I. RÓWNOWAGI W ROZTWORACH ELEKTROLITÓW. KWASY I ZASADY
Pojęcia podstawowe i definicje
ELEKTROLITEM jest każda substancja, która rozpuszczając się w wodzie lub innym rozpuszczalniku polarnym ulega dysocjacji elektrolitycznej na jony: dodatnie - kationy i ujemne - aniony. |
● Elektrolitami są kwasy, zasady i sole, niezależnie od tego, czy są to związki nieorganiczne, czy też organiczne. Roztwory elektrolitów przewodzą prąd elektryczny. Przewodnictwo elektryczne roztworu zależy od rodzaju i stężenia elektrolitu oraz właściwości rozpuszczalnika.
● Roztwory nieelektrolitów nie przewodzą prądu.
ROZPUSZCZALNIKI POLARNE mają dużą przenikalność dielektryczną (ε) względem próżni, |
Rolę przenikalności dielektrycznej ośrodka (rozpuszczalnika), w którym znajdują się dwa ładunki elektryczne q1 i q2 uzasadnia prawo Coulomba. Gdy ładunki te w próżni i w ośrodku
o względnej przenikalności dielektrycznej ε dzieli taka sama odległość r, to siły ich oddziaływań elektrostatycznych są następujące:
- w próżni, (1)
- w danym ośrodku, (2)
gdzie k = 8,9867·109 Nm2/C2, oraz F1 > F2.
Dipolowy charakter cząsteczek rozpuszczalnika jest jednym z warunków solwatacji cząsteczek lub jonów substancji rozpuszczonej.
Tabela 1. Właściwości wybranych rozpuszczalników
Rozpuszczalnik
|
Względna przenikalność dielektryczna ε |
Moment dipolowy μ, D |
Oktan, C8H18 Cykloheksan, C6H12 Benzen, C6H6 Toluen, C6H5CH3 Dichlorometan, CH2Cl2 Chloroform, CHCl3 Metanol*, CH3OH Etanol*, C2H5OH Nitrobenzen*, C6H5NO2 DMSO**, (CH3)2SO Formamid*, HC(O)NH2 Ciekły amoniak*, NH3 Woda* |
1,95 2,02 2,30 2,38 8,93 4.72 32,6 24.3 34.8 46,6 109,5 16,9 78.4 |
0 0 0 0.31 1.14 1.15 1.70 1.73 4.03 3,90 1,51 1,47 1.83 |
1Debye (1D), 1D = 3,338∙10-30 C·m; *rozpuszczalniki solwatujące;
**DMSO - dimetylosulfotlenek
Charakterystyczne właściwości fizyczne wody:
Cząsteczki wody mają budowę polarną - są dipolami.
Nietypowa zmiana gęstości ciekłej wody wraz
z temperaturą - maksymalna wartość przy +40C.Gęstość lodu jest mniejsza od gęstości ciekłej wody.
Wysoka pojemność cieplna ciekłej wody.
Szeroki zakres temperatury istnienia wody
w stanie ciekłym - 1000C.Duże napięcie powierzchniowe ciekłej wody.
Właściwości solwatacyjne ciekłej wody, zwane dalej zdolnością hydratacyjną, czynią ją
najbardziej uniwersalnym rozpuszczalnikiem
różnych substancji, w tym soli i gazów.
Wszystkie wymienione cechy wody wynikają
z dipolowej natury jej cząsteczek, zdolnych do wzajemnej, silnej asocjacji za pośrednictwem wiązań wodorowych (lód, ciekła woda) i do hydratacji cząsteczek lub jonów substancji rozpuszczonych za pośrednictwem oddziaływać elektrostatycznych, wiązań koordynacyjnych i wiązań wodorowych.
O
163 pm
H H
Rys. 1. Struktura cząsteczki wody
Woda jako rozpuszczalnik łatwo rozpuszcza sole lub wodorotlenki litowców, ponieważ jonowe sieci krystaliczne tych związków ulegają zniszczeniu,
a jony w roztworze są stabilizowane przez hydratację.
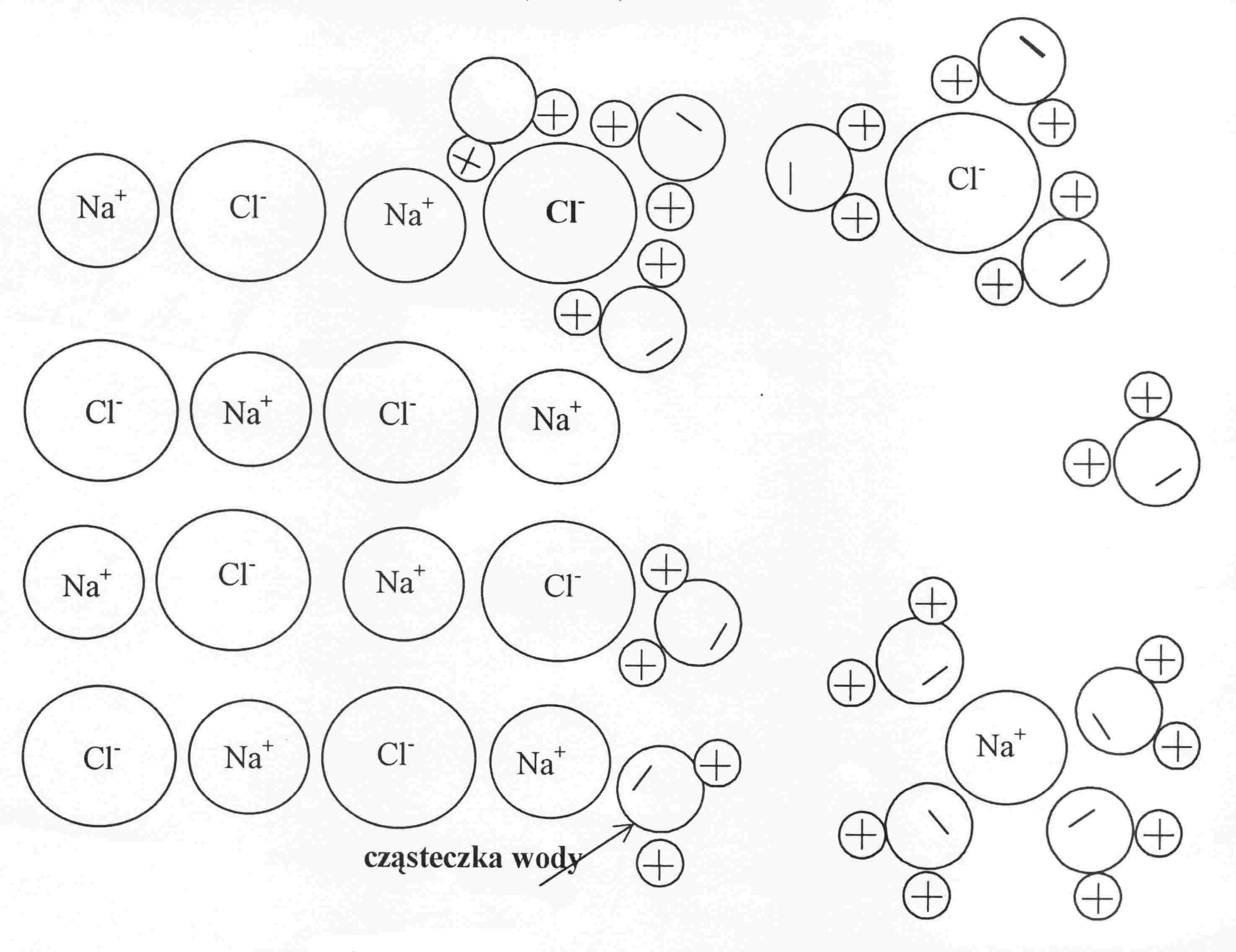
Rys. 2. Schemat rozpuszczania kryształów NaCl w wodzie
Chlorowodór, bromowodór i jodowodór zachowują się w roztworach wodnych jak elektrolity, ponieważ dipolowe cząsteczki wody ułatwiają rozerwanie spolaryzowanych wiązań σ
w cząsteczkach tych związków, co powoduje ich całkowitą dysocjację na jony stabilizowane
w roztworze przez hydratację.
Dysocjacja elektrolitów w roztworach wodnych. Elektrolity mocne i słabe
● Elektrolity zdysocjowane w rozcieńczonych
roztworach wodnych całkowicie są elektrolitami
mocnymi. Do elektrolitów mocnych zaliczamy
praktycznie wszystkie sole oraz niektóre kwasy
i zasady.
● Kwasy lub zasady, tylko częściowo zdysocjowane
na jony w roztworach wodnych, zwłaszcza
w roztworach rozcieńczonych, są elektrolitami
słabymi.
UWAGA: Podział na elektrolity mocne i słabe jest umowny, bowiem dany elektrolit dysocjujący w roztworach wodnych całkowicie, może w roztworach w innym rozpuszczalniku dysocjować tylko częściowo.
Przykłady:
Elektrolity mocne
Sole: NaCl, KI,AgNO3, CuSO4; NaCH3COO, NH4Cl;
NaCl → Na+ + Cl-
AgNO3 → Ag+ + NO3-
Kwasy: HCl, HBr, HI, HClO4, HNO3, H2SO4;
HCl → H+ + Cl-
Stężenie jonów wodorowych w roztworze mocnego kwasu HX jest równe jego stężeniu analitycznemu.
Zasady: NaOH, KOH, Ca(OH)2, Ba(OH)2; czwarto-rzędowe zasady amoniowe (CnH2n+1)4OH.
KOH → K+ + OH-
Stężenie jonów wodorotlenowych w roztworze mocnej zasady MOH jest równe jej stężeniu analitycznemu.
Elektrolity słabe
Kwasy: HNO2, HCN, H2S, HClO, kwasy karboksylowe - HCOOH i CH3COOH;
Zasady: NH4OH, Be(OH)2, Fe(OH)2, aminy - metyloamina CH3NH2, anilina C6H5NH2, pirydyna C5H5N.
HClO ↔ H+ + ClO-
NH4OH ↔ NH4+ + OH-
UWAGA:
● Równania stechiometryczne dysocjacji z pojedynczą strzałką (→) dotyczą elektrolitów mocnych. Równania ze strzałką podwójną (↔) odnoszą się do elektrolitów słabych.
● Równania stechiometryczne dysocjacji kwasów są uproszczone, ponieważ nie uwzględniają hydratacji jonów wodorowych. Taki zapis równań stosuje się jednak powszechnie, pomimo, że w rzeczywistości jony wodorowe w roztworach wodnych występują jako jony hydroniowe H3O+.
AUTODYSOCJACJA WODY i RÓWNOWAGI
w ROZTWORACH ELEKTROLITÓW SŁABYCH w UJĘCIU PRAWA DZIAŁANIA MAS
Autodysocjacja wody
Woda jest nieelektrolitem i w stanie czystym nie przewodzi prądu elektrycznego, ponieważ proces jej dysocjacji na jony (autodysocjacja) zachodzi tylko
w znikomym stopniu:
H2O ↔ H+ + OH- (1)
Uwzględniając hydratację jonów wodorowych, proces autodysocjacji możemy zapisać następująco:
2H2O ↔ H3O+ + OH- (2)
W czystej wodzie stężenia jonów wodorowych
i wodorotlenowych są sobie równe i znikomo małe, natomiast stężenie jej cząsteczek jest stałe.
Dlatego stan równowagi w reakcji dysocjacji wody zgodnie z równaniem (1) możemy na podstawie prawa działania mas scharakteryzować za pomocą stałej równowagi, Kw, zwanej iloczynem jonowym wody.
(3)
gdzie co = 1 mol/dm3 lub kmol/m3 jest tkz. stężeniem standardowym - stosowanym po to, aby iloczyn jonowy oraz inne stałe równowag, definiowane za pomocą równowagowych stężeń produktów
i substratów, były wielkościami bezwymiarowymi. Iloczyn jonowy dla czystej wody zależy tylko od temperatury. W temperaturze 25oC:
Kw = [H+][OH-]/(co)2 = 1,0·10-14, (4)
zatem
[H+] = [OH-] = 1,0·10-7 mol/dm3. (5)
Czysta woda ma odczyn obojętny.
Podobnie jak w czystej wodzie, również
w obojętnych roztworach wodnych stężenia jonów wodorowych i wodorotlenowych są sobie równe,
[H+] = [OH-]. Gdy wodne roztwory są kwaśne, to [H+] >[OH-], natomiast w roztworach zasadowych [H+] < [OH-].
W praktyce posługujemy się wartością ujemnego logarytmu iloczynu jonowego wody:
pKw ≡ - log Kw (5)
Z równań (1) i (5) wynika następująca zależność:
pKw = pcH + pcOH, (6)
gdzie pcH i pcOH są zdefiniowane wzorami
pcH = - log [H+]/co (7)
pcOH = - log [OH-]/co (8)
W roztworach wodnych wartość Kw zależy nie tylko od temperatury. Istotny jest również wpływ siły jonowej roztworu, która jest funkcją stężeń obecnych w nim elektrolitów.
Do określenia odczynu roztworu wodnego wystarczy znać jego pcH. W roztworach obojętnych pcH = 7, w kwaśnych pcH < 7 i w zasadowych
pcH > 7.
Uwaga: W odróżnieniu od pH, wyznaczanego przez pomiar pH-metryczny, wartość pcH można bardzo łatwo obliczyć. W rozcieńczonych roztworach elektrolitów pcH ≈ pH.
Dysocjacja elektrolitów słabych w roztworach wodnych
W roztworach słabych elektrolitów odpowiednie jony i niezdysocjowane cząsteczki są w stanie równowagi, którego położenie zgodnie z prawem działania mas zależy od ich równowagowych stężeń. |
Słabe kwasy lub słabe zasady mają różną zdolność do dysocjacji. Zdolność tę, określaną jako moc słabych elektrolitów, możemy ocenić i porównywać ilościowo stosując jako kryterium wartości stałych dysocjacji kwasów - Ka i zasad - Kb. Im mniejsza stała dysocjacji, tym słabszym elektrolitem jest dany kwas lub dana zasada. |
Dysocjacja słabego kwasu HA
HA ↔ H+ + A- (9)
Stężeniowa stała dysocjacji kwasu dana jest wzorem:
(10)
gdzie [H+], [A-] i [HA] są równowagowymi stężeniami jonów i cząsteczek, a co stężeniem standardowym.
Definicja stopnia dysocjacji słabego kwasu HA:
(11)
Bilans stężeń w stanie równowagi:
(12)
(13)
(14)
Gdy cHA >> [H+], to równanie (14) upraszcza się do postaci:
(15)
Równania (14) lub (15) pozwalają obliczyć stężenie jonów wodorowych w roztworze słabego kwasu
o znanym stężeniu analitycznym cHA. Stopień dysocjacji kwasu można obliczyć z definicji (11). Stopień dysocjacji słabego kwasu można również obliczyć z korzystając prawa rozcieńczeń Ostwalda:
(16)
Z równania (16) wynika, że stopień dysocjacji słabego kwasu wzrasta, gdy jego stężenie
w roztworze maleje.
Tabela 2. Stopień dysocjacji w roztworach wodnych kwasu octowego w 18o C, Ka = 1,83·10-5
Stężenie kwasu, mol/dm3 |
Stopień dysocjacji, % |
0,2 0,1 0,01 0,005 0,001 |
0,95 1,36 4,19 5,85 12,6 |
Gdy α << 1, wówczas równanie (16) upraszcza się do postaci:
(17)
Stężeniowa stała dysocjacji słabego kwasu zależy od temperatury i jest jego cechą charakterystyczną.
Na wartości stężeniowych stałych dysocjacji kwasów wpływa również siła jonowa roztworu. Stężeniowe stałe dysocjacji kwasów są podawane w tablicach jako wartości pKa:
pKa = - log Ka, (18)
wyznaczone w temperaturze 25oC przy ustalonej sile jonowej.
Dysocjacja słabej zasady BOH
BOH ↔ B+ + OH-
Stężeniową stałą dysocjacji zasady definiuje wzór:
(19)
gdzie [B+], [OH-] i [BOH] są równowagowymi stężeniami jonów i cząsteczek.
Definicja stopnia dysocjacji słabej zasady BOH:
(20)
Bilans stężeń w stanie równowagi:
(21)
(22)
(23)
Gdy cBOH >> [OH-], to równanie (23) upraszcza się do postaci:
(24)
Równania (23) i (24) pozwalają obliczyć stężenie jonów hydroksylowych w roztworze słabej zasady
o znanym stężeniu analitycznym cBOH.
Stopień dysocjacji zasady można obliczyć z definicji (20) lub prawa rozcieńczeń Oswalda:
(25)
Z równania (25) wynika, że stopień dysocjacji słabej zasady wzrasta, gdy je stężenie w roztworze maleje. Gdy α << 1, wówczas równanie (25) upraszcza się do postaci:
(26)
Stężeniowa stała dysocjacji słabej zasady zależy od temperatury i jest jej cechą charakterystyczną. Dane z tabeli 3 dotyczą dysocjacji amoniaku (NH4OH) w wodzie.
Tabela 3. Wpływ temperatury na stałe dysocjacji amoniaku
Temperatura, oC |
Kb |
0 10 25 40 |
1,4∙10-5 1,6∙10-5 1,8∙10-5 2,0∙10-5 |
Na wartości stężeniowych stałych dysocjacji zasad wpływa również wielkość I zwana siłą jonową roztworu. W tablicach stężeniowe stałe dysocjacji zasad są podawane jako wartości pKb dla 25oC
i ustalonej siły jonowej.
pKb = - log Kb (27)
Dysocjacja słabych kwasów i zasad
w obecności elektrolitów mocnych o wspólnym jonie
Słabe elektrolity często występują w roztworach zawierających elektrolity mocne dysponujące identycznym (wspólnym) jonem: słaby kwas HA
z mocnym kwasem, np. HCl, lub z własną solą, np. NaA; słaba zasada BOH z mocną zasadą, np. KOH, lub z własną sólą, np. BCl. Obecność elektrolitu mocnego o wspólnym jonie cofa dysocjację słabego elektrolitu, w wyniku czego jego stopień dysocjacji znacząco maleje.
Słaby kwas HA w obecności mocnego kwasu HX
HA ↔ H+ + A-
HX → H+ + X-
Bilans stężeń w stanie równowagi:
[H+] = cHX + [H+]HA
[A-] = [H+]HA
[HA] = cHA - [H+]HA
(28)
Gdy cHX i cHA >> [H+]HA, to:
(29)
(30)
(31)
Przykład: Porównać stopień dysocjacji 0,20 M kwasu octowego ze stopniem dysocjacji w obecności 0,050 M HCl.
pKa = 4,55, cHA= 0,20 mol/dm3, cHCl = 0,050 mol/dm3
a)
CH3COOH ↔ H+ + CH3COO-
Przyjęte założenie jest nieźle spełnione:
b)
CH3COOH ↔ H+ + CH3COO-
HCl → H+ + Cl-
Przyjęte założenia są bardzo dobrze spełnione:
.
Wniosek: W obecności 0,050 M HCl stopień dysocjacji 0,20 M HCl zmalał 21 razy.
Słaba zasada BOH w obecności mocnej zasady MOH
BOH ↔ B+ + OH-
MOH → M+ + OH-
Bilans stężeń w stanie równowagi:
[OH-] = cMOH + [OH-]BOH
[B+] = [OH-]BOH
[BOH] = cBOH - [OH-]BOH
(32)
Gdy cMOH i cBOH >> [OH-]BOH, to:
(33)
(34)
(35)
Roztwory buforowe
Roztwór zawierający słaby elektrolit oraz pochodzącą od niego sól nazywamy roztworem buforowym. pcH roztworu buforowego jako funkcja stosunku stężeń słabego elektrolitu i soli pozostaje praktycznie stałe w trakcie rozcieńczania roztworu. Ponadto, pcH roztworu buforowego zmienia się stosunkowo nieznacznie po dodaniu niewielkich liczności mocnych kwasów lub mocnych zasad, co nazywamy efektem buforowania.
STĘŻONE ROZTWORY WODNE ELEKTROLITÓW MOCNYCH
Elektrolity mocne w rozcieńczonych roztworach wodnych są całkowicie zdysocjowane, a ich jony są hydratowane przez pewną liczbę cząsteczek wody (tkz. liczba hydratacyjna). Hydratacja ma dwojaki charakter:
• oddziaływania chemiczne między cząsteczek wody
a cząsteczkami lub jonami występującymi
w roztworze prowadzą do utworzenia między nimi wiązań chemicznych, koordynacyjnych lub wodorowych;
• oddziaływania elektrostatyczne jon - dipol prowadzą do zorientowanego uporządkowania bliskiego zasięgu pewnej liczby cząsteczek wody wokół jonów występujących w roztworach. Kationy metali w roztworach wodnych występują w postaci akwakompleksów, [M(H2O)m]n+, które są również hydratowane elektrostatycznie.
Rys. 3. Mechanizm hydratacji kationów i anionów
Ze wzrostem stężenia elektrolitów w roztworach wodnych może się okazać, że liczba dostępnych cząsteczek wody nie wystarcza do pełnej hydratacji wszystkich jonów w roztworze. Ponadto, ze wzrostem stężenia elektrolitów maleją średnie odległości między jonami, a silne oddziaływania elektrostatyczne między jonami o przeciwnych znakach prowadzą do powstania par jonowych kation - anion. Przy bardzo dużych stężeniach elektrolitów w roztworze powstają nie tylko pary jonowe kation - anion, ale również trójki jonowe typu anion - kation - anion lub kation - anion - kation, a także agregaty typu czwórek jonowych.
Rys. 4. Typy par jonowych: a) całkowicie hydratowana para jonowa, kation i anion są rozdzielone przez cząsteczki wody; b) para jonowa o wspólnych cząsteczkach wody;
c) kontaktowa para jonowa.
SIŁA JONOWA. AKTYWNOŚĆ
i WSPÓŁCZYNNIKI AKTYWNOŚCI JONÓW
Występowanie trwałych agregatów jonowych typu par, trójek lub czwórek jonowych w stężonych roztworach elektrolitów powoduje, że obserwowane przewodnictwo elektryczne tych roztworów jest mniejsze od oczekiwanego na podstawie analitycznych stężeń występujących w nich jonów. Przyczyna tego zjawiska tkwi w tym, że wypadkowy ładunek elektryczny poszczególnych agregatów zależy od liczby i ładunku jonów wchodzących w ich skład. Rozpatrzmy to na przykładzie nasyconego roztworu chlorku sodu, którego stężenie w 20oC jest równe 26,5%. W roztworze tym występują zarówno swobodne, hydratowane kationy Na+ i aniony Cl-, jak i pary jonowe [Na(H2O)m]+[Cl(H2O)n]-, których wypadkowy ładunek równy jest 0. Z tego powodu pary te nie uczestniczą w procesie przewodzenia prądu elektrycznego.
Tworzenie trwałych agregatów jonowych jest przyczyną, że efektywne stężenie zwane aktywnością ai jonów i-tego rodzaju, jako nośników ładunków elektrycznych w roztworze jest mniejsze od ich rzeczywistego stężenia analitycznego ci.
Aktywność jonów jest zdefiniowana wzorem:
ai ≡ yici (36)
Bezwymiarowy współczynnik proporcjonalności yi jest współczynnikiem aktywności i-tego jonu
w roztworze. Współczynnik ten zawiera się
w granicach 0 < yi ≤ 1.
Współczynniki aktywności jonów i-tego rodzaju w roztworze zależą od stężeń i ładunków wszystkich jonów, przy czym zależność ma tym bardziej skomplikowany charakter im większe jest stężenie wszystkich elektrolitów. Z tego powodu wygodnie jest posługiwać się funkcją zwaną siłą jonową roztworu, I , zdefiniowaną wzorem:
(37)
gdzie ci jest stężeniem, a zi ładunkiem i-tego jonu
w roztworze. Gdy I → 0, to współczynnik aktywności yi → 1.
Przykład
Roztwór zawiera 0,25 M Na2SO4 0,25 M NaCl, oraz 0,50 M KNO3. Obliczyć siłę jonową tego roztworu.
Na2SO4 → 2Na+ + SO42-
NaCl → Na+ + Cl-
KNO3 → K+ + NO3-
[Na+] = 2∙0,25 + 0,25 = 0,75 mol/dm3
[K+] = 0,50 mol/dm3
[SO42-] = 0,25 mol/dm3
[Cl-] = 0,25 mol/dm3
[NO3-] = 0,50 mol/dm3
I = ½(0,75∙12 + 0,50∙12 + 0,25∙22 + 0,25∙12 + 0,50∙12) = 1,50 mol/dm3
Współczynniki aktywności jonów w roztworach rozcieńczonych o sile jonowej I≤ 0,01 można obliczyć z dokładnością do 1% korzystając z granicznego równania Debye'a - Hückela:
, (38)
gdzie współczynnik A zależy od temperatury
i rodzaju rozpuszczalnika. W temperaturze 25oC
w roztworach wodnych A = 0,509.
Przykład: W roztworze 0,010 M kwasu solnego siła jonowa I = 0,010 mol/dm3 . Stężenie jonów wodorowych jest równe 0,010 mol/dm3, z = 1. Obliczony ze wzoru (38) współczynnik aktywności yH+ = 0,889.
Dla siły jonowej roztworu I < 0,1 do obliczeń stosuje się rozszerzone równanie Debye'a - Hückela:
, (39)
gdzie B jest stałą empiryczną zależną od rodzaju rozpuszczalnika, natomiast parametr a jest efektywnym promieniem solwatowanego jonu
w roztworze. Wartości iloczynu B·a dla różnych jonów roztworach wodnych podano w tabeli 4.
W celu obliczenia współczynników aktywności przy sile jonowej I < 0,5, do równania (39) wprowadzono dodatkową poprawkę empiryczną:
(40)
Tabela 4. Iloczyn B·a dla rożnych jonów w roztworach wodnych
Jon |
Iloczyn B·a |
H+, Al3+, Fe3+, Cr3+ |
3,0 |
Be2+, Mg2+ |
2,6 |
Li+, Ca2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+ |
2,0 |
Sr2+, Ba2+, Cd2+, Hg2+, S2-, CH3COO- |
1,6 |
Na+, Pb2+, CO32-, SO42- |
1,3 |
K+, NH4+, Ag+, OH-, F-, Cl-, Br-, I-, NO3-, ClO4- |
1,0 |
Obecnie, korzystając z definicji aktywności jonów wodorowych i wodorotlenowych na podstawie wzoru (36), możemy wyjaśnić na czym polega różnica między pcH a pH i pcOH a pOH roztworu:
(41)
(42)
(43)
(44)
DYSOCJACJA KWASÓW WIELOPROTONOWYCH
Do kwasów wieloprotonowych, HmX, zwanych także kwasami wielozasadowymi, należą kwasy nieorganiczne, np. H2SO4 i H3PO4, oraz kwasy organiczne, np. kwas szczawiowy - H2C2O4. Kwasy te mają różną moc, a ich znamienną cechą jest etapowy charakter dysocjacji elektrolitycznej.
Przykłady:
Kwas siarkowy(VI) - H2SO4, mocny kwas dwuzasadowy
H2SO4 → H+ + HSO4-, pierwszy etap
HSO4- ↔ H+ + SO42-, drugi etap
Anion HSO4- jest kwasem średniej mocy, dlatego w drugim etapie dysocjacji H2SO4 ustala się stan równowagi ze stałą dysocjacji Ka2:
(45)
Bilans stężeń w stanie równowagi:
ck - analityczne stężenie kwasu siarkowego(VI)
[H+]1 = ck
[H+]2 = [SO42-]
[H+] = [H+]1 + [H+]2 = ck + [H+]2
[HSO4-] = ck - [H+]2
Aby w roztworze H2SO4 obliczyć całkowite stężenie jonów wodorowych, należy najpierw obliczyć z wyrażenia na Ka2 stężenie jonów wodorowych [H+]2 z drugiego etapu dysocjacji:
(46)
Po odpowiednim przekształceniu wyrażenia (46) uzyskuje się równanie kwadratowe zupełne względem [H+]2.
Kwas fosforowy(V) - H3PO4, trójzasadowy kwas średniej mocy, który dysocjuje w trzech etapach:
H3PO4 ↔ H+ + H2PO4- , Ka1
H2PO4- ↔ H+ + HPO42-, Ka2
HPO42- ↔ H+ + PO43-, Ka3
Ka1 >>> Ka2 >>> Ka3, ponieważ aniony H2PO4- i HPO42- są bardzo słabymi kwasami w porównaniu z cząsteczkami H3PO4. Zatem, o stężeniu jonów wodorowych w roztworze H3PO4 decyduje pierwszy etap dysocjacji.
SUPERKWASY
Stężenie i aktywność jonów wodorowych oraz skala pH są miarodajne tylko dla rozcieńczonych wodnych roztworów kwasów. W przypadku dużych stężeń kwasów w roztworach wodnych i niewodnych stosuje się powszechnie funkcję kwasowości Hammeta, H0, zdefiniowaną w oparciu o zachowanie się jednego lub kilku indykatorów (wskaźników) zasadowych B, ulegających w stężonym roztworze kwasu reakcji protonowania, czyli przyłączenia jonu wodorowego:
B + H+ ↔ BH+
Stała równowagi reakcji odwrotnej dana jest wzorem:
(47)
Funkcję kwasowości Hammeta definiuje zależność:
(48)
Jeśli wskaźnik w roztworze kwasu jest sprotonowany w 50%, to:
(49)
i
(50)
W rozcieńczonych roztworach wodnych H0 = pH.
Wartości funkcji H0 dla niektórych ciekłych kwasów w stanie czystym są podane w tabeli 5.
Tabela 5. Funkcja kwasowości H0 dla wybranych kwasów
Kwas |
- H0 |
HSO3F H2S2O7 CF3SO3H H2SO4 100% HF H3PO4 HC(O)OH |
15 15 13,8 11,9 15,1 5 2,2 |
Kwasy z ujemną wartością H0 > 6 są bardzo mocne, dlatego nadano im nazwę superkwasów.
WSPÓŁCZESNE TEORIE KWASÓW i ZASAD
Historycznie najstarsza koncepcja kwasów
i zasad to teoria Arrheniusa, sformułowana w celu wyjaśnienia ich właściwości w roztworach wodnych. Nowsze teorie biorą pod uwagę obok wody inne rozpuszczalniki lub stosują inne podejście
w pojmowaniu kwasów i zasad.
Teoria rozpuszczalnikowa
Punkt wyjścia tej teorii jest autodysocjacja rozpuszczalnika S według ogólnego schematu:
2S ↔ S1+ + S2- (51)
Kwasem w rozpuszczalniku S jest związek odszczepiający ten sam kation S1+, natomiast związek odszczepiający ten sam anion S2- jest zasadą.
Zgodnie z tą teorią, reakcja zobojętniania kwasu zasadą prowadzi zawsze do powstania soli
i rozpuszczalnika.
Przykłady
Woda:
2H2O ↔ H3O+ + OH-
H3O+Cl- + NaOH = NaCl + 2H2O
Ciekły amoniak:
2NH3 ↔ NH4+ + NH2-
NH4Cl + NaNH2 = NaCl + 2NH3
Ciekły ditlenek siarki:
2SO2 ↔ SO2+ + SO32-
SOCl2 + K2SO3 = 2KCl + 2SO2
Teoria Brønsteda - Löwry'ego
Teoria Brønsteda - Löwry'ego zakłada, że kwasami są donory protonów, a zasadami akceptory protonów. Zgodnie z tym założeniem, kwasami lub zasadami mogą być kationy, aniony lub elektroobojętne cząsteczki.
Tabela 6. Kwasy i zasady w teorii Brønsteda - Löwry'ego
Typ kwasu lub zasady |
Kwas |
Zasada |
Cząsteczkowy |
HClO4 H2SO4 CH3COOH |
NH3 C6H5NH2 H2N-NH2 * |
Kationowy |
H3O+ NH4+ H2F+ |
H2N-NH3+ * |
Anionowy |
HSO4- HS- H2PO4- |
OH- ClO4- Cl- |
*Hydrazyna, H2N-NH2, może przyłączyć kolejno dwa protony. Kation
H2N-NH3+ może przyłączyć drugi proton, dlatego jest zasadą.
Kwas oddając proton przechodzi w sprzężoną
z nim zasadę, natomiast zasada przyjmując proton przechodzi w sprzężony z nią kwas. Ogólne równanie ma postać:
kwas ↔ zasada + proton (52)
Drugie, kluczowe założenie tej teorii głosi, że produktem oddziaływań kwasowo-zasadowych nie jest sól, lecz nowa para kwas - zasada:
kwas1 + zasada2 ↔ kwas2 + zasada1 (53)
Przykłady:
Kwas1 Zasada2 Zasada1 Kwas2
HCl + H2O ↔ Cl- + H3O+
NH4+ + H2O ↔ NH3 + H3O+
H2O + CN- ↔ OH- + HCN
HSO4- + OH- ↔ SO42- + H2O
Teoria Lewisa
Według założeń teorii Lewisa akceptory par elektronowych są kwasami, a donory tych par są zasadami. Dzięki temu do kategorii kwasów można zaliczyć niektóre związki aprotonowe, np. AlCl3
i SbCl5, oraz kationy metali przejściowych tworzących z donorami par elektronowych (ligandami) związki kompleksowe. Donorowy atom zasady Lewisa ma wolną parę elektronową, dzięki czemu może utworzyć trwałe wiązanie koordynacyjne z akceptorowym atomem kwasu Lewisa.
Przykładowo, trójtlenek siarki SO3 jest kwasem Lewisa, ponieważ atom siarki w tym związku ma cztery hybrydyzowane orbitale sp3 obsadzone przez sześć elektronów. Produktem reakcji SO3 z wodą jako zasadą Lewisa jest kwas siarkowy(VI). Anion siarczanowy(VI) ma strukturę tetraedryczną, a atom siarki uzyskuje w powłoce walencyjnej trwałą, oktetową konfigurację elektronową.
Czysty, bezwodny trójchlorek glinu, AlCl3, jest katalizatorem w syntezie organicznej, ponieważ jako kwas Lewisa reaguje z organicznymi donorami par elektronowych, np. pirydyną.
Teoria twardych i miękkich kwasów i zasad
Koncepcja twardych i miękkich kwasów i zasad, wprowadzona przez Pearsona, jest jakościowym rozwinięciem teorii Lewisa. Według Pearsona do twardych kwasów należą trudno polaryzowalne akceptory par elektronowych, a łatwo polaryzowalne akceptory są miękkimi kwasami. Twardymi zasadami są trudno polaryzowalne donory par elektronowych, zaś łatwo polaryzowalne donory należą do miękkich zasad. Czysto formalną cechą, na podstawie której wprowadzono taki podział, jest fakt, że trwałe produkty powstają w reakcjach twardych kwasów z twardymi zasadami lub miękkich kwasów z miękkimi zasadami. Kwasy
i zasady Lewisa, którym nie udało się jednoznacznie przypisać cech twardości lub miękkości, zaliczono do grupy o charakterze pośrednim.
Tabela 7. Twarde i miękkie kwasy i zasady
Kwasy |
Twarde: H+, Li+, Na+, K+, Be2+, Mg2+, Ca2+, Sr2+, Al3+, Mn2+, Cr3+, Fe3+, Co3+, Cr(VI), Mn(VII), BF3, BCl3, AlH3, AlCl3, SO3, CO2 |
Pośrednie: Sn2+, Pb2+, Sb3+, Bi3+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+, Ru2+, Rh3+, Ir3+, NO+, R3C+, SO2, GaH3 |
Miękkie: HO+, Cs+, Cu+, Ag+, Au+, Tl+, Hg2+, Pt2+, Cd2+, Au3+, Br2, I2, BH3, GaCl3, GaBr3 |
Zasady |
Twarde: F-, Cl-, OH-, O2-, CH3COO-, CO32-, NO3-, SO42-, PO43-, ClO4-, H2O, ROH, R2O, NH3, N2H4, RNH2 |
Pośrednie: NO2-, SO32-, Br- |
Miękkie: H-, I-, CN-, SCN-, H-, R2S, R3P, R3As, CO, RNC, alkeny, związki aromatyczne |
WŁAŚCIWOŚCI ROZTWORÓW
Równowagi fazowe. Reguła faz Gibbsa. Wykresy fazowe dla czystego rozpuszczalnika i roztworu
W zależności od warunków zewnętrznych (ciśnienie, temperatura), czysty związek chemiczny może występować w trzech stanach skupienia: stałym, ciekłym i gazowym. Należy również pamiętać, że dany związek w stanie stałym może mieć kilka różnych odmian polimorficznych. Każdy
z tych stanów jest odrębną fazą. Liczbę faz f występujących równocześnie w zależności od liczby składników n w układzie oraz liczby stopni swobody s w zakresie zmian parametrów niezależnych, tj. ciśnienia, temperatury oraz stężeń składników, określa reguła faz Gibbsa: n + 2 = f + s
Rozpatrzmy jako układ jednoskładnikowy czystą wodę, n = 1, w którym występują dwie fazy, f = 2. Zgodnie z regułą Gibbsa mamy s = n + 2 - f = 1, czyli jeden stopień swobody. Możemy zatem zmieniać temperaturę lub ciśnienie w takim zakresie, aby przy stałej wartości drugiego parametru, nie wykroczyć poza obszar istnienia którejkolwiek
z obydwu faz występujących w układzie
i pozostających w stanie wzajemnej równowagi. Pod stałym ciśnieniem woda w temperaturze niższej od temperatury wrzenia jest w równowadze z własną parą nasyconą, a w temperaturze krzepnięcia jest w równowadze z lodem. Z kolei lód, przy ustalonym ciśnieniu i w temperaturze niższej od temperatury topnienia pozostaje w równowadze z parą nasyconą. Wszystkie trzy fazy: lód, woda i para nasycona, istnieją we wzajemnej równowadze przy ściśle określonym ciśnieniu i temperaturze, które na wykresie fazowym wody (rys. 5) wyznaczają tzw. punkt potrójny. W punkcie tym przecinają się linie równowag dwufazowych: woda - para nasycona (parowanie - skraplanie), woda - lód (krzepnięcie - topnienie) oraz lód - para nasycona (sublimacja - kondensacja).
Wodny roztwór związku chemicznego jest układem dwuskładnikowym. Gdy stężenie roztworu jest stałe, to parametrami niezależnymi są ciśnienie
i temperatura, a odpowiedni wykres fazowy (rys. 6) jest podobny do wykresu fazowego wody.
Rys. 5. Wykres fazowy wody
po = 101, 3 kPa, Tt - temp. topnienia, Tw - temp. wrzenia,
T3 - temp. w punkcie potrójnym
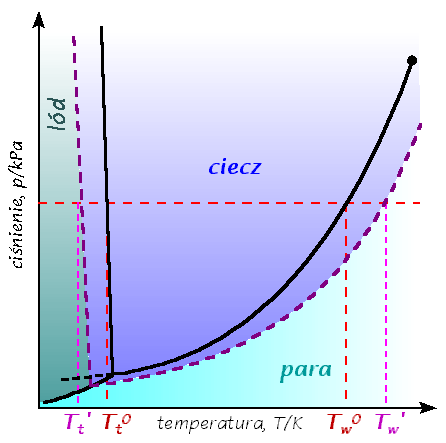
Rys. 6. Wykres fazowy roztworu wodnego (linie przerywane) na tle wykresu fazowego wody (linie ciągłe)
Dyfuzja, osmoza, ciśnienie osmotyczne
Dyfuzja jest to zdolność cząsteczek lub jonów substancji rozpuszczonej do samorzutnego przemieszczania się w roztworze aż do wyrównania jej stężenia w całej objętości roztworu. Zdolność tę wykazują również cząsteczki rozpuszczalnika. Dyfuzja cząsteczek w cieczach zachodzi znacznie wolniej niż w gazach.
Jeśli roztwór jakiejś substancji i czysty rozpuszczalnik są przedzielone porowatą membraną, nieprzenikliwą dla cząsteczek substancji rozpuszczonej i przepuszczalną dla rozpuszczalnika, to w układzie tym zachodzi jednostronna dyfuzja rozpuszczalnika przez membranę czyli osmoza. Cząsteczki rozpuszczalnika dyfundujące przez membranę do roztworu powodują jego rozcieńczenie.
Właściwości półprzepuszczalnych membran mają błony komórkowe organizmów żywych, celofan, trioctan celulozy, itp. Dawniej dla celów praktycznych stosowano błony pochodzenia naturalnego, obecnie na dużą skalę stosuje się membrany syntetyczne z tworzyw sztucznych.
Ciśnienie osmotyczne roztworu, π, jest różnicą ciśnień wywieranych równocześnie na membranę półprzepuszczalną przez roztwór i czysty rozpuszczalnik.
Ciśnienie to jest wprost proporcjonalne do temperatury bezwzględnej T i stężenia molowego c substancji rozpuszczonej:
(54)
gdzie R = 8,314 J·mol-1·K-1 jest uniwersalną stałą gazową, natomiast współczynnik i jest zależy od rodzaju substancji rozpuszczonej:
i = 1 dla nieelektrolitów,
i > 1 dla elektrolitów.
Gdy elektrolit mocny dysocjuje na k jonów, wówczas i = k., np. przypadku soli: NaCl, MgCl2 i Fe(NO3)3, wartości i są równe odpowiednio 2, 3, 4. Dla elektrolitu słabego wartości i muszą być skorygowane o stopień jego dysocjacji α:
i = 1 + (k - 1)α (55)
gdzie k jest liczbą jonów powstałych w wyniku dysocjacji, np. dla kwasu octowego k = 2.
Odwrócona osmoza polega na zmianie kierunku dyfuzji cząsteczek rozpuszczalnika od roztworu bardziej stężonego do roztworu mniej stężonego. Proces ten zachodzi wtedy, gdy ciśnienie wywierane na membranę po stronie rozpuszczalnika lub roztworu mniej stężonego jest większe od ciśnienia osmotycznego w układzie. Odwróconą osmozę przez syntetyczne membrany polimerowe stosuje się obecnie do uzdatniania wody, odsalania wody morskiej oraz oczyszczania i zatężania ścieków przemysłowych.
Efekty ebulioskopowe i krioskopowe
Roztwory mają wyższe temperatury wrzenia (efekt ebulioskopowy) oraz niższe temperatury krzepnięcia (efekt krioskopowy) niż czysty rozpuszczalnik (rys. 6). Obydwa efekty zależą od właściwości rozpuszczalnika oraz od rodzaju
i stężenia substancji rozpuszczonej.
Podwyższenie temperatury wrzenia (Δtw) oraz obniżenie temperatury krzepnięcia (Δtk) roztworu są określone wzorami:
(56)
(57)
Współczynniki E i K są odpowiednio stałą ebulioskopową i stałą krioskopową rozpuszczalnika, parametr i ma takie samo znaczenie jak w równaniu (54), zaś m jest stężeniem molalnym substancji rozpuszczonej.
Uwaga: Stężenie molalne (mol/kg) jest wyrażone stosunkiem liczności substancji rozpuszczonej i masy czystego rozpuszczalnika w kg.
Tabela 8. Stałe krioskopowe i ebulioskowe kilku rozpuszczalników
Rozpuszczalnik |
K, oC |
E, oC |
Woda Benzen Chloroform Tetrachlorometan |
1,86 1,79 4,90 29,8 |
0,513 1,83 3,802 5,3 |
STOPIONE SOLE
Elektrolity mocne, które stanie stałym mają jonową sieć krystaliczną (np. sole i wodorotlenki litowców), po stopieniu przewodzą prąd elektryczny, ponieważ w uzyskanym stopie są zdysocjowane na jony. Stopione sole wykazują szereg specyficznych właściwości. Mianowicie, w szerokim zakresie temperatur (nawet do 1200oC) cechuje je niska prężność pary, duża trwałość termiczna, niska prężność par, mała lepkość, dobre przewodnictwo cieplne i elektryczne oraz znaczna szybkość transportu masy, obserwowana w reakcjach chemicznych zachodzących w ich środowisku.
Temperatury topnienia soli nieorganicznych są zazwyczaj wysokie, dlatego w praktyce stosuje się dwu- lub trójskładnikowe mieszaniny różnych soli, które przy ściśle określonym składzie (tzw. mieszaniny eutektyczne lub eutektyki, tabela 9) mają minimum temperatury topnienia, znacznie poniżej temperatur topnienia czystych składników.
Tabela 9. Skład i temperatury topnienia niektórych mieszanin eutektycznych
Skład mieszaniny, % mol. |
Temperatura eutektyczna, oC |
44% LiNO3 - 56% KNO3 39% NaOH - 61% KOH 58% LiCl - 42% KCl 11% LiCl - 18% NaCl - 71% KCl 33% KCl - 67% MgCl2 46,5% LiF - 11,5% NaF -42% KF 50% NaCl - 50% KCl |
125 173 361 357 426 454 658 |
Stopione sole, np. Na2CO3, KHSO4, K2S2O7, znajdują zastosowanie jako topniki w chemii analitycznej materiałów mineralnych. Krzemionka (SiO2) reaguje ilościowo ze stopionym Na2CO3 dając rozpuszczalne w wodzie krzemiany. Nierozpuszczalne w wodzie tlenki, np. Al2O3 Fe2O3, ulegają w stopionym K2S2O7 ilościowej przemianie w łatwo rozpuszczalne siarczany(VI).
SiO2 + Na2CO3 = Na2SiO3 + CO2↑
Fe2O3 + 3K2S2O7 = Fe2(SO4)3 + K2SO4
Elektroliza stopionych soli, najczęściej fluorków lub chlorków, litowców oraz berylowców jest metodą otrzymywania litowców oraz berylowców w stanie czystym. Do produkcji aluminium na dużą skalę przemysłową stosuje się elektrolizę mieszaniny 15-20% Al2O3 i stopionego kriolitu, Na3AlF6. Elektroliza stopionych soli zawierających związki trudno topliwych metali rzadkich jest metodą otrzymywania tych metali w stanie czystym, np. niob można otrzymać przez elektrolizę mieszaniny NaF-KF-K2NbF7.
Ostatnio furorę robią tzw. ciecze jonowe, które są ciekłymi solami organicznymi. Sole te są złożone
z dużego kationu organicznego i anionu, zazwyczaj anionu kompleksowego, np. heksafluorofosforan(V) 1-butylo-3-metylo-imidazoliowy.
{kind=link}
CIECZE NADKRYTYCZNE
Cieczami nadkrytycznymi są płynne gazy (super critical fluids), których temperatura i ciśnienie są większe od wartości krytycznych. Reaktywne gazy są traktowane jako nadkrytyczne rozpuszczalniki niewodne. Należy do nich wspomniany wcześniej ciekły amoniak, NH3, który jako rozpuszczalnik polarny jest zdolny do autodysocjacji. Szczególną cechą ciekłego amoniaku jest zdolność do rozpuszczania metali z rodziny litowców. Uzyskane roztwory dobrze przewodzą prąd elektryczny, ponieważ zawierają jednododatnie kationy litowców oraz solwatowane amoniakiem elektrony, nadające roztworom niebieską barwę.
Kolejnym rozpuszczalnikiem nadkrytycznym jest płynny ditlenek węgla, który jako twardy kwas Lewisa ma powinowactwo do substancji zasadowych, między innymi do takich alkaloidów, jak kofeina. Ze względu na tę właściwość znalazł zastosowanie w przemyśle spożywczym do produkcji kawy bezkofeinowej
i rozpuszczalnej.
Skroplony CO2 przechowuje się w butlach gazowych ze stali. Ciekły CO2 wypływając z butli gwałtownie paruje i silnie ochładza, w wyniku czego ulega częściowemu zestaleniu (tzw. suchy lód). Stały CO2 pod ciśnieniem atmosferycznym sublimuje, co wykorzystuje się w gaśnicach śniegowych.
Specyficzne cechy fizyczne ditlenku węgla
w stanie gazowym (łatwość skroplenia), ciekłym
i stałym (sublimacja), położenie punktu potrójnego oraz parametry krytyczne są przedstawione na wykresie fazowym.
Rys. 7. Wykres fazowy ditlenku węgla
Punkt potrójny leży powyżej po = 101,3 kPa.
Tabela 10. Parametry krytyczne niektórych gazów i par
W fizyce ciecze i gazy określa się bardziej ogólnym mianem płynów. Jeśli płyn znajduje się w stanie
o parametrach przekraczających wartości krytyczne, T > Tkr i p > pkr, to jego właściwości są pośrednie między właściwościami fazy ciekłej
i gazowej.
{kind=link}
101 pm
Wyszukiwarka
Podobne podstrony:
1 Rwnowagi w roztworach elektrolitw Kwasy i zasady
Równowaga w roztworach elektrolitów, NAUKA, chemia, lab
13 Równowagi w roztworach elektrolitów słabych
6 pHelek ul, Temat: Równowaga w roztworach elektrolitów
,podstawy chemii nieorganicznej L,Równowagi w roztworach elektrolitów
004elektrolit, ELEKTROLITY, KWASY, ZASADY I SOLE
12 Elektrolity, kwasy, zasady i sole
13 Równowagi w roztworach elektrolitówid 14769 ppt
,podstawy chemii nieorganicznej L,Równowagi w roztworach elektrolitów
więcej podobnych podstron