokalny
Mikroskop zwykły
Mikroskop odwrócony
Mikroskop polaryzacyjny
Mikroskop interferencyjny
Mikroskop
kontrastująco-
fazowy
Mikroskop ciemnego pola
Mikroskop odwrócony
- stosowany dla preparatów biologicznych
wymagających mikroskopowania „od dołu”,
np. hodowli komórek w płaskich naczyniach, w których komórki osiadają na dnie, a
nad nimi jest warstwa płynu (odżywki) utrudniająca tradycyjne mikroskopowanie od
góry (uniemożliwia zbliżenie obiektywu od góry na odległość niezbędną do
wytworzenia ostrego obrazu),
-
układ optyczny jest odwrócony o 180º (źródło światła i kondensor są w górnej części
mikroskopu, a obiektyw i okular w dolnej),
- specjalne obiektywy (20 -
60 x) o dużej odległości roboczej pozwalają na obserwacje
poprzez dna naczyń wykonane ze szkła lub tworzywa sztucznego o grubości 0,2 – 1,5
mm,
-
najczęściej uzyskany obraz nie jest odwrócony co bardzo ułatwia mikromanipulację,
-
mikroskop odwrócony może posiadać dodatkowo specjalny układ optyczny
(kontrastowo-fazowy, fluorescencyjny, polaryzacyjny).

4
Mikroskop pola ciemnego
-
obserwacja w polu ciemnym umożliwia dostrzeżenie w preparacie drobnych cząstek o
wymiarach poniżej zdolności rozdzielczej układu optycznego. Wykorzystuje się zjawisko
dyfra
kcji (ugięcia) promieni świetlnych padających na obiekt o bardzo małych rozmiarach.
Przykładowo drobne cząstki kurzu w powietrzu, niewidoczne w normalnych warunkach, gdy
znajdą się w smudze światła padającej do pokoju, świecą się. Dzieje się tak, gdy patrzymy na
jasną smugę światła widoczną na ciemnym tle.
-
występuje specjalny kondensor, którego centralna część jest nieprzepuszczalna dla światła;
promienie świetlne przechodzą przez preparat pod bardzo ostrym kątem, a do obiektywu
trafiają tylko te promienie, które załamują się na krawędziach oglądanych obiektów. Obiekty
te widać jako drobne świecące punkty na ciemnym tle,
-
wykorzystując zjawisko rozpraszania światła na obserwowanych strukturach, można w polu
ciemnym rozpoznać np. włókna o grubości zaledwie 5 nm, a więc o wymiarach około 40 razy
mniejszych od wartości zdolności rozdzielczej mikroskopu świetlnego,
-
mikroskopię ciemnego pola stosuje się głównie do obserwacji mikroorganizmów w płynach
ustrojowych.
Mikroskop kontrastowo- fazowy
Siatkówka o
ka rejestruje tylko 2 z 3 parametrów fizycznych fali świetlnej – długość fali
(odbiera ją jako barwę) i amplitudę fali (odbiera jako stopień jasności). Trzeci parametr – faza
fali świetlej - nie jest rejestrowany przez siatkówkę. Faza jednak ulega zmianom przy
przechodzeniu światła przez preparat. Różne struktury obecne w preparacie powodują
przesunięcie fazy fali świetlnej. To zjawisko wykorzystano w mikroskopii kontrastowo-
fazowej.
Żywe komórki ssaków (z wyjątkiem tych zawierających pigment) są obiektami fazowymi.
Światło przechodzi przez nie jak przez przejrzyste szkło. Obserwator nie dostrzega żadnych
zmian ani w jego natężeniu ani w barwie. Jedyną zachodzącą zmianą, którą ludzkie oko nie
potrafi ocenić, jest zmiana (przesunięcie) fazy światła.
Mikroskop kontrastowo-fazowy zawiera specjalny
układ optyczny, który przekształca
przesunięcie fazy światła w zmianę jej amplitudy (czytelną dla oka), dzięki temu struktury
obecne w preparacie
widoczne są w postaci skontrastowanej, czyli jako ciemniejsze lub
jaśniejsze.
Zastosowanie:
-
powszechnie używany do badania komórek nie zabarwionych, z reguły żywych (np. z
hodowli komórkowej),
-
do wzmocnienia ogólnego kontrastu w słabo zabarwionych preparatach.
Mikroskop interferencyjny
-
działa także na zasadzie przekształcenia różnic faz fal świetlnych w różnice ich
amplitudy
-
posiada jednak odmienną konstrukcję kondensora, który dodatkowo rozdziela wiązkę
światła na dwie składowe (jedną przechodzącą przez preparat, drugą obok preparatu,
przez tło - tzw. wiązkę odniesienia, charakteryzującą się m. in. stałym
współczynnikiem załamania światła. W tubusie dochodzi do interferencji między
nimi, czyli ponownego zespolenia dwóch wiązek w jedną składową).
–
układ optyczny kontrastuje obraz oraz dzięki temu, że umożliwia obliczenie współczynnika
załamania światła promieni przechodzących przez preparat, może służyć do analizy
ilościowej suchej masy badanych struktur i do określania grubości skrawków. Istnieje
bowiem poznana zależność między stężeniem substancji wchodzących w skład komórki
(białka, cukry, lipidy) a wartością współczynnika załamania światła.
-
układ optyczny pozwala też na rozszczepianie światła białego na barwne części widma i
optyczne „wybarwienie” struktur widocznych w obrazie mikroskopowym.
5
Mikroskop z o
ptyką (kontrastem) Nomarskiego
- modyfikacja mikroskopii kontrastowo- fazowej i interferencyjnej,
-
pozwala na znaczące wzmocnienie kontrastu nie zabarwionych struktur komórkowych,
- uzyskanie plastycznego nieomal trójwymiarowego obrazu tych struktur.
Mikroskop polaryzacyjny
-
używany do badań obiektów anizotropowych w świetle spolaryzowanym,
-
posiada w układzie optycznym pryzmaty powodujące polaryzację światła (uporządkowanie
chaotycznych drgań fali świetlnej w jednej płaszczyźnie). Wszystkie obiekty biologiczne pod
względem ich wpływu na polaryzację przechodzącego przez nie światła dzielimy na
izotropowe (załamują pojedynczo światło i nie zmieniają płaszczyzny polaryzacji, nie są więc
widoczne w mikroskopie polaryzacyjnym) oraz anizotropowe (załamują podwójnie światło i
zmieniają płaszczyznę polaryzacji promieni świetlnych, są wtedy widoczne jako jasne na
ciemnym tle)
-
do obiektów anizotropowych, które można oglądać, należą substancje, w których występuje
wysoce uporządkowany układ cząsteczek, np. włókna kolagenowe, zmineralizowana
substancja międzykomórkowa tkanki kostnej, miofibryle włókien mięśniowych poprzecznie
prążkowanych, krystaliczne i parakrystaliczne wtręty wewnątrzkomórkowe
- w histopatologii mikroskop polaryzacyjny
służy do wykrywania złogów skrobiawiczych
(amyloidu) w komórkach.
Mikroskop fluorescencyjny
Fluorescencja –
emitowanie światła (widzialnego) przez substancję chemiczną pod wpływem
naświetlania jej promieniami ultrafioletowymi (UV). Własność tę posiadają niektóre
substancje obecne w komórkach i tkankach (porfiryny, witamina A, lipofuscyny, chlorofil)
oraz specjalne barwniki tzw.
fluorochromy używane w metodyce histologicznej i
histochemicznej.
Źródłem światła w mikroskopie fluorescencyjnym jest specjalna żarówka emitująca
promieniowanie UV. Dodatkowo mikroskop ten
posiada zestaw filtrów przepuszczających
jedynie widzialne UV o pożądanej długości fali, chroniąc tym samym narząd wzroku przed
szkodliwym działaniem pozostałych promieni UV.
Obrazem w mikroskopie fluorescencyjnym są widoczne na ciemnym tle świecące
(fluoryzujące) struktury komórek i tkanek.
Kolor fluorescencji zależy od rodzaju substancji fluoryzującej.
Konfokalny skaningowy mikroskop świetlny
- nowoczesna odmiana mikroskopu fluorescencyjnego, w którym
źródłem światła jest laser,
skupiany w wiązkę o średnicy zaledwie 0,1 μm, co stanowi teoretyczną zdolność rozdzielczą
tego mikroskopu,
- mikroskop ten jest
sprzężony ściśle z odpowiednim programem komputerowym,
-
wiązka lasera z dużą szybkością skanuje preparat na określonej równoległej płaszczyźnie,
komputer rejestruje obrazy serii przekrojów optycznych wszystkich warstw preparatu (np.
warstw o grubości 1-2 µm w skrawku histologicznym o grubości 20 µm), łączy je razem i
umożliwia tworzenie trójwymiarowego obrazu badanego obiektu, który ogląda się na
monitorze.
W innych typach mikroskopu świetlnego ostry obraz można uzyskać tylko z jednej
określonej głębokości – warstwy preparatu uwarunkowanej m.in. jego odległością od
obiektywu,
-
w większości przypadków mikroskopy konfokalne wykorzystują fluorescencję emitowaną
pod wpływem laserowego światła przez odpowiednio zabarwiony preparat.

6
Przygotowanie materiału biologicznego do badań w mikroskopie świetlnym.
Przygotowanie materiału biologicznego do badań w mikroskopie świetlnym to często długa i
skomplikowana procedura, która służy do uzyskania trzech podstawowych wymaganych cech
dobrego preparatu mikroskopowego:
-
przejrzystość – odpowiednia grubość preparatu, która musi umożliwić przejście przez niego
promieni świetlnych. Najlepszą jakość uzyskuje się przy preparatach jak najcieńszych,
- zabarwienie (ewentualnie skontrastowanie) –
by zróżnicować struktury występujące na
preparacie,
- zachowanie struktur komórek i tkanek w preparacie w stanie jak najbardziej
zbliżonym do
str
uktury tkanki żywej – co jest warunkiem wiarygodności obrazu mikroskopowego.
W praktyce najczęściej wykorzystuje się dwa typy preparatów mikroskopowych:
- skrawki –
uzyskane przez pokrojenie materiału biologiczne na cienkie „plasterki”,
- rozmazy – wyko
nywane z płynów ustrojowych, w których są zawieszone wolne komórki
(kre
w, szpik czerwony, płyn mózgowo-rdzeniowy, płyny wysiękowe i wydzieliny). Kroplę
płynu rozprowadza się na szkiełku tak, by zawarte w nim komórki utworzyły pojedyńczą
płaską warstwę.
TECHNIKA PARAFINOWA
1.
Pobieranie materiału
•
Pośmiertne - w czasie jak najkrótszym od ustania pracy serca (by zapobiec zmianom
rozkładowym), pobiera się wycinek o wymiarach nie większych niż 1 cm x 1 cm x 1
cm, 2 cm x 0,5 cm x 0,5 cm
(by zapewnić dobrą i szybką penetrację płynów
stosowanych w dalszych etapach –
utrwalaczy, płynów odwadniających i pośrednich)
za pomocą ostrych narzędzi
•
Przyżyciowe - w trakcie zabiegu operacyjnego lub metodą biopsji
2. Utrwalanie
• Cel - zatrzymanie procesów
życiowych w komórkach bez naruszenia ich struktury,
przede wszystkim przez denaturację enzymów. Zapobiega to zmianom autolitycznym,
czyli samostrawieniu komórek przez własne enzymy lizosomalne i wtórnym zmianom
rozkładowym i gnilnym wywoływanym przez obecne w materiale bakterie,
- zachowanie struktury tkanek i komórek tak,
aby nie uległa ona zniszczeniu w
t
rakcie dalszych etapów obróbki poprzez wstępne utwardzenie materiału i zwiększenie
jego odporności na działanie odczynników używanych w dalszych etapach procedury.
•
Najczęściej stosowane utrwalacze stabilizują tkanki i komórki poprzez wytwarzanie
nieodwracalnych wiązań krzyżowych (co usztywnia bardziej tkankę) pomiędzy
końcowymi grupami polipeptydów i białek, tworzenie mostków (np. formaldehyd –
mostki metylenowe CH
2
= CH
2
) lub wiążą się z grupami SH białek. Proces ten
zachowując integralność budowy niszczy biologiczną funkcjonalność białek
(szczególnie enzymów) poprzez denaturację białek (czyli zmiany w II, III i IV
rzędowej strukturze białka)
• Utrwalacze
– proste: formalina, alkohol, aceton
–
złożone: płyn Bouina (formalina, kwas pikrynowy,
kwas octowy)
AFA (alkohol, formalina, kwas octowy).
• Czas i temperatura utrwalania –
różne w zależności od rodzaju utrwalacza i materiału.
Sposoby utrwalania:
Immersyjne - zanurzenie wycinka w naczyniu z utrwalaczem
o objętości 50 – 100 x większej
niż utrwalanego wycinka. Część obwodowa tkanki zostaje utrwalona najszybciej, do części

7
środkowej utrwalacz dociera z opóźnieniem, co może sprzyjać powstawaniu artefaktów
(struktur nie
istniejących w żywych tkankach) związanych z procesami autolitycznymi,
szczególnie przy większych wycinkach. Czas utrwalania od 1 godziny do kilku dni.
Perfuzyjne – do otwartych
głównych naczyń tętniczych badanego narządu (lub w przypadku
małych zwierząt dosercowo) wprowadza się sól fizjologiczną (w celu wypłukania krwi) a
następnie utrwalacz, który wypływa z narządu przy otwartych żyłach. Dzięki bogatej sieci
naczyń włosowatych utrwalacz dociera natychmiast do każdego rejonu narządu. Tkanka jest
utrwalana równomiernie i szybko
od wewnątrz, co zapewnia najlepsze zachowanie jej
struktury
. Czas utrwalania perfuzyjnego może być bardzo krótki – 5-15 minut.
3. P
łukanie
•
Ma na celu usunięcie nadmiaru utrwalacza nie związanego z utrwalanym materiałem.
• Jest przeprowadzane w:
–
wodzie bieżącej - w przypadku utrwalaczy będących roztworami wodnymi
– alkoholu etylowym –
w przypadku utrwalaczy złożonych zwierających alkohol
etylowy w stężeniu ≥ 70 %.
•
Trwa najczęściej 12 – 18 godzin.
4. Odwadnianie
• Cel –
usunięcie wody z badanego materiału (wewnątrztkankowej i z resztek
utrwalacza)
i zastąpienie jej alkoholem
• Przeprowadzane
w naczyniach zawierających kolejno roztwory alkoholu etylowego o
rosnącym stężeniu: 30, 50, 60, 70, 80, 90, 96, 100%
5. P
rzeprowadzanie przez płyny pośrednie (prześwietlanie)
• Cel –
usunięcie alkoholu i resztek wody z badanego materiału, gdyż ich obecność
uniemożliwia późniejsze zatopienie materiału w substancji utwardzającej (parafinie)
•
Kąpiel w ksylenie - wycinek stopniowo staje się półprzeźroczysty. Ksylen jako
rozpuszczalnik organiczny rozpuszcza się w parafinie. Nosi nazwę płynu pośredniego,
ponieważ pośredniczy między odwadnianiem a zatapianiem. Do dokładnym
przepojeniu materiału płynem pośrednim można go zatopić.
6. Zatapianie w parafinie
• Cel – ut
wardzenie i przygotowanie materiału do krojenia.
• Przepajanie w
naczyniu z roztopioną parafiną (najpierw o niższej, a później o wyższej
temperaturze topnienia) przez kilka godzin w cieplarce w temperaturze o kilka stopni
wyższej od temperatury topnienia parafiny.
•
Używa się kilku odmian parafiny o różnej twardości i temperaturze topnienia (im
większa twardość tym wyższa temperatura topnienia, np. 45°C dla bardzo miękkiej i
60°C dla bardzo twardej)
• Utwardzenie bloczka tkankowo-parafinowego poprzez
oziębienie parafiny.
•
Każdy bloczek jest odpowiednio podpisany i skatalogowany.
7. Krojenie
Skrawki histologiczne kroi się na mikrotomach. Każdy mikrotom prócz noża krojącego (z
wysokogatunkowej stali) i stolika, do którego montuje się bloczek parafinowy z materiałem,
posiada mechanizm umożliwiający ustawienie pożądanej grubości skrawków i automatyczne
dosuwanie bloczka do noża (lub odwrotnie) o dystans odpowiadający odpowiedniej grubości
skrawka. W zależności od typu mikrotomu, częścią ruchomą jest stolik z bloczkiem lub nóż:
- mikrotom rotacyjny -
ruchomy bloczek, nieruchomy nóż
- mikrotom saneczkowy -
ruchomy nóż, nieruchomy bloczek

8
W mikroskopii świetlnej kroi się skrawki o grubości 6 – 10 μm. Im cieńsze skrawki, tym
lepsza rozdzielczość mikroskopu świetlnego i możliwość obserwowania drobniejszych
szczegółów w obrazie mikroskopowym.
Przy krojeniu bloczka parafinowego tworzy się wstęga złożona z kolejnych (seryjnych)
skrawków zlepionych ze sobą krawędziami. Skrawki te odrywa się delikatnie od siebie i
ponieważ są pofałdowane, umieszcza się na powierzchnię wody o temperaturze 40°C w celu
zmiękczenia parafiny i rozprostowania skrawków. Następnie skrawek nakłada się na
powierzchnię szkiełka podstawowego, suszy i barwi.
8. Barwienie
Metody barwienia komórek i tkanek
- barwienie
zwiększa kontrast komórek i tkanek oraz uwidacznia ich charakterystyczne
cechy, pozwala na wyr
óżnienie poszczególnych komórek i tkanek pod mikroskopem
-
umożliwia określenie rodzajów komórek i podstawowych cech ich budowy (kształt,
wielkość, główne struktury) oraz topografii komórek
W
yróżniamy barwienia rutynowe i specjalistyczne. Metody rutynowe wykorzystują
najczęściej kwaśny lub zasadowy charakter barwionych elementów (opierają się wtedy na
reakcji zobojętniania – barwniki zasadowe wiążą się ze strukturami kwaśnymi) oraz
powinowactwo struktur do pewnych substancji (np. soli metali)
P
rzykłady:
- metoda HE (hematoksyksylina, eozyna –
barwienie jąder i cytoplazmy),
- metoda Giemsy (barwienie komórek krwi i szpiku),
- metoda Azan-
Mallory (barwienie komórek i włókien tkanki łącznej na niebiesko),
- metoda srebrzenia (impregnacja azotanem srebra
, który redukuje się w tkankach do
metalicznego czarnego strątu – na obecność włókien srebrochłonnych).
Najpopularniejsze barwniki:
• Barwniki rozpuszczalne w wodzie:
–
Barwniki zasadowe (jądrowe) – wiążą się z kwaśnymi strukturami
komórkowymi –
jądrem, siateczką szorstką, rybosomami
• hematoksylina – wybarwia na kolor fioletowy
–
Barwniki kwaśne (plazmatyczne) – wiążą się z zasadowymi elementami
komórki -
z cytoplazmą i pozostałymi organellami
• eozyna –
wybarwia na kolor różowy
•
Barwniki rozpuszczalne w tłuszczach (obojętne)
• sudan –
wybarwia na kolor pomarańczowy
Barwienie HE (metoda HE):
• odparafinowywanie (ksylen 2x) – u
suwamy parafinę ze skrawka, gdyż barwnik się w
niej nie rozpuszcza
• uwadnianie (alkohol 100 % - 50 %)
•
płukanie (woda destylowana) – barwnik rozpuszczalny w wodzie wymaga dobrego
uwodnienia tkanki
•
barwienie jąder - hematoksylina
•
płukanie (woda)
•
różnicowanie (roztwór alkoholu)
•
płukanie (woda)
• barwienie cytoplazmy – eozyna
•
płukanie (woda)
• odwadnianie (alkohol 80 % - 100 %)
•
prześwietlanie (ksylen 2x)
9
9. Zamykanie –
zakrywanie zabarwionego preparatu szkiełkiem nakrywkowym i
wypełnienie przestrzeni między oboma szkiełkami żywicami - balsamem kanadyjskim lub
DPX.
Żywice te mają ten sam współczynnik załamania światła co szkło, co zapobiega
ewentualnemu rozpraszaniu i uginaniu promieni świetlnych przechodzących przez preparat w
mikroskopie świetlnym.
Tak przygotowany
preparat mikroskopowy może służyć nawet kilkadziesiąt lat.
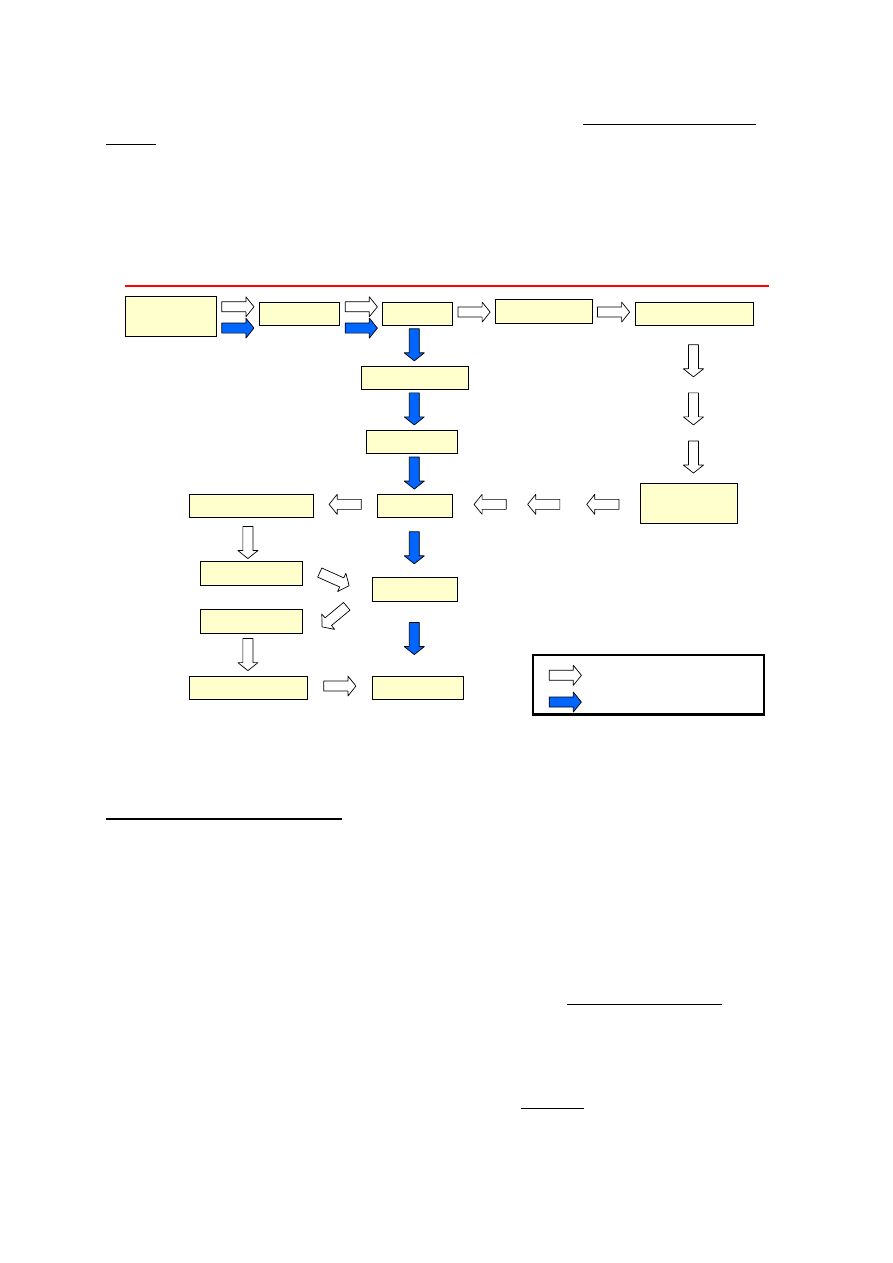
pobieranie
materiału
Techniki stosowane w mikroskopii świetlnej
utrwalanie
płukanie
odwadnianie
płyny pośrednie
krioprotekcja
zamrażanie
krojenie
zatapianie w
parafinie
odparafinowanie
nawadnianie
barwienie
odwadnianie
prześwietlanie
zamykanie
Technika parafinowa
Technika mrożeniowa
TECHNIKA MROŻENIOWA
polega na utwardzeniu materiału biologicznego przez jego zamrożenie.
technikę mrożeniową stosuje się w histochemii i immunocytochemii, gdyż zachowuje
aktywność biologiczną białek (enzymów) oraz skład chemiczny wszystkich substancji
komórki (m.in. lipidów),
ok. 70% masy materiału biologicznego stanowi woda, która podczas zamrażania
zamienia się w bardzo liczne i szybko przybierające na masie i wielkości kryształki,
które mogą uszkodzić struktury komórek. By temu zapobiec lub nawet wyeliminować
proces krystalizacji, przed zamrożeniem stosuje się krioprotekcję tkanek – czyli
przepojenie utrwalonej tkanki substancją, której wodny roztwór nie tworzy
kryształków, lecz ulega żelifikacji. Najczęściej stosuje się roztwory sacharozy lub
glicerolu. Tak przepojoną tkankę zamraża się w temperaturze minimum -25°C (przy
dłuższym czasie przechowywania nawet -80°C),
krojenie
zamrożonego materiału przy pomocy kriostatu – jest to zmodyfikowany
mikrotom rotacyjny umieszczony w komorze chłodniczej o temperaturze regulowanej
od -25 do -60 ºC
. Elementy sterujące (napęd, regulacja grubości skrawków, odległości
10
noża od materiału) są wyprowadzone na zewnętrznej obudowie komory, by zapobiec
odmrożeniu rąk pracującego.
Pojedynczy skrawek
z noża zbiera się bezpośrednio na szkiełko, na którym jest
rozmrażany, suszony i od razu barwiony (bez konieczności stosowania alkoholi i
innych rozpuszczalników organicznych).
Preparaty sporządzane techniką
mrożeniową
Preparaty sporządzane techniką
parafinową
ZALETY
•
Dobrze zachowana struktura
komórek i tkanek
•
Wysoka jakość obrazów w
mikroskopie optycznym
(poprzez możliwość uzyskania
wystarczająco cienkich
skrawków)
WADY
•
Długotrwała procedura
przygotowania
•
Wypłukane lipidy (efekt
działania rozpuszczalnika
organicznego)
•
Znacznie zmieniona biologiczna
funkcjonalność i struktura białek
(wpływ utrwalania i wysokiej
temperatury przy przepajaniu
parafiną)
ZALETY
•
Dobrze zachowany skład chemiczny
wszystkich składników komórek łącznie
z lipidami
•
Krótsza procedura przygotowania niż w
technice parafinowej
•
Zapobiega denaturacji białek,
zachowuje aktywność enzymów
WADY
•
Struktura materiału częściowo
uszkodzona na skutek
mrożenia
(proces krystalizacji)
•
Grubsze preparaty i
gorsza jakość
obrazu niż w przypadku preparatów
parafinowych
KROJENIE
MATERIAŁU NIEUTWARDZONEGO
stosowane w histochemii i immucytochemii
krojenie przy pomocy wibratomu
(nóż drga z określoną częstotliwością)
skrawki grube 50 –
100 μm, nierówne
W medycynie z
aletą krojenia na wibratomie i w kriostacie
jest wykorzystanie
tych technik np. w czasie kilkugodzinnych operacji, gdy chirurg
wycinając fragment tkanki i
przekazując do analizy, może liczyć na to, że w trakcie operacji wróci do niego wynik
histopatologiczny
. Wówczas wie dokładnie, z jaką tkanką patologiczną ma do czynienia i
odpowiednio do tego zmodyfikować przebieg zabiegu.

11
Biologia Komórki
Ćwiczenie II
Metody cytochemiczne
Zasady metod cytochemicznych :
-
pozwalają na umiejscowienie w komórkach (cytochemia) i tkankach (histochemia)
określonych substancji chemicznych oraz enzymów,
-
stanowią one połączenie metodyki histologicznej i reakcji chemicznych,
-
w odróżnieniu od barwienia histologicznego (mającego na celu skontrastowanie struktur
komórek i tkanek), reakcje cytochemiczne prowadzą do uwidocznienia w preparacie
mikroskopowym jedynie ściśle określonych substancji chemicznych będących przedmiotem
badań,
-
na szkiełku podstawowym przeprowadza się reakcję chemiczną, która przebiega pomiędzy
szukaną substancją zawartą w materiale biologicznym oraz wprowadzonym do środowiska
reakcji zwi
ązkiem chemicznym (substratem), w miejscu wykrywanej substancji powstaje
produkt reakcji cytochemicznej, którego lokalizacja i ilość jest oceniana w MŚ lub ME,
- produkt reakcji musi by
ć widoczny (barwny, fluoryzujący lub elektronowo gęsty) i
nierozpuszczalny (co zapobiega jego wypłukaniu i zatarciu pierwotnej lokalizacji) w
środowisku reakcji,
- reakcje cytochemiczne cechuje
duża specyficzność (swoistość).
Typy reakcji cytochemicznych :
- bezpo
średnia – reakcja wykrywanej substancji z substratem prowadząca do powstania
produktu wyznaczającego miejsce jej lokalizacji,
- dwu lub kilkuetapowa –
w której wstępnie dochodzi do zmiany struktury chemicznej
szukanej substancji, a potem ten zmieniony związek chemiczny jest wykrywany przez
przeprowadzenie końcowej reakcji barwnej,
- specyficzne wi
ązanie barwników na drodze oddziaływań elektrostatycznych lub
chemicznych,
- wybiórcze uwidacznianie niektórych zwi
ązków oparte na selektywnej,
rozpuszczalno
ści w nich barwników (np. wykrywanie lipidów Sudanem).
Wykrywanie związków chemicznych w cytochemii:
• białka, kw. nukleinowe i cukrowce – można badać w materiałach przygotowanych techniką
parafinową,
• aminy biogenne -
najlepiej w materiałach przygotowanych metodą liofilizacji (proces
suszenia zamrożonego materiału w próżni i w obniżonej temperaturze) –
• enzymy
, tłuszcze – w technice mrożeniowej, oprócz nich można tu badać wszystkie
pozostałe substancje chemiczne.
Przygotowanie do badań cytochemicznych w transmisyjnym mikroskopie elektronowym
(TEM):
- utrwalenie,
- pokrojenie
materiału nieutwardzonego na skrawki vibrat omowe o grubości 50-100 µm,
- reakcja cytochemiczna,
- zatapianie
w żywicy epoksydowej,
- krojenie na ultra cienkie skrawki,
- kontrastowanie i obserwacja w TEM.
Przykłady reakcji cytochemicznych
• wykr
ywanie białek- reakcja Millona,

12
• wykrywanie cukrowców - reakcja P.A.S.,
• wykrywanie lipidów -
głównie Sudan III i IV, czerń sudanowa, czerwień oleista,
• kwasów nukleinowych - reakcja Feulgena,
• wykrywanie enzymów -
oparte jest na uwidocznieniu produktów ich aktywności
(wykrywany e
nzym musi być czynny; do środowiska wprowadzamy substrat, który pod
wpływem funkcji wykrywanego enzymu ulega przemianie w barwny lub elektronowo gęsty
strąt).
Reakcje kontrolne -
pozwalają na sprawdzenie swoistości reakcji cytochemicznych, a przez to
wiarygodności uzyskanych wyników. Reakcje kontrolne przeprowadza się równolegle z
właściwymi reakcjami cytochemicznymi.
Typy reakcji kontrolnych:
- swoiste blokowanie chemiczne badanej substancji,
-
opuszczanie procedur wstępnych w przypadku reakcji wieloetapowych,
- inkubacja bez substratu ( w reakcjach enzymatycznych),
- blokowanie wykrywanych enzymów swoistymi inhibitorami.
Jeśli zastosowana metoda cytochemiczna jest prawidłowa, to w reakcjach kontrolnych
uzyskuje się wynik negatywny.
Metody immunocytochemiczne
Charakteryzują się najwyższą swoistością. Polegają na wykrywaniu i lokalizacji składników
komórek i tkanek na zasadzie reakcji antygen – przeciwcia
ło, czyli wykrywaniu substancji o
charakterze antygenowym za pomocą znakowanych przeciwciał.
Antygen - jest to substancja (zazwyczaj bia
łko proste lub złożone, rzadziej związek o niższej
masie cz
ąsteczkowej) wykazująca zdolność do wywoływania produkcji swoistych przeciwciał
przez system immunologiczny oraz swoistego wi
ązania wytworzonych przeciwciał i
tworzenia z nimi kompleksów.
Każda struktura komórkowa, w skład której wchodzą białka (lub inne substancje zdolne do
wywołania odpowiedzi immunologicznej) ma potencjalne własności antygenowe i to właśnie
wykorzystuje immunocytochemia.
Przeciwciało - białko – immunoglobulina wytwarzana przez limfocyty B, wiążąca się z
antygenem, ma k
ształt litery Y i składa się z dwóch ciężkich i dwóch lekkich łańcuchów
polipeptydowych. P
od wpływem trawienia papainą rozpada się na fragment Fc oraz dwa
fragmenty Fab, które zawierają miejsca wiążące antygen.
Przeciwciała do badań uzyskuje się w laboratoriach w wyniku immunizacji zwierząt.
Uzyskuje się generalnie dwa typy przeciwciał:
• Przeciwciała monoklonalne - zbiór przeciwciał, które wykazują jednakową swoistość
względem danego antygenu oraz takie same powinowactwo (najlepsze do badań).
• Przeciwciała poliklonalne – przeciwciała, które wiążą różne antygeny i wykazują
różne powinowactwo względem tego samego antygenu.
Przeciwciała są znakowane, by można było uwidocznić na preparacie mikroskopowym
miejsca wiązania przeciwciała z szukanym antygenem.
Znakowanie
przeciwciał:
- fluorochromami
(gdy ogląda się w mikroskopie fluorescencyjnym),
- enzymami (peroksydaza, fosfataza zasadowa) –
zarówno do MŚ i ME,
- metalami -
ferrytyną (białko zawierające żelazo) i złotem – tylko do badania w ME.
Typy reakcji immunocytochemicznych
-
reakcje bezpośrednie – dodaje się znakowane przeciwciało bezpośrednio przeciwko
szukanemu antygenowi,

13
-
reakcje pośrednie – bardziej swoiste, w I etapie przeprowadza się reakcję antygenu z
nieznakowa
nym przeciwciałem, a w II etapie stosuje się znakowane antyprzeciwciała
(przeciwciało przeciw przeciwciału) – by taka reakcja mogła zajść, antyprzeciwciała muszą
pochodzić od innego gatunku zwierzęcia, np. w I etapie używa się nieznakowane przeciwciała
kró
licze, a w II etapie znakowane antyprzeciwciała kozy przeciwko immunoglobulinom
króliczym.
- reakcja awidyna – biotyna (metoda ABC). Awidyna –
białko obecne w jajach.
Wykazuje du
że powinowactwo do biotyny (witaminy H) i jest to jedno z najmocniejszych
zaobserwowanych w przyrodzie interakcji niekowalencyjnych. Jedna cząsteczka awidyny
wiąże cztery cząsteczki biotyny. Reakcja ABC zachodzi w trzech etapach:
1) reakcja między antygenem tkankowym a przeciwciałem pierwotnym z
utworzeniem kompleksu,
2) reakcja powstałego kompleksu z przeciwciałem wtórnym związanym z biotyną
swoistym dla przeciwci
ała pierwotnego,
3) reakcja kompleksu antygen tkankowy-
przeciwciało pierwotne-przeciwciało wtórne
z kompleksem awidyna-biotyna-
peroksydaza, który ma zdolność łączenia się z biotyną
związaną z przeciwciałem wtórnym.
Przebieg tej reakcji oceni
a się na podstawie aktywności peroksydazy w reakcji
enzymatycznej.
Hybrydyzacja in situ (Hybrydocytochemia)
Wykorzystuje właściwość wiązania się komplementarnych łańcuchów kwasów
nukleinowych
poprzez wybiórcze tworzenie się par przez zasady purynowe i pirymidynowe.
Do pojedynczych łańcuchów nukleotydów rozspiralizowanego DNA lub RNA w komórce
mogą się przyłączać fragmenty łańcuchów kwasów nukleinowych pochodzących z innych
źródeł – w tym te wytworzone w wyniku preparatyki biochemicznej lub sztucznej syntezy w
laboratoriach. Prowadzi to do wiązania się odpowiednich par zasad i odtworzenia struktury
dwułańcuchowej – proces ten nosi nazwę hybrydyzacji. Może on zachodzić między
wszystkimi rodzajami kwasów nukleinowych (DNA-DNA, DNA-RNA, RNA-RNA).
W technikach
mikroskopii hybrydyzację wykorzystuje się do określenia precyzyjnej
lokalizacji badanych sekwencji nukleotydów w obrębie komórki, „in situ” – czyli w miejscu
naturalnego występowania. Stąd nazwa – hybrydyzacja in situ.
Badane sekwencje nukleotydów wykrywa
się za pomocą specjalnie przygotowanych
komplementarnych odcinków DNA lub RNA, które noszą nazwę sond, dodatkowo
znakowanych, by można było dostrzec miejsce ich związania w komórce.
Hybrydyzacja in situ umożliwia:
-
wykrywanie obcego materiału genetycznego w komórce (np. przy zakażeniach
wirusowych),
-
ocenę ekspresji poszczególnych genów w komórce poprzez wykrywanie w nich m-RNA,
-
lokalizację konkretnych genów lub ich fragmentów w obrębie poszczególnych
chromosomów (tworzenie map genowych, diagnostyka chorób dziedzicznych, wykrywanie
genów nowotworów).
Rodzaje sond
(im dłuższe sondy, tym bardziej swoiste, lecz trudniej penetrują do komórek,
więc ustalona optymalna długość sondy wynosi 100-250 nukleotydów):
• dwuniciowe sondy DNA,
• jednoniciowe sondy DNA,
• jednoniciowe sondy RNA,
• krótkie jednoniciowe sondy oligonukleotydów o dł. 25-50 nukleotydów,
Znakowanie sond:

14
• radioaktywne – do łańcucha sondy wbudowuje się izotopy promieniotwórcze
3
H,
32
P,
35
S. Sondy te są wykrywane drogą autoradiografii..
• biotyna – wykrywa się reakcją immunocytochemiczną awidyna-biotyna,
• znacznik antygenowy – połączony z nukleotydami może łączyć się z przeciwciałem,
wykrywany wybraną reakcją immunocytochemiczną.
•
Audioradiografia
Polega na lokalizacji substancji znakowanych izotopami w komórkach i tkankach (np.
radioaktywnych sond w hybrydyzacji in situ).
Substancje te wprowadza się doświadczalnie do
żywego organizmu lub żywych komórek (w hodowli), gdzie są gromadzone lub wbudowane
do znanego związku w organizmie i włączane w procesy metaboliczne (np. tymidyna
znakowana trytem wbudowuje się do DNA komórek). Wykorzystuje się fakt, że substancje
chemiczne zawierające izotopy promieniotwórcze nie są odróżniane przez organizm od
substancji nie znakowanych.
Po pewnym czasie można zbadać stopień wbudowania
znakowanego prekursora do produktu końcowego. Pobiera się narządy i tkanki zawierające
izotopy, nas
tępnie sporządza się z nich preparaty mikroskopowe (do badań w MŚ i ME),
które pokrywa się w ciemni warstwą emulsji fotograficznej. Tak sporządzony preparat leży w
ciemni kilka, kilkanaście dni (a nawet tygodni) w celu ekspozycji emulsji na działanie
promieniowania beta izotopów. W tym czasie izotopy
obecne w komórkach, emitując
elektrony,
powodują miejscowe zmiany w kryształkach bromku srebra obecnych w emulsji.
Następnie po normalnej obróbce fotograficznej emulsji (wywołaniu i utrwaleniu) powstają na
tych miejscach
strąty metalicznego srebra, widoczne w mikroskopie w formie czarnych
ziarenek.
Umożliwia to lokalizację miejsca zawierającego izotop. Skrawki pod emulsją
podbarwia się dodatkowo metodami histologicznymi.
Autoradiografia u
możliwia m.in.:
-
śledzenie szlaków metabolicznych, w które włączane są znakowane substancje chemiczne,
-
śledzenie losów populacji komórek namnażających się,
-
badanie sposobu namnażania organelli komórkowych,
-
śledzenie dróg migracji komórek przemieszczających się,
-
badanie rozmieszczenia i magazynowania związków radioaktywnych w narządach
organizmu.
Izotopy stosowane w autoradiografii do znakowania badanych substancji chemicznych to
przede wszystkim izotopy emitujące promieniowanie beta:
3
H,
14
C,
32
P,
35
S,
45
Ca,
90
Sr,
125
I.
Hodowla tkanek
- j
est metodą umożliwiającą utrzymywaniem poza organizmem czyli In vitro komórek,
fragmentów tkanek lub na
rządów przez okres dłuższy niż 24 godziny (hodowla do 24 godzin
używana w badaniach biochemicznych to inkubacja),
- t
ermin hodowla tkanek jest pojęciem szerokim i obejmuje hodowlę wycinków tkankowych,
hodowle narządowe, hodowle komórek (pierwotne, linii i szczepów komórkowych). Często
używane są dwa terminy – hodowla komórek (w odniesieniu do wyizolowanych komórek) i
hodowla tkanek (
w stosunku do hodowli narządów, hodowli przestrzennych).
Metoda pozwala na obserwowanie zachowania i
badanie żywych komórek i tkanek
pozbawionych kontrolnego i regulacyjnego wpływu organizmu.
Najczęściej stosowane typy hodowli:
- hodowla jednowarstwowa
komórek (w płaskim naczyniu)
- hodowla komórek w zawiesinie
- hodowla komórek w postaci agregatów

15
-
hodowla narządowa statyczna
-
hodowla narządowa przepływowa
Przykład techniki hodowli
Technika hodowli jednowarstwowej:
-
komórki do hodowli jednowarstwowej mogą być pobrane od zwierząt w różnym wieku,
-
pobiera się wycinki narządów lub całe narządy, których komórki izoluje się poprzez
trawienie enzymatyczne (jednostopniowe –
trypsyną lub kolagenazą i hialuronidazą lub
dwustopniowe -
kolagenazą, a następnie trypsyną), dodatkowo przygotowanie zawiesiny
komórek można wspomagać rozdrabnianiem mechanicznym tkanki
-
do zakładania i prowadzenia hodowli stosuje się pożywki (np. DMEM, HAMS F12) do
których jako suplement dodaje się surowicę cielęcą płodową, neonatalną lub surowicę
końską),
-
pożywki zapewniają sterylne środowisko wzrostu oraz dostarczają komórkom
niezbędnych związków odżywczych - węglowodany, aminokwasy, białka i peptydy, kw.
t
łuszczowe, cholesterol, lipidy, witaminy, sole mineralne; pH pożywek jest zbliżone do
naturalnego pH organizmu (utrzymywane przez występujące w pożywce bufory) i wynosi
od 6,8 do 7,2. Do pożywek dodaje się czasem antybiotyki chroniące hodowle przez
zakażeniami bakteryjnymi, grzybiczymi lub wirusowymi.
- surowice
dostarczają hormonów, czynników wzrostu i różnicowania komórek
pobudza
jących wzrost, intensyfikują różnicowanie,
-
hodowle prowadzi się w naczyniach polistyrenowych przystosowanych do adhezji komórek,
na siatkach z poliwęglanu lub na szkle,
-
przy zakładaniu hodowli umieszcza się 1-5 x 10
5
komórek na 1 cm
2
,
- hodowla prowadzona jest specjalnych inkubatorach w at
mosferze nawilżonego powietrza o
temp. 37-38ºC w przypadku komórek ssaków i 38-40ºC w przypadku komórek ptaków oraz
dodatkiem 95% tlenu i 5% d
wutlenku węgla.
- h
odowla komórkowa może by prowadzona w warunkach statycznych lub w przepływie
medium.
Mikroskop elektronowy transmisyjny (TEM)
TEM jest
niejako odpowiednikiem MŚ, gdyż strumień elektronów (podobnie jak strumień
promieni świetlnych) przechodzi przez preparat – stąd nazwa transmisyjny, w odróżnieniu od
mikroskopu elektronowego skaningowego (SEM), w którym strumień elektronów odbija się
od powierzchni preparatu.
TEM jest hermetyczną metalową kolumną w kształcie rury, w której występuje wysoka
próżnia (do -100 atmosfer) wytwarzana przez zespół pomp połączonych z mikroskopem. Do
mikroskopu podłączony jest też system transformatorów przetwarzających prąd sieciowy na
prąd o wysokim napięciu i niskim natężeniu. Na szczycie kolumny jest działo elektronowe, na
drugim końcu ekran fluorescencyjny. W środku kolumny umieszcza się siateczkę z badanym
obiektem.
Działo wytwarza elektrony, które są przyspieszane przez wysokie napięcie i
formowane przez elektromagnetyczne soczewki
działające kolejno jako kondensor, obiektyw
i okular. Obraz w TEM powstaje w wyniku rozpraszania elektronów przez jądra atomów
badanego obiektu. Rozproszone
elektrony nie docierają do ekranu fluorescencyjnego, widać
wtedy obraz ciemny. W mie
jscach, gdzie elektrony przenikają swobodnie przez preparat
widać obraz jasny.
Ponieważ pierwiastki, z których składają się żywe komórki, są lekkie, mają bardzo słabą
z
dolność rozpraszania elektronów. Ich obraz w TEM byłby jasny i słabo czytelny. Dlatego
wprowadzono specjalne techniki barwienia (kontrastowania) polegające na wprowadzeniu do
badanych obiektów soli metali ciężkich zawierających atomy o wysokiej masie. Tak
skontrastowany preparat daje wyraźny czarno-biały obraz w TEM.

16
Porównanie podstawowych cech mikroskopów -
świetlnego i elektronowego
– transmisyjnego
Cechy
Świetlny (optyczny)
Elektronowy (transmisyjny)
Powiększenie
do 2000x
1 000 000x lub więcej
Rozd
zielczość max
200 nm (0,2 µm)
0,5 nm
Źródło obrazu
promienie światła
widzialnego
wiązka elektronów
Ostrość obrazu
zapewniają
szklane soczewki
obiektywu
elektromagnetyczne soczewki
obiektywu
Obraz widziany poprzez
szklane soczewki okularu
ekran fluorescencyjny
i monitor
Obiekt umieszczony na
szkiełku podstawowym
siateczce
(miedź, nikiel)
Obiekt może być żywy
tak
nie
Obiekt wymaga specjalnej
obróbki
nie zawsze
zawsze
Otrzymany obraz jest
barwny
tak
nie
Przygotowanie materiału biologicznego do badań w mikroskopie
elektronowym transmisyjnym (TEM)
A. P
obieranie materiału
•
Przyżyciowe - w trakcie zabiegu operacyjnego lub metodą biopsji
•
Pośmiertne - w czasie 3 minut od ustania pracy serca
• w
ycinek o wymiarach nie większych niż 1 mm x 1 mm x 1 mm
• z
a pomocą bardzo ostrych narzędzi
B. Utrwalanie I
Utrwalacz = nośnik + substancje utrwalające
Nośnik - zapewnia stałe i optymalne pH (7,2- 7,4), ciśnienie osmotyczne i skład jonowy
Najczęściej stosowane nośniki to bufor fosforanowy i bufor kakodylowy.
Substancj
e utrwalające:
• parafolmaldehyd -
szybko dyfunduje do utrwalanego materiału, wiąże się luźno z
białkami (poprzez wiązania krzyżowe) i nie modyfikuje ich struktury, reaguje z
glikogenem, nie utrwala lipidów,
• aldehyd glutarowy - powoli dyfunduje do utrwalane
go materiału, wiąże się ściśle z
białkami i zmienia ich strukturę, reaguje z lipidami.
Przykład utrwalacza:
• Mieszanina 2,5 % aldehydu glutarowego oraz 1 % paraformaldehydu
w 0,1 M buforze fosforanowym o pH 7,4
Sposób wykonania: immersyjne lub perfuzyjne.
Czas trwania i temperatura: 2 godziny w temp. 4ºC lub pokojowej.

17
C. P
łukanie
•
Ma na celu usunięcie utrwalaczy nie związanych z utrwalanym materiałem.
•
Jest przeprowadzane w buforze zastosowanym jako nośnik substancji utrwalającej
(np. buforze fosforanowym)
•
Trwa najczęściej 24 godziny, ale może być wydłużone do 7 – 14 dni (transport prób)
D. Utrwalanie II (wtórne)
i wstępne kontrastowanie
• Utrwalacz
i pierwszy środek kontrastujący: 1 - 2 % roztwór czterotlenku osmu w
wodzie destylowanej lub buforze fosforanowym
• Utrwalanie lipidów
•
Różnicowanie gęstości elektronowej (wstępne kontrastowanie)
Sposób wykonania: immersyjne.
Czas trwania i temperatura: 2 godziny w temp. pokojowej.
E. Odwadnianie
W roztworach alkoholu etylowego o rosnącym stężeniu: 30, 50, 60, 70, 80, 90, 96, 100%
F. P
rzeprowadzanie przez płyny pośrednie (rozpuszczalniki organiczne)
• aceton
• tlenek propylenu
G. Zatapianie
•
Bardzo mała grubość skrawków używanych w TEM stwarza konieczność zatapiania
materiału w substancjach o wysokiej twardości.
•
Żywice syntetyczne - epoksydowe (EPON 812), poliestrowe, metakrylany. Żywice w
temperaturze pokojowej są płynne.
•
Przepajanie w mieszaninie rozpuszczalnika i żywicy przez 24 godziny.
•
Umieszczenie materiału w kapsułce z płynną żywicą.
•
Utwardzanie żywicy poprzez polimeryzację w podwyższonej temperaturze (60-80°C
przez 24-48 h).
•
Powstają bardzo twarde bloczki zawierające zatopiony materiał.
H. Trymowanie i krojenie
Ultracienkie skrawk
i są nie tylko minimalnie grube ale i bardzo niewielkie – długość
większego boku nie przekracza zazwyczaj 0,3 mm (jest mniejsza niż materiału krojonego
przy pobieraniu)
. W związku z tym przed właściwym krojeniem wstępnie przygotowuje się
płaszczyznę skrawania o takich wymiarach.
-
wpierw wykonuje się kilka tzw. skrawków półcienkich (o grubości 0,5 do 2µm)
obejmujących całą powierzchnię zatopionego materiału, barwi na szkiełku np. błękitem
toluidynowym, następnie ogląda pod MŚ w celu wybrania tego rejonu bloczka, z którego
będzie się potem kroić właściwe skrawki ultracienkie czyli o grubości 20-60 nm,
-
po ocenie skrawków półcienkich w MŚ, na ultramikrotomach pod kontrolą powiększającej
lupy lub okularów zatopiony materiał poddaje się trymowaniu – bocznemu ścinaniu w
piramidkę, której wierzchołek będzie odpowiadał wielkości skrawka ultracienkiego i
wybranego na podstawie obserwacji skrawków półcienkich w MŚ rejonu.
- ultramikrotomy –
ich noże charakteryzują się szczególną twardością i ostrością. Warunków
tych nie spełnia najlepszy nóż stalowy, w związku z tym stosuje się noże szklane lub najlepiej
diamentowe.
-
do noży montuje się (lub są wbudowane fabrycznie) wanienki z tworzywa sztucznego lub
folii metalowej, które wypełnia się wodą. Podczas krojenia ultracienkie skrawki wypływają
na powierzchnię wody, skąd za pomocą precyzyjnej pensety są przenoszone na okrągłą

18
miedzianą lub niklową siateczkę o średnicy 3 mm, która jest odpowiednikiem szkiełka
podstawowego w MŚ.
I. Kontrastowanie - r
óżnicowanie gęstości elektronowej za pomocą soli metali ciężkich.
Skrawki na siateczkach
były już wstępnie skontrastowane przy wtórnym utrwalaniu za
pomocą czterotlenku osmu, lecz kontrast ten jest stosunkowo słaby, stąd dodatkowo
kontrastuje się je innymi solami metali ciężkich:
- octanem uranylu
- cytrynianem
ołowiu
Mikroskop elektronowy skaningowy (SEM)
- cienki
strumień elektronów nie przechodzi przez preparat, lecz odbija się od niego –
bada punkt po punkcie
powierzchnię obiektu (skanuje). Wybijane z powierzchni
elektrony są zbierane i wzmacniane, a ich obraz uwidacznia się na ekranie monitora,
-
stosuje się do badań zróżnicowania przestrzeni powierzchni, kształtu i rzeźby struktur
komórkowych i organelli
-
uzyskuje się plastyczny, niemal trójwymiarowy obraz preparatu
-
technika przygotowania materiału odmienna niż w TEM
-
do pokrywania preparatu stosuje się atomy węgla i złota
P
reparaty półcienkie
• pr
eparaty o grubości 0,5 – 2 µm
•
wykonywane za pomocą ultramikrotomu
•
z materiału pobranego i przygotowanego do badań w ME
•
przeznaczone do oglądania w mikroskopie świetlnym (MŚ)
• barwione
na szkiełku podstawowym błękitem toluidyny
Wyszukiwarka
Podobne podstrony:
Mikroskopia świetlna i elektronowa w badaniach naukowych
sprawozdanie badanie mikroskopowe-teoria!!!!!!!!!!, Elektrotechnika, dc pobierane, Podstawy Nauk o m
Mikroskopia elektronowa id 3018 Nieznany
Mikroskopy, Elektrotechnika, dc pobierane, pnom wimir, PNOM, I Semestr - Materialoznawstwo - sprawoz
Mikroskopia elektronowa ETI
Mikroskop elektronowy
Mikroskopia elektronowa i konfokalna
mikrosystemy 1, Analogowe Układy Elektroniczne
mikroskopia elektronowa, 1
Mikroskopowe obserwacje metali i stopów, Elektrotechnika, dc pobierane, pnom wimir, PNOM, Materiałki
BUDOWA I ZASADA DZIAŁANIA SKANINGOWEGO MIKROSKOPU ELEKTRONOWEGO
IFPAN101210a Pierwsze swiatlo mikroskopu elektronowego
Kopia ściąga bmikroskopowe stali węglowych wyżarzonych i żeliw, Elektrotechnika, dc pobierane, pnom
Technika skaningowej mikroskopii elektronowej SEM, Uczelnia, Metalurgia
sprawozdanie badanie mikroskopowe, Elektrotechnika, dc pobierane, pnom wimir, PNOM, bad mikros
Mikroskopia elektronowa, Analityka Medyczna UMB, III, Patomorfologia, Wykłady
więcej podobnych podstron