CHAPTER 7 - CATALYSIS
B: MECHANISMS OF ENZYME CATALYSIS
BIOCHEMISTRY - DR. JAKUBOWSKI
Learning Goals/Objectives for Chapter 7B: After class and this reading, students will be able to
|
We can apply what we learned about catalysis by small molecules to enzyme-catalyzed reactions. To understand the mechanism of an enzyme-catalyzed reaction, we try to alter as many variables, one at a time, and ascertain the effects of the changes on the activity of the enzyme. Kinetic methods can be used to obtain data from which inferences about the mechanism can be made. Obviously, crystal structures of the enzyme in the presence and absence of a competitive inhibitor give abundant information about possible mechanisms. It is amazing, however, how much information about enzyme mechanism can be gained even if all you have is a blender, a stopwatch, an impure enzyme, and a few substrates and inhibiting reagents. Systematically, the kineticist and organic chemist can change:
the substrate - for example, changing the leaving group or acyl sustituents of a hydrolyzable substrate;
the pH or ionic strength - which can give data about general acids/bases in the active site;
the enzyme - by chemical modification of specific amino acids, or through site-specific mutagenesis;
the solvent - as will be discussed in the next chapter section .
We will explore in detail the mechanisms of three enzymes. For carboxypeptidase, we will study possible mechanisms for the cleavage of C-terminal hydrophobic amino acids from a peptide. For lysozyme, we will study the structural features of the enzyme and substrates along with the mechanism for cleavage of glycosidic links in bacterial peptidoglycan cell walls. For chymotrypsin, we will study experiments which vary the substrate, pH, and the enzyme and infer from this information about a mechanism consistent with the experimental data. Kinetic analyses can be used to determine the:
order of binding/dissociation of substrates and products
rate constants for individual steps
and clues to the nature of catalytic groups found in the enzyme.
CARBOXYPEPTIDASE
A peptide substrate binds at the active site of the enzyme. X-ray structures of the enzyme with and without a competitive inhibitor show a large conformational change at the active site when inhibitor or substrate is bound. Without inhibitor, several waters occupy the active site. When inhibitor and presumably substrate are bound, the water leaves (which is entropically favored), and Tyr 248 swings around from near the surface of the protein in the absence of a molecule in the active site to interact with the carboxyl group of the bound molecule, a distance of motion equal to about 1/4 the diameter of the protein. This effectively closes off the active site and expels the water. A Zn2+ ion is present at the active site. It is bound by His 69, His 196, Glu 72, and finally a water molecule as the fourth ligand. A hydrophobic pocket which interacts with the phenolic group of the substrate accounts for the specificity of the protein.
In the catalytic mechanism, Zn2+ helps polarize the labile amide bond, while Glu 270, acting as a general base, which along with Zn2+ helps promote dissociation of a proton from the bound water, making it a better nucleophile. Water attacks the electrophilic carbon of the sessile bond, with Glu 270 acting as a general base catalyst. The tetrahedral intermediate then collapses, expelling the leaving amine group, which picks up a proton from Glu 270, which now acts as a general acid catalyst. People used to believe that Tyr 248 acted as a general acid, but mutagenesis showed that Tyr 248 can be replaced with Phe 248 without significant effect on the rate of the reaction
LYSOZYME
This enzyme, found in cells and secretions of vertebrates but also in viruses which infect bacteria, cleaves peptidoglycan GlcNAc ( 1->4) MurNAc repeat linkages (NAG-NAM) in the cell walls of bacteria and the GlcNAc ( 1->4) GlcNAc (poly-NAG) in chitin, found in the cells walls of certain fungi. Since these polymers are hydrophilic, the active site of the enzyme would be expected to contain a solvent-accessible channel into which the polymer could bind. The crystal structures of lysozyme and complexes of lysozyme and NAG have been solved to high resolution. The inhibitors and substrates form strong H bonds and some hydrophobic interactions with the enzyme cleft. Kinetic studies using (NAG)n polymers show a sharp increase in kcat as n increases from 4 to 5. The kcat for NAG6 and (NAG-NAM)3 are similar. Models studies have shown that for catalysis to occur, (NAG-NAM)3 binds to the active site with each sugar in the chair conformation except the fourth which is distorted to a half chair form, which labilizes the glycosidic link between the 4th and 5th sugars. Additional studies show that if the sugars that fit into the binding site are labeled A-F, then because of the bulky lactyl substituent on the NAM, residues C and E can not be NAM, which suggests that B, D and F must be NAM residues. Cleavage occurs between residues D and E.
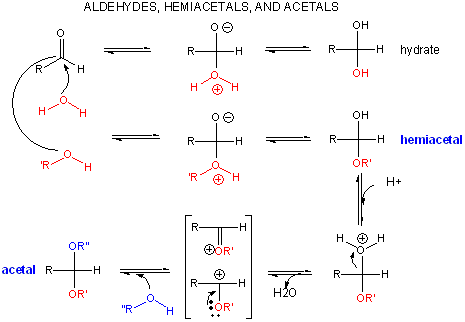
A review of the chemistry of glycosidic bond (an acetal) formation and cleavage shows the acetal cleavage is catalyzed by acids and proceeds by way of an oxonium ion which exists in resonance form as a carbocation.
Catalysis by the enzyme involves Glu 35 and Asp 52 which are in the active site. Asp 52 is surrounded by polar groups but Glu 35 is in a hydrophobic environment. This should increase the apparent pKa of Glu 35, making it less likely to donate a proton and acquire a negative charge at low pH values, making it a better general acid at higher pH values. The general mechanism appears to involve:
binding of a hexasaccharide unit of the peptidoglycan with concomitant distortion of the D NAM.
protonation of the sessile acetal O by the general acid Glu 35 (with the elevated pKa), which facilitates cleavage of the glycosidic link and formation of the resonant stabilized oxonium ion.
Asp 52 stabilizes the positive oxonium through electrostatic catalysis. The distorted half-chair form of the D NAM stabilizes the oxonium which requires co-planarity of the substituents attached to the sp2 hybridized carbon of the carbocation resonant form (much like we saw with the planar peptide bond).
water attacks the stablized carbocation, forming the hemiacetal with release of the extra proton from water to the deprotonated Glu 35 reforming the general acid catalysis.
Binding and distortion of the D substituent of the substrate (to the half chair form as shown above) occurs before catalysis. Since this distortion helps stabilize the oxonium ion intermediate, it presumably stabilizes the transition state as well. Hence this enzyme appears to bind the transition state more tightly than the free, undistorted substrate, which is yet another method of catalysis.
pH studies show that side chains with pKa's of 3.5 and 6.3 are required for activity. These presumably correspond to Asp 52 and Glu 35, respectively. If the carboxy groups of lysozyme are chemically modified in the presence of a competitive inhibitor of the enzyme, the only protected carboxy groups are Asp 52 and Glu 35.
A recent paper suggests an alternative mechanism to the Phillips mechanism above. In this case, Asp 52 acts as a nucleophilic catalysis and forms a covalent bond with NAM, expelling a NAG leaving group with Glu 35 acting as a general acid. This alternative mechanism also is consistent with other -glycosidic bond cleavage enzyme. Substrate distortion is also important in this alternative mechanism.
CHYMOTRYPSIN
Chymotrypsin, a protease, cleaves amides as well as small ester substrates after aromatic residues. The following data using different chymotrypsin substrates suggests a covalent intermediate occurs on chymotrypsin catalyzed cleavage of esters and amides
We have seen a kinetic mechanism consistent with these ideas before. The reaction equations are shown below:
In this reaction, a substrate S might interact with E to form a complex, which then is cleaved to products P and Q. Q is released from the enzyme, but P might stay covalently attached, until it is expelled. This conforms exactly to the mechanism described above. For chymotrypsin-catalyzed cleavage, the step characterized by k2 is the acylation step (with release of the leaving group such as p-nitrophenol in Lab 5). The step characterized by k3 is the deacylation step in which water attacks the acyl enzyme to release product P (free phosphate in Lab 5). In class and for homework you derived the following equation::
Equation 7: v = [(k2k3)/(k2 + k3)]EoS/[Ks(k3)/(k2+k3)] + S
As mentioned above, for hydrolysis of ester substrates, which have better leaving groups compared to amides, deacylation is rate limiting, ( k3<<k2). Then equation 7 becomes :
v = k3EoS/[Ks(k3)/(k2) + S]
Vm = k3Eo and Km = Ks(k3)/(k2)
For amide hydrolysis, as mentioned above, acylation can be rate-limiting (k2<<k3). Then equation 7 becomes:
v = k2EoS/[Ks+ S]
Vm = k2Eo and Km = Ks
Just as we saw before for the rapid equilibrium assumption (when ES falls apart to E + S more quickly than it goes to product), Km = Ks in amide hydrolysis.
changing the pH or ionic strength - which can give data about general acids/bases in the active site:
a graph of kcat as a function of pH indicates that a group of pKa of approx. 6 must be deprotonated to express activity (i.e. Vm/2 is at about pH 6). This suggests that an active site histidine is necessary, which if it must be deprotonated to express activity, must be acting as a general base.
a graph of kcat/Km shows a bell-shaped curve indicating the necessity of a deprotonated side chain with a pKa of about 6 (i.e. the same His above) and a group which must be protonated with a pKa of about 10. This turns out to be an N terminal Ile (actually at the 16 position in the inactive precursor of chymotrypsin called chymotrypsinogen, which on activation of chymotrypsinogen loses the first 15 amino acids by selective proteolysis), which must be protonated to form a stabilizing salt bridge in the protein.
pH effects on trypsin and chymotrypsin: a mathematical derivation
Note: A new theoretical computer program, called THEMATICS (theoretical microscopic titration curves) has been developed to calculate the titration curves for all ionizable groups in a protein. When performed on test proteins, those amino acids that showed anomalous curves (flattened compared to normal titration curves) where usually found in the active site of the protein. The flattened curves show that the amino acid is partially protonated over a wider range of pH then theoretically expected. The program can be used to predict active site regions on protein of known structure but unknown function, and will be useful in the emerging field of proteomics.
the enzyme - by chemical modification of specific amino acids, or through site-specific mutagenesis:
modification of chyrmotrypsin (and many other proteases) with diisopropylphosphofluoridate (DIPF) modifies one of many Ser residues (Ser 195), suggesting that it is hypernucleophilic. and probably the amino acid which attacks the carbonyl C in the substrate, forming the acyl-intermediate.
modification of the enzyme with tos-L-Phe-chloromethyl ketone inactives the enzyme with a 1:1 stoichiometry which results in a modified His
comparison of the primary sequence of many proteases show that three residues are invariant: Ser 195, His 57, Asp 102
site-specific mutagenesis show that if Ser 195 is changed to Ala 195, the enzymatic activity is almost reduced to background.
Wyszukiwarka
Podobne podstrony:
chrystus jest zyciem mym ENG
06 Kinetyka reakcji enzymatycznych
Przegląd rozwiązań konstrukcyjnych wtryskarek (ENG)
Enzymatyczna redukcja związków karbonylowych i zawierających wiązania C=C
Assembler ENG
Frequenzimetro eng 2003
PM [R2] Sylabus ENG
P000476 D Eng Main dimensions
Eurocode 3 Part 1 11 Pren 1993 1 11 (Eng)
Humulon and lupulon eng
Konwencja w sprawie zapobiegania i karania zbrodni ludobójstwa eng
Instrukcja biologia Aktywnosc enzymatyczna
Listy Biotechnologia2010-2011, Biotechnologia Enzymatyczna
Curriculum vitae Team III ENG
defekty enzymatyczne pytania biochem
kinetyka reakcji enzymatycznych I
P000722 A Eng Lower preassembly
P000718 A Eng Vertical shaft assembly
więcej podobnych podstron