
dr in
ż
. Aneta Białkowska
(pokój 322; aneta.bialkowska@p.lodz.pl)
ENZYMOLOGIA
WYKŁAD – 15 GODZIN
LABORATORIUM – 15 GODZIN (II POŁOWA
SEMESTRU)
FORMA ZALICZENIA WYKŁADU I
LABORATORIUM –
KOLOKWIUM PISEMNE

dr inż. Aneta Białkowska
LITERATURA:
1. Witwicki, Ardelt, 1989, Warszawa PWN – „Elementy enzymologii”
2. Kafarski, Lejczak, 1994, Warszawa PWN – „Chemia bioorganiczna”
3. Szlegel,1996, Warszawa PWN – „Mikrobiologia ogolna”
4. Whitaker, Voragen, Wong, 2002, CRC Press – „Handbook of food
enzymology”
5. Bommarius, Bettina, 2004, Wiley-VCH, Weinheim – „Biocatalysis”
6. Kołakowski, Bednarski, Bielecki, 2005, Wydawnictwo Akademii Rolniczej
w Szczecinie – „Enzymatyczna modyfikacja składników żywności”
7. Berg, Tymoczko, Stryer, 2009, Warszawa PWN – „Biochemia”
8. Kłyszejko-Stefanowicz, 2011, Warszawa PWN – „Ćwiczenia z biochemii”
9. Podstawy biotechnologii, 2011, Warszawa PWN
10.Methods of enzymology (wydawnictwo ciagłe, począwszy od 1955 r.)
11.Literatura oryginalna (artykuły w czasopismach)

dr inż. Aneta Białkowska
ENZYMOLOGIA
– PODSTAWOWA CZĘŚĆ BIOCHEMII
DYNAMICZNEJ, ZAJMUJĄCA SIĘ:
poznaniem relacji między strukturą a funkcją
enzymów;
sposobami ich izolowania i oczyszczania;
właściwościami i mechanizmami działania enzymów;
przebiegiem katalizowanych przez nie reakcji;
drogami biosyntezy biokatalizatorów
CO TO JEST BIOKATALIZATOR
?
ENZYM
(białko globularne)
RYBOZYMY (RNAzymy)
ABZYMY
(PRZECIWCIAŁA-
IMMUNOGLOBULINY)
MIKROZYMY
(
nowa klasa naturalnych, katalitycznych
biocząsteczek, których masy są niższe od 10 kDa)
Katalizatory: enzymy, abzymy, rybozymy mogą być stosowane w
formie:
preparatów o różnej czystości (rozpuszczalnych lub
unieruchomionych)
całych komórek (głównie drobnoustrojów) lub ich fragmentów
dr inż. Aneta Białkowska
CO TO JEST BIOKATALIZATOR
?
SYNZYMY – biokatalizatory
otrzymane na drodze syntezy
chemicznej lub modyfikacji
genetycznych
DEOKSYRYBOZYMY
(DNAZYMY) - katalityczne DNA
dr inż. Aneta Białkowska

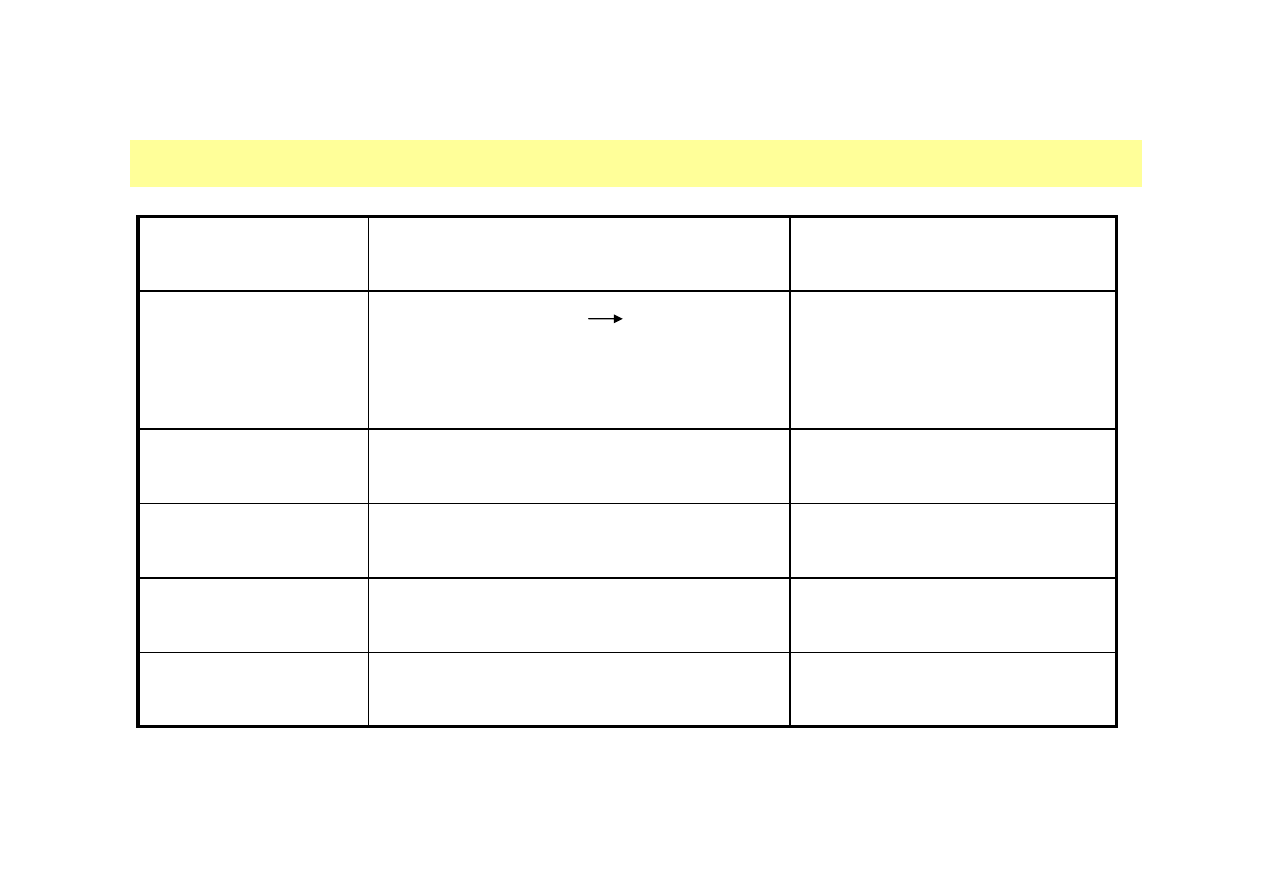
SYNZYMY UZYSKANE NA DRODZE MODYFIKACJI CHEMICZNYCH
Technologia enzymów, dr inż. Aneta Białkowska
Obniżona termostabilność w
celu łatwiejszej inaktywacji w
czasie pasteryzacji przy
produkcji sera
Utlenianie metioniny, takie jak przy
użyciu H
2
O
2
Rennina (szczepy Mucor)
Wzrost aktywności
koagulacyjnej mleka do 2 razy
Acylacja bezwodnikowa
Rennina
Wzrost aktywności do 70 razy
Acylacja N-hydroksybursztynowymi
estrami aminokwasów
Termolizyna (Bacillus
thermoproteolyticus)
Zwiększona aktywność
esterazy i wyeliminowanie
aktywności peptydazy
Acetylacja lub jodowanie tyrozyny w
aktywnym centrum
Karboksypeptydaza A
Zwiększona termostabilność
powyżej 70°C, zmniejszona
termostabilność poniżej 67°C
Acetylacja octanem p-nitrofenolu
α
-amylaza (Bacillus
subtilis)
Efekt
Modyfikacja chemiczna
Enzym
SYNZYMY UZYSKANE NA DRODZE INśYNIERII GENETYCZNEJ
Technologia enzymów, dr inż. Aneta Białkowska
Zmiany w przedziale
substratów
RM seryna
→
fenyloalanina i inne
Dehydrogenaza
Zmiany w przedziale
substratów
RM seryna
→
fenyloalanina i inne
Amidaza
Zmieniona specyficzność
substratowa
SSM glicyna
226
→
alanina
Trypsyna
Zwiększona
termostabilność
SSM izoleucyna
3
→
cysteina,
następnie chemiczne sieciowanie
Lizozym
Większa odporność na
wybielanie
Zmieniona specyficzność
substratowa
SSM metionina
222
→
alanina
SSM glicyna
166
→
kwas
asparaginowy i glutaminowy
Subtilizyna
Nowa cecha
Metoda modyfikacji
Enzym
SSM-ukierunkowana mutacja wyizolowanego materiału genetycznego
RM- przypadkowa mutacja genu in situ w organizmie producenta

wysoka specyficzność, selektywność i nietoksyczność
wytwarzanie
enancjomerycznie
czystych
produktów
lub
półproduktów
potanienie procesu
możliwość wielokrotnego wykorzystania (immobilizacja enzymów)
uzyskanie
zmodyfikowanych
produktów
spożywczych
o
udoskonalonych cechach sensorycznych i przedłużonej trwałości
podniesienie przyswajalności i walorów zdrowotnych żywności
poprzez
usunięcie
składników
szkodliwych
dla
konsumenta
nieprzyswajalnych
i/lub
utrudniających
przyswajanie
innych,
wartościowych komponentów
zastąpienie tradycyjnych procesów chemicznych, niejednokrotnie
uciążliwszych
dla
środowiska
przez
przyjemne
dlań
i
energooszczędne biotechnologie
pełniejsze wykorzystanie surowców, zmniejszenie ilości odpadów,
mogących negatywnie wpływać na środowisko naturalne
DLACZEGO WARTO STOSOWAĆ ENZYMY (KOMÓRKI)?:
dr inż. Aneta Białkowska
PROBLEMY ZWIĄZANE ZE STOSOWANIEM
ENZYMÓW (KOMÓREK)
niestabilność w zbyt wysokich temperaturach
niestabilność w ekstremalnych pH
niestabilność w rozpuszczalnikach organicznych
inhibicja przez niektóre jony metali
możliwość łatwej degradacji pod wpływem działania enzymów
proteolitycznych
wysoki koszt (np. enzymów z klasy oksydoreduktaz)
konieczność stosowania drogich kosubstratów
czynniki alergenne
dr inż. Aneta Białkowska
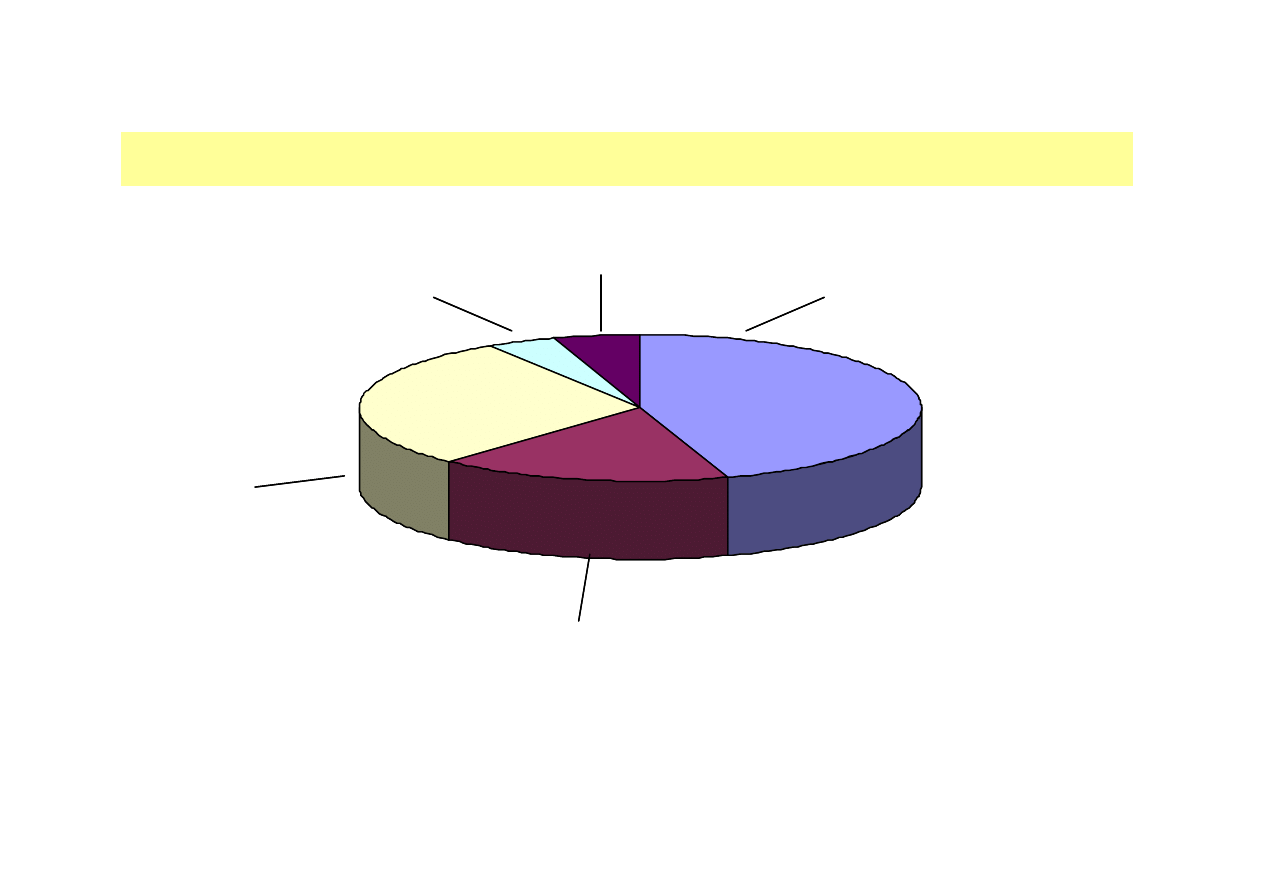
ŚWIATOWY RYNEK ENZYMÓW PRZEMYSŁOWYCH
Novozymes (DK) 45%
Others 29%
Danisco/Genencor (DK/USA)
BASF (Germany) 4%
DSM (NL) 5%
Novozymes (DK) 45%
Others 29%
Danisco/Genencor (DK/USA)
BASF (Germany) 4%
DSM (NL) 5%
Rys. Największe firmy biotechnologiczne zajmujące się produkcją enzymów
przemysłowych i ich udział w globalnym rynku tych produktów.
dr inż. Aneta Białkowska
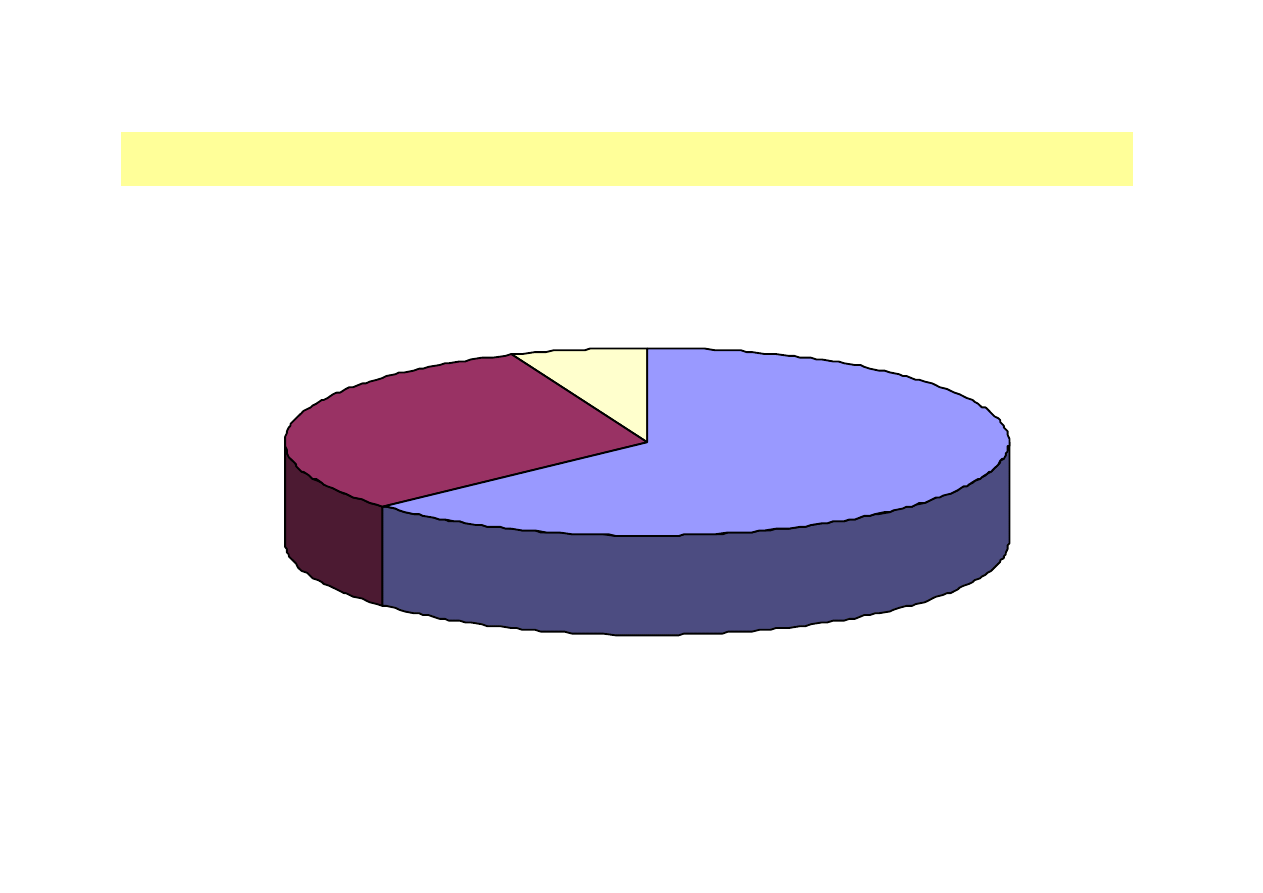
RYNEK ENZYMÓW I JEGO GŁÓWNE SEKTORY
Sektor techniczny
63%
Sektor spożywczy
31%
Sektor
paszowy 63%
dr inż. Aneta Białkowska
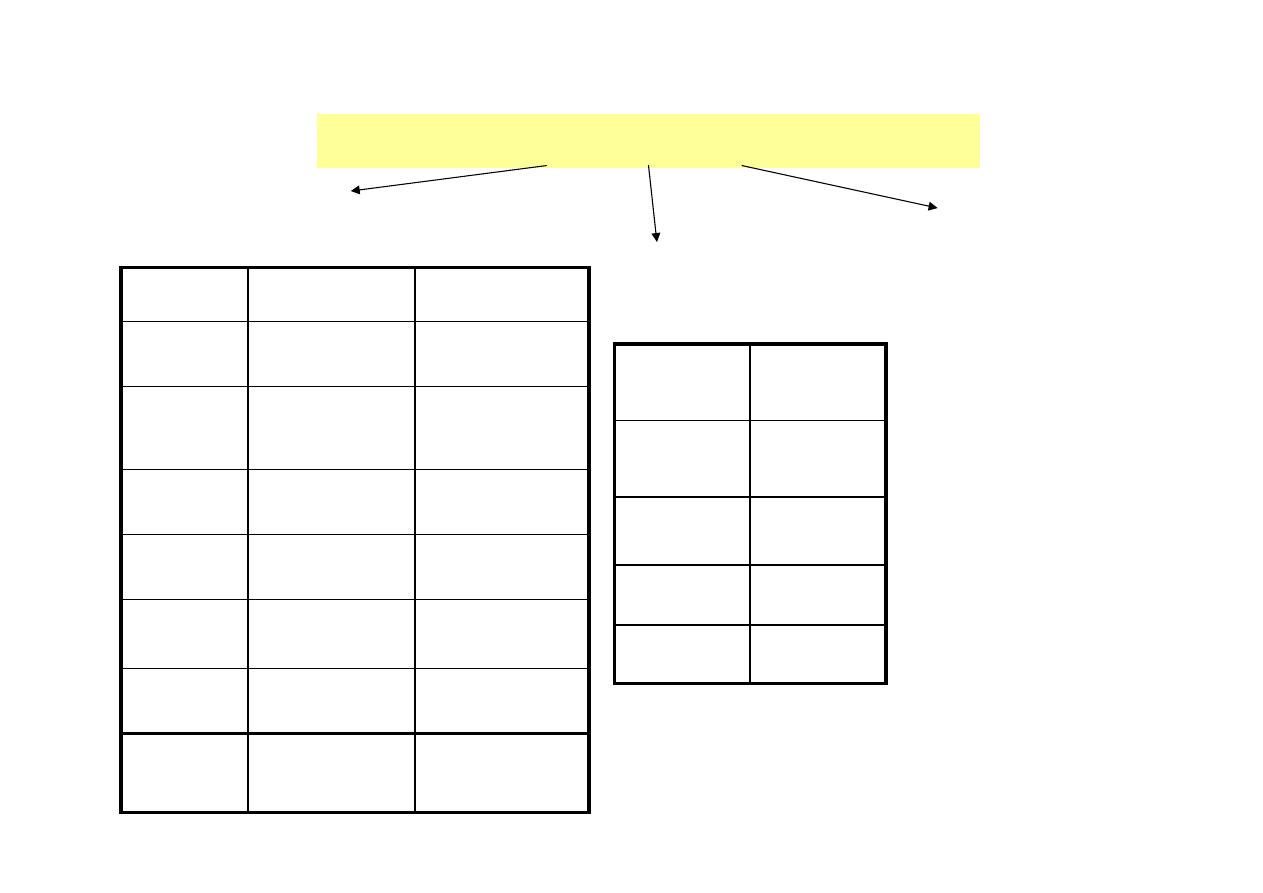
ŹRÓDŁA ENZYMÓW
izolowane z roślin
izolowane ze
zwierząt
produkowane
przez
mikroorganizmy
(bakterie,
drożdże, grzyby
strzępkowe
)
Piekarnictwo
Ficyna
Drzewo
figowe
Piekarnictwo,
produkcja syropów
maltozowych
ββββ
-amylaza
Soja,
jęczmień
Hydroliza estrów
Esteraza
Pszenica
Właściwości
przeciwzapalane
Bromelaina
Ananas
Diagnostyka
Peroksydaza
Chrzan
Piekarnictwo,
garbarstwo,
mleczarstwo,
tenderyzacja mięsa
Papaina
Papaja
Diagnostyka
Ureaza
Soja
Zastosowanie
Enzym
Źródło
Lizozym
Jaja kurze
Urokinaza
Ludzki mocz
Pepsyna,
trypsyna
Wątroby
cielęce
Podpuszczka
śołądki cieląt
Enzym
Źródło
dr inż. Aneta Białkowska

1. tańsza produkcja
2. łatwiejsza kontrola produkcji
3. większa dostępność surowców do wzrostu drobnoustrojów
(łatwe przygotowanie podłoży)
4. mniejsza zawartość potencjalnie szkodliwych substancji tj.
związków fenolowych, proteinaz, inhibitorów w komórkach
mikroorganizmów w porównaniu do tkanek roślinnych i
zwierzęcych
DROBNOUSTROJE JAKO ŹRÓDŁO ENZYMÓW:
dr inż. Aneta Białkowska

1. wydajność produkcji enzymu i szybkość jego biosyntezy
2. czystość wytwarzanych enzymów (brak ubocznych aktywności
enzymatycznych)
3. niepatogenność i brak produktów toksycznych, alergicznych
4. stabilność genetyczna i fenotypowa oraz fagooporność
5. wymagania pokarmowe oraz tolerancja na zmienne stężenie
składników podłoża
6. wielkość zapotrzebowania na tlen oraz tolerancja na zmiany
warunków natlenienia
7. wymagania w zakresie temperatury, odczynu i innych
parametrów procesu oraz stopień wrażliwości na ich zmiany
8. łatwość wydzielania enzymu
CECHY ZEZWALAJĄCE NA OCENĘ
PRZYDATNOŚCI SZCZEPÓW PRZEMYSŁOWYCH:
dr inż. Aneta Białkowska
dr inż. Aneta Białkowska
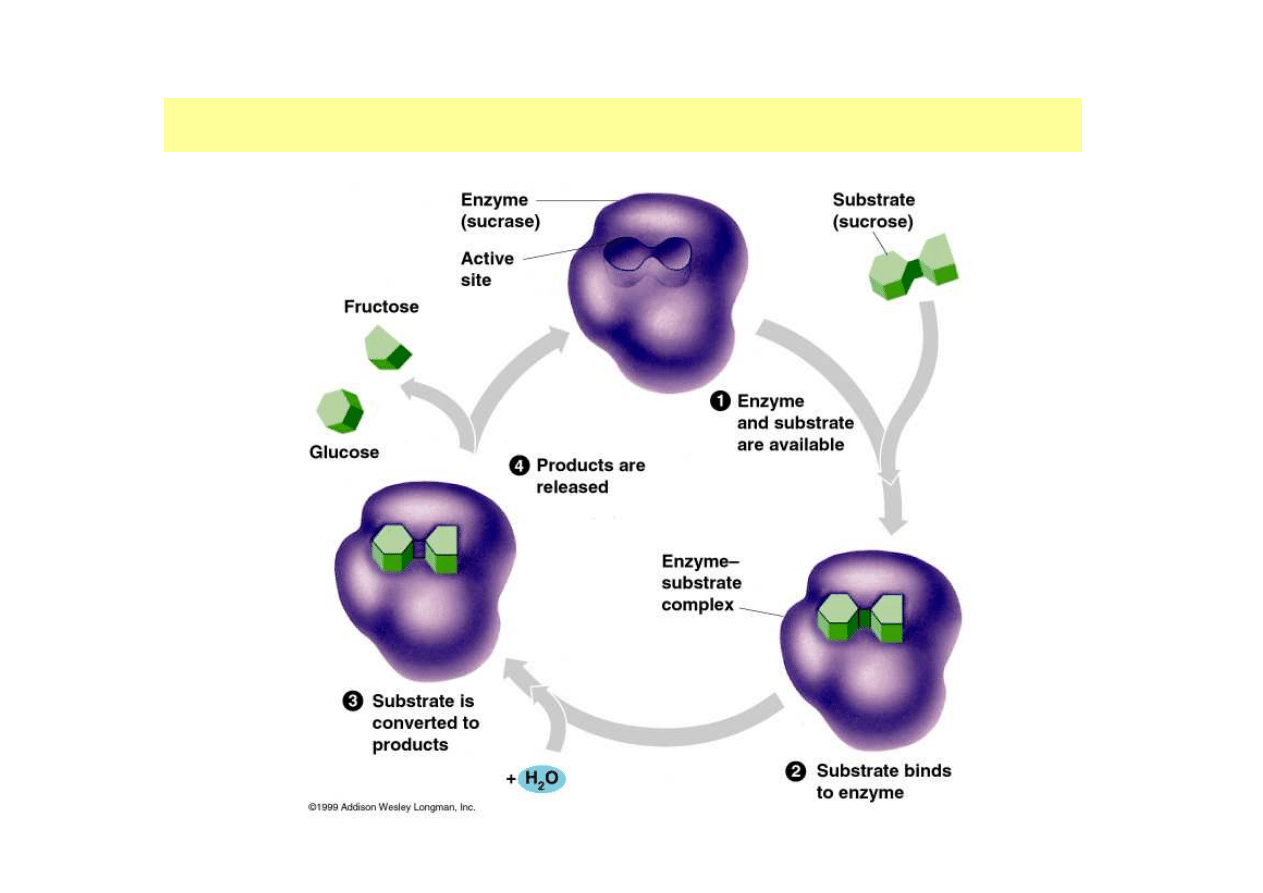
ETAPY REAKCJI ENZYMATYCZNEJ
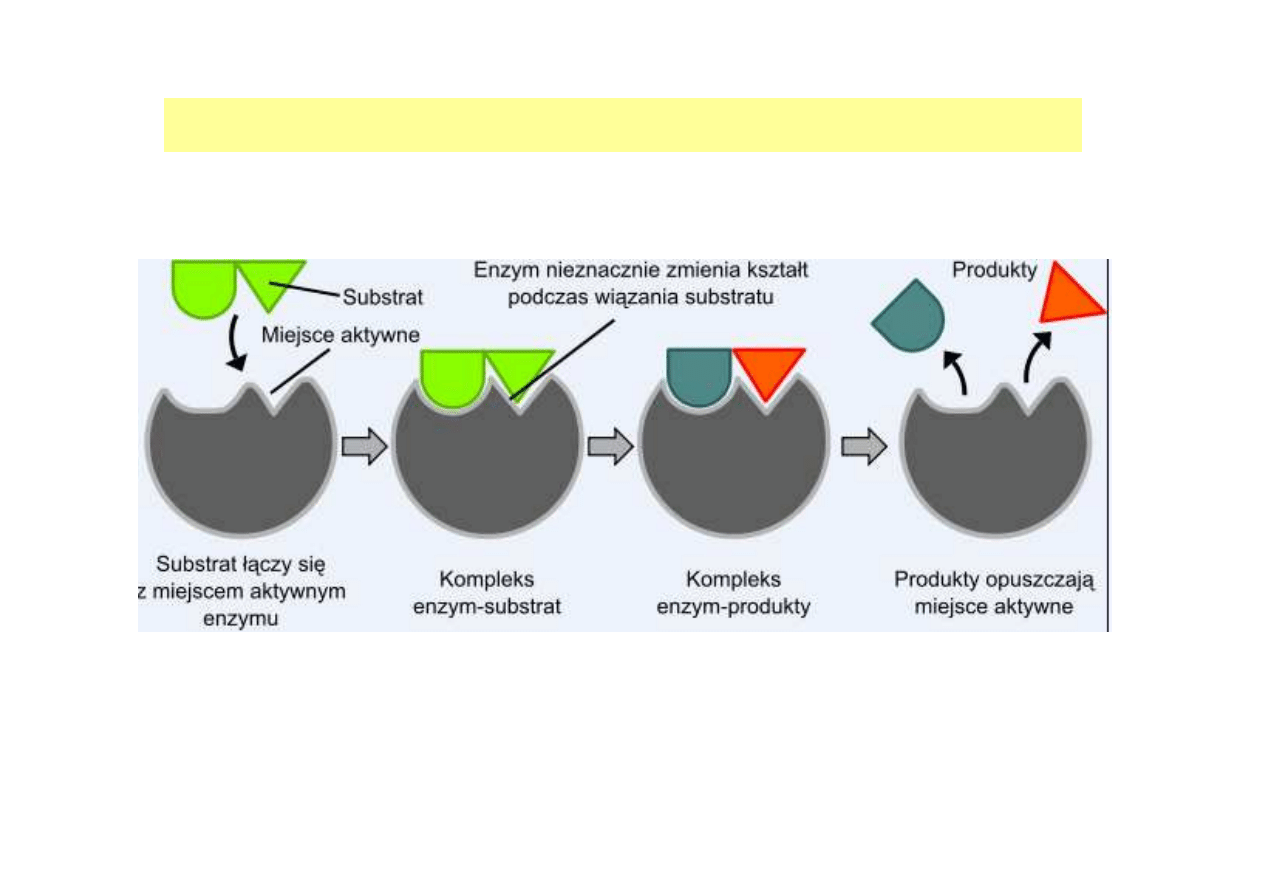
dr inż. Aneta Białkowska
ETAPY REAKCJI ENZYMATYCZNEJ

dr inż. Aneta Białkowska
ETAPY REAKCJI ENZYMATYCZNEJ
1. Rozpoznanie i zwi
ą
zanie substratu, dzi
ę
ki oddziaływaniu reszt
aminokwasów wi
ążą
cych zlokalizowanych w kieszeni katalitycznej
(etap charakteryzowany przez K
m
)
2. Konformacyjny stres enzymu, indukowany przez zmiany w aktywnym
centrum, prowadz
ą
cy do utworzenia produktu (etap entropowy,
charakteryzowany przez k
kat
)
3. Uwolnienie produktu z kompleksu z enzymem
dr inż. Aneta Białkowska
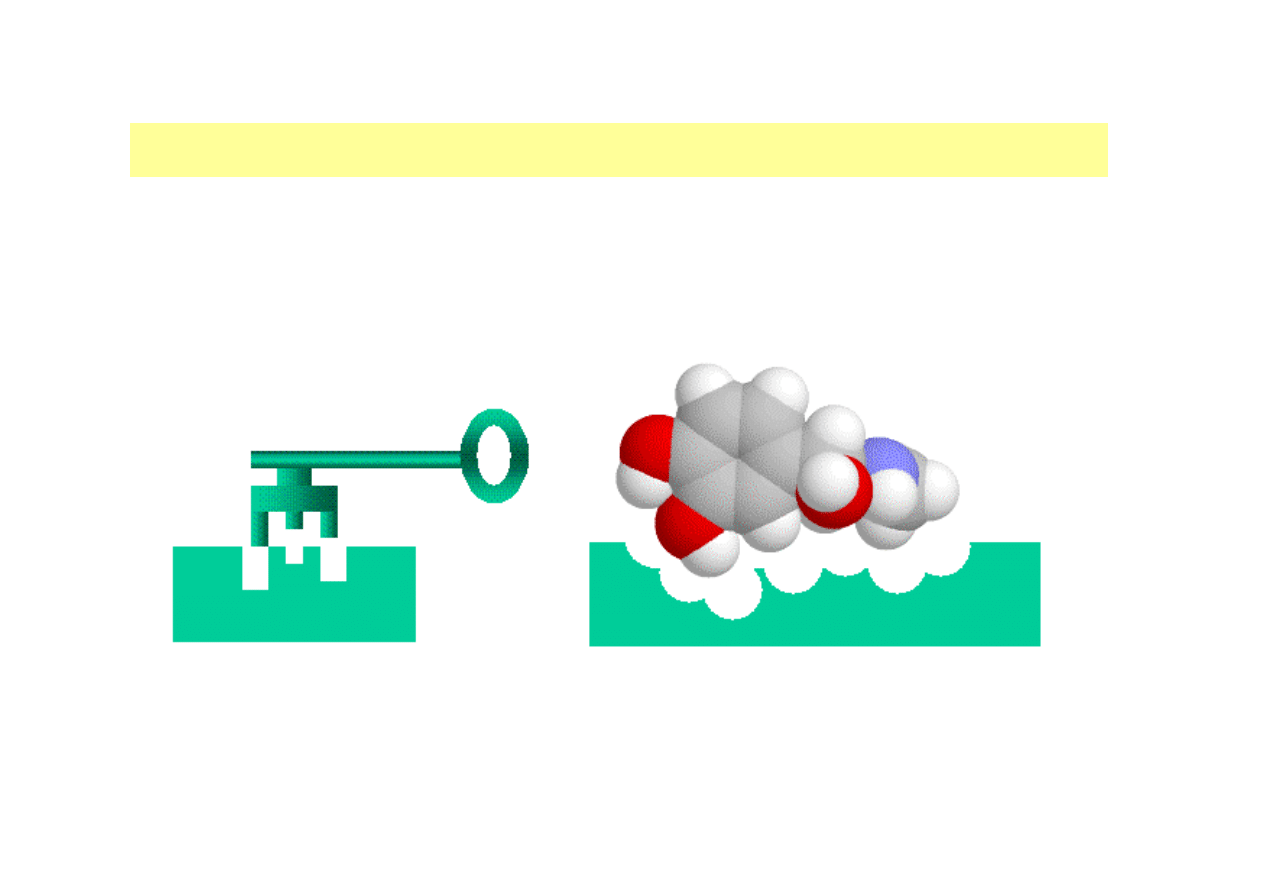
MECHANIZMY WIĄZANIA ENZYMU Z SUBSTRATEM
MODEL tzw. ZAMKA I KLUCZA
(z ang. „lock and key” – teoria
Fischera, znaczenie historyczne)
dr inż. Aneta Białkowska
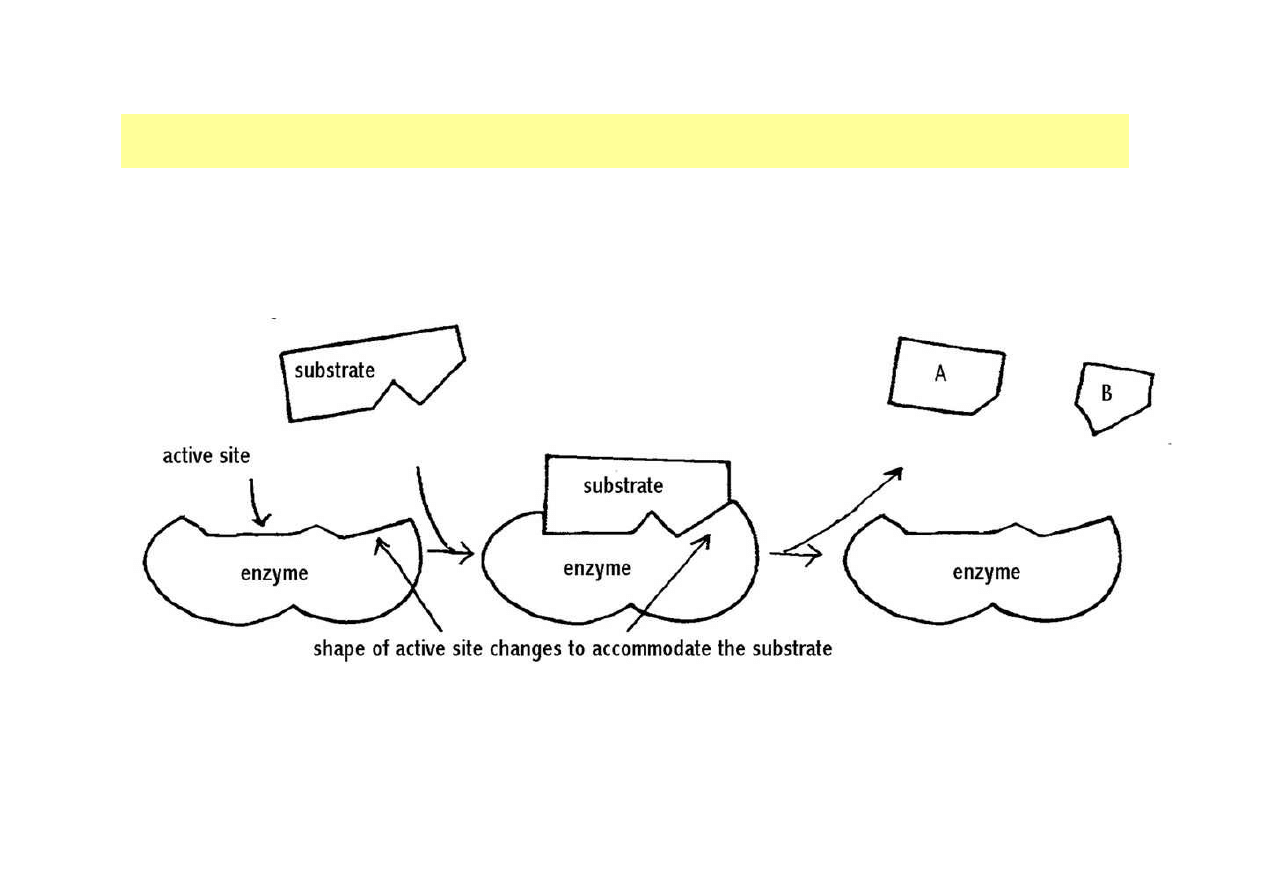
MECHANIZMY WIĄZANIA ENZYMU Z SUBSTRATEM
MODEL INDUKOWANEGO DOPASOWANIA SIĘ ENZYMU
( z ang.
induced fit model; teoria Koshlanda)
Obszar katalityczny enzymu jest elastyczny; obecność substratu indukuje
zmiany konformacyjne białka, dzięki czemu następuje właściwe ułożenie grup
katalitycznych względem grup funkcyjnych i wiązań w cząsteczce substratu.
dr inż. Aneta Białkowska
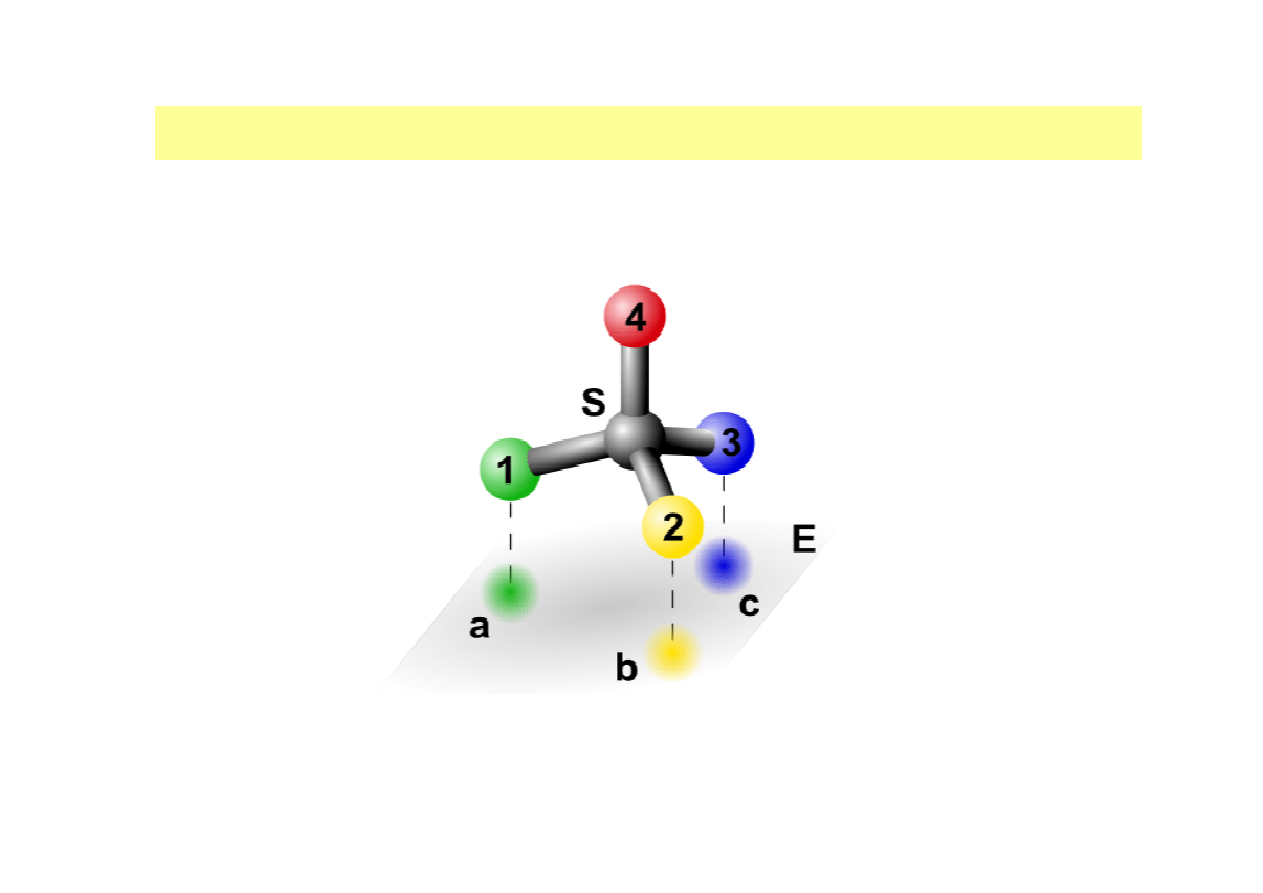
MECHANIZMY WIĄZANIA ENZYMU Z SUBSTRATEM
MODEL TRÓJPUNKTOWEGO PRZYŁĄCZENIA SUBSTRATU
(z
ang. three-point interaction model; teoria Ogstona)
Substrat przyłącza się do powierzchni enzymu w trzech punktach; położenie cząsteczki
substratu wobec enzymu jest jednoznacznie określone, a reakcja zachodzi tylko w
jednym z trzech punktów przyłączenia; wyjaśnia swoistość przestrzenną enzymów.

dr inż. Aneta Białkowska
CENTRUM KATALITYCZNE ENZYMU
1. Względnie mała część całkowitej objętości cząsteczki enzymu (od
kilku do kilkunastu reszt aminokwasowych)
2. Struktura trójwymiarowa, obejmująca zwykle odległe reszty
aminokwasów
3. Ma charakter kieszonki lub szczeliny zlokalizowanej w obszarze
hydrofobowym molekuły enzymu co wzmaga oddziaływania
polarnych reszt (Ser, Thr, Lys, His, Asp, Glu, Cys, Met, Arg)
aminokwasów na substrat
4. Ulega zmianom konformacyjnym przy współdziałaniu z
substratem – jego rozmiary mogą się więc zmieniać, choć
specyficzność wiązania substratu jest zdefiniowana określonym
położeniem atomów w centrum aktywnym
5. Współdziała z substratem przy użyciu względnie słabych
oddziaływań, np. hydrofobowych, jonowych, wodorowych, czy
van der Waalsa
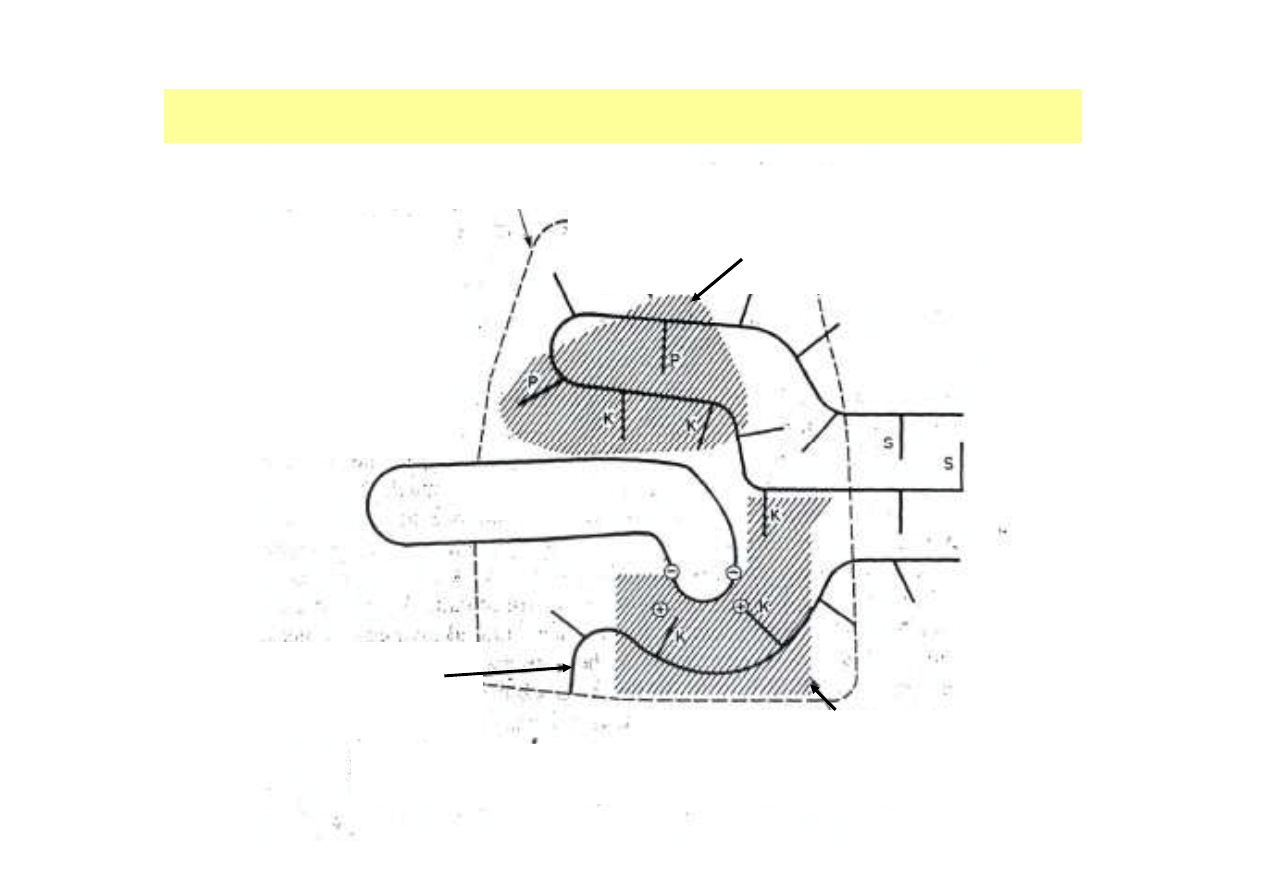
dr inż. Aneta Białkowska
CENTRUM KATALITYCZNE ENZYMU
Łańcuch polipeptydowy z
bocznymi łańcuchami
aminokwasowymi
substrat
miejsce katalityczne (odpowiedzialne za czynność
katalityczną enzymu, czyli za jego specyficzność
działania)
OBSZAR CENTRUM AKTYWNEGO (budowa wg. Daniela E. Koshlanda)
K - aminokwasy
kontaktowe (swoiste i
katalityczne)
P - aminokwasy
pomocnicze
S - aminokwasy
stabilizujące
miejsce określające swoistość, tzw.
miejsce wiążące (odpowiedzialne za
specyficzność substratową enzymu)

dr inż. Aneta Białkowska
DOWODY NA ISTNIENIE KOMPLEKSU ENZYM-
SUBSTRAT
1. Kompleksy ES zostały bezpośrednio zarejestrowane dla licznych
enzymów za pomocą mikroskopii elektronowej i badań
krystalograficznych
2. Fizyczne właściwości enzymu, np. jego rozpuszczalność i stabilność
cieplna mogą się często zmieniać podczas tworzenia kompleksu ES
3. Kompleks ES może mieć inne właściwości spektroskopowe, np.
peroksydaza wykazuje maksimum pochłaniania światła przy 400
nm, a jej kompleks z H
2
O
2
przy 415 nm
4. Współczynnik sedymentacji kompleksów ES jest wyższy w
porównaniu z wolnym enzymem
5. Kinetyka reakcji enzymatycznych potwierdza tworzenie się
kompleksu ES, o czym świadczy charakter krzywej V=f[S] przy [E] =
const i tzw. efekt nasycenia. Efekt ten nie jest obserwowany dla
reakcji nie katalizowanych przez enzymy

PODZIAŁ ENZYMÓW WZG. BUDOWY
CHEMICZNEJ
dr inż. Aneta Białkowska
1. Enzymy będące
białkami prostymi
(zbudowane
jedynie z aminokwasów)
Grupa czynna
– specyficzne ugrupowanie
aminokwasów, których nie można odszczepić
bez nieodwracalnej zmiany struktury białka
Przykłady: Amylaza, RNAza, Ureaza

PODZIAŁ ENZYMÓW WZG. BUDOWY
CHEMICZNEJ
dr inż. Aneta Białkowska
2. ENZYMY ZŁOśONE
holoenzym
- pełny układ katalityczny
apoenzym
część białkowa
holoenzymu
koenzym
przyłączona część o
niebiałkowym charakterze
HOLOENZYM = apoenzym + koenzym
holoenzym
- pełny układ katalityczny
apoenzym
część białkowa
holoenzymu
koenzym
przyłączona część o
niebiałkowym charakterze
trwale
związany
nieaminokwasowy koenzym =
grupa prostetyczna
(witaminy i
ich pochodne)
(odłączenie grupy prostetycznej
powoduje i
naktywacje
enzymu
(zazwyczaj nieodwracalną))
luźno
związany nieaminokwasowy
koenzym (kosubstrat)
którego nie można
traktować jako integralnej części enzymu
(nukleotydy i ich pochodne, jony
nieorganiczne).
(jest on łatwo i odwracalnie usuwalny bez
uszkodzenia części białkowej, ale jego
usunięcie prowadzi do inaktywacji enzymu)
PODZIAŁ ENZYMÓW WZG. BUDOWY
CHEMICZNEJ
dr inż. Aneta Białkowska
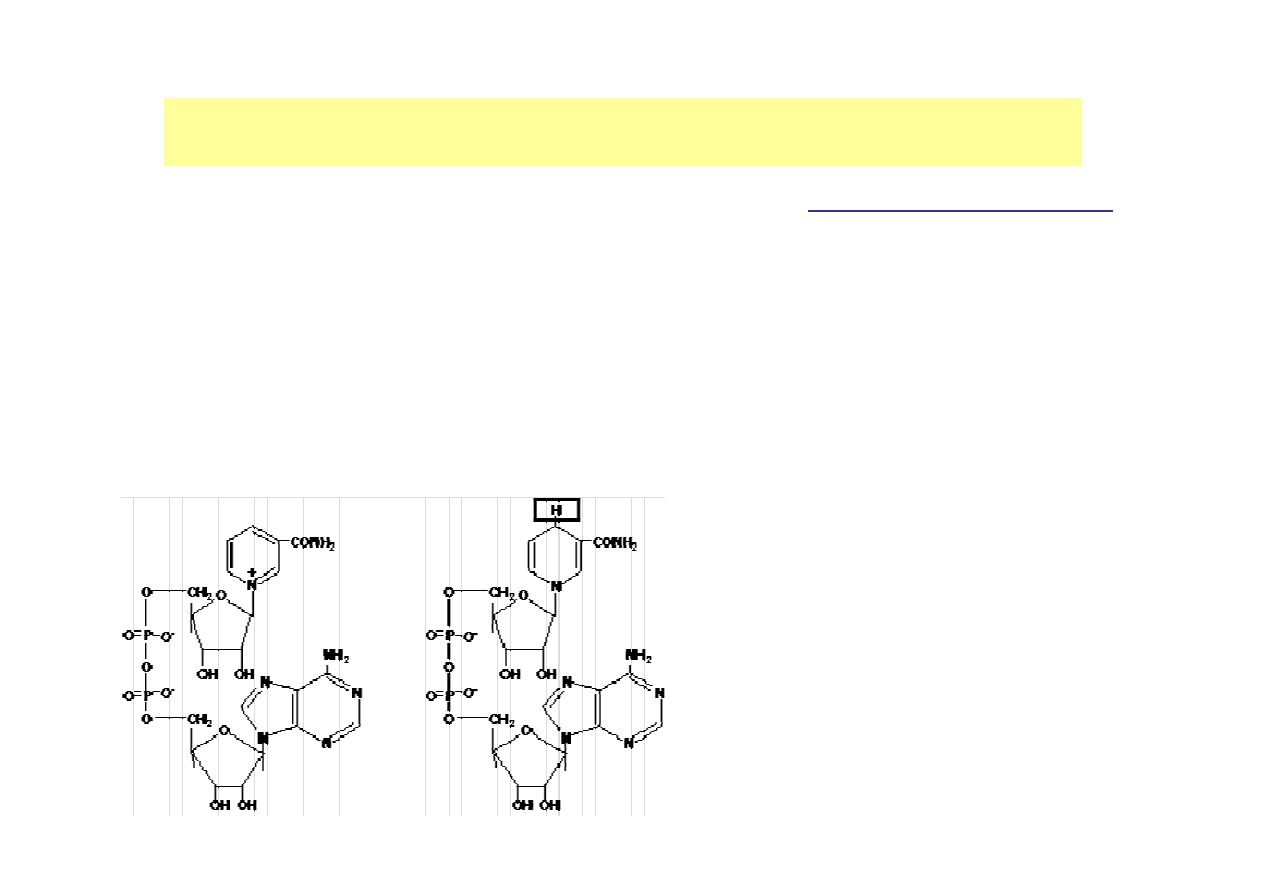
Koenzym mogą być klasyfikowane
zależnie od grupy
,
której przeniesienie ułatwiają
1. Koenzymy przenoszące H
NAD
+
, NADP
+
,
FMN, FAD,
Kwas liponowy,
Koenzym Q
2. Koenzymy przenoszące inne
grupy niż H
CoASH
Pirofosforan tiaminy
Biotyna
Koenzymy kobamidowe (B
12
)
KLASYFIKACJA KOENZYMÓW
dr inż. Aneta Białkowska

dr inż. Aneta Białkowska
KLASYFIKACJA ENZYMÓW
I Oksydoreduktazy - katalizują procesy oksydo-redukcyjne (przenoszenie elektronów i
protonów na różne akceptory, np. NAD+, NADP+, flawoproteidy).
II Transferazy - katalizują reakcje przenoszenia grup funkcyjnych z cząsteczki donora
do cząsteczki akceptora, np. metylowej -CH
3
(transmetylazy), aminowej -NH
2
(transaminazy), acylowych R-CO-(transacylazy).
III Hydrolazy - katalizują rozpad cząsteczek złożonych na prostsze przy udziale H
2
O;
innymi słowy katalizują przenoszenie grupy funkcyjnej z cząsteczki donora do
cząsteczki wody; w ten sposób dochodzi do hydrolizy np. wiązań estrowych (esterazy),
eterowych, glikozydowych (glikozydazy), peptydowych (proteazy).
IV Liazy - katalizują reakcję addycji wody, amoniaku lub CO
2
do wiązań podwójnych;
katalizują również reakcje odwrotne.
V Izomerazy - przebudowują strukturę cząsteczki bez jej rozkładu; katalizują więc
wewnątrzcząsteczkowe przegrupowanie atomów, czyli izomerię (np. izomerazy cis,
trans).
VI Ligazy - katalizują reakcje łączenia dwóch substratów, w wyniku czego powstają
wiązania C-O, C-S, C-N, C-C. Są to reakcje wymagające nakładu energii ze związków
wysokoenergetycznych, np. ATP, GTP.

dr inż. Aneta Białkowska
JEDNOSTKI AKTYWNOŚCI ENZYMATYCZNEJ
1 UNIT (1 U)
– jest to taka ilość enzymu, bądź jego aktywność, która katalizuje
przekształcenie 1
µ
mola substratu w ciągu 1 minuty, w optymalnej temperaturze i
w optymalnych warunkach pH oraz stężenia substratu.
1 KATAL (1 kat)
– jest to taka ilość enzymu, bądź jego aktywność, która katalizuje
przekształcenie 1 mola substratu w ciągu 1 sekundy, w optymalnej temperaturze i
w optymalnych warunkach pH oraz stężenia substratu. Jest to jednostka zalecana
przez Komisję Międzynarodowej Unii Biochemicznej.
AKTYWNOŚĆ MOLEKULARNA
– jest to ilość mmoli substratu przekształcona w
ciągu 1 minuty w standardowych warunkach przez 1
µ
mol enzymu (w odniesieniu
do jednego centrum aktywnego, jeżeli enzym zawiera ich więcej). Ten sposób
wyrażania aktywności można jednak stosować tylko wtedy, gdy znana jest masa
cząsteczkowa enzymu i liczba centrów aktywnych.
AKTYWNOŚĆ WŁAŚCIWA
–określa liczbę jednostek enzymu na 1 mg białka. Jest
to bardzo przydatny sposób wyrażania aktywności podczas wydzielania i
oczyszczania enzymów, ponieważ w kolejnych etapach oczyszczania aktywność
całkowita zazwyczaj maleje, a aktywność właściwa wzrasta i jest miarą stopnia
oczyszczenia enzymu.

dr inż. Aneta Białkowska
•
temperatura
•
pH
•
aktywatory i inhibitory
•
stała dielektryczna
•
siła jonowa
•
stężenie substratu, ewentualnie koenzymu
•
stężenie enzymu
•
czas reakcji
dr inż. Aneta Białkowska
CZYNNIKI WPŁYWAJĄCE NA SZYBKOŚĆ
REAKCJI ENZYMATYCZNEJ

dr inż. Aneta Białkowska
METODY OZNACZANIA AKTYWNOŚCI
ENZYMÓW
1. METODY STATYCZNE
•
dwupunktowe – porównanie ilości substratu lub produktu w 2
punktach czasowych, tj. w czasie zero i po zakończeniu inkubacji
mieszaniny reakcyjnej (należy zapewnić warunki reakcji aby
zapewnić stałą szybkość reakcji przez cały czas inkubacji)
2. METODY KINETYCZNE
•
pomiar zmian stężenia substratu lub produktu w jednostce czasu
(wymagają ciągłej rejestracji zmian stężeń badanego składnika w
czasie, czyli jest to tzw. pomiar wielopunktowy lub ciągły)

dr inż. Aneta Białkowska
METODY OZNACZANIA AKTYWNOŚCI
ENZYMÓW
1. Spektrofotometryczne (w tym kolorymetryczne)
2. Fluorymetryczne
3. Manometryczne
4. Wiskozymetryczne (np. oznaczanie aktywności enzymów
pektynolitycznych, której miarą jest spadek lepkości
mieszaniny reakcyjnej)
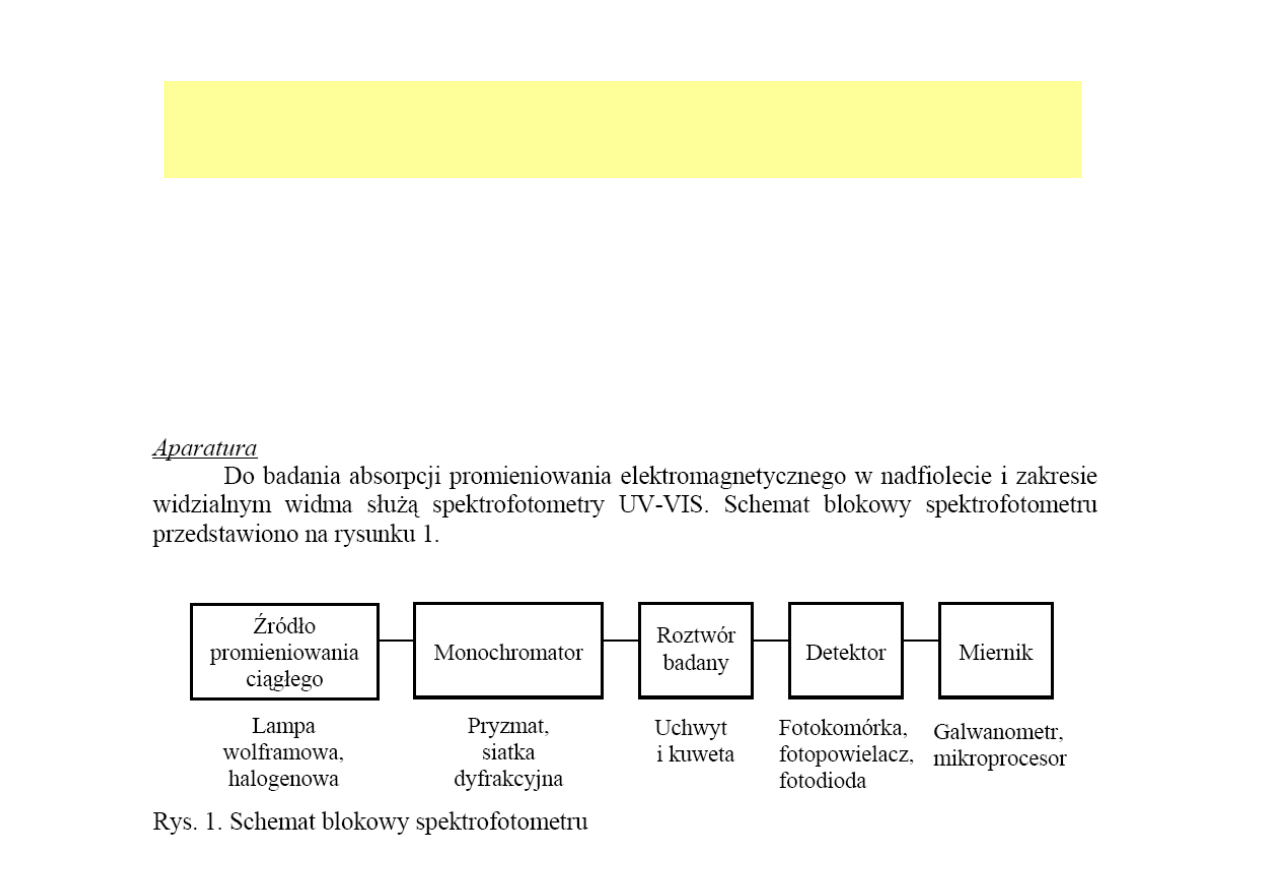
SPEKTROFOTOMETRYCZNE OZNACZANIE
AKTYWNOŚCI ENZYMÓW
dr inż. Aneta Białkowska
SPEKTROFOTOMETRIA
jest techniką instrumentalną, w której do celów
analitycznych wykorzystuje się przejścia energetyczne zachodzące w
cząsteczkach substratów lub produktów, spowodowane absorpcją
promieniowania elektromagnetycznego w zakresie nadfioletu (UV, 200-380
nm) lub widzialnym (VIS, 380 – 780 nm).
SPEKTROFOTOMETRYCZNE OZNACZANIE
AKTYWNOŚCI ENZYMÓW
dr inż. Aneta Białkowska
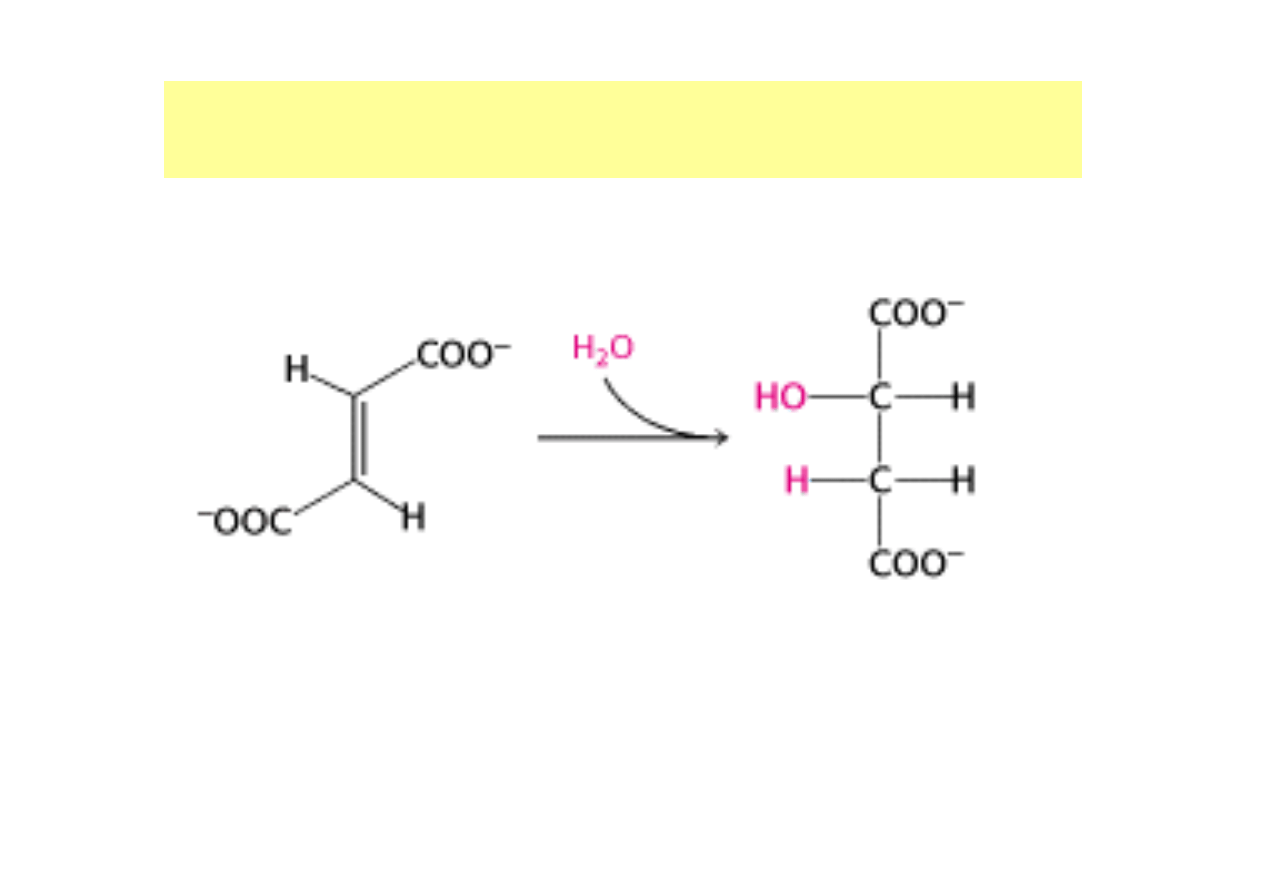
Np. Oznaczanie aktywności hydratazy fumaranowej
hydrataza
fumaranowa
Fumaran
(A
max
→
λ
= 300 nm)
L-jabłczan
(brak maksimum przy 300 nm)
SPEKTROFOTOMETRYCZNE OZNACZANIE
AKTYWNOŚCI ENZYMÓW
dr inż. Aneta Białkowska
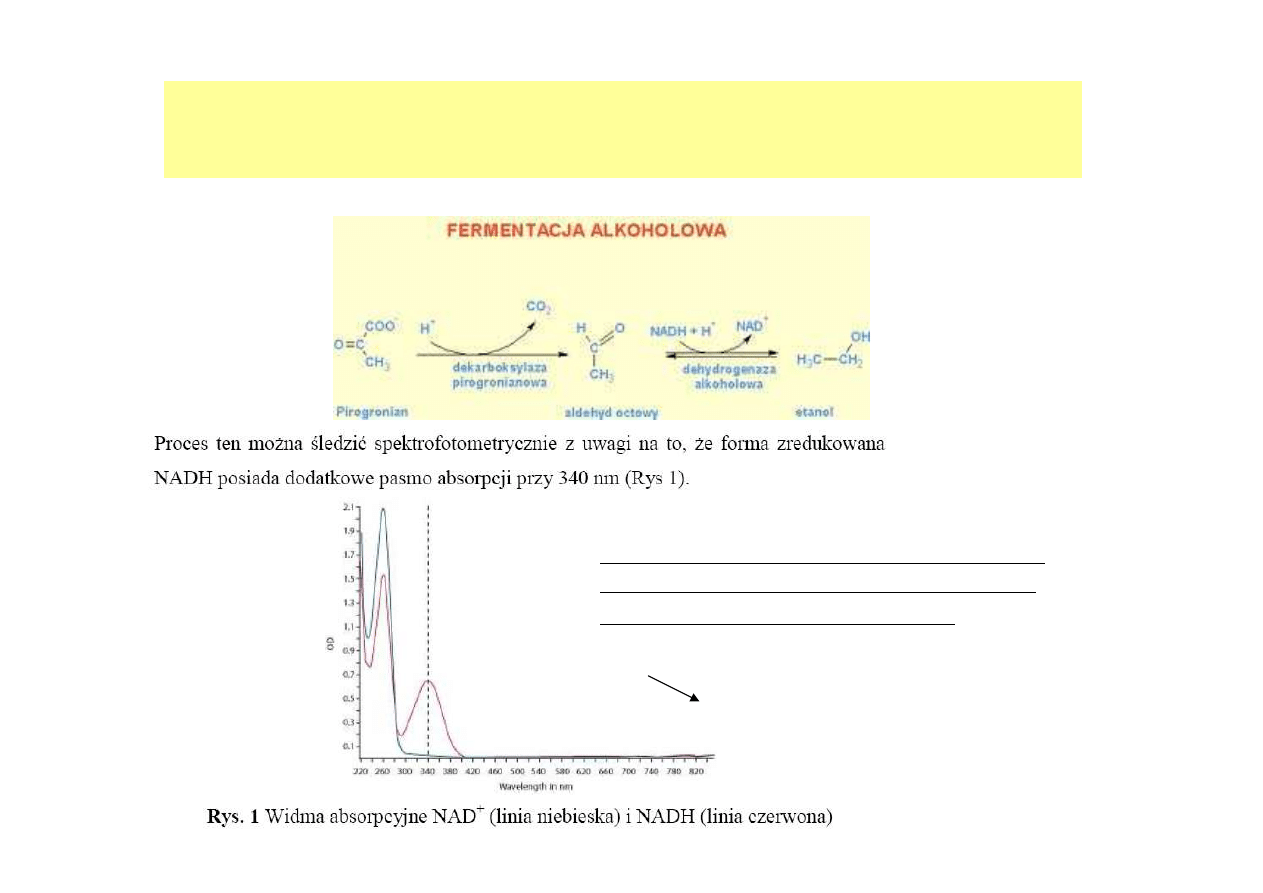
Np. Oznaczanie aktywności dehydrogenazy alkoholowej
Szybkość reakcji tworzenia się NAD+ i
etanolu można śledzić, mierząc zanik
pasma absorpcji przy 340 nm
.
A
340
SPEKTROFOTOMETRYCZNE OZNACZANIE
AKTYWNOŚCI ENZYMÓW
dr inż. Aneta Białkowska
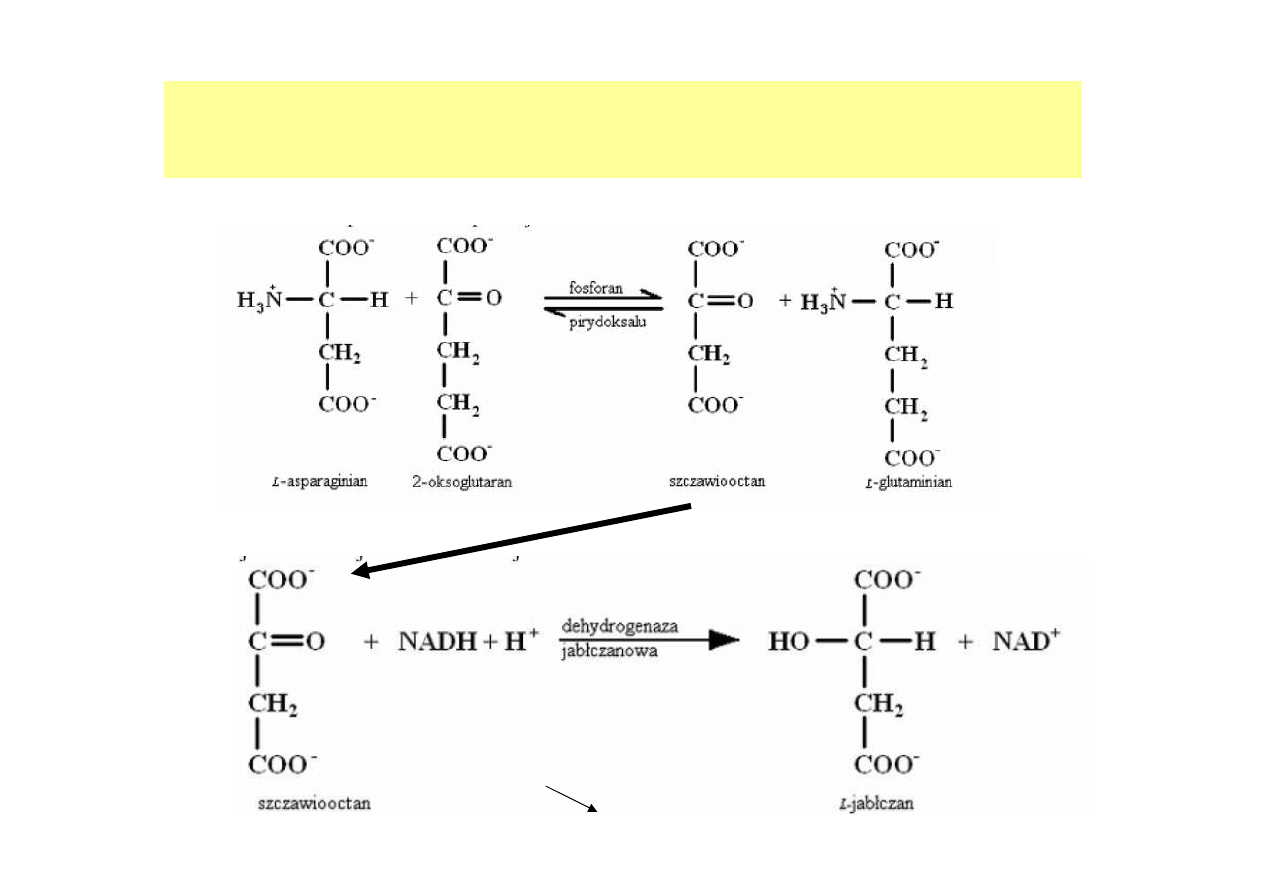
Np. Oznaczanie aktywności aminotransferazy asparaginianowej (AspAT)
reakcja wskaźnikowa
A
340
SPEKTROFOTOMETRYCZNE OZNACZANIE
AKTYWNOŚCI ENZYMÓW
dr inż. Aneta Białkowska
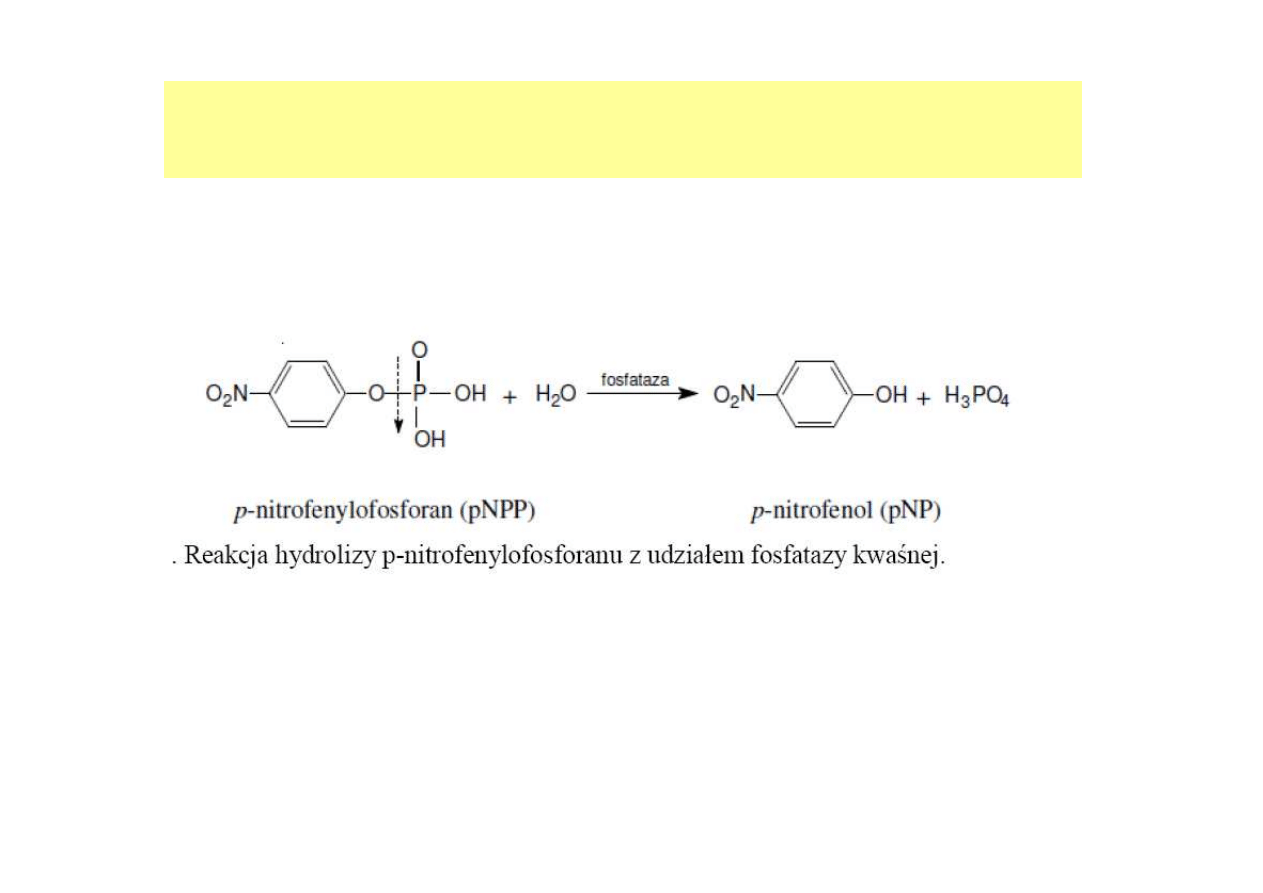
Np. Oznaczanie aktywności enzymów z użyciem substratów
syntetycznych
(w przypadku, gdy substraty czy kosubstraty nie zmieniają podczas reakcji
widma pochłaniania, jak również nie można sprzęgnąć reakcji z reakcją wskaźnikową)
Produkt reakcji, p-nitrofenol przyjmuje w środowisku zasadowym
barwę żółtą, co jest wykorzystane do oznaczania absorbancji w
próbach przy długości fali 420 nm.
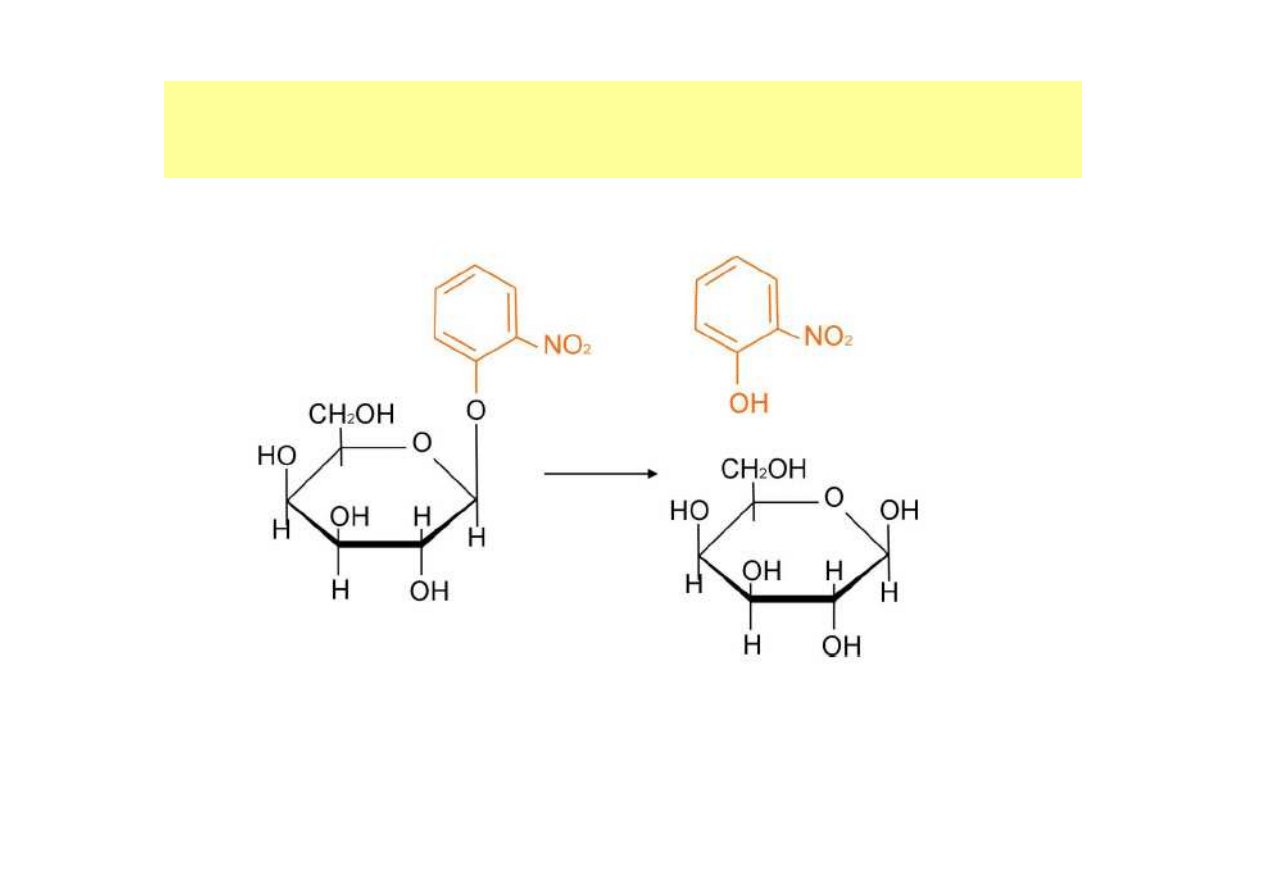
Reakcja hydrolizy o-nitrofenylo-
β
-D-galaktopiranozydu (ONPG) z udziałem
β
-galaktozydazy
SPEKTROFOTOMETRYCZNE OZNACZANIE
AKTYWNOŚCI ENZYMÓW
dr inż. Aneta Białkowska
Np. Oznaczanie aktywności enzymów z użyciem substratów
syntetycznych
Produkt reakcji, o-nitrofenol przyjmuje w środowisku zasadowym barwę żółtą, co jest
wykorzystane do oznaczania absorbancji w próbach przy długości fali 420 nm
.

FLUORESCENCYJNE OZNACZANIE
AKTYWNOŚCI ENZYMÓW
dr inż. Aneta Białkowska
Metoda fluorymetryczna opiera się na pomiarze natężenia emitowanego
promieniowania
fluorescencyjnego
powstałego
po wzbudzeniu
próbki.
Intensywność
fluorescencji
danej
substancji
jest
proporcjonalna do jej stężenia w pewnym przedziale stężeń.
Substancjami wykazującymi zjawisko fluorescencji wśród związków
biologicznie czynnych są m.in.:
1. Aminokwasy aromatyczne; tryptofan, tyrozyna i fenyloalanina
2. Zasady nukleinowe w DNA i RNA: adenina, guanina, cytozyna,
tymina i uracyl
3. Barwniki roślinne: chrofile, bakteriochlorofile i karotenoidy
4. Witaminy i hormony: np. ryboflawina
Metody te są w wielu wypadkach znacznie czulsze niż metody
spektrofotometryczne
.

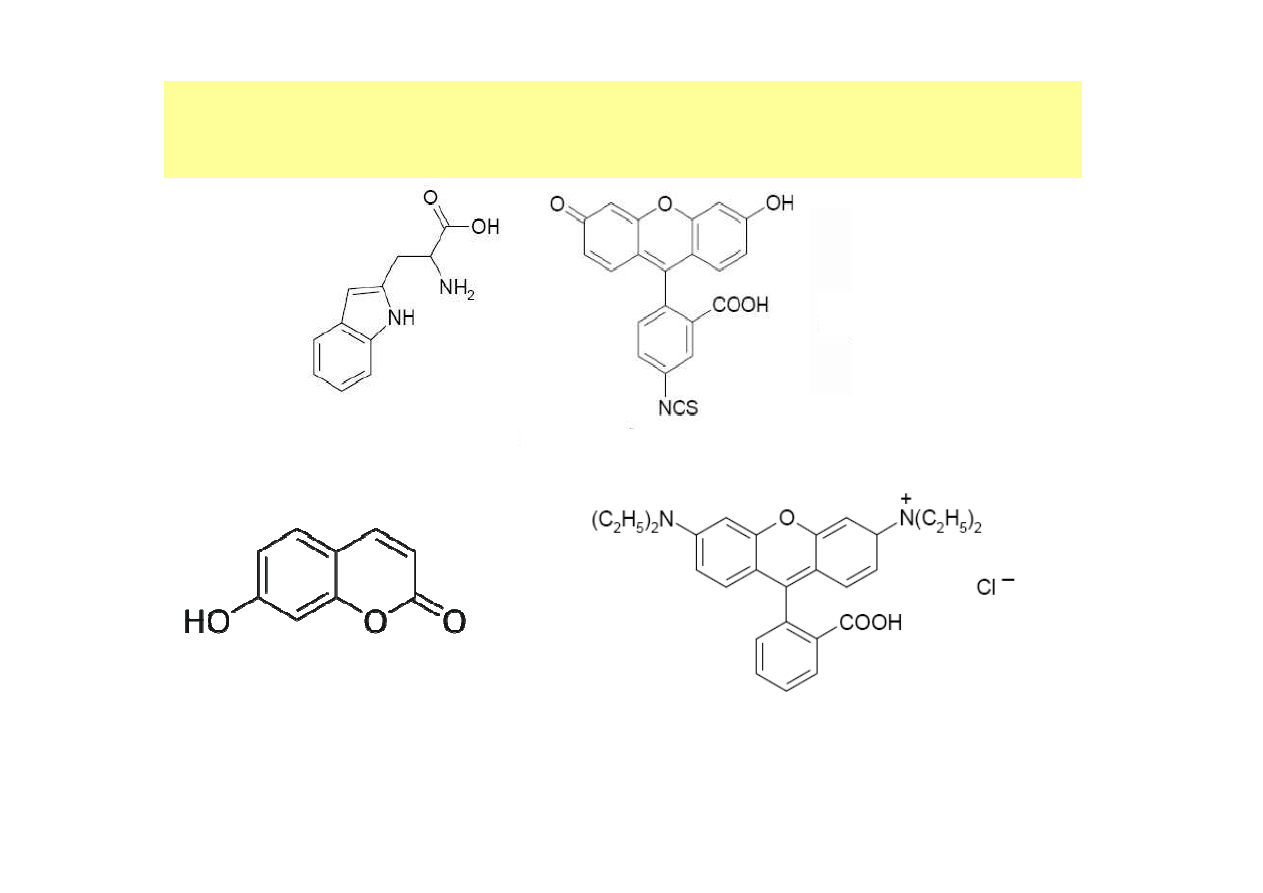
ZWIAZKI FLUORYZUJĄCE MOśNA PODZIELIĆ NA NASTĘPUJĄCE GRUPY:
•
Wewnętrzne
Tryptofan - zawierający w swojej strukturze grupę indolową. Wykazuje on
fluorescencję przy długości fali wzbudzenia 280 nm i długości fali emisji 340
nm.
•
Zewnętrzne
W przypadku, gdy badany związek nie wykazuje fluorescencji, możliwa jest
jego modyfikacja chemiczna za pomocą tzw. substancji fluoryzujących,
zwanych fluoroforami. Są to substancje selektywnie absorbujące i emitujące
światło o określonej długości fali. Jednym z przykładów jest izotiocyjanian
fluoresceiny, który reaguje z wolnymi grupami aminowymi obecnymi w
strukturach białek. Innymi mogą być np. rodamina lub umbeliferon.
FLUORESCENCYJNE OZNACZANIE
AKTYWNOŚCI ENZYMÓW
dr inż. Aneta Białkowska
FLUORESCENCYJNE OZNACZANIE
AKTYWNOŚCI ENZYMÓW
dr inż. Aneta Białkowska
umbelliferon
tryptofan
izotiocyjanian fluoresceiny
rodamina B
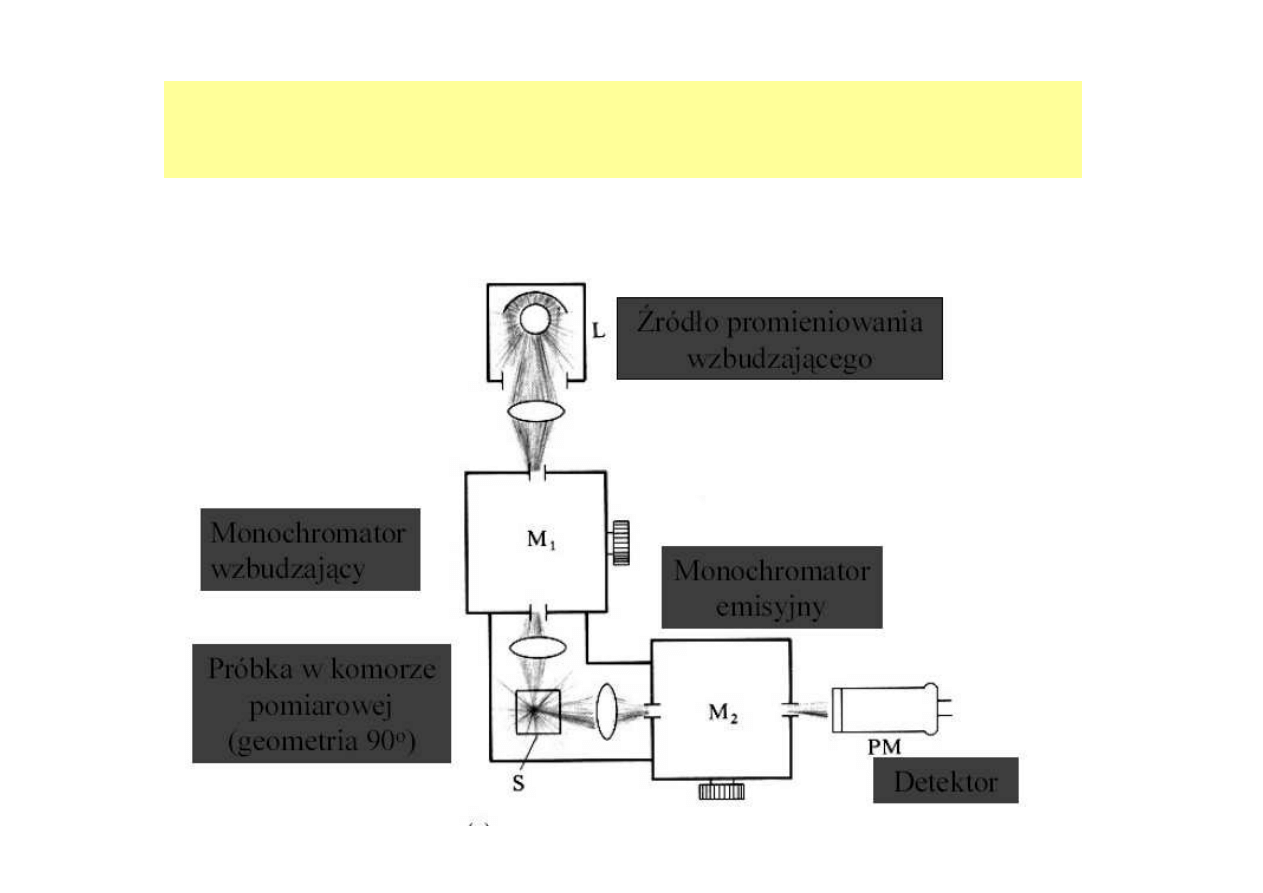
FLUORESCENCYJNE OZNACZANIE
AKTYWNOŚCI ENZYMÓW
dr inż. Aneta Białkowska
APARATURA- FLUORYMETR I SPEKTROFLUORYMETR

MANOMETRYCZNE OZNACZANIE AKTYWNOŚCI
ENZYMÓW
dr inż. Aneta Białkowska
Metody te stosuje się przede wszystkim wówczas, gdy np. jeden z
komponentów reakcji występuje w stanie gazowym i może to być substrat
(np. O
2
zużywany przez oksydazy), CO
2
(produkt wydzielany w reakcji
dekarboksylacji) lub N
2
(produkt wydzielany w reakcji deaminacji)
do oznaczania aktywności enzymów
hydrolizujących białka
GAZOMETRYCZNA METODA van SLYKE’a

MANOMETRYCZNE OZNACZANIE AKTYWNOŚCI
ENZYMÓW
dr inż. Aneta Białkowska
GAZOMETRYCZNA METODA WARBURGA
Jeżeli podczas reakcji powstaje kwas, a reakcję tę prowadzi się
w buforze dwuwęglanowym, znajdującym się w równowadze z gazową
mieszaniną, zawierającą określony procent CO
2
, wówczas stopniowe
wydzielanie się kwasu powoduje uwalnianie się odpowiednich ilości
CO
2
, który mierzy się manometrycznie. Grupy karboksylowe mogą
powstawać, np. przy hydrolizie wiązania peptydowego, eterowego,
estrowego i itp
..
OZNACZANIE STĘśENIA BIAŁKA W ROZTWORZE
dr inż. Aneta Białkowska
Metody
stosowane
do oznaczania
zawartości białka w
określonym materiale zależą od tego, czy zawarte w nim białka można w
całości rozpuścić w środowisku wodnym, a także czy badany materiał
nie zawiera substancji mających wpływ na wynik oznaczenia
.
Białka nierozpuszczalne
Białka rozpuszczalne
Metoda Kjeldahla
(polega na mineralizacji
materiału celem konwersji
związków azotu w jony
amonowe, które następnie
oznacza się miareczkowo)
Reakcja
biuretowa
Spektrofotome-
tryczny pomiar
absorbancji w
ultrafiolecie A
280
Reakcja
wykorzystująca
zdolność białek
do wiązania
specjalnego
barwnika (np.
Coomassie
Brilant Blue G-
250)
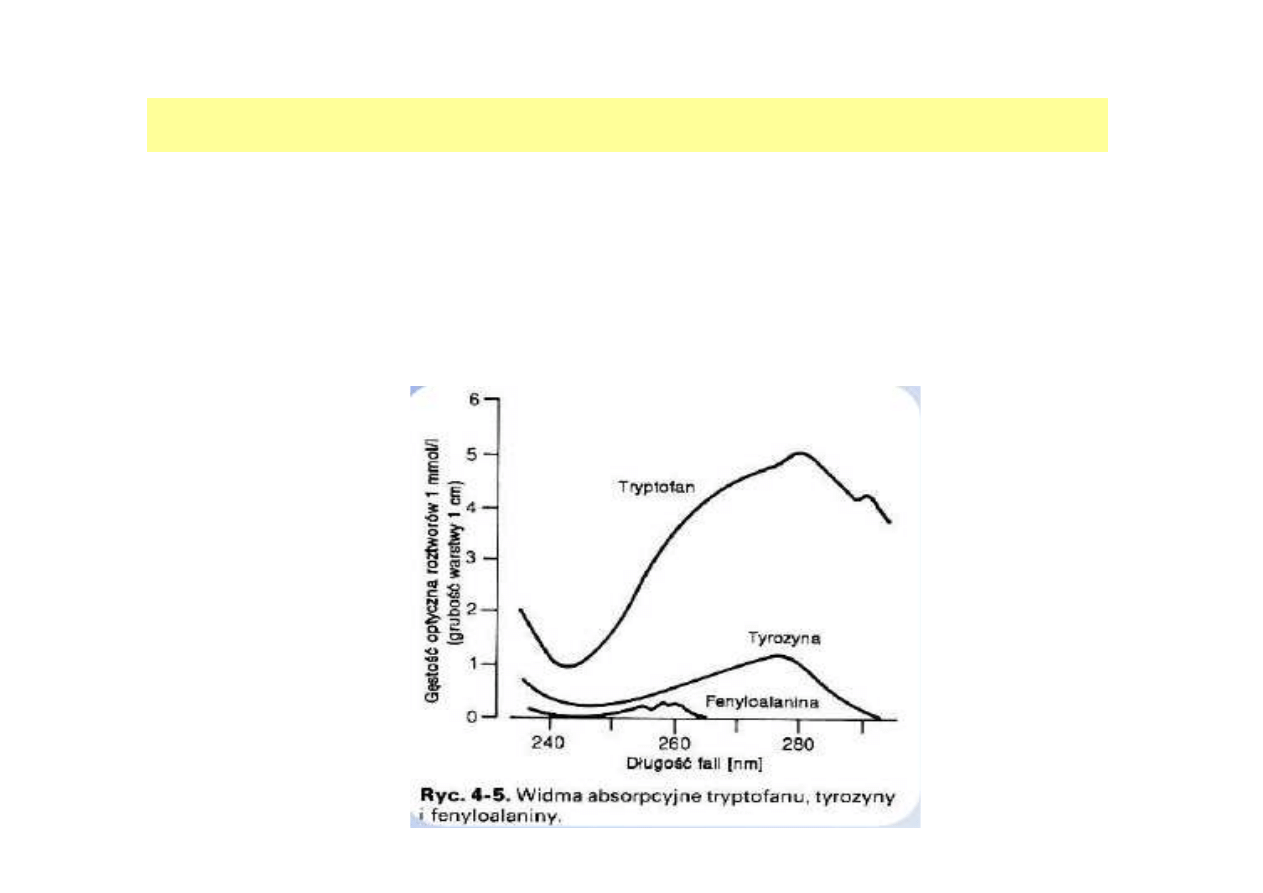
OZNACZANIE STĘśENIA BIAŁKA W ROZTWORZE
dr inż. Aneta Białkowska
Metoda pomiaru absorbancji w UV
Zasada:
Białka, dzięki obecności w swoim składzie aminokwasów
aromatycznych (np. tyrozyna, tryptofan) wykazują zdolność
absorpcji światła o długości fali 280 nm
.
OZNACZANIE STĘśENIA BIAŁKA W ROZTWORZE
dr inż. Aneta Białkowska
Zalety:
•Proste i szybkie oznaczenie
•Oszczędność materiału
Wady:
•Mała specyficzność
•Metoda nadaje się do oznaczania stężenia oczyszczonego białka (na
podstawie znanego współczynnika absorpcji). Wartość współczynnika
absorpcji zależy od % zawartości aminokwasów aromatycznych
•Obecność kwasów nukleinowych w badanym materiale przeszkadza
w oznaczeniu (max absorbcji 260 nm)
Metoda pomiaru absorbancji w UV
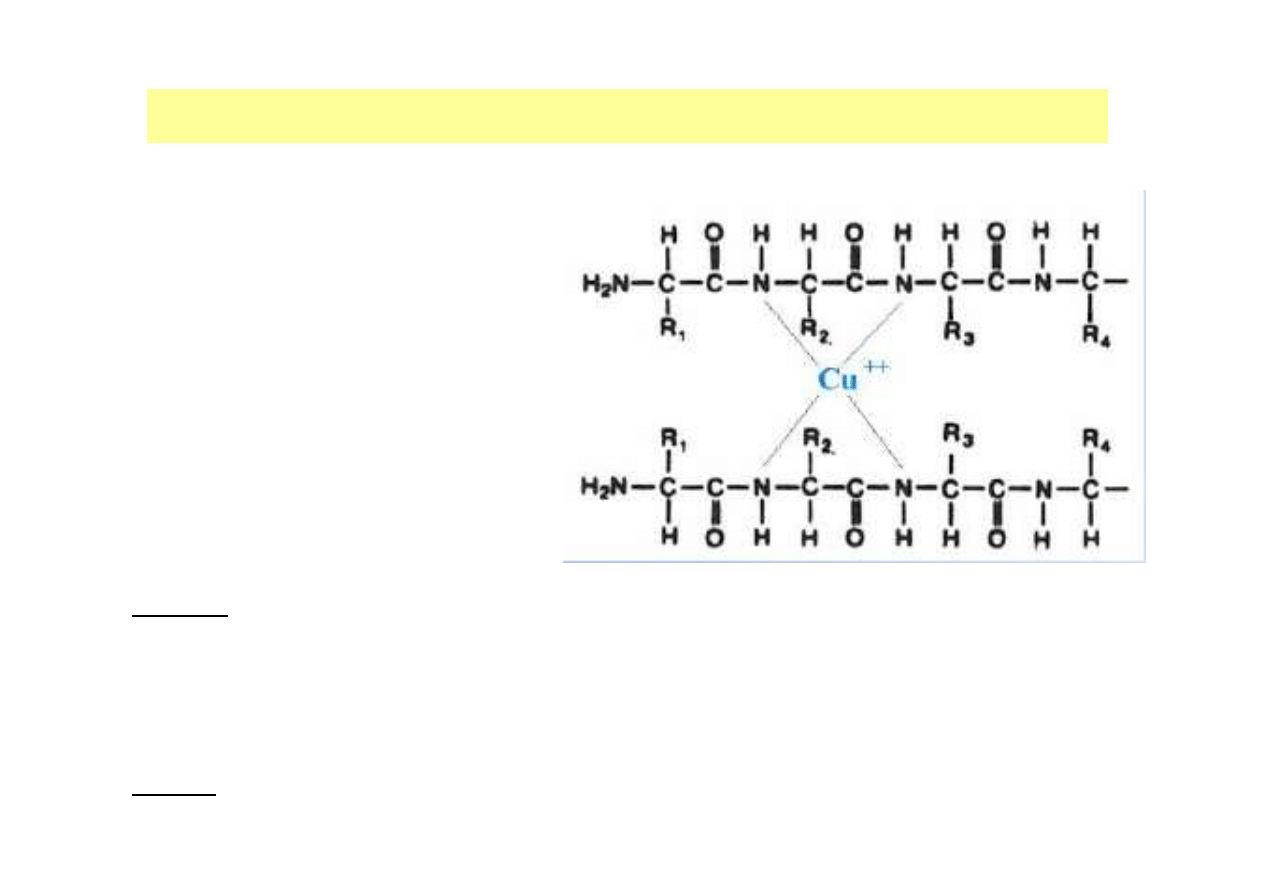
OZNACZANIE STĘśENIA BIAŁKA W ROZTWORZE
dr inż. Aneta Białkowska
Metoda biuretowa
•Oznaczenie natężenia barwy
powstałej w wyniku
utworzenia związków
kompleksowych białek z
jonami miedzi (II) w
środowisku zasadowym
•Intensywność barwy w
reakcji biuretowej jest
proporcjonalna do liczby
wiązań peptydowych
λλλλ
=530 nm
Zalety:
•Wysoka specyficzność dla wszystkich białek – absorbancja nie zależy od
rodzaju oznaczanego białka tylko od jego stężenia w próbie
•Prostota wykonania
Wady:
•Niska czułość
OZNACZANIE STĘśENIA BIAŁKA W ROZTWORZE
dr inż. Aneta Białkowska
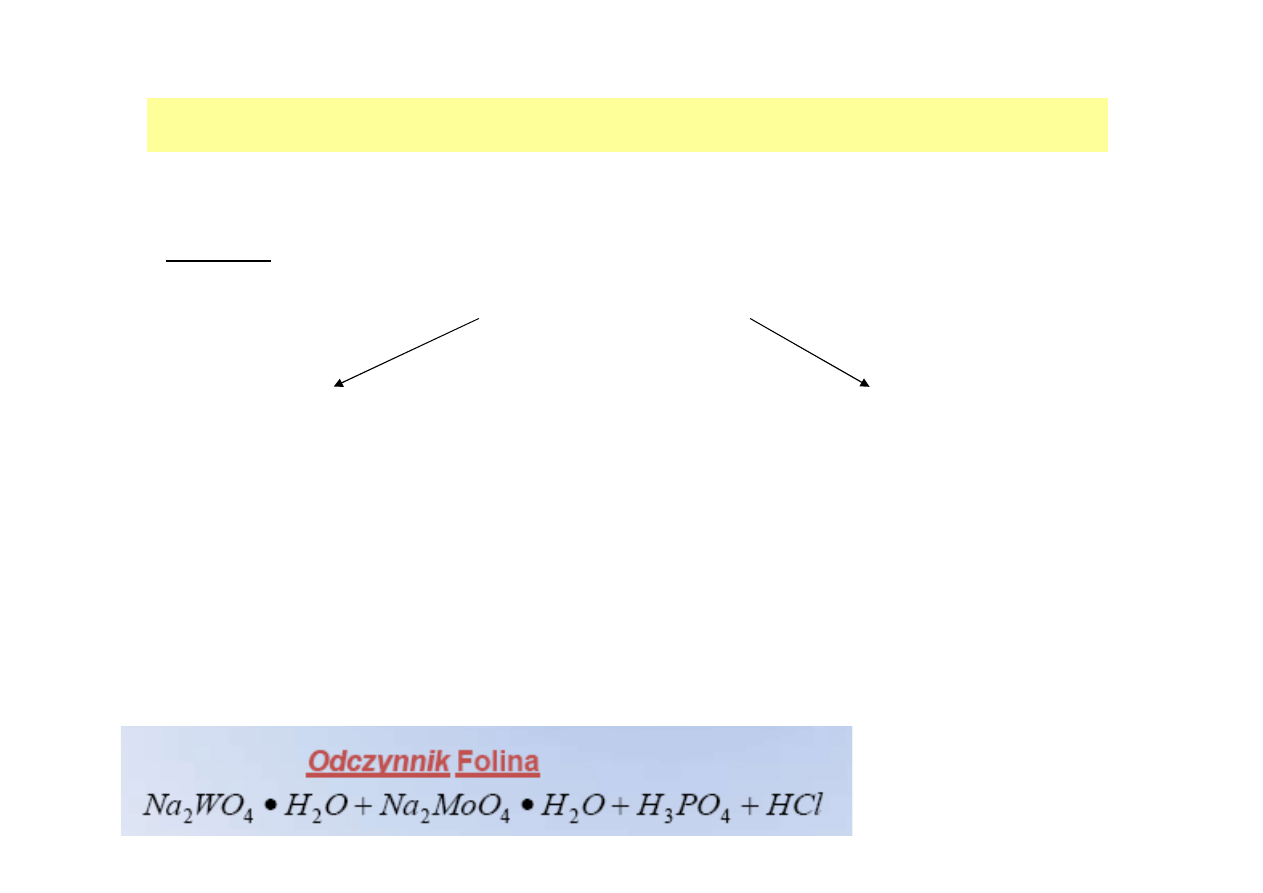
Oznaczenie stężenia białka metodą Lowry’ego
Zasada:
W metodzie tej wykorzystuje się 2 odrębne reakcje:
Reakcje aminokwasów z odczynnikiem
Folina:
Tyrozyna, tryptofan, cysteina oraz
przypuszczalnie histydyna występujące w
białkach redukują jony Mo
6+
/W
6+
zawarte w
odczynniku Folina- Ciocalteau do niższych
tlenków
Reakcję biuretową:
Jony miedzi (II) tworzą
kompleksowe połączenia z
wiązaniami peptydowymi,
uczestniczą również w redukcji
odczynnika Folina, przez co
zwiększa się czułość reakcji
KOŃCOWA BARWA JEST WYNIKIEM TYCH DWÓCH POWYśSZYCH REAKCJI
OZNACZANIE STĘśENIA BIAŁKA W ROZTWORZE
dr inż. Aneta Białkowska
Oznaczenie stężenia białka metodą Lowry’ego
Zalety:
•Bardzo wysoka czułość reakcji (ok. 70-krotnie wyższa niż w metodzie
biuretowej)
•Względna prostota wykonania
Wady:
•Wrażliwość na obecne w próbach substancje interferujące
Fenole, zasady purynowe i pirymidynowe, odczynniki sulfhydrylowe
oraz detergenty (zwiększają natężenie barwy)
Inne związki jak ZnSO
4
, siarczan amonu w stężeniach powyżej
0,1%, 1% etanol i eter obniżają intensywność barwy
•Mała specyficzność wobec różnych białek
OZNACZANIE STĘśENIA BIAŁKA W ROZTWORZE
dr inż. Aneta Białkowska
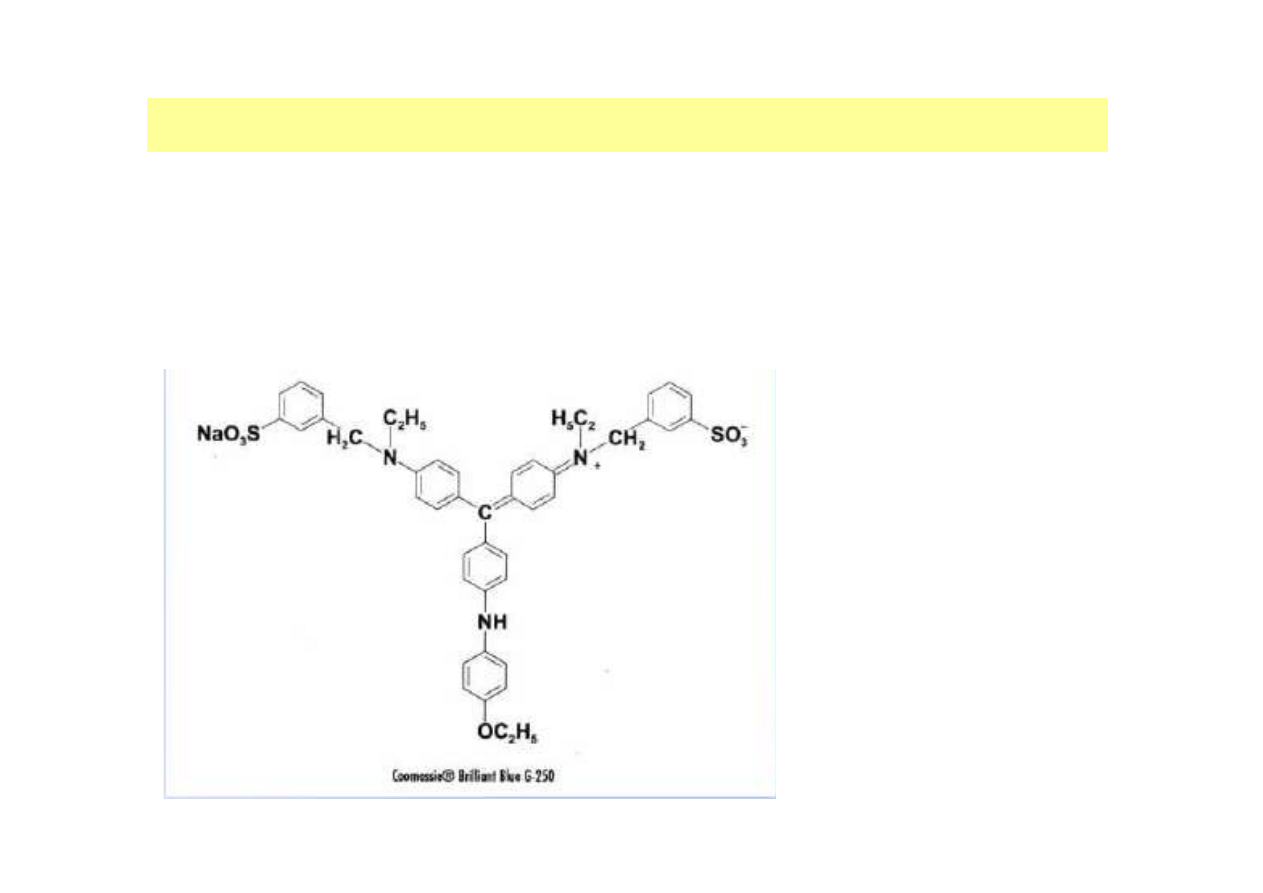
Oznaczenie stężenia białka metodą Bradforda
W metodzie wykorzystuje się zdolność wiązania barwnika błękitu
brylantowego – Coomassie Brilliant Blue G-250 (CBB G250) z grupami
aminowymi białek.
Maksimum absorpcji
λλλλ
= 595 nm
OZNACZANIE STĘśENIA BIAŁKA W ROZTWORZE
dr inż. Aneta Białkowska
Oznaczenie stężenia białka metodą Bradforda
•Wiązanie ma charakter oddziaływań elektrostatycznych
•CBB G-250 w środowisku kwaśnym ma brunatne zabarwienie, które
po reakcji z białkami zmienia się na błękitne
•Wiąże się z tym zmiana maksimum absorpcji z 465 nm na 595 nm
•Natężenie barwy jest proporcjonalne do zawartości białka w roztworze
400
450
500
550
600
650
700
0.0
0.2
0.4
0.6
0.8
1.0
1.2
odczynnik Bradforda +BSA (12
µµµµ
g/ml)
odczynnik Bradforda
A
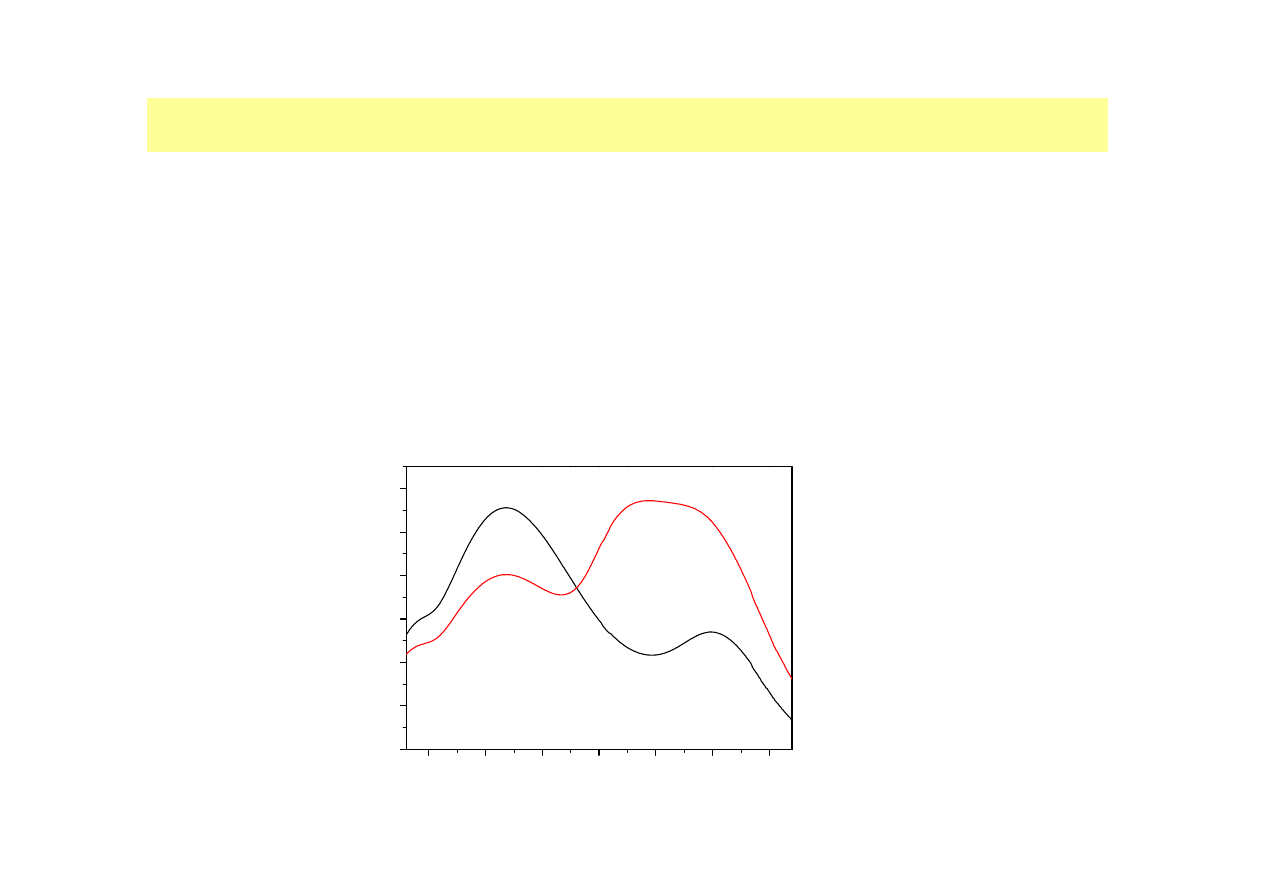
nm
Rys. Widmo absorpcji odczynnika Bradforda przed (----) i po dodaniu BSA (12
µ
g/ml)(
----
).

OZNACZANIE STĘśENIA BIAŁKA W ROZTWORZE
dr inż. Aneta Białkowska
Oznaczenie stężenia białka metodą Bradforda
Zalety:
•Prostota i szybkość oznaczenia
•Duża czułość porównywalna z metodą Lowry’ego
•Oznaczenie można wykonywać w obecności powszechnie stosowanych
buforów, EDTA, związków tiolowych, sacharozy i glicerolu
Wady:
•Mała specyficzność wobec różnych białek
•Detergenty (SDS, Triton X-100) dają dodatkowy interferujący kolor

OZNACZANIE STĘśENIA BIAŁKA W ROZTWORZE
dr inż. Aneta Białkowska
umiarkowana
Tak
0,005-0,05
Bradford
duża
Tak
0,05-0,5
Lowry’ego
bardzo mała
Tak
0,5-5,0
Biuretowa
duża
Nie
0,2-2,0
UV 280 nm
Zależność wyniku
od składu
aminokwasowego
Destrukcyjność
Zakres
oznaczalności
[mg]
Metoda
Wyszukiwarka
Podobne podstrony:
Microsoft PowerPoint Enzymologia cz IV
Microsoft PowerPoint Enzymologia cz V
Microsoft PowerPoint Enzymologia cz VI Ekstremozymy
Microsoft PowerPoint Cz II CFD
Nowy Prezentacja programu Microsoft PowerPoint 5
Rola rynku i instytucji finansowych INowy Prezentacja programu Microsoft PowerPoint
ZADANIA PiP Prezentacja Microsoft PowerPoint
Nowy Prezentacja programu Microsoft PowerPoint ppt
Microsoft PowerPoint IP5 klasyfikacje tryb zgodnosci
Microsoft PowerPoint IP tryb zgodnosci
Microsoft PowerPoint 02 srodowisko bazy danych, modele
(Microsoft PowerPoint 2 KONWENCJA WIEDENSKAid 1358 (2)
Microsoft PowerPoint IP5 bazydanych tryb zgodnosci
Microsoft PowerPoint znaki
więcej podobnych podstron