
I zasada termodynamiki
U = q + w
II zasada termodynamiki
T
dq
dS
ENERGIA WEWNĘTRZNA U
ENTALPIA H = U + pV
ENTROPIA S
Nowe funkcje termodynamiczne
Energia swobodna
Helmholtza
F =
U
– TS
TERMODYNAMICZNE FUNKCJE STANU
Entalpia swobodna
Gibbsa
G =
H
- TS
Josiah Willard Gibbs
(1839 –1903)
Hermann Ludwig Ferdinand von Helmholtz
(1821-1894)

Energia swobodna Helmholtza F = U – TS
I zasada termodynamiki : dU = q
el
+ w
el
= đq + đw
II zasada termodynamiki: đq ≤T ∙dS
dU ≤ T dS + đw
dU – T dS ≤ đw
T = const
dU – d(TS) ≤ đw
d ( U – TS) ≤ đw
dF ≤ đw
w = w
obj
+ w
nieobj
V = const
w
obj
= 0 dF ≤ đw
nieobj
T
dq
dS
ΔF = (F
k
– F
p
) ≤ w
nieobj
W warunkach izotermiczno-izochorycznych różnica pomiędzy energią swobodną Helmholtza dla
dwóch stanów układu, stanu koocowego i początkowego, określa pracę, którą układ może
wymienid z otoczeniem.
ΔF jest potencjałem termodynamicznym w procesie izotermiczno-izochorycznym.
w = w
obj
+ w
nieobj

Entalpia swobodna Gibbsa G = H - TS
Entalpia H = U + pV
dH = dU + pdV + Vdp
I zasada termodynamiki: dU = đq+ đw
II zasada termodynamiki: dq ≤T ∙dS
dU ≤ T dS + đ w
dH = dU + pdV + Vdp
dH ≤ T dS + đw + pdV + Vdp ,
p = const
dH ≤ T dS + đw + pdV
dH – T dS ≤ đw+ pdV
T= const
d(H – TS) ≤ đw + pdV
w = w
obj
+ w
nieobj
w
nieobj
= w - w
obj
dw
nieobj
= dw –(-pdV)
dG ≤ đw + pdV
dw + pdV = dw
nieobj
dG ≤ dw
nieobj
T
dq
dS
W warunkach izotermiczno-izobarycznych różnica pomiędzy entalpią swobodną Gibbsa dla dwóch
stanów układu, stanu koocowego i początkowego, określa pracę, którą układ może wymienid z
otoczeniem.
ΔG jest potencjałem termodynamicznym w procesie izotermiczno-izobarycznym.
w = w
obj
+ w
nieobj

ΔF i ΔG jako kryteria określające charakter procesu
Zdolnośd układu do wykonania pracy jest miarą tendencji do SAMORZUTNEGO przebiegu procesu .
Gdy układ nie wykonuje pracy – znajduje się w stanie równowagi
.
Procesy izotermiczno-izochoryczne ΔF ≤ w
nieobj
Ponieważ praca oddana przez układ do otoczenia w procesie samorzutnym jest ujemna, stąd
możliwe jest samorzutne przejście układu ze stanu początkowego do koocowego gdy ΔF < 0,
co oznacza, że F
k
< F
p
w procesach samorzutnych energia swobodna Helmholtza maleje
.
Proces jest odwracalny (układ znajduje się
w stanie równowagi
) gdy F
k
= F
p
, czyli gdy nie ma
zmiany energii swobodnej Helmholtza
ΔF=0
.
Procesy izotermiczno-izobaryczne ΔG ≤ w
nieobj
Ponieważ praca oddana przez układ do otoczenia w procesie samorzutnym jest ujemna, stąd
możliwe jest samorzutne przejście układu ze stanu początkowego do koocowego gdy ΔG < 0,
co oznacza, że G
k
< G
p
w procesach samorzutnych entalpia swobodna Gibbsa maleje.
Proces jest odwracalny (układ znajduje się
w stanie równowagi
) gdy G
k
= G
p
, czyli gdy nie ma
zmiany entalpii swobodnej Gibbsa
ΔG=0
.

Wpływ temperatury i objętości na energię swobodną F
F = U – TS
F= f(T,V)
dV
V
F
dT
T
F
dF
T
V
dF = dU – TdS – SdT
dF = dq + dw –TdS – SdT
Gdy dq =TdS (proces odwracalny) i
dw = -pdV (możliwe jest wykonanie tylko pracy objętościowej)
dF= - SdT
– pdV
Z porównania różniczek funkcji otrzymujemy zależnośd:
p
V
F
oraz
S
T
F
T
V

Wpływ temperatury i ciśnienia na entalpię swobodną G
G= H – TS
G= f(T,p)
dp
p
G
dT
T
G
dG
T
p
dG = dH – TdS – SdT
dG = dU + pdV + Vdp –TdS –SdT
dG = dq + dw + Vdp – TdS - SdT
Gdy dq =TdS (proces odwracalny) i
dw = -pdV (możliwe jest wykonanie tylko pracy objętościowej)
dG = - SdT + Vdp
Z porównania różniczek funkcji otrzymujemy zależnośd:
V
p
G
oraz
S
T
G
T
p
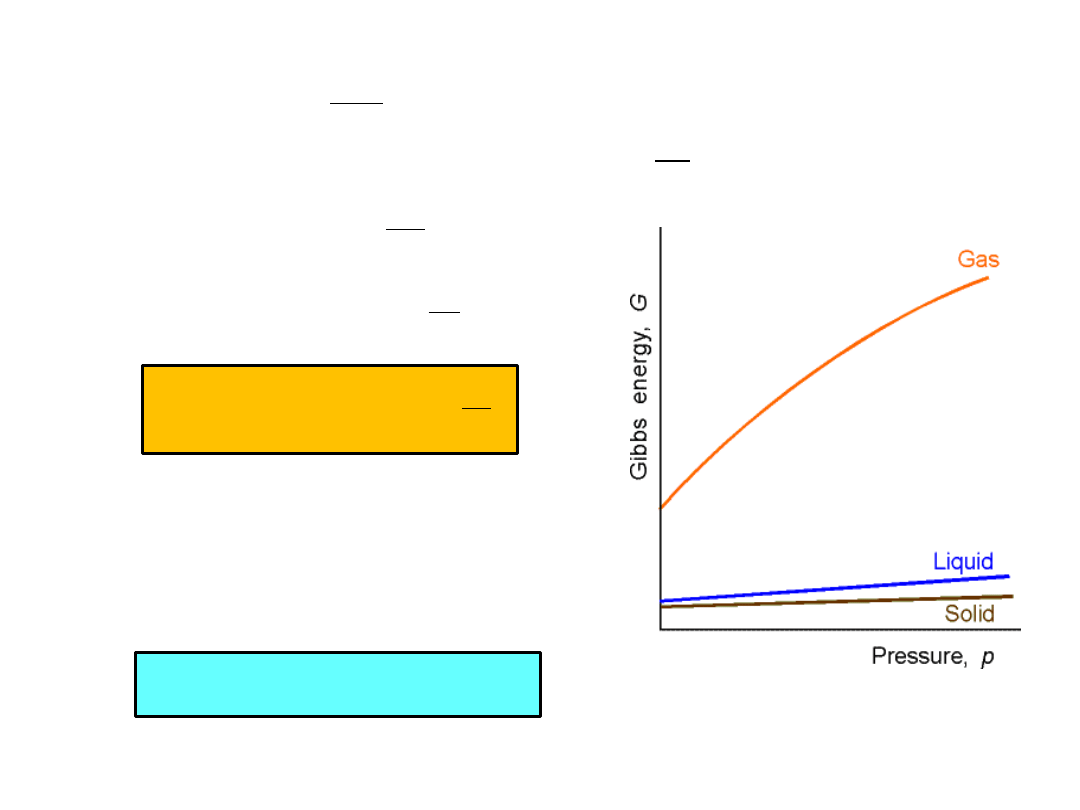
Wpływ ciśnienia na molową entalpię swobodną Gibbsa (T=const)
m
T
m
V
p
G
Gdy gaz spełnia równanie stanu gazu doskonałego
p
RT
V
m
dp
p
RT
dp
V
dG
m
m
2
1
p
p
1
m
2
m
p
dp
RT
)
(p
G
)
(p
G
1
2
1
m
2
m
p
p
ln
RT
)
(p
G
)
(p
G
dla cieczy i ciał stałych V
m
const.
)
p
(p
V
)
(p
G
)
(p
G
1
2
m
1
m
2
m
Entalpia swobodna wzrasta ze wzrostem ciśnienia
2
1
p
p
m
1
m
2
m
dp
V
)
(p
G
-
)
(p
G
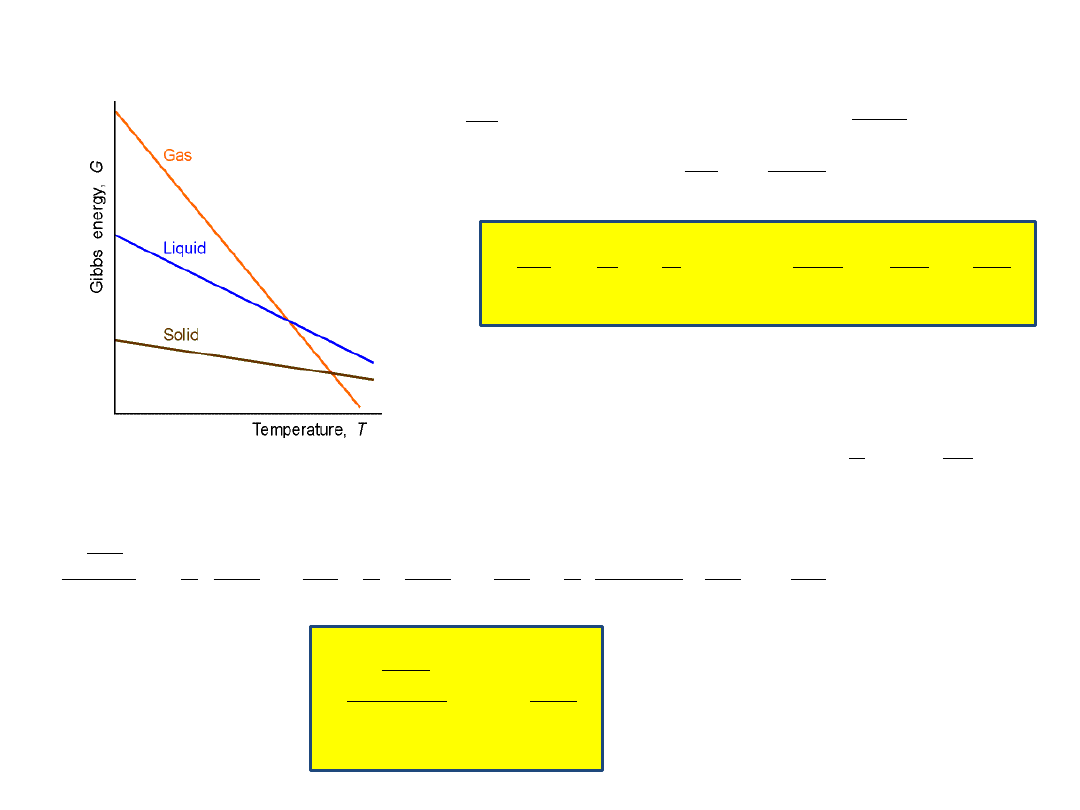
Wpływ temperatury na entalpię swobodną Gibbsa (p=const):
S
T
G
p
G = H
– T S
S
T
H
G
T
H
G
T
G
p
T
ΔH
T
ΔG
T
ΔG
lub
T
H
T
G
T
G
p
p
W procesie izobarycznym (p= const) zmiana entalpii
swobodnej Gibbsa zależy tylko od zmian temperatury
Równania (
) można zapisad w innej formie, wprowadzając nową wielkośd:
(
)
T
ΔG
lub
T
G
Pod stałym ciśnieniem zmiany tych wielkości są skutkiem zmian temperatury, stąd
2
p
2
p
p
T
ΔH
T
ΔG
T
ΔH
ΔG
T
1
T
ΔG
T
ΔG
T
1
T
ΔG
T
ΔG
T
1
T
T
ΔG
2
p
T
ΔH
T
T
ΔG
Równanie Gibbsa- Helmholtza

Funkcje termodynamiczne
Energia wewnętrzna U I zasada termodynamiki dU = q
el
+ w
el
Entalpia H
H = U + pV
Entropia S
II zasada termodynamiki
Energia swobodna F F = U - TS
Entalpia swobodna G G = H - TS
T
dq
dS
U
H
F
G
+ pV
- T S
- T S
+ pV
G = H – TS = U + pV – TS = F + pV

Różniczki zupełne funkcji termodynamicznych
Energia wewnętrzna U
dU = q
el
+ w
el
U = f(T,V) dU = c
V
dT
– pdV
U = f(S,V) dU = TdS - pdV
Entalpia H
H = U + pV
H = f(T,p) dH = c
p
dT + Vdp
H = f(S,p) dH = TdS + Vdp
Entropia S
S = f(T,V)
S = f(T,p)
T
dq
dS
Energia swobodna F
F = U - TS
F = f(T,V) dF= -SdT
– pdV
Entalpia swobodna G
G = H - TS
G = f(T,p) dG = -SdT + Vdp
dV
V
R
dT
T
c
dS
V
dp
p
R
dT
T
c
dS
p

Funkcje termodynamiczne układu wieloskładnikowego
W układzie wieloskładnikowym funkcje termodynamiczne zależą od ciśnienia,
temperatury, objętości i ilości składników tworzących układ
(składu układu)
.
F = f ( V, T,
n
1
, n
2
, .........n
k
); G = f ( p, T,
n
1
, n
2
, ...........n
k
)
Zmiana liczby moli składników powoduje zmianę wartości funkcji termodynamicznej:
...
dn
n
F
dT
T
F
dV
V
F
dF
1
,...
n
,
n
T,
V,
1
,...
n
V,
,..
n
T,
3
2
1
1
.
..
dn
n
G
dT
T
G
dp
p
G
dG
1
,...
n
,
n
T,
p,
1
,...
n
p,
,...
n
T,
3
2
1
1
dF = – pdV – SdT
układ jednoskładnikowy
dG = + Vdp – SdT
układ jednoskładnikowy
Entalpia swobodna układu wieloskładnikowego
)
n
T,
p,
i
i
n
G
μ
.
..
dn
n
G
dT
T
G
dp
p
G
dG
1
,...
n
,
n
T,
p,
1
,...
n
p,
,...
n
T,
3
2
1
1
Potencjał chemiczny składnika
określa zmianę entalpii swobodnej
G układu,
gdy zmieniamy ilośd tego składnika w układzie w warunkach stałego składu innych składników
oraz p = const i T = const
i
i
i
dn
μ
SdT
Vdp
dG
Różniczka zupełna entalpii swobodnej układu wieloskładnikowego
V -S
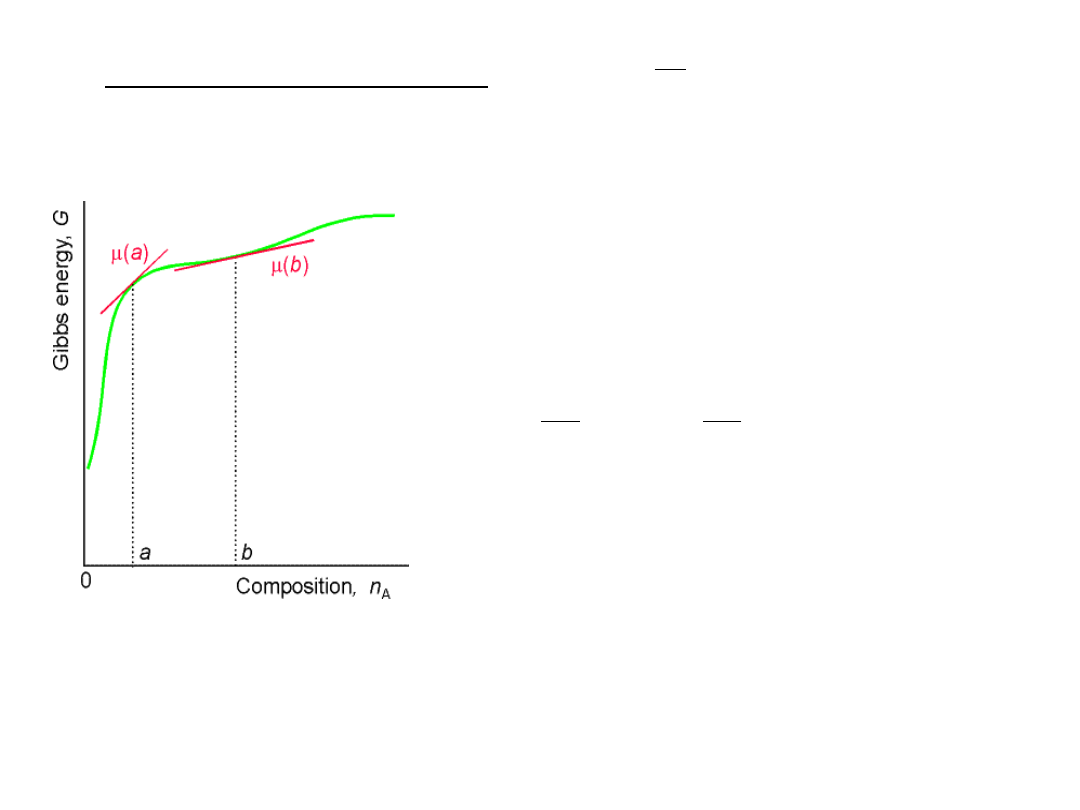
Potencjał chemiczny składnika układu
)
n
T,
p,
i
i
n
G
μ
jest cząstkową molową entalpią swobodną
tzn. jest nachyleniem wykresu entalpii swobodnej jako funkcji ilości
składnika,( np. A) gdy p i T oraz ilości pozostałych składników są stałe
B
B
A
A
B
n
T,
p,
B
A
n
T,
p,
A
dn
μ
dn
μ
dn
n
G
dn
n
G
dG
A
B
W warunkach izotermiczno-izobarycznych zmiana
entalpii swobodnej układu dwuskładnikowego
spowodowana zmianą składu układu wynosi:
całkowita entalpia swobodna dwuskładnikowej mieszaniny
w warunkach izotermiczno-izobarycznych
G = n
A
A
+ n
B
B
Potencjał chemiczny substancji w mieszaninie jest wkładem jaki substancja ta wnosi do
całkowitej entalpii swobodnej układu
T = const
p = const
Równanie Gibbsa- Duhema
i
i
i
dn
μ
SdT
Vdp
dG
W układzie wieloskładnikowym zmianę entalpii swobodnej Gibbsa może spowodowad
zmiana temperatury, ciśnienia lub składu mieszaniny
i
i
i
dn
μ
dG
W warunkach
izotermiczno-izobarycznych entalpia swobodna Gibbsa
zależy tylko od
składu: od rodzaju składników i ich ilości, czyli
jest funkcją potencjałów chemicznych i liczby
moli
:
i
i
i
μ
n
G
Entalpię swobodną Gibbsa można zmieniad
zmieniając albo potencjały chemiczne
składników, albo liczbę moli składników
i
i
i
i
i
i
d
μ
n
dn
μ
dG
0
d
μ
n
i
i
i
0
d
μ
n
SdT
Vdp
i
i
i
Równanie Gibbsa-Duhema wiąże zmiany
temperatury, ciśnienia i potencjałów
chemicznych składników
gdy p,T = const
W warunkach izotermiczno-izobarycznych, potencjały
chemiczne składników są od siebie uzależnione.
Potencjał chemiczny jednego ze składników mieszaniny nie
może się zmieniad w sposób niezależny od potencjałów
chemicznych pozostałych składników
.
POTENCJAŁ CHEMICZNY składnika w stanie gazowym
)
n
T,
p,
i
i
n
G
μ
Potencjał chemiczny jest funkcją termodynamiczną określającą zmianę entalpii swobodnej
G układu,
gdy dodajemy do niego substancji w warunkach p=const i T=const
dla substancji czystych
potencjał chemiczny jest
molową entalpią swobodną
i
= (G
m,
)
i
,
określa pracę, jaką może wymienid z otoczeniem 1 mol składnika w warunkach izotermiczno-izobarycznych.
potencjał chemiczny gazu A wynosi:
i
0
- standardowy potencjał chemiczny gazu – potencjał chemiczny czystego
gazu w warunkach standardowych czyli pod ciśnieniem p
0
= 1 bar
1
2
1
m
2
m
p
p
ln
RT
)
(p
G
)
(p
G
Zależnośd molowej entalpii swobodnej gazu doskonałego
od ciśnienia
Jeżeli przyjmiemy jako stan początkowy gazu(A) stan standardowy, czyli p
1
= p
0
, a p
2
= p
A
,
to molowa entalpia swobodna gazu (A) pod ciśnieniem p
A
wynosi
0
A
0
m
m
p
p
ln
RT
(A)
G
A
G
0
A
0
A
A
p
p
ln
RT
μ
μ
0
i
0
i
i
p
p
ln
RT
μ
μ
Potencjał chemiczny substancji (składnika) „i”
w stanie gazowym

Entalpia swobodna reakcji chemicznej
aA + bB ⇌ cC + dD
Zmiana entalpii swobodnej dla reakcji entalpia swobodna reakcji
∆
r
G = ∆G
prod
- ∆G
substr
b
0
B
a
0
A
d
0
D
c
0
C
p
p
p
p
p
p
p
p
Q
Potencjał chemiczny każdego reagenta wyraża zależnośd:
standardowa entalpia swobodna reakcji
)
b
μ
(a
μ
d
μ
c
μ
G
Δ
B
A
D
C
r
0
i
0
i
i
p
p
ln
RT
μ
μ
0
B
o
B
0
A
o
A
0
D
o
D
0
C
o
C
r
p
p
bRTln
b
μ
p
p
aRTln
a
μ
p
p
dRTln
d
μ
p
p
cRTln
c
μ
G
Δ
b
0
B
a
0
A
d
0
D
c
0
C
o
B
o
A
o
D
o
C
r
p
p
p
p
p
p
p
p
RTln
b
μ
a
μ
d
μ
c
μ
G
Δ
RTlnQ
G
Δ
G
Δ
o
r
r
o
B
o
A
o
D
o
C
o
r
b
μ
a
μ
d
μ
c
μ
G
Δ
Iloraz reakcji Q
Reakcja zachodzi w fazie gazowej, a
wszystkie reagenty zachowują się jak gaz
doskonały

Stała równowagi reakcji chemicznej
Gdy reakcja chemiczna osiąga stan równowagi ∆G = 0,
a
ciśnienia cząstkowe reagentów gazowych przyjmują wartości równowagowe
b
r
0
B
a
r
0
A
d
r
0
D
c
r
0
C
p
p
p
p
p
p
p
p
RTln
0
r
ΔG
0
r
0
B
r
0
A
r
0
D
r
0
C
p
p
,
p
p
,
p
p
,
p
p
aA + bB ⇌ cC + dD
b
r
0
B
a
r
0
A
d
r
0
D
c
r
0
C
p
p
p
p
p
p
p
p
p
K
Stała równowagi reakcji chemicznej
wyrażona za pomocą ciśnieo
cząstkowych reagentów
∆G
r
0
= - RT ln K
p
związek między standardową entalpią
swobodną reakcji chemicznej
i stałą równowagi reakcji
.

RT
ΔG
p
o
r
e
K
Jeśli znamy
standardową entalpię swobodną reakcji ΔG
0
r
– możemy obliczyd stałą równowagi reakcji K
p
Stała równowagi reakcji chemicznej
∆G
r
0
= - RT ln K
p

substraty
o
i
i
produkty
o
j
j
o
r
ΔH
ν
ΔH
ν
H
Δ
substraty
o
i
i
produkty
o
j
j
o
r
S
ν
S
ν
S
Δ
o
r
o
r
o
r
S
Δ
T
H
Δ
G
Δ
I sposób
obliczamy:
standardową entalpię reakcji
r
H
o
standardową entropię reakcji
r
S
o
standardową entalpię swobodną reakcji
G
r
o
korzystając z tablic termochemicznych
Δ
r
G
0
można obliczyd dwoma sposobami
:
0
i
tw
substr
i
0
j
tw
prod.
j
0
r
G
Δ
ν
G
Δ
ν
G
Δ
II sposób
Na podstawie wartości ∆
tw
G
0
reagentów można obliczyd
wartośd standardowej entalpii swobodnej dowolnej reakcji ∆
r
G
0

Standardowa ENTALPIA SWOBODNA tworzenia
analogicznie do ENTALPII
tw
H tworzenia związku chemicznego wprowadza się pojęcie
ENTALPII SWOBODNEJ tworzenia
( potencjału termodynamicznego tworzenia) związków chemicznych
Standardowa ENTALPIA SWOBODNA tworzenia ∆
tw
G
0
jest to
zmiana ENTALPII SWOBODNEJ towarzysząca reakcji tworzenia związku chemicznego z
pierwiastków w ich stanach podstawowych ( stabilnych )
Standardowy potencjał termodynamiczny G
0
trwałej odmiany pierwiastka wynosi 0.
Na podstawie wartości ∆
tw
G
0
z tabel termodynamicznych można obliczyd
wartośd standardowej entalpii swobodnej dowolnej r-cji ∆
r
G
0
0
i
tw
substr
i
0
j
tw
prod.
j
0
r
G
Δ
ν
G
Δ
ν
G
Δ
W tablicach fizyko-chemicznych zamieszczone są
standardowe entalpie swobodne tworzenia reagentów
tw
G
0

RÓWNOWAGA CHEMICZNA W FAZIE GAZOWEJ - OBLICZENIA
Zadanie
Standardowe entalpie i entropie reakcji
CO
(g)
+ H
2
O
(g)
CO
2 (g)
+ H
2 (g)
w temperaturze 300 K i 1200 K są następujące:
H
0
300K
= - 41,16 kJ/mol;
H
0
1200K
= - 32,93 kJ/mol
S
0
300K
= - 42.4 J/mol K;
S
0
1200K
= - 29,6 J/ mol K
W jakim kierunku reakcja będzie przebiegad samorzutnie a) w temp.300K i b) w temp.1200K, jeżeli
ciśnienie mieszaniny w stanie równowagi wynosi 1 x 10
5
N/m
2
.Obliczyd wartości K
p
dla podanej
reakcji w każdej z tych dwóch temperatur.
Obliczenie
G
0
G
0
T
=
H
0
T
– T
S
0
T
G
0
300
= - 28440 J/mol
G
0
1200
= 2590 J/mol
G
0
300
< 0 r-cja przebiega samorzutnie w prawo powstaje CO
2
i H
2
G
0
1200
> 0 r-cja przebiega samorzutnie w lewo powstaje CO i H
2
O
Obliczenie K
p
G
0
= - RT ln K
p
ln K
p300
= -
G
0
300
/ RT = 11,4
K
p300
= 8,95 x 10
4
ln K
p1200
= -
G
0
1200
/ RT = -0,26 K
p1200
= 0,77
Zadanie
Standardowa molowa entalpia spalania fenolu C
6
H
5
OH
(s)
w 25
0
C wynosi -3054 kJ
mol
-1
, a jego
standardowa molowa entropia jest równa 146 J
K
-1
mol
-1
. Obliczyd
standardową molową entalpię
swobodną
spalania fenolu w temperaturze 25
0
C .
Rozwiązanie:
C
6
H
5
OH
(s)
+ 7 O
2(g)
= 6CO
2(g)
+ 3H
2
O
(c)
∆
r
G
0
= ∆
r
H
0
- T
∆
r
S
0
∆
r
S
0
sp
(obliczamy na podst. tablic)=-89,80 JK
-1
,
Odp. ∆G
0
= -3027 kJ
mol
-1
Zadanie
Obliczyd
entalpię swobodną
dla reakcji spalania gazowego propanu w temperaturze 298K i pod
ciśnieniem 1 atm.
C
3
H
8(g)
+ 5O
2(g)
= 3CO
2(g)
+ 4H
2
O
(c)
Rozwiązanie:
I metoda:
∆
r
G
0
= ∆
r
H
0
- T
∆
r
S
0
∆S
0
(obliczamy z tablic)=-374,74J
K
-1
, ∆H
0
odczytujemy z tablic = -2220kJ; ∆G
0
= -2108,3 kJ.
II metoda: Obliczmy na podstawie ∆G
0
tw
wziętych z tablic. ∆G
0
= -2108kJ
RÓWNOWAGA CHEMICZNA W FAZIE GAZOWEJ - OBLICZENIA
0
im
substraty
i
0
j m
produkty
j
0
S
ν
S
ν
S
r

Stałe równowagi reakcji K
p
, K
x
b
r
0
B
a
r
0
A
d
r
0
D
c
r
0
C
p
p
p
p
p
p
p
p
p
K
K
p
stała równowagi wyrażona za pomocą ciśnieo
cząstkowych reagentów gazowych p
i
(ciśnieniowa stała równowagi).
aA + bB
⇌ cC + dD
p
0
- ciśnienie standardowe =1bar
Jeżeli reagenty spełniają prawo Daltona to ich ciśnienia cząstkowe są proporcjonalne do
ułamka molowego reagenta x
i
(p- ciśnienie całkowite w układzie)
p
i
= x
i
p
Δn
p
p
b
r
)
(x
a
r
)
(x
d
r
)
(x
c
r
)
(x
b
r
0
p
p
x
a
r
0
p
p
x
d
r
0
p
p
x
c
r
0
p
p
x
0
B
A
D
C
B
A
D
C
p
K
Δ n
0
x
p
p
p
K
K
gdzie ∆n = ( c + d ) – ( a + b )
jest zmianą liczby moli reagentów gazowych
K
x
stała równowagi chemicznej wyrażona za
pomocą ułamków molowych reagentów
b
r
B
a
r
A
d
r
D
c
r
C
x
x
x
x
x
K
Stałe równowagi reakcji K
p
, K
c
aA + bB
⇌ cC + dD
p V = nRT
jeżeli założymy że reagenty spełniają założenia modelu gazu doskonałego
p = (n/V) RT
p
i
= c
i
RT
Δn
0
p
RT
b
r
)
B
(c
a
r
)
A
(c
d
r
)
D
(c
c
r
)
C
(c
b
r
0
p
RT
B
c
a
r
0
p
RT
A
c
d
r
0
p
RT
D
c
c
r
0
p
RT
C
c
p
K
b
r
c
c
a
r
c
c
d
r
c
c
c
r
c
c
K
0
B
0
A
0
D
0
C
c
K
c
stała równowagi chemicznej wyrażona
za pomocą stężeo molowych c
i
c
0
= 1 mol dm
-3
stężenie standardowe
Δ n
0
0
c
p
p
RT
c
K
K
Stałe równowagi chemicznej K
p
, K
c
i K
x
są liczbami bez miana
Zależności pomiędzy K
p
, K
c
i K
x
dotyczą
układów doskonałych
lub takich, które w
przybliżeniu zachowują się jak doskonałe ( gazy lub roztwory)

Termodynamiczna stała równowagi
W przypadku układów rzeczywistych należy stosowad termodynamiczną stałą równowagi, w której
ilośd reagentów wyraża się za pomocą
aktywności
.
Aktywnośd
określa efektywne stężenie reagenta
jest proporcjonalna do jego stężenia
a
i
=
i
x
i
- współczynnik aktywności
c
0
= 1 mol/L, m
0
=1 mol/kg – stężenia normalizujące
,
aktywnośd jest wielkością bezwymiarową
aA + bB ⇌ cC + dD
b
r
B
a
r
A
d
r
D
c
r
C
a
a
a
a
a
K
Dla gazów rzeczywistych
aktywnośd ciśnieniowa nazywana
jest lotnością f
i
0
i
i
i
m
m
γ
a
i
0
i
i
i
f
p
p
γ
a
0
i
i
i
c
c
γ
a
RÓWNOWAGA CHEMICZNA W FAZIE GAZOWEJ - OBLICZENIA
Zadanie
Stopieo dysocjacji jodowodoru w temperaturze 1500 K wynosi 0,33 pod ciśnieniem 1,013x10
5
Pa. Obliczyd K
p
dla reakcji:
1). HJ
(g)
½ H
2 (g)
+ ½ J
2(g)
odp K
p1
=
/2(1-
)=0,25
2). 2 HJ
(g)
H
2 (g)
+ J
2(g)
odp. K
p2
= (K
p1
)
2
= 0,0625
3). H
2 (g)
+ J
2(g)
2HJ
(g)
odp. K
p3
= 1/K
p2
= 16
0
HI
0
I
1/2
0
H
p1
p
p
p
p
p
p
K
2
2
2
1/
2
2
0
HI
0
I
0
H
p2
p1
2
2
K
p
p
p
p
p
p
K
p2
0
H
0
I
2
0
HI
p3
K
1
p
p
p
p
p
p
K
2
2
Wyszukiwarka
Podobne podstrony:
Ch w6 19.11, Studia (Geologia,GZMIW UAM), I rok, Chemia
P w6 19.11, Studia (Geologia,GZMIW UAM), I rok, Paleontologia ze Stratygrafią, 1. PALEONTOLOGIA WYKŁ
Listopad 15 11 19 11
MO 11 15, A,B0009
wiczenia Makroekonomia 19.11, SEMESTR I, makroekonomia
MO 11 15, A,B0005
MO 11 15, A,B0006
MO 11 15, A,B0013
FINANSE PUBLICZNE - 19.11.2013 (wersja rozszerzona), Wykłady(4)
przetwórstwo projekt 19 01 15
2015 08 20 08 19 11 01
PRAWO ADMINISTRACYJNE wyklad 19 11 2011 id 386058
enzymologia 11-15, BIOTECHNOLOGIA POLITECHNIKA ŁÓDZKA, ENZYMOLOGIA
TPL PRAC 13 11 15 Emulsje
IS 2011 12 wyklad 11 15 12 2011 MDW
AK Instrukcja nr 3 PS 7 2011 11 15
MO 11 15, A,B0002
19 11 10
więcej podobnych podstron