

Leki po wchłonięciu się z miejsca
zastosowania i przedostaniu się do
krwiobiegu ulegają tu częściowemu związaniu
z białkiem osocza krwi, głównie z
albuminami.
Stopień wiązania zależy przy tym od budowy
cząsteczki leku oraz od stężenia białka.
Wiązanie leku z białkiem jest jednym z
podstawowych czynników warunkujących siłę
i czas trwania działania farmakologicznego
danego leku.

Część leku związana z białkiem staje się
biologicznie nieczynna.
W takiej postaci lek nie może przenikać przez
bariery ustrojowe, a tym samym wchodzić w
reakcje z miejscami receptorowymi w
odpowiednich narządach.
Nie metabolizuje się, ani też w tej postaci nie
jest wydalany z organizmu.
Proces wiązania z białkiem przedłuża
natomiast działanie farmakologiczne na skutek
stopniowego uwalniania leku z kompleksu.

Wiązanie leku z białkiem osocza jest na
ogół reakcją odwracalną, podlegającą
prawu zachowania mas.
Trwałość kompleksu lek-białko zależy od
typu wiązania zachodzącego między
cząsteczką białka a cząsteczką leku. Znane
są różnego typu wiązania: kowalencyjne,
tworzenie chelatów i wiązania jonowe.

Kompleks lek-białko może ulec rozerwaniu
pod wpływem różnych czynników a
uwolniona frakcja czynnego leku może
wiązać się z odpowiednim receptorem,
inicjując swoistą dla siebie reakcję.
Liczba miejsc w cząsteczkach białek, które
są zdolne do wiązania leków, jest
ograniczona, dlatego też jedynie określona
ilość wprowadzonego leku może je wysycić.

Teoria kompartmentowa rozmieszczania
leków w organizmie zakłada podział
organizmu na 2 główne kompartmenty:
wewnątrzkomórkowy i pozakomórkowy.
W kompartmencie pozakomórkowym
wyróżnia się ponadto kompartment
wewnątrznaczyniowy i śródmiąższowy.
Pod pojęciem kompartmentu centralnego
określa się rozmieszczenie leku we krwi,
natomiast obwodowego - w tkankach.

Ważnym czynnikiem wpływającym na
rozmieszczenie leku w organizmie i
przenikanie go do tkanek jest stopień
ukrwienia danej tkanki.
Leki przenikają szybciej do tkanek silniej
ukrwionych niż do tkanek ukrwionych
słabo.

Przenikanie leku z krwi do mózgu jest
regulowane przez kilka barier:
barierę krew-mózg,
krew-płyn mózgowo-rdzeniowy
płyn mózgowo-rdzeniowy-mózg

Proces przenikania leku przez barierę
naczyniowo-mózgową odbywa się według
ogólnych zasad fizykochemicznych kierujących
przenikaniem przez błony komórkowe.
Oznacza to, że związki małocząsteczkowe
niezwiązane z białkiem, niezjonizowane i
rozpuszczalne w tłuszczach pokonują barierę
naczyniowo-mózgową łatwiej niż związki o
dużej cząsteczce, związane z białkiem,
zjonizowane i rozpuszczalne w wodzie.

W związku z tym stężenie niektórych
związków, np. alkoholu etylowego i
tiopentalu, osiąga w mózgu bardzo szybko
wartości stężenia w osoczu, podczas gdy
dla innych związków, np. insuliny, kwasu
sulfanilowego, metotreksatu, stosunek
stężenia w mózgu i w osoczu nigdy nie jest
większy od 0,1.

Dzieląc całkowitą wchłoniętą dawkę leku
przez jego stężenie we krwi uzyskuje się
wartość mającą wymiar objętości.
Jeśli lek jest rozmieszczony równomiernie
we wszystkich płynach ustrojowych, to
wielkość ta opowiada objętości, w której
jest rozpuszczony lek, a więc rzeczywistej
objętości dystrybucji.

Jednak rozmieszczenie leku jest z reguły
nierównomierne, więc uzyskana wielkość
zwykle nie odpowiada rzeczywistej
objętości dystrybucji i jest wartością
fikcyjną.
Mimo to ten parametr, jako tzw. pozorna
objętość dystrybucji może udzielić
informacji o sposobie rozmieszczenia leku
w organizmie.

Pozorną objętość dystrybucji można
określić również jako objętość płynu, w
której należałoby rozpuścić wchłoniętą
dawkę leku, aby uzyskać takie stężenie jak
we krwi. Objętość tę można wyrazić w
litrach, w litrach na kilogram masy ciała,
jako odsetek ogólnej objętości płynów
ustrojowych bądź jako odsetek masy ciała.

W tym ostatnim przypadku można posłużyć
się wzorem:
V
d
[%]
=
Dawka leku [g] x 100
Stężenie w surowicy [g/l] x masa
ciała [kg]
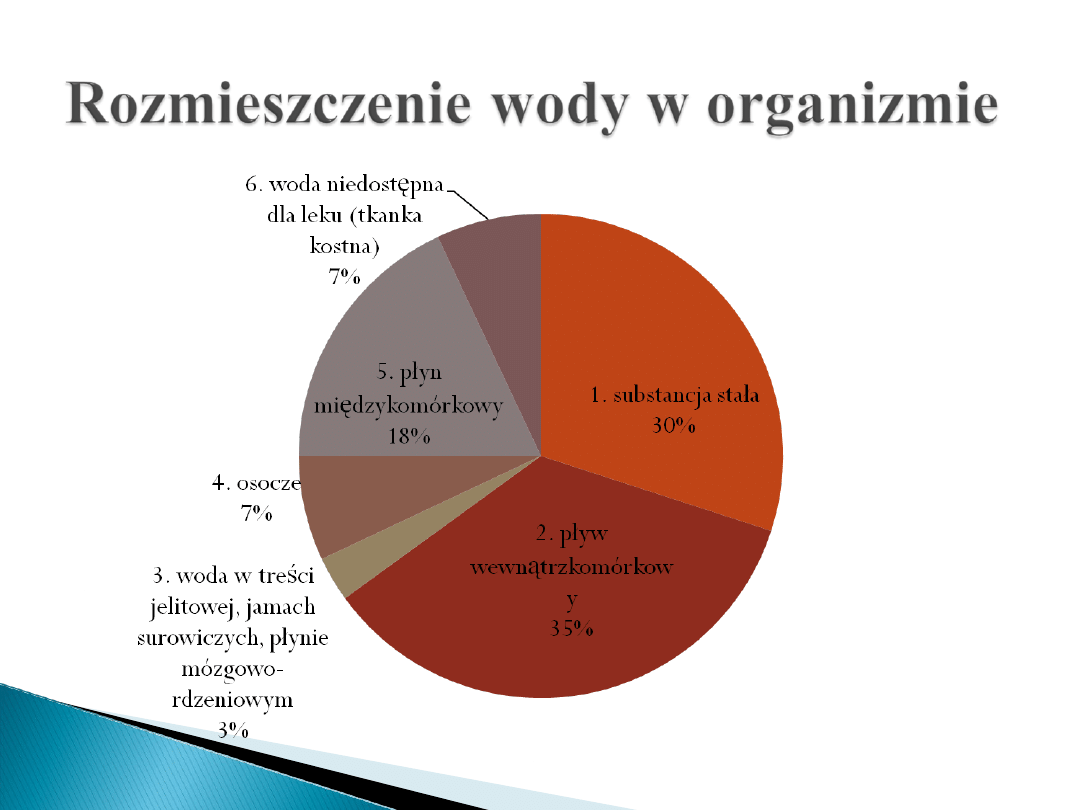

Jeśli objętość dystrybucji wyniesie ok. 7%
masy ciała, to można przypuszczać, że lek
nie jest w znaczących ilościach
transportowany poza łożysko naczyniowe.
Podobnie, objętość dystrybucji zbliżona do
25 lub 75% wskazuje, że lek jest
rozmieszczony we wszystkich płynach
ustrojowych.

Pozorna objętość dystrybucji może
przekraczać (nieraz bardzo znacznie)
całkowitą objętość płynów ustrojowych.
Sytuacja taka występuje wówczas, gdy lek
jest gromadzony w jakimś kompartmencie,
np. w tkance tłuszczowej.

Wydalanie przez nerki
Wydalanie pozanerkowe

Szybkość wydalania leków z moczem zależy
od
◦
wielkości klirensu nerkowego,
◦
stopnia wiązania leku z białkami osocza,
◦
szybkości procesów biotransformacji,
◦
zakresu wydalania leku drogami pozanerkowymi i
jego rozmieszczenia w organizmie.

W kłębuszkach ulegają przesączeniu
wszystkie znajdujące się w osoczu leki
niezwiązane z białkiem, niezależnie od ich
właściwości fizykochemicznych.
Do przesączu mogą przenikać przez pory
śródbłonka naczyń włosowatych nawet
duże cząsteczki związków, o m.cz. do 60
000.

Z żółcią
Ze śliną
Przez błonę śluzową jelit
Przez płuca
Przez skórę
Z potem
z mlekiem

Proces wydalania leków z żółcią odbywa się
częściowo wskutek dyfuzji prostej, częściowo
wskutek przenoszenia czynnego.
W ten sposób przenoszone są z krwi do żółci
niektóre antybiotyki, np. penicylina, tetracykliny,
makrolidy, chloramfenikol, środki cieniujące.
Osiągają one zazwyczaj bardzo duże stężenie w
żółci, przekraczające znacznie ich stężenie we krwi.
Leki lipofilowe są wydalane z żółcią do jelit, skąd
zostają ponownie wchłonięte do krwi. Proces ten
może się odbywać do chwili, aż lek ulegnie, w
wyniku biotransformacji, przemianom
umożliwiającym wydalanie go z organizmu.

Wydalanie leków ze śliną zależy od wielkości
cząsteczki, stopnia jonizacji, rozpuszczalności w
tłuszczach.
Przez gruczoły ślinowe są wydalane w znikomych
ilościach związki jodu, ołowiu, rtęci, bizmutu. Mogą
one drażnić w zatruciu przewlekłym błonę śluzową
jamy ustnej, tworząc ciemny osad na brzegu dziąseł.
Ze śliną są ponadto wydalane związki salicylowe i
niektóre alkaloidy.
Wydalanie leków ze śliną ma raczej większe
znaczenie dla toksykologii, zwłaszcza w przypadku
związków metali ciężkich, niż dla farmakologii.

Przez błonę śluzową jelit są wydalane
związki metali ciężkich (żelazo, rtęć,
bizmut), które z kałem zostają usunięte z
organizmu.
Morfina zresorbowana z jelita cienkiego
dostaje się z krwi do żołądka, a stąd do
jelita cienkiego, gdzie ponownie ulega
wchłonięciu.

Przez płuca są wydalane głównie związki
lotne, jak alkohol, etery, chloroform i jego
homologi, aceton, kwas octowy, fenol,
kamfora, olejki eteryczne, kreozot,
gwajakol.

Szybkość wydalania zależy od różnicy
ciśnienia cząsteczkowego gazu w
pęcherzykach płucnych i we krwi.
Związki trudno rozpuszczalne we krwi, jak
benzen, disiarczek węgla i inne, są wydalane
szybko, w ciągu 1 lub kilku godzin, natomiast
dobrze rozpuszczalne, jak alkohole, są
wydalane powoli, nawet do kilku dni.
Duży wpływ na wydalanie przez płuca ma
również wielkość przepływu krwi przez płuca i
stopień wentylacji płuc.

Przez skórę leki są wydalane głównie z
potem - do nich należą związki bromu, fenol i
inne.
Powodują one przy tym podrażnienie skóry,
wywołując niekiedy stany zapalne skóry,
rumień, trądzik, wysypki, swędzenie.
Objawy powyższe, jak również nieżyt błon
śluzowych, występują po zastosowaniu
wymienionych związków w większych
ilościach (zatrucia) lub na tle uczuleniowym.

Z potem jest wydalana w dużym stopniu
witamina B, i jej metabolity, nadając potowi
charakterystyczny zapach drożdży.

Wydalanie leków z mlekiem ma duże
znaczenie ze względu na stosunkowo łatwe
ich przechodzenie przez nabłonek
pęcherzyków gruczołu sutkowego, który
odgrywa rolę błony lipidowej dla wielu leków
i związków szkodliwych przenikających z
krwi do mleka.
Związki o charakterze słabych zasad łatwiej
przenikają do mleka, osiągając stężenie
nawet większe niż w osoczu, natomiast leki
kwaśne przenikają do mleka gorzej.

Leki nieulegające jonizacji są rozdzielane
równomiernie między krew a mleko.
Leki przechodzą do mleka stosunkowo
szybko, zależnie od sposobu podania,
właściwości leku i innych czynników
pojawiają się w mleku po kilkunastu
minutach do godziny.
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
Wyszukiwarka
Podobne podstrony:
2 c Dystrybucja leków w organizmie
(5) Dystrybucja leków w organizmie(1)
Losy leków w organizmie
6 LOSY LEKÓW W ORGANIZMIE
Molekularne aspekty działania leków Wchłanianie i dystrybucja leków
(3) Zaburzenia biotransformacji leków w organizmie
LOSY LEKÓW W ORGANIZMIE
Losy leków w organizmie
Nauka o lekach Działanie leków na organizm
2 WŁAŚCIWOŚCI LEKÓW I RODZAJE REAKCJI ORGANIZMU NA ICH DZIAŁANIE
Czynniki ograniczajace rozmieszczenie organizmow
13, Kanały dystrybucji są to zbiory wzajemnie zależnych od siebie różnych organizacji, instytucji i
więcej podobnych podstron