
WYKŁAD 7
• Potencjał chemiczny
• Równowaga fazowa – współczynnik podziału Nernsta
• Kataliza, reakcje enzymatyczne
Zakład Farmakokinetyki i Farmacji Fizycznej, 2004

POTENCJA
POTENCJA
Ł
Ł
CHEMICZNY
CHEMICZNY
]
mol
J
[
1
i
j
n
p,
T,
i
i
n
G
μ
CZĄSTKOWA MOLOWA ENTALPIA SWOBODNA
Potencjał chemiczny substancji (
i
) przedstawia udział
wnoszony
przez tę substancję w całkowitą entalpię swobodną
układu.

G = f (T, p, n)
Dla układu jednofazowego dwuskładnikowego (np. roztwór składający
się z rozpuszczalnika A i substancji rozpuszczonej B):
G = f (T, p, n
1
, n
2
)
A
B
n
p,
T,
A
A
μ
n
G
G
B
A
n
p,
T,
B
B
μ
n
G
G
Całkowita entalpia swobodna roztworu:
G = n
A
G
A
+ n
B
G
B
G = n
A
A
+ n
B
B
A ,
B
-
potencjały chemiczne przy danym składzie mieszaniny

Dla układu dwuskładnikowego równanie:
dG = V dp - S dT
przybiera postać:
dG = V dp - S dT +
A
dn
A
+
B
dn
B
maks.
e,
W
dG
Jeżeli T, p = const
dG =
A
dn
A +
B
dn
B
maks.
e,
W
Zatem:
=
A
dn
A
+
B
dn
B
Praca nieobjętościowa może być spowodowana zmianą składu
chemicznego układu.

Dla mieszaniny dwuskładnikowej:
G = n
A
A
+ n
B
B
ponieważ potencjały chemiczne zależą od składu, więc gdy skład
ulega zmianie:
dG =
A
dn
A
+
B
dn
B
+ n
A
d
A
+
n
B
d
B
Jeżeli T, p = const to:
dG =
A
dn
A +
B
dn
B
A
dn
A
+
B
dn
B
+ n
A
d
A
+
n
B
d
B
=
A
dn
A +
B
dn
B
n
A
d
A
+
n
B
d
B
= 0
RÓWNANIE GIBBSA – DUHEMA
RÓWNANIE GIBBSA – DUHEMA
0
μ
d
n
i
i
i
Potencjał chemiczny jednego ze składników mieszaniny
nie może się zmieniać w sposób niezależny od
potencjałów chemicznych pozostałych składników.
A
B
A
B
μ
d
n
n
-
μ
d

Potencjał chemiczny gazu doskonałego
Potencjał chemiczny gazu doskonałego
pod ci
pod ci
ś
ś
nieniem
nieniem
p:
p:
Θ
Θ
p
p
ln
T
R
μ
μ
μ
Θ
- standardowy potencjał chemiczny, czyli
molowa entalpia swobodna czystego gazu
pod ciśnieniem 1 atm ( 10
5
Pa )
= f (T, p)
μ
Θ
Dla mieszaniny gazów A i B:
A
Θ
A
A
p
ln
T
R
μ
μ
B
Θ
B
B
p
ln
T
R
μ
μ
A ,
B
-
potencjały chemiczne składników A i B w mieszaninie lub roztworze

Potencjał chemiczny składnika mieszaniny ciekłej:
Potencjał chemiczny składnika mieszaniny ciekłej:
= f (T, p) - standardowy potencjał chemiczny
(a = 1)
i
Θ
i
a
ln
T
R
μ
μ
i
μ
Θ
i
i
Θ
i
f
X
ln
T
R
μ
μ
i
i
i
Θ
i
f
ln
T
R
X
ln
T
R
μ
μ
i
Ponieważ a
i
= X
i
f
i
X
ln
T
R
μ
μ
i
Θ
i
i
roztwór idealny (f
i
= 1):
c
ln
T
R
μ
μ
i
Θ
i
i
roztwór rozcieńczony:
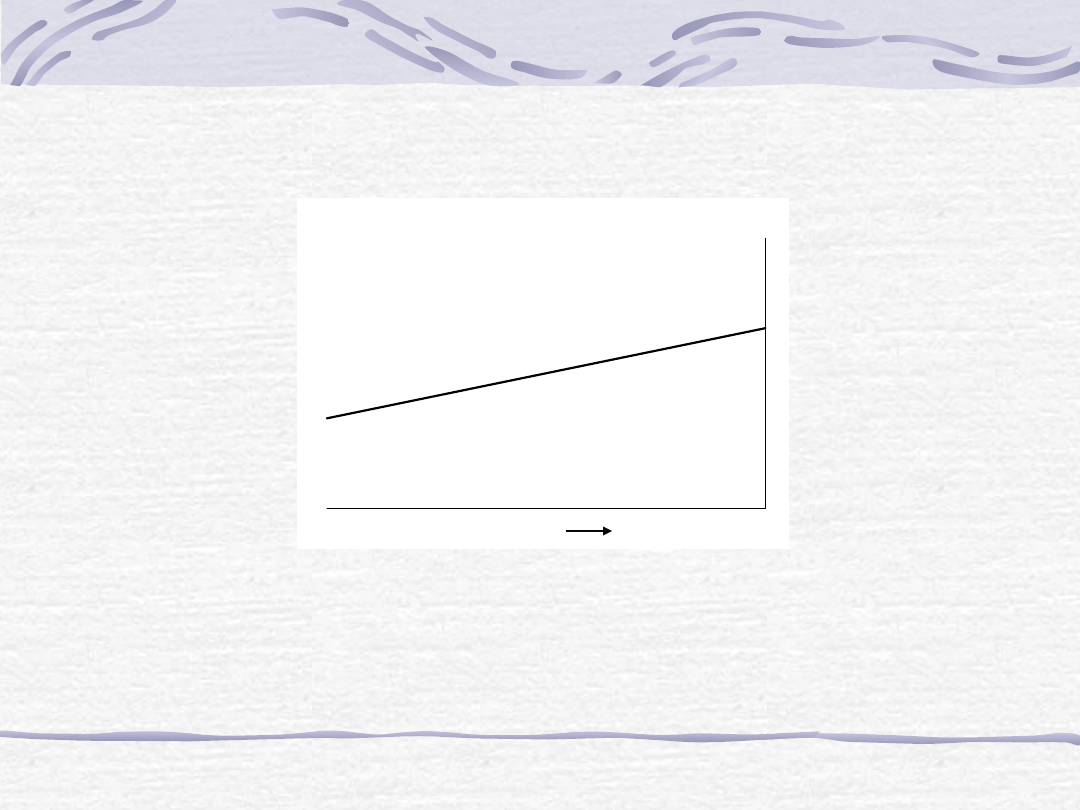
Potencjał
chemiczny
składnika
roztworu
doskonałego (
i
) jako funkcja jego ułamka
molowego X
i
lnX
i
μ
Θ
i
i
μ
0
X
ln
T
R
μ
μ
i
Θ
i
i

Potencjał chemiczny charakteryzuje zdolność danego składnika układu
do opuszczania jednej z faz układu.
W samorzutnie zachodzących przemianach fazowych
(parowanie, krzep-
nięcie, krystalizacja), przy powstawaniu roztworów i w
przebiegu reakcji
chemicznych określony składnik układu może przejść
jedynie z fazy układu o wyższym potencjale chemicznym do
fazy o niższym potencjale.
W stanie równowagi potencjały chemiczne każdego składnika
układu
są we wszystkich fazach jednakowe.
W procesie samorzutnym:
i
· dn
i
< 0
natomiast w stanie równowagi:
i
· dn
i
= 0
gdzie: dn
i
– przyrost liczby moli składnika układu

RÓWNOWAGA FAZOWA W UKŁADACH
RÓWNOWAGA FAZOWA W UKŁADACH
WIELOSKŁADNIKOWYCH, WIELOFAZOWYCH
WIELOSKŁADNIKOWYCH, WIELOFAZOWYCH
TERMODYNAMICZNE KRYTERIUM RÓWNOWAGI
W stanie równowagi potencjał chemiczny substancji w całym
układzie jest taki
sam, bez względu na liczbę faz.
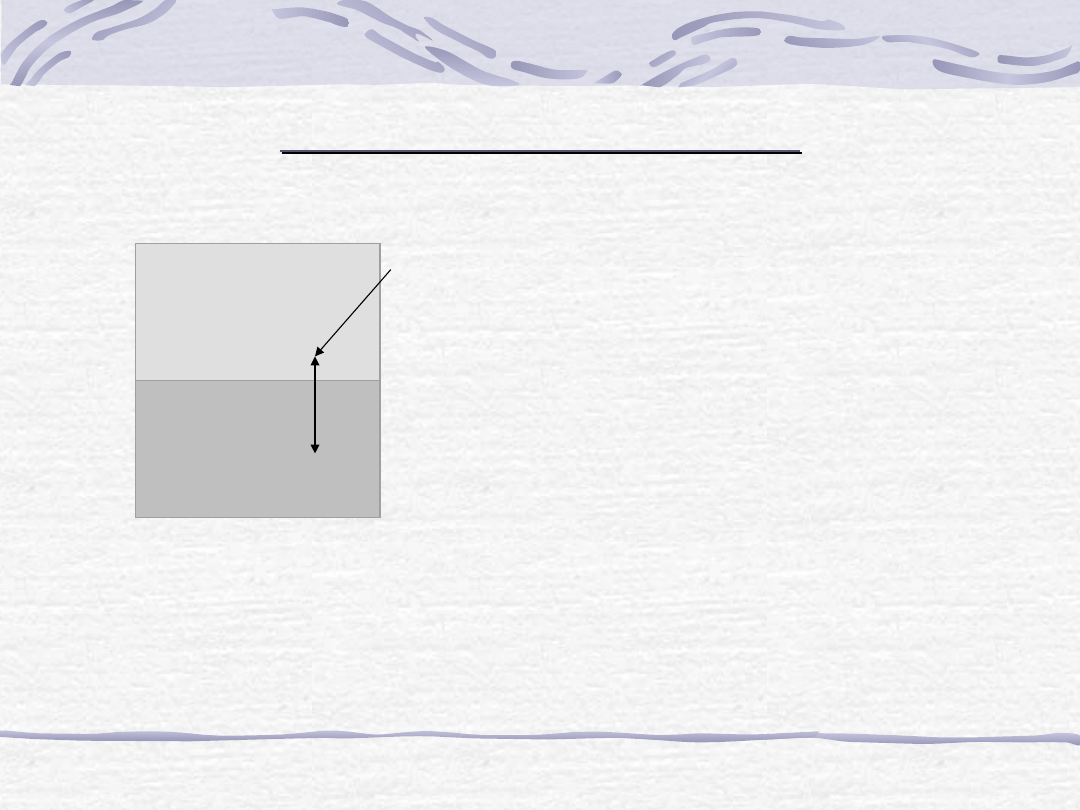
faza ciekła
faza ciekła
PRAWO PODZIAŁU NERNSTA
PRAWO PODZIAŁU NERNSTA
opisuje rozdział substancji między dwie nie mieszające się ze sobą fazy ciekłe.
substancja
( i )
W stanie równowagi fazowej, dla T, p =
const:
- standardowy potencjał chemiczny
składnika i
odpowiednio w fazie i
- aktywność składnika i odpowiednio
w fazie i
β
i
α
i
μ
μ
α
i
α
Θ
i
α
i
a
ln
T
R
μ
μ
β
i
β
Θ
i
β
i
a
ln
T
R
μ
μ
μ
,
μ
β
i
α
i
a
,
a
β
i
α
i

=
β
i
α
i
μ
μ
α
i
α
Θ
i
a
ln
T
R
μ
β
i
β
Θ
i
a
ln
T
R
μ
μ
-
μ
a
ln
T
R
-
a
ln
T
R
α
Θ
i
β
Θ
i
β
i
α
i
T
R
μ
-
μ
a
ln
-
a
ln
α
Θ
i
β
Θ
i
β
i
α
i
T
R
μ
-
μ
a
a
ln
α
Θ
i
β
Θ
i
β
i
α
i
T
R
μ
-
μ
β
i
α
i
α
Θ
i
β
Θ
i
e
a
a
W warunkach T, p = const:
czyli:
const
T
R
μ
-
μ
α
Θ
i
β
Θ
i
K
const
e
T
R
μ
-
μ
α
Θ
i
β
Θ
i
K
a
a
β
i
α
i
a
a
K
β
i
α
i
β
/
α

Roztwór idealny (f
i
= 1):
X
X
K
i
i
/
WSPÓŁCZYNNIK PODZIAŁU NERNSTA JEST RÓWNY
ILORAZOWI STĘŻEŃ SUBSTANCJI ROZPUSZCZONEJ W DWÓCH
NIEMIESZAJĄCYCH SIĘ FAZACH CIEKŁYCH, W STANIE
RÓWNOWAGI FAZOWEJ, W STAŁEJ TEMPERATURZE.
c
c
K
β
i
α
i
β
/
α
Roztwór rozcieńczony:

Równania słuszne gdy:
ciecze nie mieszają się ( lub mieszają się ograniczenie )
stężenie substancji rozpuszczonej w obu fazach nie jest duże
( roztwory rozcieńczone )
nie zachodzi dysocjacja ani asocjacja cząsteczek substancji
rozpuszczonej w żadnej z faz
stała temperatura
Wartość współczynnika podziału ( K ):
zależy od rodzaju składników układu (cieczy i substancji) oraz od
temperatury
nie zależy od ilości substancji rozpuszczonej i objętości cieczy

WSPÓŁCZYNNIK PODZIAŁU OLEJ / WODA
WSPÓŁCZYNNIK PODZIAŁU OLEJ / WODA
(
(
K
K
O/W
O/W
)
)
c
c
K
w
o
w
/
o
C
o
- stężenie substancji w fazie olejowej (niepolarnej)
C
w
- stężenie substancji w fazie wodnej (polarnej)
dysocjacja cząsteczek substancji w fazie wodnej:
- stopień dysocjacji
α
-
1
c
c
K
w
o
w
/
o
asocjacja cząsteczek substancji w fazie olejowej:
n - stopień asocjacji
c
c
K
w
n
o
w
/
o
α
-
1
c
c
K
w
n
o
w
/
o
dysocjacja cząsteczek substancji w fazie wodnej i asocjacja w
fazie olejowej:

ZASTOSOWANIE WSPÓŁCZYNNIKA PODZIAŁU NERNSTA
ZASTOSOWANIE WSPÓŁCZYNNIKA PODZIAŁU NERNSTA
• określanie właściwości substancji
> 1
substancja ma charakter lipofilny (c
o
>
c
w
)
(większe powinowactwo do fazy olejowej)
< 1
substancja ma charakter hydrofilny (c
o
< c
w
)
(większe powinowactwo do fazy wodnej)
w
/
o
K
w
/
o
K
• QSAR (Quantitative Structure-Activity Relationships)
• proces ekstrakcji
(przewidywanie najkorzystniejszych warunków ekstrakcji - dobór
odpowiedniego rozpuszczalnika, przewidywanie stopnia rozdziału
składników)
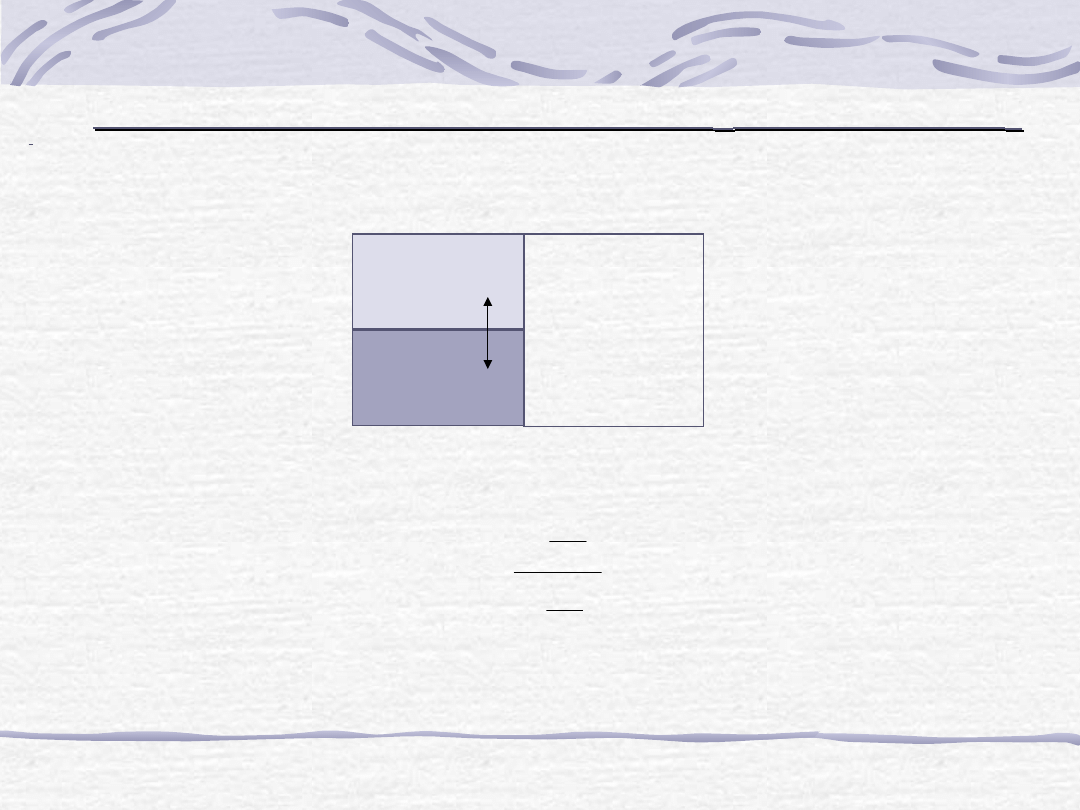
Ekstrakcja substancji z fazy ciekłej
Ekstrakcja substancji z fazy ciekłej
do fazy ciekłej
do fazy ciekłej
m
o
– początkowa masa substancji w fazie
Zgodnie z prawem podziału Nernsta:
m
,
m
–
masa substancji zawarta odpowiednio w fazie
i (w stanie
równowagi fazowej)
v
,
v
– objętość fazy i
β
β
α
α
β
/
α
v
m
v
m
K
stan równowagi
faza ciekła
m
v
faza ciekła
m
v

Po jednorazowej ekstrakcji:
m
1
– masa substancji w fazie
(m
0
- m
1
) - masa substancji w fazie
po przekształceniach:
Po kolejnych ekstrakcjach w fazie
pozostaje m
2,
m
3
... m
n
gramów substancji.
K
v
m
-
m
v
m
β
/
α
β
1
ο
α
1
β
α
β
/
α
α
β
/
α
ο
1
v
v
K
v
K
m
m
v
v
K
v
K
m
m
n
β
α
β
/
α
α
β
/
α
ο
n
Ilość przeprowadzonej substancji z fazy do zależy od liczby
powtarzanych
operacji
(ekstrakcji) oraz od objętości użytych cieczy. Większą wydajność ekstrakcji
osiąga się przy wielokrotnie powtarzanym wytrząsaniu małymi porcjami
fazy .
Masa substancji pozostała w fazie
po n – krotnej
ekstrakcji:
EKSTRAKCJA
EKSTRAKCJA
to
to
proces wyodrębniania substancji z
proces wyodrębniania substancji z
jednej fazy do
jednej fazy do
drugiej
drugiej
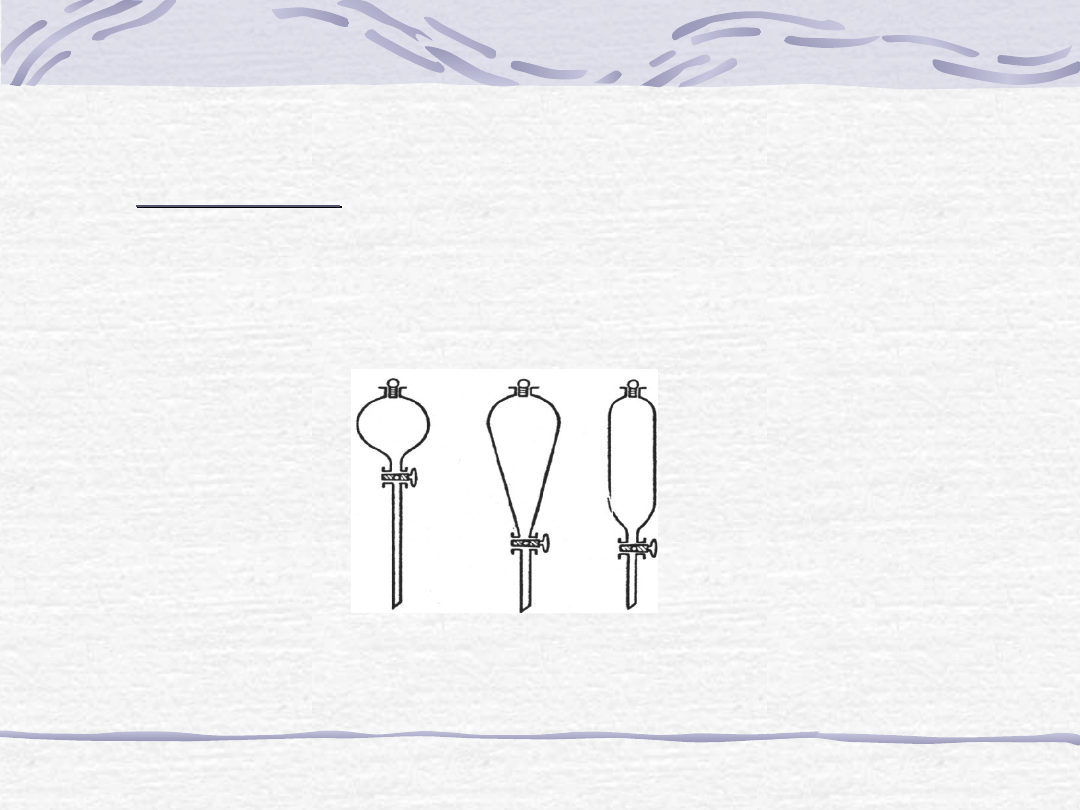
Ekstrakcja prosta (w rozdzielaczach):
Różne typy rozdzielaczy
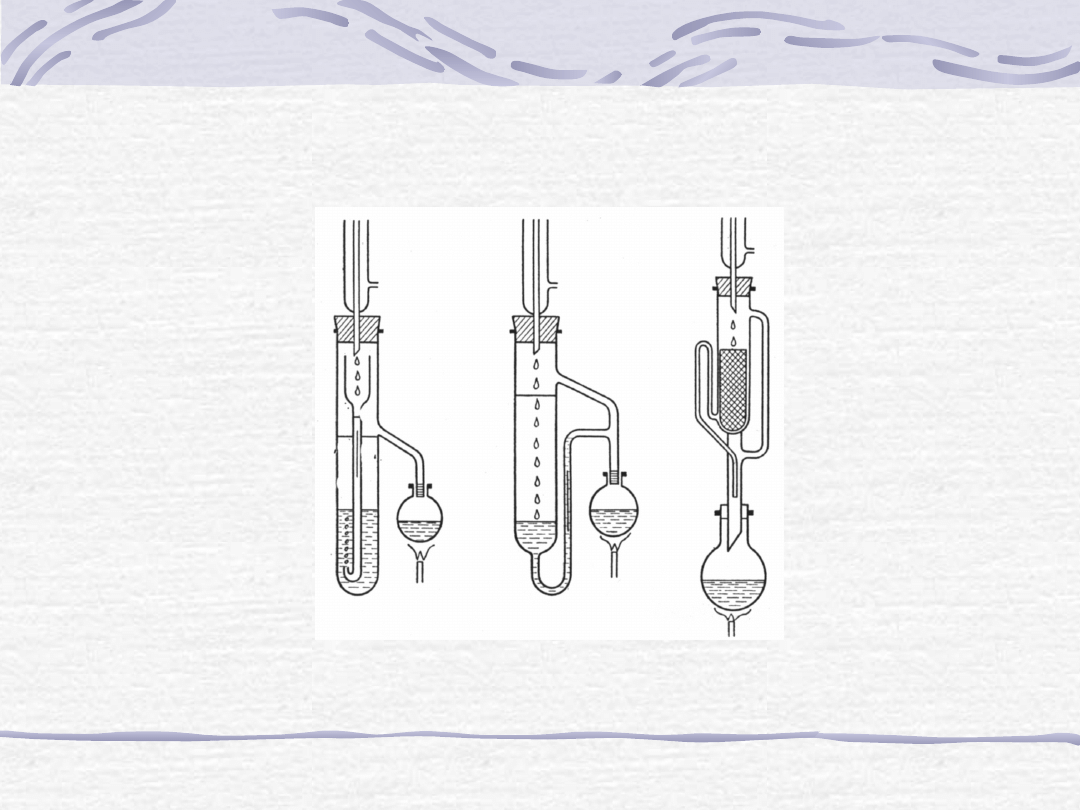
Ekstrakcja ciągła (w ekstraktorach):
Aparat do ekstrakcji ciągłej za pomocą cieczy lżejszej od wody (a), cieczy cięższej od wody (b)
oraz aparat Soxhleta do ekstrakcji substancji z ciał stałych (c).
a
b
c

KATALIZA
KATALIZA
W uk
W uk
ł
ł
adzie homogenicznym
adzie homogenicznym
(gdy katalizator oraz substraty i produkty reakcji stanowi
(gdy katalizator oraz substraty i produkty reakcji stanowi
ą
ą
jedną fazę
jedną fazę
np. utlenianie SO
np. utlenianie SO
2
2
wobec tlenków azotu, powstawanie eteru
wobec tlenków azotu, powstawanie eteru
etylowego
etylowego
wobec H
wobec H
2
2
SO
SO
4
4
)
)
W uk
W uk
ładzie heterogenicznym
ładzie heterogenicznym
(gdy katalizator stanowi odrębną fazę, przy czym
(gdy katalizator stanowi odrębną fazę, przy czym
najczęściej jest on ciałem stałym i nosi nazwę kontaktu np.
najczęściej jest on ciałem stałym i nosi nazwę kontaktu np.
czerń platynowa podczas ulteniania SO
czerń platynowa podczas ulteniania SO
2
2
, żelazo w procesie
, żelazo w procesie
konwersji gazu wodnego)
konwersji gazu wodnego)
W układzie mikroheterogenicznym
W układzie mikroheterogenicznym
(gdy heterogeniczny katalizator znajduje się w stanie
(gdy heterogeniczny katalizator znajduje się w stanie
rozdrobnienia koloidalnego np. enzym)
rozdrobnienia koloidalnego np. enzym)

Apoenzym + koenzym = holoenzym (E)
E + S
ES P + E
k
1
k
3
k
2
v
1
=k
1
[E-ES][S]
v
2
=k
2
[ES]
v
3
=k
3
[ES]
Ponieważ [ES] tworzy się z szybkością v
1
, a rozpada z z szybkością v
2
+v
3
to w stanie równowagi:
v
1
= v
2
+v
3
k
1
[E-ES][S] = (k
2
+k
3
) [ES]
m
2
K
k
k
k
[ES]
ES][S]
-
[E
1
3
gdzie K
m
to stała Michaelisa
v = v
3
= k
3
[ES]
O szybkości całej reakcji enzymatycznej decyduje v
3
, czyli:
Gdy [S] jest tak duże, że wszystkie cząsteczki E wezmą udział w tworzeniu ES,
czyli gdy [ES] = [E], wówczas:
V
maks
= k
3
[E]
m
K
[ES]
ES][S]
-
[E
Jeśli wyliczymy [ES] i [E] i wstawimy je do równania:
otrzymamy:
[S]
K
[S]
V
v
m
maks
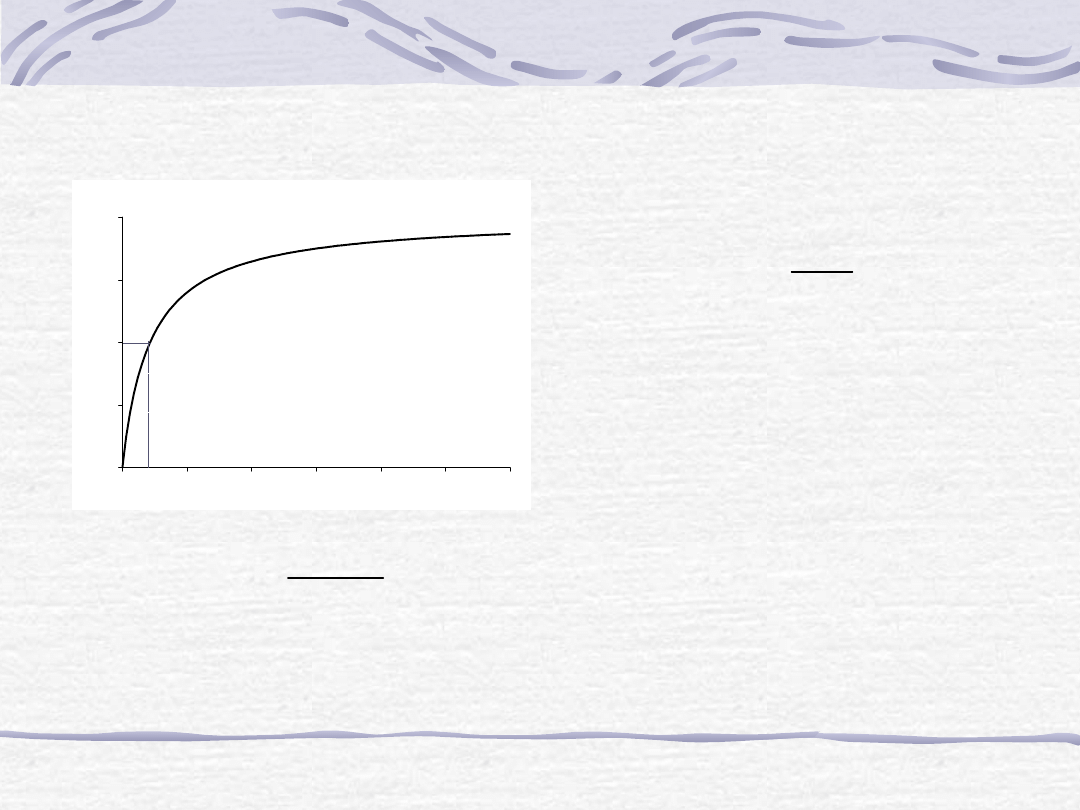
S
K
S
V
v
m
maks
Równanie Michaelisa-Menten
S (mol/l)
0.0
0.1
0.2
0.3
0.4
0.5
0.6
v
(m
m
o
l/l
m
in
)
0.0
0.5
1.0
1.5
2.0
v – szybkość reakcji katalizowanej przez enzym
V
maks
- maksymalna szybkość reakcji
S – stężenie substratu
K
m
– stała Michaelisa, czyli takie [S], przy którym v=1/2V
maks
V
max
=2 mmol/l·min
K
m
=0.043 mol/l
Je
żeli S<<K
m
:
czyli reakcja enzymatyczna przebiega
zgodnie z kinetyką I-go rzędu
Je
żeli S>>K
m
:
jest to reakcja 0-go rzędu, w której szybkość
nie zależy od stężenia
S
K
V
v
m
maks
V
v
maks
K
m
Równanie Lineweavera-Burka
-40
-20
0
20
40
60
80
100
0
1
2
3
maks
maks
m
V
1
S
1
V
K
v
1
maks
V
1
m
K
1
1
/v
(
l·
m
in
/m
m
o
l)
1/S (l/mol)

Czynniki wp
Czynniki wp
ł
ł
ywaj
ywaj
ą
ą
ce na szybko
ce na szybko
ść
ść
reakcji enzymatycznej
reakcji enzymatycznej
• ilość substratu
• temperatura
• pH środowiska
• obecność aktywatorów i inhibitorów enzymów
gdzie: I – stężenie inhibitora
K
i
– stała dysocjacji kompleksu enzym-inhibitor
S
K
S
V
v
m
maks
K
I
1
i
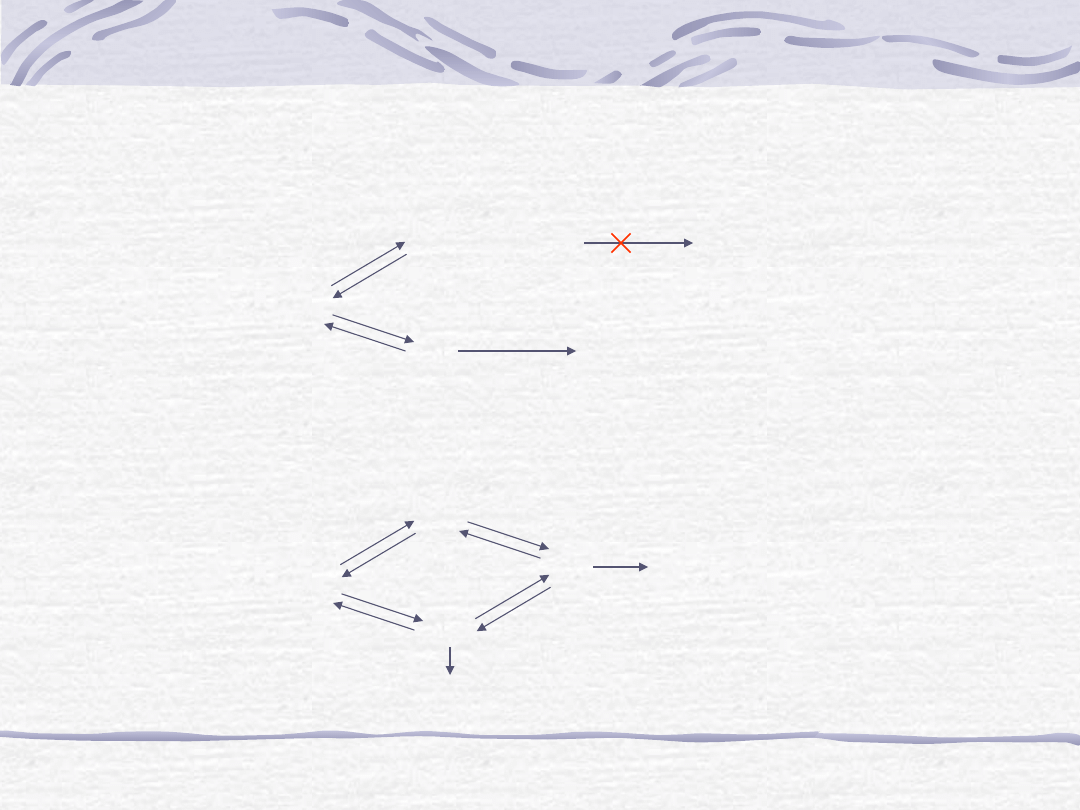
Hamowanie kompetycyjne
E
EI (nieaktywny)
ES
E + P
E + P
S
I
Hamowanie niekompetycyjne
E
EI
ES
E+P
EIS
EI + S
I
S
S
I
Inhibitory enzym
Inhibitory enzym
ów
ów
Inhibitory kompetycyjne
Inhibitory kompetycyjne
Inhibitory niekompetycyjne
Inhibitory niekompetycyjne
Przykłady: allopurynol, azaseryna, neostygmina,
sulfonamidy, metotreksat, 5-fluorouracyl
Przykłady: związki fosforoorganiczne, metale
ciężkie, cyjanki, azydki, związki arsenu
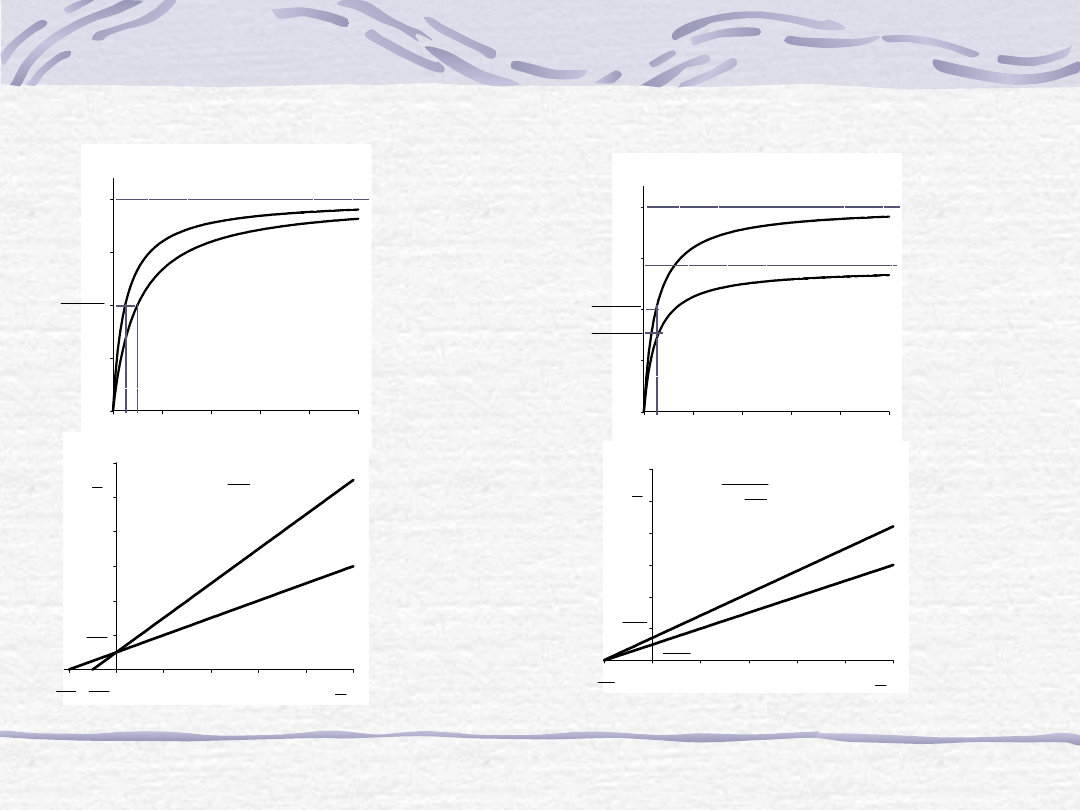
K
m1
K
m2
E
E+I
k
maks
V
E
E
E
E+I
nk
E+I
nk
E+I
k
maks1
V
maks2
V
K
m
)
K
]
I
[
(1
K
K
i
m1
m2
i
maks1
maks2
K
]
I
[
1
V
V
2
maks1
V
2
maks2
V
2
maks
V
m1
K
1
m2
K
1
maks
V
1
m
K
1
maks1
V
1
maks2
V
1
v
v
v
1
v
1
S
S
S
1
S
1
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
Wyszukiwarka
Podobne podstrony:
chfwyklad 2
chfwyklad 8
chfwyklad 4
chfwyklad 3
chfwyklad 2
więcej podobnych podstron