
Sylwia Ojdowska
Analityka medyczna

Szpiczak mnogi jest klonalną chorobą
rozrostową z linii B – komórkowej, która
charakteryzuje się proliferacją komórek
plazmatycznych. Komórki te znajdują się
głównie w szpiku i z reguły wydzielają
paraproteiny.
Szpiczak związany jest ze zmianami
osteolitycznymi w układzie kostnym, rozlaną
osteoporozą a także upośledzeniem
wytwarzania immunoglobulin.
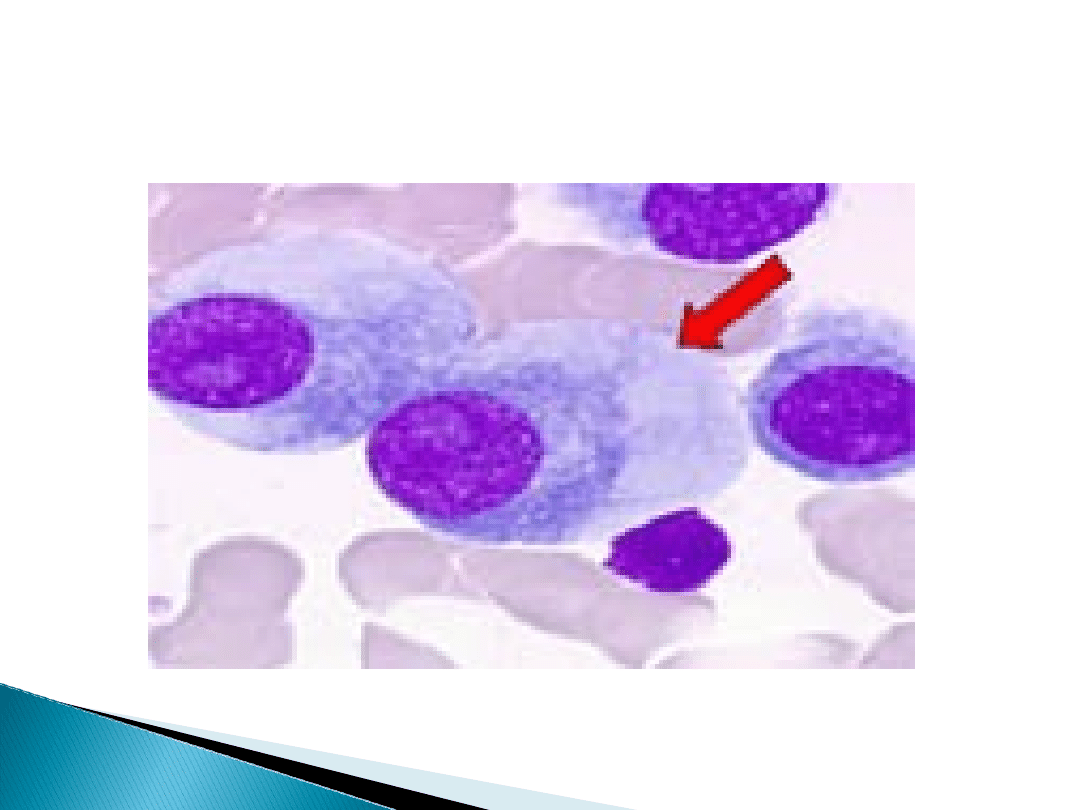

Szpiczak plazmocytowy niewydzielający
Szpiczak typu IgD
Szpiczak typu IgM
Szpiczak typu IgE

Szpiczak plazmocytowy stanowi :
10% hematologicznych chorób rozrostowych
1% wszystkich chorób nowotworowych
Średnia wieku wynosi 65 lat i znacznie
częściej chorują mężczyźni.
Istnieje związek między zachorowaniem a
ekspozycją na promieniowanie, benzen,
pestycydy i pracą w rolnictwie.

Powstaje z komórek z linii B, które wywodzą
się z ośrodka rozmnażania (znajdujących się
prawdopodobnie w węzłach chłonnych lub
śledzionie)
Komórki te zostały poddane procesowi
selekcji antygenowej, rekombinacji VDJ,
somatycznej hipermutacji regionów
zmiennych, jak również rekombinacji genów
IgH.
Sekrecja IL-6, IL –1,TNF-α oraz ligandu
RANK pobudza proliferację szpiczakową.

Rozwój szpiczaka mnogiego jest powolny.
Początkowo niezauważalny, ponieważ
praktycznie bezobjawowy, a objawy są mało
charakterystycze.
Objawem jest ogólne osłabienie,
zmniejszenie sprawności fizycznej, a także
postępującą utrata masy ciała i narastającą
niedokrwistością.

Bóle kostne
Wynikają z ubytków osteolitycznych, jak
również z patologicznych złamań, którym
często towarzyszy osteoporoza.
W postaci bardziej zaawansowanej choroby
występują patologiczne złamania żeber i
kości długich.
Spowodowane jest to tym, że sekrecja IL-6,
IL-1, TNF-α oraz ligand RANK pobudzają
również aktywację osteoklastów, które
powodują destrukcję kostną
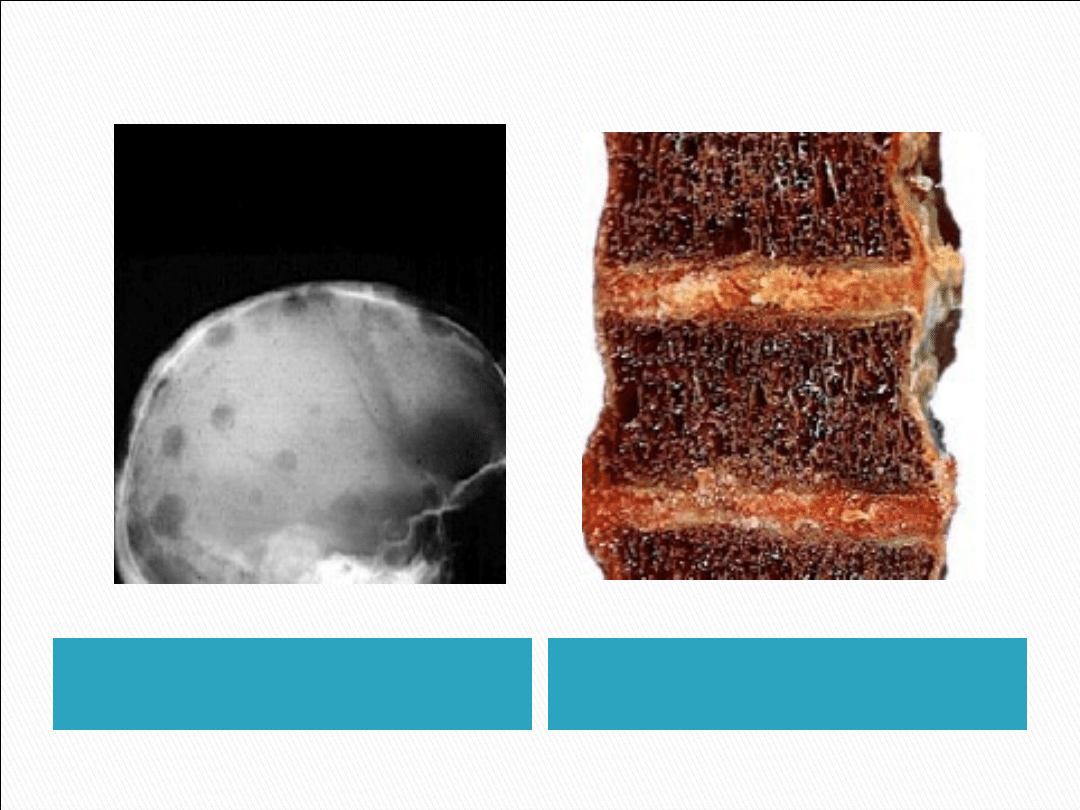
Czaszka – obraz RTG
Kręgosłup – zmiany
osteolityczne

Nawracające infekcje, które związane są z
małym stężeniem prawidłowych
immunoglobulin. Jest to spowodowane tym,
że produkowane przez plazmocyty szpiczaka
immunoglobuliny są białkami patologicznymi.
Niedokrwistość jest jednym z najczęstszych
objawów klinicznych. W momencie
rozpoznania choroby stwierdza się u 40 –
75% chorych. Jest następstwem infiltracji
szpiku przez nowotworowe plazmocyty, ale
także skutkiem przyspieszonej apoptozy
erytroblastów indukowanej cytokinami
takimi jak TNF i interleukine 1.

Zespół nadlepkości związany jest z
hiperproteinemią i tworzeniem
wielocząsteczkowych polimerów przez
monoklonalną immunoglobulinę
wytwarzaną przez nowotworowe
plazmocyty.
Charakteryzuje się krwawieniami
śluzówkowymi, zaburzeniami świadomości,
zaburzeniami widzenia.
Znacznie częściej występuje w szpiczaku
plazmocytowym IgG niż IgA

Zespół krioglobulinemii, występuje gdy
produkowane białko patologiczne jest
krioglobuliną. Objawem jest nadmierna
reakcja na obniżenie temperatury, tak więc
bóle, zsinienie, zatory, zakrzepy, drętwienie
itp.
Amyloidoza powodująca wtórne uszkodzenie
narządów miąższowych, np. nerek
spowodowana odkładaniem się amyloidu.
Krwawienia z dziąseł, nosa, wybroczyn
na skórze lub krwotoków wewnętrznych
w następstwie zaburzeń syntezy lub wiązania
przez białko monoklonalne prawidłowych
osoczowych czynników krzepnięcia krwi.

Neuropatia, której przyczyną jest ucisk
guza nowotworowego na rdzeń kręgowy
bądź też na nerwy czaszkowe
Polineuropatia, która jest częścią
wieloobjawowego zespołu tzw POEMS
Okołoneuronowe i okołonaczyniowe
odkładanie się amyloidu, który może
wyzwalać polineuropatie.
Zaburzenia mózgowe w zaawansowanych
okresach leukodystrofii i leukoencefalopatii.

Nefropatia szpiczakowa z zaburzeniami
czynności nerek, jest spowodowana :
Odkładaniem w nich amyloidu
Zespołem nerczycowym powstałym w wyniku
amyloidozy bądź precypitacją w kanalikach i
cewkach zbiorczych patologicznych białek
Hiperkalcemią i hiperkalciurią, które
prowadzą do wielomoczu
Śródmiąższowym zapaleniem nerek,
wynikająca z odkładania paraproteiny
szpiczakowej, głównie łańcuchów lekkich

Hiperkalcemia, która jest obserwowana u
25% chorych. Towarzyszy zmianom
litycznym kości lub też osteroporozą.
Może dawać objawy polidypsji, polinurii i
zaparcia.

We krwi obwodowej:
Niedokrwistość, która jest spowodowana
wyparciem układu erytropoetycznego przez
proliferujące plazmocyty w szpiku kostnym
małopłytkowość, uwarunkowaną podobnie
jak niedokrwistość, a także postępowaniem
terapeutycznym czy w początkach choroby
Wybitna rulonizacja spowodowana
obecnością białka monoklonalnego M w
surowicy

Liczbę leukocytów prawidłową lub obniżoną
i pojedyncze plazmocyty w polu widzenia
Wyraźnie zwiększona liczba plazmocytów w
schyłkowym okresie choroby, która daje
obraz białaczki plazmatycznokomórkowej.

Wzrost liczby plazmocytów o co najmniej
15%
Występowanie plazmocytów w skupiskach
Obecność cech parytypii komórek
plazmatycznych, jak np. podwójne lub
poliploidalne jądra, wodniczki lub wtręty
komórkowe
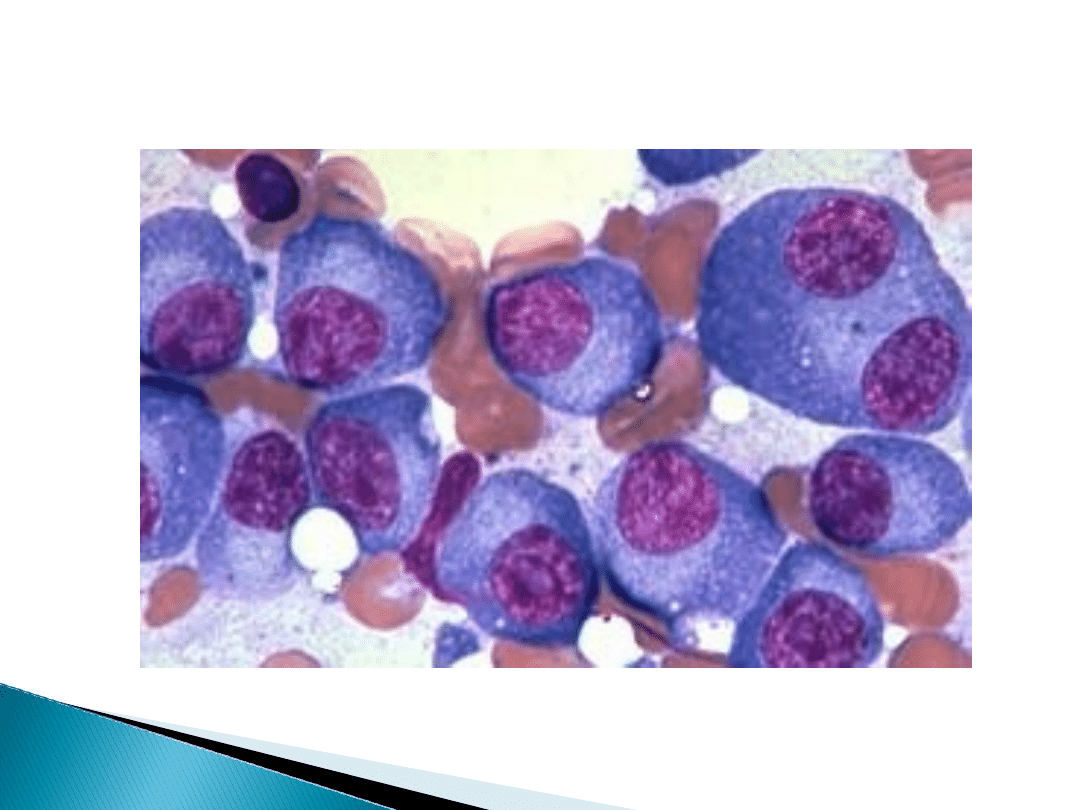

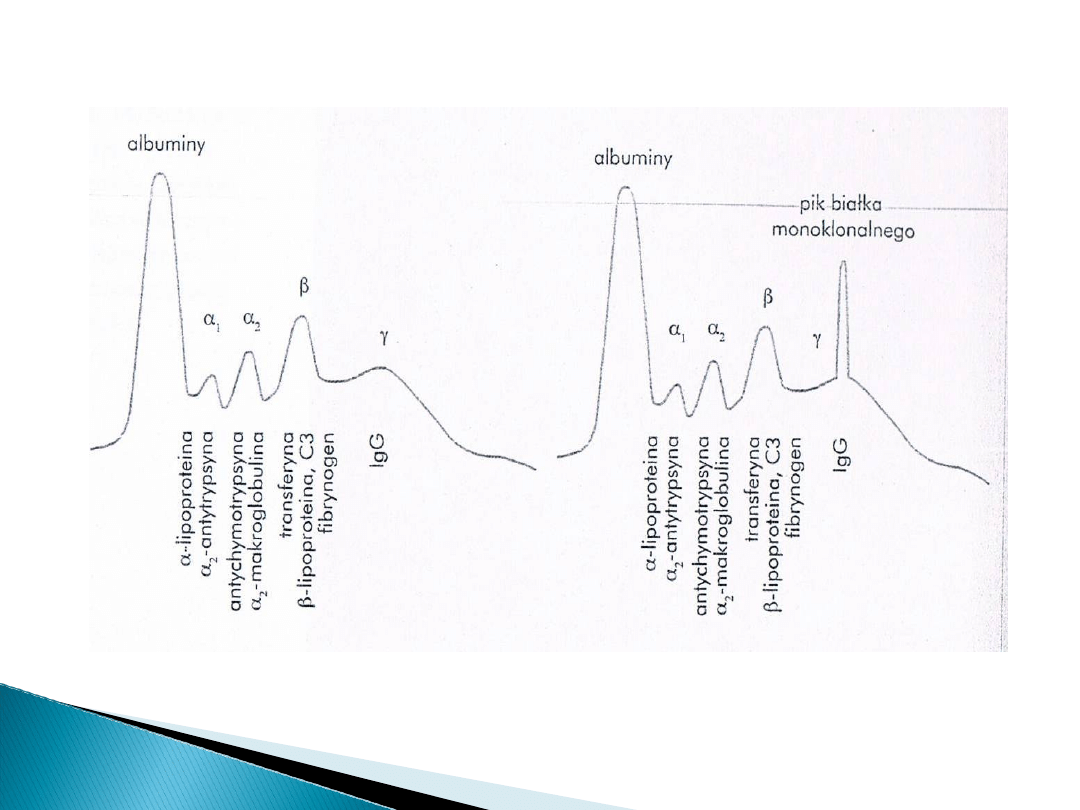
Obecność paraeproteiny szpiczakowej M
Hiperkalcemia
Wzrost stężenia białka ogólnego w surowicy
Wzrost poziomu mocznika i kreatyniny –
występuje w przypadku uszkodzenia nerek
Wzrost stężenia immunoglobulin, głównie
IgG z wyjątkiem IgM i IgD
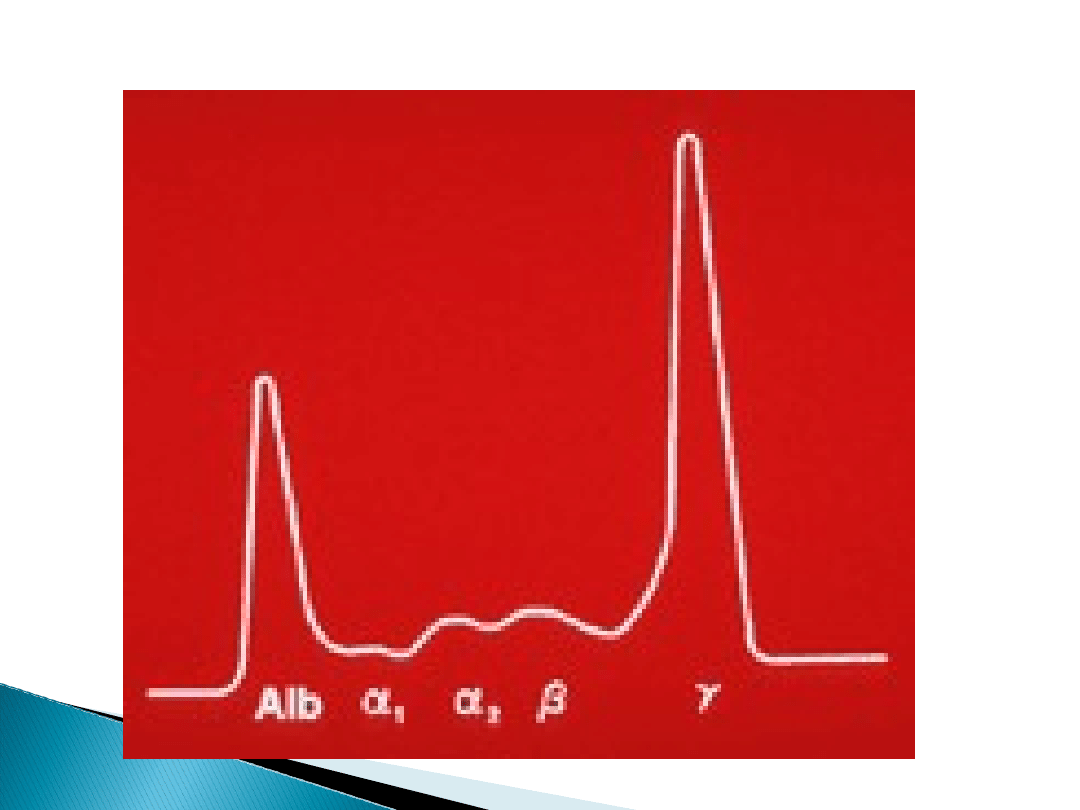

W badaniach obserwuje się również bardzo
wysokie OB
Podwyższone CRP pogarsza rokowanie
pacjenta
Rutynowe badanie moczu może wykazać
obecność proteinurii

Kryteria większe
Kryteria mniejsze
Plasmocytoma w biopsji tkanki
Infiltracja szpiku kostnego
powyżej 30% plazmocytów
Pik monoklonalnych globulin w
elektroforezie (IgG>35g/l,
IgA>20g/l)
Łańcuchy lekkie w moczu powyżej
1g/24h.
Infiltracja szpiku kostnego –
10-30% plazmocytów
Paraproteinemia poniżej wartości
w kryteriach większych
Obecność zmian litycznych w
kościach
Prawidłowe IgM<0,5g/l, IgA<1g/l,
IgG<6g/l

stopień
parametry
Mediana
przeżycia
1
Β
2
- mikroglobulina <3,5mg/l i
albuminy>35g/l
62 miesiące
2
Β
2
- mikroglobulina <3,5mg/l i
albuminy<35g/l
lub
Β
2
- mikroglobulina 3,5 – 5,5 mg/l
44 miesiące
3
Β
2
- mikroglobulina>5,5 mg/l
29 miesięcy

Opanowanie bólu – dobieranie
indywidualnych dawek prostych
analgetyków oraz leków opioidowych, a
także stosowanie niesteroidowych leków
przeciwzapalnych, miejscowa radioterapia
Opanowanie niewydolności nerek –
nawodnienie dużą ilością płynów a także
intensywne wyrównywanie hiperkalcemii,
opanowanie infekcji i hiperurykemii. Należy
unikać podawania leków nefrotoksycznych.

Hiperkalcemia – nawodnienie organizmu,
podawanie diuretyków pętlowych,
dożylnych bisfosforanów jak również
włączenie chemioterapii
Uszkodzenie układu kostnego – konieczna
jest radioterapia na okolice objętą
nasilonymi, ograniczonymi dolegliwościami
bólowymi
Infekcje – leczenie antybiotykowe, coroczna
immunizacja przeciwko grypie

Niedokrwistość – przetaczanie krwi w
przypadkach jawnych niedokrwistości.
Zespół nadlepkości – wymiana 3 l osocza
oraz szybko włączana chemioterapia
Ucisk rdzenia kręgowego – pilna miejscowa
radioterapia, podawanie doustne
deksametazonu

Melfalan i prednizolon – 4- dniowe cykle
podawane w przerwach 4-6 tygodniowych
co pozwala uzyskać 50% zmniejszenie
stężenia paraprotein u ok. 50% chorych.
ABCM, VMCP/VBAC oraz VBMCP wykazują
całkowitą remisję u 10% chorych
VAD pozwala uzyskać lepsza odpowiedź na
leczenie a także większy wskaźnik remisji
choroby.

Interferon α wydłuża czas trwania
odpowiedzi organizmu a także ma niewielki
wpływ na okres przeżycia pacjenta.
Cyklofosfamid stosowany jest u osób, które
nie toleruję melfalanu. Są to osoby z
przewlekłą cytopenią
Bifosforany, które hamują aktywację
osteoklastów, a co za tym idzie mniej bólów
kostnych jak i mniej uszkodzeń kostnych i
złamań.

„Uniwersalny przewodnik po metodach
leczenia nowotworów” M. Dollinger, E.
Rosenbaum, G. Cable; 2000r
„Podstawy hematologii” W.S. Nowak, A.B.
Skotnicki; 2011r.
„Hematologia” K. Janicki; 2001r.
„Hematologia” Antczak, Myśliwiec,
Pruszczyk; 2011r.
„Hematologia kliniczna” D. Provan, C.R.J.
Singer, T. Baglin, J. Lilleyman; 2004r.
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
Wyszukiwarka
Podobne podstrony:
2 Objawy kliniczne w szpiczaku plazmocytowym – patomechanizm objawów 2id 19582 ppt
2 Objawy kliniczne w szpiczaku plazmocytowym – patomechanizm objawów 1
2 Objawy kliniczne w szpiczaku plazmocytowym – patomechanizm objawów 2id 19582 ppt
2 Patomechanizm objawów klinicznych w ostrej białaczce szpikowejid 19599 ppt
Szpiczak mnogi – historia badań nad chorobą, epidemiologia, patofizjologia, objawy kliniczne oraz na
OBJAWY KLINICZNE AIDS zmiany ogólne i w obrębie jamy ustnej
Objawy kliniczne i postępowanie w stanie padaczkowym(2), stany zagrożenia życia
pediatria 2, MÓZGOWE PORAZENIE DZIECIĘCE.objawy kliniczne, MÓZGOWE PORAZENIE DZIECIĘCE
Patogeneza, objawy kliniczne i diagnostyka przy nadczynności tarczycy kotów
W 1 27.02.2009 Patomechanizm objawów uszkodzenia układu nerwowego, studia, Neurologia
1 Hemochromatoza – patogeneza i objawy kliniczne
Objawy kliniczne COPD (PP vs?)
szpiczak plazmocytowy
OBJAWY KLINICZNE AIDS zmiany ogólne i w obrębie jamy ustnej
The Merck Manual Objawy kliniczne Praktyczny przewodnik diagnost Kaminski Bogdan Klin Zuzanna Ksiad
więcej podobnych podstron