
International Conference
Diagnosis & Treatment of Inner Ear
Disorders
Genetics of deafness
Lech Korniszewski
The Medical University of Warsaw
Institute of Physiology and Pathology of
Hearing

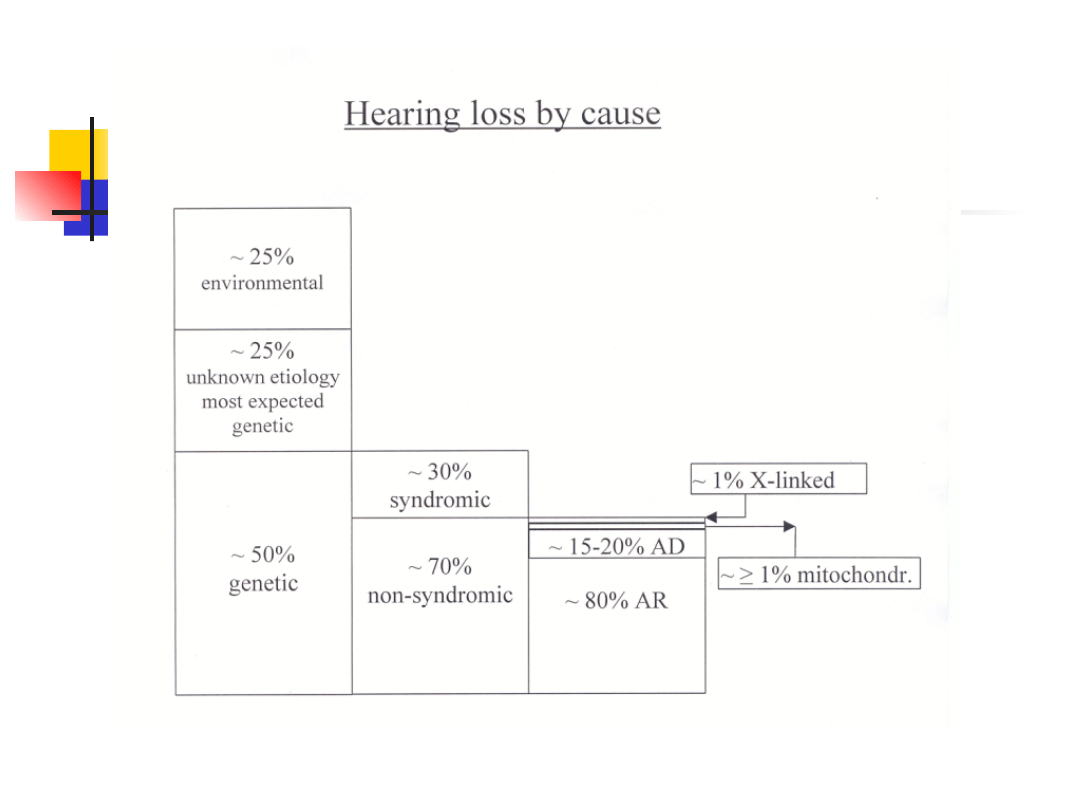
Hearing loss – incidence:
6-8% of population – when all
causes are combined hearing
loss – most common birth defect
1 in 1000 newborns are deaf
1 in 300 children are affected with
congenital hearing loss of a lesser
degree additional 1 in 1000 become
profoundly hearing impaired before
adulthood

Genetic hearing loss
approximately 1% of all human genes are
involved in the hearing process
inheritance: autosomal recessive
autosomal dominant
X-linked
mitochondrial
allelic mutatione in some genes can cause recessive
and dominant hearing loss
mutations in the same gene may cause syndromic or
nonsyndromic hearing loss
recessive hearing loss may be caused by a combination
of two mutations in differrent genes from the same
functional group
Syndromic hearing loss
Over 400 syndromes have been described
in which hearing loss is a component part.
There are many factors that make specific
syndrome diagnosis difficult:
*
The rarity of most of these syndromes (lack
personal experience)
*
Variability of clinical expression
*
Genetic heterogeneity (a single phenotype may
be result of different genes mutations)
*
Pleiotropy (single gene may cause many
different phenotypic effects)
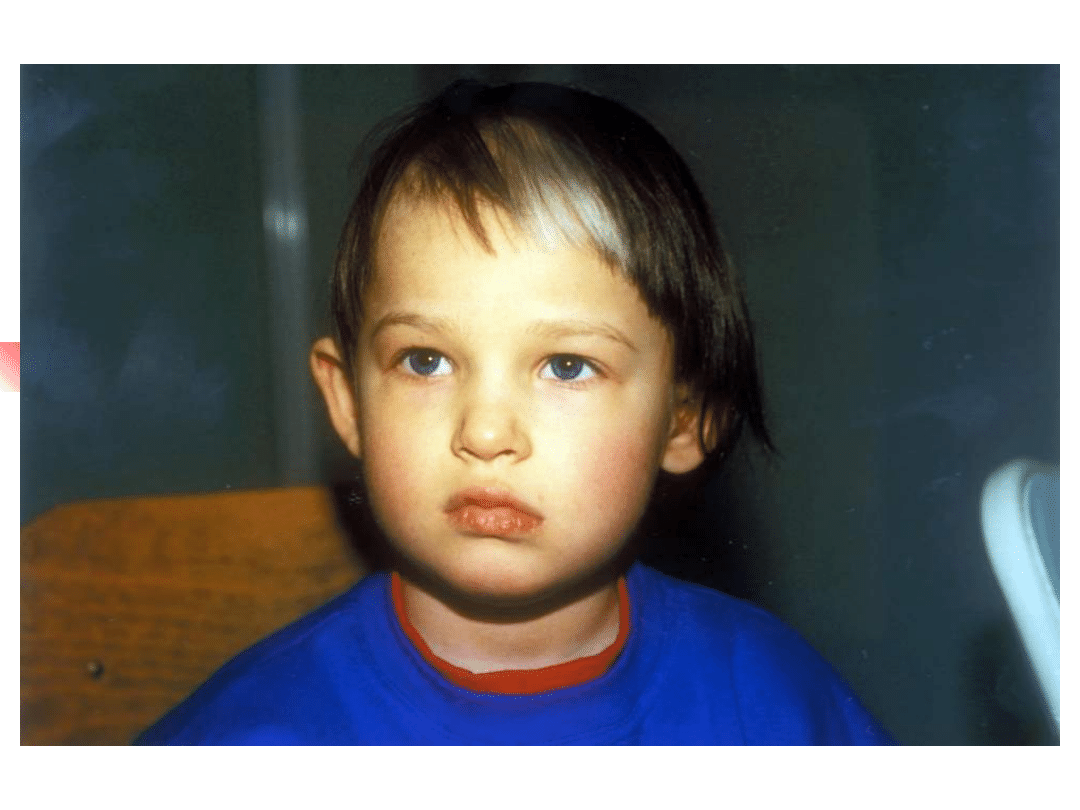
Waardenburg syndromes
–
Bilateral or unilateral sensorineural
hearing loss in association with defects in
tissues derived from neural crest cells
–
pigmentary abnormalities hair, skin and
eyes
–
hearing loss is due to defective migration of
melanocytes info the intermediate layer of
the stria vascularis
–
genetically heterogeneous; inheritance AD
–
four clinical subtypes
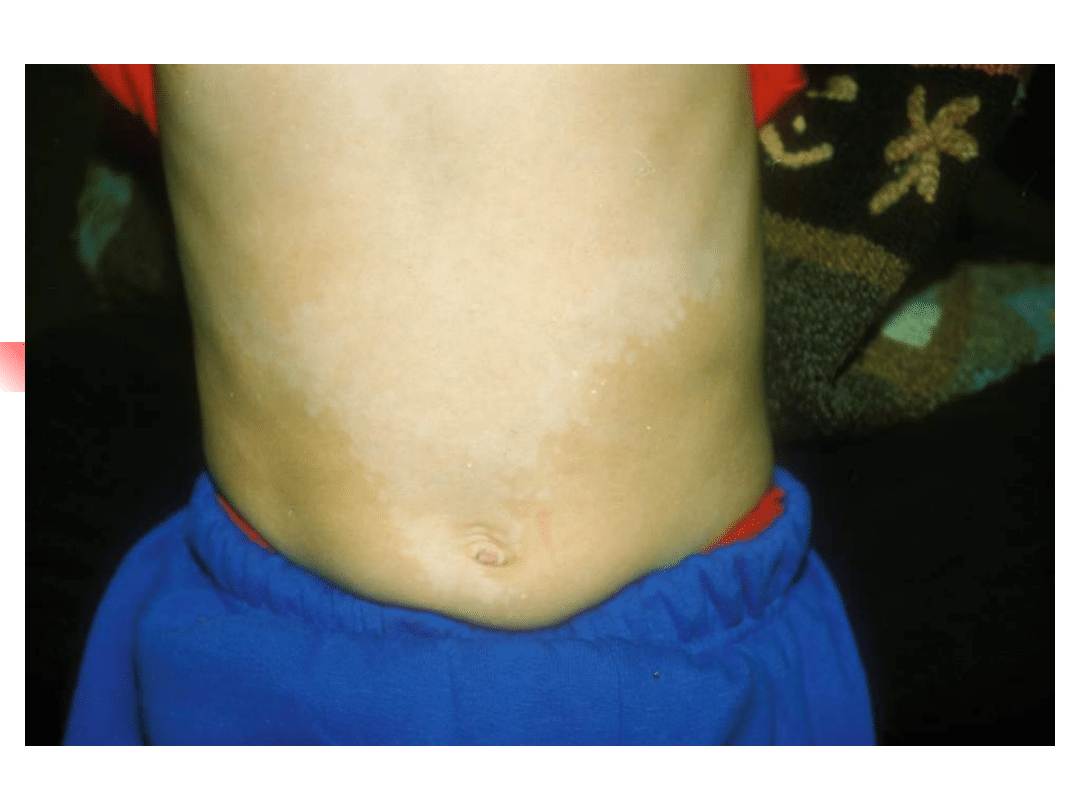
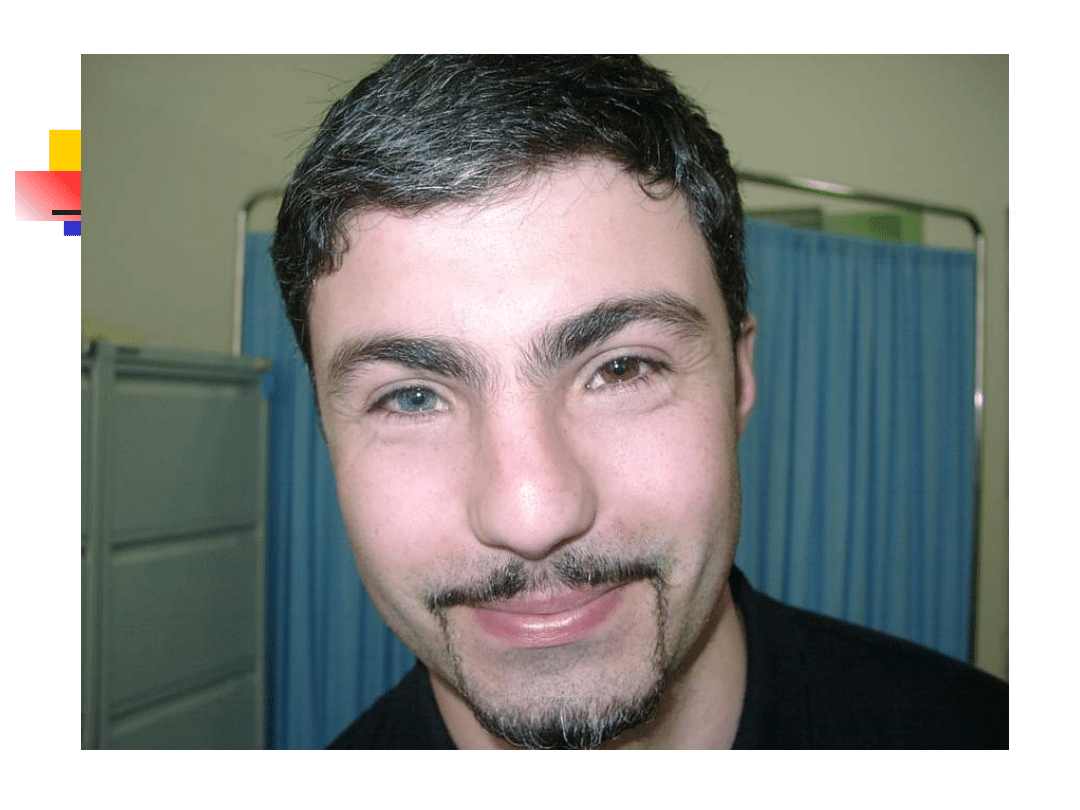
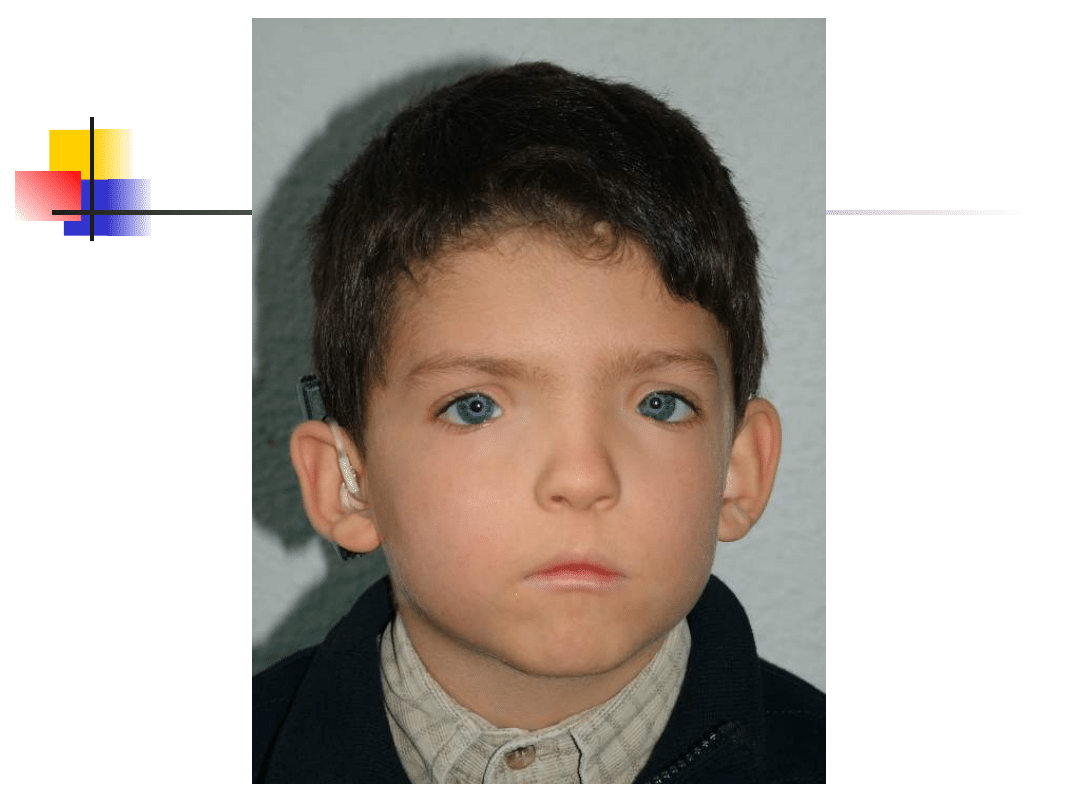


Waardenburg syndromes
Typ
e
Gene
Protein/func
tion
Clinical features
WS1
PAX3
transcription
factor
Abnormal pigmentation of hair,
eyes and skin. Dystopia
canthorum, short philtrum,
synophrys. Deafness in 20%
(unilateral or bilateral)
WS2
MITF
transcription
factor
Abnormal pigmentation of hair,
eyes and skin. Deafness in 40%
(unilateral or bilateral). No
dysmorphic features
WS3
PAX3
transcription
factor
Features of WS1 with limb
anomalies
WS4
EDN3
EDNRB
SOX10
endothelin ligand
endothelin
receptor
transcription
factor
Abnormal pigmentation of hair,
eyes and skin with Hirschprung
disease
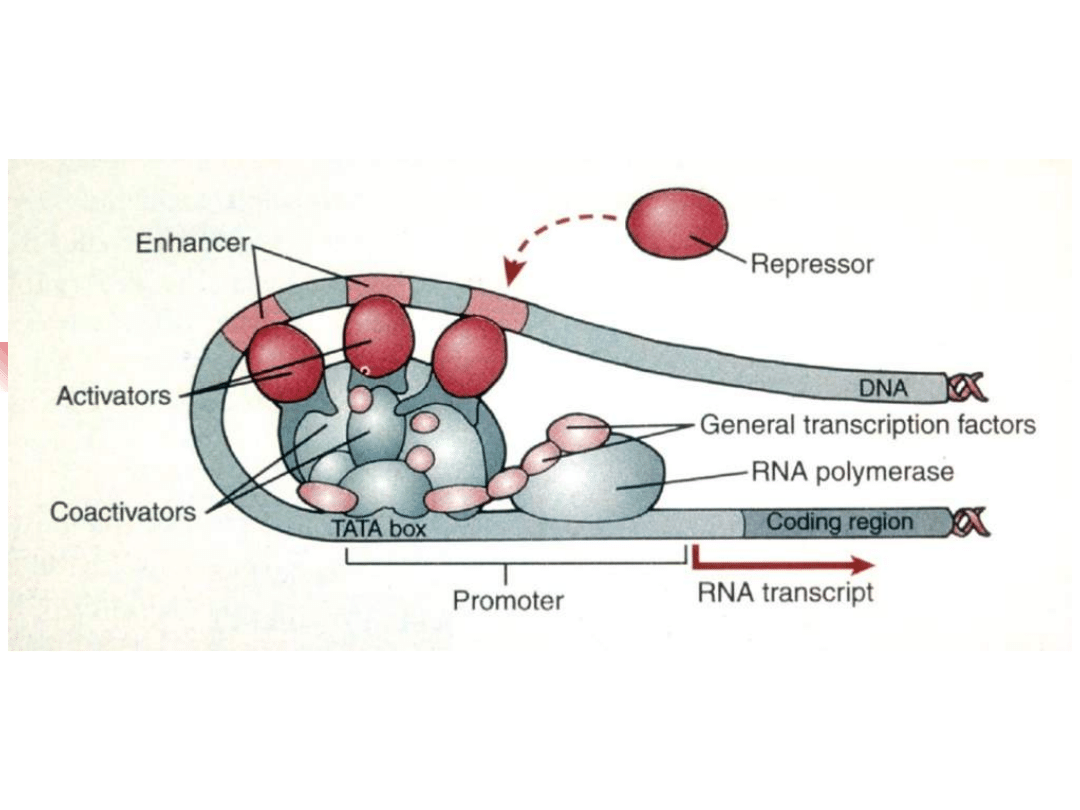
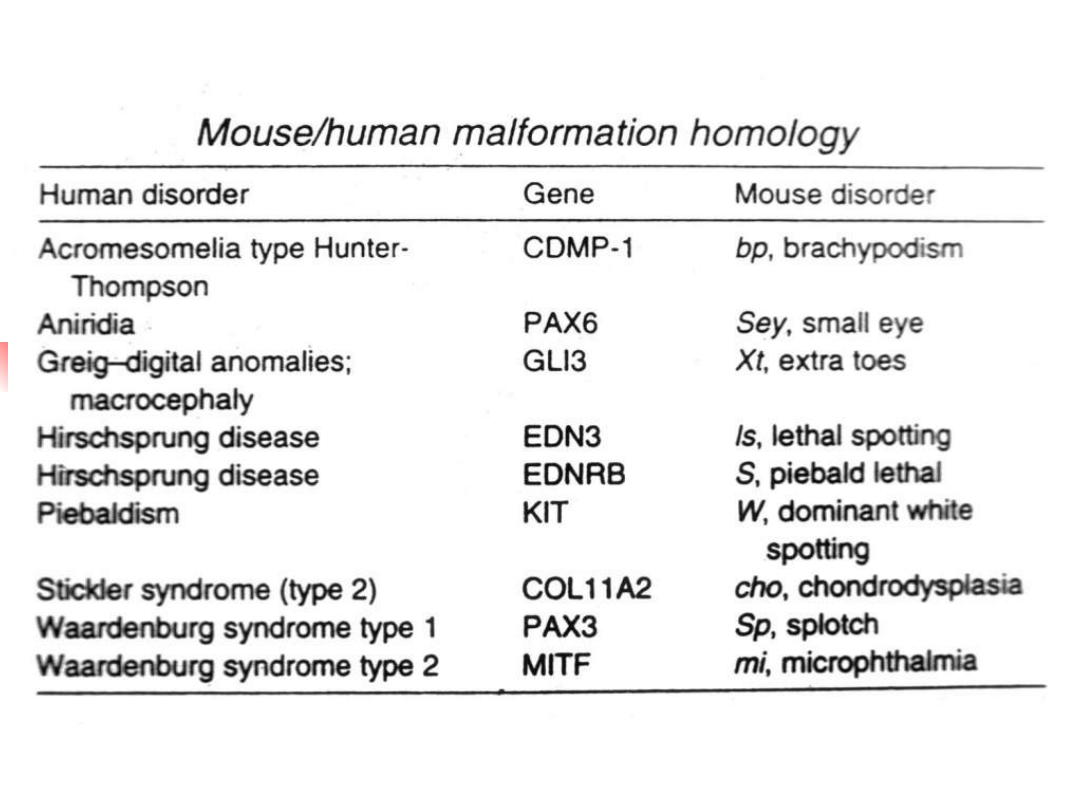
EDN
3
EDNRB
3
WS
1
WS
3
PAX
3
SOX
10
WS 4
transactivat
ion
WS 2
MITF
melanocyte
tyrosinase
transactivatio
n
Transcription factor hierarchy in Waardenburg syndrome:
regulation of MITF expression by SOX10 and PAX3
Branchio-oto-renal
syndrome
Hearing loss conductive, sensorineural or
mixed;
Branchial cysts and fistulae, external ear
malformations, renal dysplasia or
hypoplasia. Some patients also eye
anomalies
Gene EYA1 on 8q13.3; encoded molecule –
transcription factor.
Inheritance autosomal dominant. Genetically
heterogenous (second BOR locus on 1p31)
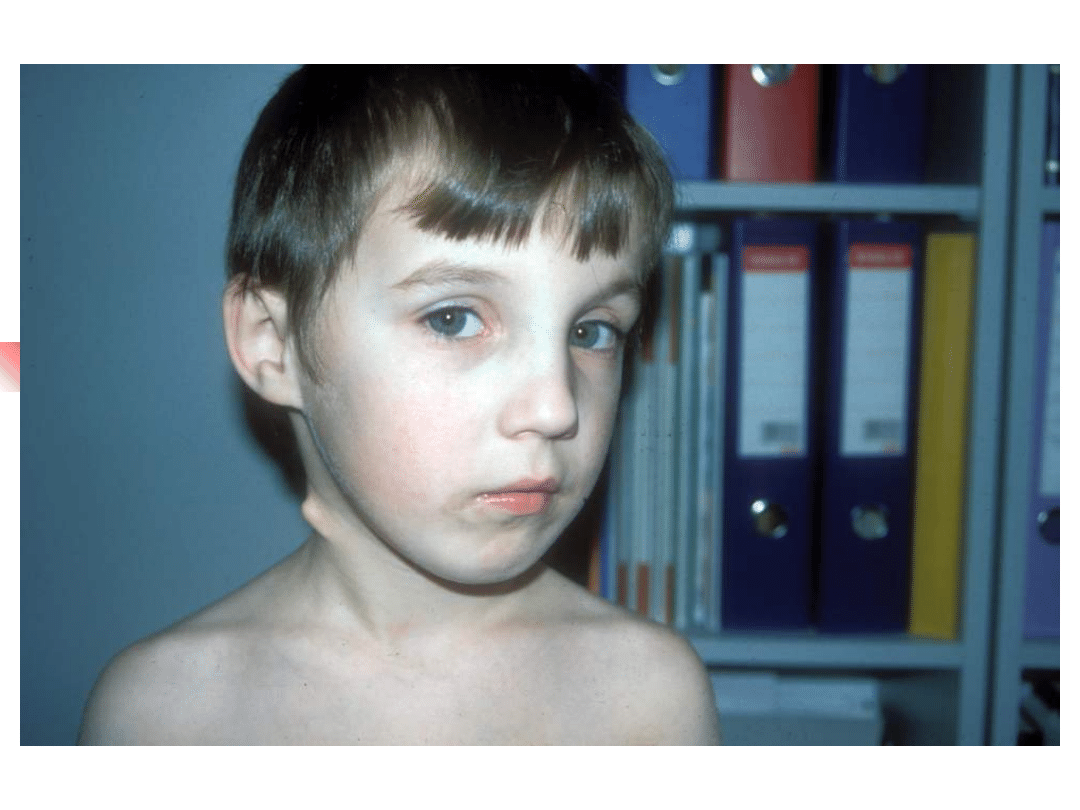
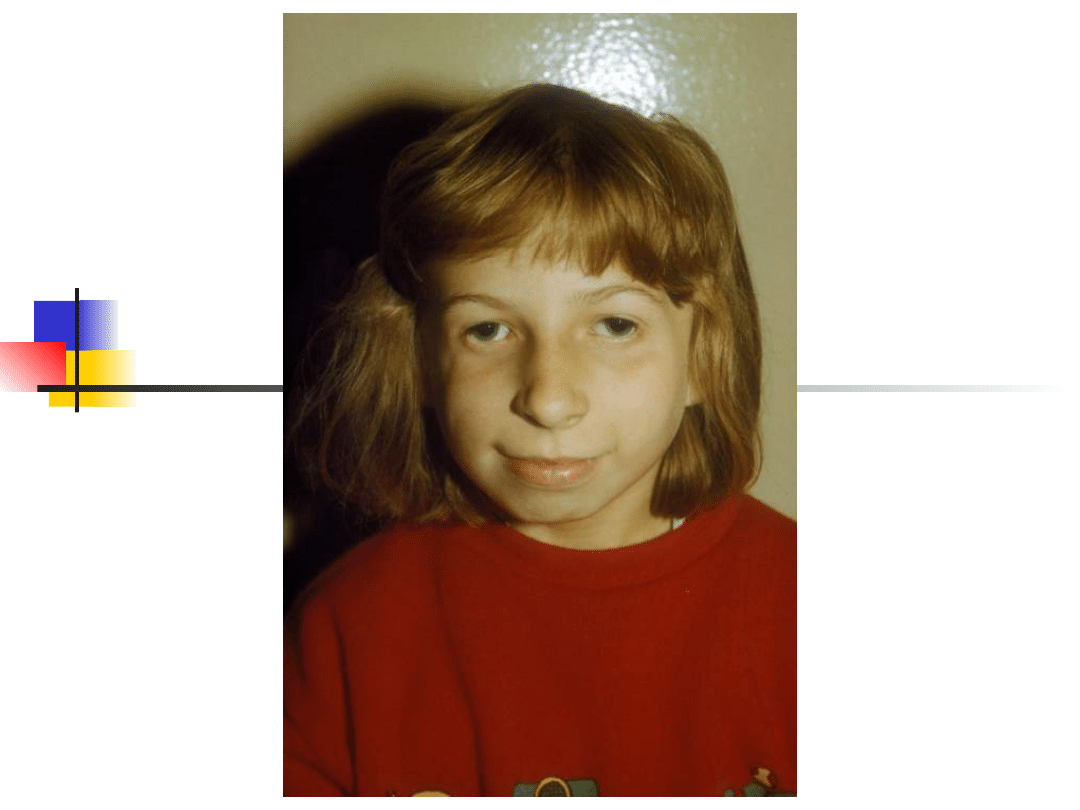
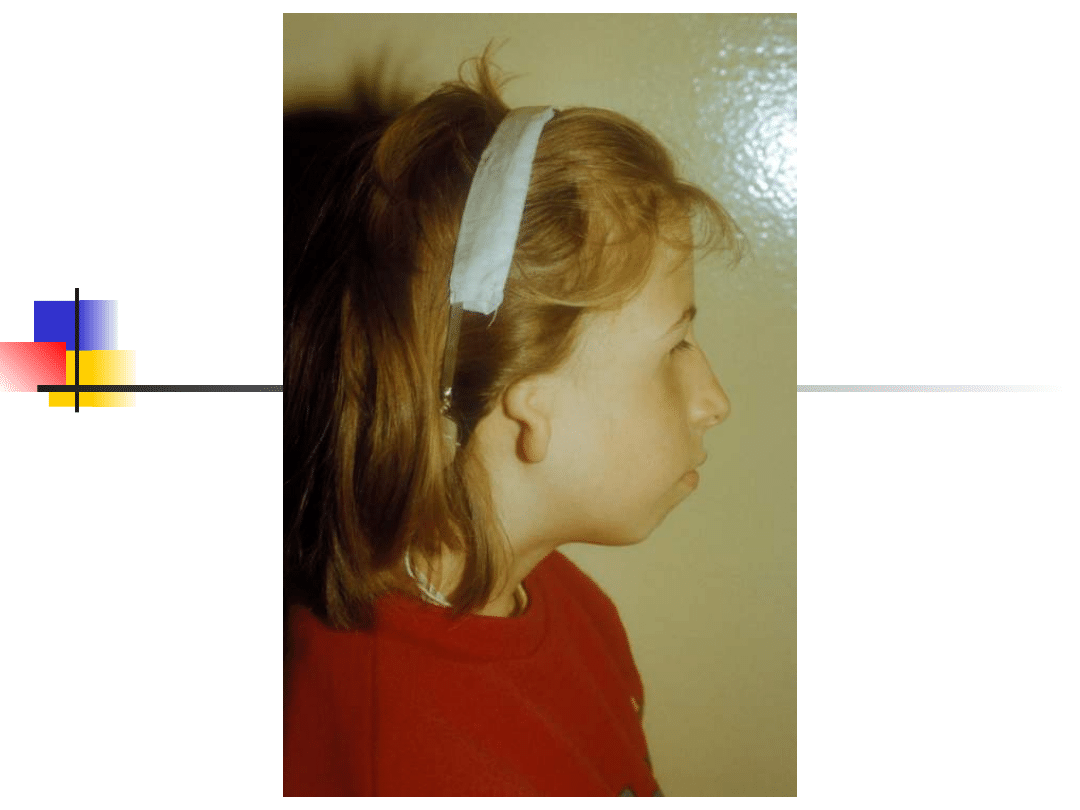
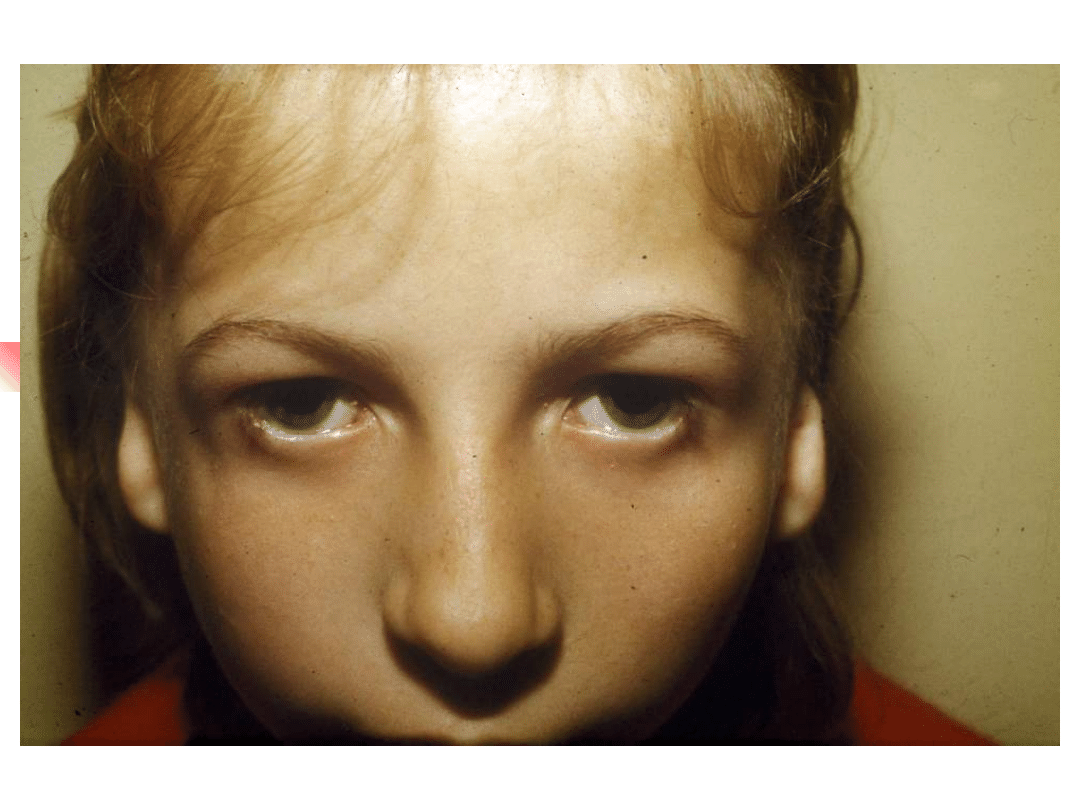
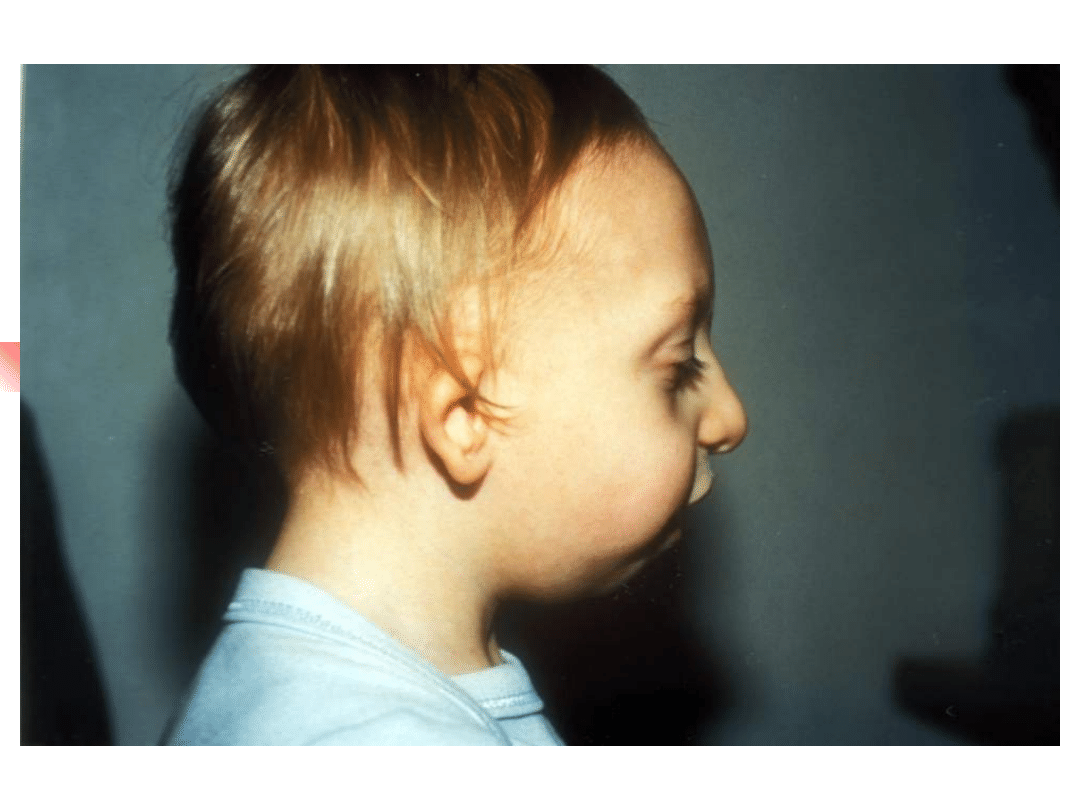
Treacher-Collins syndrome
Hearing loss conductive, sensorineural or
mixed;
Clinical features: down-slanting palpebral
fissures, malformation of external and middle
ears, sparse lower eyelashes and colobomata of
lower eyelids, malar hypoplasia.
Gene TCOF; encoded nuclear cytoplasmic
transport protein
Inheritance autosomal dominant

Usher syndromes
*
Syndromic association of hearing loss with retinitis
pigmentosa
*
Accounts 2-4% of all cases of profound deafness and
50% of the deaf-blind population
*
Inheritance autosomal recessive.
Genetic heterogeneity high – more than 12 loci
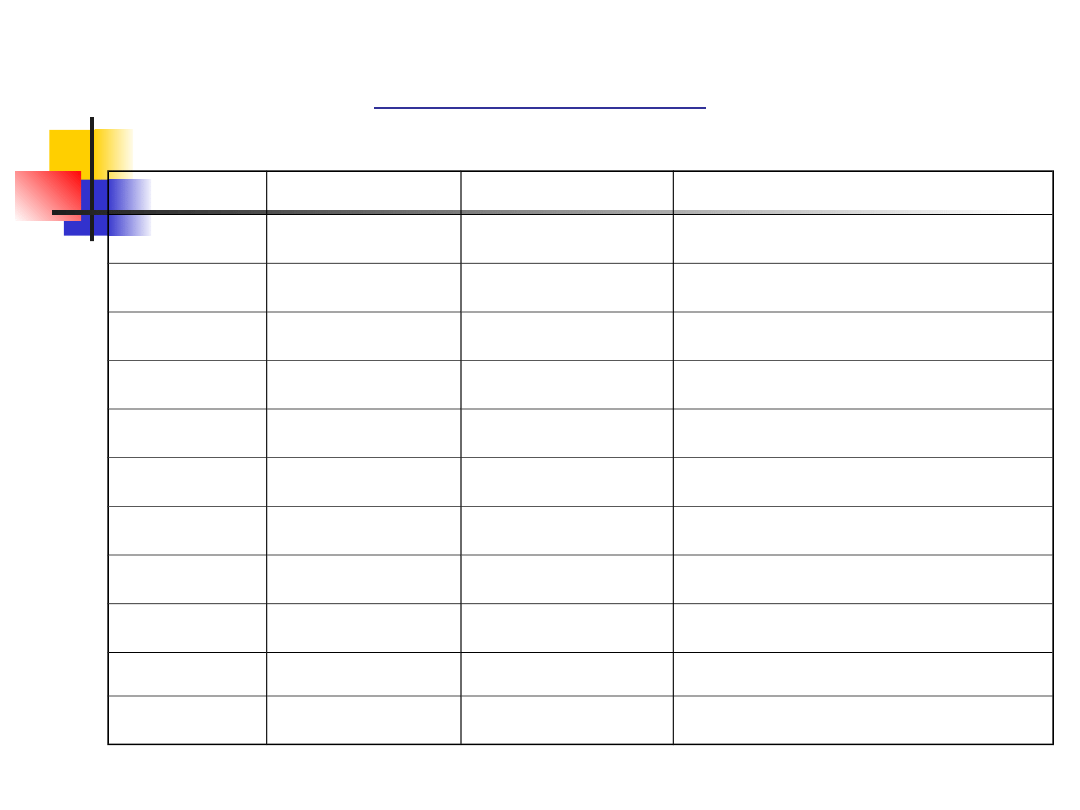
Clinically three main types:
TYPE
HEARING LOSS
VESTIBULAR
RESPONSE
ONSET OF
REINITIS PIGM.
I
Profound from
birth
Absent
1st decade
II
Moderate from
birth
Normal
1st or 2nd decade
III
Progressive
Variable
Variable
Usher syndrome
Type
Locus
Gene
Protein
USH1A
14q32
-
-
USH1B
11q13.5
MYO7A
myozyn VIIA
USH1C
11p15.1
USH1C
harmonin
USH1D
10q21
CDH23
cadherina 23
USH1E
21q21
-
-
USH1F
10q21-22
PCDH15
protocadherin15
USH1G
17q24-25
USH1G
SANS
USH2A
1q41
USH2A
usherin
USH2B
3p23-24.2
-
-
USH2C
5q14.3-21.3
-
-
USH3A
3q21-25
USH3A
clarin 1

Usher syndromes
Usher
syndrome
type
Gene
Molecule
encoded/function
clinical features
1B
MYO7A
myosin 7A (motor
molecule)
profound congenital deafness,
retinitis pigmentosa, vestibular
areflexia
1C
USH1C
harmonin
- " - - " -
1D
CDH23
cadherin 23
progfound congenital deafness,
variable retinitis pogmentosa and
variable vestibular function
1F
PCDH15
protocadherin 15
profound congenital deafness,
retinitis pigmentosa, vestibular
dysfunction
2A
USH2A
usherin (extracellular
matrix protein)
congenital moderate to severe
sensorineural hearing loss (normal
vestibular function) retinitis
pigmentosa
3A
USH3A
clarin 1 (trans-
membrane protein)
Progressive sensorineural hearing
loss, normal or absent vestibular
function, retinitis pigmentosa
Nonsyndromic deafness: DFNA11 (dominant) and DFNB2 (recessive) results
from other alleles of MYO7A; DFNB18 results from different harmonin mutation.

Pendred syndrome
Sensorineural deafness, goiter and malformation of the
inner ear
Hearing loss is most frequently profound, variable in its
onset, rapidly progressive
Goiter results from a specific defect in the organification
of iodine (abnormal release of iodine trapped by thyroid
after administration of perchlorate)
Malformation of the inner ear in 86% of cases:
dilatation of the vestibular aqueduct and endolymphatic
sacs, Mondini malformation
Inheritance autosomal recessive
Mutation of SLC26A4 gene encoding pendrin – protein
primarily involved in transport of chloride and iodide
ions.
Nonsyndromic deafness DFNB4 also result from
mutation in the SLC26A4 gene.
Jervell and Lange-Nielsen
syndrome
*
Congenital sensorineural hearing loss and
prolongation of the QT interval on
electrocardiogram
*
Hearing loss initially involves the high frequencies
and progress to become a profound
*
Prolongation of QT reflect a defect in cardiac
repolarization. This can lead to recurrent attacks of
syncope, ventricular arrhythmia and possible
sudden death.
*
Mutation in genes KCNQ4, KCNE1 coding potassium
chanels (K
+
active transport in outer hair cells)
*
Inheritance autosomal recessive

Alport syndrome
Association of sensorineural high frequency
hearing loss with progressive nephritis. Anterior
lenticonus, macular flecks, cataracts
Gene mutation: COL4A5, COL4A3, COL4A4 coding
tissue specific polypeptide subunits of collagen
The subunits are expressed in the basilar
membrane, spiral ligament and basement
membranes of the stria vascularis
Genetically heterogeneous. Inheritance X-linked
dominant and autosomal recessive

Stickler syndrome
–
sensorineural hearing loss, high
frequency, progressive
–
Myopia, retinal detachment
–
Arthropathy
–
Mid-face hypoplasia, cleft palate,
micrognathia
–
Gene defect: COL2A1, COL11A1,
COL11A2
–
Inheritance autosomal dominant
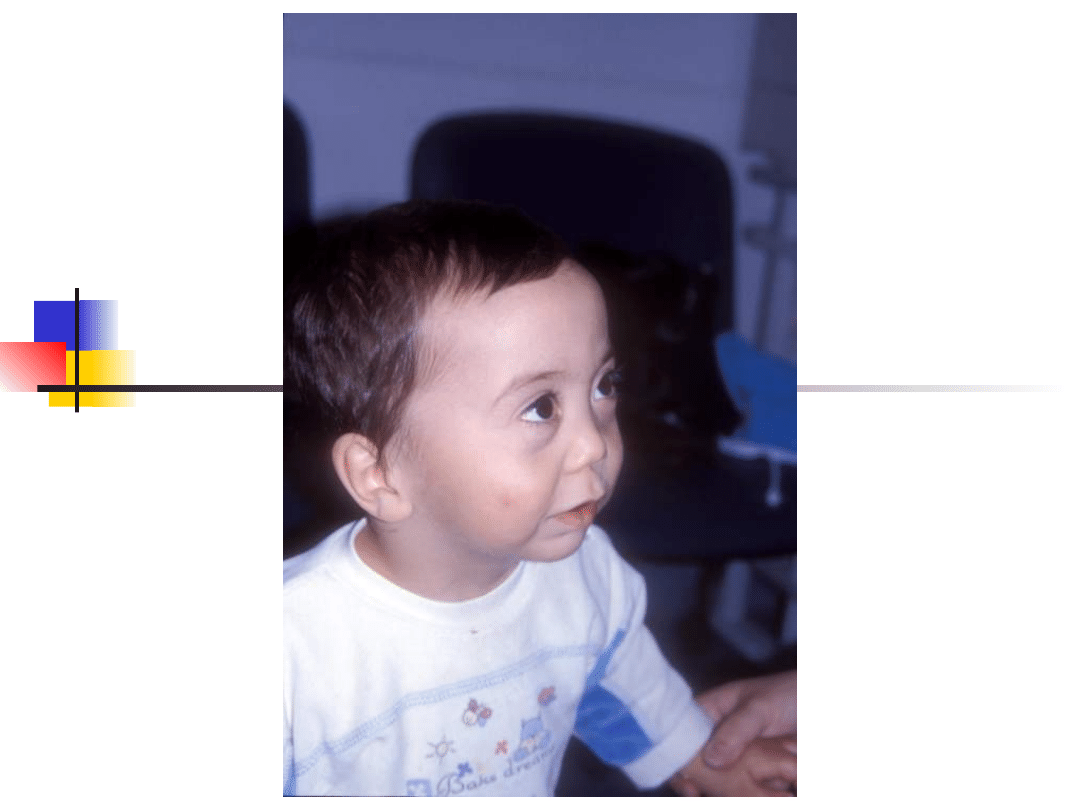

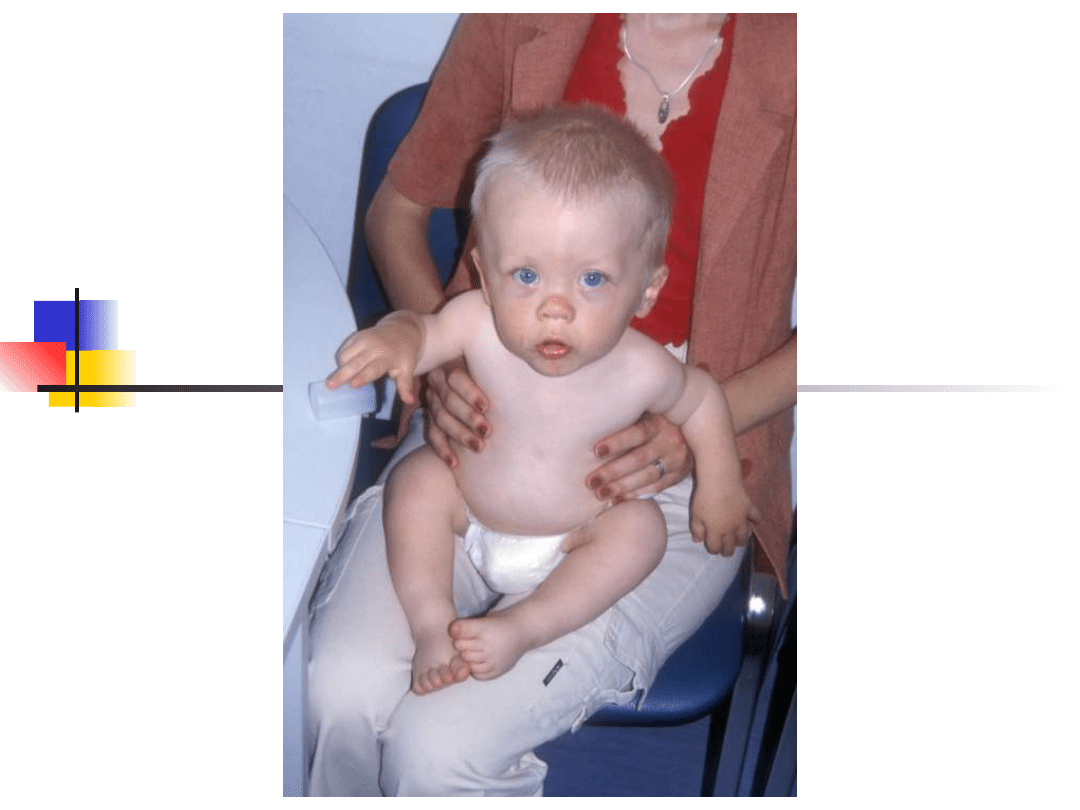
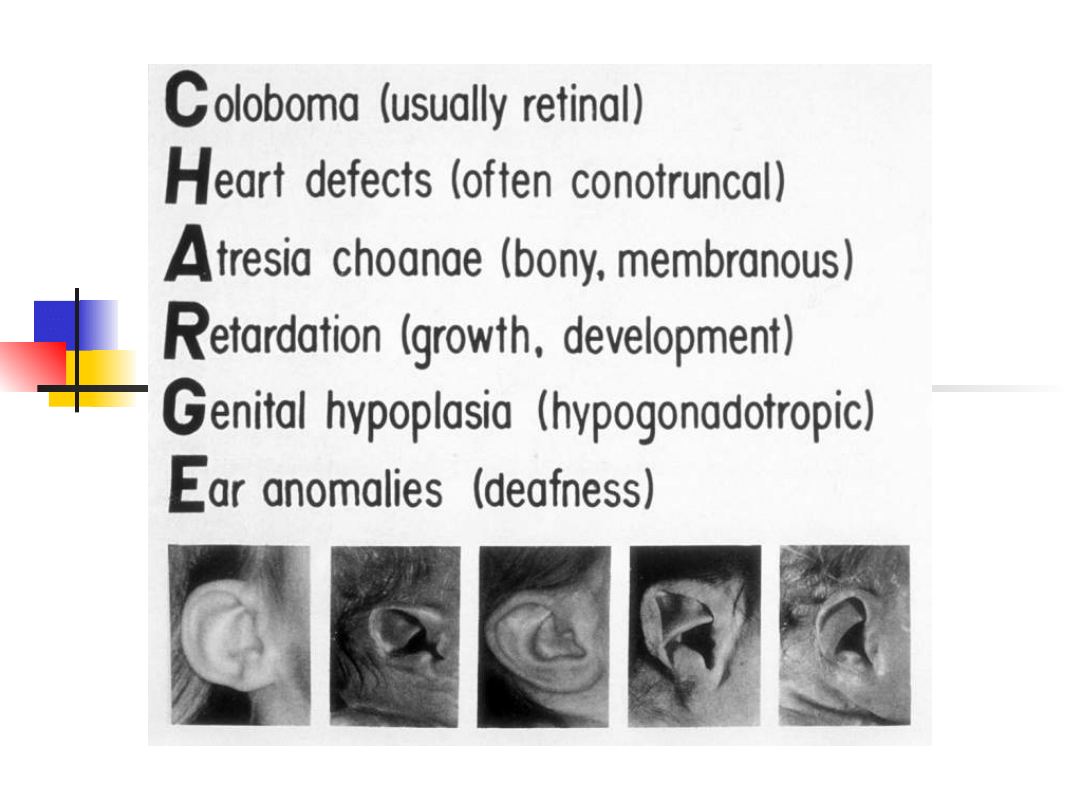
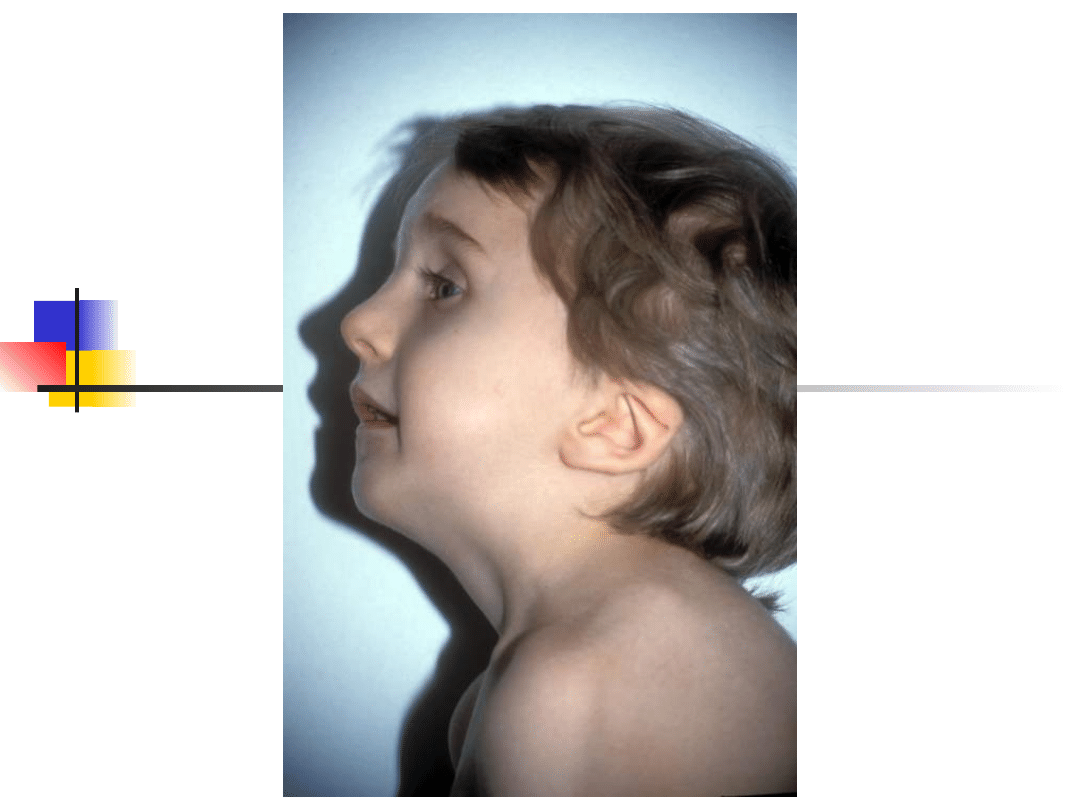
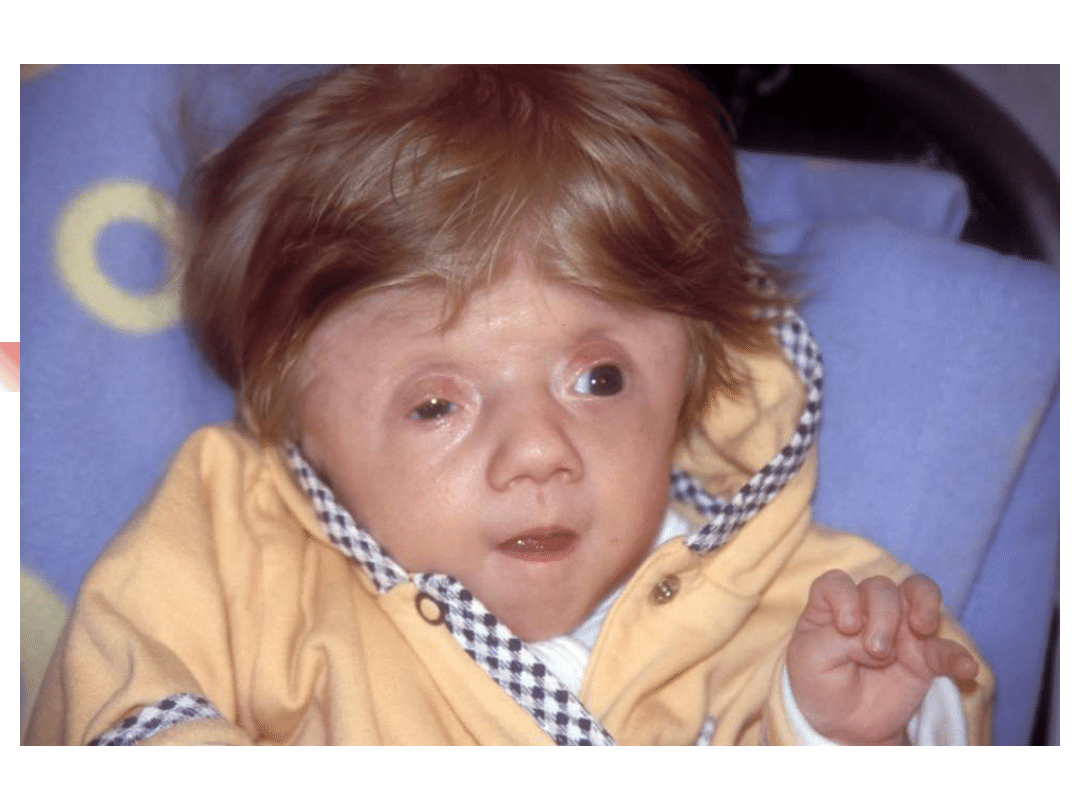
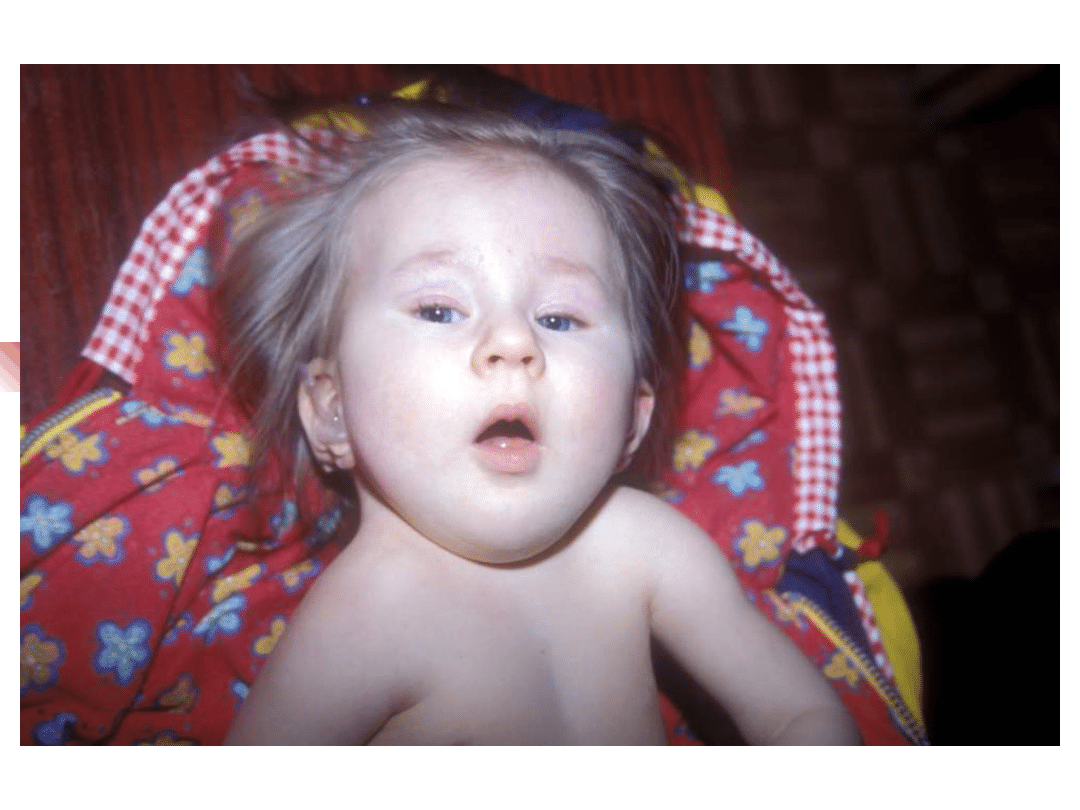
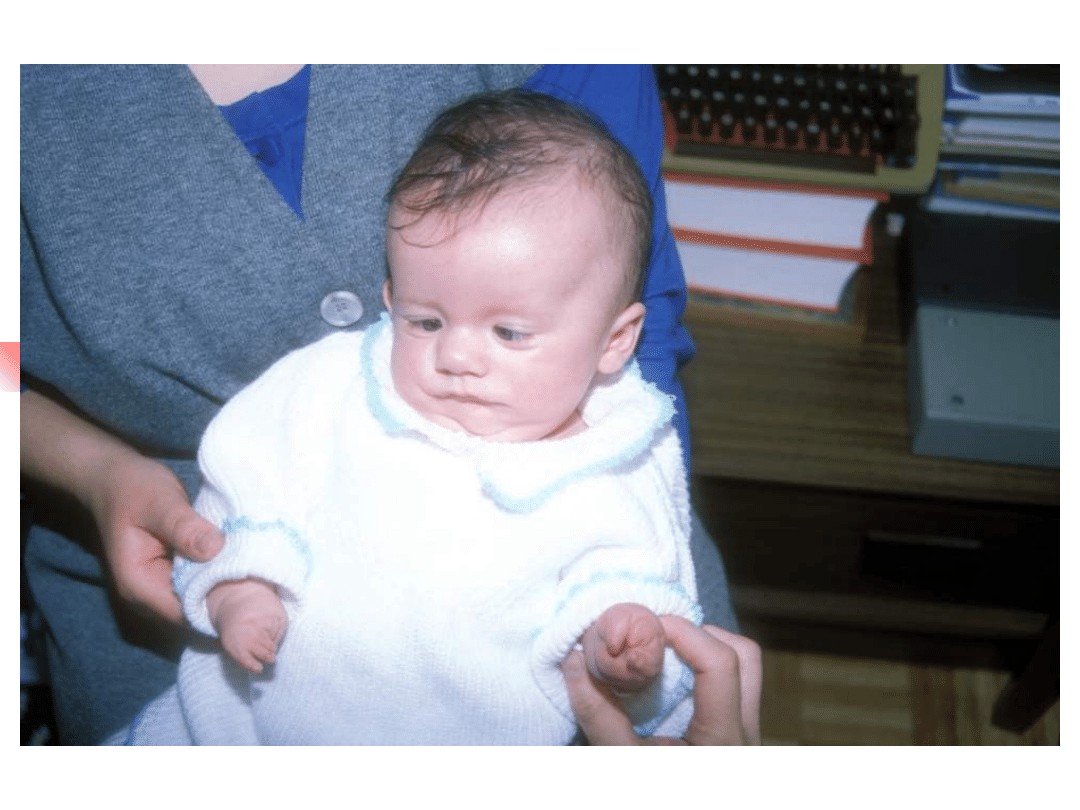
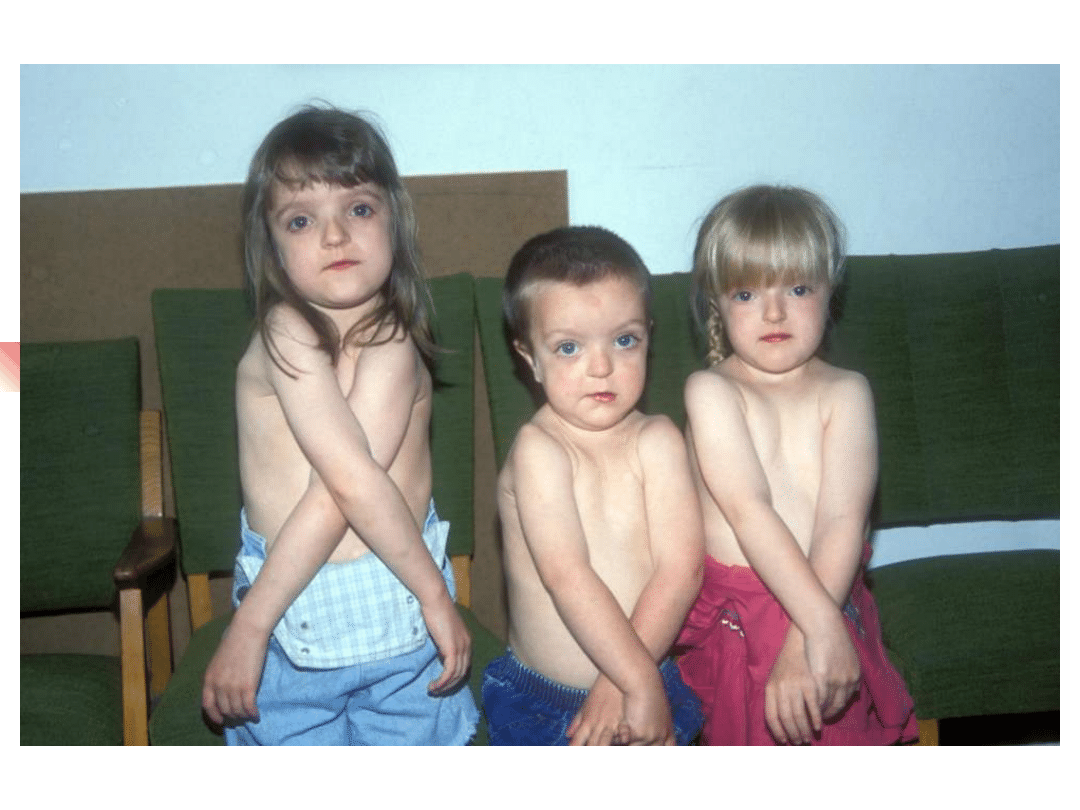
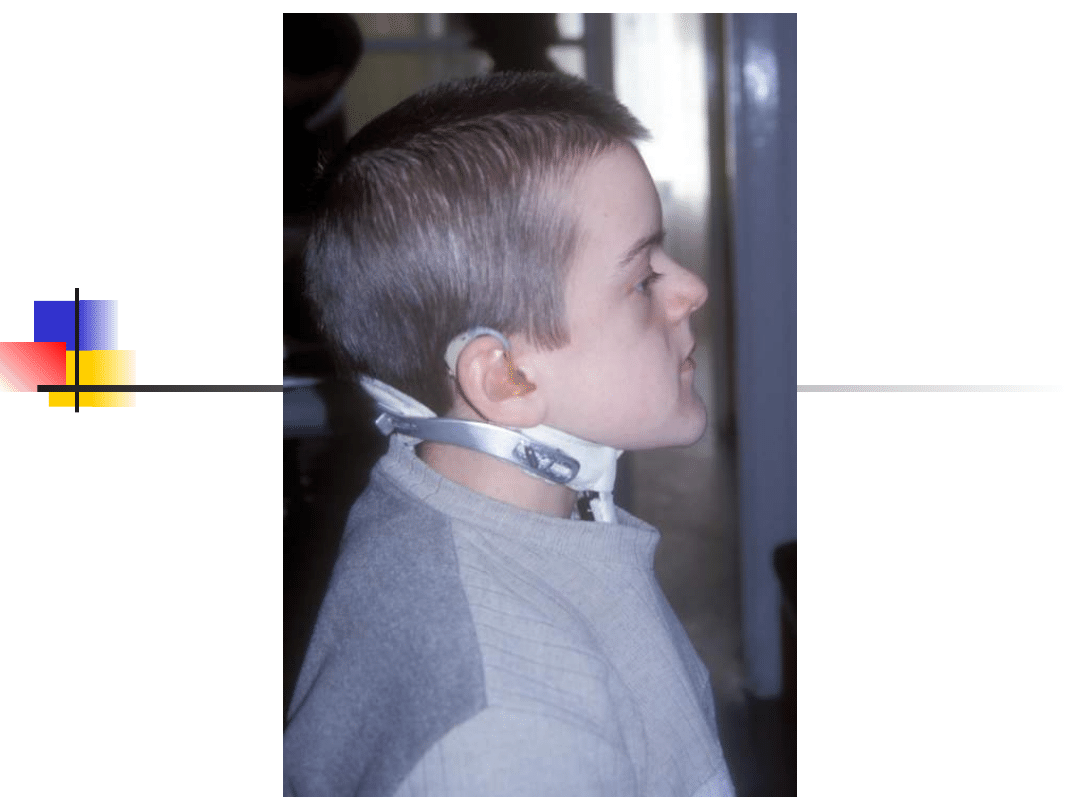
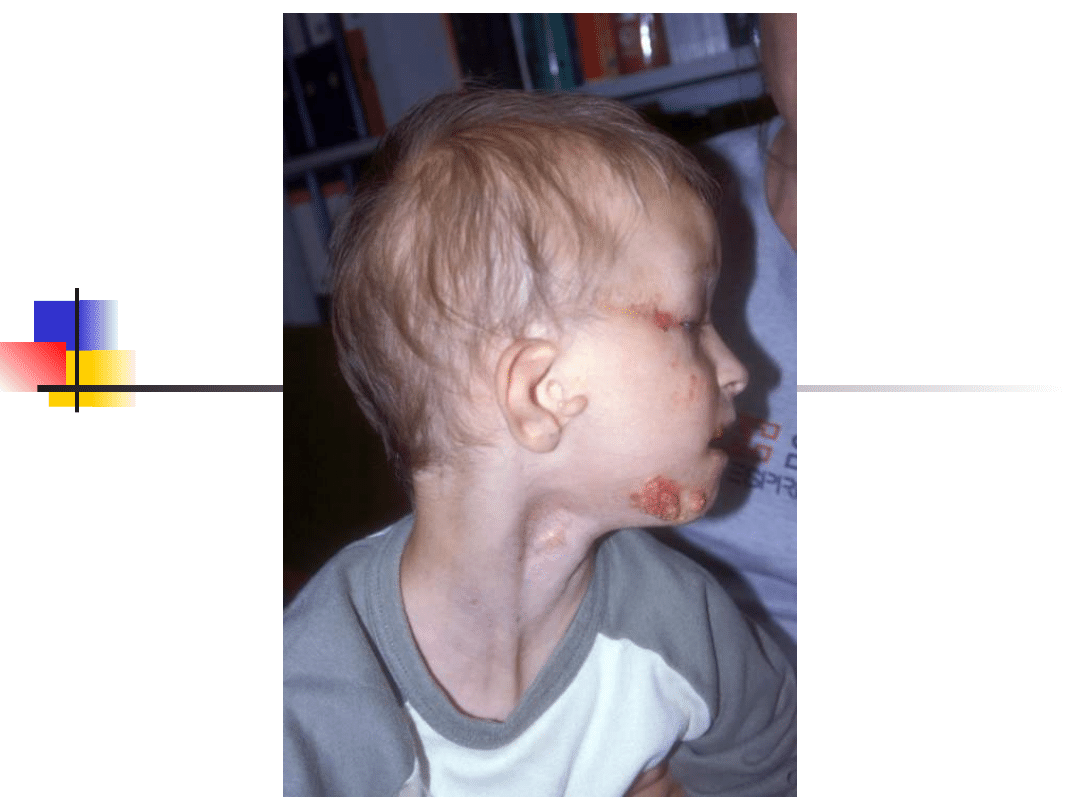
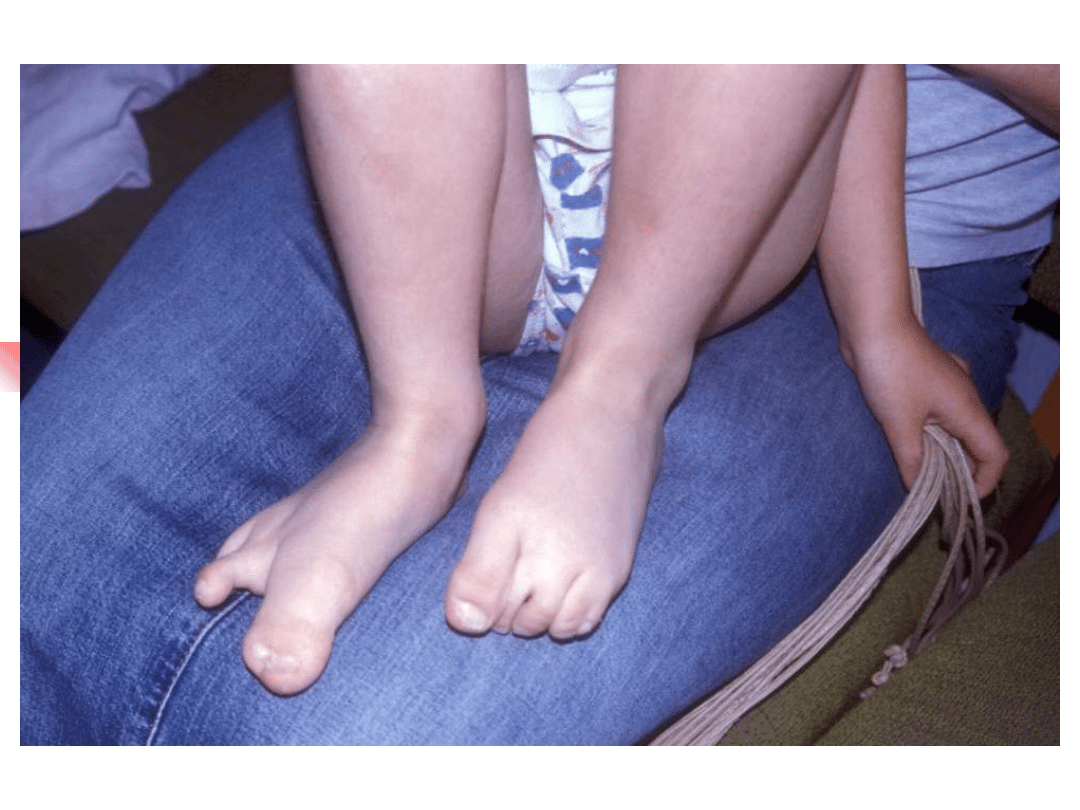
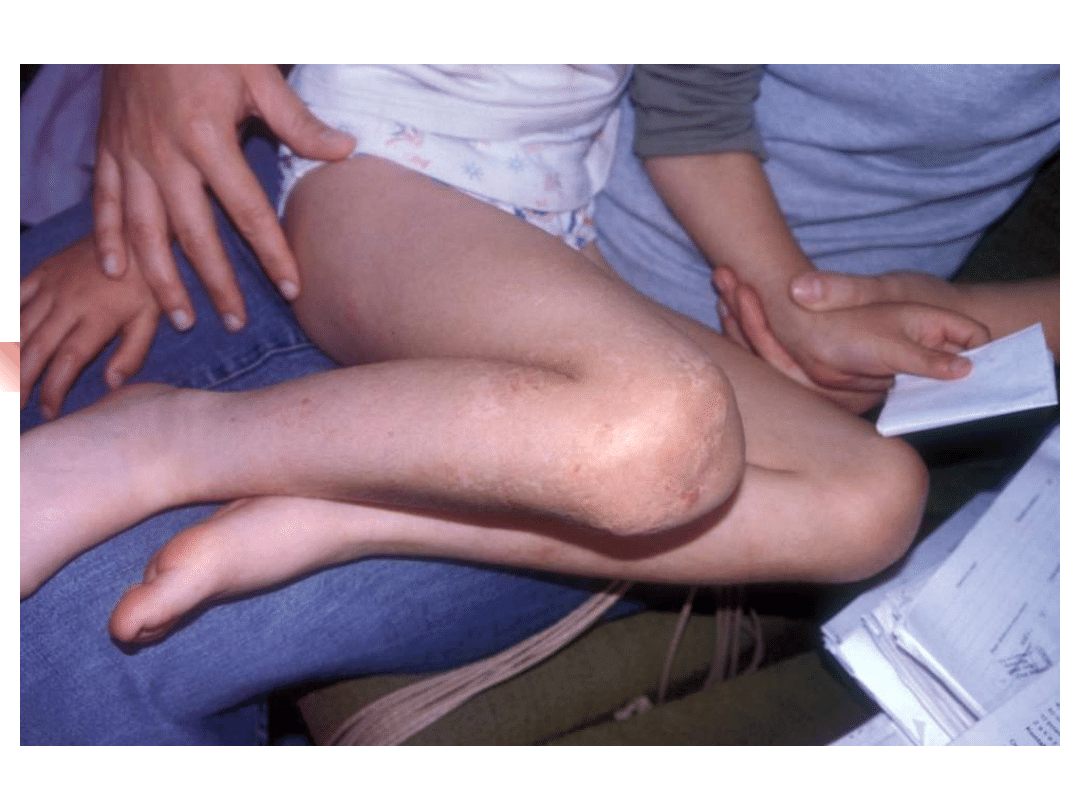
Most important genes involved in non-syndromic hearing
loss
Chromosom
al location
Locus
/mutation
Gene
symbol
Inheritance Protein
Function
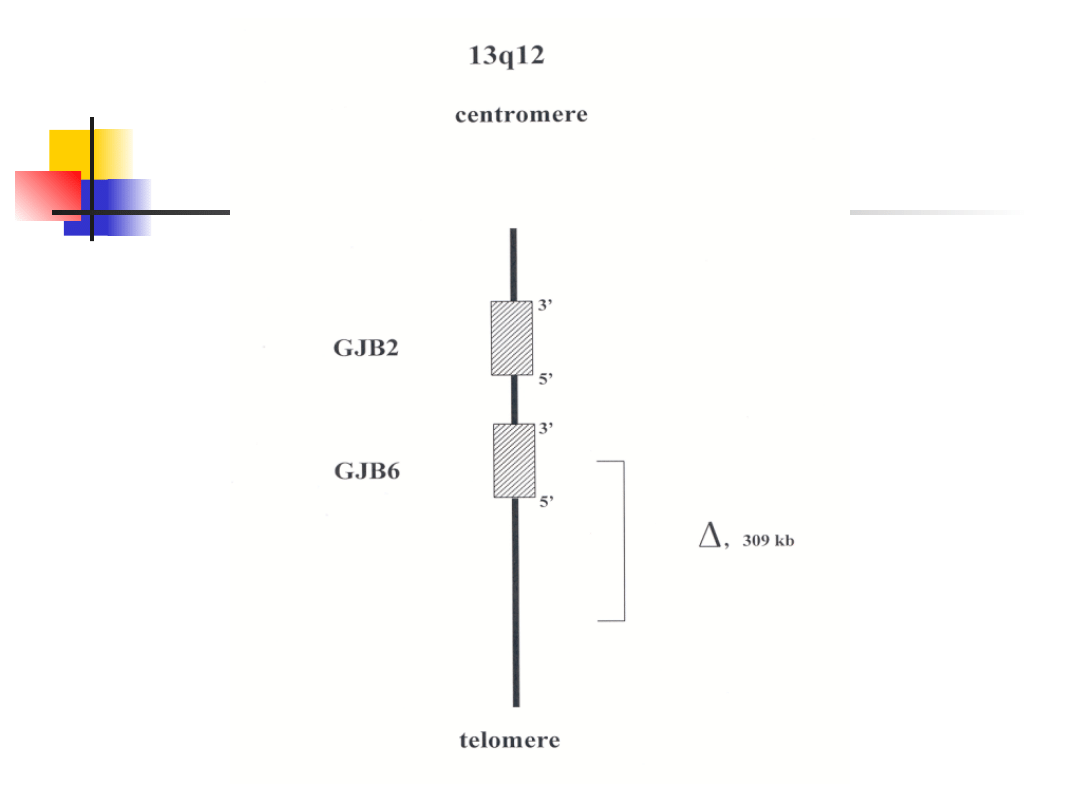
13q11-12
DFNB1/DFNA
3
GJB
2
AR/AD
Conexin
26
Gap junction
GJB
6
AR/AD
Conexin
30
Gap junction
7q31
DFNB
4
SLC26A
4
AR
pendrin
Anion transporter
14q12-13
DFNA9
COCH
AD
cochlin
Extracellular
matrix protein
mitochondri
um
1555A>G
MTRNR1 Mitoch.
12SrRNA
7445A>G
MTTs1
tRNA serine
7472insC
7511T>C
Xq21.1
DFN3
POU3F4
XL
domain
class 3
Pou
Transcription
factor
4p16.1
DFNA6/14/38 WFS1
AD
wolframin
ER transmembrane
protein

Hearing loss caused by mutation
in GJB2
(connexin deafness)
–
most common cause of hearing loss in many populations
–
deafness usually stable, onset is nearly always prelingual (but not
necessarily congenital); hearing may be normal at birth and
hearing loss progress rapidly during first few month of life (some
babies may pass neonatal hearing screening but become deaf
during infancy)
–
GJB2 encodes a gap junction protein – connexin 26
–
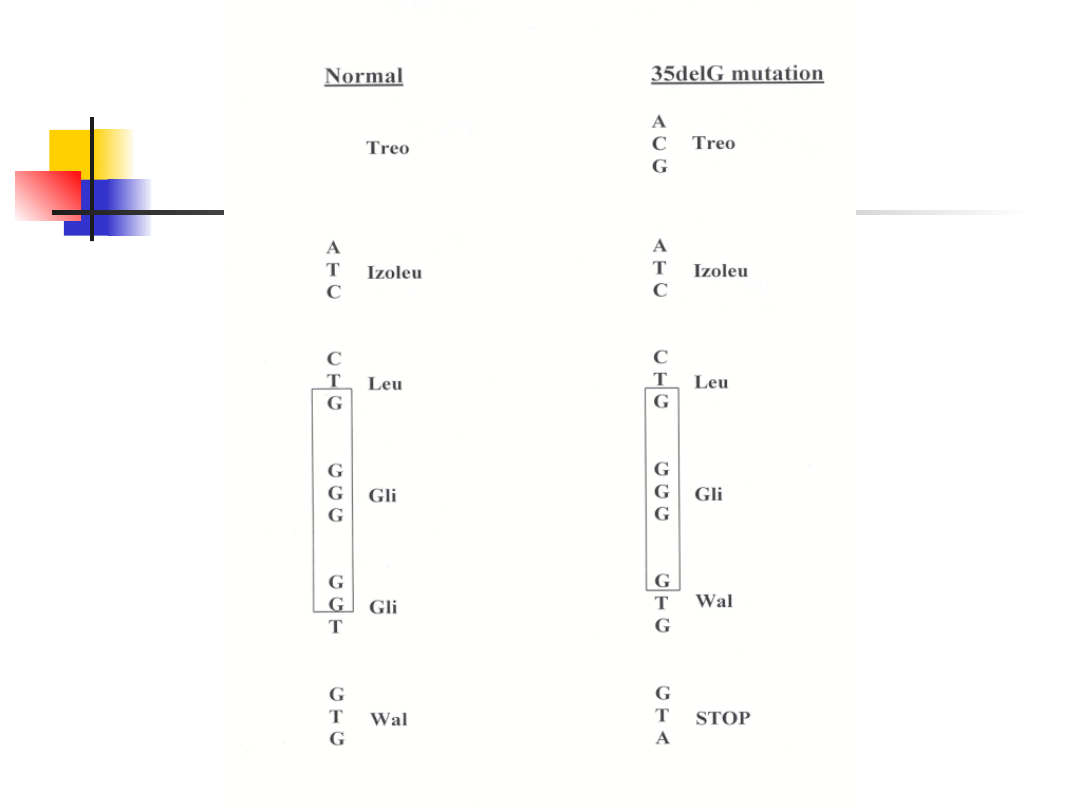
most common mutation is a deletion of single guanine – 35delG
(70% mutant alleles, carrier frequency 2-3%)
–
mutation 35delG in thought rather a founder effect not hot-spot
deletion
–
GJB2 mutations may also be a rare cause of autosomal dominant
deafness – syndromic and nonsyndromic (DFNA3).
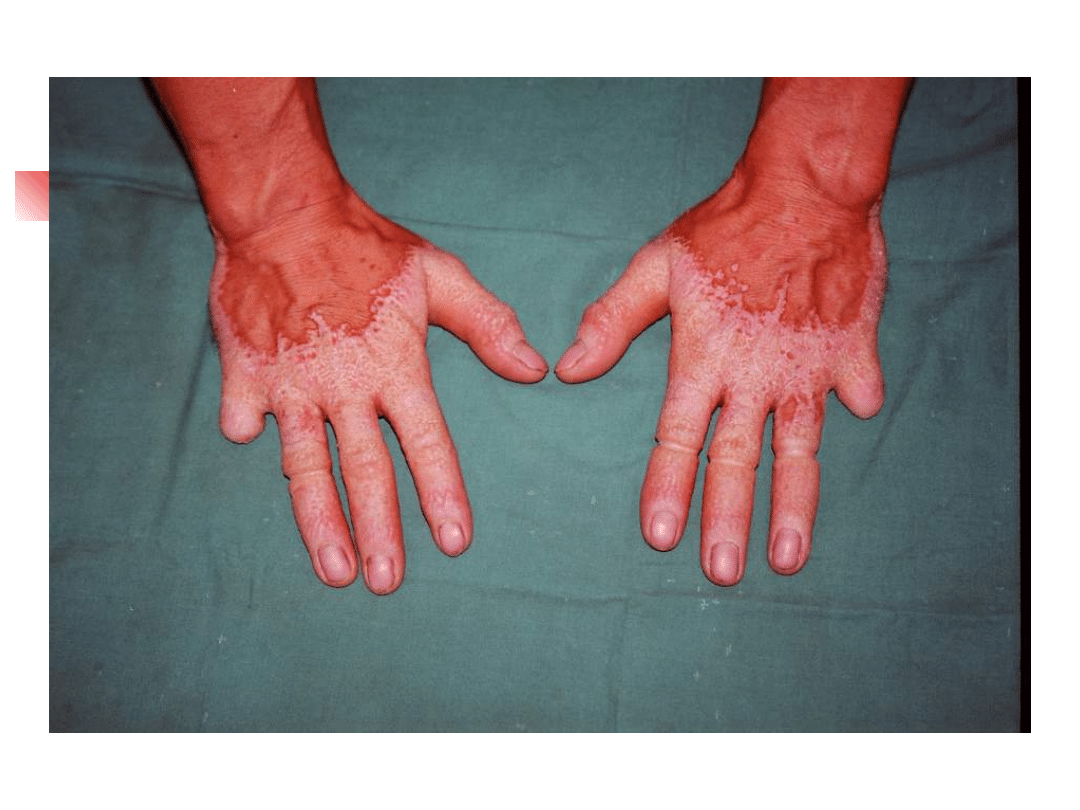
Specific mutation:
- hyperkeratosis palmoplantaris
-
mutilating keratoderma – (Vohwinkel sy.)
- keratoderma – ichthyosis – deafness (KID sy.)
Screening GJB2 should be offering as part
of the routine work-up in the diagnosis of
all cases of non-syndromic deafness of
unknown cause.
Rationale: - common cause of hearing impairment
- phenotype unremarkable and variable
- small coding region
- common mutations in some populations
- enables accurate genetic information to
be given to families
disadvantages: counselling difficult with missense
and
heterozygous mutation

Mitochondrial hearing loss
–
Sensorineural hearing loss is present in 40-70% patients with
mitochondrial disorders and can be syndromic or non-syndromic.
–
Mitochondrial mutations are transmitted exclusively through the
maternal line and demonstrate complete (or nearly complete)
homoplasmy.
–
Up to 20% patients receiving aminoglycosides experience
hearing impairment. 50% of those carry the 12S ribosomal RNA
mutation 1555A>G.
–
Mitochondrial hearing loss may be syndromic: Kearns-Sayre sy.,
MELAS, maternally inherited diabetes and deafness, and others
–
Pathogenesis of mitochondrial hearing loss is based on high ATP
requirement in the cochlear hair cells. A reduction of available
ATP caused by dysfunction of the mitochondrial oxidative
phosphorylation results in disturbances of the ionic gradient in
the inner ear.
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
Wyszukiwarka
Podobne podstrony:
J Hauser Genetyka zaburzenia lękowe
Gunia Terapia logopedyczna dzieci z zaburzeniami słuchu i mowy
GENETYKA KLINICZNA V - seminarium Genetyka zaburzen roznicow, VI rok, Genetyka, Genetyka, Egzamin
Choroby genetyczne. Zaburzenia w liczbie chromosomów płci, Biologia
Jak można sklasyfikować zaburzenia słuchu, logopedia, ćw. mowy, języka słuchu fon
konsekwencje zaburzeń słuchu, Fizjoterapia, Rehabilitacja osób ze złożoną niepełnosprawnością
Zaburzenia słuchu fonematycznego, logopedia, ćw. mowy, języka słuchu fon
Choroby genetyczne. Zaburzenia w liczbie chromosomów płci, Genetyka
Otolaryngologia Zaburzenia słuchu i mowy u dzieci
UWARUNKOWANIA GENETYCZNE ZABURZEŃ W ZESPOLE
więcej podobnych podstron