
CHEMIA MATERIAŁÓW
2006/2007
„
We all agree […..] that the emphasis in synthetic research
is shifting
toward the synthesis of properties, and not just
compounds”.
Albert Eschenmoser, 1996.
Profesor Chemii Organicznej
Swiss Federal Institute
of Technology
Współczesna nauka o materiałach koncentruje się nie tylko
na produktach lecz także procesach prowadzących do ich
otrzymania

Zakres wykładu
1. Metody
1.1 Zasady „Zielonej Chemii”
1.2 Chemia i kataliza metaloorganiczna
1.3 Katalizatory heterogenizowane
1.3 Kataliza enzymatyczna
2. Materiały
2.1 Opto-elektronika
2.2 Nowe polimery i metody ich syntez
CHEMIA MATERIAŁÓW
2006/2007

1.
Lepiej zapobiegać powstawaniu produktów odpadowych niż je
przetwarzać i utylizować
2. Metody syntez powinny być tak projektowane, aby finalny
produkt zawierał maksymalną ilość wprowadzonych substratów
3. Zawsze, jeśli to jest możliwe, metody syntetyczne powinny
wykorzystywać substancje o niskiej toksyczności, lub całkowicie
bezpieczne dla ludzi i środowiska naturalnego
4. Produkty chemiczne powinny zachować funkcjonalną
skuteczność i właściwości przy zredukowanej toksyczności
5. Należy dążyć do wyeliminowania substancji pomocniczych,
takich jak rozpuszczalniki, lub wykorzystywać substancje
nieszkodliwe
6. Należy ograniczyć zużycie energii i dążyć do opracowania
metod syntetycznych prowadzonych w temperaturze otoczenia
i pod normalnym ciśnieniem
Zasady „zielonej chemii”

7. Jeżeli jest to technologicznie i ekonomicznie możliwe należy
wykorzystywać surowce ze źródeł odnawialnych
8. Należy unikać metod syntezy obejmujących dodatkowe reakcje
pośrednie (grupy blokujące, protekcja/deprotekcja)
9. Reagenty katalityczne (selektywne) są korzystniejsze w porównaniu
z reagentami stechiometrycznymi
10. Produkty chemiczne powinny być tak projektowane, aby po ich
wykorzystaniu nie stanowiły obciążenia dla środowiska i ulegały
degradacji do nieszkodliwych substancji
11. Należy rozwijać techniki analityczne tak aby nie dopuszczać do
powstawania substancji szkodliwych, monitorując proces w czasie
rzeczywistym
12. Substancje stosowane w procesach chemicznych powinny być
tak dobierane aby eliminować potencjalne eksplozje, przenikanie do
środowiska, pożar, etc.
Zasady „zielonej chemii”

Chemia
metaloorganiczna
1963 -K.Ziegler i G.Natta.
1973 -E.O.Fisher i G.Wilkinson-katalizatory homogeniczne wielu procesów
przemysłowych.
1976 –W.N. Lipscomb –struktura i wiązania boranów
1979 -H.C.Brown i G.Wittig-zastosowanie organoboranów w syntezie
organicznej.
1981 -R.Hoffmann i K.Fukui-badania teoretyczne (półempiryka) nad strukturą
i
reaktywnością związków metaloorganicznych.
1983 –H. Taube –mechanizm reakcji z przeniesieniem elektronu związków
koordynacyjnych
2000 –A.J. Heeger, A.G. MacDiarmid i H. Shirehawa –polimery przewodzące
2005 –Y. Chauvin, R.H. Grubbs i R.R. Schrock-badania nad reakcją metatezy
NAGRODY
NOBLA
Co
Ile [tony/r]
Silikony
700 000 z tendencją rosnącą
Dodatki antystukowe
600 000 z tendencją malejącą, bo są
coraz doskonalsze
Aluminoksany (głównie MAO)
50 000
Polimery cynoorganiczne i
cynoorganiczne dodatki do farb
35 000
Związki litoorganiczne
900
Co
Ile [tony/r]
Polipropylen
7 700 000
Produkty hydroformylowania
5 000 000
Acetaldehyd
2 200 000
Kwas octowy
1 000 000
PRODUKCJA ZWIĄZKÓW METALOORGANICZNYCH
Chemia metaloorganiczna
SiO
2
+ C
Elektrolityczna piroliza
Si + CO
2
Piasek Wegiel
Si + RCl
Cu
R
x
SiCl
4-x
R=H, Me, Et, Ph, itp
(najczesciej H i Me)
Rochow (USA, 1943)
Si
R
R
O
m
Anionowo
Kationowo
Si
R
R
O
n
n = do 100 000
silikony "II generacji"
Podstawa przemysłu silikonowego,
z roczną produkcją ok. 700 000 ton
GE-Bayer Silicones, 3M, Shin-Etsu, Dow
Corning, Rhone-Poulenc, Goldsmith,
Zakłady Silikonowe „Nowa Sarzyna”
n
Si
H
O
R
+
R'
[Pt]
n
Si O
R
CH
2
CH
2
R'
R = Me, Ph, Et
R' = niemal dowolna
grupa organiczna
silikony modyfikowane
I gen: Oleje
hydrauliczne i grzejne,
środki
smarujące, kity i
lepiszcza, tanie
elastomery
II gen: transparente
elastomery, śr.
powierzchniowo czynne
i powłokotwórcze,
izolatory, nabłyszczacze
inne.
Modyfikowane:
hydrożele, ciekłe
kryształy, śr. pow.
czynne, dodatki do
kosmetyków, śr.
homogenizujące i wiele,
wiele innych.
R
2
SiCl
2
(+ czasami R
3
SiCl)
Si
R
R
HOR
2
SiO
O SiR
2
OH
n
Si
R
R
O
m
+
n = 3-100 m = 3-11
silikony "I generacji"
H
2
O
H
+
/ OH
-
HIGH - TECH
1.Nanoszenie idealnie czystych nanowarstw metalicznych (elektronika, mechanika
precyzyjna kataliza):
Związki metaloorganiczne są często lotne i
łatwo ulegają rozkładowi do czystego
metalu z wydzieleniem łatwo lotnych
związków organicznych
[Cu(HCOO)
2
]
620-720 K
Cu + 2 HCOOH
H
2
[Co
2
(CO)
8
]
500 K
2 Co + 8 CO
2. Produkcja półprzewodników, materiałów ceramicznych i magnetycznych:
GaEt
3
+ P 600 K GaP (fosforek galu stosowany w mikroprocesorach)
4 Fe(CO)
5
+ 3 O
2
2 Fe
2
O
3
+ 20 CO (do pokrywania tasm magentycznych)
3. Materiały z anizotropowym przewodnictwem prądu:
[IrI(CO)
3
]
n
Ir
OC
CO
I
CO
Ir
OC
CO
I
CO
Ir
OC
CO
I
CO
kompleks "kwadratowy" z wiazaniami
metal-metal, tworzacy kolumny przewodzace
CH=C
Fe
n
I
2
CH=C
Fe
+
CH=C
Fe
+
I
3
-
m
suchy elektrolit na bazie
poli(ferocenoacetylenu)
4. Ciekłe kryształy i inne materiały „samoporządkujące się”:
Pd
Y
N
X
Cl
Pd
Y
N
X
Cl
Pd
Y
N
X
Cl
Pd
Y
N
X
Cl
Struktura
"otwartej ksiazki"

Chemia metaloorganiczna
Ni
Cu
Cr
But
But
But
But
Związki metaloorganiczne stanowią zarówno cenne substraty jak i
materiały finalne, w tym katalizatory.
(Me
3
Si)
3
- Mn-
C(SiMe
3
)
3
CO
CO
Fe
OC
CO
CO
CO
OC
CO
Mo
OC
CO
CO
RNC
CNR
Mo
CNR
CNR
CNR
RNC
RNC
2+
Re
Me
Me
Me
Me
Me
Me
Me
Me
2
_
Chemia metaloorganiczna to chemia połączeń zawierających
wiązanie
metal – węgiel
.
W zakres chemii metaloorganicznej włącza się obszar pochodnych
organicznych metaloidów – np. boru, krzemu, germanu
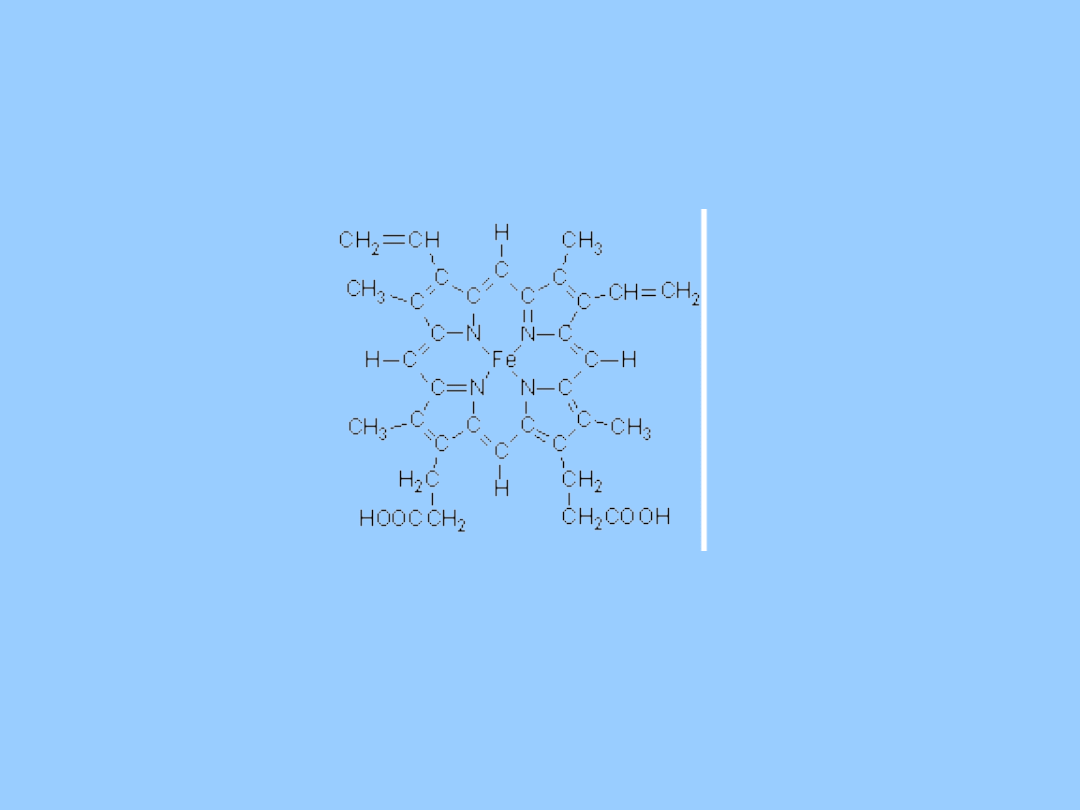
Czy hem jest związkiem metaloorganicznym?
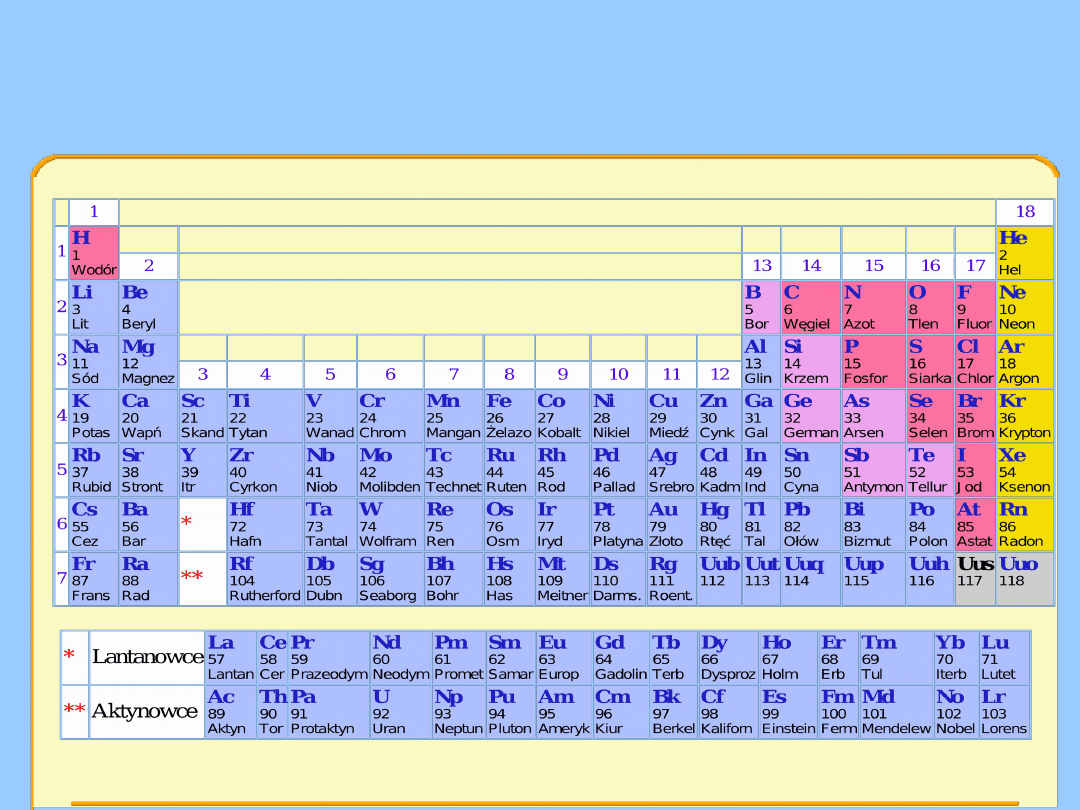
Różnorodność chemii metaloorganicznej jest rezultatem istnienia 99
pierwiastków metalicznych i metaloidów
Kataliza metaloorganiczna - które związki są dobrymi
katalizatorami?
Kataliza metaloorganiczna - które związki są dobrymi
katalizatorami?
Wnioski:
1. Katalizator musi posiadać wolne miejsca koordynacyjne, lub być zdolny do takiego
przekształcenia swojej struktury, aby te wolne miejsca pojawiały się w trakcie reakcji.
2. Katalizator nie może jednak wiązać substratów zbyt trwale - bo wtedy reakcja
zatrzyma się na etapie produktów przejściowych.
Z drugiej jednak strony: Zbyt niestabilne związki są trudne w praktycznym stosowaniu.
W warunkach przemysłowych katalizator trzeba transportować, przechowywać i
dozować do dużych reaktorów. Ponadto substraty są zwykle bardziej lub mniej
zanieczyszczone i katalizator nie może zbyt łatwo być dezaktywowany przez te
zanieczyszczenia.
W sumie: Idealny katalizator powinien być stabilny w warunkach „pokojowych”, ale
reaktywny w warunkach reakcji. Powinien być zdolny łączyć się z substratami, ale nie
reagować z innymi związkami obecnymi w środowisku reakcji. Powinien łączyć się z
substratami w miarę mocnymi wiązaniami ale nie zbyt mocnymi.
Jak to się osiąga: Poprzez dobór takich metali centralnych i ligandów aby dostosować
moc powstających wiązań między substratem i katalizatorem. Często ważna jest też
geometria całego układu katalitycznego.
Podstawowy fakt:
Katalizator działa zawsze poprzez tworzenie przejściowych kompleksów z
substratami.

Wiązania
Metale grup głównych (np. cyna) wykorzystuje swoje 4 elektrony
walencyjne do utworzenia 4 wiązań σ (wiązania 2-elektronowe –
2-centrowe); rezultatem jest np. trwała cząsteczka SnMe
4
. Trwałość
SnMe
4
i brak trwałości SnMe
3
można przewidzieć w oparciu o
regułę oktetu
.
W wypadku metali przejściowych ujawnia się olbrzymia
różnorodność
chemii metaloorganicznej. Poza orbitalami
s
i
p
pierwiastki te
posiadają powłokę orbitali
d
, które mogą nakładać się z orbitalami
π
nienasyconych cząsteczek organicznych.
Udział tych orbitali w tworzeniu wiązań determinuje liczbę
koordynacyjną i geometrię cząsteczek związków
metaloorganicznych
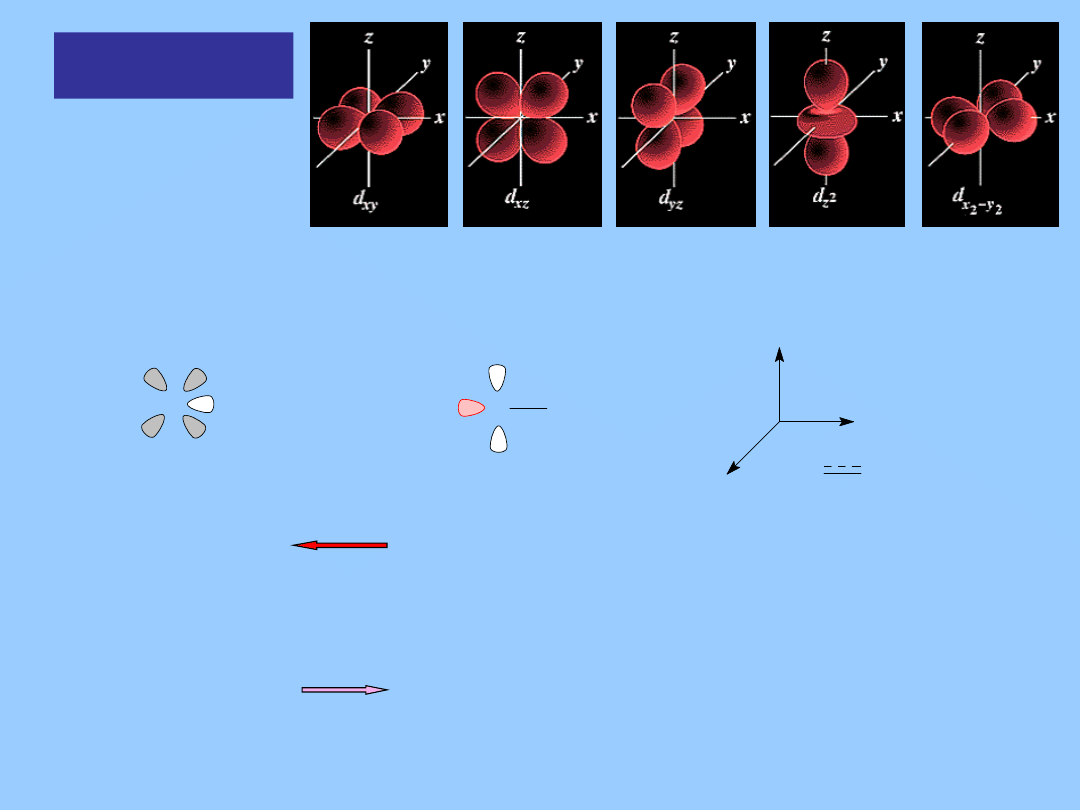
Orbitale d
W wypadku metali przejściowych orbitale (n-1)d, ns i np muszą być
traktowane jako orbitale wiążące
M
L
M L
x
y
z
s, p
z
, d
z
2
- OA
hybrydy
niezapełnione
s, p
z
- OA
hybrydy
σ
*_
OM
zapełnione
wiązanie
σ -
donorowe
p
x
, p
y
, d
xy
, d
yz
– OA
hybrydy
zapełnione
p
x
, p
y
, d
xz
, d
yz
–
OA
hybrydy
π
* _
OM
niezapełniony
wiązanie
π - akceptorowe

Reguła 18
elektronów
Sidgwick (1927)
„Stabilne termodynamicznie związki metaloorganiczne tworzą się gdy
suma elektronów d metalu i elektronów dostarczanych przez ligandy
wynosi 18.”
W związku z tym metal uzyskuje konfigurację elektronową
odpowiedniego gazu szlachetnego (nie uwzględniamy lantanowców i
aktynowców – orbitale f).
Na przykład 0 wartościowy nikiel w Ni(CO)
4
posiada 10 elektronów d
A cztery ligandy CO wiążą się poprzez wolne pary elektronowe węgla
Suma elektronów wynosi:
d
10
+ 4 x 2 = 18
Podobnie w wypadku Fe(CO)
5
d
8
+ 5 x 2 = 18
Choć podręczniki często podają konfigurację elektronową zero wartościowych metali
takich jak Ti, jako [Ar]3d
2
4s
2
i żelazo jako [Ar]3d
6
4s
2,
to w tworzeniu wiązań poziom 4s
jest zawsze wyższy energetycznie niż 3d. Stąd Ti(0) ma konfigurację [Ar]3d
4
a Fe(0)
[Ar]3d
8
.

Reguła 18
elektronów
Konwencje obliczania elektronów:
1. Podział elektronów wewnątrz cząsteczki nie może zmieniać całkowitego
ładunku kompleksu.
Zwykle wygodnie jest traktować anionowe (i kationowe) ligandy jako
rodniki a metal jako 0-wartościowe centrum.
Alternatywnie można obliczać elektrony walencyjne „realistycznie”,
traktując metale jako kationy a ligandy jako aniony.
Fe
Konwencja kowalencyjna Konwencja jonowa
C
5
H
5
* 5e
-
C
5
H
5
-
6e
-
Fe
0
d
8
Fe
2+
d
6
C
5
H
5
* 5e
-
C
5
H
5
-
6e
-
18e
-
18e
-

Reguła 18
elektronów
Mn
Mn
CO
CO
CO
OC
CO
CO
OC
CO
OC
OC
Mn
CO
CO
CO
OC
OC
.
Wiązania metal-metal wnoszą po jednym elektronie do każdego
centrum
metalicznego.
d
7
+ 5 x 2 = 17 d
7
+ 5 x 2
+ 1 = 18
Ligandy związane wiązaniem σ mogą wykorzystać wolną parę
elektronową
do dodatkowych 2-elektronowych oddziaływań donorowych.
Brom jako ligand 1-elektronowy Brom jako ligand 3-elektronowy
Mn
CO
CO
CO
OC
OC
Br
CO
CO
OC
OC
Br
Br
Mn Mn
CO
CO
CO
CO
d
7
+ 5 x 2 + 1 = 18 d
7
+ 4 x 2 + 1 + 2 = 18
Reguła 18 elektronów
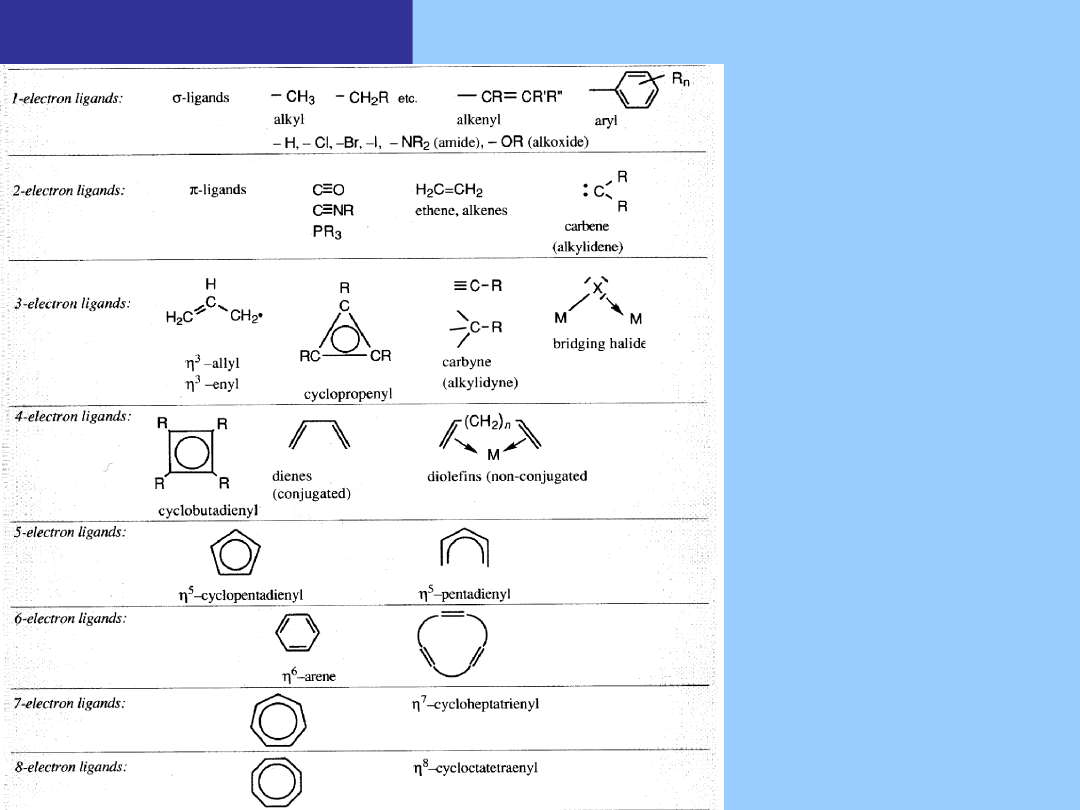
Klasyfikacja
ligandów
Liczba elektronów
dostarczanych do
centrum
metalicznego
Specyficzne
oznaczenia
W wypadku nienasyconych ligandów konieczne jest określenie liczby
atomów, z którymi oddziałuje centrum metaliczne η
n
(„n” oznacza liczbę takich
atomów węgla. Symbol μ
k
określa ligand mostkujący „k” atomów węgla
Reguła 18
elektronów
Większość kompleksów metali przejściowych spełnia regułę 18
elektronów. Wyjątek stanowią kompleksy o konfiguracji d
8
– Rh(I), Ir(I), Ni(II),
Pd(II), które wykazują silną preferencję do tworzenia kwadratowych,
planarnych kompleksów 16-elektronowych. Przyczyną jest wzrost trwałości
powłoki elektronowej d w miarę wzrostu liczby atomowej i na przykład orbital
d
z
2
nie uczestniczy w tworzeniu wiązań z ligandami.
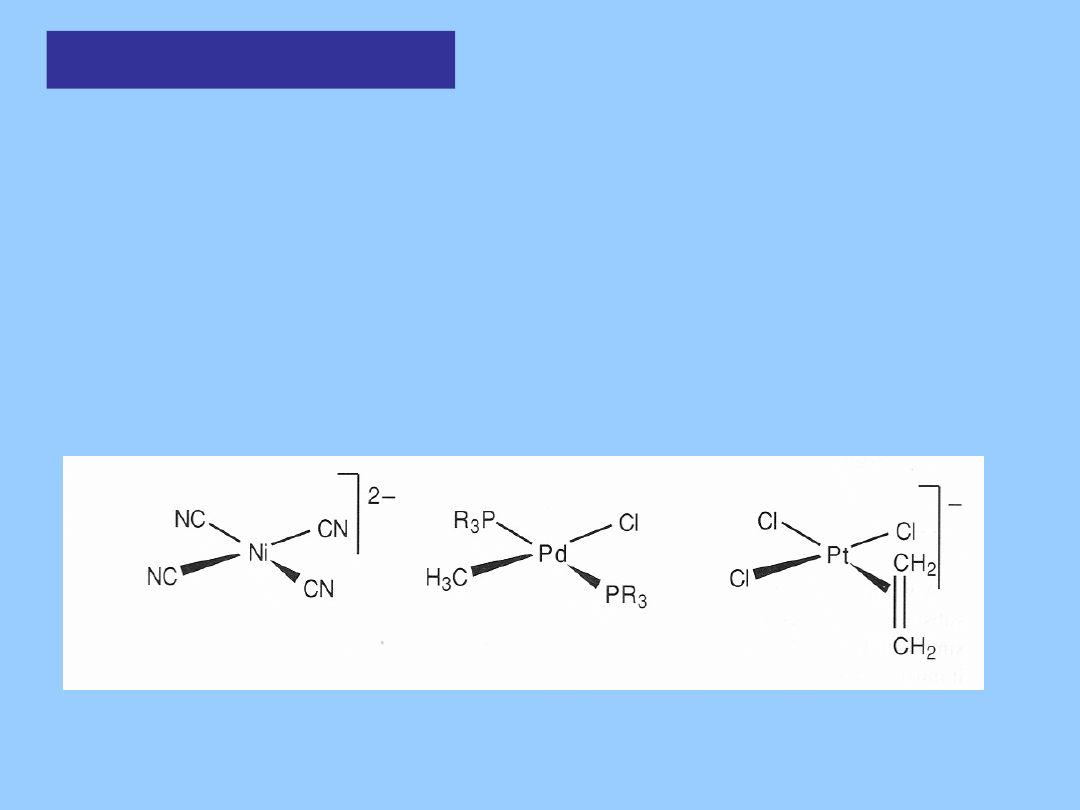
Przykłady typowych kompleksów typu d
8
:

Karbonylki
metali
Karbonylki są to związki metali przejściowych z tlenkiem węgla. Znajdują
zastosowanie w wielu procesach katalitycznych oraz syntezie organicznej.
Otrzymywanie:
Z metalu
1 bar, 25
o
C
Ni + 4CO Ni(CO)
4
bezbarwna
ciecz, t.w. 34
o
C
Z soli metali
300 bar CO, C
6
H
6
CrCl
3
+ Al Cr(CO)
6
+ AlCl
3
bezbarwny
CO wiąże się trwale z hemoglobiną i uniemożliwia
transport tlenu.
Karbonylki metali, szczególnie lotne, są przeto bardzo
toksyczne.
Ni(CO)
4
jest substancją rakotwórczą.
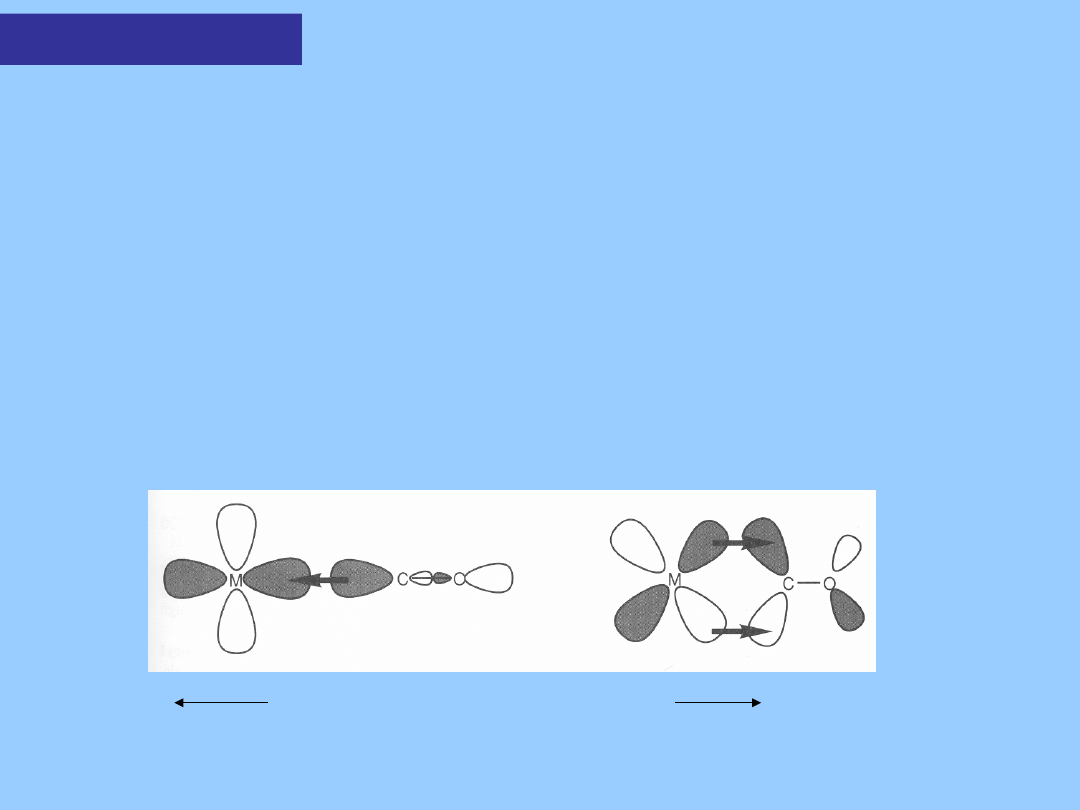
Wiązanie Metal-
CO
Wolna para elektronowa węgla oddziałuje z nie zapełnionym
orbitalem metalu
(oddziaływanie donorowe – najistotniejszy wkład w tworzenie
wiązania).
Natomiast orbital π* posiada idealną symetrię do przyjęcia
elektronów od
zapełnionego orbitalu d metalu. To silne oddziaływanie akceptorowe
dodatkowo stabilizuje wiązanie M-CO.
CO jest silnym kwasem typu π.
Nienasycone ligandy organiczne zachowują się generalnie jako
kwasy typu π,
choć niewiele z nich ma moc akceptorową charakterystyczną dla CO.
Zjawisko delokalizacji elektronów z udziałem orbitalu π* określane
jest
mianem „back boding”.
M(σ) CO(σ) M(d) CO(π*) wiązanie π
Wzmocnienie wiązania M-C Wzmocnienie wiązania M-C
Osłabienie wiązania C-O
Reakcje
podstawienia
Ligandy CO ulegają podstawieniu przez inne ligandy na drodze termicznej lub
fotochemicznej.
Podstawienie w 18 elektronowych kompleksach karbonylkowych obejmuje
proces dysocjacyjny, który generuje układ koordynacyjnie nienasycony.
L
n
M-CO L
n
M L
n
M-L'
- CO, k
1
L', k
2
+ CO, k
-1
Kompleksy metali przejściowych drugiego rzędu Układu
Okresowego zwykle
reagują szybciej niż ich analogi z pierwszego i trzeciego rzędu.
Stąd też
Najaktywniejsze katalizatory homogeniczne to zwykle kompleksy
metali
drugiego rzędu.
Reakcje
podstawienia
Podstawienie fotochemiczne pozwala na stosowanie
łagodniejszych
warunków reakcji i umożliwia otrzymanie bardziej reaktywnych
produktów.
Na przykład reaktywne kompleksy z THF czy ligandami
alkenowymi
stanowią korzystne syntony w chemii metaloorganicznej
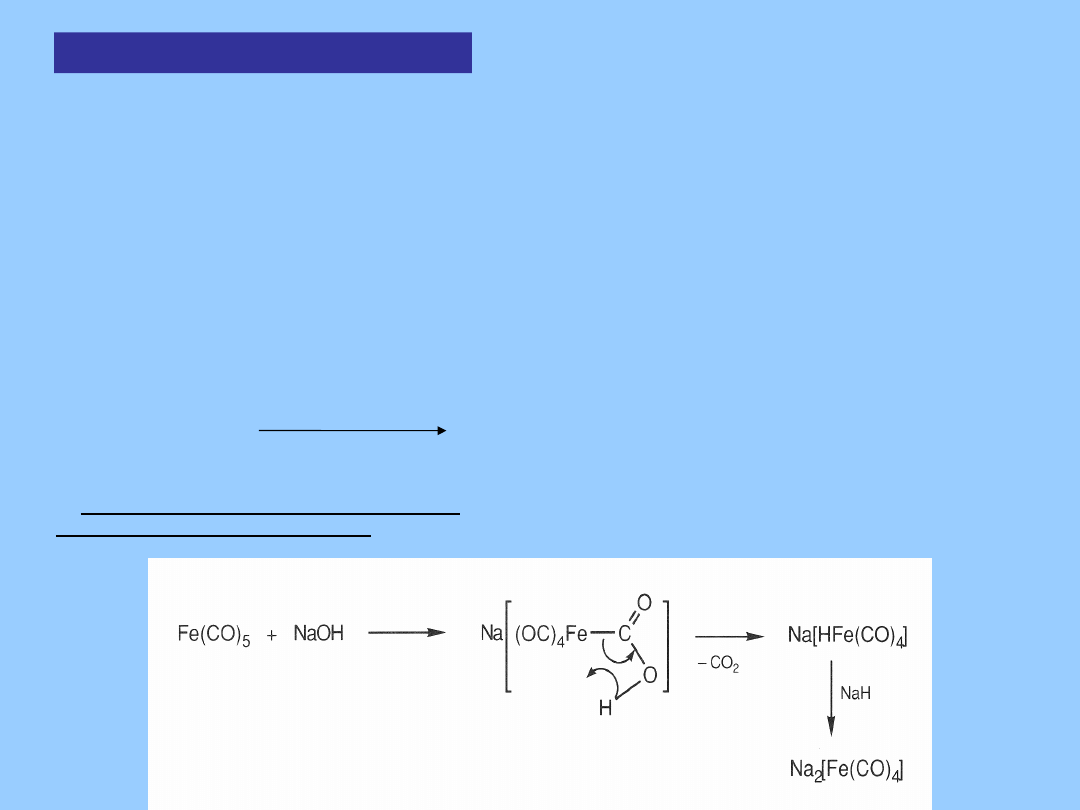
Aniony karbonylków
metali
Reakcje redukcji karbonylków prowadzą do kompleksów
anionowych. Są
one bardzo reaktywne, wrażliwe na atak elektrofilowy, mogą
ulegać
utlenianiu, lub dalszej redukcji dając kompleksy wodorkowe,
alkilowe i
acylowe.
Można je otrzymywać w wyniku dysproporcjonowania, atak
nukleofilowy
lub redukcję:
Dysproporcjonowanie:
3 Mn
2
(CO)
10
Pirydyna,
120
o
C
2 [Mn(Py)
6
]
2+
[Mn(CO)
5
-
]
2
Atak nukleofilowy:

Aniony karbonylków
metali
Otrzymywanie w wyniku redukcji
Co
2
(CO)
8
+ 2
Na
THF
2
Na[Co(CO)
4
]
ZrCl
2
(THF)
2
+ 6 KC
10
H
8
15-crown-5
THF, CO 1 bar
[K(15-crown-5)]
2
[Zr(CO)
6
]
Z silnymi czynnikami redukcyjnymi można otrzymać aniony o formalnym
stopniu utlenienia do (-IV).
Na[V(CO)
6
] +
2 Na
NH
3
ciekły
Na
3
[V(CO)
5
] + CO
Na[Mn(CO)
5
+ 2Na Na
3
[Mn(CO)
4
] + CO
K[Co(Co)
4
+ 2K K
3
[Co(CO)
3
] + CO
NH
3
ciekły
HMPA, NH
3,
c.
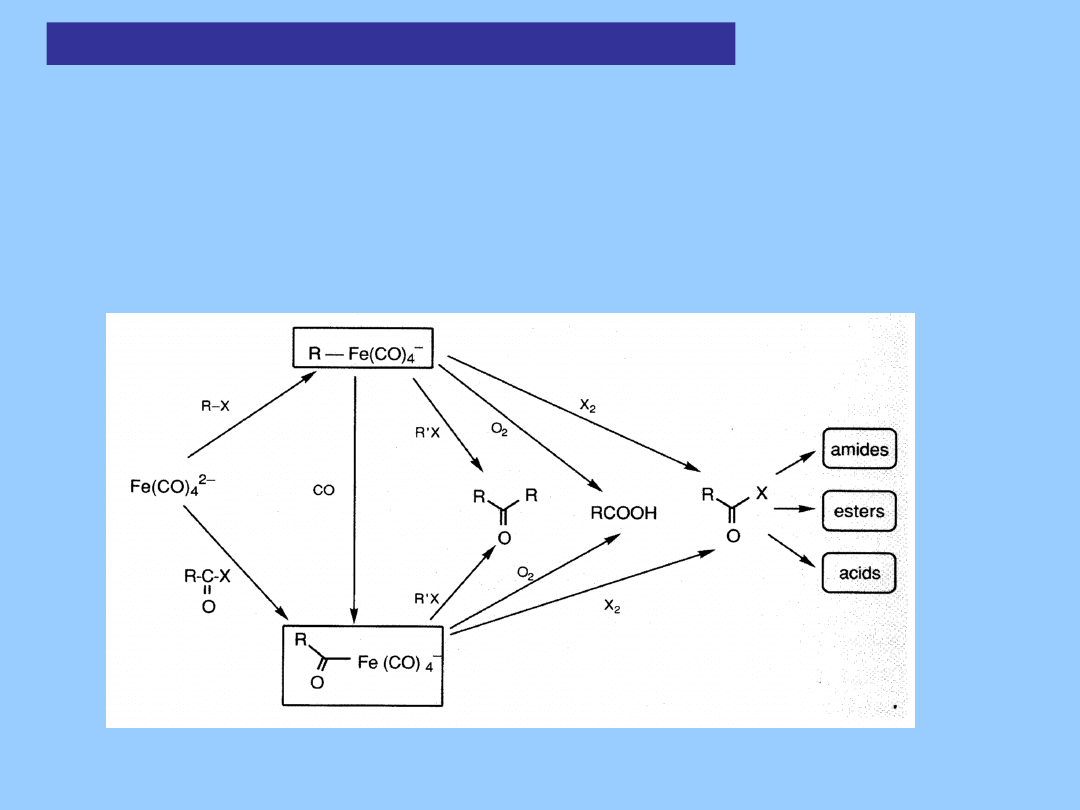
Aniony karbonylków metali w syntezie
organicznej
Aniony karbonylkowe metali, jako czynniki nukleofilowe, reagują
z organicznymi elektrofilami tworząc kompleksy alkilowe, arylowe i acylowe.
Szczególnie dogodny jest łatwo dostępny Na
2
[Fe(CO)
4
]; jego alkilowe
Pochodne znalazły szerokie zastosowanie syntetyczne podobne do
związków Grignarda (związki Collmana)

Hydrokarbonyl
ki
Otrzymywanie
1. Protonowanie: Co(CO)
4
-
+ H
+
HCo(CO)
4
HFe(CO)
4
-
+ H
+
H
2
Fe(CO)
4
Hydrokarbonylki są lotnymi cieczami, trwałymi w atmosferze CO i
w niskiej temperaturze. Bez CO rozkładają się do karbonylków
metali z
wydzieleniem wodoru. Odporność termiczna rośnie dla cięższych
metali - w dół grupy.
Obojętne kompleksy karbonylkowe ulegają protonowaniu przez
silne
kwasy dając kationy wodorkowe:
Fe(CO)
3
(PPh
3
)
2
+ H
2
SO
4
[HFe(CO)
3
(PPh
3
)
2
]
+
[HSO
4
]
-
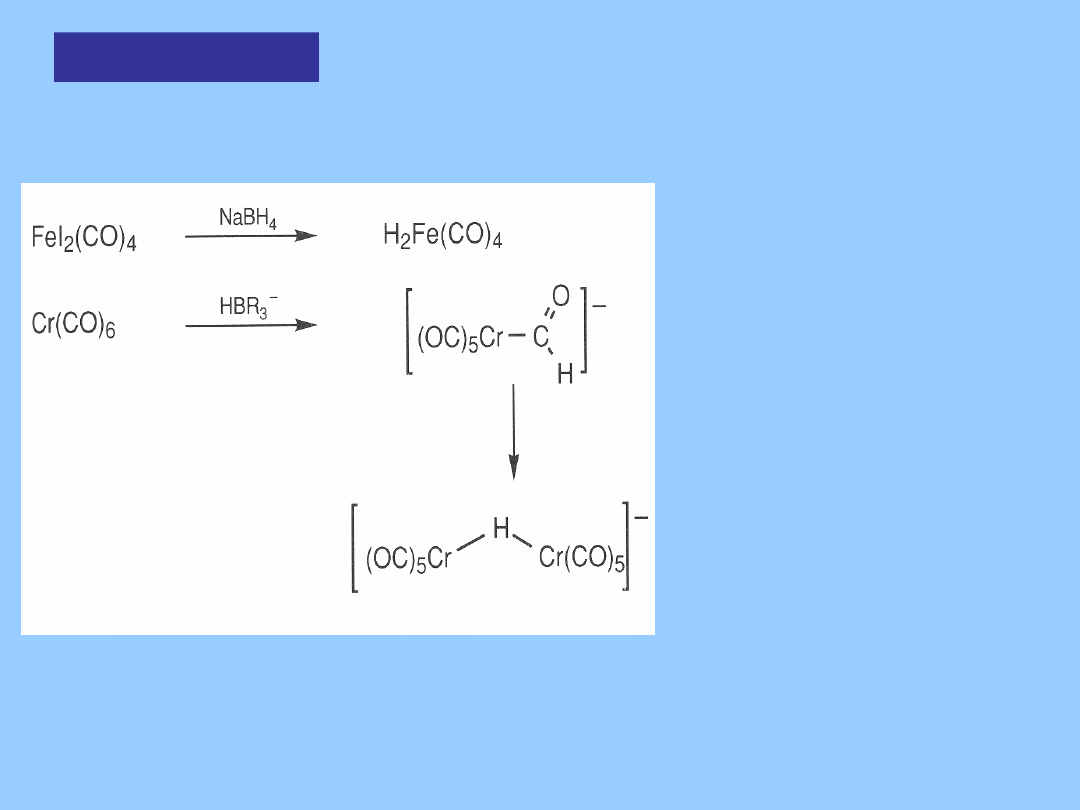
Hydrokarbony
lki
Anionowy kompleks
formylowy
jako nietrwały produkt
pośredni
2. Redukcja
HCr
2
(CO)
10
-
oraz jego Mo i W analogi stanowią rzadkie przykłady
zgiętych mostków M-H-M, nie wspomaganych wiązaniem M-M
Hydrokarbonyl
ki
3. Z dihydrogenu
Kompleksy metali przejściowych unikalnie reagują z cząsteczkowym
wodorem dając reaktywne wiązania M-H, choć entalpia wiązania H-H wynosi
450 kJ mol-1.
Mn
2
(CO)
10
+ H
2
2 HMn(CO)
5
Co
2
(CO)
8
+ H
2
2 HCo(CO)
4
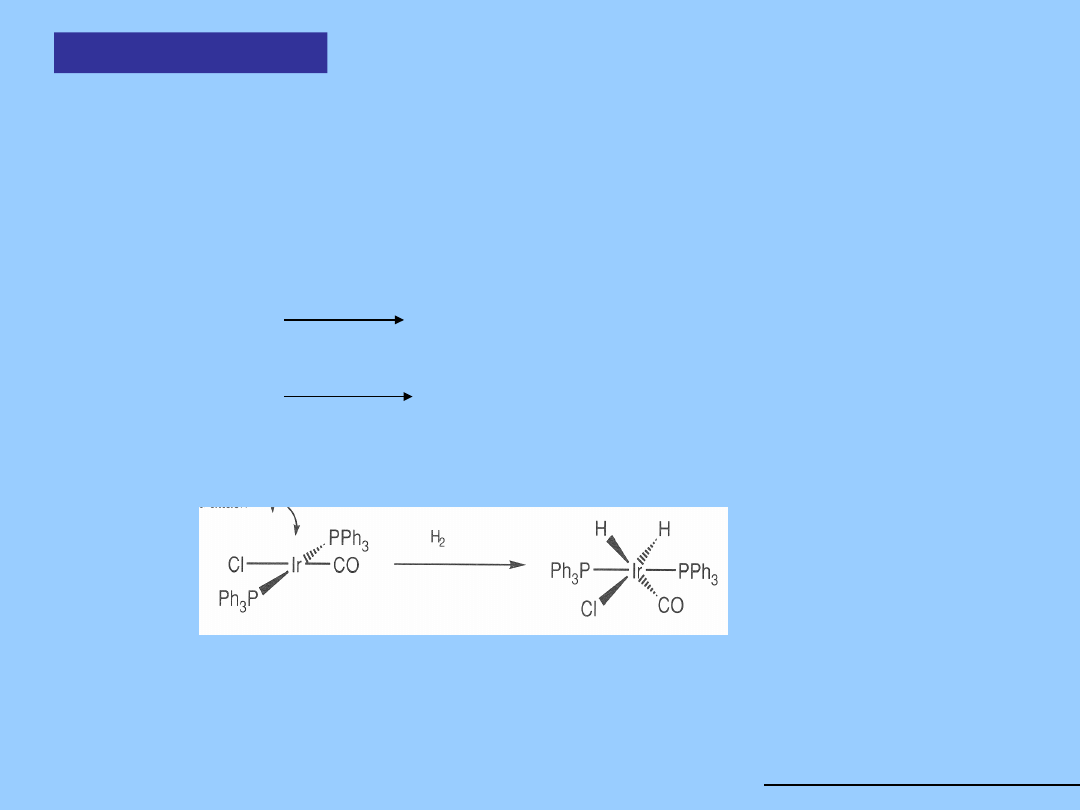
Miejsce ataku ligandu
Ir(I) 16 EV, kwadratowy Ir(III) 18 EV, oktaedryczny
Metal zmienia swój formalny stopień utlenienia o 2 jednostki. Aktywacja
wodoru jest przykładem ważnego typu przemiany – utleniającej addycji
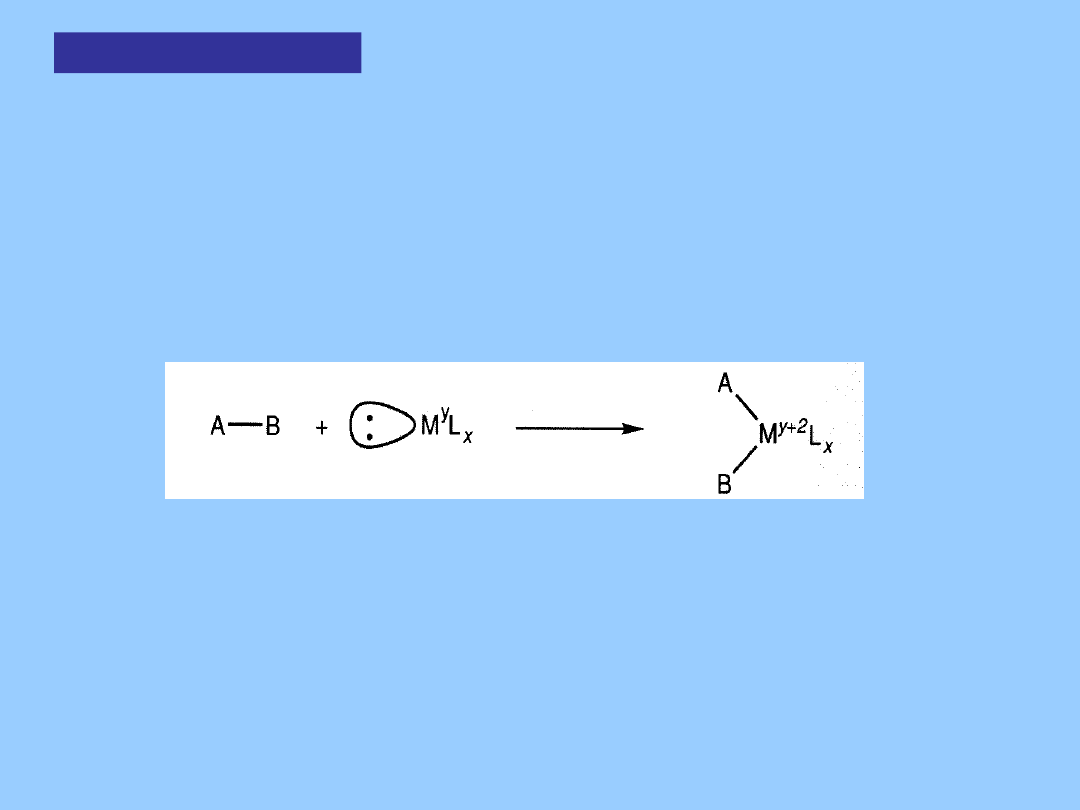
Utleniająca
addycja
Utleniająca addycja może przebiegać, gdy kompleks metalu
zachowuje się
jednocześnie jako kwas i zasada Lewisa. Prowadzi to do rozbicia
cząsteczki A-B i utworzenia nowych wiązań A-M i B-M:
Zwiększa to liczbę koordynacyjną centrum metalicznego o 2
a ponieważ A i B uważa się za bardziej elektroujemne niż metal, formalny
stopień utlenienia M wzrasta o dwie jednostki.
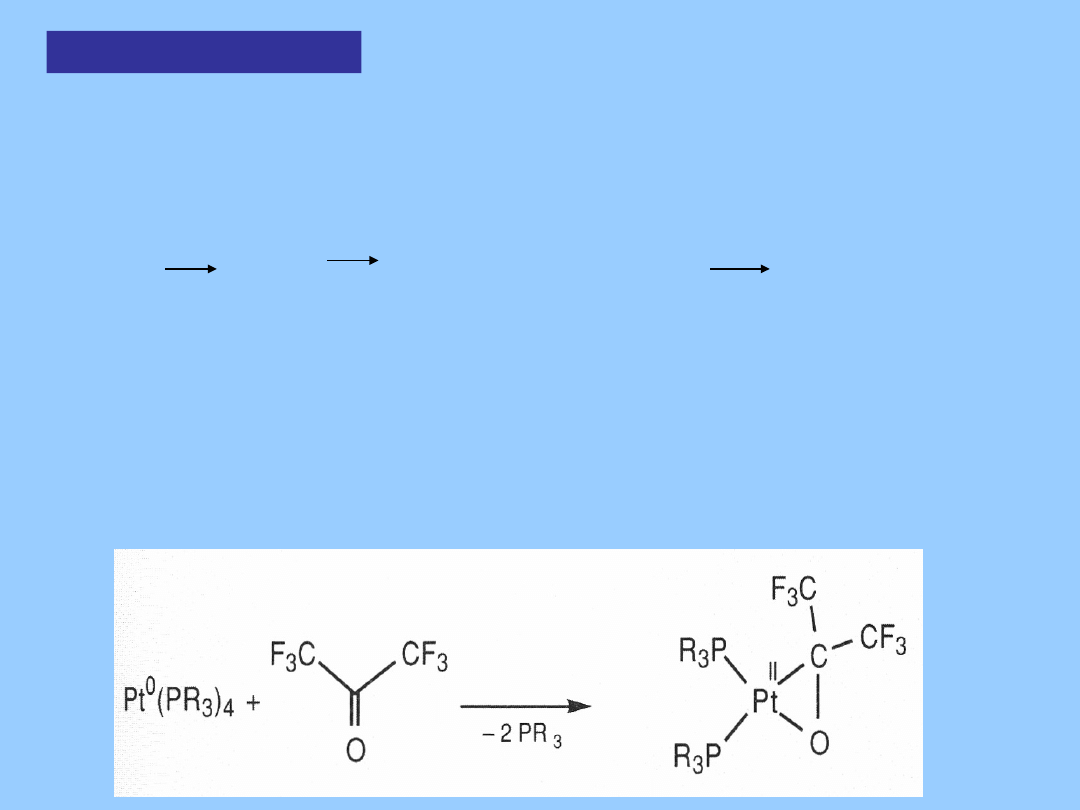
Utleniająca
addycja
Utleniająca addycja przebiega łatwo, jeśli:
-M
y
L
x
jest koordynacyjnie nienasycony. Przykłady: kwadratowe kompleksy d
8
i d
10
metali: Rh
I
, Ir
I
, Ni
0
, Pd
0
, Pt
II
i Pt
0
.
-Metal charakteryzuje się energetycznie dostępnym stanem energetycznym
M
y+2
.
Ni
0
Ni
II
i Pt
II
Pt
IV
są dostępne, zaś Ni
II
Ni
IV
nie.
Obok H
2
wiele substratów ulega utleniającej addycji: HCl, Cl
2
oraz inne
halogeny, RCOOH, HSiR
3
, halogenki alkilowe, arylowe, winylowe i benzylowe,
związki acylowe RC(O)Cl, i O
2
. Substraty z wiązaniem podwójnym A=B,
zwykle ulegają addycji do metali z zachowaniem pojedynczego wiązania.
Na przykład aldehydy, ketony, alkeny i alkiny, szczególnie z podstawnikami
elektrono-akceptorowymi ulegają utleniającej addycji do metalu:
Utleniajaca
addycja
Reaktywność zależy od metalu i ligandów. Podstawniki
elektronoakceptorowe takie jak CO deaktywują metal a elektrono-donorowe,
np. PMe
3
, podnoszą energię niewiążących par elektronów (podwyższając
zasadowość metalu) co dramatycznie zwiększa reaktywność
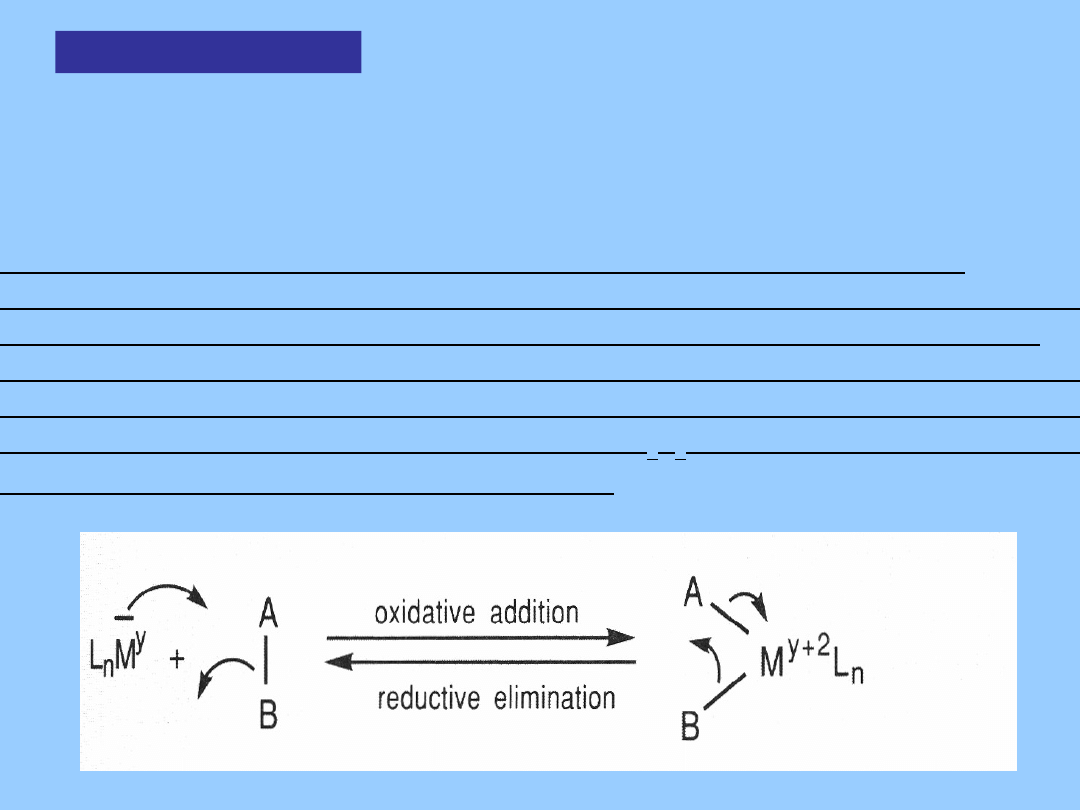
Fundamentalną cechą utleniającej addycji jest jej stereochemia.
Nowe ligandy wiązą się w położeniu cis względem siebie (czasami może
zachodzić przegrupowanie). Wynika to z zasady mikroodwracalności.
Reakcja odwrotna redukująca eliminacja A-B z utlenionego kompleksu
może zachodzić tylko wtedy, gdy A i B są w polożeniu cis. W konsekwencji
dialkilowe kwadratowe kompleksy trans MR
2
L
2
nie ulegają łatwo redukującej
eliminacji w odróżnieniu od analogów cis.

Właściwości
hydrokarbonylków
Ogólne
trendy
1. Zastąpienie CO silnie elektrono-donorowymi ligandami (Cp,
fosfiny)
drastycznie obniża kwasowość wiazania M-H. Na przykład
HCo(PMe
3
)
3
jest bardzo silną zasadą.
2. Trwałość termiczna wodorków metali zmienia się w kierunku:
pierwszy szereg << drugi szereg < trzeci szereg
3. Kwasowość wodorków metali zmienia się w kierunku:
pierwszy szereg > drugi szereg > (~) trzeci szereg. Wodorki
mostkujące
są bardziej kwaśne niż wodorki terminalne M-H
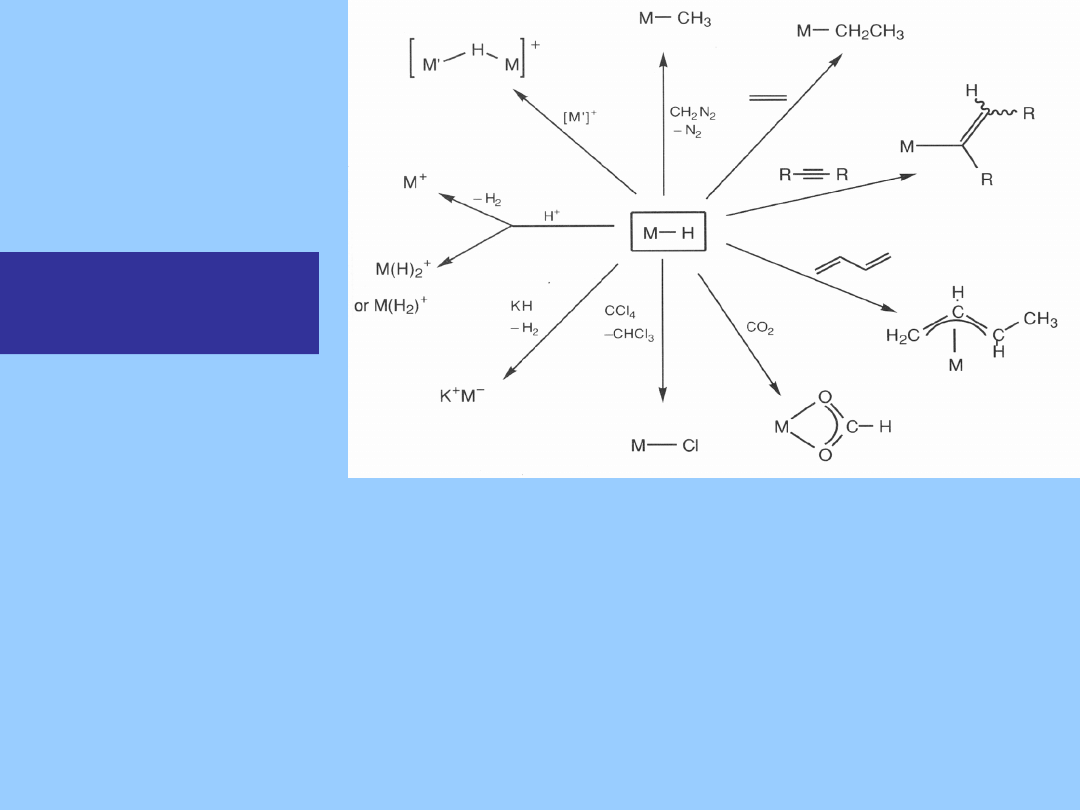
Reakcje
hydrokarbonylkó
w
Wodorki są bardzo reaktywne, umożliwiają insercję alkenów
i alkinów, ulegają protonowaniu i deprotonowaniu a kwaśne
wodorki reagują z diazometanem, podobnie jak kwasy
karboksylowe, dając połączenia alkilowe.
Reagują także z halogenkami (CCl
4
) dając w tym wypadku
chlorki. Reakcja ta jest dobrym testem hydrokompleksów
metali a często dogodną drogą stabilizacji niemożliwych do
wydzielenia nietrwałych związków pośrednich.

Kompleksy
z wiązaniem
metal-alkil
Tworzenie i rozrywanie wiązania σ metal-węgiel odgrywa ważną rolę w chemii
metaloorganicznej i jest kluczowe w jej wykorzystaniu w katalizie.
Zawsze przy syntezie alkanów, alkenów, alkinów a także ich uwodornieniu,
polimeryzacji czy ich funkcjonalizowaniu powstają związki pośrednie
z wiązaniami metal-węgiel. Ocenia się że ¾ wszystkich produktów
przemysłu chemicznego obejmuje w którymś etapie proces katalityczny.
Entalpia wiązania M – C (siła wiązania)
maleje
ze wzrostem liczby atomowej
pierwiastków grupy głównej ale
rośnie
w triadach metali przejściowych.
Entalpia wiązania M – C pochodnych alkilowych metali przejściowych
(150 – 300 kJ mol
-1
) jest taka sama jak pochodnych grupy głównej
i porównywalna z siłą wiązania węgiel – jod.
Termodynamicznie alkilki metali przejściowych powinny być trwalsze niż
na przykład PbEt
4
, a jest dokładnie odwrotnie. PbEt
4
jest trwalszy kinetycznie.
niż np. TiEt
4
. Ma on zapełnione wszystkie orbitale, w tym 5d i ulegać może tylko
homolitycznemu rozpadowi o stosunkowo wysokiej energii.
Δ
PbEt
4
PbEt
3
* + Et*
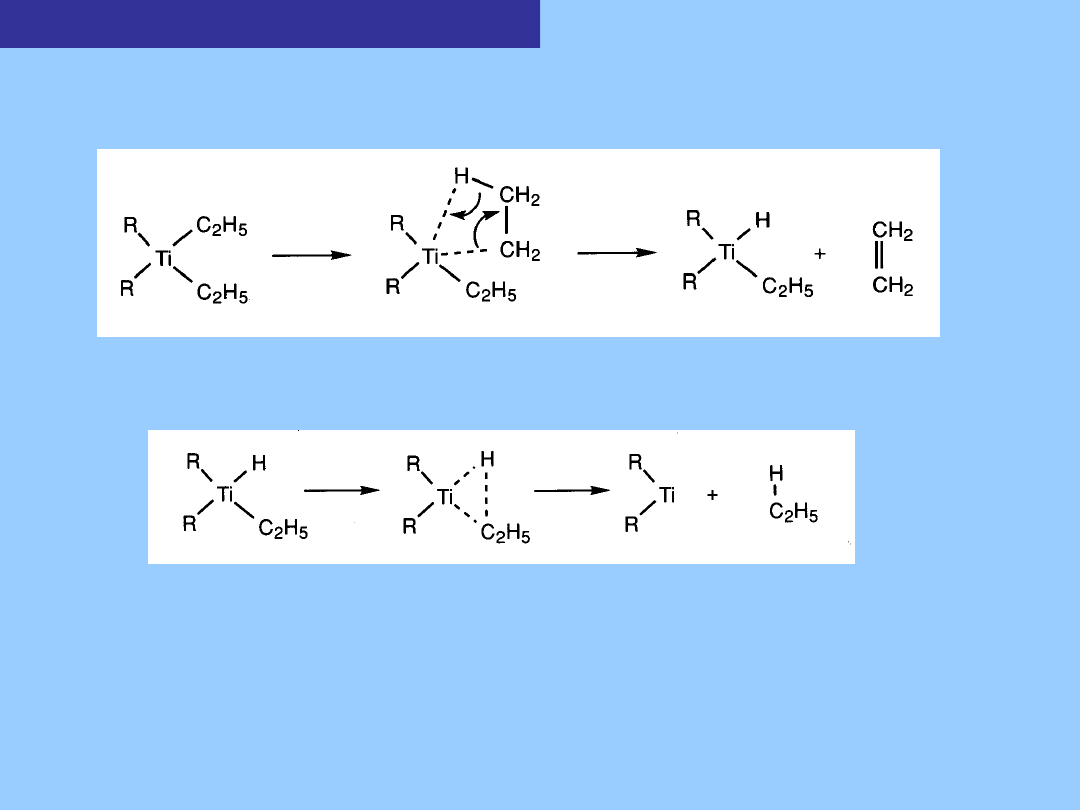
Metale przejściowe, np. tytan posiada niezapełnione orbitale d
(3d), które dają
łatwą drogę rozkładu w wyniku oddziaływania z wiązaniem C- H
ligandów.
Kompleksy
z wiązaniem
metal-alkil
Powstający związek tytanu jest nietrwały i ulega rozkładowi:
Takie uwolnienie alkanu związane jest z redukcją metalu o dwie jednostki –
jest to etap
redukującej eliminacji
. Jest to proces „uzgodniony” (jednoczesny)
i może przebiegać jedynie gdy ligandy (wodorowy i alkilowy) są w położeniu
cis. W wyniku przebiegu procesu β-eliminacji w równo-molowych ilościach
powstaje alken i alkan.
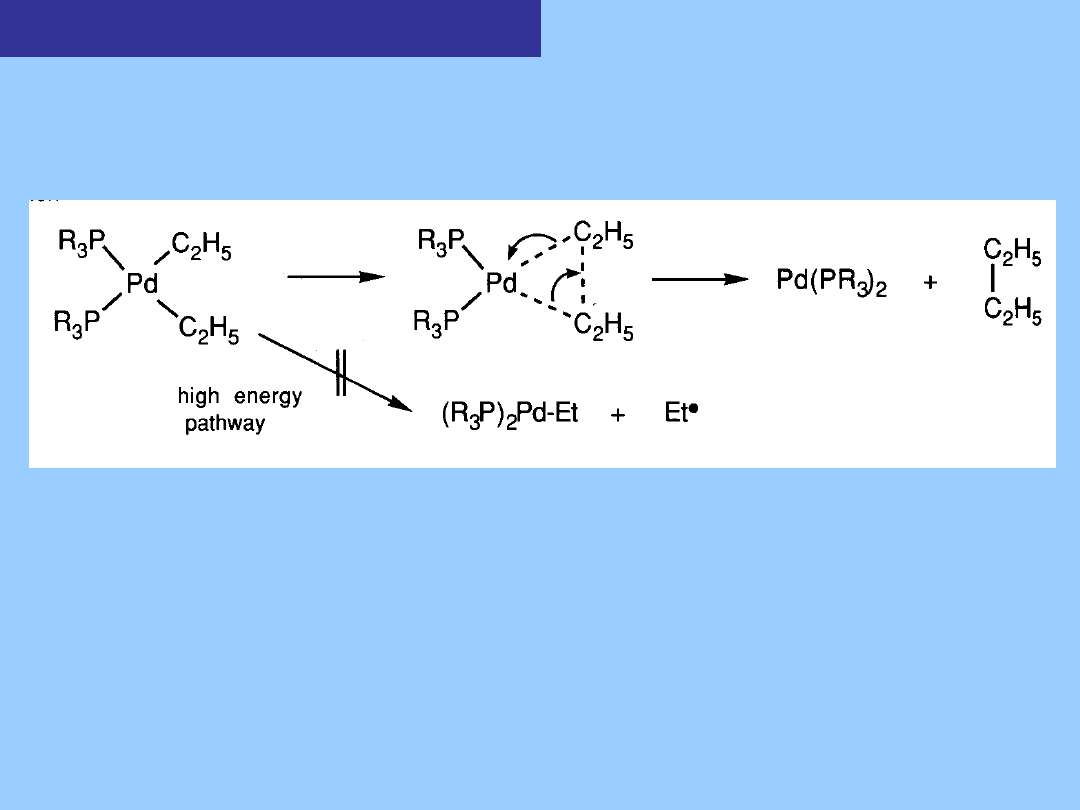
Kompleksy z wiązaniem metal
alkil
Redukująca eliminacja bez oderwania β-H jest uprzywilejowana
drogą rozkładu
niektórych pochodnych dialkilowych, np. palladu.
W jego rezultacie powstają alkany o podwójnej ilości atomów węgla
w stosunku
liganda alkilowego. Nie powstają alkeny.
Dwa ligandy mające ulegać sprzęganiu muszą być w położeniu cis
Proces eliminacji jest uzgodniony i nie obejmuje homolizy
wiązania M-C (jak w
wypadku PbEt
4
)
Synteza trwałych pochodnych alkilowych obejmuję dwie strategie;
1. Blokowanie miejsc koordynacji przez ligandy elektronodonorowe,
np. difosfinę
Me
2
PCH
2
CH
2
PMe
2
(dmpe). TiMe
4
rozkłada się w – 50
o
C a
TiMe
4
(dmpe)
w temperaturze pokojowej.
2. Stosowanie ligandów alkilowych nie ulegających β-eliminacji: -CH
3
,
-CH
2
Ph,
-CH
2
CMe
3
, -CCR, -CH
2
SiMe
3
, etc.

Kompleksy z wiązaniem metal
alkil
Trwałość wiązania M – C, generalnie, rośnie ze wzrostem
charakteru-
s
wiązania
M – C:
M-CR
3
< M-aryl, M-winyl < M-alkenyl
sp
3
sp
2
sp
siła wiązania/ elektrono-akceptorowy charakter liganda
alkilowego
Typowe geometrie wiązań metal - alkil
Ligandy metylowe mogą przyjąć zaskakującą ilość sposobów
koordynacji
w zależności elektronowych wymagań stawianych przez centrum
metaliczne
z którym są one związane, z włączeniem η
3
-CH
3
i trójkątny,
planarny węgiel.
C
M
H H
H
M
M
M
C
H
H
H
M C
H
H
H
M'
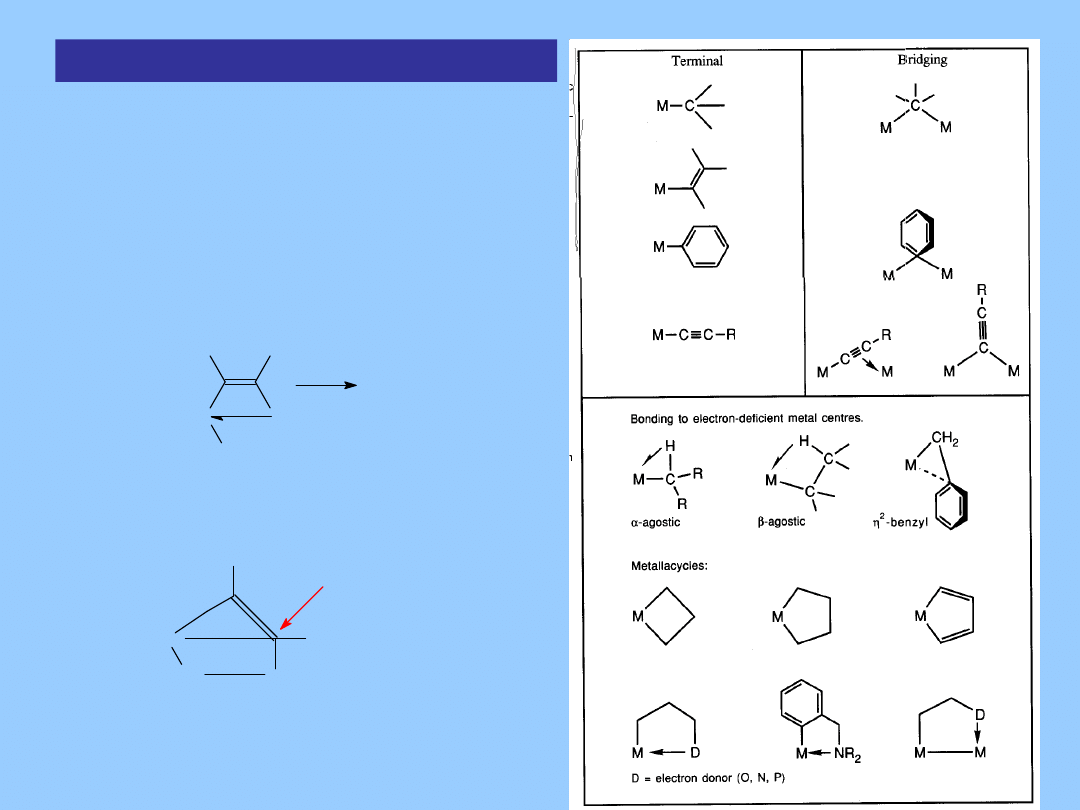
Kompleksy z wiazaniem metal -
alkil
Istnienie węgla w geometrii planarnej -
kwadratowej jest przedmiotem dyskusji
Ostatnio stwierdzono, że w kompleksie
cyrkonu suma kątów wokół atomu
węgla wynosi 360
o
C.
Me
3
Si
Cp
2
Zr H
Ph
Cl
Al Me
3
-
CH
4
SiMe
Cp
2
Zr
Ph
AlMe
2
Cl
planarny
kwadratowy C
Kompleksy z wiązaniem metal -
alkil
Syntez
a
1. Alkilowanie halogenków metali
Jest to najszerzej stosowana
metoda
syntezy, tak dla
homoleptycznych
jak i dla kompleksów
stabilizowanych
donorowymi ligandami.
Kompleksy homoleptyczne
zawierają
jednolity typ ligandów, np.
MMe
4
,
M(PMe
3
)
4
TiCl
4
+ 2
Mg(CH
2
SiMe
3
)
4
-78
o
C – RT -2 MgCl
2
Ti(CH
2
SiMe
3
)
4
Alkilowanie może zmienić stan
utlenienia metalu
Me
2
Mg
Rh
II
2
(OAc)
4
Rh
III
Me
3
(PMe
3
)
3
PMe
3

Kompleksy z wiazaniem metal
- alkil
Synte
za
2. Utleniająca addycja
Kompleksy metali o niskiej wartościowości, szczególnie kompleksy Ir
I
, Ni
0
,
Pd
0
, Pt
0
,
stabilizowane ligandami fosfinowymi ulegają utleniającej addycji w reakcji
z halogenkami
alkilowymi, alkenylowymi, benzylowymi i arylowymi. Reakcja przebiega
szybko, gdy są
to kompleksy nienasycone koordynacyjnie (kwadratowe planarne) lub
posiadają łatwo
dysocjujące ligandy (etylen, fosfiny).
Ir
I
Cl (CO)(PPh
3
)
2
+ MeI Ir
III
Me(I)(Cl)
(CO)(PPh
3
)
2
PEt
3
Ni(PEt
3
)
3
+ Cl- Cl Ni
- PEt
3
PEt
3
Jeżeli ograniczona jest liczba ligandów, które mogą się znaleźć się w
sferze
koordynacyjnej metalu, powstaje kompleks jonowy:
CpCo(PMe
3
)
2
+ MeI
Co
Me
Me
3
P
Me
3
P
I
-

Kompleksy z wiązaniem metal-alkilSynte
za
3.
Reakcje addycji do wodorków metali
Alkeny i alkiny łatwo wbudowują się w wiązanie M-H. Jest to to
reakcja odwrotna do
omawianej wcześniej β-eliminacji. Reakcje tego typu – tworzenie różnych
izomerów
termicznie nietrwałych izomerów alkilków kobaltu stanowią ważne etapy
procesów
katalitycznych.
(OC)
4
Co-H + +
(OC)
4
Co
Co(CO)
4
4. Atak nukleofilowy na skoordynowane ligandy
Polarne, nienasycone ligandy, szczególnie CO, są wrażliwe na atak nukleofilowy
Fe(CO)
5
+ LiR Li
+
[ (OC)
4
Fe-C-R]
-
O
Kompleksy z wiązaniem
metal-alkil
Synteza
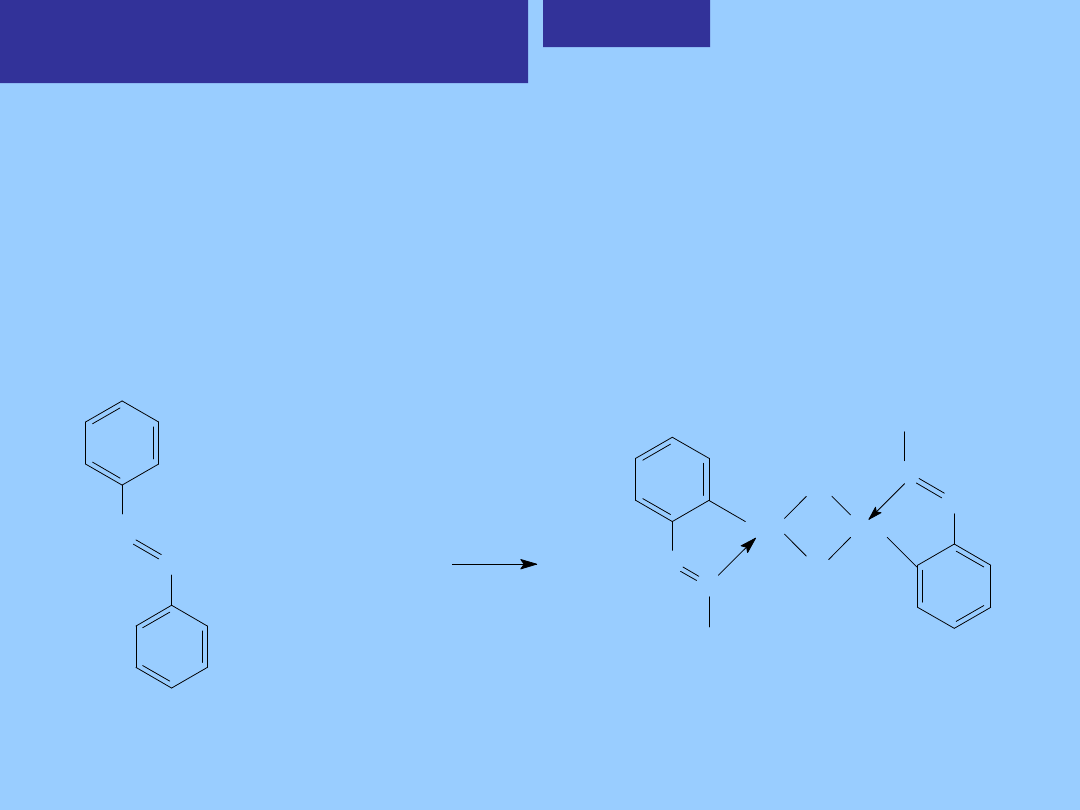
4. Orto-metalowanie
Orto-metalowanie polega na zerwaniu wiązań C- H w położeniu orto grup arylowych i
utworzeniu wiązań M-C. Reakcja przebiega łatwo wówczas, gdy ugrupowania arylowe
jednego z reagentów związane są z atomami o właściwościach elektro-donorowych,
takich jak N lub P a drugi reagent zawiera „bogaty” elektronowo metal na niskim stopniu
utlenienia, który może ulegać utleniającej addycji.
N
N
+
PdCl
2
NEt
3
-
HNEt
3
Cl
N
N
Pd
Ph
Cl
Cl
Pd
N
N
Ph
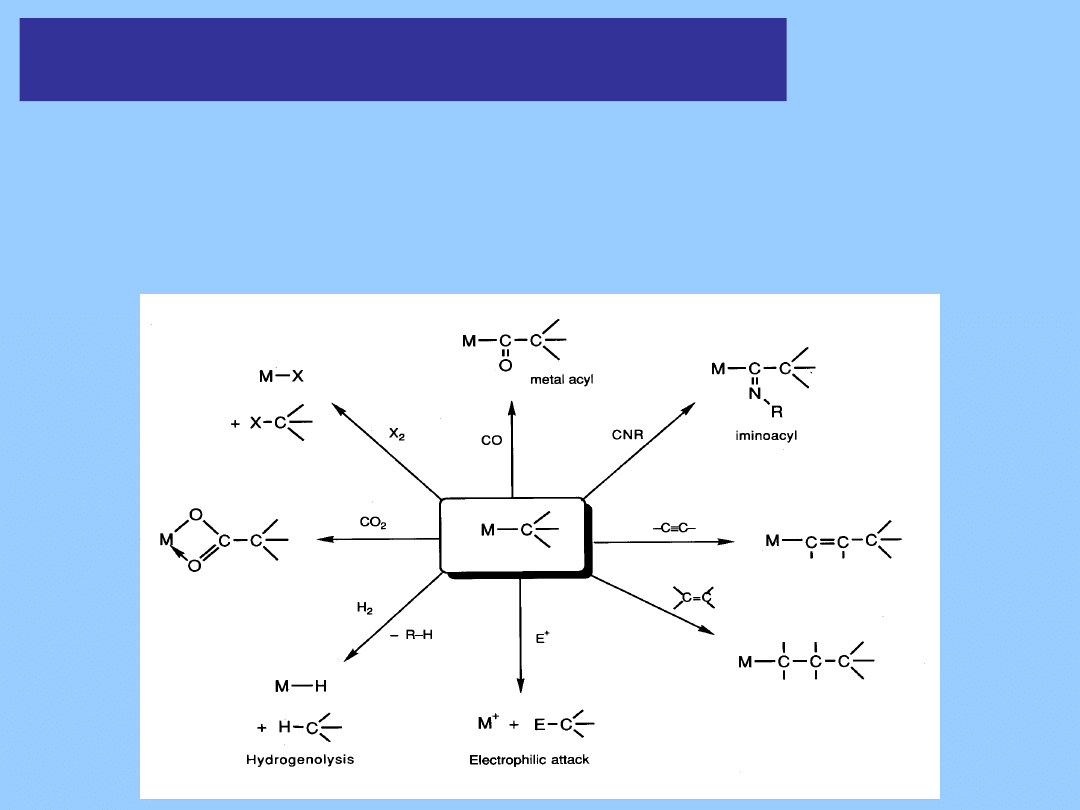
Reaktywność kompleksów z wiązaniem
alkil-metal
Większość
pochodnych z wiązaniem alkil metal jest bardzo reaktywna. Najważniejsze rekcje
jakim ulegają to:
1. Reakcje prowadzące do rozpadu wiązania M-C: hydrogenoliza wiązania M-C; reakcje z
elektrofilami, takimi jak CO
2
, H
+
, I
2
2. Reakcje insercji z udziałem nienasyconych związków organicznych CO, izonitryli, alkenów
i alkinów
3. Reakcje redukującej eliminacji: na przykład sprzęganie ligandów alkilowych z nukleofilami

Reaktywność kompleksów z wiązaniem alkil-metal,
przykłady reakcji
Rozbicie wiązania M-C
1. Reakcje z elektrofilami
Ogólnie, wiązania σ metal – węgiel reagują gwałtownie z elektrofilami HX, dając alkany
i kompleksy L
n
MX
Zr(CH
2
Ph)
4
Zr(CH
2
Ph)
3
(OAr) Zr(CH
2
Ph)
2
(OAr)
2
ArOH ArOH
- toluen - toluen
Cp(OC )
2
Fe R Br
2
Cp(OC)
2
Fe
+
+
Br
R Br
Halogeny są często stosowane do rozbicia wiązania M-C. Reakcja jest
stosowana do
otrzymywania funkcjonalizowanych pochodnych alkilowych, z
wykorzystaniem
kompleksów metaloorganicznych
Dwutlenek węgla może dać produkty podwójnego i pojedynczego wbudowania.
Hydroliza powstających kompleksów prowadzi do otrzymania odpowiednich kwasów
karboksylowych. Reakcje te są podobne do reakcji związków Grignarda.
Cp
2
TiMe
2
+ 2 CO
2
Cp
2
Ti(O
2
CMe)
2
L
3
Rh R + CO
2
L
3
Rh-O-C-R
O
R = Me, Ph
L = PPh
3

Reaktywność kompleksów z wiązaniem alkil-metal, przykłady
reakcji
2. Reakcje z H
2
Reakcje z wodorem cząsteczkowym zachodzą w wielu reakcjach katalitycznych –
uwodornienie, hydrosililowanie. Stanowią także łatwą metodę syntezy wodorków metali.
Cp
2
ZrMe
2
Cp
2
ZrH
2
+ 2 CH
4
(OC)
4
Co-R (OC)
4
Zr-H + R-H
H
2
, cisnienie
H
2
, cisnienie
Reakcje insercji do wiązania M-C
1. Insercja CO
Jest to jedna z najszerzej stosowanych w przemyśle reakcji kompleksów
alkilowych metali przejściowych, prowadząca do powstania kompleksów
acylowych.
Mechanizm reakcji, został ustalony przy zastosowaniu znaczonego
węglem
13
C tlenku węgla.
Ugrupowanie acylowe powstaje ze skoordynowanego już z metalem CO a
znaczony ligand zajmuje położenie cis do grupy acylowej.
Mn
C
OC
OC
C O
CO
C O
H
H
H
OC
OC
Mn
CO
C
O
CH
3
CO
OC
OC
Mn
CO
CO
CO
13
C
O
CH
3

Reaktywność kompleksów z wiązaniem alkil-metal, przykłady
reakcji
Trzy ważne cechy procesu to:
Insercja CO do wiązania M-C polega na
migracji liganda alkilowego do już
skoordynowanego liganda CO
Migracja polega na
wewnątrz-cząsteczkowym ataku nukleofilowym
grupy
alkilowej na koordynacyjnie nienasycony elektrofil
Migracja grupy alkilowej jest
odwracalna
2. Insercja izonitryli
Izonitryle podobnie jak CO ulegają łatwo reakcjom insercji. W ich wypadku
możliwe są reakcje wielokrotnej insercji. Kompleksy niklu, np. są
katalizatorami polimeryzacji do liniowych polimerów iminoacylowych
Cl Ni R + Bu
t
NC
L
L
L = PMe
3
N
Bu
t
L
2
ClNi C C R
N
Bu
t
L
Cl
C
R
N
Ni
Bu
t
L
2
ClNi C R
N
Bu
t
Insercja tlenku węgla w katalizie
1. Hydroformylowanie
Jednym z najbardziej znanych procesów jest hydroformylowanie olefin,
opracowane po raz pierwszy w BASF (1938) i znane jako „proces oxo”.
Stosowano wówczas katalizator kobaltowy – Co
2
(CO)
8
.
Hydroformylowanie wobec karbonylku kobaltu wymaga jednak
drastycznych warunków (temperatura, ciśnienie CO/H
2
). Takie prekursory
katalityczne, jak HRh(CO)(PPh
3
)
3
wyparły obecnie układ kobaltowy.
HRh(CO)(PPh
3
)
3
18
VE
1
HRh(CO)(PPh
3
)
2
- (+) PPh
3
16
R
(PPh
3
)
2
(CO)Rh
R
H
2
18
EV
3
R
CO
2
(PPh
3
)
2
(CO)Rh
16
EV
R
CO
18
EV
3
R
O
C
(PPh
3
)
2
(CO)Rh
H
2
4
R
C
O
Rh
H
OC
Ph
3
P
Ph
3
P
5
VE
H
R
C
O
H
16
18
EV
EV
(PPh
3
)
2
(CO)Rh

Insercja tlenku węgla w
katalizie
HRh(CO)(PPh
3
)
3
18
VE
1
HRh(CO)(PPh
3
)
2
- (+) PPh
3
16
R
(PPh
3
)
2
(CO)Rh
R
H
2
18
EV
3
R
CO
2
(PPh
3
)
2
(CO)Rh
16
EV
R
CO
18
EV
3
R
O
C
(PPh
3
)
2
(CO)Rh
H
2
4
R
C
O
Rh
H
OC
Ph
3
P
Ph
3
P
5
VE
H
R
C
O
H
16
18
EV
EV
(PPh
3
)
2
(CO)Rh
1
Oddysocjowanie liganda – generowanie koordynacyjnie nienasyconego
produktu
pośredniego
2
Związanie substratu w miejscu wakancji
3
Insercja
4
Utleniająca addycja
5
Redukująca eliminacja
1. Hydroformylowanie
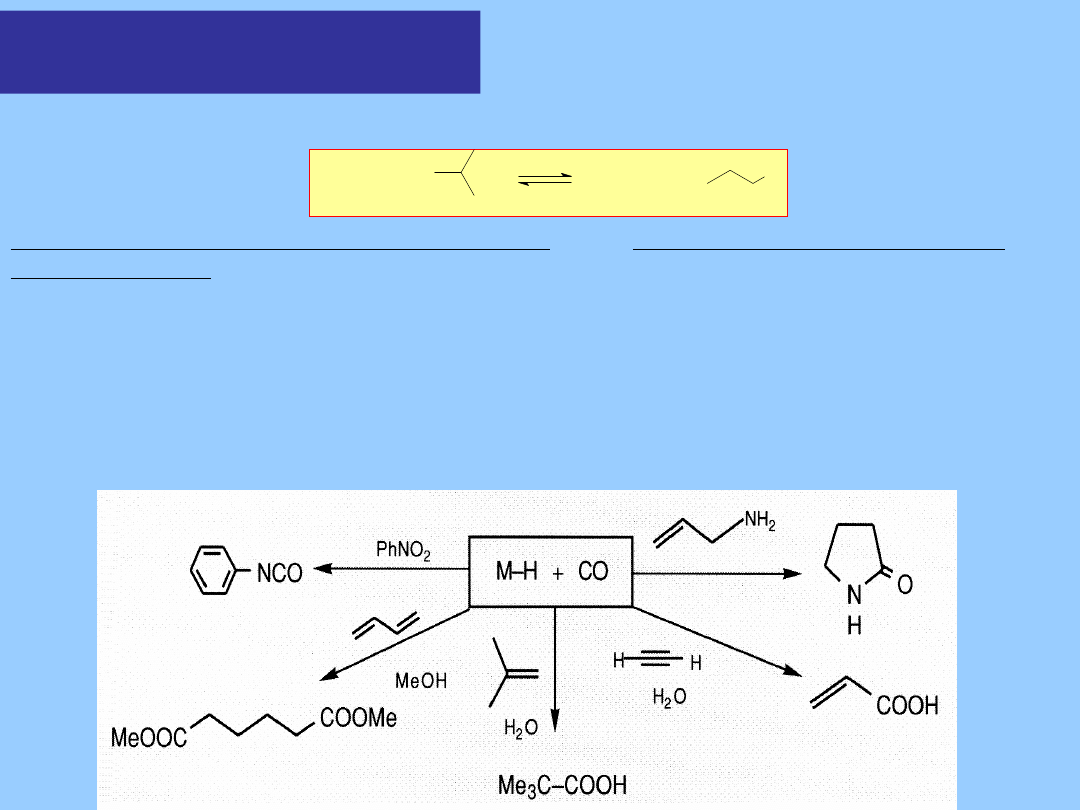
Insercja tlenku węgla w
katalizie
1.Hydroformylowanie
Insercja terminalnej olefiny do wiązania M-H może zachodzić zgodnie, lub
niezgodnie
regułą Markownikowa
(OC)
4
Co
R
(OC)
4
Co
R
Produkt zgodny z regułą Markownikowa Produkt niezgodny z regułą
Markownikowa
W wypadku stosowania katalizatorów kobaltowych tworzą się liniowe i
rozgałęzione aldehydy. Aldehydy stanowią surowiec do produkcji alkoholi i
dalej detergentów, których struktura powinna być liniowa. Rozgałęzione
aldehydy są niepożądane. Katalizatory rodowe dają niemal wyłącznie
produkty liniowe i stąd, obok wyeliminowania drastycznych warunków
hydroformylowania wobec kompleksów kobaltu, ten właśnie element
decyduje o stosowaniu katalizatorów rodowych mimo ich wyższej ceny. Wodór
można zastąpić innymi
Elektrofilami uzyskując szereg produktów karbonylowania.
Insercja
tlenku węgla w katalizie
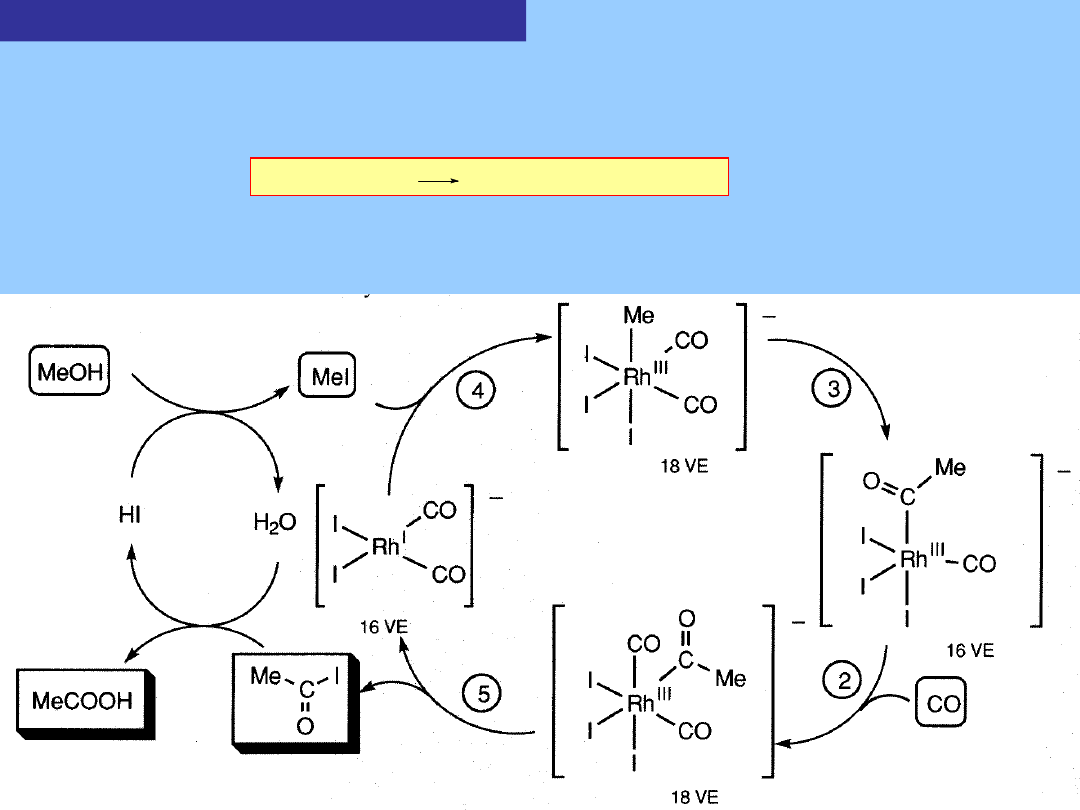
2. Karbonylowanie metanolu (proces Monsanto – synteza kwasu octowego)
Reakcje karbonylowania nie są ograniczone do olefin, np. sole kobaltu
wobec jodu katalizują przekształcenie metanolu w mieszaninę kwasu
octowego i octanu metylu:
Reakcja jest ponownie bardziej efektywna i selektywna jeżeli stosowane są
katalizatory rodowe. Reakcja składa się z dwóch sprzężonych cykli: cykl
jodkowy, przekształcający nie reaktywny metanol w jodek metylu oraz cykl
karbonylowania wobec rodu
MeOH + CO
CoI
2
MeCOOH + MeCOOMe
Insercja
alkenów
Jeden z najważniejszych katalitycznych procesów przemysłowych oparty jest
na zdolności insercji alkenów do wiązania metal–alkil – polimeryzacja olefin.
Ni
R
Ni CH
2
CH
2
R
C
2
H
4
50
bar
Ni
CH
2
CH
2
R
Przykład modelowy:
Mechanizm:
M
R
H
2
C
CH R
M
R
CH
2
R C
H
M CH
2
CH
'
'
R
R'
M CH
CH
2
R
R'
anty - Markownikow Markownikow
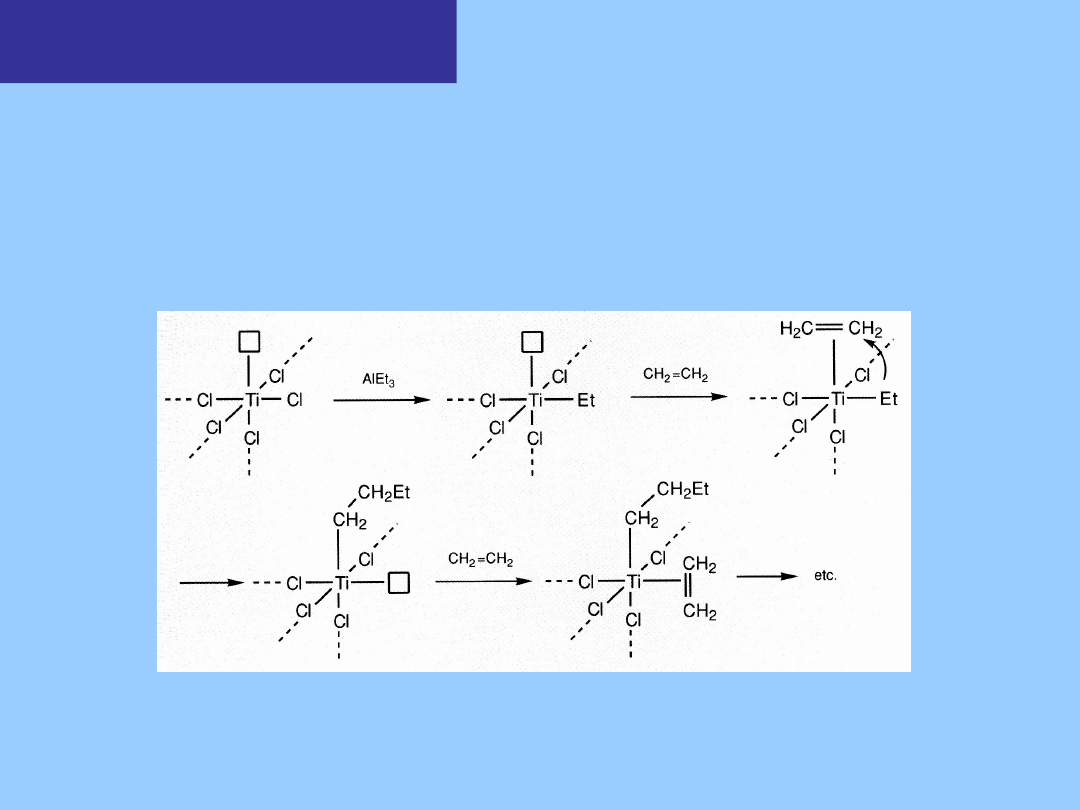
Polimeryzacja Zieglera -
Natty
(Nagroda Nobla 1963)
Mechanizm reakcji wobec heterogenicznego katalizatora TiCl
4
/AlEt
3.
Efekt
katalityczny ma miejsce w obszarach defektów sieci krystalicznej, gdzie
metal jest koordynacyjnie nienasycony. Struktura powierzchni jest
prawdopodobnie odpowiedzialna za analogiczny proces stereoregularnej
polimeryzacji propylenu (propenu), ze względu na sposób w jaki monomer
może koordynować do metalu w rezultacie ograniczeń sterycznych.

Polimeryzacja Zieglera -
Natty
Dihalogenki metalocenów
Cp
2
MCl
2
( M = Ti, Zr, Hf )
wobec alkilowych
pochodnych glinu dają wysoce reaktywne homogeniczne katalizatory
Zieglera – Natty.
Najbardziej efektywnym katalizatorem jest metyloalumoksan ( MAO,
[MeAlO]
n
)
polimer, zawierający przeciętnie jedną grupę metylową na jeden
atom glinu.
Me Me
Cp
2
MR
2
+ - - Al-O-Al-O- -
[Cp
2
MR]
+
- -Al-O-Al-O- -
Me Me
R
Rolą pochodnej glinu jest:
-alkilowanie metalu przejściowego
-spełnienie roli kwasu Lewisa i utworzenie
wakancji przez przyłączenie liganda
chlorkowego lub alkilowego od metalu
przejściowego
-działanie „czyszczące” poprzez usuwanie
zanieczyszczeń z monomeru i środowiska
reakcji

Polimeryzacja Zieglera - Natty
Kationowe kompleksy 14-elektronowe typu [Cp
2
M-R]
+
zostały
zidentyfikowane jako cząstki aktywne. Takie same kompleksy można
uzyskać bez udziału alkilków glinowych z dialkilowych metalocenów w
wyniku protonowania lub oderwania R
-
za pomocą CPh
3
+
.
Wobec słabo koordynujących przeciwjonów, jak na przykład B(C
6
H
5
)
4
-
, te
kationowe katalizatory wykazują bardzo wysoką aktywność.
Otrzymano szereg kompleksów stabilizowanych ligandami [Cp
2
MR(L)]
+
,
gdzie
L = THF, RCN, PR
3
, które katalizują polimeryzację alkenów, jeśli ligand L
łatwo oddysocjowuje.
Cp
2
MR
2
+ [HNMe
2
Ph]
+
X
-
+ CPh
3
+
- NMe
2
Ph
- RCPh
3
[Cp
2
M-R]
+
X
-
M = Ti, Zr, Hf
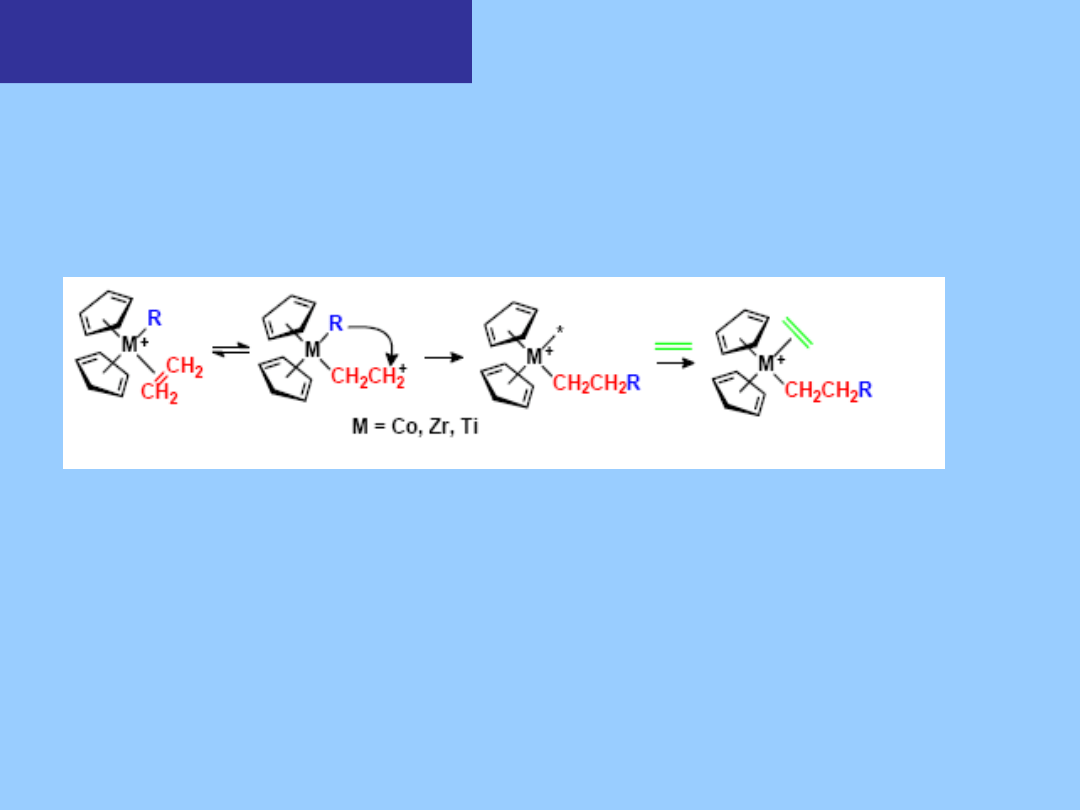
Polimeryzacja Zieglera -
Natty
Mechanizm propagacji łańcucha jest
podobny do mechanizmu dla katalizy
heterogenicznej. W obydwu wypadkach potrzebne są dwa miejsca
koordynacyjne – jedno dla grupy alkilowej a drugie do związania
nienasyconego substratu.
Aktywność katalizatora rośnie w
kierunku M = Ti << Hf < Zr
Istotnym czynnikiem jest struktura geometryczna kompleksu. Podstawniki
cyklopentadienylowe umożliwiają atak olefiny tylko z jednej strony.

Insercja
alkinów
Insercja alkinów do wiązania M-C może być poprzedzona substytucją
ligandów.
Ni
O
O
CH
3
PPh
3
PhC CPh
-
PPh
3
Ni
O
O
CH
3
C
C
Ph
Ph
PPh
3
C
C
Ph
H
3
C
Ni
O
O
PPh
3
Ph
Kompleksy alikilidenowe i
alkilidynowe
Najszerzej stosowaną metodą syntezy kompleksów alkilidenowych jest
wykorzystanie karbonylków metali w reakcji z nukleofilami.
Kompleks karbenowy
Fischera
Reakcja kompleksu karbenowego Fischera z halogenkami boru i glinu
prowadzi do oderwania podstawnika zawierającego heteroatom na atomie
węgla. Układ M-C-C jest liniowy.
(OC)
5
W-CO (OC)
5
W=C
LiR
O
-
Li
+
R
Me
3
O
+
BF
4
-
- LiBF
4
(OC)
5
W=C OMe
R
(OC)
5
W=C OMe
R
Kompleks karbynowy Fischera
+ BX
3
(OC)
5
W=C
X
R
X W C R
CO
CO
OC
OC
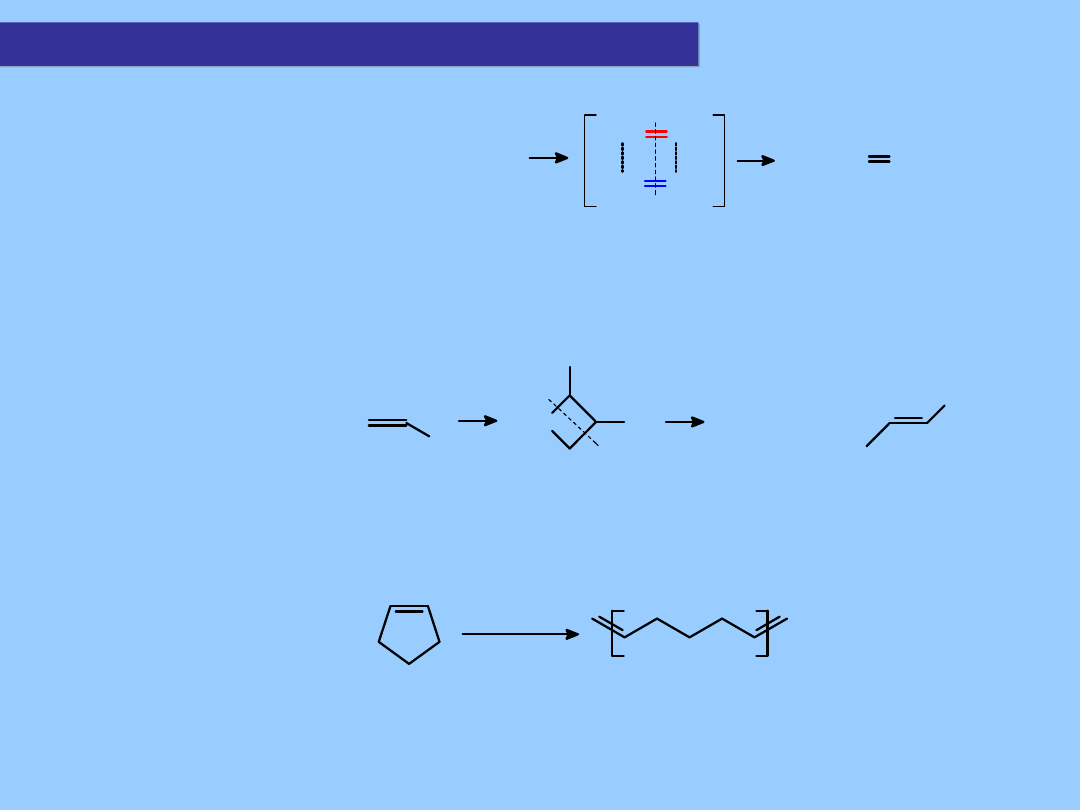
Metateza olefin – a kompleksy alkilidenowe:
Metateza olefin – a kompleksy alkilidenowe:
Reakcja metatezy:
RCH=CHR
+
R'CH=CHR'
RCH CHR
R'CH CHR'
RCH
CHR'
2
Rozwój chemii połączeń alkilidenowych miał fundamentalne znaczenie dla
poznania reguł tej raczej niezwykłej reakcji.
Mechanizm (Chauvin) z użyciem katalizatorów alkilidenowych – zachodzi przez
układy metalacykliczne:
L
n
M=CH
R
+
R'
L
n
M
R
R'
L
n
M=CH
2
+
R'
R
Dobre zrozumienie tego mechanizmu w latach ’70 umożliwiło opracowanie
katalizatorów alkilidenowych do syntezy poliolefin metodą polimeryzacji z
otwarciem pierścienia (ROMP – ring opening metathesis polymerisation):
[cat.] = [Mo(=CHBu
t
)(NAr)(OR)
2
]
[cat.]
n
(Schrock)

Metateza
Kompleksy alkalidenowe molibdenu (np. Mo(NAr)(OR)
2
) (Schrock) okazały
się bardzo aktywne jako katalizatory ROMP. Druga grupa pochodnych to
kompleksy rutenu (Grubbs).
Ru
Cl
Cl
PPh
3
PPh
3
Ph
Ph
Winylidenowy
kompleks rutenu
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
Wyszukiwarka
Podobne podstrony:
Chemia materiałów V
zadania - stężenia, Notatki i materiały dodatkowe, Chemia, materiały od Romka
Chemia materiałów budowlanych, Studia e Liceum, Chemia, Materiałów budowlanych
mater. - wskaźniki, chemia, materiały do lekcji
masalski,chemia materiałow, Materiały światłoczułe
chemia materiałów zagadnienia
chemia material cwiczeniowy 2013 pr model
masalski, chemia materiałów, GALWANO TECHNIKA
zadania powtrkowe- procesy redoks elektrochemia, Chemia, Materiały do korepetycji w liceum - p. rozs
CHEMIA Z MATERIAŁOZNASTWEM wyk1
POWŁOKI NIKLOWE, Chemia materiałów
Opracowanie pytań do wykładu o warstwach, PWR, Chemia Materiałów Inżynieria Materiałowa
korki chemia materiały, redox zadania
Chemia materiałów III
Chemia materiałów II
Chemia materialow 10L
korki chemia materialy reakcje Nieznany
mater. - zadania z gęstości 1d wsp, chemia, materiały do lekcji
ściąga na chemie [Jasiorski], Mechanika i Budowa Maszyn PWR MiBM, Semestr I, Chemia materiałów
więcej podobnych podstron