CHEMIA ORGANICZNA
A. Kołodziejczyk Gdańsk 2010. 02.
1. WIĄZANIA CHEMICZNE
1.1 WPROWADZENIE
Chemia organiczna obok chemii ogólnej, nieorganicznej, analitycznej i fizycznej należy do głównych, klasycznych nauk chemicznych.
Zajmuje się chemią związków węgla z wyłączeniem węgla pierwiastkowego i tak typowych nieorganicznych pochodnych tego pierwiastka, jak CO, CO2, H2CO3, węglany - sole, mocznik czy węgliki.
Nazwę chemia organiczna wprowadził J. J. Berzelius w 1808 r. Początkowo była to nauka badająca substancje wytwarzane przez świat ożywiony, czyli organiczny. Mocznik, jako składnik moczu był zaliczany do związków organicznych, tzn. wytwarzanych przez organizmy żywe.
Natomiast przedmiotem chemii nieorganicznej były substancje pochodzenia mineralnego, wydobywane z minerałów (ze skał) i innych składników świata nieożywionego (powietrza czy wody). Ropa naftowa, dzisiaj typowy surowiec organiczny, w wieku XVIII była uważana za substancję nieorganiczną, ponieważ wypływała ze skał; nazwano ją olejem mineralnym - ang. mineral oil lub olejem skalnym - ang. petroleum. Podobnie było z węglem kopalnym, zarówno torf, węgiel brunatny czy kamienny były dawniej uznawane za związki mineralne. Obecnie wiadomo, że pomimo wydobywania z ziemi stanowią one mieszaninę różnorodnych, bardzo skomplikowanych związków węgla, wodoru, tlenu, azotu i siarki, a więc należą do substancji organicznych.
Koks, a tym bardziej grafit i diament, jako węgiel pierwiastkowy są substancjami nieorganicznymi.
W czasach Berzeliusa węglan potasu, amoniak i fosforan wapnia zaliczano do związków organicznych, ponieważ ten pierwszy wydobywano z popiołu roślin, a kolejne były produktami zwierzęcymi. Bursztyn jako typowy minerał pasował do substancji nieorganicznych.
Dzisiaj związki klasyfikuje nie z uwagi na pochodzenie, ale skład.
Kiedy rodził się podział na chemię nieorganiczną i organiczną uważano, że do wytworzenia substancji organicznych potrzebna jest nadprzyrodzona siła, zwana vis vitalis, którą dysponowały wyłącznie organizmy żywe. Stąd produkowany przez ssaki mocznik był zaliczany do substancji organicznych.
W 1828 r. F. Wöhler otrzymał mocznik w wyniku ogrzewania cyjanianu amonu, czyli typowego związku nieorganicznego.
Ta synteza nie tylko obaliła teorię vis vitalis, ale dała początek nowoczesnej chemii organicznej, którą definiuje się jako chemię związków węgla z wyjątkami, jak powyżej. Mocznik nie jest zaliczany do związków organicznych.
Początkowo osiągnięcie Wöhlera związane z laboratoryjną syntezą „organicznego” mocznika nie wzbudziło większego zainteresowania ówczesnych chemików. silniejszym echem odbiło się opublikowanie w 1845 r. informacji o otrzymania z pierwiastków, a więc z substancji niewątpliwie nieorganicznych, tak typowo organicznego związku, jakim jest kwas octowy. Autorem tej laboratoryjnej syntezy kwasu octowego był H. Kolbe.
Z biegiem czasu synteza związków organicznych stała się nie tylko ciekawostką naukową obalającą teorie filozoficzne, ale ważnym sposobem otrzymywania produktów o dużym znaczeniu praktycznym, a synteza mocznika przez Wöhlera została uznana za początek chemii organicznej, jako odrębnej nauki.
W miarę przybywania syntetycznie otrzymywanych związków węgla do związków organicznych pochodzenia organicznego (naturalnego) dołączono te produkty syntetyczne zawierające atom (atomy) węgla, które nie były zaliczane do substancji nieorganicznych. Znane są, np. pochodne kwasu węglowego, w tym analogi mocznika, które są substancjami organicznymi. Wystarczy jeden lub więcej atomów wodoru w H2CO3 lub NH2CONH2 zastąpić resztami zawierającymi atom (atomy) węgla, żeby otrzymać związki organiczne.
Uzasadnieniem dalszego utrzymywania podziału na chemię organiczną i nieorganiczną jest to, że poznano wielokrotnie więcej związków organicznych niż nieorganicznych. Ta granica zaciera się jednak, ponieważ coraz częściej chemicy łączą cząsteczki typowo nieorganiczne z organicznymi. Podział na chemię organiczną i nieorganiczną jest utrzymywany w dalszym ciągu między innymi w celach dydaktycznych. Nazwa chemia organiczna została zachowana z tego względu, że większość składników żywych organizmów stanowią substancje organiczne.
Dodatkowo za oddzieleniem chemii organicznej od chemii nieorganicznej przemawia fakt, że żaden inny pierwiastek nie jest zdolny do tworzenia tak wielu różnorodnych związków, w tym polimerów zawierających łańcuchy węglowe, które mogą być poprzeplatane innymi atomami (heteroatomami). Zdarza się, że takie łańcuchy zbudowane są z tysięcy, milionów a nawet miliardów atomów (np. kwasy polinukleinowe). Ponadto łańcuchy węglowe i węglowo-heteroatomowe mogą ponadto zamykać się w różnej wielkości pierścienie.
W skład większości związków organicznych wchodzi wodór, z tego też powodu w 1889 r. K. Schorlemmer określił chemię organiczną jako chemię węglowodorów i ich pochodnych. W związkach organicznych powszechnie występuje też tlen. Zawierają często także takie pierwiastki jak azot, siarka, fosfor i halogeny. W cząsteczkach związków organicznych mogą być obecne również inne pierwiastki, np. krzem, a także metale, w tym magnez, lit, kadm, potas, sód i inne.
Chemia organiczna początkowo była nauką typowo opisową, tzn. zajmowała się charakterystyką związków organicznych, ich właściwościami fizycznymi, chemicznymi, tzn. reaktywnością, sposobami ich otrzymywania, oczyszczania i zastosowaniem.
Z czasem zainteresowanie chemii organicznej jako nauki skupiło się w znacznej części na przemianach chemicznych, czyli na prawach, jakie decydują o powstawaniu nowych związków organicznych i o zjawiskach, jakie towarzyszą przekształcaniu jednych w drugie.
Poznano sposoby wprowadzania do istniejącej cząsteczki różnych atomów lub ich grup (reakcje addycji i substytucji), jak również odrywania określonych elementów (reakcje eliminacji) czy też łączenia dwóch lub więcej cząsteczek razem (reakcje polimeryzacji).
Spośród jednolitej chemii organicznej zaczęły się wyodrębniać nauki pokrewne, w tym fizyczna chemia organiczna, chemia badająca mechanizmy reakcji, chemia spektroskopowa, technologia chemiczna, chemia polimerów, chemia związków naturalnych, chemia medyczna, a także chemie tak specjalistyczne, jak np. chemia cukrów, aminokwasów, lipidów, alkaloidów, terpenów, itp.
1.2 WIĄZANIA CHEMICZNE
W podstawowym kursie chemii organicznej obok chemii opisowej, podaje się informacje o mechanizmach reakcji. Duży nacisk kładzie się na zrozumienie zasad rozrywania i tworzenia nowych wiązań chemicznych.
Zmiany zachodzące w wiązaniach chemicznych, czyli przemieszczanie się elektronów walencyjnych jest podstawą przemian chemicznych, tj. otrzymywania nowych związków, zaś rozmieszczenie (konfiguracja) tych elektronów w cząsteczce decyduje o właściwościach fizycznych i chemicznych związków organicznych.
Wiązanie chemiczne tworzone jest przez parę elektronów walencyjnych. Wiązanie, w którym oba elektrony pochodzą od jednego z łączonych atomów nazywane jest wiązaniem jonowym, bowiem łączy jony:
K+ A- wiązanie jonowe (K+ = kation, A- = anion)
Występują one w solach, zarówno nieorganicznych jak i organicznych, np.
Na+Cl- czy CH3COO-Na+ (octan sodu)
Elektrony tworzące wiązanie kowalencyjne pochodzą od obu związanych atomów. Mogą one w jednakowym stopniu należeć do obu łączonych atomów, wówczas takie wiązanie nazywa się wiązaniem kowalencyjnym niespolaryzowanym; w wiązaniu kowalencyjnym spolaryzowanym elektrony są przesunięte w stronę bardziej elektroujemnego atomu:
Cl :Cl H : Cl
wiązania kowalencyjne: niespolaryzowane spolaryzowane
W związkach organicznych dominują wiązania kowalencyjne. W tej samej cząsteczce mogą równocześnie występować wiązania kowalencyjne niespolaryzowane, spolaryzowane, jak i jonowe:
Zadanie: wskaż w powyższym wzorze 3-chloropropanianu sodu wiązanie jonowe oraz wiązania kowalencyjne spolaryzowane i niespolaryzowane
Wiązania kowalencyjne niespolaryzowane powstają pomiędzy atomami o takiej samej lub zbliżonej elektroujemności, np. występują w cząsteczkach Cl-Cl, H-H czy w łańcuchach poliwęglowych: -C-C-C-C-. Atomy niezbyt mocno różniące się elektroujemnością powiązane są wiązaniami kowalencyjnymi spolaryzowanymi. Wiązania takie obserwuje się w następujących układach: -C-Cl, H-O-H, -N-H czy -C-O-. Jeżeli różnica elektroujemności pomiędzy atomami jest znaczna to dochodzi do jonizacji i utworzenia wiązań jonowych, np. w cząsteczkach Na+Cl-, Li+F-, K+Br- czy CH3COO-Na+. Wartości elektroujemności atomów wg skali zaproponowanej przez Paulinga dla wybranych pierwiastków podane są w tabeli 1.1.
Elektroujemność wybranych pierwiastków, wg skali Paulinga Tabela 1.1
Związki chemiczne zbudowane są z atomów połączonych wiązaniami chemicznymi. W każdym atomie wyróżnia się jądro złożone z protonów i neutronów, przy czym liczba protonów decyduje o liczbie atomowej, a suma protonów i neutronów odpowiada liczbie masowej. W obojętnym atomie wokół jądro rozlokowane są elektrony w liczbie równej liczbie protonów. Jeżeli liczba elektronów przewyższa liczbę protonów w jądrze to mamy do czynienia z jonami ujemnymi (anionami), a kiedy jest niższa to są to jony dodatnie (kationy).
Zgodnie z teorią E. Schrödingera ruch elektronów wokół jądra opisany jest funkcją falową. Przestrzeń, w której porusza się elektron nazywana jest orbitalem. Zwykle pojęcie orbitalu zawęża się do przestrzeni, w której prawdopodobieństwo obecności elektronu jest największe. Wielkość tego prawdopodobieństwa można założyć, np. równą 95%. Przestrzeń wypełnioną przez elektrony nazywa się często chmurą elektronową.
Orbitale różnią wielkością zajmowanej przestrzeni i kształtem przestrzeni wokół jądra. Wielkość ta zależy od odległości orbitali atomowych od jądra. Odległości dla poszczególnych powłok są skwantowane, tzn. przyjmują określone wielkości (nie ma wartości pośrednich). W zależności od oddalenia powłoki od jądra oznacza się je kolejnymi liczbami naturalnymi 1, 2, 3...lub literami K, L, M ....
W obrębie poszczególnych powłok występują orbitale (s, p, d i f) różniące się kształtami.
Na każdej powłoce elektronowej jest jeden bezkierunkowy orbital s
Na powłokach począwszy od 2. znajdują się po 3 orbitale p. Mają one kształt ósemki obrotowej (hantli) i są ukierunkowane w przestrzeni. Ich osie wzdłużne są ułożone względem siebie tak, jak osie rzędnych (x, y i z). Energia elektronów zlokalizowanych na orbitalach p tego samego poziomu jest identyczna; orbitale o jednakowej energii określa się jako zdegenerowane.
Na powłokach elektronowych od 3. w górę znajdują się po cztery orbitale d o kształcie rozety (koniczynki przestrzennej).
Na powłokach elektronowych od 4. w górę znajdują się orbitale f. Ich kształty są bardziej skomplikowane niż orbitali już omówionych.
1.3 KONFIGURACJA ELEKTRONÓW, ORBITALE ATOMOWE
Elektrony w stanie podstawowym (nie wzbudzonym) zajmują kolejne orbitale zaczynając od tego o najniższej energii - pierwszy elektron obsadza orbital s na powłoce 1; tę pozycje oznaczamy 1s. Tak jest w atomie wodoru:
Konfiguracja elektronowa atomu H - 1s
Kolejność obsadzania elektronami równorzędnych orbitali regulowany jest zakazem Pauliego, który brzmi: na jednym orbitalu mogą znajdować się co najwyżej dwa elektrony, a ich spisy muszą mieć przeciwne kierunki. Elektrony o przeciwnie skierowanych spinach nazywane są elektronami sparowanymi. Elektrony niesparowane zajmują oddzielne orbitale.
Drugi elektron atomu helu obsadza także orbital 1s, ale przyjmuje przeciwny kierunek spinu. W ten sposób zostaje zapełniona powłoka elektronowa 1.
Konfiguracja elektronowa atomu He - 1s2.
W indeksie górnym s2 podana jest liczba elektronów na orbitalu s.
Kolejny, trzeci elektron zajmuje orbital s na powłoce 2. Tak jest w atomie litu.
Konfiguracja elektronowa atomu Li - 1s22s; atomu berylu - 1s22s2
Piąty elektron zajmuje pierwszy wolny orbital, a więc 2p, przyjmuje się, że jest to 2px.
Konfiguracja elektronowa atomu B - 1s22s22px.
Sposób zajmowania przez elektrony kolejnych orbitali odbywa się zgodnie z regułą Hunda:
niecałkowicie zapełnione orbitale o tej samej energii zajmowane są kolejno przez pojedyncze elektrony o spinach równoległych, aż do całkowitego zapełnienia sąsiadujących z sobą orbitali.
W atomie węgla znajduje się 6 elektronów i jego konfiguracja elektronowa jest następująca - 1s22s22px2py.
Po zapełnieniu pojedynczymi elektronami orbitali p, następuje ich parowanie elektronami o przeciwnych spinach, tak więc konfiguracja elektronowa kolejnych atomów wygląda następująco:
N - 1s22s22px2py2pz O - 1s22s22px22py2pz F - 1s22s22px22py22pz Ne - 1s22s22px22py22pz2
W trzecim okresie układu okresowego, zaczynając od atomu Na elektrony zapełniają kolejne orbitale w podobny sposób, jak powłokę drugą; konfigurację elektronową atomu Na zapisujemy - 1s22s22px22py22pz23s.
W tabeli 1.1 przedstawiono zebrane razem konfiguracje elektronowe atomów pierwiastków pierwszych dwóch okresów układu okresowego.
Konfiguracja elektronowa pierwiastków I i II okresu Tabela 1.2.
Pier- wiastek |
Konfiguracja elektronowa |
Liczba elektronów walencyjnych |
Pier- wiastek |
Konfiguracja elektronowa |
Liczba elektronów walencyjnych |
H |
1s1 |
1 |
C |
1s22s22px12py1 |
4 |
He |
1s2 |
2 |
N |
1s22s22px12py12pz1 |
5 |
Li |
1s22s1 |
1 |
O |
1s22s22px22py12pz1 |
6 |
Be |
1s22s2 |
2 |
F |
1s22s22px22py22pz1 |
7 |
B |
1s22s22px1 |
3 |
Ne |
1s22s22px22py22pz2 |
8 |
Zadanie: podaj konfigurację elektronową atomów Na, P i S
Konfigurację elektronową można zapisywać inaczej, np.za pomocą strzałek; dla atomu C:
Konfigurację elektronową przedstawia się również za
pomocą strzałek w kwadracikach; np. dla atomu O:
1.4 ORBITALE CZĄSTECZKOWE
W wyniku nałożenia się na siebie dwóch orbitali atomowych pochodzących od dwóch atomów powstają orbitale cząsteczkowe, dzięki czemu między tymi atomami tworzy się więź natury elektronowej, zwana inaczej wiązaniem chemicznym. Orbitale cząsteczkowe (molekularne, MO) obejmują jądra obu atomów związanych z sobą.
Liczba powstających orbitali cząsteczkowych jest równa liczbie nakładających się orbitali atomowych, tzn. po nałożeniu się dwóch orbitali atomowych powstają dwa orbitale cząsteczkowe, przy czym jeden o niższej energii nazywa się orbitalem wiążącym, a drugi o wyższej energii - orbitalem niewiążącym.
Najprostsze wiązanie chemiczne występuje w cząsteczce H2.
Rys. 1. 1. Poziomy energetyczne elektronów na orbitalach atomowych s atomów wodoru oraz orbitalach
cząsteczkowych, wiążących sigma (σ) i antywiążących (σ*) cząsteczki wodoru H2
W stanie podstawowym jedynie orbital wiążący jest obsadzony elektronami tworzącymi wiązanie, po wzbudzeniu (pobraniu kwantu energii) mogą one przejść na orbital niewiążący (antywiążący).
Energia wiązania zależy między innymi od stopnia nałożenia się orbitali atomowych - im stopień nałożenia orbitali większy, tym energia wiązania większa.
Wiązania sigma (σ) są najpopularniejszymi wiązaniami występującymi w cząsteczkach związków organicznych. Charakteryzują się one symetrią cylindryczną (osiową wzdłużną).
W tworzeniu wiązań cząsteczkowych biorą udział elektrony walencyjne, tj. elektrony z ostatniej powłoki; w atomie węgla są to 2s22px2py. Atomy pierwiastków przejściowych mogą również tworzyć wiązania za pomocą elektronów z orbitalu d przedostatniej powłoki.
Na walencyjnej powłoce elektronowej atomu węgla znajdują się 4 elektrony, ale jedynie dwa z nich, te niesparowane są zdolne do wytworzenia wiązań - dwóch wiązań. Dlaczego, mimo to, atom węgla jest zwykle czterowiązalny? Ponieważ przekształca się on w atom czterowiązalny po przeniesieniu jednego elektronu z orbitalu 2s na orbital 2pz.
Przejście elektronu na wyższy orbital nazywa się wzbudzeniem:
Pośród wzbudzonych elektronów jeden zajmuje orbital s, a trzy orbitale p. Wiązania utworzone przez tak skonfigurowane elektrony byłyby nierównocenne; jedno stałoby się bezkierunkowe (z orbitalu s), a z trzech orbitali p powstałyby wiązania pod kątem 90o - ich osie odpowiadałyby osiom współrzędnych (x,y,z).
1.5. HYBRYDYZACJA
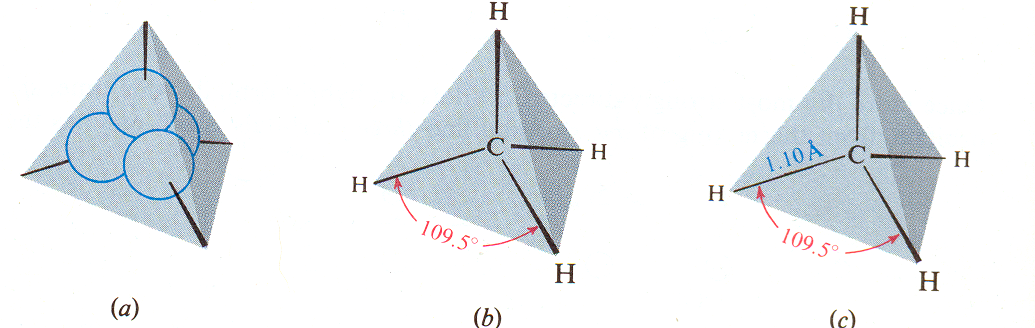
W czterowiązalnych pochodnych węgla, np. w metanie wiązania są równocenne i rozmieszczone w przestrzeni podobnie jak osie wychodzące ze środka tetraedru w kierunku naroży - pod kątem tetraedrycznym, czyli 109,5o:
Cząsteczka metanu jest wpisana w tetraedr
Rys. 1. 2 ułożenie atomów cząsteczki metanu w przestrzeni, wg Organic Chemistry, R.T. Morrison, R.N. Boyd, New
York University, Prentice Hall, 6th ed., 1992
Takie wzajemne ułożenie atomów jest możliwe dzięki hybrydyzacji (wymieszaniu) wzbudzonych orbitali walencyjnych atomu węgla: z jednego 2s i trzech 2p powstają cztery równocenne, zhybrydyzowane orbitale sp3.
Rys. 1. 3. Hydrydyzacja sp3
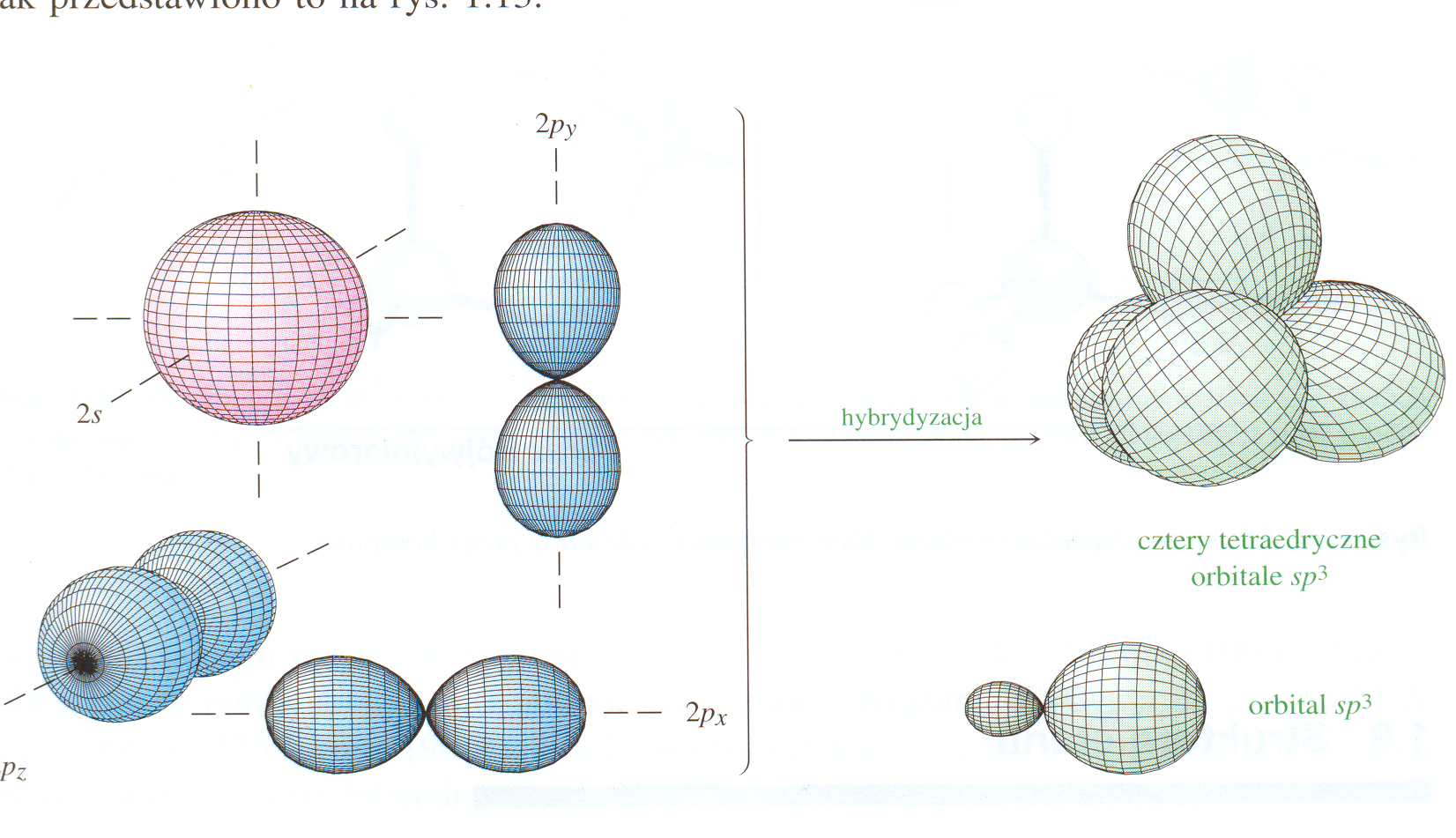
Każdy nowo powstały orbital sp3 ma w ¼ właściwości orbitalu s i w ¾ właściwości orbitalu p.
Rys. 1. 4. Orbitale atomowe atomu węgla przed i po hybrydyzacji, wg. J. McMurry Chemia Organiczna PWN Warszawa 2000
1.6 CZĄSTECZKA METANU
Schemat reakcji tworzenia cząsteczki metanu jest następujący:
Hybrydyzacja sp3 elektronów walencyjnych atomu C zapewnia utworzenie czterech wiązań, które są maksymalnie oddalone od siebie.
Wiązania pomiędzy atomami H i atomem C powstały w wyniku nałożenia się 4 orbitali s czterech atomów wodoru z 4 orbitalami sp3 jednego atomu węgla.
Rys. 1. 5. Orbitale cząsteczkowe metanu tworzą się poprzez nałożenie się na siebie orbitali atomowych H i C
Rys. 1. 6. W wyniku nałożenia się 4 orbitali atomowych s wodoru i 4 orbitali atomowych sp3 węgla powstało 8 orbitali cząsteczkowych (4 + 4) (MO, ang. molecular orbital), z czego 4 wiążące σ i 4 antywiążące σ*
W cząsteczce metanu w stanie podstawowym obsadzone są orbitale cząsteczkowe wiążące - σ, po wzbudzeniu elektrony przechodzą na wyższy poziom energetyczny - orbitale antywiążące σ*.

Związki organiczne przedstawiane są często za pomocą modeli, mogą być prętowe, prętowo-kulowe lub czaszowe. Cząsteczka metanu prezentowana za pomocą modeli wygląda następująco:
model prętowo-kulowy model prętowy model czaszowy
Rys. 1. 7. Modele związków organicznych wg. R.T. Morrison & R.N. Boyd Organic Chemistry, sixth edition, Prentice Hall, 1992
1.7 CZĄSTECZKA ETANU
Do powstania cząsteczki etanu dochodzi poprzez utworzenie wiązania cząsteczkowego pomiędzy dwoma atomami C−C i sześciu wiązań cząsteczkowych C−H:
Zgodnie z teorią orbitali molekularnych w wyniku połączenia się dwóch atomów węgla i sześciu atomów wodoru (sześć orbitali s atomów H, osiem orbitali sp3 atomów C) powstało siedem wiązań cząsteczkowych wiążących σ i tyle samo antywiążących.
↑
Rys. 1. 8. Orbitale molekularne etanu wiązanie σ sp3− sp3
1.8 Swobodny obrót wokół pojedynczego wiązania, rotamery
Stopień nakładania się orbitali sp3 dwóch atomów C tworzących wiązanie σ sp3− sp3 jest niezależny od obrotu atomów węgla wokół osi wiązania. Oznacza to, że energia tego wiązania nie zmienia się podczas obrotu osiowego tych atomów węgla; możliwy jest zatem swobodny obrót wokół pojedynczego wiązania C−C.
Rys. 1. 9. Swobodny obrót wokół pojedynczego wiązania C−C
Znane są inne sposoby przedstawiania obrotu wokół pojedynczego wiązania C−C i równoczesnej prezentacji powstających konformerów rotacyjnych (rotamerów). Obrót o 120o nie prowadzi do żadnych zauważalnych zmian, obrót o inny kąt zmienia wzajemne położenie atomów wodoru względem siebie, np. z położenia naprzeciwległego do naprzemianległego przy obrocie o 60o.
Rotamerów może być nieskończenie dużo, ale wyróżnia się jedynie graniczne.
Rys. 1. 10. Konformery rotacyjne etanu w projekcji Newmana
Rotamery etanu różnią się trwałością - konformer naprzemianległy jest o około 3 kcal/mol (12 kJ/mol) trwalszy od naprzeciwległego. Bariera energetyczna przejścia jednego rotameru w drugi jest pokonywana bez trudu przez cząsteczki w temperaturze pokojowej, a nawet znacznie niższej. Z tego powodu konformerów nie można rozdzielić; one zbyt łatwo przechodzą jeden w drugi.
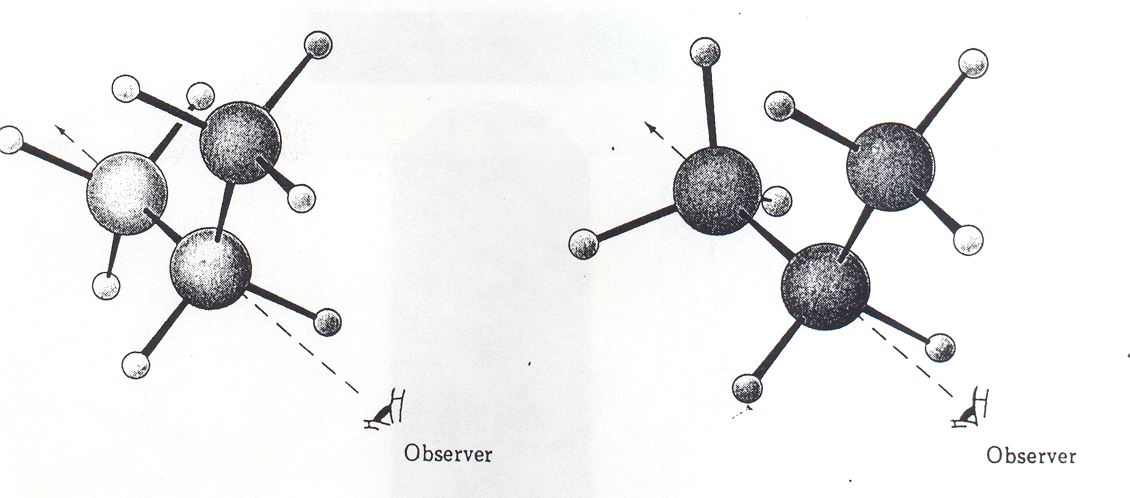
Konformery etanu przedstawione na modelach:




konformer naprzeciwległy konformer naprzemianległy
widok od frontu widok z boku widok od frontu widok z boku
Rys. 1. 11. Konformery etanu, wg. R.T. Morrison & R.N. Boyd Organic Chemistry, sixth edition, Prentice Hall, 1992
Udział konformerów w populacji wszystkich cząsteczek zależy od ich energii, im niższą energię mają, tym jest ich więcej.
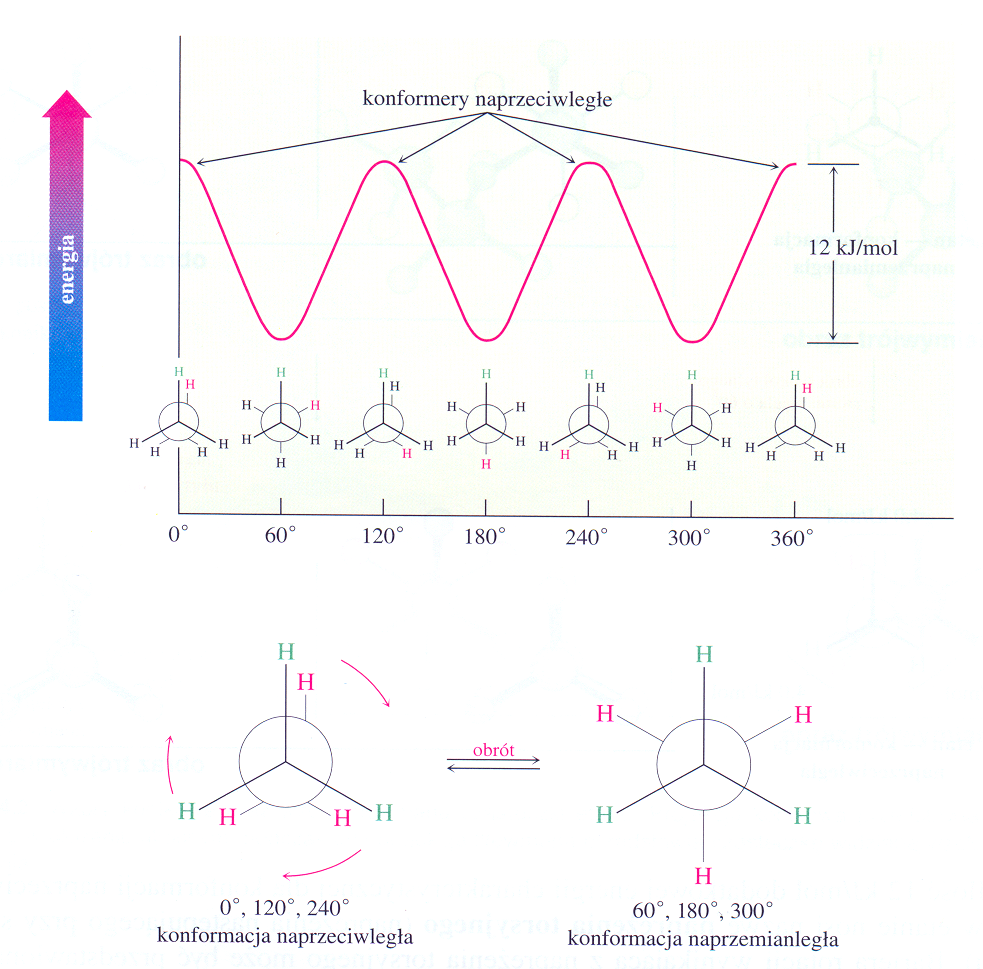
Wykres zmiany energii cząsteczek konformerów obrotowych etanu Wykres 1. 1.
(zmiana energii w trakcie obrotu wokół C−C), wg. J. McMurry Chemia Organiczna PWN Warszawa 2000
Różnica trwałości konformerów etanu wynika z tego, że w ustawieniu naprzeciwległym atomy wodoru zaczynają się odpychać, natomiast w położeniu naprzemianległym znajdują się one w większej odległości od siebie i nie ma tego oddziaływania.
W cząsteczce propanu różnica energetyczna pomiędzy najbardziej trwałym i najmniej trwałym rotamerem wynosi 3,4 kcal/mol (14 kJ/mol)
Rys. 1. 12. Konformery rotacyjne propanu na modelach
Rys. 1. 13. Konformery rotacyjne propanu przedstawione za pomocą wzorów
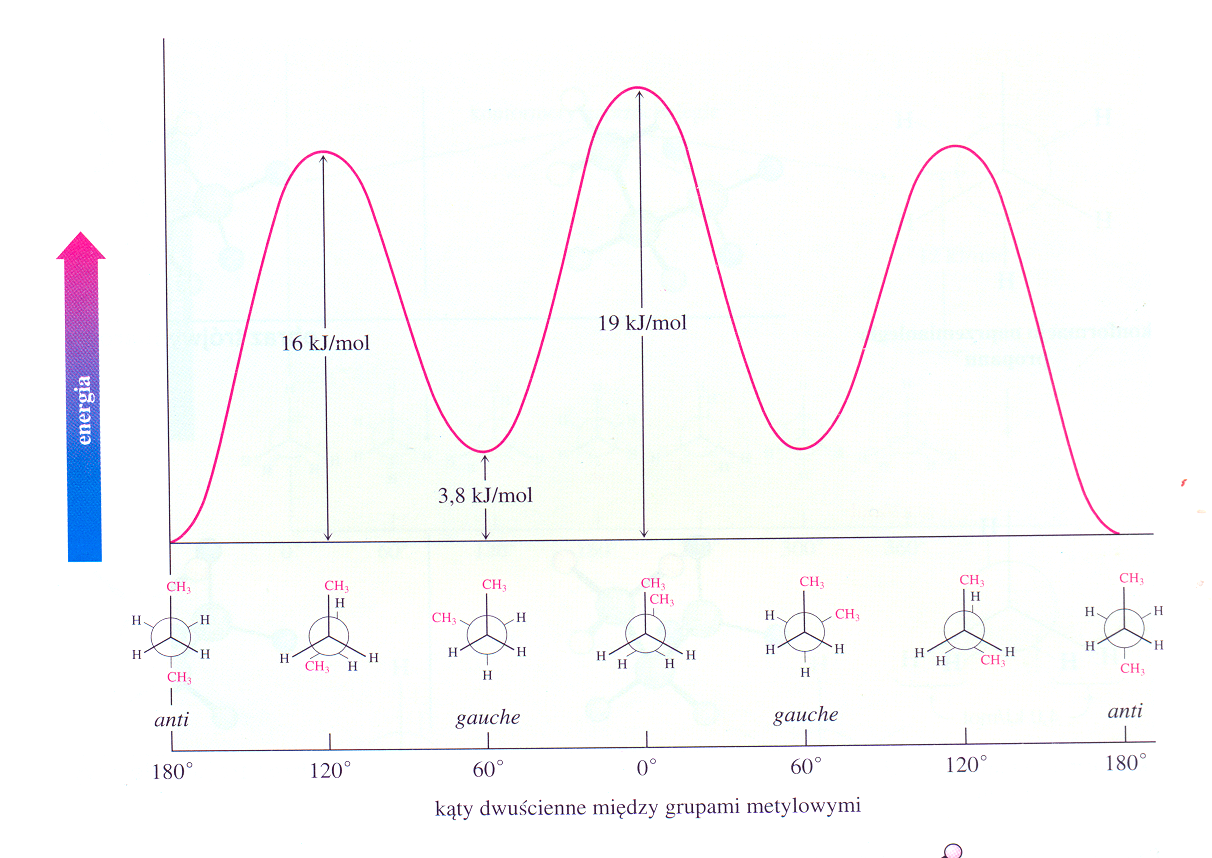
W butanie są trzy graniczne konformery rotacyjne.
Różnica energetyczna pomiędzy najbardziej stabilnym (naprzemianległym, anti lub antiperiplanarnym), a najmniej stabilnym (naprzeciwległym, synperiplanarnym)) 5,8 kcal/mol (19 kJ/mol) jest większa niż w propanie, ponieważ efekt odpychania się dwóch grup metylowych jest większy niż H z CH3.
Rys. 1. 14. Konformery butanu
Rys. 1. 15. Konformery butanu przedstawione w inny sposób
Zmiana energii cząsteczek konformerów butanu, wg. J. McMurry Chemia Organiczna PWN Warszawa 2000 Wykres 1. 2.
1.8 WIĄZANIA KOWALENCYJNE SPOLARYZOWANE, MOMENT DIPOLOWY
Wiązania kowalencyjne (atomowe) niespolaryzowane powstają pomiędzy atomami o takiej samej lub zbliżonej elektroujemności, np. występują w cząsteczkach Cl-Cl, H-H czy w łańcuchach poliwęglowych: -C-C-C-C-. Atomy różniące się elektroujemnością tworzą wiązania kowalencyjne spolaryzowane. Wiązania takie obserwuje się w takich układach, jak np. -C-Cl, H-O-H, -N-H czy -C-O-. Jeżeli różnica elektroujemności pomiędzy atomami jest duża to dochodzi do jonizacji i utworzenia wiązań jonowych, np. w cząsteczkach Na+Cl-, Li+F-, K+Br- czy CH3COO-Na+. Wartości elektroujemności atomów wg skali zaproponowanej przez Paulinga dla wybranych pierwiastków podane są w tabeli 1.1. Różnica wartości elektroujemności pomiędzy atomami tworzącymi wiązanie kowalencyjne spolaryzowane wynosi, 0,3 dla C i H; 0,7 dla C i Cl, a dla wiązań jonowych przekracza 2: 2,3 dla Na i Cl; 3,2 dla K i F.
W wiązaniach kowalencyjnych niespolaryzowanych ładunki elektryczne są rozmieszczone równomiernie pomiędzy połączonymi atomami, zaś w wiązaniach kowalencyjnych spolaryzowanych gęstość elektronów tworzących wiązanie nie jest symetrycznie rozłożona - większa jest zgromadzona wokół atomu bardziej elektroujemnego (δ+), a mniejsza wokół atomu mniej elektroujemnego (δ-). Natomiast para elektronów tworzących wiązanie jonowe zlokalizowana jest przy atomie elektroujemnym, przez co zostaje on obdarzony ładunkiem ujemnym (-), a atom elektrododatni zyskuje ładunek dodatni (+).
Rozkład gęstości elektronowej w wiązaniach:
kowalencyjnych niespolaryzowanych kowalencyjnych spolaryzowanych jonowych
Symbol δ oznacza częściowy ładunek elektryczny
Dla przedstawienia stopnia spolaryzowania wiązania wprowadzono pojęcie momentu dipolowego (), który jest równy iloczynowi ładunku Q (w kulombach) zgromadzonego na jednym z atomów i odległości r (w m) dzielącej środki atomów tworzących wiązanie.
m = Q . r
Wartości momentu dipolowego podaje się w debajach (D), przy czym 1D = 3,336 . 10-30 C.m.
Moment dipolowy dla rozdzielonych ładunków jednostkowych (jeden elektron od jednego protonu) na odległość 1Å (rząd długości wiązania kowalencyjnego) wynosi 4,80 D. Moment dipolowy jest wartością wektorową skierowaną od ładunku dodatniego do ujemnego. W tabeli 1.2 podane są wartości momentu dipolowego najbardziej charakterystycznych wiązań występujących w związkach organicznych.
Wartości momentów dipolowych () wybranych wiązań Tabela 1.3
Wiązanie |
[D] |
Wiązanie |
[D] |
|
0 |
|
0,22 |
|
0,3 |
|
0,86 |
|
1,31 |
|
1,29 |
|
1,53 |
|
1,51 |
|
2,4 |
|
1,48 |
|
3,6 |
|
1,56 |
Moment dipolowy cząsteczki jest wypadkową momentów dipolowych występujących w niej wiązań. Wartość momentu dipolowego cząsteczki o symetrycznej budowie jest równa 0, pomimo tego, że poszczególne wiązania są polarne. To stwierdzenie dotyczy, np. ditlenku węgla, metanu czy tetrachlorku węgla.
Rys. 1.16 Wartość momentu dipolowego cząsteczek symetrycznych jest równa 0
Wartość momentu dipolowego cząsteczek niesymetrycznych jest równy sumie wektorów momentu poszczególnych wiązań. W tabeli 1.3 podane są wartości momentu dipolowego wybranych cząsteczek. Im wyższa wartość momentu dipolowego, tym większa polarność cząsteczki.
Wartości momentu dipolowego cząsteczek niektórych związków Tabela 1.3
Nazwa związku |
Wzór |
[D] |
Nazwa związku |
Wzór |
[D] |
chlorek sodu |
|
9,0 |
chlorek metylu |
CH3Cl |
1,9 |
woda |
HOH |
1,9 |
chlorek metylenu |
CH2Cl2 |
1,6 |
metanol |
CH3OH |
1,6 |
chloroform |
CHCl3 |
1,0 |
|
|
|
|
|
|
nitrometan |
|
3,5 |
aceton |
|
2,8 |
diazometan |
|
1,5 |
metyloamina |
CH3NH2 |
1,6 |
Zadanie: wytłumacz, dlaczego wartość momentu dipolowego chlorku metylu jest większa niż chloroformu;
oceń wartość momentu dipolowego etanu (CH3CH3) i benzenu
Momenty dipolowe cząsteczek oznacza się doświadczalnie za pomocą pomiaru refrakcji molowej lub wyznaczania zależności stałej dielektrycznej od temperatury; można je także obliczyć na podstawie informacji o wielkości rozdzielonych ładunków elektrycznych i długości wiązań. I odwrotnie, znając moment dipolowy i długość wiązania można obliczyć wielkość rozdzielonego ładunku. W ten sposób obliczono, że cząstkowy ładunek ujemny na atomie chloru w chlorku metylu jest równy -0,2 ładunku elektronu, a na atomie węgla występuje deficyt elektronu tej samej wielkości (+0,2); w obliczeniach pominięto oddziaływanie H-C.
Polaryzowalność
Wiązania kowalencyjne pod wpływem czynników zewnętrznych, takich jak oddziaływanie sąsiadujących cząsteczek (w tym rozpuszczalnika), jonów lub pola elektromagnetycznego ulegają odkształceniu, polegającym na zmianie długości wiązania i wielkości ładunków elektrycznych. To zjawisko nosi nazwę polaryzowalności (zdolności do polaryzacji). Wiązania w zależności od tego, w jaki sposób i jakie łączą atomy ulegają odkształceniu łatwiej lub trudniej, a tym samym są bardziej lub mniej podatne na atak odpowiednich reagentów; innymi słowy od polaryzowalności wiązań zależy reaktywność cząsteczek. Wiązania mało podatne na polaryzowanie trudno jest rozerwać, a tym samym są mało reaktywne. Atomy lub grupy atomów połączone wiązaniem łatwo polaryzowalnym są podatne na wymianę na inne atomy lub grupy. Wiązanie z większym atomem łatwiej ulega polaryzacji (elektrony takiego atomu są luźniej upakowane), a z mniejszym atomem trudniej (elektrony są ściśle upakowane). Przykładem mało reaktywnych związków są fluorki alkilowe. Wiązanie C-F, chociaż stosunkowo wysokopolarne jest słabo polaryzowalne (sztywne) i dlatego trudne do rozerwania. W przeciwieństwie do pochodnych fluorkowych jodki alkilowe są bardzo reaktywne (jod można łatwo wymienić na inny atom lub grupę), ponieważ wiązanie C-I chociaż mniej polarne, jest łatwo polaryzowalne.
H3C-F H3-I
Wiązanie C-F jest bardzo polarne ale słabo polaryzo- wiązanie C-I jest mniej polarne ale łatwiej polaryzo-
walne i dlatego fluorek metylu jest mało reaktywny walne i dlatego jodek metylu jest bardziej reaktywny
1.9 Siły (wiązania) międzycząsteczkowe
Cząsteczki chemiczne, oddziałują na siebie w ten sposób, że w pewnych określonych odległościach przyciągają się, a kiedy odległość pomiędzy nimi zmniejsza się do wartości sumy promieni atomów (cząsteczek) zaczynają się silnie odpychać. W ciałach stałych cząsteczki zbliżone są do siebie maksymalnie, tzn. przyciągają się najsilniej, jak to jest tylko możliwe, ale nieznaczne dalsze zbliżenie wywołuję znacznie silniejsze odpychanie. W cieczach cząsteczki mają trochę więcej swobody, niemniej ciecze też są mało ściśliwe, co oznacza, że ich cząsteczki znajdują się w odległości zbliżonej do tej, w której zaczynają działać siły odpychające. Gazy są znacznie bardziej ściśliwe, ponieważ odległości pomiędzy cząsteczkami są znacznie większe od tej, która wyzwala siły odpychające.
1.9.1 Przyciąganie dipol-dipol
Przyczyny przyciągania się cząsteczek są różne. W związkach o budowie jonowej jony przyciągane są siłami elektrostatycznymi. Cząsteczki obojętne skupiają się po wpływem oddziaływań dipol-dipol, sił dyspersyjnych i wiązań wodorowych. Cząsteczki polarne, czyli te, które mają momentem dipolowym większy od zera, gromadzą na swoich biegunach ładunek cząstkowy i ich bieguny dodatnie przyciągają się z biegunami ujemnymi z siłami proporcjonalnymi do wielkości ładunków. Przyciąganie wywołane oddziaływaniem dipol-dipol jest słabsze niż jonów, ponieważ przyciągają się ładunki cząstkowe, a nie pełne. Z tego powodu związki polarne mają zwykle niższe temperatury topnienia niż sole o zbliżonej masie molowej. Efekt wzajemnego przyciągania się cząsteczek zależy nie tylko od wielkości ładunków zgromadzonych na biegunach, ale również od kształtu i rozmiarów cząsteczek. Przyciągające oddziaływanie dipol-dipol zostało pokazane schematycznie na rysunku 1.16. Cząsteczkę można przedstawić w uproszczeniu jako elipsoidę obrotową z rozdzielonymi ładunkami na jej końcu.
Rys. 1.16 Cząsteczki polarne przyciągają się odmiennie naładowanymi końcami
1.9.2 Siły dyspersyjne - van der Waalsa (Londona)
Pomiędzy cząsteczkami niepolarnymi, w których nie ma na stałe rozdzielonych ładunków (moment dipolowy jest równy zero), też występują siły przyciągające. Ich źródłem są pojawiające się chwilowe ładunki elektryczne, jako wynik drgania elektronów. Nawet niewielkie przesunięcie się elektronów tworzących wiązania lub wolnych par elektronowych tworzy doraźne bieguny elektryczne w różnych częściach cząsteczki. Te bieguny indukując ładunek elektryczny o przeciwnym znaku w cząsteczce sąsiadującej wyzwalają siły przyciągające. Taki efekt trwa krótko, ale w następnej chwili ładunki pojawiają się w innej częściach cząsteczek, podtrzymując przyciąganie. Takie przyciąganie się cząsteczek zwane siłami dyspersyjnymi jest znacznie słabsze od oddziaływań dipol-dipol i w jeszcze większym stopniu zależy od kształtu i rozmiarów cząsteczek. Im cząsteczki są większe i większymi powierzchniami mogą się do siebie zbliżać, tym silniej będą się wzajemnie przyciągać. Oczywiście w cząsteczce polarnej występują też siły van der Waalsa, zwykle jednak ich udział w sumarycznym przyciąganiu się cząsteczek jest mniejszy od oddziaływań dipol-dipol, ale rośnie wraz ze wzrostem rozmiarów cząsteczek.
Rys. 1.17 Ładunki dyspersyjne pojawiające się w sposób ciągły w różnych miejscach cząsteczek powodują wzajemne przyciąganie się cząsteczek. Płaskie cząsteczki benzenu mogą przylegać do siebie na większej powierzchni niż w n-heksanie (łańcuchowym), więc oddziaływanie sił dyspersyjnej jest większe, a tym samym temperatura topnienia benzenu (5,5oC) znacznie przewyższa temperaturę topnienia n-heksanu (-94oC), podobnie jest z temperaturą wrzenia, odpowiednio - 80oC i 69oC
1.9.3 Wiązania wodorowe
Pomimo nazwy, wiązania wodorowe nie są typowymi wiązaniami chemicznymi pomiędzy atomami tworzącymi cząsteczkę. Ich natura jest zbliżona do oddziaływania typu dipol-dipol, ale specyficznym, ponieważ tworzonym pomiędzy atomem wodoru związanym z silnie elektroujemnym atomem (głównie F, O lub N), a innym elektroujemnym atomem z sąsiedztwa, posiadającym wolne pary elektronowe. Pośród związków organicznych, tego typu wiązania występują w alkoholach i kwasach karboksylowych (grupa -O-H), a także w aminach, w które zawierają wiązanie -N-H. Chociaż energia wiązania wodorowego rzędu 5 kcal/mol (20 kJ/mol) jest wielokrotnie mniejsza od typowego wiązania kowalencyjnego (C-H około 100 kcal/mol), to jego skutki są dobrze widoczne. Przejawiają się przede wszystkim w podwyższeniu temperatury wrzenia związków, w których występuje; alkohol etylowy wrze w temperaturze o ponad 100oC wyższej, niż jego izomer - eter dimetylowy.
H3CCH2OH etanol, tw. 78oC H3COCH3 eter dimetylowy, tw. -25oC
Wiązania wodorowe w alkoholach: ROH......O są znacznie silniejsze niż w aminach: RNH......NR, ponieważ atom tlenu jest bardziej elektroujemny od atomu azotu.
Rys. 1.18 Wiązania wodorowe w etanolu i w etyloaminie
16
Wyszukiwarka
Podobne podstrony:
Wyklad 4 Wiazania chemiczne w cialach stalych
Wiązania chemiczne (II)
6 wykad WiĄzania chemiczne[F]
Wykład 1, budowa atomu, wiązania chemiczne
2 Atom i cząstka Wiązania chemiczne klucz
2 Atom i cząstka Wiązania chemiczne
7 układ okresowy pierwiastków, wiązania chemiczne
Budowa atomu i wiązania chemiczne test odpowiedzi
wypisać i wymienić rodzaje wiązań chemicznych
WIĄZANIA CHEMICZNE
Atom, cząsteczka, wiązanie chemiczne
ściąga rodzaje wiązań chemicznych
Wiązania chemiczne
Chemia wiązania chemiczne
wiązania chemiczne wstęp(1)
Wiązania chemiczne
Rodzaje wiązań chemicznych
10 wiązania chemiczne
więcej podobnych podstron