
„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
MINISTERSTWO EDUKACJI
NARODOWEJ
Stefania Kubik
Wykonywanie dokumentacji aparatury medycznej
322[18].Z3.03
Poradnik dla ucznia
Wydawca
Instytut Technologii Eksploatacji – Państwowy Instytut Badawczy
Radom 2007

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
1
Recenzenci:
dr hab. n. med. Ewa Marzec
dr inż. Krystian Rudzki
Opracowanie redakcyjne:
mgr inż. Beata Organ
Konsultacja:
mgr inż. Gabriela Poloczek
Poradnik stanowi obudowę dydaktyczn ą programu jednostki modułowej 322[18].Z3.03,
„Wykonywanie dokumentacji aparatury medycznej”, zawartego w modułowym programie
nauczania dla zawodu technik elektroniki medycznej.
Wydawca
Instytut Technologii Eksploatacji – Państwowy Instytut Badawczy, Radom 2007

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
2
SPIS TREŚCI
1. Wprowadzenie
4
2. Wymagania wstępne
7
3. Cele kształcenia
8
4. Materiał nauczania
9
4.1. System dyrektyw europejskich i norm zharmonizowanych. Definicja
wyrobu medycznego oraz zasady klasyfikacji wyrobów medycznych
9
4.1.1. Materiał nauczania
9
4.1.2. Pytania sprawdzające 11
4.1.3. Ćwiczenia 11
4.1.4. Sprawdzian postępów 12
4.2. Przepisy regulujące warunki używania aparatury medycznej
13
4.2.1. Materiał nauczania
13
4.2.2. Pytania sprawdzające 21
4.2.3. Ćwiczenia 21
4.2.4. Sprawdzian postępów 24
4.3. Wykonywanie dokumentacji i statystyki badań
25
4.3.1. Materiał nauczania
25
4.3.2. Pytania sprawdzające 30
4.3.3. Ćwiczenia 30
4.3.4. Sprawdzian postępów 31
4.4. Zasady tworzenia metryk dla aparatury medycznej
32
4.4.1. Materiał nauczania
32
4.4.2. Pytania sprawdzające 33
4.4.3. Ćwiczenia 34
4.4.4. Sprawdzian postępów 35
4.5. Wymagania dotyczące instrukcji użytkowania wyrobu medycznego
36
4.5.1. Materiał nauczania
36
4.5.2. Pytania sprawdzające 37
4.5.3. Ćwiczenia 37
4.5.4. Sprawdzian postępów 38
4.6. Instrukcja dotycząca uruchamiania i serwisu wyrobu medycznego
39
4.6.1. Materiał nauczania
39
4.6.2. Pytania sprawdzające 40
4.6.3. Ćwiczenia 41
4.6.4. Sprawdzian postępów 41
4.7. Postępowanie na stanowisku pracy
42
4.7.1. Materiał nauczania
42
4.7.2. Pytania sprawdzające 43
4.7.3. Ćwiczenia 43
4.7.4. Sprawdzian postępów 44
4.8. Instrukcja postępowania awaryjnego
46
4.8.1. Materiał nauczania
46
4.8.2. Pytania sprawdzające 47
4.8.3. Ćwiczenia 48
4.8.4. Sprawdzian postępów 48

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
3
4.9. Dokumentacja eksploatacyjna wyrobu medycznego
49
4.9.1. Materiał nauczania
49
4.9.2. Pytania sprawdzające 50
4.9.3. Ćwiczenia 51
4.9.4. Sprawdzian postępów 52
5. Sprawdzian osiągnięć
53
6. Literatura
58

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
4
1.
WPROWADZENIE
Niniejszy „Poradnik dla ucznia” jest związany z nauczaniem w zakresie jednostki
modułowej „Wykonywanie dokumentacji aparatury medycznej” 322[18].Z3.03, w szkole
kształcącej w zawodzie technik elektroniki medycznej. Sposób podania materiału
przewidzianego kształceniem służy przygotowaniu ucznia do przyszłej pracy na stanowisku
technicznym w zakładzie produkcyjnym lub w zakładzie opieki zdrowotnej. Mam nadzieję, że
Poradnik będzie stanowił dla Ciebie pomoc w
przygotowaniu się do ćwiczeń oraz
w powtórkach materiału przekazanego podczas zajęć dydaktycznych.
Poradnik zawiera:
−
wymagania wstępne – wykaz umiejętności, jakie powinieneś posiadać i wiedzy, jaką
musisz sobie przyswoić przed przystąpieniem do nauki w zakresie objętym tym
Poradnikiem,
−
cele kształcenia – wykaz umiejętności, jakie powinny zostać ukształtowane, gdy będziesz
sobie przyswajać materiał objęty tym Poradnikiem,
−
materiał nauczania – czyli wybrane wiadomości dotyczące wyrobów medycznych,
wymagań, jakim takie wyroby podlegają, zasad tworzenia dokumentacji towarzyszącej
wyrobom medycznym w procesie wytwarzania, wprowadzania ich na rynek, użytkowania
i serwisowania,
−
wykaz materiałów, z jakich możesz korzystać podczas nauki,
−
zestawy pytań, które pomogą Ci sprawdzić, czy opanowałeś podane w
materiale
nauczania treści,
−
ćwiczenia, które umożliwią Ci nabycie umiejętności praktycznych,
−
sprawdzian postępów,
−
wykaz literatury uzupełniającej.
Zamieszczony w Poradniku materiał nauczania zawiera podstawowe pojęcia
i wyjaśnienia. Poradnik nie może być dla Ciebie jedynym źródłem wiedzy. Istotnym
elementem nauczania jest udział w zajęciach przewidzianych w programie jednostki
modułowej 322[18].Z3.03. Szerszy zakres informacji, razem z wyjaśnieniami, powinny
dostarczyć zajęcia prezentacyjno-dyskusyjne i
wykonywane ćwiczenia przewidziane
programem tej jednostki modułowej. Ponieważ całościowe i odpowiednio ukierunkowane
opracowania na objęte tym modułem kształcenia tematy praktycznie nie istnieją, autorka
odwołuje się głównie do aktów prawnych, norm zharmonizowanych i stron www jako do
źródeł materiałów pomocniczych. Pochodzące z nich definicje i wymagania zostały opatrzone
pewnym komentarzem. Do zajęć dodano dużą ilość interaktywnych ćwiczeń, które powinny
Ci pozwolić na przyswojenie tak dużej ilości niełatwych pojęć i sprawić, że będziesz umiał
posłużyć się właściwym językiem w sprawach dotyczących wyrobów medycznych.
Ponieważ celem kształcenia jest doprowadzenie do wykształcenia umiejętności
samodzielnego tworzenia różnego rodzaju dokumentacji pomocniczej potrzebnej na
stanowisku pracy w zakładzie produkcyjnym lub w zakładzie opieki zdrowotnej, powinieneś
dobrze znać pojęcia dotyczące takich wyrobów, wymagania dotyczące ich badania,
serwisowania i nadzorowania oraz podstawowe wymagania prawne związane z używaniem
wyrobów medycznych. Powinieneś również umieć znaleźć i przełożyć na potrzeby
postawionego Ci zadania, wymagania zawarte w dokumentacji technicznej wyrobu oraz
w dokumentacji systemu zarządzania jakością obowiązującego w Twoim przyszłym miejscu
pracy.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
5
W czasie większości ćwiczeń uczniowie będą pracować w niewielkich zespołach. Praca
zespołowa pozwoli Wam na przedyskutowanie postawionego problemu, wzajemne
uzupełnianie informacji, dostrzeganie różnych punktów widzenia sytuacji problemowych.
Z rozdziałem „Pytania sprawdzające” dla każdego tematu możesz się zapoznać:
−
przed przystąpieniem do rozdziału „Materiał nauczania” – poznając przy tej okazji
wymagania wynikające z potrzeb zawodu, a po przyswojeniu wskazanych treści,
odpowiadając na te pytania sprawdzisz stan swojej gotowości do ćwiczeń,
lub:
−
po zapoznaniu się z rozdziałem „Materiał nauczania”, aby sprawdzić stan swojej wiedzy,
która będzie Ci potrzebna do ćwiczeń.
Ważnym sposobem nabywania umiejętności przewidzianych jako rezultat kształcenia
w zakresie modułu, którego dotyczy Poradnik, jest Twój aktywny udział w wykonywaniu
ćwiczeń, których celem jest uzupełnienie oraz utrwalenie pozyskanych w czasie wykładu
wiadomości.
Zawarty
w
końcowej części każdego rozdziału Poradnika test „Sprawdzian postępów”
pozwoli Ci na sprawdzenie stopnia przygotowania do testu sprawdzającego, jaki jest
przewidziany na koniec kształcenia wg tej jednostki modułowej.
Przystępując do rozwiązania testu „Sprawdzian postępów” należy:
−
przeczytać pytania i odpowiedzieć na nie,
−
ocenić wynik testu wstawiając „x” w odpowiednie miejsce:
Odpowiedzi „NIE” wskazują na luki w Twojej wiedzy i informują Cię o tym, jakich
zagadnień tutaj omawianych jeszcze dobrze nie poznałeś. Oznacza to także, że powinieneś
wrócić do treści, które nie są przez Ciebie dostatecznie opanowane.
Poznanie przez Ciebie w toku zajęć przewidzianego materiału, będzie stanowiło dla
nauczyciela podstawę przeprowadzenia sprawdzianu poziomu przyswojonych wiadomości
i ukształtowanych umiejętności. W tym celu nauczyciel posłuży się zestawem zadań
testowych, zawierającym różnego rodzaju pytania. W rozdziale 5 tego Poradnika jest
zamieszczony przykładowy zestaw zadań testowych, zawiera on:
−
instrukcję, w której omówiono tok postępowania podczas przeprowadzania sprawdzianu,
−
przykładową kartę odpowiedzi, w której, w przeznaczonych miejscach uczeń powinien
wpisać odpowiedzi na pytania.
„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
6
322[18].Z3
Obsługa i nadzorowanie aparatury
medycznej
322[18].Z3.01
Instalowanie i uruchamianie aparatury
medycznej
322[18].Z3.02
Badanie i naprawa aparatury medycznej
322[18].Z3.03
Wykonywanie dokumentacji
aparatury medycznej
Schemat układu jednostek modułowych

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
7
2.
WYMAGANIA WSTĘPNE
Przystępując do realizacji programu jednostki modułowej powinieneś umieć:
−
wyjaśniać ogólne zasady działania i bezpiecznego użytkowania podstawowych maszyn
i urządzeń elektrycznych,
−
rozróżniać podstawowe parametry maszyn i urządzeń elektrycznych,
−
rozróżniać instalacje elektryczne i ich osprzęt,
−
rozróżniać zabezpieczenia stosowane w instalacjach elektrycznych,
−
znać zasady pomiaru podstawowych parametrów maszyn i urządzeń elektrycznych,
−
znać zasady bhp i ochrony od porażeń prądem elektrycznym podczas pracy przy
urządzeniach elektrycznych,
−
odczytywać i wykonywać schemat funkcjonalny urządzenia,
−
odczytywać z rysunku technicznego dane istotne w procesie wykonywania lub serwisu
wyrobu,
−
odczytywać dokumentację techniczno-ruchową, konstrukcyjną, technologiczną oraz
zinterpretować zamieszczone w nich oznaczenia,
−
wykonywać zadaną dokumentację z wykorzystaniem narzędziowego oprogramowania
komputerowego.
−
dobierać właściwie narzędzia, przyrządy i materiały do wykonywanych zadań,
−
identyfikować znaczenie nieprawidłowego wykonania obróbki dla zgodności
wykonywanego wyrobu,
−
korzystać z dokumentacji technicznej wyrobu i instrukcji urządzeń (narzędzi),
wykorzystywanych w pracy,
−
charakteryzować budowę, zasadę działania, określać zastosowanie, właściwości
i parametry wskazanego urządzenia medycznego na podstawie jego instrukcji użytkowania
i bardziej szczegółowo na podstawie instrukcji serwisowej lub normy zakładowej,
−
charakteryzować sposób odbioru lub podania sygnału w aparacie oraz znać objawy
nieprawidłowego podania lub odbioru sygnału,
−
interpretować, pod względem technicznym podstawowe wyniki badań diagnostycznych
(określić prawdopodobne przyczyny niezgodności – rodzaj uszkodzenia),
−
stosować sposoby eliminowania zakłóceń wpływających na pracę aparatury medycznej,
nie powodujące nieuprawnionej ingerencji w konstrukcję urządzenia,
−
identyfikować działania w toku eksploatacji aparatury mogące mieć negatywny wpływ na
wyniki jej pracy i trwałość aparatury,
−
znać metody dokonywania prostych napraw, konserwacji, pomiarów i montażu aparatury
medycznej,
−
określać, na podstawie dokumentacji wyrobu, zakres testów i pomiarów aparatury
medycznej w celu określenia jej sprawności po naprawie,
−
określać odpowiednie dla danej aparatury materiały eksploatacyjne oraz sposób ich
wymiany i likwidacji materiałów zużytych.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
8
3.
CELE KSZTAŁCENIA
W wyniku realizacji programu jednostki modułowej powinieneś umieć:
–
określić przepisy regulujące warunki używania aparatury medycznej,
–
wykonać dokumentację i statystykę badań oraz statystykę związaną z zarządzeniem
jakością,
–
wykonać specyfikację metryk aparatury medycznej,
–
wskazać konieczne elementy instrukcji użytkownika i instrukcji serwisowej,
–
skorzystać z instrukcji serwisowej i instrukcji użytkownika,
–
opracować na podstawie dokumentów firmowych instrukcję postępowania na określonym
stanowisku,
–
opracować instrukcję postępowania awaryjnego,
–
utworzyć i wypełnić dokumentację eksploatacyjną urządzenia medycznego,
–
opracować instrukcję postępowania przy wykonywaniu testów eksploatacyjnych.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
9
4.
MATERIAŁ NAUCZANIA
4.1.
System dyrektyw europejskich i norm zharmonizowanych.
Definicja wyrobu medycznego oraz zasady klasyfikacji
wyrobów medycznych
4.1.1.
Materiał nauczania
Czym są dyrektywy europejskie zwane Dyrektywami Nowego Podejścia?
Prawa i reguły postępowania przyjęte w Unii Europejskiej zakładają dużą swobodę
gospodarczą, usunięcie barier handlowych wobec wytwórców (przede wszystkim z krajów
unijnych) oraz zapewnienie możliwości swobodnego przemieszczania wyrobów na całym
obszarze Unii.
Środkiem przyjętym po 1985 roku, który miał na celu usunięcie przeszkód w korzystaniu
z wyrobów pochodzących z jednych państw unijnych w innych państwach członkowskich,
było nowe podejście (New Approach) do harmonizacji (możliwie daleko idącego
upodobnienia) wybranych przepisów prawa obowiązujących na obszarze całej Unii.
Dokonano tego w oparciu o dyrektywy zwane Dyrektywami Nowego Podejścia.
Koncepcja „Nowego Podejścia” składa się z następujących zasad:
−
Harmonizacja techniczna zostaje ograniczona do zasadniczych (podstawowych)
wymagań.
−
Podstawowe wymagania są określone w Dyrektywach i odnoszą się głównie do
zapewnienia bezpieczeństwa (dodajmy: dość szeroko pojętego).
−
Przyjęto, że wyrób spełnia wymagania zasadnicze, jeśli spełnia wymagania podane
w odpowiednich dla niego, ze względu na właściwości techniczne i przeznaczenie,
normach zharmonizowanych.
−
Wyroby wytworzone zgodnie z normami zharmonizowanymi korzystają z domniemania
zgodności.
−
Wytwórca odpowiada za swój wyrób. Jeśli jednak nie jest to wytwórca z obszaru Unii,
musi ustanowić swojego autoryzowanego przedstawiciela w jednym z krajów Unii, który
staje się „podmiotem odpowiedzialnym” w odniesieniu do wyrobów tego wytwórcy
wprowadzanych na rynek unijny.
Koncepcja ta leży u podstaw zasady swobodnego przepływu towarów w Unii
Europejskiej. Zasada ta polega na tym, że dowolny wyrób, legalnie wprowadzony do obrotu
w jednym z krajów członkowskich, może być sprzedawany we wszystkich pozostałych
państwach członkowskich. Wyrób taki powinien być oznaczany prawnie chronionym znakiem
CE (będącym wzorem zastrzeżonym i własnością Unii Europejskiej) – patrz
rozporządzenie [6]. Zamieszczenie znaku CE na produkcie i/lub informacji o produkcie (np.
na ulotce) oznacza, że sprawdzono, zgodnie z przyjętymi metodami, zgodność wyrobu
z przyjętymi w Unii Dyrektywami, którym produkt ten podlega.
Dyrektywy unijne są wprowadzane do narodowych systemów prawnych w krajach Unii,
w określonym terminie i zakresie, lecz zgodnie z panującymi w tych krajach zasadami.
Wyrób
medyczny to narzędzie, przyrząd, aparat, sprzęt, materiał lub inny artykuł,
stosowany samodzielnie lub w połączeniu, włączając oprogramowanie niezbędne do
właściwego stosowania wyrobu, przeznaczone przez wytwórcę do stosowania u ludzi w celu:
a) diagnozowania, zapobiegania, monitorowania, leczenia lub łagodzenia przebiegu chorób,

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
10
b) diagnozowania, monitorowania, leczenia, łagodzenia lub kompensowania urazów lub
upośledzeń,
c) badania, zastępowania lub modyfikowania budowy anatomicznej lub prowadzenia
procesu fizjologicznego,
d) regulacji poczęć,
który nie osiąga swojego zasadniczego zamierzonego działania w ciele lub na ciele ludzkim
środkami farmakologicznymi, immunologicznymi lub metabolicznymi, lecz którego działanie
może być przez nie wspomagane.
(Definicja wg Ustawy o wyrobach medycznych).
Wyposażenie wyrobu medycznego do różnego przeznaczenia to przedmioty, które nie
będąc wyrobami medycznymi, są przeznaczone do stosowania z tymi wyrobami,
umożliwiając ich używanie zgodnie z zastosowaniem przewidzianym przez wytwórcę wyrobu
medycznego.
Wyposażenie wyrobu medycznego do diagnostyki in vitro to przedmioty, które nie będąc
wyrobami medycznymi, są przeznaczone do stosowania z tymi wyrobami, umożliwiając ich
używanie zgodnie z przewidzianym przez wytwórcę wyrobu medycznego zastosowaniem,
z wyjątkiem inwazyjnych wyrobów medycznych do pobierania próbek oraz wyrobów
medycznych bezpośrednio stosowanych na ludzkim ciele, w celu pobrania próbek.
Wyrób medyczny do diagnostyki in vitro to:
a) wyrób medyczny będący odczynnikiem, produktem z odczynnikiem, kalibratorem,
materiałem kontrolnym, zestawem, przyrządem, aparatem, sprzętem lub systemem
stosowanym samodzielnie lub w połączeniu, przeznaczonym przez wytwórcę do
stosowania in vitro, w celu badania próbek pobranych z organizmu ludzkiego, w tym
próbek krwi lub tkanek dawcy, wyłącznie lub głównie celem dostarczenia informacji:
–
o stanie fizjologicznym lub patologicznym,
–
odnoszących się do wad wrodzonych,
–
do ustalenia bezpieczeństwa i zgodności z potencjalnym biorcą,
–
umożliwiających nadzorowanie działań terapeutycznych,
b) pojemniki typu próżniowego na próbki i inne przeznaczone przez wytwórcę do
bezpośredniego przechowywania oraz konserwacji próbek pochodzących z ciała
ludzkiego do badania diagnostycznego in vitro,
c) sprzęt laboratoryjny ogólnego stosowania, jeżeli ze względu na jego właściwości jest on
specjalnie przeznaczony przez wytwórcę do badań diagnostycznych in vitro;
Aktywny wyrób medyczny do implantacji to wyrób medyczny, którego prawidłowe
funkcjonowanie zależy od źródła energii elektrycznej lub jakiegokolwiek źródła zasilania
innego niż energia generowana bezpośrednio przez organizm ludzki lub przez siłę ciężkości,
przeznaczony do umieszczania w ciele ludzkim częściowo lub w całości, za pomocą zabiegu
chirurgicznego lub medycznego, lub w naturalnych otworach ciała za pomocą zabiegu
medycznego, celem pozostawienia go w tym ciele.
Wymienione wyżej trzy zasadnicze typy wyrobów medycznych są przedmiotem rozważań
trzech dyrektyw nowego podejścia dotyczących wyrobów medycznych i jednocześnie
„Ustawy o wyrobach medycznych”, która przenosi te dyrektywy do prawa polskiego.
Wyrobami medycznymi nie są między innymi:
−
produkty lecznicze,
−
produkty biobójcze,
−
kosmetyki,
−
środki ochrony indywidualnej.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
11
Dalsza część materiału dotyczy głównie wyrobów medycznych nazwanych w Ustawie
o wyrobach medycznych wyrobami do różnego przeznaczenia (Dyrektywa 93/42/EWG).
Co to są normy zharmonizowane?
Dyrektywy nowego podejścia, których stosowanie jest obowiązkowe, zawierają jedynie
wymagania zasadnicze. Zharmonizowane z nimi normy europejskie (oznaczone symbolem
EN i odpowiednim numerem), zawierają szczegółowe wymagania techniczne i/lub metody
badania wyrobów, odnoszące się w dużym stopniu do bezpieczeństwa, ale również inne
wymagania, których spełnienie powoduje, że spełniające je wyroby są na podobnym
poziomie technicznym. Ich harmonizacja z odpowiednią dyrektywą oznacza, że spełniając
wymagania normy, wyrób spełnia również wymagania tej dyrektywy. Spełnianie norm
zharmonizowanych nie jest obowiązkowe (inaczej: obligatoryjne). Każda norma
zharmonizowana zawiera informację (zwykle w Przedmowie) z jaką dyrektywą/dyrektywami
jest zharmonizowana. Wykaz norm zharmonizowanych z poszczególnymi dyrektywami
nowego podejścia można znaleźć na stronie internetowej Polskiego Komitetu Normalizacji
oraz na stronach http://europa.eu.int lub www.NewApproach.org/directives.asp oraz na wielu
innych stronach polskich i innych krajów europejskich.
4.1.2.
Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do ćwiczeń.
1.
Co to są Dyrektywy Nowego Podejścia?
2.
Co to jest wyrób medyczny?
3.
Czy leki są wyrobami medycznymi?
4.
Co to są normy zharmonizowane?
4.1.3.
Ćwiczenia
Ćwiczenie 1
Określ, czy wskazane 2 produkty są wyrobami medycznymi. Jakie elementy za tym
przemawiają? Wypisz cechy za i przeciw.
Sposób wykonania ćwiczenia:
Aby wykonać ćwiczenie, powinieneś:
1)
porównać cechy wskazanych wyrobów z definicją wyrobu medycznego,
2)
przygotować odpowiedź w formie pisemnej razem z opisem ewentualnych wątpliwości,
3)
przedstawić wynik pracy nauczycielowi do oceny.
Wyposażenie stanowiska pracy:
−
krótki opis techniczny i funkcjonalny dwóch wyrobów medycznych,
−
poradnik dla ucznia,
−
materiały biurowe.
Ćwiczenie 2
Jak sprawdzić, czy istnieje norma dotycząca danego wyrobu medycznego, czy jest to
norma zharmonizowana z dyrektywą, której wyrób podlega? Co powinna zawierać
dokumentacja techniczna wyrobu medycznego, jeśli nie ma norm odpowiednich dla danego
wyrobu?

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
12
Sposób wykonania ćwiczenia:
Aby wykonać ćwiczenie, powinieneś:
1)
znaleźć normy, tematycznie związane z wyrobem wśród szkolnego zbioru norm,
2)
sprawdzić ich aktualność w elektronicznych katalogach norm,
3)
sprawdzić, czy są to normy zharmonizowane z dyrektywą 93/42.EEC (93/42/EWG),
4)
przygotować odpowiedź w formie pisemnej razem z opisem ewentualnych wątpliwości.
Wyposażenie stanowiska pracy:
−
krótki opis techniczny i funkcjonalny dwóch wyrobów medycznych,
−
poradnik dla ucznia,
−
szkolny zbiór norm,
−
komputer z dostępem do Internetu,
−
materiały biurowe.
4.1.4.
Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1)
określić, czym charakteryzuje się tzw. Nowe Podejście?
!
!
2)
posługując się dokumentacją produktu sprawdzić, czy masz do czynienia
z wyrobem medycznym?
!
!
3)
znaleźć interesującą Cię normę w katalogach internetowych?
!
!
4)
określić, co to jest norma zharmonizowana, odróżnić normę
zharmonizowaną od innej?
!
!

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
13
4.2.
Przepisy i zasady regulujące warunki używania aparatury
medycznej
4.2.1.
Materiał nauczania
Struktura polskich instytucji i aktów prawnych związanych z
wyrobami
medycznymi oraz ich rola w systemie nadzorowania wyrobów medycznych
W Polsce instytucją nadzorującą rynek wyrobów medycznych jest Urząd Rejestracji
Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych w Warszawie
(URPLWMiPB). Działalność tej instytucji obejmuje przede wszystkim rejestrację wytwórców
i wyrobów medycznych (także pozostałych produktów wymienionych w jej nazwie), ocenę
incydentów medycznych i podejmowanie odpowiednich środków zapobiegających ich
wystąpieniu w przyszłości, ale również kontakty z innymi podobnymi urzędami w krajach
Unii i nadzór nad rynkiem m.in. wyrobów medycznych.
Ważną rolę nadzorowania wyrobów medycznych odgrywają laboratoria akredytowane –
prowadzące badania wyrobów zgodnie z
ustalonymi i kontrolowanymi procedurami
badawczymi, w dużej części opartymi na normach międzynarodowych. Laboratoria takie
podlegają nadzorowi jednostki akredytującej, a wyniki ich badań są uznawane przez jednostki
notyfikowane. Polską jednostką akredytującą laboratoria jest Polskie Centrum Akredytacji.
Jednostki notyfikowane są instytucjami, których zadaniem jest udział w ocenie zgodności
wyrobów. Jeśli wynik oceny jest pozytywny – jednostka wydaje certyfikat zgodności na okres
3 do 5 lat, po którym to okresie następuje tzw. recertyfikacja. W czasie trwania okresu
ważności certyfikatu jednostka notyfikowana przeprowadza coroczne audity kontrolne –
wyrywkowe sprawdzenia, czy w przedsiębiorstwie jest utrzymywany właściwy poziom
zgodności wyrobów i systemu zarządzania jakością. Same jednostki notyfikowane podlegają
systemowi auditowemu ze strony jednostek akredytujących oraz ze strony organów
notyfikujących. Udzielenie notyfikacji oznacza potwierdzenie uprawnień jednostki do
wydawania potwierdzeń zgodności ze wskazaną w zakresie tej notyfikacji dyrektywą
i
rodzajem wyrobów. Np. jednostka może być notyfikowana w zakresie dyrektywy
93/42/EWG dla medycznych urządzeń elektrycznych i elektronicznych. Nie oznacza to
jednak, że dla wszystkich takich urządzeń. Jednostki notyfikowane są zazwyczaj
akredytowane zarówno w zakresie certyfikacji wyrobów podlegających danej dyrektywie jak
i w zakresie odpowiednich systemów zarządzania, co oznacza, że mogą wydawać certyfikaty
zgodności z systemu zarządzania jakością z normą, a nie tylko z dyrektywą.
Najważniejsze wymagania prawa dotyczące wyrobów medycznych
Ustawa o wyrobach medycznych przenosi do prawa polskiego wymagania dyrektyw
medycznych Unii Europejskiej. Zasadą jest, by narodowy akt prawny stawiał co najmniej
takie wymagania jak dyrektywa, którą przenosi. W tekście każdej polskiej ustawy są
wypisane wszystkie dyrektywy, których wymagania stawia dana ustawa. Trzeba przy tym
pamiętać, że prawie każdej ustawie towarzyszą tzw. akty wykonawcze – rozporządzenia
ministrów i dopiero taka całość pozwala na pełne zapoznanie się z przepisami
i wymaganiami. Np. wymagania zasadnicze są przekazane właśnie w postaci rozporządzenia.
Ustawa o wyrobach medycznych zajmuje się następującymi zagadnieniami:
−
definiuje pojęcia używane w ustawie,
−
podaje zasady wprowadzania wyrobów medycznych do obrotu i do używania,
−
podaje zasady klasyfikacji i kwalifikacji wyrobów medycznych,
−
określa wymagania zasadnicze i metody prowadzenia oceny zgodności wyrobów
medycznych z wymaganiami zasadniczymi,

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
14
−
określa zadania, obowiązki i uprawnienia jednostek notyfikowanych,
−
określa zasady przygotowania oceny klinicznej wyrobów medycznych do różnego
przeznaczenia i aktywnych wyrobów medycznych do implantacji,
−
opisuje rejestr wyrobów medycznych i podmiotów odpowiedzialnych za ich
wprowadzenie do obrotu i do używania i zadania URPLWMiPB,
−
określa postępowanie w przypadku wystąpienia incydentu medycznego,
−
określa sposób prowadzenia nadzoru nad wyrobami medycznymi wprowadzonymi do
obrotu i do używania,
−
określa przepisy karne w odniesieniu do wymagań jakie wprowadza.
Przytoczymy tu kilka zasadniczych wymagań omawianej ustawy, które pozwolą
zrozumieć na czym polega stworzony przy jej pomocy system nadzoru wyrobów
medycznych:
−
Do obrotu i używania mogą być wprowadzone tylko wyroby medyczne spełniające
wymagania ustawy.
−
Wyrób medyczny powinien być właściwie dostarczony, prawidłowo zainstalowany
i
konserwowany oraz używany zgodnie z przewidywanym przez wytwórcę
zastosowaniem. Z tego złożonego wymagania wynika szereg obowiązków dla wytwórcy,
dostawcy i użytkownika – w tym również obowiązek odpowiedniego dokumentowania
spełnienia tych wymagań, ponieważ wytwórca podlega kontroli nadzoru rynku wyrobów
medycznych, a w razie wystąpienia incydentu medycznego również użytkownik musi się
umieć wykazać spełnieniem wymagań dotyczących właściwego używania wyrobu.
−
Ustawa mówi jeszcze wyraźniej: użytkownicy wyrobów są zobowiązani do zachowania
należytej staranności w zakresie doboru, instalowania, przeprowadzania przeglądów
i
konserwacji, a przede wszystkim przestrzegania instrukcji użytkowania wyrobu
dostarczonej przez wytwórcę.
−
Instrukcja wyrobu medycznego nie może wprowadzać użytkownika w błąd co do ryzyka
związanego ze stosowaniem wyrobu, nie może mu przypisywać właściwości, funkcji lub
działania, jakich wyrób nie posiada, nie może sugerować zastosowania innego niż
wynika z przeprowadzonej oceny zgodności.
−
Wyroby medyczne do różnego przeznaczenia są klasyfikowane, a wyroby do diagnostyki
in vitro są kwalifikowane w zależności od ryzyka, jakie stwarzają dla otoczenia.
−
Wytwórca wyrobu jest zobowiązany do przeprowadzenia oceny zgodności wyrobu
z odpowiednimi dla niego wymaganiami zasadniczymi.
−
Poprzez odpowiednie rozporządzenia ustawa wprowadza teksty wymagań zasadniczych
oraz szczegółowe wskazania sposobów przeprowadzania oceny zgodności.
−
Ustawa, omawiając jednostki notyfikowane i ich udział w procesie oceny zgodności,
ustala obowiązki i postępowanie takiej jednostki wobec wytwórcy wyrobów medycznych
oraz wobec organów państwa.
−
Ustawa przewiduje badania kliniczne wyrobów medycznych na ludziach jedynie
w sytuacji, kiedy nie można udowodnić na podstawie procesu walidacji wyrobu (testy
eksploatacyjne) oraz poprzez analizę publikacji i stosowania innych wyrobów, że wyrób
spełnia swoją funkcję z medycznego punktu widzenia. Wymagana jest wtedy zgoda
komisji bioetycznej, która analizuje ryzyko związane z prowadzeniem takich badań.
−
Ustawa opisuje budowę rejestru wyrobów medycznych i podmiotów odpowiedzialnych,
czyli przedsiębiorców, którzy odpowiadają prawnie za spełnianie wymagań ustawy przez
wyrób medyczny, a przede wszystkim za bezpieczeństwo jego używania.
Bezpieczeństwo w przypadku wyrobów medycznych jest rozumiane szeroko: wobec
pacjenta, użytkownika (np. personelu medycznego), osób trzecich i środowiska. Chodzi
o bezpieczeństwo techniczne, medyczne i higieniczne.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
15
−
Rejestr powinien ułatwić zorientowanie się dowolnej osobie, czy wyrób jaki posiada lub
zamierza kupić spełnia wymagania zasadnicze. Szczególnie dotyczy to wyrobów klasy I,
gdzie w ocenie zgodności nie uczestniczy jednostka notyfikowana. Lista
zarejestrowanych wyrobów jest publikowana przez Urząd Rejestracji w Biuletynach, na
stronie internetowej Urzędu.
−
Podmioty odpowiedzialne są zobowiązane zgłaszać Urzędowi wszelkie zmiany
wprowadzone w wyrobie lub dokumentacji przekazywanej użytkownikowi.
−
Urząd Rejestracji jest również powołany do nadzorowania incydentów medycznych
i właściwego postępowania w przypadku ich wystąpienia. Opisy incydentów
medycznych w niektórych krajach Europy są publikowane na stronach internetowych
podobnych urzędów. Ustawa opisuje kto ma obowiązek zgłoszenia wystąpienia
incydentu, w jakiej formie, oraz jaka jest sekwencja dalszych działań celem usunięcia
skutków zdarzenia lub dla zapobieżenia takim zdarzeniom w przyszłości.
−
Do obowiązków Urzędu Rejestracji należy również nadzór nad różnymi działaniami
związanymi z wyrobami medycznymi – od projektowania do wprowadzenia do obrotu
lub używania oraz wobec wyrobów używanych, jeśli zachodzi uzasadnione podejrzenie,
że wyrób stanowi zagrożenie.
Przed wprowadzeniem wyrobu podlegającego dyrektywie i oznaczanego znakiem CE do
obrotu lub do używania, wytwórca jest zobowiązany osiągnąć zgodność wyrobu
z wymaganiami zasadniczymi tej dyrektywy (lub kilku dyrektyw). Dotyczy to nie tylko
wyrobów medycznych. Proces osiągania tej zgodności jest zależny od rodzaju wyrobu i jego
przeznaczenia. Jest on nazywany procesem oceny zgodności. Na proces ten składają się:
−
budowanie odpowiednich mechanizmów systemu zarządzania jakością u wytwórcy,
−
odpowiednie prowadzenie procesu projektowania wyrobu,
−
zarządzanie ryzykiem związanym z używaniem wyrobu,
−
przygotowanie dokumentacji technicznej,
−
udokumentowanie spełniania wymagań bezpieczeństwa (przeprowadzenie badań
technicznych), a w przypadku wyrobu medycznego – również ocena kliniczna wyrobu
(analiza literaturowa lub bezpośrednie badania kliniczne),
−
przygotowanie produkcji i walidacja procesów krytycznych.
Proces oceny zgodności, czyli proces zbierania dowodów na spełnienie wymagań
zasadniczych, może być prowadzony różnymi ścieżkami – patrz dokumenty [2 i 4].
W przypadku wyrobów medycznych w procesie tym często uczestniczy jednostka
notyfikowana, mająca uprawnienia w odpowiednim zakresie.
Ocena
zgodności wyrobu medycznego polega na wykazaniu spełnienia wymagań Ustawy
o wyrobach medycznych przez wyrób, sposób jego wytwarzania, wprowadzania do obrotu
lub do używania, oraz sposób jego nadzorowania przez wytwórcę w całym okresie „życia”
wyrobu. Wygodnym sposobem pokazania jak spełniono wymagania zasadnicze jest lista
kontrolna wymagań zasadniczych, w której na każde wymaganie odpowiada się wpisując
posiadane dokumenty lub opisuje się sposób spełnienia tego wymagania.
Ocenę zgodności prowadzi się różnie, w zależności od klasy ryzyka wyrobu:
−
Ocenę dla wyrobu klasy I – przeprowadza wytwórca samodzielnie.
−
Ocenę dla wyrobów klasy I, sterylnych lub z funkcją pomiarową – przeprowadza
wytwórca, a w zakresie sterylizacji i/lub funkcji pomiarowej wyrobu w procesie tym
bierze udział jednostka notyfikowana.
−
Ocenę dla wyrobów klasy IIa, IIb i III przeprowadza wytwórca przy udziale jednostki
notyfikowanej.
Proces
kończy się wystawieniem przez wytwórcę deklaracji zgodności, w której osoba
upoważniona do reprezentowania przedsiębiorstwa poświadcza spełnienie wymagań

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
16
zasadniczych, udowodnione poprzez przeprowadzenie odpowiedniego procesu oceny
zgodności. Jeśli wyrób jest zgodny z wymaganiami norm – wytwórca może się tym
pochwalić. W deklaracji wpisuje się również dane jednostki notyfikowanej, jeśli taka
jednostka brała udział w procesie oceny zgodności.
Zasady klasyfikacji wyrobów medycznych
Wyroby medyczne, w zależności od ryzyka związanego z ich stosowaniem, dzieli się na
klasy (wyroby medyczne do różnego przeznaczenia) lub grupy (wyroby do diagnostyki in
vitro). Ponieważ wyroby medyczne do różnego przeznaczenia (podlegające dyrektywie
93/42/EWG) stanowią około 90% wyrobów medycznych, w niniejszym kursie przedstawiono
sposób klasyfikowania tych właśnie wyrobów.
W klasyfikacji wyrobu bierzemy na wstępie pod uwagę 4 kryteria:
−
stopień inwazyjności,
−
to, czy wyrób jest aktywny,
−
jakie jest miejsce stosowania wyrobu na ciele lub w ciele człowieka,
−
jaki długi czasowo jest kontakt pacjenta z wyrobem.
Definicje każdego z tych pojęć podaje rozporządzenie [3].
Sposób klasyfikacji składa się z 18 reguł, które pozwalają zakwalifikować wyrób do
jednej z czterech klas ryzyka: I, IIa, IIb lub III.
Zasady postępowania przy klasyfikowaniu wyrobu są następujące:
−
Określamy cechy klasyfikowanego wyrobu przy pomocy 4 wymienionych wyżej
kryteriów.
−
Przechodzimy kolejno od pierwszej do ostatniej reguły i przy każdej zaznaczamy klasę
wyrobu, jaka z niej wynika dla klasyfikowanego wyrobu oraz wypisujemy cechy
wyrobu, które decydują, że podjęliśmy taką a nie inną decyzję.
−
Wybieramy klasę najwyższą ze wskazanych w postępowaniu opisanym powyżej – jest to
klasa ryzyka naszego wyrobu.
−
Opracowujemy kartę klasyfikacji wyrobu zawierającą również uzasadnienie klasyfikacji.
Dzisiejsze wymagania w odniesieniu do wyrobów medycznych mówią już nie tylko
o konieczności przeprowadzenia analizy ryzyka dla wyrobu, ale o zarządzaniu ryzykiem, co
oznacza systematyczne stosowanie określonych procedur i polityki związanych z analizą,
oceną oraz nadzorowaniem ryzyka jakie niesie stosowanie wyrobu.
Szkoda – jest tu rozumiana jako uraz fizyczny lub uszczerbek na zdrowiu lub mieniu.
Ryzyko (zagrożenie) to potencjalna szkoda, jaka może być wynikiem używania wyrobu,
potencjalne źródło szkody.
Zarządzanie ryzykiem oznacza podejmowane decyzje i wprowadzane środki celem
obniżenia poziomów zagrożeń lub utrzymania ich na określonym poziomie.
Podejmuje się je w celu:
−
określenia potencjalnych szkód i ich przyczyn na możliwie wczesnym etapie życia
wyrobu,
−
uszeregowania zagrożeń według poziomu ryzyka,
−
doskonalenia projektu wyrobu i procesów,
−
wykrycia i wskazania dróg usunięcia zagrożeń, a przez to zwiększenie bezpieczeństwa
pacjenta, użytkownika i innych osób,
−
zwiększenia bezpieczeństwa środowiska,
−
wskazania potrzeb w zakresie rodzaju informacji i szkoleń potrzebnych użytkownikom,
−
ukierunkowania programu rozwoju wyrobu,
−
spełnienia wymagań prawnych,
−
spełnienia wymagań systemu zarządzania jakością.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
17
Zarządzanie ryzykiem powinno obejmować cały proces realizacji wyrobu –
projektowanie, rozwój, zakupy, dostawę, procesy produkcyjne, instalowanie, serwisowanie,
używanie.
Proces analizy ryzyka, będący elementem zarządzania ryzykiem, powinien być
wielokrotny i odbywać się na różnych etapach życia wyrobu (założenia, projektowanie,
produkcja, używanie). Na każdym z tych etapów z innych miejsc mogą pochodzić dane
o wyrobie i mogą pojawić się elementy, na które wcześniej nie zwrócono uwagi.
Analizę ryzyka wykonuje się według zasad opisanych w normie [17] i przeprowadzają:
−
jednoosobowo specjalista analizy ryzyka,
−
jednoosobowo konstruktor prowadzący tego wyrobu,
lub
−
zespół osób znających wyrób lub podobne wyroby i sytuacje, jakie się z nim wiążą –
pracą takiego zespołu kieruje moderator.
Sesja analizy ryzyka powinna się składać z następujących elementów:
−
zapoznania jej uczestników z wyrobem,
−
przyjęcia metodyki oceny zagrożeń,
−
identyfikacji zagrożeń (ryzyk) i ich przyczyn,
−
oceny możliwych skutków związanych z poszczególnymi zagrożeniami.
−
jeśli to możliwe – podania środków zaradczych możliwych do zastosowania.
−
podsumowania analizy ryzyka.
W efekcie analizy ryzyka powinny być przeprowadzone odpowiednie działania
doskonalące, mające na celu eliminację lub zmniejszenie zagrożeń do poziomu przyjętego za
dopuszczalny.
Dla
każdego wyrobu medycznego ważną sprawą jest określenie krytycznych elementów
składowych i procesów krytycznych, tzn. takich, które decydują o bezpieczeństwie
użytkowania wyrobu lub warunkują odpowiednie wypełnianie funkcji, do jakich wyrób został
przeznaczony. Zarówno wyroby jak usługi związane z listą elementów i procesów
krytycznych muszą podlegać szczególnemu nadzorowi.
Kwalifikowanie
części, materiałów i procesów do kategorii krytycznych zależy od
projektanta i osoby odpowiedzialnej za wdrożenie wyrobu do produkcji. Kwalifikowanie
części, materiałów i procesów do kategorii krytycznych powinno wynikać przede wszystkim
z analizy ryzyka i powinno gwarantować producentowi minimalizację kłopotów związanych
z uzyskaniem bezpiecznego i sprawnego wyrobu.
Zbiór dokumentacji technicznej tworzy się dla wyrobu lub rodziny wyrobów
medycznych narastająco, dokładając kolejne dokumenty w miarę postępowania prac i zdarzeń
występujących na każdym etapie „życia” wyrobu.
W skład dokumentacji technicznej powinny wejść:
−
dane wejściowe (założenia),
−
plan projektu,
−
plan zarządzania ryzykiem,
−
zapisy z przeglądów wykonywanych zgodnie z planem projektu – (zazwyczaj: po
wykonaniu założeń, prototypu, serii próbnej),
−
dokumentacja projektu wstępnego służąca do wykonania prototypu,
−
zapisy z weryfikacji prototypu i jego badań konstruktorskich,
−
zapisy z kolejnych sesji analizy ryzyka i ich podsumowanie,
−
norma zakładowa,
−
zapisy dotyczące serii próbnej,
−
dokumentacja z weryfikacji serii próbnej (sprawdzenie właściwości i protokoły z badań
przeprowadzonych w laboratoriach akredytowanych),

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
18
−
dokumentacja z walidacji wyrobu:
a)
oceny kliniczne (ocena przydatności medycznej),
b)
sprawozdania z próbnego użytkowania w warunkach eksploatacyjnych,
c)
laboratoryjne badania spełniania funkcji odpowiadających przewidzianemu
zastosowaniu,
d)
lista kontrolna zgodności z ustawą o wyrobach medycznych.
−
dokumentacja odnosząca się do:
a)
postaci (wzorce, programy komputerowe),
b)
materiałów,
c)
sposobu wykonania (instrukcje procesowe, wskazówki technologiczne),
d)
oceny jakości i odbioru (postępowanie z wyrobami niezgodnymi, kryteria odbioru),
e)
użytkowania (instrukcja użytkowania, serwisowa, montażu, instalacji, materiały
szkoleniowe dla serwisów autoryzowanych, dla handlowców i dla użytkowników).
Zbiór dokumentacji technicznej powinien także zawierać informacje o zamierzeniach
rozwojowych w odniesieniu do wyrobu.
Część dokumentacji wyrobu – tzw. dokumentacja EC, czyli minimalna dokumentacja
techniczna wymagana przez dyrektywy Unii Europejskiej, musi być w każdej chwili gotowa
do okazania władzom, jako dowód zgodności wyrobu z ustawą lub odpowiednimi
dyrektywami. Jej posiadanie jest prawnym obowiązkiem podmiotu odpowiedzialnego za
wyrób medyczny. Ze względu na tak szeroką dostępność dokumentacja ta nie powinna
zawierać tajemnic firmy ani osób trzecich. Dyrektywy mówią o informacji, a nie
o dokumentach! Oto typowy skład takiej dokumentacji:
−
instrukcja użytkowania
(ogólny opis wyrobu lub wyrobu z wariantami
,
opis przewidywanego zastosowania,
sposobu użycia, wskazanie materiałów zużywalnych, lista wyposażenia, etykiety i notki,
określenie czasu życia lub przydatności, przestrogi, ostrzeżenia i wskazówki, wynikające
przede wszystkim z analizy ryzyka)
−
dokument ustalenia klasy wyrobu,
−
wyniki analizy ryzyka (podsumowanie),
−
wyniki sprawdzeń, badań i prób dotyczących bezpieczeństwa używania wyrobu,
−
wyniki walidacji,
(ocen klinicznych, laboratoryjnych ocen działania, raportów z doświadczeń
z użytkowania wyrobu)
−
lista kontrolna zgodności z dyrektywą lub ustawą,
(powinna
zawierać wykaz norm zharmonizowanych, zastosowanych w całości lub
częściowo, z dokumentacji powinno wynikać, gdzie je zastosowano)
−
krótkie omówienie procesu wytwarzania i sterylizacji (jeśli proces ten ma znaczenie dla
wyrobu).
Zalecenia dotyczące zawartości poszczególnych składników dokumentacji technicznej:
Opis wyrobu ma pozwalać zrozumieć konstrukcję wyrobu, jego możliwości i różnice
między wariantami, jeśli istnieją oraz pomocne w tej materii rysunki, zdjęcia i diagramy. Opis
powinien zawierać:
−
W zakresie czynności wykonywanych w związku z produktem powinny się w nim
znaleźć: instalacja, przygotowanie do użycia, sprawdzenie przed użyciem, konserwacja,
kalibracja i serwis – odpowiednio do rodzaju wyrobu.
−
Jeśli producent powołuje się na normy zharmonizowane, to dokumentacja techniczna
powinna zawierać wyraźne wskazania, co zostało zrobione dla uzyskania zgodności
z odpowiednimi wymaganiami tych standardów
.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
19
−
Dokumentacja ma udowodnić zgodność wyrobu i postępowania wytwórcy
z wymaganiami dyrektywy, co nie znaczy, że dokładnie określa się jej formę.
Jeśli wyrób zawiera substancję medyczną dokumentacja powinna opisywać cel
wprowadzenia tej substancji i sposób jej działania. Dotyczy to tylko tych przypadków, gdzie
substancja ta ma wpływ na ciało (pacjenta lub operatora), wspomagając działanie urządzenia.
Opis procesu wytwarzania powinien być na tyle szczegółowy, żeby udowodnić, że
zamierzone parametry i właściwości urządzenia są osiągane. Należy również opisać specjalne
procesy technologiczne (formowanie, sterylizacja), tzw. procesy krytyczne oraz istotne
warunki środowiskowe.
W opisie użycia wyrobu z akcesoriami, interfejsami lub innymi wyrobami wymagany jest
krótki opis wystarczający do zrozumienia, które parametry tych elementów są ważne dla
bezpieczeństwa całości oraz znane niekompatybilności, które mogą być zaznaczone przy
pomocy etykietek, uwag w instrukcji itp.
Kiedy normy zharmonizowane są używane dla osiągnięcia zgodności z wymaganiami
zasadniczymi – wszystko, co trzeba zrobić, to udowodnić spełnienie wymagań tych norm.
Kiedy
używa się innych metod dla wykazania zgodności wyrobu z wymaganiami
zasadniczymi, producent musi uzasadnić, że:
−
stosowane metody dokładnie odpowiadają wymaganiom zasadniczym,
−
wyrób spełnia te wymagania.
Dowód
zgodności wyrobu z wymaganiami norm może mieć formę raportu
z przeprowadzonych
badań. Badania takie powinno przeprowadzić laboratorium
akredytowane, mające odpowiedni zakres akredytacji (patrz strona internetowa PCA).
Producent jest zobowiązany do dołączenia do wyrobu ulotki, a jeśli wyrób jest bardziej
skomplikowany, to instrukcji użytkowania. Musi też uwzględniać wszelkie zmiany, jakie
następują podczas życia wyrobu.
Na dane kliniczne składają się dane pochodzące z doświadczeń rynkowych na temat tego
lub podobnego urządzenia i informacje z literatury naukowej. Producent musi ocenić jak
dalece dane literaturowe odnoszą się do jego wyrobu.
Deklaracja
zgodności jest dokumentem, w którym wytwórca wyrobu (lub szerzej:
podmiot odpowiedzialny) potwierdza i deklaruje zgodność wyrobu z obowiązującymi dla
niego wymaganiami (w Polsce – z Ustawą o wyrobach medycznych). Deklaracja jest
potwierdzeniem świadomości wytwórcy co do jego odpowiedzialności za wyrób medyczny,
którego deklaracja dotyczy. Deklaracja oznacza między innymi, że została przygotowana
dokumentacja techniczna, wyrobu pozwalająca stwierdzić jego zgodność oraz, że
przeanalizowano zgodność wyrobu i całego procesu jego powstawania, z wymaganiami
zasadniczymi. Jest to również deklaracja, że wytwórca ma ustanowione procedury
nadzorowania wyrobu znajdującego się na rynku oraz procedury doskonalenia wyrobu.
Deklaracja jest oświadczeniem imiennym osoby upoważnionej do reprezentowania
podmiotu odpowiedzialnego. Musi zawierać:
−
nazwę i adres wytwórcy (podmiotu odpowiedzialnego),
−
nazwę wyrobu lub grupy wyrobów (może być wystawiona dla konkretnej, oznaczonej
serii wyrobu,
−
określenie klasy ryzyka wyrobu,
−
oświadczenie, jakie spełnia wymagania (np. symbol dyrektywy i dane Ustawy
o wyrobach medycznych),
−
oświadczenie, jakie normy wyrób spełnia,
−
informacja o certyfikatach posiadanych przez podmiot odpowiedzialny,
−
dane i numer identyfikacyjny jednostki notyfikowanej, jeśli w ocenie zgodności jest
wymagany udział takiej jednostki,

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
20
−
data wystawienia deklaracji,
−
identyfikowalny podpis osoby deklarującej.
Deklaracja jest ostatnim krokiem w procedurze oceny zgodności. Jeśli wymagany jest
udział jednostki notyfikowanej w ocenie zgodność wyrobu, to nie można jej wystawić przed
uzyskaniem zatwierdzenia systemu jakości przez taką jednostkę, a w przypadku urządzenia
klasy III, również przed sprawdzeniem przez nią procesu projektowania. Można jednak
przygotować projekt takiej deklaracji i przedstawić go jednostce notyfikowanej.
Certyfikat, jaki otrzymuje wytwórca dla wyrobu medycznego, jest dokumentem
potwierdzającym zgodność wyrobu z odpowiednimi dla niego dyrektywami i ustawami.
Certyfikat taki może wystawić jedynie jednostka notyfikowana w odpowiednim zakresie, tzn.
mająca uprawnienia do przeprowadzania oceny zgodności dla tego rodzaju wyrobu, którego
certyfikat dotyczy. Jeśli nawet, zgodnie z wymaganiami dyrektywy (ustawy), wytwórca ma
obowiązek posiadać odpowiedni system zarządzania jakością, to nie ma on obowiązku
posiadania dodatkowo certyfikatu zgodności systemu z normą międzynarodową. Jednak
certyfikat zgodności z normą [16] w łatwy sposób potwierdza tę zgodność.
Certyfikat
zgodności jest zwykle wydawany jako potwierdzenie, że system wytwórcy
spełnia wymienione w certyfikacie wymagania i jako potwierdzenie zdolności wytwórcy do
prowadzenia określonych procesów (nie zawsze jest to pełny proces od projektowania po
sprzedaż). Do certyfikatu większość jednostek notyfikowanych dodaje załącznik, w którym
podaje, jakich wyrobów dotyczyła przeprowadzona i zatwierdzona ocena zgodności.
Wyroby certyfikowane, ich instrukcja użytkowania, opakowania handlowe i opakowania
gwarantujące sterylność wyrobu powinny być oznakowane znakiem CE (wg odpowiedniego
wzoru) z unikalnym numerem identyfikacyjnym jednostki notyfikowanej.
Wytwórca powinien określić dla wyrobu sposób i czas jego przechowywania (również
poza używaniem), mając na względzie zachowanie parametrów użytkowych wyrobu. To
samo dotyczy transportu wyrobu do miejsca przeznaczenia. W niektórych wypadkach
warunki transportu (np. opakowanie, temperatura, środek transportu) muszą być bardzo
dokładnie określone tak, aby ochronić wyrób, osoby z jego otoczenia i środowisko np. przed:
−
zanieczyszczeniami bądź skażeniem,
−
zagrożeniem ze strony substancji, które mogłyby wyciekać z wyrobu,
−
przedostaniem się substancji niszczących wyrób do jego wnętrza,
−
utratą sterylności przez wyrób.
W roku 2005 została wprowadzona ustawa o zużytym sprzęcie elektrycznym
i elektronicznym (12).
Ustawa ta jest przeniesieniem wymienionej w niej dyrektywy
europejskiej do prawa polskiego. Na wytwórcy medycznego wyrobu elektrycznego lub
elektronicznego spoczywa cały szereg obowiązków takich jak:
−
obowiązek zarejestrowania się w Głównym Inspektoracie Ochrony Środowiska,
−
ponoszenie corocznych opłat z tytułu wprowadzania wyrobów na rynek,
−
zawarcia umów z instytucjami zbierającymi i przetwarzającymi zużyty wyrób,
−
sprawozdawczość miesięczna, kwartalna i roczna dotycząca wprowadzania podlegających
ustawie wyrobów na rynek polski,
−
dostarczenia dla każdego wyrobu instrukcji postępowania z wyrobem dla instytucji
odzysku zajmującej się likwidacją wyrobu.
Zgodnie z ustawą użytkownik nie może sam demontować sprzętu elektromedycznego ani
wyrzucać go do śmieci. Powinien przeprowadzić procedurę dezynfekcji wyrobu i przekazać
go zgodnie z instrukcją użytkowania wyrobu lub przekazać go do przedsiębiorstwa mającego
odpowiednie uprawnienia organizacji odzysku, jeśli brak innych wskazań w dokumentacji
wyrobu. Idea tej ustawy jest szczytna – chodzi o zmuszenie projektantów do ostrożnego
stosowania substancji niebezpiecznych w swoich wyrobach oraz do zmniejszenia zużycia
cennych surowców.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
21
Każdy wytwórca jest zobowiązany do ustalenia procedury postępowania w odniesieniu
do swoich wyrobów znajdujących się na rynku (sprzedanych). Powinien zbierać sygnały
i opinie odnoszące się do ich działania oraz uwzględniać reklamacje i analizować przyczyny
pojawiających się usterek (szczególnie, jeśli występują często).
Szczególnie
ważne jest szczegółowe ustalenie sposobu postępowania w przypadku
wystąpienia incydentu medycznego, gdyż terminy ustawowe nie pozostawiają zbyt wiele
czasu na reakcję, a musi ona być precyzyjna i skuteczna. Incydent medyczny to:
−
Każde wadliwe działanie, ale też pogorszenie cech lub działania wyrobu medycznego
i każde wprowadzające w błąd oznakowanie wyrobu lub zapis w instrukcji użytkowania,
mogące doprowadzić do śmierci lub pogorszenia stanu zdrowia pacjenta, użytkownika
lub osoby trzeciej.
−
Techniczna lub medyczna nieprawidłowość dotycząca wyrobu, która doprowadziła do
wycofania wyrobu z ryku z przyczyn określonych wyżej.
Wystąpienie incydentu związanego z wyrobem medycznym należy zgłosić Prezesowi
Urzędu Rejestracji PL, WM i PB. Obowiązek zgłaszania dotyczy podmiotów
odpowiedzialnych, ale również instytucji i personelu używających wyrobu.
Zgłaszanie incydentu i procedura postępowania w następstwie zgłoszenia, są
sformalizowane. Opis postępowania zawarto w ustawie [2] i w rozporządzeniu [7].
4.2.2.
Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń.
1.
Jakie są obowiązki wytwórcy przed wprowadzeniem wyrobu medycznego na rynek
(do obrotu lub do używania)?
2.
Co to jest proces oceny zgodności wyrobu?
3.
Jak przeprowadza się ocenę zgodności dla wyrobu medycznego? Od czego zależy sposób
jej przeprowadzenia?
4.
Co to jest zarządzanie ryzykiem? Jak powinna przebiegać analiza ryzyka dla wyrobu
medycznego?
5.
Co składa się na dokumentację techniczną wyrobu medycznego?
6.
Do czego służy deklaracja zgodności? Jakie informacje musi zawierać?
7.
W jakim stopniu zużyty wyrób elektromedyczny staje się odpadem niebezpiecznym?
8.
Co to jest incydent medyczny? Czy zawsze „winnym” jest wyrób medyczny (czyli jego
wytwórca)?
4.2.3.
Ćwiczenia
Ćwiczenie 1
Określ, czy wskazane 2 produkty są wyrobami medycznymi. Jakie elementy za tym
przemawiają? Wypisz cechy za i przeciw.
Sposób wykonania ćwiczenia:
Aby wykonać ćwiczenie, powinieneś:
1)
omówić rodzaj i kolejność działań, jakie musi wykonać wytwórca przed wprowadzeniem
do obrotu lub użytkowania wyrobu medycznego określonego w zadaniu,
2)
skorzystać przy tym z materiałów pomocniczych (ustawy, rozporządzenia) lub innych
materiałów, jeśli takie przygotowaliście w toku wcześniejszych zajęć (lista kontrolna
zgodności, schemat postępowania w ocenie zgodności wyrobów, schemat klasyfikacji
wyrobu medycznego),

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
22
3)
ustalić, co wiesz o wyrobie z dostarczonego do zadania opisu, określić klasę wyrobu
medycznego, wybrać dla niego ścieżkę oceny zgodności i opisać kolejne kroki procesu,
którego wykonanie jest konieczne przed wprowadzeniem wyrobu medycznego do obrotu
lub użytkowania.
Wyposażenie stanowiska:
−
pełny zbiór aktów prawnych dotyczących wyrobów medycznych lub dostęp do Internetu
(www.sejm.gov.pl),
−
stanowisko komputerowe,
−
Poradnik dla ucznia.
Ćwiczenie 2
Jak sprawdzisz, czy istnieje norma dotycząca danego wyrobu medycznego, czy jest to
norma zharmonizowana z dyrektywą, której wyrób podlega? Co powinna zawierać
dokumentacja techniczna wyrobu medycznego, jeśli nie ma norm odpowiednich dla danego
wyrobu?
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
znaleźć normy, tematycznie związane z wyrobem wśród szkolnego zbioru norm,
2)
sprawdzić ich aktualność w elektronicznych katalogach norm,
3)
sprawdzić, czy są to normy zharmonizowane z dyrektywą 93/42.EEC (93/42/EWG).
4)
przygotować odpowiedź w formie pisemnej razem z opisem ewentualnych wątpliwości.
Wyposażenie stanowiska:
−
pełny zbiór aktów prawnych dotyczących wyrobów medycznych lub stanowisko
komputerowe z dostępem do Internetu (www.sejm.gov.pl),
−
opis zawierający: rodzaj i klasę wyrobu, kraj pochodzenia i zestaw dokumentów
przedstawianych kupującemu,
−
poradnik dla ucznia,
−
materiały biurowe.
Ćwiczenie 3
Scharakteryzuj przebieg oceny zgodności wskazanego wyrobu medycznego – ustalić
klasę wyrobu, podać działania wymagane przez prawo. Wskaż, które procedury postępowania
muszą być określone w przedsiębiorstwie nawet, jeśli nie ma ono certyfikowanego systemu
zarządzania jakością?
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
ustalić klasę wyrobu (uzasadnić) – przygotować kartę klasyfikacyjną,
2)
wybrać ścieżkę oceny zgodności odpowiednią dla danego przedsiębiorstwa (uzasadnić),
3)
wymienić pozostałe działania wymagane przez prawo (zakres udziału JN, przygotowanie
deklaracji zgodności, uporządkowanie dokumentacji technicznej – z podziałem na część
A i B).

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
23
Wyposażenie stanowiska pracy:
−
szkolny zbiór aktów prawnych (szczególnie rozporządzenie [4]) zawierające wymagania
zasadnicze) i norm dotyczących wyrobów medycznych,
−
dostęp do Internetu, 1 komputer na grupę ćwiczeniową,
−
krótki opis wyrobu medycznego i wytwórcy (rodzaj przedsiębiorstwa, spodziewana
wielkość produkcji) pozwalający na klasyfikację wyrobu i wybranie ścieżki oceny
zgodności,
−
poradnik dla ucznia.
Ćwiczenie 4
Przeprowadź analizę ryzyka wybranego wyrobu elektromedycznego zgodnie
z wymaganiami normy [17]. Wykonaj podsumowanie analizy ryzyka.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
identyfikować przyczyny i skutki zagrożeń metodą ”burzy mózgów”,
2)
ocenić poszczególne zagrożenia – uzgadnianie oceny w toku dyskusji.
Wyposażenie stanowiska pracy:
−
formularze do analizy ryzyka w postaci elektronicznej,
−
komputer z oprogramowaniem pozwalającym na obsługę formularzy,
−
rzutnik multimedialny,
−
tabele oceny wielkości zagrożenia (egzemplarz dla każdego ucznia i egzemplarz
elektroniczny),
−
tabela oceny ryzyka (egzemplarz dla każdego ucznia i egzemplarz elektroniczny).
−
poradnik dla ucznia.
Ćwiczenie 5
Sprawdź kompletność przedstawionej dokumentacji technicznej wybranego wyrobu
elektromedycznego.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
sprawdzić kompletność posiadanego zestawu dokumentów uwzględniając dane z opisu
wyrobu,
2)
wypisać jakich dokumentów brakuje i które są niekompletne.
Wyposażenie stanowiska pracy:
−
opis techniczny i funkcjonalny wyrobu medycznego z podaną klasą wyrobu
i producentem (dokładny adres),
−
lista kontrolna dokumentacji technicznej sporządzona wg NB–MED 2.5.1 Rec.
5
i wymagań zasadniczych dla wyrobów medycznych,
−
lista kontrolna wymagań zasadniczych,
−
zestaw dokumentów dotyczących konkretnego wyrobu medycznego,
−
poradnik dla ucznia,
−
materiały biurowe.
Listy kontrolne powinny być wynikiem wcześniej wykonywanych prac.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
24
Ćwiczenie 6
Przeprowadź analizę przykładowych sytuacji określonych jako incydent medyczny.
Oceń, czy odpowiedzialnym za incydent jest producent wyrobu medycznego, który był
używany podczas tego zdarzenia. Przedstaw i uzasadnij pisemnie swoją opinię.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
przypomnieć sobie definicję incydentu medycznego z ustawy [2],
2)
zapoznać się z opisem zdarzenia i przedyskutować ze swoją grupą wszystkie jego
aspekty,
3)
ocenić rolę wyrobu medycznego w zdarzeniu i uzasadnić opinię grupy.
Wyposażenie stanowiska pracy:
−
tekst ustawy [2] (definicja incydentu medycznego – określenie kiedy wyrób medyczny
jest przyczyną incydentu medycznego),
−
opis wydarzenia,
−
poradnik dla ucznia,
−
materiały biurowe.
4.2.4.
Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1)
określić, co powinien zrobić wytwórca wyrobu medycznego zanim
wprowadzi wyrób na rynek?
!
!
2)
określić, od czego zależy wybór ścieżki oceny zgodności?
!
!
3)
wyszukać w instrukcji użytkowania wyrobu elementy wynikające
z podsumowania analizy ryzyka przeprowadzonej dla wyrobu?
!
!
4)
wskazać najważniejsze elementy deklaracji zgodności?
!
!
5)
wskazać części i materiały wchodzące w skład wyrobów
elektromedycznych, które sprawiają, że wyroby te muszą być
likwidowane pod nadzorem?
!
!
6)
określić, co powinien zrobić użytkownik, gdy podczas pracy z wyrobem
medycznym wystąpi incydent medyczny?
!
!

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
25
4.3.
Wykonywanie dokumentacji i statystyki badań
4.3.1.
Materiał nauczania
Zaprezentowane
niżej metody mogą być stosowane do analizy danych liczbowych lub
nieliczbowych, bądź związków przyczynowo-skutkowych wpływających na procesy lub
działania.
Jednym z istotnych działań, gdzie metody te mogą być zastosowane jest uzupełnienie
dokumentacji przeprowadzenia założonej serii badań wyrobu (wykonywanych w toku
produkcji lub badań końcowych) o zestawienie wyników tych badań – wykonanie statystyki
wyników. Takie uzupełnienie dokumentacji złożonej z kart pomiarowych pozwala na lepsze
wykorzystanie danych i umożliwia ich szybszy ogląd, np. w celu znalezienia przyczyny
niedoskonałości lub niezgodności i wykonania odpowiednich działań doskonalących.
Niżej zaprezentowano kilka prostych w stosowaniu i skutecznych metod:
Lista kreskowa
Lista kreskowa służy do analizy częstości występowania określonej cechy lub wartości
cechy. Metoda jest przydatna w sytuacjach, gdy nie ma możliwości automatycznego
gromadzenia danych.
Przygotowanie
formularza
−
Określić dane potrzebne do sporządzenia formularza. Mogą to być cechy rozpatrywanego
procesu lub klasy wyodrębnione z danej cechy (zazwyczaj w liczbie 6 do 12).
−
Zaprojektować formularz do zapisywania danych.
Formularz może być zaprojektowany w ten sposób, aby po jego wypełnieniu otrzymać
uproszczoną postać histogramu. Listę kreskową (Tabela 1) buduje się w następujący sposób:
−
Zbierane dane muszą dotyczyć tego samego źródła i procesu.
−
Dane zbieramy dostępnymi metodami. Przy każdorazowym wystąpieniu danej
cechy/klasy zaznaczany to za pomocą znacznika (kreski ukośnej), w odpowiednim
wierszu formularza.
−
Każdy piąty znacznik powinien być kreską poziomą, przekreślającą cztery poprzednie.
−
Po ustalonym czasie lub zebraniu odpowiedniej liczby danych sumujemy znaczniki dla
każdej z cech/klas i określamy ich udział procentowy w ogólnej liczbie wykonanych
pomiarów.
Analiza listy kreskowej polega na:
−
Ocenie, czy zebrana ilość danych jest wystarczająca dla założonych celów.
−
Stwierdzeniu, czy przyjęty do analizy zestaw cech lub liczba klas jest właściwa.
−
Zidentyfikowaniu cech/klas o największym udziale procentowym.
−
Właściwej analizie zbadanego zjawiska pod kątem spełnienia oczekiwań i potrzeby
podjęcia ewentualnych działań korekcyjnych lub korygujących.
−
Wyniki analizy listy kreskowej zapisujemy na formularzu, na którym wykonywana była
lista. Przykładowy formularz listy kreskowej przedstawia Tabela 1.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
26
Tabela 1. Przykład listy kreskowej
od do
Liczba pomiarów w przedziale
Razem
12,51 12,53
0
12,54
12,56
///
3
12,57
12,59
//// /
6
12,60
12,62
//// //// //// ////
19
12,63
12,65
//// //// //// //// /
21
12,66
12,68
//// //// //// //// //// /
26
12,69
12,71
//// //// /
11
12,72
12,74
//// ///
8
12,75
12,77
//// 5
12,78
12,80
/
1
12,81 12,83
0
Razem 100
Histogram
Histogram służy do wizualnego prezentowania i analizy częstości występowania wartości
określonej cechy w zakresie jej zmienności. Histogram, to seria przylegających do siebie
prostokątów o szerokości równej rozpiętości przedziału zmienności badanej cechy
i wysokości równej częstości występowania.
Przygotowanie i wykonanie histogramu.
a)
Zbieranie i przygotowanie danych.
−
Na bazie specyfiki danego procesu ustalić cechy, które będą poddane analizie.
−
Zebrać dostępnymi metodami (np. pomiary, obserwacje, itp.) wartości danych do
histogramu. Przy przygotowywaniu histogramu zalecane jest posłużenie się Listą
kreskową.
−
Ustalić rozstęp danych poprzez odjęcie najmniejszej wartości w zbiorze danych od
wartości największej:
Rozstęp = x
max
– x
min
−
Ustalić liczbę k przedziałów (klas) histogramu (w
przybliżeniu równą pierwiastkowi
kwadratowemu z liczby danych n).
−
Podzielić rozstęp przez liczbę przedziałów k w celu określenia szerokości każdego
przedziału.
−
Ustalić granice poszczególnych przedziałów (klas).
b) Wykreślenie histogramu.
−
Zaznaczyć na osi poziomej granice przedziałów (klas).
−
Zaznaczyć na osi pionowej skalę częstości występowania wartości w
danym
przedziale (liczba lub procent obserwacji).
−
W każdym przedziale wrysować prostokąt o podstawie równej szerokości przedziału
i wysokości równej ilości obserwacji (lub procentowi obserwacji), które przypadają
na ten przedział.
c) Analiza histogramu
−
Na podstawie kształtu uzyskanego histogramu możemy wyciągnąć wnioski
dotyczące przebiegu procesu, jego zmienności, a dalej, przyczyn pojawiających się w
nim problemów.
−
Wyniki analizy formułujemy w postaci wniosków pod wykonanym histogramem.
Przykład takiego histogramu pokazano na rysunku 1.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
27
Rys.1. Przykład histogramu
Analiza Pareto–Lorentza
Analiza Pareto–Lorentza stosowana jest jako metoda służąca do dotarcia do
najistotniejszych przyczyn wybranego zjawiska. Narzędziem tej analizy jest wykres Pareto
z krzywą Lorentza.
Wykres Pareto stosuje się do uszeregowania i graficznego przedstawienia badanych
czynników, mających wpływ na określone zjawisko, z uwzględnieniem ich znaczenia.
Pozwala to na wyodrębnienie z całego szeregu różnych czynników tych, które mają
decydujące znaczenie. Metody tej można używać do analizy przyczyn występowania
najróżniejszych zjawisk.
Przygotowanie i wykonanie wykresu
a)
Zbieranie i przygotowanie danych.
−
Zebrać dostępnymi metodami (np. poprzez pomiary, obserwacje, itp.) komplet
danych o analizowanym procesie (wyrobie, zjawisku) w formie charakterystyki
interesujących nas cech np. przyczyn występowania problemu (rodzaju wad),
częstości ich występowania, kosztu, itp.
−
Przygotować dane do analizy poprzez:
−
wybór czynników, które mają być poddane analizie (ilość poszczególnych
zdarzeń, koszt, czas trwania procesu, itp.),
−
ustalenie przedziału czasowego, dla którego dokonywana jest analiza,
−
uszeregowanie czynników od najbardziej do najmniej znaczących,
−
obliczenie wartości skumulowanej w procentach, dla uszeregowanych
czynników.
b)
Wykreślenie wykresu Pareto.
−
Zaznaczyć na osi poziomej poszczególne czynniki, w równych odstępach,
w porządku malejącym co do znaczenia, od lewej do prawej strony. Czynniki
o bardzo małym udziale można zsumować razem i ująć w ostatniej kategorii pod
nazwą „Inne” lub „Pozostałe”.
−
Narysować z lewej strony osi poziomej oś pionową, wyskalowaną w przyjętej
jednostce miary, natomiast z prawej strony drugą oś pionową wyskalowaną
w procentach (0 do 100%).
Wartość cechy
Liczba
zdarzeń
„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
28
−
Dla każdego czynnika narysować prostokąt (słupek) o wysokości odpowiadającej
wielkości miary. Jest to wykres Pareto.
−
Zaznaczyć punkty odpowiadające wartościom skumulowanym i połączyć je linią
łamaną (tzw. linią Lorentza).
c) Analiza
wykresu.
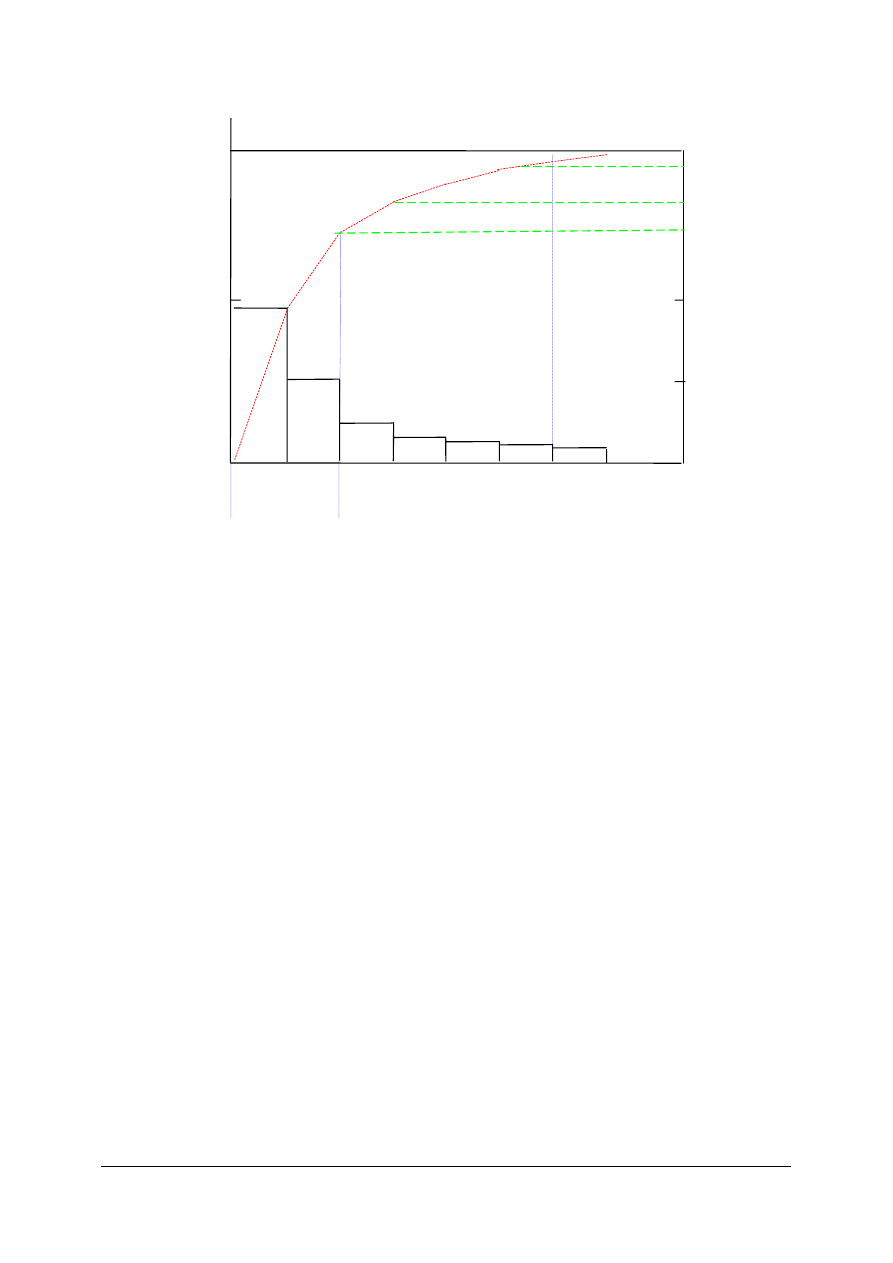
Analizy wykresu Pareto można dokonywać wykorzystując tzw. zasadę ABC, według
której:
−
20 % przyczyn powoduje ok. 80 % skutków (obszar A),
−
30 % przyczyn powoduje kolejne 15 % skutków (obszar B),
−
50 % przyczyn powoduje 5 % skutków ( obszar C).
Jeśli analiza Pareto dotyczyła problemu, dla którego można sformułować działania, jakie
należy podjąć w celu jego rozwiązania lub poprawy istniejącej sytuacji, postępujemy według
następujących zasad:
−
w pierwszej kolejności określamy i wykonujemy działania wynikające z czynników
(przyczyn) należących do obszaru A (przyczyn głównych),
−
w drugiej kolejności analizujemy i podejmujemy działania dotyczące czynników
z obszaru B,
−
przyczyny zgrupowane w obszarze C możemy pominąć, gdyż jak wynika z doświadczeń,
usunięcie przyczyn głównych z reguły powoduje także eliminację wielu przyczyn
drobniejszych, pomimo iż związki pomiędzy nimi nie zawsze są dostrzegalne,
−
po podjęciu wymienionych działań i upływie określonego czasu, po którym można
spodziewać się efektów wprowadzonych zmian, powtórnie analizujemy problem.
Wyniki analizy formułujemy w postaci wniosków pod wykresem Pareto. Oto przykład:
Tabela 2. Dane wejściowe
Lp. Przyczyna
Ilość
obserwacji
% obserwacji
1 Przyczyna
„a”
8
5
2 Przyczyna
„b”
2
1,3
3 Przyczyna
„c”
10
6
4 Przyczyna
„d”
2
1,3
5 Przyczyna
„e”
76
48
6 Przyczyna
„f”
38
24
7 Przyczyna
„g”
16
10
8 Przyczyna
„h”
7
4
9 Przyczyna
„i”
1
0,4
Łącznie
160 100
Tabela 3. Dane po uporządkowaniu
Lp.
Przyczyna Ilość
obserwacji
% obserwacji
% sumarycznie
1
przyczyna „e”
76
48
48
2
przyczyna „f”
38
24
72
3
przyczyna „g”
16
10
82
4
przyczyna „c”
10
6
88
5
przyczyna „a”
8
5
93
6
przyczyna „h”
7
4
97
7
przyczyny „inne” (b+d+i)
5
3
100
Łącznie
160 100
„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
29
„e” „f” „g” „c“ „a“ „h“ „inne“
Rys. 2. Wykres Pareto
Tabela 1 przedstawia dane wejściowe, Tabela 2 – dane wejściowe po uporządkowaniu
(malejąco). Wykres (Rys. 2) wyraźnie pokazuje, gdzie należy upatrywać głównych przyczyn
interesującego nas zjawiska (przyczyny e, f, g). Charakterystycznym jest, że metoda ta
wskazuje przyczyny wiodące, powodujące istotnie przeważającą ilość zdarzeń. W ten sposób
skupiamy siły i środki na działaniu, które da największy efekt.
Podstawowym pojęciem statystyki jest populacja, czyli zbiór elementów podlegających
badaniu lub szacowaniu. Elementy te są badane lub szacowane ze względu na pewną cechę
ilościową lub jakościową (np. kolor). Cechę jakościową możemy zawsze zamienić sztucznie
na cechę ilościową przypisując poszczególnym jej wartościom wybraną liczbę (np. kolor
żółty to 0, a czerwony, to 1). Podobnie cechę ilościową możemy zawsze zamienić sztucznie
na cechę jakościową (np. jeśli badamy wytrzymałość prętów metalowych na rozerwanie, to
ustalamy pewną wartość siły rozrywającej, poniżej której pręt jest złej jakości, a pozostałe są
dobrej jakości).
W statystyce interesują nas nie własności poszczególnych elementów populacji ale
własności populacji traktowanej jako całość. Np. w przypadku wymienionych wyżej prętów
może nas interesować jak duża ich część nie spełnia wymagań jakościowych i dalej: jaka
może być tego przyczyna. W takich przypadkach stosujemy jedną z metod opisanych w tym
rozdziale.
Oprócz tego mogą nas również interesować pewne parametry liczbowe obrazujące
populację, takie jak:
a) Wartość średnia wybranej cechy liczbowej x:
m =
(
Σ
x
k
)/
N,
gdzie k=1,....N, N – liczność populacji, x
k
- wyniki pomiaru cechy x
a)
Wariancja tej cechy:
υ
2
=
[
Σ
(
x
k
–m)
2
]
/
N
gdzie k = 1,....N, N – liczność populacji, m – wartość średnia, x
k
- wyniki pomiaru cechy x
24%
10%
6%
5%
4% 3%
80
48%
Linia
Lorentza
N
160
skutek
100
50
Obszar A
Obszar B
Obszar C
%
>80
28%
przyczyna

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
30
Parametr ten jest miarą rozrzutu wartości cechy w stosunku do jej wartości średniej.
b)
Rozstęp, czyli różnica między największą a najmniejszą wartością cechy.
Badania statystyczne przeważnie wykonuje się na próbie (części populacji), a wnioski
wyciąga się dla całej populacji. Trzeba jednak pamiętać, że takie wnioskowanie jest
obarczone błędem tym większym im mniej reprezentatywna (czyli odzwierciedlająca
rzeczywistą strukturę populacji) jest próba.
Aby wypełniony formularz z graficznym przedstawieniem wyników stał się użytecznym
dokumentem powinien być zaopatrzony w
następujące informacje: czego dotyczą
przedstawione dane, jakie dokumenty bazowe posłużyły do wykonania statystyki, kiedy,
gdzie i w jaki sposób dane zostały zebrane, kto zebrał wyniki, wnioski.
4.3.2.
Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń.
1.
Jakiemu celowi może służyć statystyczne opracowywanie wyników?
2.
Na czym polega tworzenie listy kreskowej?
3.
Na czym polega analiza metodą tworzenia histogramu?
4.
Na czym polega analiza Pareto-Lorentza? Do czego może służyć?
5.
Jakie znasz parametry statystyczne charakteryzujące populację?
4.3.3.
Ćwiczenia
Ćwiczenie 1
Na podstawie rzeczywistej dokumentacji wyrobu medycznego:
a)
wskaż dokumenty, z których można odczytać dane służące do kontroli procesu
wytwarzania wyrobu i do kontroli końcowej,
b)
wykonaj formularz/formularze karty kontrolnej wyrobu do kontroli przebiegu procesu
produkcyjnego i sprawdzenia końcowego zgodności wyrobu z
wymaganiami
dokumentacyjnymi,
c)
przygotuj spis narzędzi i urządzeń potrzebnych do przeprowadzenia omawianych badań,
d)
przeprowadź zadaną serię badań egzemplarzy wyrobu i opracuj ich statystykę pod kątem
spełnienia wymagań jakościowych w procesie produkcji.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
znaleźć i spisać podstawowe cechy wyrobu i jego punkty kontrolne na podstawie
posiadanej dokumentacji,
2)
przygotować formularz karty kontrolnej wyrobu,
3)
znaleźć i spisać metody kontroli i pomiaru cech wyrobu kontrolowanych podczas, oraz
po zakończeniu produkcji na podstawie posiadanej dokumentacji,
4)
określić (spis), jakie narzędzia i przyrządy pomiarowe będą konieczne do badań podczas
całego procesu produkcji wyrobu, a jakie podczas kontroli końcowej,
5)
wykonać wyznaczoną ilość wskazanego rodzaju badań wyrobu elektromedycznego
i zrobić statystykę wyników.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
31
Wyposażenie stanowiska pracy:
−
zestawy dokumentacji technicznej różnych wyrobów dla różnych grup,
−
przykładowa, wypełniona karta kontrolna wyrobu elektromedycznego,
−
wyrób – przedmiot badań – jeśli to możliwe,
−
stanowisko do badań – jeśli to możliwe,
−
poradnik dla ucznia,
−
materiały biurowe.
4.3.4.
Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1)
wykonać statystykę wyników pomiarów metodą listy kreskowej?
!
!
2)
opisać na czym polega metoda analizy danych Pareto–Lorenza?
!
!
3)
Wwykonać analizę metodą Pareto–Lorenza i wyciągnąć na jej podstawie
wnioski?
!
!
4)
określić, czym jest zbiór wykonanych pomiarów określonego parametru
produktu?
!
!

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
32
4.4.
Zasady tworzenia metryk dla aparatury medycznej
4.4.1.
Materiał nauczania
Wyroby medyczne muszą być odpowiednio oznaczone tak, aby same wyroby i ich
ewentualne części składowe były łatwo identyfikowalne.
Wymóg ten jest związany bezpośrednio z bezpieczeństwem użytkowania wyrobów oraz
z
systemem nadzorowania wyrobów medycznych ustalonym w dyrektywach Unii
Europejskiej, a tym samym w Ustawie o wyrobach medycznych i w przepisach określających
sposób działania Urzędu
Rejestracji. Jednym z najważniejszych elementów wyrobu
medycznego jest z tego powodu tzw. metryczka wyrobu medycznego (etykieta, tabliczka
znamionowa).
Metryczka wyrobu medycznego stanowi jeden z dowodów w przypadku kontroli oraz
w przypadku postępowania podejmowanego po wystąpieniu incydentu medycznego. Wyrób
powinien być oznakowany w sposób czytelny i niezmywalny. Na wyrobach
elektromedycznych jest to na ogół tabliczka, naniesiona w sposób możliwie trwały na
obudowę urządzenia, na zewnątrz wyrobu. Jeśli wyrób jest zbyt mały aby można było na nim
zmieścić wszystkie wymagane informacje – wytwórca musi znaleźć inny sposób spełniający
wymagania tak, aby użytkownik mógł wykazać, że wyrób, którego używa jest wyrobem
którego dotyczą określone informacje.
Wszystkie informacje i oznaczenia zamieszczane na metryczce urządzenia muszą być
albo zgodne z symbolami określonymi w normach zharmonizowanych (np. EN 980 Symbole
graficzne do stosowania w oznakowaniu wyrobów medycznych) albo ich opisy muszą
znajdować się w dokumentacji dostarczonej użytkownikowi wraz z wyrobem medycznym.
Wszystkie jednostki miar użyte na metryczce muszą być jednostkami układu SI lub
innymi, jeśli ich użycie jest zgodne z Dyrektywą Rady Europy 80/181/EEC.
Na tyle na ile jest to praktyczne i właściwe, informacja potrzebna do stosowania wyrobu
w sposób bezpieczny powinna być podana na samym wyrobie lub jego opakowaniu.
Metryczka (etykieta) wyrobu powinna zawierać następujące informacje:
−
nazwę lub nazwę handlową wyrobu,
−
nazwę i adres producenta
−
nazwę i adres podmiotu odpowiedzialnego, jeśli jest to inny podmiot niż wytwórca
wyrobu,
−
kod partii, najlepiej poprzedzony międzynarodowym symbolem LOT lub numer seryjny
wyrobu – poprzedzony symbolem SN,
−
wskazanie daty (w formacie rok, miesiąc), do której wyrób może być bezpiecznie
używany, jeśli to odpowiednie w odniesieniu do wyrobu,
−
jeśli wytwórca nie podaje daty z poprzedniego opisu – należy podać rok produkcji
wyrobu,
−
jeśli to właściwe – zaznaczenie, że wyrób jest do jednorazowego użytku,
−
oznakowanie CE odpowiednie do klasy ryzyka wyrobu, wykonane zgodnie z wzorcem
podanym w dokumentach prawnych.
Szczególną cechą wyrobu jest jego jałowość. Tę cechę mogą posiadać również wyroby
elektromedyczne (tzw. aktywne implanty). Ponieważ mogą być obecne na rynku wyroby,
nawet pochodzące od tego samego producenta, a sprzedawane zarówno w stanie jałowym jak
i nie jałowym wymaga się, aby metryka (etykieta) wyrobu medycznego umożliwiała
rozróżnienie takich wyrobów. Wskazaniem jest oznaczenie wyrobów udostępnianych
w stanie sterylnym z metryczką zawierającą słowo STERILE, zrozumiałe bez dodatkowych
wyjaśnień we wszystkich językach Unii.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
33
Dodatkowe wskazania dotyczą wyrobów implantowanych. Dla takich wyrobów
wymagane jest bezwzględnie, aby wyrób był opatrzony kodem, dzięki któremu wyrób i jego
producent mogą być jednoznacznie zidentyfikowane bez interwencji chirurgicznej. Konieczne
informacje, jakie powinny być możliwe do uzyskania poprzez odczytanie metryczki lub
zamieszczonego na niej kodu to:
−
wytwórca wyrobu,
−
typ wyrobu,
−
rok lub data produkcji wyrobu.
Inne dane, jakie mogą zawierać metryczki wyrobów medycznych lub inne oznaczenia
umieszczane na wyrobie przez jego wytwórcę to np.:
−
obecność w wyrobie substancji niebezpiecznych,
−
konieczności zachowania ostrożności w stosunku do opakowania wyrobu (wyrób
dostarczany w stanie sterylnym).
W razie, gdy mamy do czynienia z wyrobem złożonym (systemem lub zestawem
medycznym) – każda jego część musi być tak oznaczona, aby było wiadomo, że jest częścią
określonego egzemplarza wyrobu, jeśli inne podłączenie mogłoby stworzyć zagrożenie
podczas używania wyrobu. To samo dotyczy odłączalnych części wyrobów medycznych.
W przypadku wyrobów elektromedycznych istnienie części odłączalnych to bardzo częsty
przypadek. Zasadą jest takie oznaczenie poszczególnych części wyrobu tam, gdzie to tylko
możliwe, aby uniemożliwić przypadkowe połączenie części różnych wyrobów niekoniecznie
nawet tego samego typu oraz uniemożliwić niewłaściwe podłączenie (np. do niewłaściwego
gniazda).
Dodatkowo oznacza się wyroby medyczne wykonywane tylko na zamówienie, tzn.
przeznaczone dla konkretnego pacjenta. Dokładną definicję takiego wyrobu podaje ustawa
[2].
Inny przypadek stanowi wyrób przeznaczony do badań klinicznych, na którym musi się
znaleźć napis „wyrób wyłącznie do badań klinicznych”.
Podstawowe zasady tworzenia metryk i znakowania wyrobów medycznych zawarte są
przede wszystkim w wymaganiach zasadniczych rozporządzenia [2]. Zagadnienia te
omawiają także szczegółowo normy zharmonizowane [13, 14]. Norma [14] podaje
wskazówki dotyczące prawidłowego przeniesienia wymagań zasadniczych na oznaczenie
wyrobu medycznego.
Ważne jest również rozporządzenie [6], które podaje wzór oznakowania wyrobu
medycznego znakiem CE. Znak ten, oprócz tego, że musi mieć kształt określony w tym
dokumencie, powinien być naniesiony ma wyrób lub opakowanie w sposób trwały.
4.4.2.
Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń.
1.
Jakie dane powinna zawierać tabliczka znamionowa (inaczej: etykieta lub metryka)
wyrobu medycznego?
2.
Jakie normy i przepisy prawne regulują rodzaj danych umieszczanych na metryce
i sposób ich przedstawiania?
3.
Kiedy wyrób medyczny należy oznakować znakiem CE z numerem? Co oznacza ten
numer?
4.
Jak muszą być oznakowane elementy wyrobu medycznego składającego się z kilku
odłączalnych części?

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
34
4.4.3.
Ćwiczenia
Ćwiczenie 1
Opracuj tabliczkę znamionową (etykietę, metrykę) dla wskazanego wyrobu
elektromedycznego, zgodnie z wymaganiami prawa i norm zharmonizowanych.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
wypisać jakiego rodzaju informacje muszą się znaleźć na metryczce wyznaczonego dla
grupy wyrobu,
2)
znaleźć te informacje w posiadanym zbiorze dokumentacji wyrobu,
3)
wypisać te informacje, których brakuje w dokumentacji wyrobu, a powinny się znaleźć
na projekcie metryczki,
4)
zaprojektować tabliczkę.
Wyposażenie stanowiska pracy:
−
tekst normy PN–EN 980 „Symbole graficzne do stosowania w oznakowaniu wyrobów
medycznych”,
−
tekst normy PN–EN 1041 „Informacja dostarczana przez producenta wraz z wyrobem
medycznym”,
−
różne części dokumentacji wyrobu zawierające potrzebne informacje ale nie zawierające
projektów tabliczek i etykiet,
−
stanowisko komputerowe z oprogramowaniem umożliwiającym zaprojektowanie
metryczki,
−
poradnik dla ucznia.
Ćwiczenie 2
Opracuj komplet tabliczek znamionowych (etykiet, metryk) dla wskazanego zestawu lub
systemu, będącego wyrobem elektromedycznym, zgodnie z wymaganiami prawa i norm
zharmonizowanych, w oparciu o dokumentację techniczną wyrobu.
Sposób wykonania ćwiczenia
Aby wykonać to ćwiczenie powinieneś:
1)
wypisać jakiego rodzaju informacje muszą się znaleźć na metryczkach poszczególnych
części składowych wyrobu wyznaczonego dla grupy,
2)
znaleźć te informacje w posiadanym zbiorze dokumentacji wyrobu,
3)
wypisać te informacje, których brakuje w dokumentacji wyrobu, a powinny się znaleźć
na projektach metryczek,
4)
zaprojektować metryczki.
Wyposażenie stanowiska pracy:
−
tekst normy PN–EN 980 „Symbole graficzne do stosowania w oznakowaniu wyrobów
medycznych”,
−
tekst normy PN–EN 1041 „Informacja dostarczana przez producenta wraz z wyrobem
medycznym”,
−
różne części dokumentacji wyrobu (zestawu lub systemu) zawierające potrzebne
informacje ale nie zawierające projektów tabliczek i etykiet,

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
35
−
stanowisko komputerowe z oprogramowaniem umożliwiającym zaprojektowanie
metryczek,
−
poradnik dla ucznia.
4.4.4.
Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1)
zidentyfikować jednostkę notyfikowaną, która brała udział w ocenie
zgodności wyrobu?
!
!
2)
sprawdzić prawidłowość oznakowania wyrobu medycznego?
!
!
3)
sprawdzić, czy wyrób medyczny został prawidłowo wprowadzony do
obrotu?
!
!
4)
sprawdzić czy wszystkie części składowe należą do tego systemu
medycznego?
!
!
5)
określić, jaka jednostka notyfikowana brała udział w ocenie zgodności
na podstawie metryczki lub etykiety wyrobu medycznego?
!
!

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
36
4.5.
Wymagania dotyczące instrukcji użytkowania wyrobu
medycznego
4.5.1.
Materiał nauczania
Instrukcja
używania lub użytkowania wyrobu medycznego jest dokumentem jaki musi
być dostarczony użytkownikowi razem z wyrobem medycznym. W zależności od rodzaju
wyrobu, zagrożenia jakie stwarza i stopnia komplikacji funkcjonalnej wyrobu może to być
dokument wydany w formie podręcznika, broszury lub ulotki – rozszerzonej etykiety wyrobu.
Niezależnie od wielkości tego dokumentu musi on być opracowany tak, aby zawarte
w nim informacje spełniały wymagania prawa. Instrukcja musi podawać dane identyfikujące
wytwórcę i podmiot odpowiedzialny, dane identyfikujące instrukcję jako dokument (np.
nr edycji, data wydania) i odpowiednie oznaczenie znakiem CE. Instrukcja ta powinna
zawierać dane pozwalające na identyfikację wersji i wyrobu, którego dotyczy.
Instrukcja musi przedstawiać:
−
ogólny opis wyrobu lub wyrobu z wariantami i sposób ich rozróżniania oraz listę
wyposażenia,
−
określenie materiałów zużywalnych,
−
dane techniczne wyrobu,
−
w sposób jasny zasadę działania wyrobu oraz to, czego należy się spodziewać jako
skutku jego użycia w określony sposób lub wprowadzenia określonych danych.
−
rodzaj pacjentów, warunki medyczne, dla jakich wyrób jest przeznaczony oraz rodzaj
użytkownika, dla jakiego wyrób jest przewidziany; rodzaj użytkownika jest tu bardzo
istotny ze względu na sposób przedstawienia poszczególnych zagadnień. Inaczej bowiem
będzie pisana instrukcja dla wyszkolonego personelu medycznego, a inaczej dla
przeciętnego użytkownika, który może nie być zaznajomiony z pojęciami medycznymi.
Tak więc język instrukcji i sposób przekazywania informacji muszą być dostosowane do
poziomu osób, dla których wyrób jest przeznaczony,
−
wszystkie informacje potrzebne do sprawdzenia, czy wyrób jest właściwie zainstalowany
i może działać prawidłowo i bezpiecznie,
−
wzór metryk, etykiety i innych oznaczeń jakimi oznaczono wyrób (w instrukcji powinna
się znaleźć informacja, gdzie każda z tych etykiet i opisów powinna się znajdować),
−
określenie czasu życia lub przydatności do stosowania, przestrogi, ostrzeżenia
i wskazówki, wynikające przede wszystkim z podsumowania przeprowadzonych przez
wytwórcę analiz ryzyka,
−
cel wprowadzenia substancji medycznej i sposób jej działania, jeśli wyrób zawiera taką
substancję (dotyczy to jednak tylko tych przypadków, gdzie substancja ta ma wpływ na
ciało pacjenta lub operatora, wspomagając działanie urządzenia),
−
jeśli wyrób wymaga sterylizacji instrukcja ta powinna podawać dopuszczalne metody
sterylizacji.
Instrukcja
użytkowania ma stanowić źródło informacji o wyrobie dla użytkownika
wyrobu. W przypadku bardziej na ogół skomplikowanych wyrobów elektromedycznych
instrukcja ta powinna stanowić podstawowe źródło informacji dla personelu medycznego
i
technicznego zatrudnionego w ośrodku świadczenia usług medycznych. Zgodnie
z wymaganiami Ustawy o wyrobach medycznych, skierowanymi do użytkowników tych
wyrobów, instrukcja powinna pozwolić na pozyskanie informacji pozwalających na
wypełnienie wymagań.
Szczególnie w odniesieniu do wyrobu elektromedycznego instrukcja powinna zatem
również zawierać informacje, które umożliwią użytkownikowi:

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
37
−
prawidłowe przygotowanie urządzenia do pracy,
−
prawidłową obsługę i interpretację rodzaju podawanych wyników,
−
stwierdzenie nieprawidłowej pracy urządzenia,
−
opracowanie właściwego sposobu konserwacji i wykonywania przeglądów wyrobu,
−
wybranie właściwego sposobu postępowania ze zużytym wyrobem.
Zgodnie z Dyrektywą dokument ten powinien być dostarczony użytkownikowi w jego
języku narodowym, chociaż prawo polskie dopuszcza inny język, jeśli użytkownik wyrazi na
to zgodę. Instrukcja użytkowania musi być dokumentem jak najbardziej zrozumiałym dla
użytkownika. Źle przygotowana instrukcja użytkowania wyrobu medycznego może
spowodować wskazanie wyrobu, jako przyczyny wystąpienia incydentu medycznego
i spowodować poważne konsekwencje dla wytwórcy wyrobu lub podmiotu
odpowiedzialnego.
Jeśli wyrób medyczny, którego dotyczy instrukcja użytkowania, jest wyrobem
elektrycznym lub elektronicznym – instrukcja powinna zawierać również numer rejestracyjny
nadany podmiotowi odpowiedzialnemu przez Główny Inspektorat Ochrony Środowiska
i wskazówki wytwórcy dotyczące postępowania ze zużytym wyrobem.
Na podstawie instrukcji użytkowania powinno być możliwe opracowanie instrukcji
stanowiskowej, w której oprócz wymagań wytwórcy wyrobu medycznego mogą się znaleźć
dodatkowe wymagania i wskazówki dla personelu medycznego, wynikające z systemu
zarządzania jakością obowiązującego w ośrodku świadczącym usługi medyczne lub z innych
przesłanek. Jeśli informacje zawarte w instrukcji użytkowania wydają się niekompletne –
należy złożyć u wytwórcy zamówienie na odpowiednie szkolenie.
W skrajnych wypadkach można zgłosić, w ramach gwarancji, reklamację dotyczącą złej
jakości dokumentacji dostarczanej z wyrobem medycznym.
Podstawowe zasady budowy instrukcji użytkowania wyrobów medycznych zawarto
w wymaganiach zasadniczych (rozporządzenie [4]). Zagadnienia te omawia także norma
zharmonizowana [14]. Podaje ona wskazówki dotyczące prawidłowego przeniesienia
wymagań zasadniczych na zawartość instrukcji użytkowania. Należy również przypomnieć
sobie wiadomości z punktu 4.2.3 niniejszego poradnika.
4.5.2.
Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń.
1.
Jaki dokument prawny zawiera wymagania dotyczące instrukcji użytkowania wyrobu
medycznego?
2.
Jakie elementy powinna zawierać instrukcja użytkowania?
3.
Czy instrukcja użytkowania powinna zawierać informacje dotyczące likwidacji wyrobu
medycznego?
4.5.3.
Ćwiczenia
Ćwiczenie 1
Opracuj „szkielet” instrukcji użytkowania dla wskazanego wyrobu elektromedycznego,
zgodnie z wymaganiami prawa i odpowiednich norm.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
zapoznać się z otrzymanymi materiałami dotyczącymi wyrobu medycznego,
2)
przypomnieć sobie wymagania zasadnicze dotyczące tych zagadnień,

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
38
3)
utworzyć szkielet instrukcji, tzn. stronę tytułową, tytuły kolejnych rozdziałów
i dane wynikające z posiadanych dokumentów lub z innych przesłanek i wymagań.
Wyposażenie stanowiska pracy:
−
zestaw dokumentów prawnych,
−
karta katalogowa i deklaracja zgodności wyrobu,
−
stanowisko komputerowe z edytorem tekstu,
−
poradnik dla ucznia.
4.5.4.
Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1)
wyszukać informację o przeznaczeniu wyrobu medycznego?
!
!
2)
wskazać, czego brakuje w instrukcji użytkowania wyrobu medycznego?
!
!
3)
wymienić, jakie informacje powinna zawierać instrukcja użytkowania?
!
!
4)
uzasadnić dlaczego przy tworzeniu instrukcji użytkowania należy wziąć
pod uwagę rodzaj użytkownika, dla którego jest przeznaczony wyrób?
!
!

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
39
4.6.
Instrukcja dotycząca uruchamiania i instrukcja dotycząca
serwisu wyrobu medycznego
4.6.1.
Materiał nauczania
Ważnym elementem dokumentacji wyrobu są instrukcja uruchamiania i instrukcja
serwisowa dla określonego wyrobu medycznego lub rodziny wyrobów. Dokumenty te
uzupełniają się wzajemnie, toteż może to być jeden dokument, jeśli służy wyłącznie
wytwórcy. W przypadku, gdy wyrobem zajmują się również serwisy autoryzowane,
dokumenty są zwykle rozdzielone.
Dokument ten może być różnie nazywany w różnych przedsiębiorstwach – niemniej
jednak zawsze stanowi on przeniesienie wymagań normy zakładowej, czyli szczegółowych
wymagań technicznych dla wyrobu medycznego, na praktyczne wymagania dla realizatorów
procesu produkcyjnego oraz dla pracowników serwisu wytwórcy.
Instrukcja uruchamiania i instrukcja serwisowa muszą być przygotowane tak, aby
zawierały wszystkie ważne informacje dotyczące procesu produkcyjnego i kontrolowania
jego wyników oraz były przewodnikiem dla osób wykonujących naprawy i przeglądy wyrobu
medycznego, którego dotyczą.
Instrukcja uruchamiania i instrukcja serwisowa powinny zawsze mieć oznakowanie
wersji, datę zatwierdzenia i podpis osoby upoważnionej do jej zatwierdzania, powinna
powoływać się na dokumenty, z których czerpie dane i wymagania, cytując oznakowanie
tych dokumentów i datę ich zatwierdzenia. Powinny również zawierać określenie wyrobu lub
rodziny wyrobów, których dotyczą.
To obwarowanie związane jest z potrzebą zabezpieczenia się przed użyciem
nieprawidłowych informacji i danych lub zastosowaniem niewłaściwych metod pomiarowych
lub urządzeń pomiarowych czy narzędzi. Trzeba bowiem brać pod uwagę możliwość
wprowadzenia zmian w konstrukcji wyrobu lub zmiany wskazań i zaleceń na skutek
podjętych działań doskonalących w procesie produkcyjnym.
Niżej podano przykładowy opis struktury takiego dokumentu, łączącego obie funkcje, dla
wyrobu elektromedycznego. Dokument powinien zawierać:
−
schemat blokowy wyrobu,
−
opis działania wyrobu sporządzony w oparciu o jego schemat blokowy,
−
opis działania zespołów ilustrowany w miarę potrzeb schematami blokowo-ideowymi
wybranych układów,
−
kolejność oraz sposób montażu, uruchamiania, programowania i strojenia
poszczególnych zespołów i wyrobu,
−
określenie wymaganych wartości kontrolowanych parametrów we wskazanych punktach
wyrobu lub podzespołu wraz z opisem sposobu wykonania pomiaru i dopuszczalnego
odchylenia poszczególnych wartości.
−
wytyczne projektowe do opracowania urządzeń specjalizowanych przewidywanych do
zastosowania w procesie uruchamiania i kontroli, jeśli istnieje taka potrzeba,
−
projekty ideowe stanowisk technologicznych do uruchamiania i serwisu, pozwalające na
skonfigurowanie stanowiska przy wykorzystaniu aparatury kontrolno-pomiarowej i/lub
urządzeń lub programów specjalizowanych (tj. symulatorów, testerów, programatorów,
wyposażenia specjalnego itp.),
−
wykaz niezbędnego wyposażenia do uruchomienia lub testowania wyrobu, z podaniem
wymaganych parametrów metrologicznych i użytkowych,
−
wskazówki związane z montażem bądź demontażem mechanicznym zespołów i wyrobu,
jeśli są potrzebne.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
40
−
opis instalacji urządzenia lub systemu wraz z instalacją oprogramowania, jeśli ma to
zastosowanie dla wyrobu.
Instrukcja powinna również zawierać wzory kart pomiarowych, podlegających rejestracji
na stanowisku uruchamiania lub serwisu. Karta powinna zawierać:
−
nazwę podzespołu i wyrobu,
−
identyfikator podzespołu i wyrobu (zgodny z oznaczeniami używanymi
w przedsiębiorstwie),
−
odwołanie do wymagania, zgodnie z którym jest wykonywany pomiar,
−
identyfikatory przyrządów pomiarowych lub narzędzi, przy pomocy których
wykonywano poszczególne pomiary,
−
wyniki wykonanych pomiarów z zaznaczeniem czy zostało przekroczone dopuszczalne
odchylenie od wartości pożądanej,
−
nazwisko i podpis wykonującego zadanie,
−
datę wykonania pomiarów, miejsce na podsumowanie wyniku badania podzespołu lub
wyrobu.
Instrukcja serwisowa przygotowywana dla zewnętrznego serwisu autoryzowanego
powinna z pewnością zawierać pewne elementy instrukcji uruchamiania. Nie będzie jednak
na ogół cytowała jej w całości ze względu na możliwą potrzebę zachowania tajemnicy
technologicznej w pewnym zakresie. Wytwórca może ograniczyć zakres prac, jaki może
wykonywać serwis autoryzowany i pewne rodzaje napraw lub przeglądów zachować do
wyłącznej kompetencji serwisu producenta. Zewnętrzna instrukcja serwisowa powinna wtedy
wyraźnie podawać jakich napraw lub przeglądów te zastrzeżenia dotyczą.
Dobrym uzupełnieniem wykonywania prac na stanowiskach związanych
z
uruchamianiem wyrobów lub też dokonywaniem ich przeglądów czy napraw, jest
wykonywanie co jakiś czas podsumowania wyników w postaci analizy statystycznej. Analizę
taką można wykonać dla określonych podzespołów lub dla wyrobu, jako całości, za określony
okres czasu. Nawet proste metody analizy danych podane w Rozdziale 4.3 niniejszego
Poradnika pozwalają na wykonanie podsumowania i wyciągnięcie wniosków co do jakości
poszczególnych etapów procesu produkcyjnego.
Takiej ocenie szczególnie powinny być poddawane podzespoły stanowiące element
krytyczny dla wyrobu oraz elementy i podzespoły będące wynikiem procesów krytycznych.
W przypadku stwierdzenia niepokojących wyników kontroli parametrów podzespołów lub
wyników procesów istotną rolę może odegrać zastosowanie metody Pareto-Lorenza. Metoda
ta pozwala na zawężenie obszaru przyczyn niepożądanych zjawisk, a więc na najbardziej
skuteczne ich opanowanie. Z tego powodu instrukcja uruchamiania i instrukcja serwisowa
powinny odwołać się do instrukcji zawierającej metody statystycznej obróbki danych
stosowane w przedsiębiorstwie lub zawierać opis tych metod.
4.6.2.
Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń.
1.
Jak i dlaczego powinna być oznakowana instrukcja uruchamiania i serwisu odnosząca się
do danego wyrobu?
2.
Skąd czerpiemy dane do sporządzania takiej instrukcji?
3.
Dla kogo instrukcja jest przeznaczona i w jakim celu jest sporządzana?
4.
Jakie elementy powinna zawierać instrukcja uruchamiania i serwisu?

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
41
4.6.3.
Ćwiczenia
Ćwiczenie 1
Opracuj „szkielet” instrukcji serwisowej dla wskazanego wyrobu elektromedycznego,
zgodnie z wymaganiami prawa i odpowiednich norm.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
zapoznać się z otrzymanymi materiałami dotyczącymi wyrobu medycznego,
2)
przypomnieć sobie wymagania zasadnicze dotyczące tych zagadnień i wiadomości
z poradnika dla ucznia,
3)
utworzyć „szkielet” instrukcji, tzn. stronę tytułową, tytuły kolejnych rozdziałów i dane
wynikające z posiadanych dokumentów lub z innych przesłanek i wymagań.
Wyposażenie stanowiska pracy:
−
zestaw dokumentów prawnych – szczególnie Rozporządzenie [4].,
−
karta katalogowa (ulotka) i deklaracja zgodności wyrobu,
−
stanowisko komputerowe z edytorem tekstu,
−
poradnik dla ucznia.
4.6.4.
Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1)
wymienić podstawowe informacje jakie powinny się znaleźć w
instrukcji uruchamiania i instrukcji serwisu?
!
!
2)
uzasadnić do czego są potrzebne poszczególne dane?
!
!
3)
opisać, co powinna zawierać karta pomiarowa wypełniana przy
uruchamianiu lub serwisowaniu podzespołu lub wyrobu medycznego?
!
!
4)
określić, dlaczego metody statystyczne mogą być przydatne jako
narzędzie podsumowujące wyniki prac związanych z uruchamianiem lub
serwisowaniem wyrobu medycznego?
!
!

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
42
4.7.
Postępowanie na stanowisku pracy
4.7.1.
Materiał nauczania
Wymagania dotyczące instrukcji postępowania na stanowisku pracy:
Omawiane
postępowanie na stanowisku pracy może dotyczyć:
−
stanowiska dla personelu medycznego, wyposażonego w dany wyrób medyczny
(urządzenie będące wyrobem medycznym),
−
stanowiska serwisowego w ośrodku świadczącym usługi zdrowotne,
−
stanowiska serwisowego u wytwórcy, lub też
−
stanowiska produkcyjnego bądź kontrolnego u wytwórcy.
Jeśli pracodawca zdecyduje, że któraś z tych instrukcji jest potrzebna dla danego
stanowiska pracy instrukcję tę należy opracować starannie, gdyż od tej pory staje się ona nie
tylko dokumentem przekazującym wymagania pracodawcy pracownikowi, ale również może
się stać podstawą ewentualnych roszczeń pracownika.
Osoba
podejmująca pracę na stanowisku medycznym wyposażonym w wyrób
(urządzenie) medyczne powinna się dowiedzieć na podstawie instrukcji:
−
z jakich części składa się wyrób i jaką funkcję one pełnią,
−
jak sprawdzić, czy urządzenie lub urządzenia stanowiące wyrób medyczny, są zdatne do
użytku i sprawne,
−
jakie materiały są potrzebne do wykonywania pracy na tym stanowisku,
−
jak przygotować stanowisko do pracy,
−
jakie czynności i, jeśli to odpowiednie w jakiej kolejności należy wykonywać,
uwzględniając przygotowanie pacjenta do badania lub terapii,
−
jak prawidłowo zakończyć pracę urządzenia,
−
jakie czynności porządkowe lub konserwacyjne należą do obowiązków personelu
medycznego pracującego na tym stanowisku i w jaki sposób je wykonać,
−
jaka dokumentacja i w jakiej formie powinna powstać jako dokumentacja wykonanego
badania lub terapii,
−
jaka dokumentacja i w jakiej formie powinna powstać jako dokumentacja prawidłowości
użytkowania urządzenia w wyniku pracy na stanowisku,
−
jakie obowiązują zasady bhp przy wykonywaniu tej pracy,
−
jakie zagrożenia niesie praca na tym stanowisku dla pracownika medycznego,
−
jakie zagrożenia stwarza stanowisko dla pacjenta, osób trzecich, środowiska i samego
wyrobu,
−
jak prawidłowo reagować na sytuacje awaryjne (wystąpienie zagrożenia).
Osoba
podejmująca pracę na stanowisku serwisowym związanym z określonym
wyrobem (urządzeniem) medycznym powinna się dowiedzieć na podstawie instrukcji:
−
jaki rodzaj pracy jest wykonywany na tym stanowisku,
−
jakie urządzenia i/lub narzędzia oraz materiały są potrzebne do wykonywania pracy na
tym stanowisku,
−
jak sprawdzić, czy urządzenia pomiarowe i/lub narzędzia będące wyposażeniem
stanowiska są sprawne i dopuszczone do użytku,
−
jak przygotować stanowisko do pracy,
−
jakie czynności i, jeśli to odpowiednie w jakiej kolejności należy wykonywać,
−
jak prawidłowo zakończyć pracę,
−
jakie czynności porządkowe lub konserwacyjne należą do obowiązków osoby pracującej
na tym stanowisku i w jaki sposób je wykonać,

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
43
−
jaka dokumentacja i w jakiej formie powinna powstać jako dokumentacja prawidłowego
wykonania pracy,
−
jakie obowiązują zasady bhp przy wykonywaniu tej pracy,
−
jakie zagrożenia niesie praca na tym stanowisku dla pracownika,
−
jakie zagrożenia stwarza stanowisko dla osób trzecich i środowiska,
−
jak prawidłowo reagować na sytuacje awaryjne (wystąpienie zagrożenia).
Być może w niektórych okolicznościach listy te okażą się niepełne. Pozostałe dwa
rodzaje instrukcji będą miały strukturę bardzo zbliżoną do drugiego z przedstawionych
wzorców.
Jak
widać w ostatnich punktach obu tych instrukcji wykorzystano elementy instrukcji
postępowania awaryjnego. Jest ona nierozerwalnie związana z instrukcjami opisującymi pracę
na poszczególnych stanowiskach. Może jednak być znacznie szersza (np. dotyczyć całego
oddziału w ośrodku zdrowia lub wydziału w firmie produkcyjnej), a wskazane wyżej
elementy mogą być jedynie jej fragmentem.
Źródłem informacji do skonstruowania instrukcji stanowiskowych mogą być różne
dokumenty w zależności od tego, z jakiego rodzaju stanowiskiem mamy do czynienia.
Stanowisko medyczne i techniczne w ośrodku świadczącym usługi zdrowotne, musi
bazować na dokumentach dostarczonych z wyrobem medycznym i informacjach pozyskanych
w ramach szkolenia związanego z zakupem wyrobu.
W przypadku producenta lub serwisu autoryzowanego istnieje szerszy dostęp do
dokumentacji wyrobu, w tym nawet dostęp do instrukcji uruchamiania i serwisowej wyrobu,
którą można w całości lub w części umieścić w instrukcji stanowiskowej.
Bez
względu na to czy pracodawca w ośrodku świadczącym usługi zdrowotne zdecyduje
się na przygotowanie instrukcji stanowiskowej czy nie, musi przy zakupie urządzenia
będącego wyrobem medycznym zwrócić uwagę, czy instrukcja użytkowania zawiera
wszystkie wymagane i potrzebne z punktu widzenia ośrodka informacje. Brak potrzebnych
informacji należy zgłosić dostawcy wyrobu medycznego.
W przypadku instrukcji stanowiskowej w przedsiębiorstwie wytwórcy wyrobu
medycznego powinny być uwzględnione wymagania obowiązującego tam systemu
zarządzania jakością.
4.7.2.
Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń.
1.
Jakiemu celowi służy instrukcja stanowiskowa?
2.
Jakie informacje powinna zawierać w zależności od stanowiska, jakiego dotyczy?
3.
Dlaczego instrukcja konserwacji wyrobu medycznego jest taka ważna?
4.
Skąd wziąć dane do napisania instrukcji konserwacji wyrobu medycznego?
5.
Co wolno robić personelowi szpitala w ramach konserwacji lub naprawy wyrobu, a co
może robić tylko autoryzowany serwis?
4.7.3.
Ćwiczenia
Ćwiczenie 1
Posługując się instrukcją użytkowania wyrobu i mając do dyspozycji wyrób
elektromedyczny:
a)
opisz (w formie instrukcji stanowiskowej) sposób przygotowania stanowiska pracy dla
personelu medycznego, do używania wskazanego wyrobu medycznego i
sposób
przygotowania wyrobu medycznego do użycia, lub
b)
przygotuj instrukcję konserwacji wyrobu.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
44
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
przygotować wybrany typ instrukcji posługując się instrukcją użytkowania wyrobu
i opisem rodzaju ośrodka medycznego i pracowni, w której jest stanowisko pracy,
2)
wypisać te elementy instrukcji, których nie udało się ustalić, a powinny się tam znaleźć.
Wyposażenie stanowiska pracy:
−
instrukcja użytkowania wyrobu,
−
wyrób elektromedyczny lub jego dokumentacja zdjęciowa z opisem,
−
opis rodzaju ośrodka, w którym użytkowany jest wyrób elektromedyczny z opisem
pracowni, w której jest stanowisko pracy,
−
stanowisko komputerowe z edytorem tekstu (np. WORD) i edytorem graficznym (do
przygotowania ewentualnych schematów blokowych),
−
cyfrowy aparat fotograficzny i skaner do przygotowania zdjęć oraz przeniesienia
rysunków z instrukcji użytkowania wyrobu i zamieszczenia ich w przygotowywanych
instrukcjach,
−
poradnik dla ucznia.
Ćwiczenie 2
Posługując się instrukcją użytkowania i instrukcją serwisową określonego wyrobu
elektromedycznego przygotuj dokumentację opisującą stanowisko pracy potrzebne do
przeprowadzenia serwisu i pełnego, autoryzowanego przeglądu tego wyrobu.
Sposób wykonania ćwiczenia
Aby wykonać to ćwiczenie powinieneś:
1)
opracować dokumentację opisującą stanowisko pracy potrzebne do przeprowadzenia
serwisu i pełnego, autoryzowanego przeglądu przydzielonego wyrobu, biorąc pod uwagę
wymagania zamieszczone w rozporządzeniu [4],
2)
zaprezentować wykonane ćwiczenie.
Wyposażenie stanowiska pracy:
−
instrukcja użytkowania i instrukcja serwisowa określonego wyrobu elektromedycznego,
−
tekst Rozporządzenia [4].,
−
stanowisko komputerowe z edytorem tekstu i oprogramowaniem umożliwiającym
przygotowywanie schematów i rysunków,
−
skaner pozwalający uczniom przenieść rysunki z oryginalnych instrukcji wyrobu,
−
poradnik dla ucznia.
4.7.4.
Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1)
stwierdzić, na podstawie dokumentacji dotyczącej wyrobu medycznego
(w tym dokumentów instalacyjnych), jakie są uprawnienia
użytkowników i obsługi technicznej w ośrodku zdrowia w odniesieniu
do konserwacji i naprawy tego wyrobu?
!
!

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
45
2)
określić, co powinna zawierać instrukcja stanowiskowa dla stanowiska
medycznego?
!
!
3)
określić, co powinna zawierać instrukcja stanowiskowa dla stanowiska
serwisowego w ośrodku zdrowia?
!
!
4)
opisać, czym różni się serwis w ośrodku zdrowia od serwisu
autoryzowanego w odniesieniu do wyrobów medycznych?
!
!

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
46
4.8.
Instrukcja postępowania awaryjnego
4.8.1.
Materiał nauczania
Wymagania dotyczące instrukcji postępowania awaryjnego:
Instrukcja
postępowania awaryjnego powinna odnosić się do wszystkich niepożądanych
zjawisk stwarzających zagrożenie dla zdrowia lub życia osób znajdujących się lub
pracujących w miejscu, którego instrukcja dotyczy oraz dla środowiska.
Jeśli odniesiemy rozważania do stanowisk pracy wymienionych w Rozdziale 4.7 tego
Poradnika, to:
−
w przypadku stanowiska dla personelu medycznego, wyposażonego w urządzenie będące
wyrobem medycznym, instrukcja powinna brać pod uwagę niebezpieczeństwa grożące:
a)
pacjentowi,
b)
użytkownikowi,
c)
osobom trzecim,
d)
środowisku,
e)
czy wreszcie samemu wyrobowi medycznemu (może on utracić swoje istotne cechy
warunkujące jego prawidłową pracę i stać się z kolei sam źródłem zagrożenia),
−
w przypadku stanowisk serwisowych lub stanowisk produkcyjnych zagrożenia są
podobne w ośrodku świadczącym usługi zdrowotne oraz u wytwórcy, czy w serwisie
autoryzowanym: instrukcja powinna brać pod uwagę niebezpieczeństwa grożące:
a)
pracownikom,
b)
osobom trzecim,
c)
środowisku,
d)
wyrobowi medycznemu, który może ulec uszkodzeniu, a wtedy, tak jak w poprzednim
wypadku – może utracić swoje istotne cechy warunkujące jego prawidłową pracę
i stać się z kolei sam źródłem zagrożenia.
Zbierając materiały do przygotowania instrukcji postępowania awaryjnego należy
uwzględnić wszystkie możliwe ryzyka istniejące lub mogące się pojawić w otoczeniu, dla
którego przygotowywana jest instrukcja. Trzeba wziąć pod uwagę np.:
−
zagrożenia płynące ze środowiska, a w tym np.:
a)
wynikające z rozmieszczenia stanowisk pracy,
b)
wynikające z wyposażenia stanowisk pracy,
c)
zdarzenia losowe, takie jak uderzenie pioruna lub wybuch pożaru,
−
wynikające z konstrukcji wyrobu (np. to, że wyrób zawiera niebezpieczną substancję,
która może wydostać się na zewnątrz podczas prowadzenia prac),
−
będące wynikiem błędu lub niekompetencji pracowników,
−
wynikające z zachowania się osób postronnych.
W wyniku analizy ryzyka powinno się otrzymać także następujące informacje:
−
określenie potencjalnych szkód i ich przyczyn (co często umożliwia określenie i podjęcie
odpowiednich środków zaradczych),
−
uszeregowania zagrożeń według poziomu ryzyka,
−
wykrycie i wskazanie dróg usunięcia zagrożeń lub ograniczenia ich skutków,
−
zwiększenia bezpieczeństwa osób, środowiska i wyrobu medycznego,
−
wskazania potrzeb w zakresie rodzaju potrzebnych informacji i szkoleń personelu.
Analizę ryzyka można wykonać według zasad podobnych do przedstawionych dla
wyrobu medycznego.
Najbardziej prawidłowym wydaje się w tym przypadku przeprowadzenie analizy ryzyka
przez zespół osób znających przedsiębiorstwo lub ośrodek świadczący usługi zdrowotne, jego

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
47
wyposażenie i metody pracy, przepisy bhp, kodeks pracy, a poza tym charakteryzujące się
dużą elastycznością myślenia i zdolnością przewidywania.
Prowadząc analizę ryzyka należy:
−
zapoznać jej uczestników sesji z miejscem, dla którego ma być przeprowadzona analiza,
jego wyposażeniem, stosowaną technologią lub rodzajem pracy, rodzajem pracującego
w tym otoczeniu personelu oraz możliwością pojawienia się innych osób.
−
zidentyfikować wszystkie możliwe w danym wypadku źródła zagrożeń, zdarzenia, jakie
mogą zaistnieć i możliwe konsekwencje tych zdarzeń, tworząc łańcuchy:
przyczyna – zdarzenie(a) – skutek
UWAGA: należy tu także wziąć pod uwagę sugestie zawarte w przepisach bhp
związanych z danym miejscem lub rodzajem pracy.
−
uporządkować łańcuchy przyczynowo – skutkowe od najgroźniejszych w skutkach do
najmniej groźnych,
−
dla każdego łańcucha opracować odpowiedź na pytania:
a)
Co należy zrobić, aby wyeliminować możliwość zaistnienia przyczyny lub w jak
największym stopniu ograniczyć możliwość jej zaistnienia? Jakie mogą być
konsekwencje zastosowania takich środków lub metod?
b)
Co należy zrobić, aby wyeliminować możliwość zaistnienia niepożądanego zdarzenia?
Jakie mogą być konsekwencje zastosowania takich środków lub metod?
c)
Co należy zrobić, jeśli mimo podjętych środków nastąpi zdarzenie powodujące
niepożądany skutek?
W efekcie analizy ryzyka powinny być przeprowadzone odpowiednie działania
doskonalące, mające na celu eliminację lub zmniejszenie zagrożeń do poziomu przyjętego za
dopuszczalny. Ponieważ w większości przypadków ryzyko wynika z samej metody pracy,
stosowanej niebezpiecznej technologii, wyposażenia lub miejsca, w którym wszystko się
dzieje można przyjąć, że ryzyko jest dopuszczalne, jeśli korzyść z zastosowania określonego
działania wyraźnie przewyższa zagrożenie, albo inna metoda jest niemożliwa do
zastosowania, lub nie istnieje.
Oczywiście nie wszystkie ryzyka można wyeliminować i dla takich trzeba określić jasno
tryb postępowania w instrukcji postępowania awaryjnego.
Osoby, których dotyczy instrukcja należy uczulić przede wszystkim na zdarzenia
skutkujące najbardziej niepożądanymi konsekwencjami i dla tych zdarzeń opracować
najbardziej szczegółowy sposób postępowania mający na celu możliwie duże ograniczenie
niepożądanych skutków zdarzeń. Trzeba tu wziąć pod uwagę element psychologiczny
w przypadku wystąpienia awarii – możliwość pojawienia się paniki.
Trzeba
pamiętać, że typowo rozumiany termin „awaria” może tu być przyczyną,
zdarzeniem lub skutkiem.
4.8.2.
Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń.
1.
Jak identyfikujemy awarie jakie mogą wystąpić w danym miejscu?
2.
Co robimy w odniesieniu do zdarzeń, których nie jesteśmy w stanie wyeliminować?
3.
Jak można zwiększyć moc oddziaływania zapisów zamieszczanych w
instrukcji
postępowania awaryjnego na osoby, których instrukcja dotyczy?

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
48
4.8.3.
Ćwiczenia
Ćwiczenie 1
Na podstawie rzeczywistej dokumentacji opracuj instrukcję postępowania awaryjnego dla
stanowiska medycznego (użytkownika wyrobu) lub dla stanowiska serwisowego.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
opracować sposób postępowania dla różnych możliwych rodzajów awarii danego
urządzenia elektromedycznego lub dla różnych rodzajów awarii związanych z rodzajem
wykonywanej pracy i budową stanowiska pracy,
2)
uwzględnić zagrożenia dla pacjenta (na stanowisku medycznym), pracownika, osób
trzecich, wyrobu medycznego i środowiska w każdym z tych przypadków.
Wyposażenie stanowiska pracy:
Do opracowania instrukcji postępowania awaryjnego dla stanowiska medycznego
(użytkownika wyrobu):
−
instrukcja użytkowania wyrobu elektromedycznego,
Do opracowania instrukcji postępowania awaryjnego dla stanowiska serwisowego:
−
rzeczywista dokumentacja wybranego wyrobu elektromedycznego (instrukcja
użytkowania, instrukcja serwisowa, dokumentacja analizy ryzyka),
−
opis stanowiska i rodzaju wykonywanej na nim pracy,
−
stanowisko komputerowe z dostępem do Internetu, z edytorem tekstu i oprogramowaniem
umożliwiającym przygotowywanie schematów i rysunków,
−
Poradnik dla ucznia.
4.8.4.
Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1)
określić czym jest awaria?
!
!
2)
określić, co powinna zawierać instrukcja postępowania awaryjnego?
!
!
3)
przeprowadzić analizę stanowisk pod kątem sytuacji awaryjnych, jakie
mogą na nich wystąpić?
!
!
4)
określić, na jakie pytania powinna odpowiedzieć instrukcja
postępowania awaryjnego?
!
!

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
49
4.9.
Dokumentacja eksploatacyjna urządzenia medycznego
4.9.1.
Materiał nauczania
Możliwe są następujące rodzaje dokumentacji eksploatacyjnej:
−
dokumentacja testów eksploatacyjnych wykonywanych przez producenta w ramach oceny
zgodności wyrobu (procesu walidacji wyrobu),
−
dokumentacja testów eksploatacyjnych przewidzianych przez wytwórcę do wykonania
przez użytkownika celem sprawdzenia poprawności działania urządzenia medycznego,
−
dokumentacja eksploatacji wyrobu tworzona przez użytkownika celem wykazania
właściwego i zgodnego z prawem używania wyrobu.
Dokumentacja testów eksploatacyjnych wykonywanych przez producenta w ramach
oceny zgodności wyrobu (procesu walidacji wyrobu):
Testy eksploatacyjne, jakie przeprowadza się w ramach oceny zgodności wyrobu, wykonuje
się celem wykazania:
−
poprawności działania wszystkich założonych w projekcie wyrobu funkcji wyrobu,
−
prawidłowego działania wyrobu w ramach przewidywanego zastosowania.
Testy takie powinny obejmować:
−
sprawdzenie wszystkich funkcji pomiarowych wyrobu i poprawności wykonywanych
pomiarów,
−
sprawdzenie poprawności sterowania urządzeniami, jeśli jest ono funkcją wyrobu,
−
sprawdzenie poprawności zbierania i gromadzenia danych,
−
wszystkie możliwe do przewidzenia zachowania użytkownika i pacjenta, włączając w to
zachowania niezgodne z instrukcją użytkowania.
W
każdym przypadku wykonujący testy eksploatacyjne powinien odnotować
w określony w przedsiębiorstwie sposób rodzaj wykonanego testu, ilość powtórzeń
i zachowanie się wyrobu oraz ocenić, czy jest ono prawidłowe czy nie.
Materiał z takich testów powinien być podstawą do podjęcia ewentualnych działań
doskonalących i, jeśli zajdzie taka potrzeba – ponownej analizy ryzyka oraz wprowadzenia
koniecznych na danym etapie zmian konstrukcyjnych w wyrobie.
Dokumentacja testów eksploatacyjnych przewidzianych przez wytwórcę do wykonania
przez użytkownika celem sprawdzenia poprawności działania urządzenia medycznego:
Niektóre testy eksploatacyjne mogą być przewidziane również do samodzielnego
wykonywania przez użytkownika celem sprawdzenia poprawności działania wyrobu. Testy
takie, w zależności od stopnia ich komplikacji, wykonuje personel medyczny lub osoba
specjalnie przeszkolona w tym celu. Sposób wykonania testów, potrzebne oprzyrządowanie
i sposób interpretacji wyników powinny być w takim przypadku opisane w instrukcji
użytkowania wyrobu medycznego.
Dokumentacja eksploatacji wyrobu tworzona przez użytkownika celem wykazania
właściwego i zgodnego z prawem używania wyrobu:
Opracowanie sposobu prowadzenia dokumentacji eksploatacyjnej przez użytkownika
może być jedyną metodą na wykazanie zgodności postępowania ośrodka świadczącego usługi
zdrowotne zgodnie z Ustawą o wyrobach medycznych (Rozdz. 2, Art. 4). Ustawa mówi
bowiem o obowiązku:
−
używania wyrobu zgodnie z przewidzianym przez wytwórcę zastosowaniem określonym
w instrukcji użytkowania (lub używania),

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
50
−
zachowania należytej staranności w zakresie doboru, instalowania, uruchamiania,
dokonywania przeglądów i konserwacji wyrobu medycznego,
−
przestrzegania instrukcji użytkowania dostarczonej przez wytwórcę z wyrobem.
Instrukcja
użytkowania wyrobu powinna precyzować np.:
−
rodzaj, częstotliwość kontrolnych sposób przeprowadzania testów kontrolnych wyrobu
medycznego oraz sposób interpretowania wyników tych testów,
−
sposób codziennej i okresowej konserwacji wyrobu,
−
rodzaj i częstotliwość przeglądów zalecanych przez producenta oraz
−
okres przez jaki wyrób może być używany tzw. czas życia wyrobu, po którym powinien
on być poddany likwidacji we wskazanej przez producenta, uprawnionej do tego firmie.
Producent powinien również wskazać punkty serwisowe upoważnione przez niego do
prowadzenia napraw i kompleksowych przeglądów (serwisy autoryzowane).
Jeśli personel komórki technicznej ośrodka medycznego nie został przeszkolony
i upoważniony przez wytwórcę do prowadzenia wybranych prac serwisowych – nie wolno mu
wykonywać żadnych napraw ani przeglądów poza wskazanymi w instrukcji użytkowania jako
możliwymi do wykonania przez użytkownika i w sposób tam opisany. Osoba upoważniona
musi posiadać odpowiedni, imienny dokument potwierdzający to upoważnienie
poświadczony przez wytwórcę wyrobu lub podmiot odpowiedzialny.
Użytkownik powinien zwrócić uwagę na odpowiednie przekazanie wyrobu medycznego
do użytku (instalacja wyrobu, dokumentacja, przeszkolenie personelu). Sposób
dokumentowania eksploatacji wyrobu powinien być zdefiniowany i prowadzony w formie
nadzorowanych zapisów dla każdego używanego w ośrodkach zdrowia urządzenia będącego
wyrobem medycznym, gdyż inaczej kierownictwu ośrodka może być trudno wykazać
spełnienie wymagań Ustawy o wyrobach medycznych w razie kontroli lub wystąpienia
incydentu medycznego.
Zbiór takiej dokumentacji powinien zawierać:
−
instrukcję użytkowania wyrobu oraz inne dokumenty, jeśli zostały dostarczone
z wyrobem (jak np. deklaracja zgodności, dowód zgłoszenia do rejestru wytwórców
i wyrobów medycznych),
−
dowody odbycia szkoleń i uzyskania uprawnień przez personel, jeśli ma to zastosowanie,
−
zapisy, będące dowodem na spełnienie wymagań określonych przez wytwórcę wyrobu,
−
zapisy dotyczące nieprawidłowego zachowania się wyrobu i złożonych reklamacji oraz
odpowiedzi dostawcy wyrobu lub autoryzowanego serwisu,
−
dokumenty dotyczące przeprowadzonych napraw, łącznie z dowodami potwierdzającymi
prawidłowość ponownego uruchomienia lub instalacji, jeśli ma to zastosowanie w danym
przypadku.
Wszystkie zapisy powinny być opatrzone datą i podpisem osoby upoważnionej do ich
sporządzania.
4.9.2.
Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń.
1.
Jakie znasz rodzaje dokumentacji eksploatacyjnej wyrobu medycznego?
2.
Jakiemu celowi służą testy eksploatacyjne wykonywane w ramach oceny zgodności
wyrobu medycznego?
3.
Jakiemu innemu celowi niż ocena zgodności wyrobu, mogą służyć testy eksploatacyjne?
4.
Jakie cechy powinna mieć dokumentacja eksploatacyjna wyrobu medycznego
dokumentująca prawidłowe postępowanie z wyrobem przez użytkownika?

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
51
4.9.3.
Ćwiczenia
Ćwiczenie 1
Zgodnie z instrukcją użytkowania i instrukcją serwisową wyrobu elektromedycznego
przygotuj dokumentację eksploatacyjną dla jednostki służby zdrowia.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
zaprojektować kartę eksploatacji wyrobu medycznego w oparciu o instrukcje opracowane
w ćwiczeniu 1 z rozdziału 4.7,
2)
sprawdzić, czy prawidłowo wypełniana karta eksploatacji wyrobu medycznego z p.1)
może być dowodem na to, ze prawidłowo (zgodnie z zaleceniami producenta i ogólnymi
wymaganiami bezpieczeństwa) eksploatowano wyrób elektromedyczny – np.
w przypadku postępowania podjętego po wystąpieniu incydentu medycznego.
Wyposażenie stanowiska pracy:
−
wyniki ćwiczenia 1 z rozdz. 4.7 i prac domowych z tego ćwiczenia,
−
stanowisko komputerowe z edytorem tekstu i oprogramowaniem umożliwiającym
przygotowywanie schematów i rysunków,
−
poradnik dla ucznia.
Ćwiczenie 2
Na podstawie rzeczywistej dokumentacji technicznej wyrobu elektromedycznego,
opracuj instrukcję wykonywania testów eksploatacyjnych z uwzględnieniem sprawdzenia
poprawności funkcjonalnej wyrobu (poprawność pomiarowa, poprawność wykonywania
funkcji opisanych w instrukcji użytkowania) i sprawdzenie konsekwencji niewłaściwego
użycia wyrobu.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
znaleźć w dokumentacji wyrobu elementy, które powinny być wykorzystane w testach
eksploatacyjnych,
2)
utworzyć listę testów i opisy ich przeprowadzenia (potrzebne narzędzia – np. symulatory
sygnałów biomedycznych, rodzaj wprowadzanych danych, kolejne kroki i sposób
dokumentowania przeprowadzonego testu oraz jego wyniku),
3)
utworzyć listę testów symulujących zachowanie użytkownika niezgodne z instrukcją
użytkowania i sposób dokumentowania ich przeprowadzenia,
4)
dla każdego testu opisać kto, w jakich okolicznościach i jak często powinien go
przeprowadzać,
5)
uwzględnić wyniki testów z p. 3) w dokumencie podsumowującym wykonane testy
eksploatacyjne (np. formularz będący załącznikiem do instrukcji) przy pomocy którego
informacja zostanie przekazana producentowi lub konstruktorowi,
6)
opracować instrukcję w docelowym kształcie.
Wyposażenie stanowiska pracy:
−
dokumentacja wyrobu medycznego: instrukcja użytkowania, instrukcja serwisowa
(dokument producenta) lub instrukcja konserwacji wyrobu (dokument ośrodka
medycznego),

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
52
−
stanowisko komputerowe z edytorem tekstu i
oprogramowaniem umożliwiającym
przygotowywanie schematów i rysunków,
−
poradnik dla ucznia.
4.9.4.
Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1)
określić, co powinno się znaleźć w instrukcji wykonywania testów
eksploatacyjnych przeznaczonych do oceny zgodności wyrobu
medycznego?
!
!
2)
określić, co powinno się znaleźć w instrukcji wykonywania testów
eksploatacyjnych przeznaczonej dla użytkownika wyrobu medycznego?
!
!
3)
określić. co powinno być dokumentowane przez użytkownika wyrobu
medycznego, aby mógł on wykazać prawidłowe postępowanie
z używanym przez siebie wyrobem?
!
!
4)
wymienić obowiązki jakie nakłada na użytkownika wyrobu medycznego
Ustawa o wyrobach medycznych?
!
!
5)
wymienić skład przykładowego zbioru dokumentacji eksploatacyjnej
używanego wyrobu?
!
!

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
53
5.
SPRAWDZIAN OSIĄGNIĘĆ
INSTRUKCJA DLA UCZNIA
1.
Przeczytaj uważnie instrukcję.
2.
Podpisz imieniem i nazwiskiem kartę odpowiedzi.
3.
Zapoznaj się z zestawem zadań testowych.
4.
Test zawiera 20 zadań wielokrotnego wyboru o różnym stopniu trudności. Tylko jedna
odpowiedź jest prawidłowa.
5.
Udzielaj odpowiedzi tylko na załączonej karcie odpowiedzi.
Prawidłową odpowiedź zaznacz X (w przypadku pomyłki należy błędną odpowiedź
zaznaczyć kółkiem, a następnie ponownie zakreślić odpowiedź prawidłową),
6.
Test składa się z dwóch części o różnym stopniu trudności: I część – poziom
podstawowy, II część – poziom ponadpodstawowy.
7.
Pracuj samodzielnie, bo tylko wtedy będziesz miał satysfakcję z wykonanego zadania.
8.
Kiedy udzielenie odpowiedzi będzie Ci sprawiało trudność, wtedy odłóż jego
rozwiązanie na później i wróć do niego, gdy zostanie Ci czas wolny. Trudności mogą
przysporzyć Ci zadania: 16–20, gdyż są one na poziomie trudniejszym niż pozostałe.
Przeznacz na ich rozwiązanie więcej czasu.
9.
Na rozwiązanie testu masz 45 min.
Powodzenia
ZESTAW ZADAŃ TESTOWYCH
1.
W jakim celu wprowadzono Dyrektywy Nowego Podejścia?
a)
aby przepisy prawa były jednakowe we wszystkich krajach Unii Europejskiej.
b)
aby umożliwić swobodny i bezpieczny dla klienta i użytkownika przepływ towarów
między krajami Unii.
c)
aby towary i usługi można było sprzedawać bez kontroli.
d)
aby wyroby z różnych krajów miały te same parametry techniczne.
2.
Jaka cecha wyrobu decyduje o tym, czy jest on wyrobem medycznym?
a)
wskazane przez wytwórcę przeznaczenie wyrobu.
b)
oznakowanie wyrobu znakiem CE.
c)
klasa ryzyka.
d)
zgłoszenie wyrobu do rejestracji w Urzędzie Rejestracji Produktów Leczniczych,
Wyrobów Medycznych i Produktów Biobójczych.
3.
Co to jest proces oceny zgodności wyrobu?
a)
sprawdzenie zgodności parametrów wyrobu z wymaganiami norm
zharmonizowanych.
b)
proces zbierania dowodów na spełnienie odpowiednich dla wyrobu wymagań
zasadniczych.
c)
sprawdzenie czy wyrób jest zgodny z założeniami konstrukcyjnymi.
d)
proces oceny bezpieczeństwa wyrobu dla pacjentów, użytkowników i osób trzecich.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
54
4.
Co to są elementy krytyczne w wyrobie medycznym?
a)
Elementy, które warunkują odpowiednie wypełnianie funkcji, do jakich wyrób został
przeznaczony.
b)
Elementy zawierające substancje niebezpieczne.
c)
Elementy realizujące funkcje pomiarowe wyrobu.
d)
Elementy, które decydują o bezpieczeństwie użytkowania wyrobu lub warunkują
odpowiednie (zgodne z założeniami) wypełnianie funkcji, do jakich wyrób został
przeznaczony.
5.
Jakie dokumenty musi dostarczyć wytwórca (lub podmiot odpowiedzialny) razem
z wyrobem medycznym?
a)
deklarację zgodności.
b)
dowód zgłoszenia wyrobu do rejestru wyrobów medycznych.
c)
instrukcję użytkowania wyrobu.
d)
ulotkę reklamową.
6.
Kto może przeprowadzić pełny serwis wyrobu medycznego nie zmieniając
odpowiedzialności wytwórcy wyrobu?
a)
wytwórca wyrobu lub serwis autoryzowany przez wytwórcę wyrobu,
b)
dział techniczny placówki służby zdrowia,
c)
użytkownik,
d)
dowolny punkt serwisowy wyrobów medycznych.
7.
Producent wykonuje testy eksploatacyjne wyrobu elektromedycznego
a)
aby sprawdzić poprawność działania wszystkich funkcji wyrobu i prawidłowość jego
działania w ramach przewidywanego zastosowania wyrobu.
b)
aby sprawdzić, czy wyrób został wyprodukowany zgodnie z dokumentacją.
c)
aby sprawdzić spełnienie założeń konstrukcyjnych wyrobu.
d)
aby sprawdzić możliwość użycia wyrobu zgodnie z jego przeznaczeniem.
8.
Czy Ustawa o wyrobach medycznych stawia bezpośrednie wymagania użytkownikowi
wyrobu medycznego?
a)
stawia wymagania tylko wytwórcom i podmiotom odpowiedzialnym.
b)
tak.
c)
nie stawia żadnych.
d)
tak, dotyczą one konserwacji wyrobu.
9.
Jak powinna być przygotowana metryczka wyrobu medycznego?
a)
powinna być zamontowana w widocznym miejscu i być czytelna.
b)
powinna zawierać wymagane informacje.
c)
powinna być czytelna, niezmywalna i zawierać wymagane informacje.
d)
powinna być naniesiona w sposób nieusuwalny na wyrobie.
10.
Instrukcja stanowiskowa
a)
opisuje sposób przygotowania narzędzi i urządzeń do pracy.
b)
przekazuje pracownikowi wymagania bezpieczeństwa i higieny pracy.
c)
przekazuje pracownikowi wymagania systemu zarządzania jakością w odniesieniu do
stanowiska pracy.
d)
przekazuje pracownikowi podstawowe zasady pracy na stanowisku i podstawowe
ostrzeżenia.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
55
11.
Jaki akt prawny jest w Polsce podstawowym dla wyrobów medycznych?
a)
ustawa o wyrobach medycznych,
b)
ustawa o wyrobach medycznych wraz z rozporządzeniami,
c)
rozporządzenia Ministra Zdrowia w sprawie wymagań zasadniczych dla wyrobów
medycznych do różnego przeznaczenia, dla wyrobów do diagnostyki in vitro, dla
aktywnych wyrobów medycznych do implantacji,
d)
ustawa o ocenie zgodności wraz z rozporządzeniami.
12.
Instrukcja użytkowania wyrobu medycznego nie musi zawierać
a)
deklaracji zgodności.
b)
nazwy i adresu wytwórcy wyrobu.
c)
danych na temat rodzaju materiałów używanych razem z wyrobem medycznym.
d)
opisu przeznaczenia wyrobu.
13.
Instrukcja postępowania awaryjnego powinna opisywać
a)
wszystkie zjawiska, które mogą zagrozić ludziom lub ważnym elementom otoczenia
oraz sposób postępowania w przypadku ich wystąpienia.
b)
możliwe do przewidzenia zjawiska, które mogą zagrozić ludziom lub ważnym
elementom otoczenia oraz sposób postępowania w przypadku ich wystąpienia.
c)
możliwe do przewidzenia zjawiska, które mogą zagrozić ludziom oraz sposób
postępowania w przypadku ich wystąpienia.
d)
wszystkie zjawiska, które mogą zagrozić ludziom oraz sposób postępowania
w przypadku ich wystąpienia.
14.
Kiedy w ocenie zgodności wyrobu medycznego nie bierze udziału jednostka
notyfikowana?
a)
jeśli wyrób medyczny jest klasy I.
b)
jeśli wyrób medyczny jest klasy IIa, IIb lub III.
c)
jeśli wyrób medyczny jest klasy I i nie ma funkcji pomiarowej ani nie jest wyrobem
sterylnym.
d)
jeśli wyrób medyczny jest klasy I lub IIa.
15.
Co może być celem analizy wykonywanej metodą Pareto–Lorenza?
a)
analiza krzywej Pareto.
b)
graficzna prezentacja danych pomiarowych.
c)
dotarcie do najistotniejszych przyczyn analizowanego zjawiska.
d)
znalezienie wszystkich przyczyn analizowanego zjawiska.
16.
Jaka norma podaje metodykę prowadzenia analizy ryzyka dla wyrobów medycznych?
a)
EN ISO 9001.
b)
EN 980.
c)
EN 1041.
d)
EN ISO 14971.
17.
Efektem testów eksploatacyjnych, jeśli wykryto podczas nich nieprawidłowości
w działaniu wyrobu elektromedycznego, powinno być?
a)
analiza przyczyn wystąpienia nieprawidłowości i, jeśli to możliwe, wprowadzenie
koniecznych zmian w wyrobie.
b)
ponowne przeprowadzenie testów celem sprawdzenia wyników.
c)
usunięcie funkcji działających nieprawidłowo.
d)
analiza przyczyn wystąpienia nieprawidłowości i poprawienie jakości wykonania
wyrobu.

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
56
18.
Dokumentacja EC wyrobu medycznego to
a)
ulotka informacyjna zawierająca dane techniczne i deklaracja zgodności.
b)
część dokumentacji technicznej wymagana przez dyrektywy Unii Europejskiej.
c)
pełna dokumentacja wyrobu medycznego.
d)
część dokumentacji technicznej wymagana przez dyrektywy Unii Europejskiej,
gotowa zawsze do okazania władzom, jako dowód zgodności wyrobu.
19.
Dlaczego zakłady opieki zdrowotnej powinny prowadzić dokumentację eksploatacyjna
dla używanych w nich urządzeń medycznych?
a)
ponieważ dokumentacja taka może być jedyną metodą na wykazanie zgodności
postępowania ośrodka zdrowia z wymaganiami Ustawy o wyrobach medycznych.
b)
ponieważ wymaga tego Ustawa o wyrobach medycznych.
c)
ponieważ wymaga tego specjalne zarządzenie Ministra Zdrowia.
d)
ponieważ wymagają tego producenci wyrobów medycznych.
20.
O wyborze ścieżki oceny zgodności wyrobu medycznego decyduje
a)
klasa ryzyka wyrobu.
b)
wytwórca wyrobu, w zależności od klasy ryzyka wyrobów, oraz możliwości
przedsiębiorstwa i struktury produkcji.
c)
jednostka notyfikowana.
d)
wytwórca wyrobu, w zależności od swoich aktualnych możliwości finansowych.
„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
57
KARTA ODPOWIEDZI
Imię i nazwisko ucznia ..............................……………………………………………………..
Wykonywanie dokumentacji aparatury medycznej
Zakreśl poprawną odpowiedź.
Nr
zadania
Odpowiedź Punkty
1
a
b c d
2
a
b c d
3
a
b c d
4
a
b c d
5
a
b c d
6
a
b c d
7
a
b c d
8
a
b c d
9
a
b c d
10
a
b c d
11
a
b c d
12
a
b c d
13
a
b c d
14
a
b c d
15
a
b c d
16
a
b c d
17
a
b c d
18
a
b c d
19
a
b c d
20
a
b c d
Razem:

„
Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
58
6.
LITERATURA
1. Dyrektywa Rady Europy 93/42/EWG
2. Ustawa o wyrobach medycznych z dnia 20 kwietnia 2004, (Dz. U. 2004.93.896)
3. Rozporządzenie Ministra Zdrowia z dnia 30 kwietnia 2004 r. w sprawie klasyfikacji
wyrobów medycznych do różnego przeznaczenia (Dz. U. 2004.100.1027)
4. Rozporządzenie Ministra Zdrowia z dnia 3 listopada 2004 r. w sprawie wymagań
zasadniczych dla wyrobów medycznych do różnego przeznaczenia (Dz. U.
2004.251.2514)
4a. Rozporządzenie Ministra Zdrowia z dnia 3 listopada 2004 r. w sprawie wymagań
zasadniczych dla wyrobów medycznych do diagnostyki in vitro (Dz. U. 2004.251.2515)
4b. Rozporządzenie Ministra Zdrowia z dnia 3 listopada 2004 w sprawie wymagań
zasadniczych dla aktywnych wyrobów medycznych do implantacji (Dz. U.
2004.251.2516)
5. Rozporządzenie Ministra Zdrowia z dnia 30 kwietnia 2004 r. w sprawie wzorów
formularzy zgłoszeniowych do Rejestru wyrobów medycznych i podmiotów
odpowiedzialnych za ich wprowadzanie do obrotu i do używania oraz sposobu
przekazywania danych objętych formularzem zgłoszeniowym; (Dz. U. 2004.100.1028
z późn. zmianami)
6. Rozporządzenie Ministra Zdrowia z dnia 30 kwietnia 2004 w sprawie wzoru
oznakowania wyrobów medycznych znakiem CE (Dz. U. 2004.104.1111)
7. Rozporządzenie Ministra Zdrowia z dnia 30 kwietnia 2004 r. w sprawie sposobu
zgłaszania incydentów medycznych oraz dalszego postępowania po ich zgłoszeniu
(Dz.U. 2004.125.1316)
8. Rozporządzenie Ministra Zdrowia z dnia 3 listopada 2004 w sprawie wymagań
zasadniczych dla wyrobów medycznych do różnego przeznaczenia (Dz. U.
2004.251.2514)
9. Ustawa o ocenie zgodności z dnia 30 sierpnia 2002 r. (Dz. U. 2002.166.1360)
10. „ISO 9001 dla małych firm – metody postępowania”, PKN, Warszawa, 2003
11. „Zarządzanie jakością w praktyce”, Wiedza i Praktyka, Warszawa, 2006
12. Ustawa z dnia 29 lipca 2005 r. o zużytym sprzęcie elektrycznym i elektronicznym (Dz.U.
2005.180.1495)
13. PN-EN 980 Symbole graficzne do stosowania w oznakowaniu wyrobów medycznych
14. PN-EN 1041 Informacja dostarczana przez producenta wraz z wyrobem medycznym
15. NB-MED 2.5.1 Rec. 5
16. EN-ISO 13485:2003 Wyroby medyczne – Systemy zarządzania jakością – Wymagania
do celów przepisów prawnych.
17. PN-EN ISO 14971:2004 Wyroby medyczne –– Zastosowanie zarządzania ryzykiem do
wyrobów medycznych
18. Roszuk W.: Zarządzanie ryzykiem w wytwarzaniu wyrobów medycznych. Analizy
ryzyka; szkolenie dla pracowników Instytutu Techniki i Aparatury Medycznej ITAM,
styczeń 2004.
19. www.sejm.gov.pl
20. www.pkn.pl
21. www.urpl.gov.pl
22. www.pca.gov.pl
23. http://europa.eu.int
24. ec.europa.eu/enterprise/newapproach/nando
25. www.NewApproach.org/directives.asp
Wyszukiwarka
Podobne podstrony:
08 Wykonywanie dokumentacji aparatury medycznej
ETP2013 - ELEKTRONICZNA APARATURA MEDYCZNA 1, dokumenty, elektroniczna aparatura medyczna
08.Gorsety, Dokumenty szkoła, Skrypt
podbielska,elektroniczna aparatura medyczna, Elektrokardiograf Charakterystyka bloków
podbielska,elektroniczna aparatura medyczna, elektrokardiografia holterowska
35 Wykonywanie dokumentacji kon Nieznany
08 Wykonywanie połączeń w urządzeniach precyzyjnych
08 Wykonywanie izolacji wodochronnych z materiałów bitumicznych
33 Elektroniczna aparatura medyczna
podbielska,elektroniczna aparatura medyczna, Wpływ prądu stałego na tkanki
podbielska,elektroniczna aparatura medyczna, przetworniki spirometryczne
713[05] Z1 08 Wykonywanie posad Nieznany
08 Wykonywanie i remontowanie o Nieznany
podbielska,elektroniczna aparatura medyczna, Pomiar objętości zalegającej w płucach
podbielska,elektroniczna aparatura medyczna, Sposób pomiaru stężenia CO2 w gazie oddechowymx
39 Wykonywanie dokumentacji techniczno technologicznej
08 Wykonywanie pomiarow warszta Nieznany (2)
08. Monitorowanie podczas znieczulenia, MEDYCZNE, ANASTEZOLOGIA i PIEL ANSTZJOLOGICZNE
więcej podobnych podstron