Inżynieria genetyczna
Najważniejsze fakty z historii rozwoju genetyki i biologii molekularnej, definicja inżynierii genetycznej, omówienie znaczenia badań z tego zakresu.
Pojęcia genetyczne (krótki słownik)
Mechanizmy regulacji ekspresji genów u prokariotów regulacja natywna (operon laktozowy u E.coli, operon tryptofanowy, regulacja pozytywna,.
Kataboliczna represja glukozowa.
Izolowanie i oczyszczanie DNA z drobnoustrojów (genomowy DNA, plazmidowy DNA- ultrawirowanie w CsCL-EtBr).
Endonuklezay restrykcyjne (występowanie, rodzaje i zastosowanie).
Ligazy DNA z bakteriofaga T4 i E.coli.
Pozochromosomalne elementy genetyczne u drobnoustrojów (mitochondrialny DNA [mtDNA], plazmidy bakteryjne [rodzaje, cechy w nich kodowane, sposób działania antybiotyków i mechanizmy oporności]).
Plazmidowe systemy wektorowe, opis konstrukcji wektora pBR322.
Omówienie budowy i charakterystyka właściwości dobrego palzmidu wektorowego.
Klonowanie z wykorzystaniem insercyjnej inaktywacji (defosforylacja DNA przy użyciu fosfotaz CIP i BAP, jej zastosowanie).
Omównie grupy wektorów wywodzących się od pBR322.
Omówienie rodziny wektorów plazmidowych typu pUC.
Wektory niskokopiowe i typu „run-away”.
Wektory do identyfikacji sygnałów transkrypcyjnych (promotorów i terminatorów).
B. subtilis jako organizm-gospodarz, wektory do klonowania tych bakteriach.
Wektory bifunkcjonalne (wahadłowe, shuttle, wektor pv33).
Plazmid drożdżowy 2 um, wektory stosowane do klonowania w drożdżach (np.pYAC) wektory bifunkcjonalne E.coli uzasadnienie celowości klonowania w E.coli.
Niestabilność plazmidów (segregacyjna i strukturalna)
Definicja wirusa, bakteriofaga lambda, możliwości zastosowania jako wektora klonującego.
Fagowe wektory klonujace wtrąceniowe i wymienialne (zasada in vitro upakowania rekombinowanego DNA w wirusowych otoczkach białkowych).
Biologiczne zabezpieczenia przed ucieczką rekombinowanego DNA z laboratorium (mutacje nonsens w wektorach).
Charakterystyka bakteriofaga M13, jego cykl rozwoju, wektory konstruowane w oparciu o tego bakteriofaga.
Fagemidowe systemy wektorowe, charakterystyka i zastosowanie.
Charakterystyka wektorów kosmidowych (klonowanie kosmidami trawionymi dwoma enzymami restrykcyjnymi, chramoidy)
Łączenie DNA (terminalna transferaza, dorabianie homopolimerowych „ogonów”, linkery, adaptory).
Biblioteki genów, czynniki warunkujace ich wielkość , równanie Clarka_Carbona.
Metody transformacji E.coli (z CaCl2 , PEG-DMSO, elektrotransformacja)
Metody transformacji B.subtilis (komrek komplementarnych, tarnsformacja protoplastów)
Metody bezpośredniej selekcji i skriningu rekombinantów (ekspresja genów warunkujących antybiotykową oporność, ekspresja genów enzymów szlaków biosyntez związków koniecznych dla rozwoju komórek-aminokwasów, nukleotydy, witaminy-, ekspresja geów enzymów sprawdzalnej aktywności (alfa-amylaza, proteinaza, fenolo-liaza tyrozynowa).
Metody identyfikacji klonów w banku genów (metody immunologiczne i immunochemiczne w przypadku ekspresji genów, metody hybrydyzacyjne w przypadku banku ekspresji genów (sondy DNA , sondy RNA)
Charakterystyka aktywności właściwej polimerazy DNA I, fragment Klenowa oraz zastosowanie tego enzymu.
Enzymatyczna synteza genów na matrycy mRNA (polimeraza DNA zależna od RNA [odwrotna tarnskryptaza], zalety otrzymannia cDNA w odniesieniu do genów eukariotycznych).
Chemiczna synetza oligonukleotydów-ogólny schemat, automatyzacja, klonowanie chemicznie syntetyzowanego genu samalostatyny.
Metoda chemiczna określania sekwencji nukleotydów Maxama-Gilberta.
Metoda enzymatycza określania sekwencji nukleotydów Sangera-Coulsona (znakowanie radioaktywnością i fluorescencją, automatyzacja procesu0
Reakcja PCR, zasada metody oraz jej wykorzystanie.
Zalety zastosowania termostabilnych polimeraz w technice PCR.
Reakcja PCR w czasie rzeczywistym (Real_Time PCR) zasada metody oraz jej wykorzystanie.
5. Izolowanie i oczyszczanie genomowego i plazmidowego DNA z komórek bakteryjnych.
Kwas DNA genomowy (chromosomalny) występuje u bakterii w postaci pojedynczej, kolistej cząsteczki DNA o skomplikowanej konformacji. DNA plazmidowe - kolista, dwuniciowa cząsteczka DNA, która może ulegać replikacji niezależnie od genomu komórki. Wielkość plazmidów nie przekracza 8% DNA chromosomalnego. DNA plazmidowe można wypreparować z komórki nieuszkodzone natomiast nie da się wypreparować chromosomalnego DNA gdyż cząsteczka ta jest zbyt duża(następuje zniszczenie struktury natywnej konformacji)
W preparatyce kwasu nukleinowego wyróżniamy 4 etapy:
Hodowla organizmu, z którego chcemy wyizolować DNA (otrzymanie odpowiedniej ilości komórek bakteryjnych do preparatyki DNA)
Dezintegracja komórki aby uwolnić DNA (zniszczenie struktury osłon zewnętrznych)
Oczyszczanie wyizolowanego kwasu (oddzielenie innych białek)
Uzyskanie odpowiedniego stężenia (zatężanie)
Plazmid do analiz można uzyskać w małej ilości przez izolację z kilku ml hodowli bakteryjnej, taki proces zwany jest jako izolacja plazmidu z małej ilości pożywki lub miniizolacja. Kilka ml pożywki bakteryjnej inokuluje się szczepem bakterii zawierającym pożądany plazmid. Hodowlę prowadzimy na podłożach płynnych (hodowla wgłębna): o zdefiniowanym składzie tzn. zawartość wszystkich składników jest dokładnie ustalona (Na2HPO4, KH2PO4, NaCl, NH4Cl, MgSO4, glukoza, CaCl2 - podłoże dostosowane do organizmów prototroficznych, zdolnych do syntezy wszystkich potrzebnych składników z podstawowych związków) lub podłożach niezdefiniowanych np. LB (trypton jako źródło aminokwasów i peptydów, ekstrakt drożdżowy - suszony preparat komórek drożdżowych, źródło C w formie polisacharydów, NaCl). Po całonocnej hodowli, gdy bakterie osiągną pożądaną fazę S (stacjonarną) osadza się komórki bakteryjne przez wirowanie.
Najczęściej stosowaną metodą oczyszczania plazmidowego DNA z chromosomalnego DNA i innych komponentów komórkowych to liza alkaliczna. Osad komórek zawieszany jest w buforze, który zawiera lizozym niszczący ścianę komórkową bakterii. Następnie dodaje się roztwór SDS (detergent dodecylosiarczan sodu) w roztworze wodorotlenku sodu. SDS niszczy błony komórkowe i denaturuje białka. Dalej roztwór jest zobojętniany przez dodanie stężonego KOH o pH 5,0 w celu wytracenia zdenaturowanych białek i chromosomalnego DNA oraz większości detergentu. Próbę powtórnie wiruje się a uzyskany supernatant (lizat) zawiera plazmidowy DNA, który łatwo reneturuje po zobojętnieniu. Oprócz plazmidowego DNA lizat zawiera RNA i trochę białek.
Inne metody dezintegracji komórek można podzielić na:
Mechaniczne - bezpośrednie rozrywanie ścian komórkowych przez mielenie, rozcieranie, homogenizację (ucieranie w moździerzu z piaskiem kwarcowym - w przypadku preparatyki DNA chromosomalnego następują uszkodzenia co nie jest wskazane)
Fizyczne - np. szok osmotyczny, gwałtowna dekompresja, zamrażanie i rozmrażanie (powstające kryształy mogą rozrywać ścianę kom.), działanie ultradźwiękami. Jeśli zależy nam na preparatyce natywnego DNA metody fizyko-chemiczne nie są zalecane.
Chemiczne - niszczenie błon komórkowych przy użyciu rozpuszczalników, detergentów kwasów (stosowany czynnik nie może niekorzystnie wpływać na produkt, który chcemy uzyskać)
Biochemiczne - rozkład ściany komórkowej na drodze lizy enzymatycznej np. przy wykorzystaniu lizozymu, który rozkłada wiązanie β-1,4-glikozydowe powodując dezintegrację ściany komórkowej. Lizozym +EDTA (chelator czyli związek kompleksujący jony magnezu występujące w ścianie komórkowej przez co wspomaga działanie lizozymu). Po dezintegracji ściany komórkowej otrzymujemy sfero- lub protoplasty, które można zniszczyć stosując zmiany ciśnienia
Począwszy od tego etapu izolacji istnieje niezliczona ilość metod otrzymywania czystego plazmidowego DNA. Wiele z tych metod wykorzystuje selektywne wiązanie DNA do żeli lub błon i wymywanie ich bez białek i RNA. Klasyczna metoda polega na ekstrakcji lizatu fenolem lub mieszaniną fenol-chloroform. Fenol miesza się z fazą wodną ale po energicznym mieszaniu z nią i ponownym rozdzieleniu denaturuje pozostające białka, które tworzą osad na granicy faz. Przed użyciem czynnika chemicznego odbiałczanie można wspomóc przez zastosowanie enzymów proteolitycznych specyficznych o dużej czystości (preparaty te nie mogą zawierać nukleaz). Np. proteaza E, proteinaza K. Enzymy te specyficznie odcinają białka od kompleksów z kwasem.
DNA i RNA pozostające w fazie wodnej są zagęszczane przez wytracanie etanolem. Gdy chcemy uzyskać czysty preparat to pomijamy etap wytrącania etanolem. Usuwamy zanieczyszczenia preparatu poprzez dializę (fenol i chloroform przechodzą przez pory woreczka dializacyjnego do roztworu a DNA zostaje.
Metoda lizy alaklicznej może być stosowana także do izolacji plazmidu na dużą skalę i uzyskania nawet mg plazmidowego DNA. W tym przypadku można zastosować wirowanie w gradiencie gęstości CsCl jako końcowy etap oczyszczania. Jeżeli gotowy lizat jest frakcjonowany na gradiencie CsCl w obecności bromku etydyny można zobaczyć rozdzielone wszystkie jego składnik. Superhelikalny DNA wiąże mniej bromku etydyny niż linowa czy kolista forma DNA, ponieważ ma większa gęstość (jest mniej uszkodzony). Stąd w jednym etapie można izolować dużą ilość superhelikalnego plazmidu i uwolnić od zanieczyszczeń białkiem, RNA i chromosomowym DNA. Frakcję zawierającą prążek plazmidowego DNA pobiera się z probówki do wirowania i ewentualnie zagęszcza przez wytracenie etanolem po usunięciu bromku etydyny.
6. Endonukleazy restrykcyjne - rodzaje, sposób działania i własności.
Wprowadzenie do biologii molekularnej enzymów restrykcyjnych spowodowało, w latach 70-tych, szybki rozwój badań nad struktura DNA.
Endonukleazy restrykcyjne są enzymami bakteryjnymi, które rozcinają (hydrolizują) DNA na zdefiowane i powtarzalne fragmenty. Rozcinaja symetrycznie obydwie nici DNA w obrębie krótkich poliandrowych (symetrycznych) sekwencji zwykle o długości 4, 5 lub 6 nukleotydów, stanowiących rozpoznane przez nie miejsca restrykcyjne. Pozostawiają one grupę fosforanową na końcu 5' a grupę OH na końcu 3' cząsteczki DNA.
Istnieją 3 różne sposoby rozcinania DNA:
nierównomierne rozcięcie "na ukos" pozostawiające niesparowany koniec 5' (tzn. na końcu 5' pozostaje krótki jednoniciowy odcinek wysunięty przed koniec 3' rejonu dwuniciowego). Powstają wtedy końce kohezyjne (lepkie), które umożliwiają dwóm dowolnym fragmentom DNA, uzyskanym z użyciem tego samego enzymy restrykcyjnego, utworzenie komplementarnych par zasad. Końce takie mogą być następnie połączone kowalencyjnie przez ligazę (mówimy, że nastąpiła ligacja). Nową cząstkę DNA, utworzoną przez połączenie fragmentów DNA nazywamy cząsteczką zrekombinowanego DNA.
nierównomierne rozcięcie pozostawiające niesparowany koniec 3'
rozcięcie obu nici w tym samym miejscu z utworzeniem tępych końców rozciętego DNA. Cząsteczki o tępych końcach można łączyć za pomocą ligazy, ale reakcja ta przebiega trudniej niż w przypadku łączenia lepkich końców.
Enzymy restrykcyjne izoluje się z bakterii, gdzie ich rola polega na ochronie komórki gospodarza przed wprowadzeniem do niej obcego DNA. Ten mechanizm obronny składa się z dwóch elementów. Pierwszym jest endonukleaza restrykcyjna, która rozpoznaje sekwencję DNA i przecina szkielet każdej nici DNA w specyficznym miejscu w obrębie tej sekwencji. Obcy DNA jest zatem degradowany do krótkich fragmentów. Drugim etapem jest metylaza, która dodaje grupę metylową do zasady C lub A . Ta modyfikacja sprawia ze DNA gospodarza staje się odporny na degradację przez endonukleazę.
Są 3 klasy enzymów restrykcyjnych:
I klasa, enzymy, które przecinają DNA w dowolnym miejscu i nie charakteryzują się rozpoznawaniem określonej sekwencji (są mało specyficzne). Wymagają Mg2+, adenozylometioniny, ATP do działania. Są one mało przydatne.
II klasa, enzymy rozpoznają określoną sekwencję nukleotydową, przecinają obie nici DNA albo w obrębie tej rozpoznawanej sekwencji albo w dokładnie określonym miejscu w pewnej odległości od sekwencji rozpoznawanej. Do działania wymagają Mg2+.
III klasa, enzymy rozpoznające sekwencje nukleotydowe ale dokonują przecięcia (hydrolizy wiązania fosfodiestrowego) w nieokreślonym miejscu. Mało przydatne. Wymagają Mg2+ , ATP do działania.
Działanie endonukleaz restrykcyjnych przedstawia rysunek:
wystające lepkie końce od strony 5':
EcoR1 5'-GAATTC--3' __→ --GOH PAATTC--
3'-CTTAAG--5' --CTTAAP + OHG--
wystające lepkie końce od strony 3':
Pst 1 5'-GAATTC--3' __→ --GAATTOH PC--
3'-CTTAAG--5' --CP + OHTTAAG--
tępe końce :
Sma 1
5'-CCCGGG--3' __→ --CCCOH PGGG--
3'-GGGCCC--5' --GGGP + OHCCC--
Mniej specyficzne restryktazy
Bgl I GCCNNNNNGGN
Sekwencja rozpoznawana jest przecięta 5 dowolnymi nukleotydami
Hga I
5'-CACGNNNNN NNNNNNN---3'
3'-CACGNNNNNNNNNN NN---5'
Miejsce przecięcia jest dokładnie określone na nici górnej 5 za sekwencją rozpoznawaną a na dolnej 10.
Izoschizomery (czyli enzym rozpoznający identyczne sekwencje nukleotydowe pochodzące z różnych mikroorganizmów)
Wszystkie enzymy restrykcyjne stawiają wysokie wymagania pod względem środowiska działania (buforu restrykcyjnego). Dla każdej grupy tych enzymów trzeba opracować specjalne roztwory buforowe zawierające jony Mg2+, o pH ok. 7 (czasami alkaliczne) i odpowiednim stężeniu soli (stężenie to jest regulowane przez dodatek KCl, CH3COOK i innych).jak ważne jest steżniee soli pokazuje przykład restryktazy Eco R1.
W warunkach normalnych (bufor zawiera dużo soli) EcoR1 rozpoznaje sekwencje 6 nukleotydowe. Gdy działa on w buforze o niskiej zawartości soli to wykazuje aktywność z * (tzn. nie rozpoznaje 6 nukleotydowej sekwencji ale 4 nukleotydową a więc znacznie częściej będzie przecinał łańcuch DNA).
EcoR1 5'-GAATTC--3' EcoR1* 5'-NAATTN--3'
3'-CTTAAG--5' 3'-NTTAAN--5'
Enzymy restrykcyjne znalazły zastosowanie w identyfikacji mutacji i markerów polimorficznych, sporządzaniu map fizycznych cząstek DNA, wyodrębnianiu oraz klonowaniu fragmentów DNA.
7. Ligazy DNA: z bakteriofaga T4, z E. coli, sposób działania i własności
Ligaza DNA odpowiedzialna jest za kowalencyjne łączenie się cząstek DNA. Enzym ten normalnie naprawia pęknięcia w jednej nici dwuniciowego DNA gdy na jej końcu 5' znajduje się grupa fosforanowa. Do swego działania potrzebuje czynnika adenylujacego aby zaktywować grupę fosforanową i upodatnić ją na atak grupy 3' OH. Są one wykorzystywane w każdym żywym organizmie w procesie replikacji. Ligaza nie może łączyć lub zamykać w formę kolistą jednoniciowych fragmentów DNA. Wymaganie ligaz: na końcu 3' musi znajdować się grupa OH a na 5' grupa fosforanowa, łączone odcinki DNA muszą znajdować się w obrębie dwuniciowej helisy. Energia potrzebna do procesu ligacji pochodzi z ATP.
Najczęściej używane ligazy to enzymy z bakterii E.coli oraz faga T4 (przy czym ligazy faga T4 wykorzystywane są częściej niż E. Coli, ze względu na wyższą wydajność).
Ligaza DNA z faga T4 naprawia pęknięcia w rdzeniu curowo-fosforanowym dwuniciowego DNA i może kowalencyjnie łączyć hybrydyzowane końce, co stanowi odwrócenie reakcji trawienia restyrkcyjnego. Ligaza ta łączy fragmenty DNA mające tępe końce ze znacznie niższą wydajnością niż lepkie końce. Aby ligaza łączyła tępe końce z większą wydajnością należy podwyższyc stężenie soli oraz dodać PEG (glikol polietylenowy).
Ligaza DNA z E.coli używa kofaktora NAD+ jako nietypowego źródła energii. NAD+ jest rozcinany do mononukleotydu nikotynoamidowego NMN i monofosforanu adenozyny AMP, po czym AMP jest kowalencyjnie przyłączony do końca 5' fragmentu Okazaki przez wiązanie 5'-5'pirofosforanowe. Ligaza z E. coli nie jest w stanie tworzyć hybrydowych cząsteczek RNA : DNA w przeciwieństwie do ligazy z faga T4.
Mechanizm działania ligaz:
Dzięki ATP (lub NAD+) tworzy się kompleks enzym-AMP, w którym AMP połączony jest z enzymem wiązaniem fosfoamidowym poprzez resztę aminową lizyny. Jednocześnie uwalniany jest pirofosforan. Zaktywowana reszta AMP jest następnie przenoszona z reszty lizyny na koniec 5' łańcucha i powstaje kompleks adenylan-DNA. Kolejny etap to atak nukleofilowy grupy 3'OH na zaaktywowany koniec 5'. Ten ciąg reakcji jest napędzany hydrolizą pirofosforanu, uwalnianego podczas tworzenia kompleksu enzym - adenylan.
8.Pozochromosomalne elementy genetyczne u drobnoustrojów (mitochondrialny DNA [mtDNA], plazmidy bakteryjne [rodzaje, cechy w nich kodowane, sposób działania antybiotyków i mechanizmy oporności]).
Plazmidy - dziedziczne pozachromosomalne elementy genetyczne (u bakterii superskręcone, koliste cząsteczki zamknięte).Plazmidy bakteryjne w optymalnych warunkach produkty zakodowane w plazmidach nie są konieczne do rozwoju organizmu (dodatkowa informacja).
Cechy fenotypowe zakodowane w plazmidach:
oporność na działanie antybiotyków takich jak: ampicylina, kanamycyna, tetracyklina, streptomycyna, chloramfenikol, sulfonamidy. Oporność jest łatwo przekazywana między mikroorganizmami.
Produkcja antybiotyków (gdy w środowisku jest konkurencja)
Degradacja związków organicznych (gdy nie ma skażenia środowiska nie ma potrzeby)
Fermentacja cukrów
Odporność na działanie metali ciężkich
Produkcja enzymów restrykcyjnych i modyfikujących ( ta cecha może i zazwyczaj jest zakodowana w chromosomie)
Produkcja colicyn (substancje bakteriostatyczne wpływające na ograniczenie w środowisku konkurencji)
Produkcja enterotoksyn.
Ze względu na fenotyp, jaki plazmidy występujące u Enterobacteriaceae nadają komórkom gospodarza, wyróżnia się 3 grupy plazmidów:
A') czynnik płciowy F - komórki bakteryjne z plazmidem F są dawcami genów plazmidowych i chromosomalnych
B') czynniki R nadają komórkom oporność na leki i sole metali ciężkich a także na jeden lub kilka antybiotyków. Ze względu na łatwość rozprzestrzeniania się w populacji mają one duże znaczenie epidemiologiczne
C') czynniki kolicynogenne Col - warunkują syntezę substancji antybiotycznych, kolicyn
Plazmidy mogą być:
Pod ścisłą kontrolą - replikacja tych plazmidów zachodzi równocześnie z replikacją chromosomalnego DNA. Są one zazwyczaj dużych rozmiarów (ich masa jest większa niż 45 kbp). Trudno je wypreparować ze względu na duże rozmiary i małą ilość kopii w cząsteczce.
Pod luźną kontrolą - ich replikacja przebiega niezależnie od chromosomalnego DNA (masa ich <10 kpb)
Ilość kopii plazmidów jest związana z mechanizmem kontroli. Plazmidy pod ścisłą kontrolą występują w niskiej liczbie kopii gdyż są duże i replikują z chromosomalnym DNA. Ich liczba 5 na komórkę. Plazmidy pod luźną kontrolą są wielokopiowe, ich liczba może sięgać kilkuset kopii na komórkę. Gdy dodamy antybiotyku do pożywki to mikroorganizm nie będzie się namnażać ale liczba kopii plazmidów pod luźną kontrolą będzie coraz większa bo replikacja zachodzi niezależnie od DNA chromosomalnego.
Istnieją też plazmidy kryptyczne, które nie wyróżniają się żadną łatwo rozpoznawalną cechą fenotypową. Możemy stwierdzić ich istnienie bo pojawia się pasek na obrazie elektroforetycznym ale obecność tych plazmidów nie demonstruje się charakterystyczną cechą.
Ze względu na zdolność przenoszenia się do innych bakterii plazmidy możemy podzielić na: infekcyjne i nie infekcyjne. Plazmidy infekcyjne przenoszą własny materiał genetyczny, mobilizują także chromosom gospodarza lub inne plazmidy pozbawione własności płciowych. Plazmidy nieinfekcyjne i niektóre czynniki R do swojego przenoszenia wymagają obecności innego plazmidu o właściwościach płciowych.
Plazmidy koniugacyjne - wyposażone są w system przenoszenia informacji tra (transfer factor) między komórkami na drodze koniugacji.
Antybiotyk |
Sposób działania |
Mechanizm oporności |
Ampicylina Ap+, amp+ |
Pochodna penicyliny zabija rosnące komórki zakłócając końcowy etap syntezy ściany komórkowej, hamuje działanie licznych enzymów błony cytoplazmatycznej |
Gen oporności bla koduje peryplazmatyczny enzym β-laktamazę, który przecina pierścień β-laktamowy w antybiotyku. Enzym ten jest wydzielany poza komórkę do podłoża hodowlanego w wyniku czego tworzy się strefa wokół rosnącej kolonii. |
Tetracyklina tc+, tet+ |
Związek bakteriostatyczny, który hamuje namnażanie drobnoustrojów. Uniemożliwia on syntezę białek łącząc się z podjednostką 30S rybosomów. |
Gen oporności tet koduje białko modyfikujące bakteryjną membranę co uniemożliwia transport antybiotyku do komórki. Białko spełnia funkcje blokujące - antybiotyk nie wchodzi do środka (a tylko tam działa). |
Kanamycyna |
Związek bakteriobójczy. Łączy się z rybosomami i powoduje błędne odczytywanie mRNA. |
Gen oporności kan koduje enzym fosfotransferazę modyfikujący antybiotyk uniemożliwiając jego łączenie z rybosomami ze względu na inhibicję aktywnego transportu. |
Chloramfenikol cm |
Związek bakteriobójczy. Zakłóca on syntezę bakteryjnych białek przez łączenie się z podjednostką 50S rybosomów. Uniemożliwia tworzenie wiązania peptydowego. |
Gen oporności cat koduje acetylotransferazę chloramfenikolową, która inaktywuje antybiotyk poprzez jego acetylację. |
Streptomycyna |
Związek bakteriobójczy. Łączy się z podjednostką 30S rybosomów powodując błędny odczyt mRNA. |
Gen oporności str koduje enzym fosfotransferazę modyfikujący antybiotyk uniemożliwiając jego połączenie z rybosomami ze względu na inhibicję aktywnego transportu. |
9. Plazmidowe systemy wektorowe, opis konstrukcji wektora pBR322.
Wektor - cząsteczka DNA służąca do przenoszenia fragmentów DNA pomiędzy organizmami i umożliwiająca wprowadzenie DNA do innego organizmu. Jednym z pierwszych opracowanych wektorów plazmidowych został nazwany pBR322.Zarówno on jak i jego pochodne mają dwa geny oporności na atybiotyki: ampicylinę i tetracyklinę.
pBR322 - jest dobrym wektorem gdyż ma małe rozmiary, dwa markery selekcyjne, zawiera szereg pojedynczych miejsc restrykcyjnych ma miejsce inicjacji replikacji ori od którego zależy ilość kopii. Obecność sekwencji ori rozpoznawanej przez polimerazę DNA E. coli oraz fakt, iż replikacja plazmidu jest uniezależniona od syntezy białka, umożliwiają intensywne namnażanie cząsteczek wektora w komórkach, których wzrost i rozmnażanie został zahamowane np. chloramfenikolem. Inne części, pochodzące z naturalnych plazmidów Co1E1, R1, pMB1 i pSC101, zostały zreorganizowane przy udziale odpowiednich nukleaz restrykcyjnych. Wektor pBR322 zawiera 20 miejsc rozpoznawanych przez różne restryktazy, zlokalizowanych zarówno w obrębie dwóch genów oporności - na ampicylinę i tetracyklinę, jak i poza tymi genami. Część enzymów rozpoznaje i przecina tylko 1 sekwencję nukleotydową, co umożliwia precyzyjne włączenie obcego DNA w zaplanowane miejsce. Geny oporności na antybiotyki, zachowane z plazmidów naturalnych służą jako markery przy klonowaniu genów, zarówno podczas selekcji klonów, jak i identyfikacji miejsc wstawki obcego DNA. W odniesieniu do lokalizacji wstawki, utrata oporności nastąpić może w stosunku do 1 z ww antybiotyków lub też zostaje zachowana oporność na obydwa. W tym drugim przypadku wstawka znajduje się poza genami oporności. Jeżeli obcy fragment DNA został wrekombinowany, np. w obrębie genu oporności na ampicylinę, wówczas selekcję rekombinantów prowadzi się na podłożu z tetracykliną (na tym podłożu wyrosną także komórki zawierające wyjściowy wektor bez klonowanego genu a więc te, które wyrosły na podłożu z tetracykliną przesadza się na podłoże z ampicyliną i tu wyrosną tylko te, które nie przyjęły wstawki).
10.Omówienie budowy i charakterystyka właściwości dobrego palzmidu wektorowego.
Cechy dobrego wektora klonującego:
autonomiczna replikacja - plazmid w komórce pod luźną kontrolą (replikacja zachodzi niezależnie od replikacji chromosomów bakteryjnych). Autonomiczna replikacja umożliwia uzyskanie dużej ilości powtórzeń.
mały rozmiar - im mniejszy rozmiar tym więcej kopii, im więcej kopii tym więcej matryc dla transkrypcji mRNA. Łatwiej wprowadzić małe cząstki. Wydajność transformacji jest odwrotnie proporcjonalna do wielkości cząsteczki wprowadzanej. Wektor powinien posiadać tylko tyle aby mógł się replikować wraz z zawartą informacją w komórce bakteryjnej.
Markery selekcyjne - cechy fenotypowe, które są zakodowane w cząsteczce i umożliwiają łatwą selekcję cząstek, które przyjęły wstawkę. Mogą być inne niż oporność na jakiś związek np. antybiotyk. Mogą to być geny kodujące enzymy np. znoszące efekt mutacji auksotroficznych.
Pojedyncze miejsca dla enzymów restrykcyjnych - stosując enzym restrykcyjny rozpoznający tylko 1 miejsce otrzymujemy liniową cząsteczkę (otwarcie cząstki kolistego DNA na żelu elektroforetycznym uzyskujemy 1 prążek). W regionie inicjacji replikacji DNA nie ma miejsc rozpoznawanych przez enzymy restrykcyjne (nie może tu nastąpić przecięcie i/lub wstawienie fragmentu gdyż nie zaszłaby replikacja). Miejsca restrykcyjne powinny być w obrębie oporności na antybiotyki (w tym wypadku).
Silne promotory - ich siła polega na tym, że polimeraza RNA łatwo rozpoznaje ten region. Silne promotory są najłatwiej rozpoznawane przez polimerazę RNA zależną od DNA a im łatwiej się ona przyłącza tym proces szybciej zachodzi. Od silnych promotorów zależy ekspresja.
Duża liczba kopii - duża liczba genów, transkryptów i duża wydajność biosyntezy białka.
Ważne jest aby miejsca działania wektorów restrykcyjnych znajdowały się w obrębie genów markerów selekcyjnych.
Klonowanie z wykorzystaniem insercyjnej inaktywacji (defosforylacja DNA przy użyciu fosfotaz CIP i BAP, jej zastosowanie).
Technika insercyjnej inaktywacji polega na uszkodzeniu markera selekcyjnego poprzez włączenie wstawki. Klonowanie tą metodą jest możliwe gdy są co najmniej dwa markery. Wygodnym plazmidem do klonowania jest pBR322. Zawiera on geny oporności na tetracyklinę i ampicylinę. Aby klonować w wektorze plazmidowym, najpierw rozcina się kolisty plazmid za pomocą restryktazy, dla której istnieje w cząsteczce wektora pojedyncze miejsce restrykcyjne. Uzyskuje się w ten sposób liniową cząsteczkę plazmidu z kohezyjnymi końcami. Następnie obce DNA rozcinamy tymi samymi enzymami (można użyć innych enzymów ale muszą one tworzyć jednakowe końce kohezyjne). Obcy DNA i liniowy plazmid miesza się. Kohezyjne końce obcego DNA i liniowego plazmidu odnajdują się, ulegają sparowaniu i następnie zostają kowalencyjnie połączone z użyciem ligazy. Zrekombinowany DNA plazmidowy zostaje wprowadzony do komórek bakterii dzięki zwiększeniu przepuszczalności błon poprzez odpowiednie traktowanie solami. Podczas namnażania bakterii zrekombinowane cząsteczki plazmidów ulegają replikacji.
Dokonujemy w pBR322 przecięcia w obrębie genu oporności na tetracyklinę - stosujemy ograniczone trawienie enzymem BamH1 (możemy zastosować także Sau3A i Nbo I gdyż tną one w tych samych miejscach co BamH1 ale rozpoznającymi sekwencje 4 - nukleotydowe). Pojemność wektora jest ograniczona więc wstawiany fragment musi być w miarę krótki. Obcym DNA jest DNA wypreparowany z Bacillus subtilis (gen α-amylazy chcemy wstawić). Liniowy wektorowy DNA poddajemy działaniu fosfatazy, która odcina grupy fosforanowe z 5' końca - zmniejsza to prawdopodobieństwo zamknięcia przez ligazę pustego wektora. Fragmenty chromosomalnego DNA są donorami grup fosforanowych dla wektorów podczas recyrkulacji. Po wymieszaniu liniowych fragmentów wektorów z fragmentami wstawianymi w wyniku działania ligazy (wymaga ona grupy fosforanowej na 5' końcu i OH na 3') możemy otrzymać zamknięte wektory zawierające wstawkę lub nie. Po ligacji przygotowujemy podłoże z ampicyliną (w obrębie tego genu nic nie robiliśmy). Na podłożu tym wyrastają tylko te kolonie, które przyjęły wektor z wstawką lub wektor pusty. Aby spośród wyrosłych transformantów wyłonić odpowiednie dokonujemy repliki (stosujemy stemple np. plusz na walcu) na 2 płytkach, jednej z ampicyliną i drugiej z tetracykliną. Na 1 powinno wyrosnąć tyle samo koloni co na płytce, z której robiliśmy odbitkę a na 2 wyrośnie tylko część. Porównujemy obrazy na dwóch płytkach. Gdy zauważamy wzrost tylko na płytce z ampicyliną to znaczy, że nastąpiło przerwanie ciągłości w obrębie genu tetracyklinowego (wstawienie fragmentu). Insercja fragmentu DNA w miejscu rozcięcia poza obrębem genów oporności na antybiotyki nie wpływa na ich ekspresję. Jednakże wstawienie fragmentu DNA w obrębie genu oporności powoduje inaktywację oporności na dany antybiotyk i taki zjawisko nazywamy insercyjną inaktywacją. Komórki z insertem w jednym z miejsc restrykcyjnych w obrębie genu oporności są oporne na ampicylinę ale wrażliwe na tetracyklinę, co umożliwia ich łatwą selekcję.
12. Przykłady wektorów wywodzących się od pBR322 i ich własności.
Na podstawie pBR322 skonstruowano następujące systemy wektorowe:
pAT153 -(3.7 kb) -powstał w 1980 roku. W porównaniu z pBR322 mniejszy o ok. 700 par zasad (wycięto 1 niekodujący fragment) dzięki czemu liczba kopii w komórce zwiększyła się trzykrotnie a także stało się niemożliwe niekontrolowane przekazywanie tego wektora między komórkami (np. na drodze koniugacji). Zabezpieczenie przed rozprzestrzenianiem się na drodze koniugacji uzyskano poprzez usunięcie fragmentu pU2. Problem niekontrolowanego przekazywania wektora między komórkami istniał w przypadku pBR322. pAT posiada większą niż pBR322 pojemność.
pBR325 -(5.4 kb) - posiada dodatkowo trzeci marker selekcyjny (gen oporności w stosunku do chloramfenikolu). Zwiększył się rozmiar wektora bo dodany został gen oporności, zmniejszyła się też pojemność.
pBR328 -(4.9 kb) -nastąpiła rearanżacja wektora. Ma on trzy markery (oporność w stosunku do ampicyliny, tetracykliny i chloramfenikolu). Powstał przez skrócenie pBR32 o 650 par zasad.
pMK2004 -(5.2) - dodany gen oporności na kanamycynę (ale chyba brak na chloramfenikol)
pACYC177 -(3.7) - mały wektor. Zawiera gen oporności w stosunku do ampicyliny i kanamycyny.
13. Rodzina wektorów pUC, ich budowa oraz właściwości ułatwiające klonowanie fragmentów DNA i selekcję rekombinantów.
Wektory pUC zostały stworzone na podstawie pBR322. Ich wielkość ok. 270 bp. Występują w dużej liczbie kopii. Wektory te zawierają tylko 1 gen oporności. Ten system wektorowy pozwala po pierwszej nocy inkubacji wybrać transformanty ze wstawką. Wektory pUC są najczęściej używane do transformacji E. coli. Wektor pUC19 zawiera:
sekwencje odpowiedzialne za inicjację replikacji, pochodzące z plazmidu ColE1
gen oporności na ampicylinę
fragment genu β-galaktozydazy uzupełniający uszkodzony gen tego enzymu u szczepów bakterii E. coli przygotowanych specjalnie do transformacji tym wektorem. Komórki E coli wykorzystywane w tym systemie wektorowym mają mutację w obrębie genu β-galaktozydazy i jej nie produkują (normalnie E. coli mają sprawnie działający gen β-galaktozydazy i cały operon laktozowy).Gdy do E. coli wstawimy pUC bez wstawki obcego DNA to komórki odzyskują zdolność produkcji β-galaktozydazy (wektor zawiera 146 nukleotydowy fragment β-galaktozydazy, który uzupełnia uszkodzony gen).
Sekwencje kodujące fragment enzymu w plazmidzie znajdują się pod kontrolą promotora operonu laktozy i ich ekspresja może być indukowana przez syntetyczny induktor, izopropylotiogalaktozyd (IPTG). Fragment genu β-galaktozydazy zawiera specyficzne sekwencje (tworzące polilinker - krótki oligonukleotyd tak zaprojektowany aby dostarczał pojedynczych miejsc restrykcyjnych. Wstawienie polilinkera do fragmentu β-galaktozydazy nie zakłóca α-komplementacji i nie wyłącza aktywności genu) rozpoznawane i trawione przez kilkanaście nukleaz restrykcyjnych, tych samych, które mogą być używane do przygotowania fragmentów DNA do klonowania. Włączenie obcego fragmentu DNA w polilinker wektora pUC19 uniemożliwia syntezę właściwego polipeptydu kodowanego przez fragment genu β-galaktozydazy, znosząc tym samym komplementację uszkodzonego genu. Obecność obcej wstawki DNA wykrywana jest na podstawie zabarwienia kolonii bakteryjnych rosnących na odpowiednio skomponowanej pożywce agarowej. W podłożu selekcyjnym z ampicyliną, induktorem IPTG oraz syntetycznym analogiem substratu - x-Gal, kolonie zawierające plazmid pUC19 bez wstawki obcego DNA wytwarzają komplementujący fragment β-galaktozydazy, dzięki czemu w następstwie wiązania się enzymu z chromoforowym substratem zastępczym x-Gal, wybarwiają się na kolor niebieski. Kolonie bakterii posiadających wektor z obcą wstawką sklonowanego DNA (np. chromosomalny DNA kodujący α-amylazę) nie mogą syntetyzować brakującego fragmentu β-galaktozydazy i pozostają nie wybarwione. Powstałe w wyniku ekspresji białko ma oprócz sekwencji właściwej fragment β-galaktozydazy, nazywa się fuzyjnym i z reguły działa. W przypadku przyłączenia fragmentu do białka nie przeszkadza i produkowany enzym jest czynny. W przeciwnym wypadku należy się pozbyć fragmentu N-końcowego. Na etapie wektora klonującego w wektorze możemy umieścić fragment DNA kodujący peptyd łączący się trwale z czymś (np. powinowactwo do materiału kolumny chromatograficznej) dzięki czemu uzyskujemy łatwiejsze oddzielenie białka, na którym na zależy. Obecnie stosuje się fragmenty 6-histydynowe, które wiążą się z niklem (białko związane z niklem na kolumnie wymywamy na końcu). Inny przykład: zaszczepiamy białymi koloniami fermentor. Każda komórka tej koloni zawiera wstawkę z genem β-galaktozydazy oraz od strony N-terminalnej NBP (białko wykazujące powinowactwo do maltozy). Stosujemy kolumnę ze złożem amylozowym (amyloza jest nierozpuszczalna i na końcu łańcucha ma maltozę). Po przepuszczeniu produktu przez kolumnę zostaną zatrzymane białka fuzyjne. Na końcu białka fuzyjnego jest dołączona sekwencja kodująca 4 cząsteczki kwasu asparaginowego i lizynę. Enterokinaza (proteinaza) przecina wiązanie peptydowe za lizyną gdy przed nią są 4 cząsteczki kwasu asparaginowego. Białko fuzyjne związane z kolumną wymywamy roztworem maltozy. Po wymyciu mamy białko fuzyjne (nasze białko i NPB). Oddzielamy teraz te dwa składniki.
14. Wektory niskokopiowe i plazmidy typu "run away", ich wykorzystanie do klonowania.
Ważną cechą wektora jest jego występowanie w dużej liczbie kopii. Czasami jednak klonujemy geny, których ekspresja u E. coli jest dla mikroorganizmu szkodliwa (każdy obcy DNA ulegający ekspresji w nowej komórce jest dla niej w mniejszym bądź większym stopniu szkodliwy). Wektory niskokopiowe stosujemy więc gdy gen, który ulega ekspresji jest bardzo szkodliwy dla E. coli (np. produkt ekspresji klonowanego genu hamuje namnażanie kolonii). Zależy nam na tym aby przynajmniej w fazie logarytmicznego wzrostu poziom ekspresji tego szkodliwego genu był niski. Wskazane jest więc stosowanie systemu wektorowego, który zapewni nam niską dawkę genu czyli plazmidów niskokopiowych. Są systemy, w których regulacja genów jest pod ścisłą kontrolą i te właśnie systemy wykorzystujemy do tworzenia wektorów niskokopiowych. Przykłady:
plazmid pochodzący od pMF3 (11kbp). Występuje w 1-2 kopii na komórkę. Jako marker selekcyjny ma gen β-laktamazy, który nadaje oporność w stosunku do ampicyliny.
PMSG415 wektor pochodzący od plazmidu pSC101 (7kbp). Występuje w ok. 5 kopiach na komórkę (chromosom bakteryjny). Zawiera 3 markery selekcyjne: geny oporności na Amp. I Kan oraz chloramfenikol.
Przy stosowaniu systemów niskokopiowych występuje szereg problemów m.in. ze względu na małą ilość kopii trzeba je izolować z dużej ilości komórek bakteryjnych. Problem może także stanowić niestabilność takich systemów (plazmidy mogą być "gubione" przy podziałach komórkowych. Po osiągnięciu stanu nasycenia (tzn. fazy stacjonarnego wzrostu) staramy się osiągnąć wyższy poziom ekspresji. W przypadku bakteriofaga λ możemy wpływać na liczbę kopii danego wektora przez podwyższenie temperatury (gen warunkujący przejście na wyższy poziom ekspresji jest indukowany wyższą o 2-3 stopnie C temperaturą). Początkowa temperatura hodowli 300C (w tej temperaturze występuje niska ilość kopii co nie jest szkodliwe dla komórki). W fazie wzrostu stacjonarnego podwyższamy temperaturę co powoduje uruchomienie systemu i wzrost liczby kopii do kilkuset na komórkę. Tego typu rozwiązanie nosi nazwę "run away". Zmianą temperatury można regulować liczbę kopii a także uruchamiać ekspresję poszczególnych genów (gdy np. nie chcemy aby produkt genu zakłócał procesy podziału). Uruchomienie mechanizmu niekontrolowanej replikacji wektora wraz z wstawką. Zwiększenie ilości kopii - wydajniejsza transkrypcja, translacja. Systemy takie pobieramy często z wirusów. Mechanizm niekontrolowanej replikacji (w fazie stacjonarnego wzrostu) może być też uruchamiany dodatkiem jakiegoś inhibitora. Aby uzyskać lepszą ekspresję możemy zwiększać ilość kopii lub też zastosować wektor zależny od temperatury (indukcja promotora poprzez wzrost temp.) lub dodatku substancji.
15. Wektory do określania sygnałów transkrypcyjnych - identyfikacja regionów promotorowych i regulatorowych.
8bp
EcoR1
Aby sprawdzić czy klonowany fragment zawiera sekwencję regulacyjną (promotor, operator) stosujemy następujące systemy:
działanie jego jest oparte na genie nadającym oporność w stosunku do tetracykliny. Wstawiamy 8 nukleotydową sekwencję, w której 6 nukleotydów stanowi sekwencję rozpoznawaną przez EcoR1. P - promotor genu oporności w stosunku do tetracykliny. Wstawienie 8 nukleotydów było wystarczające aby uniemożliwić ekspresję genu oporności do tetracykliny. Jeśli wprowadzimy taki wektor do E. coli to nie możemy oczekiwać wzrostu na podłożu z tetracykliną. Jeżeli jednak w tym miejscu (po rozcięciu EcoR1) wstawimy fragment obcego DNA zawierającego sekwencję promotorową to może zostać uruchomiona transkrypcja genu oporności w stosunku do tetracykliny, zajdzie translacja i zostanie wytworzony produkt genu. Jeśli otrzymamy wzrost na podłożu zawierającym tetracyklinę to możemy stwierdzić, że fragment włączony zawiera sekwencję promotorową. W ten sposób można też kontrolować wydajność promotora. Stosując różne dawki tetracykliny można zbadać siłę promotora, poziom ekspresji tego genu. Przy wyższej ekspresji można oczekiwać, że bakterie otrzymają wyższe dawki tetracykliny (?)
niekiedy zależy nam na obecności sekwencji promotorowych i terminatorowych
z operonu laktozy genu
HincII KpnI β-galaktozydazy
HindIII HpaII SmaI
Promotor, operator arabinozowy
Pomiędzy regionami regulacyjnymi wstawiamy dłuższy oligonukleotyd zawierający kilka miejsc restrykcyjnych. Sekwencja oligonukleotydu, wstawiona pomiędzy sekwencje regulacyjne a gen struktury jest tak dobrana aby nie przerywała ciągłości genu (polilinker). Ekspresja może następować pod wpływem sekwencji regulatorowych z arabinozy. Dodatek arabinozy uruchamia ekspresję. β-galaktozydaza nie będzie syntetyzowana dopóki w podłożu nie będzie arabinozy (łatwo można to sprawdzić na podstawie barwy. Ten system musi współpracować ze specjalnie przygotowanymi szczepami E. coli (normalnie E. coli mają operon β-galaktozydazowy). Otrzymujemy barwne kolonie. Chcemy sprawdzić czy sekwencja promotorowa jest we włączonym fragmencie. Transformanty na podłożu z arabinozą i x-Galem jeśli mają promotor to rosną w postaci niebieskich kolonii (uruchomiona transkrypcja β-galaktozydazy). Można sprawdzić obecność promotora wysiewając je na podłożu nie zawierającym induktora. Jeśli zachodzi transkrypcja to znaczy, że dodany został promotor i nastąpiła konstytutywna ekspresja niezależna od induktora. Jeżeli jest wstawiony także terminator to wysiane komórki na podłoże z arabinozą pozostaną bezbarwne gdyż nie nastąpi ekspresja β-galaktozydazy. W przypadku sekwencji terminatorowych nawet w obecności arabinozy nie będzie ekspresji.
Wektory bifunkcjonalne (wahadłowe, shuttle, wektor pv33).
Plazmid drożdżowy 2 um, wektory stosowane do klonowania w drożdżach
(np.pYAC) wektory bifunkcjonalne E.coli uzasadnienie celowości klonowania w E.coli.
Znacznie atrakcyjniejszymi organizmami od Bacillus subtilis są drożdże (np. produkcja insuliny). U drożdży udało się zlokalizować plazmid nazwany 2 μm ze względu na małe rozmiary. Plazmid ten występuje w dwóch konformacjach (rys Krzysiek str.6b). Występuje on w liczbie 70 - 100 kopii/komórkę. Jest to plazmid kryptyczny tzn. taki, który nie wyróżnia się żadną łatwo rozpoznawalną cechą fenotypową. Możemy stwierdzić jego istnienie bo pojawia się pasek na obrazie elektroforetycznym ale obecność tego plazmidu nie demonstruje się charakterystyczną cechą. Obecność sekwencji ars, warunkującej replikację plazmidu niezależnie od genomu drożdży, umożliwia wykorzystanie go do konstruowania wektorów do manipulacji genetycznych u drożdży (i innych grzybów). W komórkach drożdżowych nie działają geny oporności w stosunku do antybiotyków (a więc nie można u nich oddzielać komórek transformowanych od nietransformowanych na podstawie oporności w stosunku do antybiotyków). Wykorzystano więc drożdże, u których wystąpiła mutacja auksotroficzna - uszkodzono gen Leu2 kodujący enzym dehydrogenazę 3-izopropylojabłczanową niezbędną do produkcji leucyny. Prototrofy, które z prostych źródeł mogą syntetyzować wszystkie potrzebne do wzrostu związki mogą rosnąć na podłożu dość ubogim. Na podłożu minimalnym po rozsiewie wyrosły tylko prototrofy natomiast na podłożu z leucyną wyrosły zarówno prototrofy jak i mutanty auksotroficzne. Do wektora wprowadzono gen Leu 2, dzięki czemu wektor ma marker i tylko komórki które przyjęły ten wektor mogły rosnąć na podłożu ubogim. W ten sposób można było wyróżnić transformanty. Niestety nie można było odróżnić komórek, które przyjęły gen z wstawką, a które bez wstawki (bo tylko 1 marker). Skonstruowano wektor hybrydowy mogący funkcjonować u drożdży i u E. coli - (Apr,Ter). Izolację interesującego nas genu możemy przeprowadzić u E. coli i jeśli znajdziemy kolonię z interesującą nas wstawką, to wektor taki możemy wprowadzić do komórek drożdżowych, gdzie będzie funkcjonował. Są trzy rodzaje wektorów dla drożdży:
wektory episomalne - niezależnie replikują
wektory integracyjne - w budowie ich są sekwencje DNA homologiczne do regionów DNA chromosomalnego (krzyżowa rekombinacja) - mogą być integrowane z chromosomem. Są stabilniejsze od poprzednich, bo materiał jest przekazywany z chromosomem
sztuczne chromosomy drożdżowe (YAC) - mają nawet 11,4 kpb. Wektor YAC zawiera wszystkie istotne cech chromosomu, które są potrzebne do jego powielania w komórkach drożdży, łącznie z miejscem początku replikacji, centromerem zapewniającym segregację chromosomów siostrzanych w komórkach i telomerami stabilizującymi końce chromosomu. Wektory YAC mogą przyjąć bardzo duże cząsteczki DNA, o długości kilkuset kpz. Mają podobne markery selekcyjne do Leu2 (TRP1, URA3, SUP4 - kolonie rosną na czerwono). Po trawieniu BamH1 otrzymujemy liniową cząsteczkę. Po trawieniu SnaB1 otrzymujemy lewe i prawe ramie. SnaB1 rozcina w miejscu SUP4. Po wstępnym przygotowaniu dodajemy interesujące nas fragmenty DNA. Po dodaniu DNA prowadzimy ligację z tępymi końcami. Ligaza może połączyć 2 ramiona lewe i 2 prawe (tandemy). Otrzymujemy różne kombinacje, ale najlepsza jest ze wstawką między prawym a lewym ramieniem. Wysiewamy zawiesinę na podłoże minimalne. Na takim podłożu nie wyrosną komórki drożdżowe, które nie przyjęły wstawki, nie wyrosną też LL i PP. Wyrosną komórki, które mają wprowadzony wektor w postaci L wstawka P (kolor biały) lub L pusty P (kolor czerwony)
Problemem u S. cerevisiae jest tendencja do rekombinacji pomiędzy plazmidowym wektorem a rodzimym plazmidem 2μm co utrudnia kontrolę procesu klonowania i analizę plazmidów izolowanych z transformantów. W celu uniknięcia tych niedogodności szczep gospodarza powinien być pozbawiony plazmidu naturalnego. Dla ułatwienia procesu transformacji, komórki drożdży trawi się enzymami litycznymi i jako biorców klonowanego DNA używa się sferoplastów pozbawionych fragmentów ściany komórkowej. Proces prowadzi się w środowisku stabilizowanym osmotycznie, zawierającym glikol polietylenowy oraz jon Ca2+. W celu uzyskania lepszej ekspresji klonowany gen musi być poprzedzony właściwą dla gospodarza sekwencją promotorowa, a ulegający transkrypcji odcinek DNA zakończony sekwencją terminatorową.
19. Rodzaje niestabilności plazmidów, sposoby zapobiegania.
Plazmid - dwuniciowa kolista cząsteczka DNA naturalnie występująca w niektórych bakteriach; ich wielkość mieści się w granicach od 2 do kilkuset kilozasad. Są nośnikami genów służących do inaktywacji antybiotyków, do wytwarzania toksyn lub do rozkładu naturalnych produktów.
Informacja wprowadzana do komórek mikroorganizmów jest obca więc chcą one się jej pozbyć i dlatego zrekombinowane cząsteczki plazmidowe są niestałe. Istnieją dwa rodzaje niestabilności:
niestabilność segregacyjna - mamy tu do czynienia z utratą plazmidu przy przejściu z generacji na generację (jest gubiony). Utrata ta często wynika z uszkodzenia w plazmidzie genu, który jest odpowiedzialny za rozdział plazmidów w następnych generacjach. Tego rodzaju regionu nie posiada pBR322 więc można się spodziewać, że będzie on niestabilny w następnych populacjach. Prawdopodobieństwo przejścia plazmidu z generacji na generację wynosi: P = 2*(0.5)Nd gdzie Nd - ilość kopii w komórce bakteryjnej. Par - jest to gen odpowiedzialny za rozdział plazmidów między generacjami. Geny te zdolne są do stabilizowania plazmidów - tworzą drugorzędowe łańcuchowe połączenia. W rejonie par znajdują się sekwencje, które inicjują procesy transkrypcji i translacji i mogą one odgrywać istotną rolę w rozdzielaniu plazmidów w następnych populacjach.
Niestabilność strukturalna - następują zmiany w strukturze cząsteczki plazmidowej ( insercja, delecja, rearanżacja - fragment plazmidu może zostać usunięty, dodany, może zajść wymiana nukleotydu - mutacja punktowa). Efektem jest często wyłączenie ekspresji.
Niestabilności zapobiega się ograniczając ilość generacji hodowanych bez presji selekcyjnej (np. obecności antybiotyku - w skali przemysłowej dodatek antybiotyku jest jednak zbyt kosztowny). Prowadzi się proces w mniejszym fermentorze w obecności antybiotyku i dopiero wprowadzamy biomasę do fermentora właściwego (plazmidy są utrzymywane a końcowe generacje prowadzone są wobec braku presji selekcyjnej). Inną przyczyną niestabilności jest wydatek energetyczny na ekspresję dodatkowych genów. Zapobiegać temu można przez zastosowanie wektorów typu "run away" - ekspresja jest tu uruchamiana jakimś bodźcem zewnętrznym np. systemy wektorowe, w których ekspresja genów w plazmidzie jest wyłączona i uruchamiana np. wysoką temperaturą. Taką hodowlę prowadzimy do momentu osiągnięcia fazy stacjonarnej (nie jest uruchomiona ekspresja genów). Osiągamy stan nasycenia hodowli tzn., że więcej komórek już nie przybędzie i wtedy podnosimy temperaturę co wywołuje ekspresję białka. Ekspresję określonego genu można indukować (fragment operonu laktozowego; obecność laktozy lub IPTG inicjuje ekspresję genu). Można to wykorzystać do stabilizacji plazmidu - bez induktora brak transkrypcji, translacji, komórka nie jest obciążona - wykorzystanie mechanicznych induktorów i represorów. Novo opracowało system wektorowy u B. subtilis zapewniający stabilność. W ścianie komórkowej bakterii G+ występuje polipeptyd, w którym zawarta jest D-alanina. Aby D-Ala mogła być wbudowywana do tego polipeptydu mikroorganizmy muszą mieć enzym racemazę, który przekształca formę L w D. Otrzymano szczep bakterii z uszkodzonym genem racemazy. Jednocześnie wyizolowano ten gen z bakterii i wstawiono do systemu wektorowego. Komórki bakteryjne z uszkodzonym genem są wybitnie zainteresowane utrzymaniem plazmidu, który dostarcza im ten gen. Można więc do tego genu wstawić interesujący nas inny gen, który komórki też będą utrzymywać. W ten sposób bez dodatku czegokolwiek do podłoża możemy stabilizować plazmid. Tego rodzaju rozwiązanie nie zapobiega niestabilności strukturalnej.
W przypadku niestałości strukturalnej trudno jest znaleźć skuteczną metodę zapobiegania. Możemy zastosować "run away" aby ograniczyć wydatek energii na ekspresję tego genu do momentu osiągnięcia fazy stacjonarnej. Opracowano systemy wektorów integracyjnych, które wbudowują się do chromosomu organizmu gospodarza. Może to polegać na homologicznej integracji (gdy zawierają komplementarne zasady).
20. Definicja wirusa, bakteriofag λ, jego cykle życiowe i możliwości wykorzystania jako wektora klonującego.
Wirusy to kompleksy nukleinowo-białkowe , niezdolne do replikacji, tarnskrypcji oraz translacji poza komórką gospodarza. Cząstki wirusowe(wirony) zasadniczo składają się z kwasu nukleinowego oraz białkowego kapsydu. Niektóre z wirusów mają otoczkę lipoproteinową a także inne białka niestrukturalne niezbędne do transkrypcji lub replikacji tuż po infekcji.
Wirusy mogą mieć genom składający się z jednoniciowego albo dwuniciowego DNA lub RNA. Wśród jednoniciowych genomów można wyróżnić pozytywne (zawierające nić +) negatywne (zawierające nić -) oraz mieszane (definiowane w odniesieniu do mRNA) Genomy różnią się wielkością i replikują się wykorzystując kombinację enzymów wirusowych i komórkowych. Dualistyczna natura wirusów sprowadza się do tego że poza organizmem zachowują sięone jak twory materii nieozywionej, natomiast w komórce i za pomocą jej metabolizmu wykazują cechy materii ożywionej a w tym zmienność genetyczną. Współczesna systematyka biologiczna nie kwalifikuje wirusów do organizmów twrktuje je jako formy (struktury)
Bakteriofagi czyli fagi są wirusami infekującymi bakterie. Mogą charakteryzować się prostym, litycznym cyklem życia lub bardziej złożonym ściśle uregulowanym wykorzystującym integrację z genomem gospodarza.
Bakteriofag λ jest jednym z najbardziej poznanych bakteriofagów.Pochodne tego faga są powszechnie używane jako wektory do klonowania DNA. Wirion bakteriofaga składa się z ikozaedralnej główki zawierającej liniowy genom dsDNA oraz długiego giętkiego ogonka. Fag wiąże się do specyficznych receptorów w zewnetrznej błonie E.coli i genom wirusowy jest wstrzykiwany przez ogonek fagowy do wnetrza komórek.
5'-koniec każdego łańcucha tego liniowego DNA zawiera jednoniciowy odcinek o długości 12 nukleotydów. Sekwencje te nazywamy końcami adhezyjnymi (końcami „cos”), są one bowiem wzajemnie komplementarne, a więc mogą ze sobą parować.
Cykl lizogenny. Komplementarne końce cos łączą się wewnątrz komórki tworząc kolisty genom, który jest naprawiany przez ligazę DNA. Kolisty DNA łączy się z chromosomem w procesie prostej, wzajemnej rekombinacji. DNA faga, który stał się częścią chromosomu E. coli nazywamy profagiem, zaś zawierającą go komórkę E. coli określamy jako bakteria lizogenna. Komórka lizogenna nie może być już zainfekowana. Zintegrowany wirusowy DNA ulega replikacji wraz z DNA dzielących się komórek. Ekspresji podlega tylko gen represor λ (w formie nie profaga), zapobiega on ekspresji wszystkich innych genów bakteriofaga λ. Ekspresji podlega tylko jego własny gen.
Uruchomienie cyklu litycznego. Indukcja cyklu tego może nastąpić m.in. dzięki podwyższeniu temperatury hodowli. Następuje wycięcie genu, uruchamiany jest cykl lityczny. Dochodzi do transkrypcji w celu wytworzenia wirusowych białek potrzebnych do replikacji wirusowego DNA i pakowania DNA w kapsyd. Zachodzi też synteza wielu cząstek białka tworzącego kapsyd. Wirusowy DNA ulega replikacji i poszczególne jego kopie po wniknięciu do nowych kapsydów, tworzą nowe cząstki fagowe. Liza komórki umożliwia następnie uwolnienie kilkuset potomnych wirusów. Infekcję lityczną charakteryzują 4 etapy: adsorpcja, penetracja, replikacja i uwolnienie. Cykl uruchamiany jest też podczas naświetlania UV. W genomie bakteriofaga λ też jest enzym, który powoduje lizę komórki bakteryjnej. Cykl lityczny wymaga około 35 min i prowadzi do uwolnienia ok. 100 cząstek wirusa.
Główka bakteriofaga jest w stanie przyjąć do swojego wnętrza DNA o wielkości od 75% (ok. 36 tys. Par zasad) do 105% naturalnej długości bakteriofaga. Fragment wstawiamy w rejon nieistotny. Rejon nieistotny ma 2 końce cos. Poddajemy go trawieniu enzymami restrykcyjnymi i uzyskujemy fragmenty DNA. Następnie przeprowadzamy ligację z obcym DNA. To co otrzymaliśmy pakujemy w główce faga i w rezultacie otrzymujemy infekcyjną cząsteczkę faga.
21. Fagowe wektory klonujace wtrąceniowe i wymienialne (zasada in vitro upakowania rekombinowanego DNA w wirusowych otoczkach białkowych).
Możemy otrzymywać wektory:
Wtrąceniowe - do DNA bakteriofaga wstawiamy obcy DNA. Niewielka pojemność można tylko ok. 5% wstawić.
Obcy DNA
ligaza
Wymienialne - usuwamy część nieistotną dla cyklu litycznego
Wektory fagowe mają większą pojemność niż wektory plazmidowe
Na podstawie sekwencji faga λ opracowano szereg różnych wektorów zwanych wektorami wymienialnymi których przykładami mogą być EMBL3 lub λDASH. Wprawdzie czysty kolisty DNA faga λ albo jego pochodne można wprowadzić do komórek komplementarnych na drodze transformacji jednak właściwości infekcyjne cząstek fagowych stanowią pewną zaletę szczególnie przy tworzeniu bibliotek DNA. Do pakowanie liniowego zligowanego DNA do kapsydów białkowych faga λ in vitro można użyć mieszaniny zawierającej fagowe białka płaszcza oraz enzymy biorace udział w procesie. Ekstrakt pakujący jest przygotowany z dwóch szczepów bakteryjnych, z których każdy zainfekowany jest innym, niezdolnym do pakowania DNA mutantem faga λ, dzięki czemu białka biorące udział w pakowaniu występują w nich w dużych ilościach. Zmieszane ze sobą ekstrakty zawierają wszystkie białka potrzebne do pakowania DNA. Przygotowane w ten sposób cząstki fagowe można użyć do infekcji hodowli normalnych komórek E.coli. Zligowane ramiona faga, które nie zawierają insertu lub zawierają fragmenty DNA których długość znacznie odbiega od optymalnej są zbyt małe lub zbyt duże by mogły zostać spakowane do kapsydu fagowego.
22. Biologiczne zabezpieczenia przed ucieczką rekombinowanego DNA z laboratorium (mutacje nonsens w wektorach).
23. Charakterystyka bakteriofaga M13 i wektorów wywodzących się od niego.
Bakteriofag M13 jest fagiem łagodnym, który nigdy nie prowadzi do lizy komórki. Ten filamentowy wirus ma długość 900 nm i szerokość 9 nm. Jednoniciowy, kolisty DNA tego faga jest chroniony przez okrywę białkową zbudowaną z 2710 identycznych podjednostek białkowych. M13 wnika do komórki E. coli przez bakteryjny pilus białkowy. Jednoniciowy DNA występujący w cząstce wirusowej w zainfekowanej komórce ulega replikacji poprzez kolistą dwuniciową formę replikacyjną RF zawierającą nici + i - (forma RF może się replikować na zasadzie toczącego się koła). Do nowych cząstek wirusowych pakowana jest jednak tylko nić +. Cechą charakterystyczną tego faga jest to, że nie zabija on bakterii będącej gospodarzem, co umożliwia wyhodowanie dużych ilości fagów M13 i łatwe ich izolowanie.
Cykl życiowy faga M13:
wnikanie fagowego DNA do komórki
jednoniciowy DNA przekształca się w dwuniciową formę replikacyjną RF
replikacja DNA - potomne RF - 100 nukleotydów
synteza jednołańcuchowego DNA
jednoniciowy DNA pakowany jest w osłonki wirusowe i dojrzałe cząstki fagów są wydalane z komórki.
Fag ten jest atrakcyjny ze względu na występowanie w formie jednoniciowej. Komórki, które przyjmą faga M13 będą się wolniej namnażać (powstaną na płytkach z podłożem stałym koliste obszary o spowolnionym wzroście). W obszarze wektorowego DNA faga jest niewiele do usunięcia. DNA poddano trawieniu Pst I rozpoznającym 10 miejsc restrykcyjnych i stwierdzono 1 miejsce rozpoznawane przez enzym restrykcyjny w obszarze międzygenowym (wstawienie obcego fragmentu nie narusza funkcjonowania wektora). W obszar międzygenowy (507 nukleotydów, które nie kodują niczego) wstawiono fragment zawierający początek genu β-galaktozydazy (promotor i operon) z polilinkerem (identyczny jak w pUC18 i 19)
Wstawka DNA
Transfekcja E. coli
Zrekombinowany wektor M13
(dwuniciowy DNA)
uwolnienie fagów
DNA
Otoczka
białkowa
Zrekombinowany fag M13
Jednoniciowy DNA
Po wysianiu na podłoże z x-Galem i induktorem obserwujemy na tle gęstego wzrostu miejsca wzrostu zubożonego w kolorze niebieskim. Semiłysinki bezbarwne są dla nas atrakcyjne gdyż w tych komórkach nastąpiło przerwanie ciągłości genu β-galaktozydazy (enzym nie może być produkowany). Komórki zainfekowane pustym wektorem rosną w postaci niebieskich kolonii.
Przygotowanie wektora M13 do klonowania polega na rozcięciu w jednym miejscu jego dwuniciowej kolistej formy replikacyjnej za pomocą enzymu restrykcyjnego. Rozcina się go w obrębie polilinkera (tj. odcinka DNA zawierającego graniczące ze sobą miejsca restrykcyjne dla kilku różnych enzymów, przy czym każde z nich występuje w wektorze tylko raz). Następnie z rozciętym wektorem łączy się (reakcja ligacji) dwuniciowej fragmenty obcego DNA uzyskane po trawieniu tym samym enzymem, którego użyto do rozcięcia wektora. Obcy DNA może zostać wbudowany do wektora w dwóch przeciwnych położeniach, ponieważ oba końce cząsteczki DNA łączonej z wektorem są jednakowe. Dzięki temu połowa nici +, zawartych w potomnych cząstkach wirusowych, zawiera jeden z dwóch łańcuchów obcego DNA a połowa drugi. Insert obcego DNA zawarty w fagu M13 można łatwo selekcjonować. Jako odcinek starterowy do sekwencjonowania używa się oligonukleotyd hybrydyzujący z rejonem przyległym do polilinkera. Oligonukleotyd taki nazywamy uniwersalnym odcinkiem starterowym (primerem) do sekwencjonowania, ponieważ można go stosować do sekwencjonowania zrekombinowanego DNA, ale nie nadaje się do jego długotrwałego rozmnażania, ponieważ inserty dłuższe niż ok. 1 kz nie są stabilne.
M13mp18/M13mp19 różnią się tylko orientacją polilinkera jak w przypadku pUC. Są różne odmiany różniące się ilością miejsc restrykcyjnych w polilinkerze. Z połączenia wektora fagowego z plazmidowym powstały fagemidy.
24. Fagemidowe systemy wektorowe, charakterystyka i zastosowanie
Fagemidy powstały z połączenia wektora fagowego z plazmidowym.
M13 (475 bp)
lac2
polilinker
amp+ lac1
Do sporej części pochodzącej od pBR322 wstawiono nukleotydy pochodzące od M13. W tym fagemidzie występuje fragment operonu laktozowego z polilinkerem w obrębie kodującym
β-galaktozydazę. Zachowuje się on jak wektor plazmidowy gdyż replikuje dwuniciowe DNA. Nie ma w tym wektorze żadnego genu kodującego białka osłonki a więc nie może powstać posiadający jednoniciowy DNA fag nitkowy. Infekujemy kolonię posiadającą ten wektor fagiem wspomagającym M13K07 (zmodyfikowany M13) aby dostarczyć informacji o białkach otoczkowych. Pewien rejon w fagemidzie jest rozpoznawany przez enzymy restrykcyjne i fag wspomagający jest przekształcany w otoczkę białkową bakteriofaga M13 (jego dwuniciowe DNA RF przechodzi w jednoniciowe). Zaletą systemów fagemidowych jest to, że możemy włączyć dłuższe sekwencje (do pewnego momentu działamy z nim jak z wektorem pUC. Staje się on fagemidem dopiero po infekcji. Wektory te są dość stabilne.
25.Charakterystyka wektorów kosmidowych (klonowanie kosmidami trawionymi dwoma enzymami restrykcyjnymi, chramoidy)
Wektory kosmidowe łączą cechy właściwe plazmidom i fagowi λ. Kosmidy posiadają wszystkie typowe cechy występujące w plazmidach łącznie z MCS i genami oporności na antybiotyki, a także sekwencje występujące w fagu λ, zwane sekwencjami cos. Znajdują się one na każdym końcu cząsteczki DNA λ i są odpowiedzialne za wprowadzenie jej do fagowego kapsydu. Obecność sekwencji cos w kosmidach umożliwia proces ich pakowania w kapsydach fagowych. Klonowanie przy pomocy kosmidów łączy metody związane z użyciem jako wektora zarówno faga λ jak i plazmidów. Kosmidowy DNA trawi się enzymem restrykcyjnym i liguje z obcym DNA. Zrekombinowane kosmidy są następnie pakowane w kapsydy λ, którymi infekuje się E. coli. Kosmidy nie zawierają genów λ i dlatego po infekcji nie tworzą łysinek. Zainfekowane komórki hoduje się na pożywce agarowej z antybiotykiem, na której będą rosły kolonie zawierające zrekombinowane kosmidy. Można je powielić w taki sam sposób jak plazmidy. Zaletą kosmidów jest ich zdolność do przyjęcia bardzo dużych wstawek. Ponieważ kapsydy λ mogą przyjąć do 52 kpz a kosmidy są bardzo małe (zwykle 8 kpz lub mniej) można klonować w nich wstawki wielkości do 44 kpz.
(rys str 319)
26. Łączenie DNA (terminalna transferaza, dorabianie homopolimerowych „ogonów”, linkery, adaptory).
Synteza drugiej nici DNA wymaga startera. Najlepszą droga przygotowania pełnej długości DNA jest wydłużenie końca 3' pierwszej nici i następnie użycie startera komplementarnego do wydłużonego końca pierwszej nici. Terminalna teransferaza nie wymaga matrycy i dodaje nukleotydy do końca 3' jedno- i dwuniciowych kwasów nukleinowych. Jeśli w mieszaninie reakcyjnej znajduje się tylko jeden rodzaj dNTP to enzym ten utworzy homopolimerowy ogon reszt C na końcu 3' dwuniciowego hybrydu mRNA-cDNA. Po zniszczeniu nici mRNA w drodze hydrolizy alkalicznej do syntezy drugiej nici cDNA jako starter może być użyty oligo(dG). Starter ten jest następnie wydłużany z pomocą odwrotnej transkryptazy albo fragmentu Klenowa polimarazy DNA E.coli. Produktem tej reakcji jest dwuniciowy cDNA, ale końce cząsteczki mogą być nieodpowiednie do klonowania. Koniec 3' pierwszej nici może wystawać poza koniec 5' drugiej nici w zależności od długości poli ogona i startera oligo. Dlatego końce DNA trzeba przygotować w sposób umożliwiający jego klonowanie.
W celu uzyskania kohezyjnych końców wygodnych do klonowania zwykle dodaje się do DNA specjalne sekwencje łącznikowe tzw. linkery kwasów nukleinowych. Ponieważ dodaje się je w nadmiarze reakcja przyłączenia linkerów do tępych końców cDNA zachodzi stosunkowo dobrze. Do każdego ufosforylowanego końca 5' cDNA zostaje dołączony jeden linker (jeśli końce linekra nie są ufosforylowane) lub wiele takich sekwencji (jeśli linker ma końce ufosforylowane). W końcu trawienie enzymem EcoR1 wytworzy w cDNA lepkie końce gotowe do ligacji z wektorem. Istnieje kilka alternatywnych sposobów przygotowania końców cDNA z udziałem terminalenj transferazy jednoniciowych homopolimerowych ogonów, komplemantarnych do odpowiednich ogonów utworzonych w wektorze do klonowania. Można też użyć cząsteczek adoptorowych które mają wcześniej przygotowane lepkie końce co umożliwia unikniecie metylacji cDNA.
27. Konstrukcja biblioteki genów, czynniki warunkujące jej wielkość, równanie Clarka&Carbona.
Biblioteka genów - zestaw klonów np. bakteryjnych lub cząsteczek DNA plazmidowych, fagowych, w którym to zestawie reprezentowany w formie fragmentów jest cały genom lub cały zestaw genów. W zależności od zastosowanego systemu wektorowego banki genów mogą być różnej wielkości. Na wielkość b.g. wpływa pojemność klonująca systemu wektorowego .B.g. jest tym większy im bardziej skomplikowany jest organizm. Jest on konstruowany w sposób dość przypadkowy. Wzór Clarka&Carbona jest wykorzystywany do określania liczby wymaganych klonów:
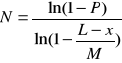
gdzie:
N- wymagana liczba rekombinantów
P - prawdopodobieństwo z jakim jesteśmy w stanie znaleźć w danym banku dany gen
X - wielkość szukanego genu
L - długość klonowanego fragmentu (zależy od pojemności klonującej danego systemu)
M - wielkość genomu
Im większe L tym mniejsza liczba klonów reprezentujących dany gen przy pewnym prawdopodobieństwie.
Bibliotekę DNA stanowi kolekcja fragmentów DNA klonowanych w jakimś wektorze, którą można przeszukać w celu znalezienia fragmentu DNA będącego przedmiotem zainteresowania. Jeśli celem pracy jest izolacja określonej sekwencji genu, to wykorzystuje się dwa rodzaje bibliotek:
Biblioteki genomowego DNA, które przygotowuje się z genomowego DNA danego organizmu. Np. genomową bibliotekę myszy można przygotować trawiąc DNA myszy enzymem restrykcyjnym tak, by uzyskać dużą liczbę fragmentów o identycznych końcach kohezyjnych. Fragmenty DNA należy następnie kowalencyjnie połączyć (poddać ligacji) z linearyzowanymi (rozciętymi w 1 miejscu) cząsteczkami wektora plazmidowego lub wirusowego. Biblioteka taka powinna zawierać wszystkie sekwencje jądrowego DNA myszy i można ją będzie przeszukiwać w celu wyłowienia z niej i następnie zbadania mysiego genu, będącego obiektem naszych zainteresowań. Każdy klon w takiej bibliotece jest nazywany klonem genomowego DNA. Nie każdy klon genomowego DNA zawiera kompletny gen, ponieważ w wielu przypadkach enzym restrykcyjny co najmniej 1 raz rozetnie DNA w obrębie genu.
Biblioteki cDNA- przygotowuje je się wykorzystując odwrotną transkryptazę retrowirusa do zsyntetyzowania DNA komplementarnego do całkowitego mRNA komórki. Jednoniciowy cDNA jest następnie przekształcany w dwuniciowy DNA i wprowadzany do wektora. Każdy klon w tak przygotowanej bibliotece to klon cDNA. W przeciwieństwie do poprzedniej ta biblioteka zawiera tylko te sekwencje DNA, które ulegają ekspresji jako mRNA. Z różnych tkanek zwierzęcych, w których dochodzi do ekspresji odmiennych zestawów genów, uzyskuje się różne biblioteki cDNA.
28. Metody transformacji Bacillus subtilis.
Atrakcyjnym gospodarzem jest obok E. coli Bacillus subtilis. (G+). Większość syntetyzowanych białek jest wydzielana poza komórkę (ułatwiony down stream procesing).
Metoda Anagnostopulos'a
Polegała ona na otrzymaniu kompetentnych komórek na drodze dwuetapowej kontrolowanej hodowli na podłożach ubogich w składniki odżywcze dzięki czemu komórki nie wytwarzały do końca składników ściany komórkowej. Metoda ta nie dawała jednak dobrych efektów gdy mieliśmy do czynienia z monomerycznym plazmidowym DNA (pojedynczy odcinek DNA w formie kolistej). Wydajność transformacji w tym wypadku była bardzo niska lub komórki w ogóle nie otrzymywano transformantów. Przyczyny problemów związanych z tą metodą:
jeśli wprowadzamy monomeryczny plazmidowy DNA to ulegnie on absorbcji na powierzchni komórki, do wnętrza wchodzi tylko jedna nić natomiast druga jest degradowana więc nie może zachodzić replikacja.
Forma dimeryczna (lub trimeryczna) Do cząsteczki wchodzą: jednoniciowy DNA z jednego meru i jeden jednoniciowy z drugiego. Odcinki te są do siebie komplementarne i dzięki temu mogą być włączone i może zajść replikacja i synteza nowych plazmidowych DNA.
Gdy wewnątrz komórki gospodarza jest plazmid lub region w chromosomalnym DNA komplementarny do DNA wchodzącego to nastąpi rozpoznanie i połączenie komplementarnych nici i zajdzie replikacja (nawet gdy wchodząca cząsteczka to monomeryczny DNA).
Inną metodę transformacji protoplastów Bacillus subtilis zaproponowali Chang i Cohen. Bakterie były hodowane do środkowej fazy logarytmicznego wzrostu po czym odwirowywano komórki i zawieszano w roztworze 0.5 molowym sacharozy z lizozymem [2mg/ml] (enzym izolowany z białka jaja kurzego działający na wiązania 1,4-β-glikozydowe w β-glikanie występującym w warstwie peptydoglikanu). Po inkubacji powstawały koliste formy - protoplasty. Roztwór sacharozy miał zabezpieczać komórki przed pęknięciem. Pomimo zastosowania stabilizatora szkło laboratoryjne musiało być bardzo czyste gdyż nawet ślady detergentów powodowały pękanie protoplastów. Po odwirowaniu przenosi się protoplasty do pożywki stabilizowanej roztworem sacharozy, dodaje się od 1pikograma do 5 mikrogramów /ml plazmidowego DNA po czym niewielką objętość tej zawiesiny przenosi się do glikolu polietylenowego o stopniu polimeryzacji 6000 (40%).Gdy brak jest zewnętrznej osłony obcy DNA może bez przeszkód wniknąć do środka. Po inkubacji rozsiewamy protoplasty na podłoża regeneracyjne, na których następuje odbudowanie osłon komórkowych a następnie na podłoża selekcyjne. Wydajność tej metody wg jej autorów 108 cfu /μg DNA. Pękanie protoplastów podczas procesu powoduje zwiększanie lepkości roztworu i powstawanie aglomeratów nieuszkodzonych protoplastów (a nam zależy aby pojedyncze kolonie przenosić na podłoża selekcyjne). Aby temu zapobiec stosuje się czasami dodatek DNA - zy, która nie zdegraduje plazmidów w protoplastach ale zdegraduje zwiększający lepkość roztworu chromosomalny DNA. Powyższa metoda nie wymaga wprowadzania multimerycznej formy DNA. W przypadku wprowadzania form monomerycznych uzyskujemy wyższą wydajność gdyż do plazmidu wchodzą całe wektory koliste.
Inną metoda to elektrotransformacja. Polega ona na uzyskaniu kompetentnych komórek dzięki elektropermeabilizacji ściany komórkowej pod wpływem napięcia. Komórki żywych organizmów pod wpływem wysokiego napięcia (dużej różnicy potencjałów) ulegają perforacji. Metoda ta dobrze sprawdza się w przypadku bakterii. Napięcie zależy od rodzaju drobnoustroju (12.5 - 25 kV/cm - odległość pomiędzy elektrodami). Naczynia do przeprowadzenia tej metody są bardzo wąskie. Komórki przygotowuje się tak jak poprzednio tzn. hodowla komórek do odpowiedniej fazy wzrostu, odwirowanie i przechowywanie z glicerolem w suchym lodzie do momentu elektrotransformacji. Przenosimy komórki do łaźni lodowej dodajemy DNA, przenosimy do kuwety z elektrodami. Stosujemy wysokie napięcie (wytwarzane przez generatory), bardzo krótkie impulsy rzędu milisekund. Czynniki decydujące o optymalizacji warunków:
siła pola (pochodna napięcia)
czas każdego impulsu (2.5 - 10 ms)
częstotliwość impulsów
stężenie DNA
Po przeprowadzeniu elektroperforacji rozsiewa się transformanty na podłoże selekcyjne. Jest to metoda wydajniejsza niż metody chemiczne (10 - 20 razy). Wydajność 1010 cfu /μg DNA. Zaletą tej metody jest też to, iż można wprowadzać duże fragmenty DNA (do 130000 par zasad). Jest ona wykorzystywana głównie do G+ i kom. org. wyższych.
Transdukcja - komórki inkubujemy w roztworze zawierającym jony Ca2+ (rozluźnienie struktury zewnętrznej osłony komórkowej co ułatwia proces transdukcji). Po rozsianiu komórek transdukowanych otrzymujemy łysinki.
29. Metody transformacji E. coli
Transformacja - stała, dziedzicząca się zmiana właściwości komórki spowodowana przez wprowadzenie nowej sekwencji DNA pozyskanej z otoczenia komórki lub przez mutację.
Zjawisko transformacji komórek bakteryjnych - przyjęcie "nagiego DNA" (bez żadnych otoczek) przez komórkę organizmu gospodarza. Komórka musi być w określonym stanie fizjologicznym aby móc przyjąć obce DNA do swojego wnętrza. Komórki w tym stanie nazywamy kompetentnymi. Komórki kompetentne muszą być w odpowiedniej fazie rozwoju (faza logarytmiczna). Nagi DNA adsorbuje się na powierzchni komórki bakteryjnej dzięki obecności specjalnych białek receptorowych. DNA wchodzi do wnętrza komórki bakteryjnej w formie dwuniciowej lub jednoniciowej a jego część może być degradowana. System restrykcji replikacji powinien być u gospodarza zlikwidowany (wyłączenie genów restrykcji replikacji poprzez odpowiednie zabiegi mutagenizacji). Do tak przygotowanych komórek możemy wprowadzić obcy DNA bez obawy, że zostanie zniszczony w komórce.
Metoda Mandel'a i Higa.
Stwierdzili oni, że aby wywołać indukcję kompetencji należy inkubować w probówce komórki E. coli w roztworze chlorku wapnia w łaźni lodowej po czym dodać roztwór plazmidowego DNA. 50 molowy CaCl2 powoduje rozluźnienie struktur komórkowych. Następnie przenosi się probówkę z łaźni lodowej do łaźni o temp. 420C (3 min.) wywołując szok termiczny. W wyniku szoku najprawdopodobniej dzięki rozluźnieniu struktury osłon komórkowych następuje wniknięcie obcego DNA do wnętrza. Przeprowadzamy inkubację w 370C w środowisku bez antybiotyku (nowe cechy wprowadzone z DNA plazmidowego mają zacząć działać). Rozsiewamy na stałe podłoże z dodatkiem antybiotyku i otrzymujemy transformanty.. Inkubacja z CaCl2 i szok termiczny sprawiają, że następuje rozluźnienie osłon komórkowych i to zjawisko jest nazywane przejściem w stan kompetencji. W wyniku metody Mandel'a i Higa osiągamy wydajność transformacji 106 jtk (cfu) z 1 mikrograma DNA [cfu/μg DNA]. Wydajność ta dotyczy cząstek plazmidowego DNA o wielkości do 5000 par zasad. Wraz ze wzrostem wielkości cząsteczki DNA wydajność transformacji spada. Gdy wykorzystujemy cząstki wektorowe z wstawionymi fragmentami (wykorzystujemy ligację) to wchodzić będzie tylko kolisty zamknięty DNA (pusty wektor lub zamknięty wektor z wstawką) a więc wydajność transformacji będzie mniejsza niż 100%. Innymi przyczynami spadku wydajności mogą być: trudniejsze wprowadzenie większej cząstki oraz zwiększenie obciążeń energetycznych komórki biorcy przez co może ona nie przeżyć. Cohen zmodyfikował tą metodę. W '93 Hanahan udoskonalił powyższą metodę poprzez wzbogacenie środowiska transformacji w jony Mg2+,Mn2+ (MgSO4, MnCl2) co zwiększyło wydajność (109). Prace nad transformacjami komórek prowadzono dalej gdyż chciano znieść konieczność przygotowywania komórek przed każdą transformacją.
Metoda Chung i Miller.
Komórki kompetentne można przechowywać w głębokim zamrożeniu. Hodowlę inokularną rozcieńcza się dziesięciokrotnie w świeżym podłożu. Hoduje się tą populację do momentu gdy gęstość optyczna osiągnie 0.3 - 0.6 przy długości fali 600 nm. Absorbancja określa nam kiedy komórki są w środku fazy logarytmicznego wzrostu. Komórki oddzielamy od podłoża hodowlanego i zawieszamy w roztworze zawierającym mieszaninę MgSO4 i MnCl2 z glikolem polietylenowym (PEG 10% o stopniu polimeryzacji 3350 a czasami i 6000 lub 8000) i 5% DMSO (sulfotlenek dimetylu). Tak przygotowany roztwór szybko zamrażamy w suchym lodzie (temp -700C) i możemy go przechowywać przez 2 lata w zamrażalniku. Gdy chcemy transformować porcjujemy bryłki zawierające komórki kompetentne i dodajemy plazmidowy DNA. W tej metodzie nie stosujemy szoku termicznego ale wszystkie operacje wykonujemy w lodzie. Później przenosimy do świeżego podłoża i inkubujemy w 370C (temperatura optymalna dla wzrostu E. coli). Wydajność 108cfu/μg DNA.
30. Metody bezpośredniej selekcji i screeningu rekombinantów.
Gdy zależy nam na wyodrębnieniu pojedynczego , konkretnego genu jakiegoś organizmu to po utworzeniu banku genów tego organizmu i stransformowaniu nim odpowiednich komórek gospodarza należy zidentyfkować klon komórek, w którym znajduje się gen. Przeszukiwanie jest zazwyczaj bardzo pracochłonne. Testowanie polega na wysiewaniu na szalki komórek gospodarza np. bakterii z plazmidami badź fagami w takim stężeniu aby możliwe było odróżnienie poszczególnych kolonii lub łysinek. Liczba tych szalek jest w najlepszym przypadku rzędu kilkaset a dochodzi nawet do kilku tysięcy. Wybór metody screeningu jest uzależniony od szeregu czynników
Selekcja:
bezpośrednia
b) pośrednia
Najłatwiej selekcjonuje się mikroorganizmy gdy interesuje nas gen kodujący oporność w stosunku do antybiotyków a trochę trudniej gdy produkt wchodzi w skład szlaku syntezy np. aminokwasów. Jeśli interesuje nas gen kodujący tryptofan to wykorzystujemy mutanty auksotroficzne a dokładniej bakterię, która dopóki nie otrzyma genu odpowiedzialnego za syntezę tryptofanu nie będzie go produkowała. Na podłożu hodowlanym nie zawierającym tryptofanu wyrosną tylko te bakterie, które przyjęły ten gen (selekcja bezpośrednia).
Wprowadzony gen podlega ekspresji (wytwarzany jest produkt), zachodzi transkrypcja, translacja a powstający produkt można w łatwy sposób zidentyfikować. Np. Wprowadzamy gen α-amylazy. Rekombinanty wysiewamy na podłoże ze skrobią. Wokół koloni zdolnych do wytwarzania enzymu powstaje strefa przejaśnienia ujawniająca się po zalaniu płytki KJ. Podobnie postępujemy w przypadku enzymów proteolitycznych. Do podłoża dodajemy kazeinę i powstają strefy przezroczyste wokół koloni zdolnych do wytwarzania proteinaz. Na użytek badań screeningowych produkowane są specjalnie modyfikowane substraty barwne np. starch azure (skrobia błękitna - trwałe połączenie z jodem), blue dextran, blue cellulose. Selekcję rekombinantów można ułatwić stosując dodatek do podłoża odpowiednich wskaźników np. drobnoustroje produkujące antybiotyki wysiewa się na płytki, na które oprócz badanej populacji wysiewa się kulturę wrażliwą na nie. Czasami mamy do czynienia z sytuacją gdy zachodzi ekspresja danego genu ale produkt nie jest łatwy do identyfikacji.
Kolonie transformantów rosnące na powierzchni agarowej
Tworzymy replikę koloni na filtrze
Przeprowadzamy częściową lizę Liza bakterii i denaturacja DNA.
Dodanie radioaktywnej sondy
RNA. Obmycie niezwiązanych sąd. komórek i dodajemy specyficzne
przeciwciała oraz radioaktywny
czynnik do detekcji związanych
przeciwciał
Autoradiografia w celu detekcji Autoradiografia w celu detekcji radioaktywnosci radioaktywności
Kilsza rtg
Przeciwciała łączą się z antygenami następuje aglutynacja (powstaje kompleks antygen- przeciwciało). Poprzez obserwację stref aglutynacji można ocenić wydzielanie produktu.
31. Immunochemiczne i immunologiczne sposoby identyfikacji klonów z biblioteki genów.
Poszukiwanie właściwego klonu może odbywać się
metodą detekcji produkcji białka z zastosowaniem specyficznych przeciwciał
metodą detekcji klonów przez hybrydyzację.
Najprościej jest gdy nowe białko jest syntetyzowane czyli następuje ekspresja np. produkcja α-amylazy. W tym wypadku hodowlę prowadzimy na podłożu ze skrobią i szukamy koloni, wokół których pojawiły się miejsca przejaśnienia. Sytuacja się komplikuje gdy następuje ekspreja genu (jest produkowany metabolit) ale trudno go oznaczyć (np. produkcja hormonu na podłożu stałym). W takim wypadku stosuje się metody immunochemiczne. Są to metody pośrednie, w których stosuje się przeciwciała specyficzne białko i dzięki temu można zidentyfikować te np. bakterie, które zawierają gen kodujący to białko. Jako sondę można w takim przypadku wykorzystać nie tylko specyficzne przeciwciało, ale każdy ligand wiążący się specyficznie z badanym białkiem. Np. do identyfikacji klonów syntetyzujących białkowy receptor hormonu jako sondę można stosować znakowany hormon oddziałujący z tym receptorem. Początkowo szczepi się zwierzęta antygenem aby uzyskać specyficzne przeciwciała. Gdy produkt ekspresji genu nie jest wydzielany poza komórkę stosujemy metodę immunochemiczną Broome'a Gilberta. W metodzie tej wykorzystujemy poliwinylowy krążek pokryty przeciwciałami (łatwo wiążącymi się z tworzywem sztucznym) a także cząsteczki immunoglobuliny IgG znaczonej J125. Żeby otworzyć komórkę możemy zainfekować ją bakteriofagiem, podziałać lizozymem lub chloroformem. Jeśli jest szukany antygen to przyłączy się on do tych przeciwciał. Filtr z koloniami dociskamy do poliwinylowego krążka i następuje wiązanie (powstaje kompleks antygen - przeciwciało). Przygotowujemy teraz immoglobulinę (przeciwciało) znakowaną jodem 125. Łączy się ona (wykazuje powinowactwo do antygenu - chyba) i powstaje trójskładnikowy kompleks. Przykładamy film rentgenowski i tam gdzie występowała radioaktywna immunoglobulina połączona z antygenem pojawiało się zaciemnienie kliszy fotograficznej.
32. Charakterystyka aktywności własciwej polimerazy DNA I, fragment Klenowa oraz zastosowanie tego enzymu.
Polimeraza DNA I jest pojedyńczym łańcuchem polipeptydowym. Katalizuje ona dodawanie jednostek deoksyrybonukleotydowych do łańcucha DNA: (DNA)n reszt +dNTP ↔ (DNA)n+1 +PPi
Do syntezy łańcucha DNA polimeraza DNA I potrzebuje:
wszystkich czterech aktywowanych prekursorów - 5'-trifosforanów deoksyrybonukleozydów: dATP, dGTP, dTTP, dCTP. Konieczna jest też obecność jonów Mg2+
Odcinka starterowego (primera) gdyż polimeraza ta dołącza deoksyrybonukleotydy do wolnej grupy 3'-hydroksylowej już istniejącego odcinka DNA
Matrycy DNA jako istotnego składnika układu. Matrycą może być jedno- lub dwuniciowy DNA. Dwuniciowy DNA jest efektywną matrycą wtedy, gdy jego rdzeń fosforanowo-rybozowy ulegnie zerwaniu co najmniej w jednym miejscu.
Reakcja wydłużania łańcucha katalizowana przez polimerazę zachodzi w wyniku nukleofilowego ataku grupy 3'-OH primera na atom fosforu trifosforanu deoksyrybonukleozydu położony najbliżej deoksyrybozy. Tworzy się wiązanie fosfodiestrowe i równocześnie zostaje uwolniony pirofosforan. Hydroliza uwolnionego pirofosforanu odbywa się przez pirofosfatazę. Elongacja łańcucha DNA odbywa się w kierunku 5'→3'. Polimeraza DNA I katalizuje tworzenie się wiązań fosfodiestrowych tylko wtedy, gdy zasada przyłączanego nukleotydu jest komplementarna do zasady w łańcuchu stanowiącym matrycę. Polimeraza DNA I jest enzymem instruowanym przez matrycę. Enzym czerpie instrukcje od matrycy i dokonuje syntezy produktu, którego sekwencja zasad jest komplementarna do sekwencji zasad matrycy. Inną właściwością polimerazy tej jest zdolność korygowania pomyłek przez eliminację nukleotydów nie dopasowanych do matrycy. Enzym ten katalizuje hydrolizę nukleotydów, które nie tworzą pary typu Watsona-Cricka. Ma ona więc aktywność egzonukleazy 3' →5'. Miejsce odpowiedzialne za tę aktywność jest różne od miejsca odpowiedzialnego za wydłużanie łańcucha. Aktywność nukleazowa 3' →5' pełni dla polimerazy funkcję redagowania. Polimeraza DNA I usuwa niesparowane nukleotydy na końcu odcinka starterowego przed rozpoczęciem polimeryzacji. Dzięki tej właściwości polimerazy DNA I replikacja DNA przebiega bardzo wiernie, a błędy pojawiają się nie częściej niż 1zasada błędna na 108zasad poprawnych. Polimeryzacja zwykle nie zachodzi dopóki para zasad nie jest dopasowane do helisy. Polimeraza DNA I sprawdza wynik każdego przyłączenia przed przystąpieniem do następnego cyklu katalitycznego. Polimeraza DNA I może także hydrolizować DNA poczynając od końca 5' łańcucha. Ta aktywność jest zupełnie odmienna od poprzedniej aktywności. W tym przypadku cięcie może zachodzić na końcowym wiązaniu fosfodiestrowym lub na wiązaniu oddalonym o kilka reszt od końca 5'. Koniec 5' może mieć grupę OH lub być ufosforylowany. Przecinane wiązanie musi znajdować się w rejonie dwuniciowym. Aktywność nukleazowa 5' →3' wzmagana jest przez towarzyszącą jej syntezę DNA. Miejsce katalityczne odpowiedzialne za tę aktywność w enzymie jest wyraźnie oddzielone od miejsca aktywnego polimeryzacji i hydrolizy 3' →5'
Polimeraza ma więc trzy różne miejsca aktywne. Aktywność egzonukleazowa 5' →3'- pełni kluczową rolę podczas replikacji ponieważ usuwa starterowy odcinek RNA a także uzupełnia aktywność
3' →5'przez naprawianie błędów innego rodzaju. Ten trójfunkcyjny enzym można podzielić przez trawienie proteolityczne na mały fragment, który wykazuje aktywność egzonukleazową 5' →3' oraz duży fragment Klenowa posiadający aktywność egzonukleazową 3' →5' (hydroliza wiązań fosfodiestrowych) ujawniającą się gdy w mieszaninie reakcyjnej brakuje właściwych substratów, czyli trideoksynukleotydów lub gdy koniec 3' fragmentu DNA występuje w formie jednoniciowej i aktywność polimerazową (5'→3'). Fragment Klenowa można otrzymać w wyniku proteolitycznego trawienia polimerazy DNA I za pomocą subtylizyny. Można go również otrzymać z hodowli szczepów bakteryjnych, w których został sklonowany odpowiedni fragment genu polimerazy DNA I.
Enzymatyczna synteza genów na matrycy mRNA (polimeraza DNA zależna od RNA [odwrotna tarnskryptaza], zalety otrzymannia cDNA w odniesieniu do genów eukariotycznych).
32. Sondy hybrydyzacyjne i rekombinacyjne do identyfikacji klonów z biblioteki genów.
Metoda Grunstein - Hogness. Metoda opierająca się na rozplataniu i splataniu się komplementarnych nici DNA. Łysinki fagowe lub kolonie bakteryjne na szalce z agarem przenosi się na stałe krążki zazwyczaj z nitrocelulozy (przykłada się je na chwilę do powierzchni szalki, kolonie przyklejają się i tworzy się na nich replika płytki). Położenie łysinek lub kolonii na nitrocelulozowe odbitce jest dokładnie taki samo jak na szalce. Krążek zanurza się następnie w odpowiednim roztworze zasadowym w celu doprowadzenia do lizy komórek bakteryjnych i denaturacji zawartego w nich DNA (dwie nici DNA w łysinkach lub koloniach ulegają rozpleceniu). DNA związany z nitrocelulozą przenosi się go do innego roztworu zawierającego pojedynczą nić DNA (lub RNA), której sekwencja jest komplementarna do jednej z nici w żadnym klonie. Ten DNA (lub RNA) jest, w celu łatwego wykrywania oznakowany (najczęściej radioaktywnym izotopem).Gdy oznakowana pojedyncza nić zwiąże się z właściwym rekombinantem na krążku, w miejscu odpowiadającym położeniu rekombinanta na oryginalnej szalce pozostaje radioaktywna plamka. Odpowiednie kolonie można następnie odzyskać z szalki agarowej. Oznakowany DNA lub RNA nazywamy sondą.
Rys.
Replika na filtr transformanty
inkubacja
usunąć filtr transformanty
kolonia z genem
1.liza komórek płytka odniesienia
2.proteinaza K
3.utrwalenie 800C
1. hybrydyzacja np. z RNA
2.autoradiografia Autoradiogram
Proteinaza K oddziela białka od kompleksów RNA i DNA
Reszta "Krótkie wykłady z biochemii"
35. Chemiczna metoda określania sekwencji nukleotydów Maxam&Gilbert
Metoda chemicznego zrywania wiązań opracowana przez Maxama&Gilberta wymaga DNA znakowanego na jednym końcu (pojedynczej nici) P32. Zwykle znakuje się koniec 5' za pomocą kinazy polinukleotydowej, która do grupy 5'hydroksylowej przyłącza P32 przenoszony przez ATP, Znakowany DNA jest następnie zrywany preferencyjnie w miejscach występowania 1 z 4 zasad. Warunki reakcji są tak dobrane, że statystycznie każda cząsteczka (łańcuch DNA) jest zrywana tylko 1 raz. W mieszaninie reakcyjnej, specyficznej ze względu na zasadę, każdy zerwany łańcuch daje radioaktywny fragment rozciągający się od miejsca przyłączenia radioaktywnego markera do miejsca występowania tej zasady w łańcuchu. Fragmenty tego typu powstają dla każdej z pozycji danej zasady. NP. jeśli mamy sekwencję 5'-P32GCTACGTA-3' to radioaktywne fragmenty wytwarzane przez specyficzne rozcięcie po stronie 5' każdej z 4 zasad w tym oligonukleotydzie byłyby następujące:
zrywanie przy A: P32-GCT P32-GCTACGT
zrywanie przy G: P32-GCTAC
zrywanie przy C: P32-G P32-GCTA
zrywanie przy T: P32-GC P32-GCTACG
Fragmenty powstające w każdej z 4 mieszanin są następnie rozdzielane elektroforetycznie w żelu poliakryloamidowym o dużej zdolności rozdzielczej (pozwalającym rozdzielić fragmenty, których długość różni się 1 nukleotydem). Następnym etapem jest odczytanie autoradiogramu. W teoretycznym przykładzie przedstawionym powyżej najniższy prążek znajdowałby się w ścieżce C, następny w ścieżce T, A. Stąd sekwencja odczytana byłaby 5'-CTA-3'. Autoradiogram z żelu, na którym rozdzielono produkty każdej z 4 mieszanin reakcyjnych zawiera obraz prążków, z którego można bezpośrednio odczytać sekwencję nukleotydów. W praktyce DNA jest specyficznie zrywany za pomocą odczynników, które modyfikują, a następnie oddzielają zasady od reszt cukru. Puryny usuwa się przez działanie siarczanem dimetylu, który metyluje guaninę przy N-7 a adeninę przy N-3. Wiązanie glikozydowe metylowanej puryny łatwo ulega zerwaniu przez ogrzewanie w obojętnym pH, co pozostawia resztę cukru bez zasady. Kolejne ogrzewanie DNA w środ. Alkalicznym prowadzi do zerwania łańcucha fosforanowo-cukrowego przy G, a traktowanie rozcieńczonym kwasem powoduje zerwanie łańcucha zarówno przy A jak i G. Cytozynę i tyminę odszczepia się od deoksyrybozy z udziałem hydrazyny. Łańcuch fosforanowo-cukrowy jest następnie zrywany w miejscach C i T przez działanie piperydyną. Hydrazynoliza prowadzona w obecności 2M NaCl ogranicza reakcję do C. Następne traktowanie DNA piperydyną prowadzi do zerwania łańcucha tylko przy C. Tak więc DNA można zerwać tylko przy G, a i G, C i T oraz tylko przy C, co umożliwia wydedukowanie kompletnej sekwencji fragmentu DNA przez porównanie 4 odpowiednich ścieżek na autoradiogramie.
36. Zasada enzymatycznej metody określania sekwencji nukleotydów -Sanger&Coulson
Do określania sekwencji nukleotydów w DNA opracowano dwie metody: metodę chemiczną (metoda Maxama&Gilberta) oraz metodę opartą na terminacji syntezy łańcuchów (metoda Sangera&Coulsona). W tej drugiej metodzie DNA przeznaczony do sekwencjonowania przygotowuje się w postaci cząsteczki jednoniciowej tak, aby mogła ona służyć jako matryca do syntezy DNA podczas reakcji sekwencjonowania. Do kopiowania tej matrycy wykorzystywana jest polimeraza DNA I E. coli. Do rozpoczęcia syntezy DNA enzym potrzebuje jednak startera, którym może być fragment restrykcyjny DNA komplementarny do jednoniciowej matrycy, albo krótki oligonukleotyd, komplementarny do matrycy, uzyskany na drodze syntezy chemicznej. Zasada metody: przygotowuje się mieszaninę inkubacyjną zawierającą matrycę w postaci jednoniciowego DNA, starter, polimerazę DNA I oraz wszystkie 4 trifosforany deoksyrybonukleozydów (dATP, dGTP, dCTP, dTTP), z których jeden jest znakowany radioaktywnie. Mieszaninę dzieli się na cztery porcje i do każdej z nich dodaje jeden z czterech trifosforanów deoksyrybonukleozydów (ddATP, ddGTP, ddCTP, ddTTP), będących analogami normalnych substratów. Podczas inkubacji polimeraza DNA I rozpoczyna kopiowanie cząsteczek matrycy, wydłużając związany z nią starter. W miarę postępu syntezy nowego DNA, za każdym razem, gdy powinien być wprowadzony np. dGTP, istnieje szansa, że zamiast niego zostanie wprowadzony ddGTP. Jeśli się to zdarzy, to dalsze wydłużanie łańcucha nie jest możliwe, ponieważ analogi dideoksy nie zawierają grupy 3'-OH, niezbędnej do utworzenia kolejnego wiązania 3',5'-fosfodiestrowego. Tak więc wydłużanie zostanie w takim miejscu zatrzymane i powstają fragmenty o różnej długości, w których dideoksyanalog znajduje się na końcu 3'. W mieszaninie inkubacyjnej kopiowaniu poddawana jest duża liczba cząsteczek matrycowych i wydłużanie każdego nowego łańcucha zatrzymuje się przypadkowo w jednym z miejsc, w którym powinna być wprowadzona G, ale zostaje wprowadzona ddG. W ten sposób każda z reszt G w matrycy wyznacza kolejny punkt terminacji syntezy nowych łańcuchów (rys krótkie wykłady z biochemii str 246). W praktyce przygotowuje się 4 mieszaniny inkubacyjne, z których każda zawiera inny analog dideoksy oraz jednakowy zestaw pozostałych składników. Po inkubacji uzyskuje się 4 zestawy łańcuchów kończących się odpowiednim analogiem dideoksy w miejscach wyznaczonych przez dystrybucję C, T, A lub G w matrycy. Produkty każdej z mieszanin inkubacyjnych poddaje się elektroforezie w czterech równoległych ścieżkach żelu poliakryloamidowego, a następnie autoradiografii. Sekwencję DNA określa się przez proste odczytanie obrazu pasm na autoradiogramie, poczynając od końca żelu di jego początku (tzn. odczytując sekwencję DNA w takim kierunku, w jakim ona powstaje poczynając od startera). Sekwencja odczytana bezpośrednio z żelu jest sekwencją łańcuchów nukleotydów nowo syntetyzowanych. Sekwencja ta jest komplementarna do sekwencji wyjściowej matrycy DNA. Obecnie DNA jest sekwencjonowany w sposób automatyczny z zastosowaniem metody dideoksy ale z zastosowaniem startera znakowanego fluoryzującym barwnikiem a nie radioaktywnie. Starter wykorzystywany do każdej z 4 mieszanin jest znakowany barwnikiem o innej fluorescencji. Po inkubacji łączy się mieszaniny i poddaje elektroforezie w jednej ścieżce żelu. Laserowy detektor rozróżnia i identyfikuje każdy z produktów w miarę ich wypływania z żelu. Kolejność w jakiej są wymywane z żelu produkty o różnej fluorescencji, stanowi sekwencję DNA. Zaletą wykrywania na podstawie fluorescencji jest eliminacja odczynników radioaktywnych oraz możliwość zautomatyzowania procedury.
37. Reakcja PCR (Polymerase Chain Reaction), zasada metody oraz jej wykorzystanie.
PCR(Polymerase Chain Reaction)-łańcuchowa reakcja polimeryzacji.
Umożliwia ona w jednej reakcji enzymatycznej, powielanie (amplifikację) w ciągu kilku godzin określonego odcinaka DNA w milionowych ilościach kopii. Kopiowanie odbywa się bez pośrednictwa wektorów. Metoda powielania DNA techniką PCR polega na cyklicznym powtarzaniu denaturacji cząsteczki DNA, przyłączaniu starterów i syntezie nowej nici DNA miedzy tymi starterami.
W pierwszym cyklu docelowe cząsteczki DNA rozdzielane są na 2 nici w wyniku ogrzewania mieszaniny do 95oC. Zazwyczaj przez około 60 sekund. Nastepnie w celu przyłączenia starterów obniża się temperaturę do ok. 55oC. (prze ok. 30 sek) Po przyłączeniu starterów podnosi się temperaturę mieszaniny do 72oC, w której zachodzi optymalna polimeryzacja z wykorzystaniem dNTP i Mg2+ zawartych w mieszaninie reakcyjnej.
Replikacja rozpoczyna się od primerów na każdej nici i zachodzi na obu niciach w dwóch przeciwstawnych kierunkach. Na nowosyntetyzowanych niciach powstają zatem nowe miejsca wiązania primerów. Po zakończeniu replikacji nici są rozdzielane (denaturacja), aby ponownie umożliwić wiązanie znajdujących się w nadmiarze primerów, syntezę DNA i kolejne rozdzielenie nici. Cykl taki powtarza się wielokrotnie, a po n cyklach otrzymuje się teoretycznie 2n dwuniciowych cząsteczek DNA będących kopiami sekwencji zawartej pomiędzy primerami.
PCR daje się stosunkowo łatwo zastosować jako technika laboratoryjna. Materiałem wyjściowym do amplifikacji jest DNA zawierający interesującą nas sekwencję. Ponieważ sekwencja ta określona jest przez primery użyte do reakcji, nie jest konieczne izolowanie samej sekwencji. Ilość DNA stosowanego do PCR jest bardzo mała (teoretycznie wystarczy jedna cząsteczka DNA). Do mieszaniny reakcyjnej, oprócz DNA, dodaje się nadmiar primerów (dwa rodzaje primerów określających miejsce startu replikacji na każdej nici), polimerazę DNA, odpowiedni bufor zawierający magnez oraz mieszaninę 4 prekursorów DNA, tj.dezoksyrybonukleotydów (dNTP). Powodzenie reakcji PCR wymaga właściwego doboru następujących parametrów:
1) starterów reakcji amplifikacji (primerów). Projektując startery reakcji PCR, należy je dobrać tak, aby:
- starter zawierał około 50% par GC,
- starter był wysoko specyficzny dla danej sekwencji,
- odległość między starterami w amplifikowanym DNA wynosiła 0.1 do 3 kb (o ile używana jest Taq polimeraza),
- długość primerów wynosiła około 20-30 zasad,a temperatura ich topnienia 50-60oC,
- startery nie powinny zawierać w 3' końcowej części fragmentów komplementarnych do siebie samych oraz do drugiego startera,
2) parametrów reakcji amplifikacji (stężenia dNTP, polimerazy, starterów, magnezu). Do reakcji PCR używa się polimerazy z Thermus aquaticus (Taq polimeraza) lub inne termostabilne polimerazy DNA (np. Pfu, Pwo, Vent). Mają one tę przewagę nad innymi polimerazami DNA, że są stabilne nawet w 94oC (optimum temperaturowe wynosi 72oC), a więc mogą być dodane jednorazowo na samym początku reakcji PCR, nie ulegając dezaktywacji w trakcie kolejnych cykli podgrzewania,
3) warunków samej reakcji PCR (temperatury, czasy trwania cykli itp).
Czasy i temperatury w dużym stopniu zależą od wielkości produktów otrzymywanych w procesie amplifikacji oraz sekwencji i długości starterów.
Reakcję prowadzi się w objętości 10-50 ul pod warstwą oleju parafinowego lub wosku, zapobiegającą parowaniu. Probówki z PCR umieszcza się w specjalnym aparacie (ang. thermal cycler), w którym programuje się czas trwania i liczbę cykli oraz temperaturę poszczególnych etapów reakcji. Zwykle stosuje się 25-35 cykli.
PCR może dostarczyć wartościowych informacji diagnostycznych w medycynie. Stosując specyficzne primery wykryć można bakterie i wirusy np. wirusa HIV-PCR jest doskonałą metodą wykrywania białaczek spowodowanych rearanżacją chromosomów. Wykrywa się tą metodą występowanie lub nieobecność częstych mutacji w znanych genach. NP. w prawidłowo powielonym segmencie DNA wykrywa zamianę pojedynczej pary zasad w genie β-globiny, która powoduje anemię sierpowatą. PCR w kryminalistyce jest też wykorzystywany. Powielanie wielu genów za pomocą PCR stosuje się by ustalić biologiczne ojcostwo w sporach prawnych. Wyniki analizy próbek krwi i nasienia metodą PCR stanowiły dowody sądowe w sprawach o zabójstwo czy gwałt. Ilość DNA w cebulce pojedynczego włosa zupełnie wystarcza do identyfikacji osoby. PCR umożliwia także dokonanie rekonstrukcji DNA z próbek pochodzących z bardzo dawnych czasów przez powielenie nielicznych ocalałych fragmentów DNA. Ostatnio odczytano fragmenty niektórych genów z mumii egipskich liczących 2400 lat oraz wykopalisk archeologicznych sprzed 7500 lat. Oznaczenie typów HLA ludzi z czasów starożytnych umożliwia wgląd w dynamikę populacji i ich wspólnot.
Zalety zastosowania termostabilnych polimeraz w technice PCR.
Do reakcji PCR stosuje się termostabilne polimerazy DNA, które zostały wyizolowane z wielu termofilnych bakterii, a ich geny skolonowane. Najbardziej popularna jest Taq polimeraza z Thermus aquaticus. Wytrzymuje ona etap denaturacji w temp 95 przez 1-2 min i w tej temperaturze ma okres półtrwania wynoszący 2 godziny. Wiadomo, że polimeraza Taq podczas kopiowania DNA wprowadza błędy (1 błąd na 250 polimeryzowanych nukleotydów).
Reakcja PCR w czasie rzeczywistym (Real_Time PCR) zasada metody oraz jej wykorzystanie.
PCR w czasie rzeczywistym polega na ilościowej reakcji amplifikacji kwasów nukleinowych i barwnych, bezinwazyjnych metodach detekcji. Procesy te łączone są poprzez wprowadzenie w reakcję barw fluorescencyjnych dla monitorowania w czasie rzeczywistym produktów amplifikacji, w trakcie ich faktycznego powstawania.
W czasie rzeczywistym = amplifikacja + detekcja (równocześnie)
Redukuje to czas oznaczenia i zagrożenie błędem przeniesienia - cały proces odbywa się w szczelnie zamkniętej probówce termocyklera.
27
P
TC
lac Z
ara C ara O ara I
pUC118/
pUC119
Wyszukiwarka
Podobne podstrony:
przykładowa prezentacja przygotowana na zajęcia z dr inż R Siwiło oceniona
Inz ogrod 2
44 OBIEKTY INż KOMUNALNEJ sem VI S1 KBI
Materialy do seminarium inz mat 09 10 czesc III
dyd inz n15
Biotechnologia inż plan studów
38 USTAWA O OC ARCH I INZ
Inz V projekt
instrukcja - HYDROLIZA SOLI, Inżynieria środowiska, inż, Semestr II, Chemia ogólna, laboratorium
10, wojtek studia, Automatyka, studia 2010, obrona inz, Pytania na obrone, brak tematu , dyplomowka
czy uC zaczyna pracę wraz z załączeniem zasilania czy potrzebny jest sygnał wyzwalający, Pierdoły, j
inzynieryjna egz.inz gospodarka, geodezja testy różne
pHmetr-instrukcja obsługi, Inżynieria środowiska, inż, Semestr II, Chemia ogólna, laboratorium
zalaczniki1, inż. BHP, V semestr
Mikroklimat TEST nr 2, inż. BHP, V semestr
Geodezja 4, BUDOWNICTWO, INŻ, semestr 2, Geodezja, Geodezja, Geodezja
Pytania kolokwium, IMiR - st. inż, sem.6 od sołtysa, III rok, energetyka, kolokwium
specjalizacyjne inż, Ogrodnictwo
więcej podobnych podstron