Metabolizm aminokwasów:
Źródła metaboliczne wolnych aminokwasów.
Większość aminokwasów to produkty rozpadu białek pokarmowych i tkankowych. Rozkład białek zachodzi pod wpływem działania enzymów proteolitycznych, które hydrolizują wiązania peptydowe. Proces ten nazywany jest proteolizą. Każdemu rozpadowi wiązania peptydowego towarzyszy odtworzenie wolnej grupy aminowej jednego aminokwasu i wolnej grupy karboksylowej drugiego aminokwasu.
Rozkład białek pokarmowych. Soki trawienne przewodu pokarmowego zawierają wiele enzymów proteolitycznych:
pepsyna w soku żołądkowym (działa w środowisku kwaśnym i rozkłada wiązania peptydowe powstałe z udziałem aminokwasów aromatycznych i kwaśnych oraz między Val i Leu);
trypsyna w soku trzustkowym (działa w środowisku alkalicznym, rozkłada wiązania powstałe z udziałem grup karboksylowych aminokwasów zasadowych - Lys i Arg);
chymotrypsyna w soku trzustkowym (hydrolizuje wiązania peptydowe powstałe z udziałem grup karboksylowych aminokwasów aromatycznych - Phe, Tyr, Trp);
elastaza w soku trzustkowym ( rozkłada wiązania peptydowe między różnymi aminokwasami obojętnymi);
karboksypeptydaza A w soku trzustkowym (odłącza od substratu różne aminokwasy, z wyjątkiem Lys i Arg);
karboksypeptydaza B w soku trzustkowym (odłącza od substratu Lys i Arg);
aminopeptydaza w soku jelitowym (odłącza od substratu różne aminokwasy N-końcowe).
Łączne działanie wspomnianych enzymów prowadzi do całkowitego rozpadu białek pokarmowych do wolnych aminokwasów. Białka pokarmowe są jedynym źródłem aminokwasów egzogennych: Phe, Ile, Leu, Met, Thr, Trp, Val, Lys.
Rozkład białek wewnątrzkomórkowych. Zachodzi pod wpływem proteaz wewnątrzkomórkowych (katepsyn), zlokalizowanych w lizosomach, wykazujących najwyższą aktywność w kwaśnym pH. W pozalizosomalnej degradacji białek biorą udział proteosomy - kompleksy wieloenzymatyczne występujące w cytozolu i w jądrze wszystkich komórek eukariotycznych.
Rozkład białek pozakomórkowych (kolagen, elastyna, glikoproteiny błon podstawowych i rdzenie białkowe proteoglikanów). Degradacja tych białek zachodzi przy udziale metaloproteinaz macierzy pozakomórkowej. Metaloproteinazy powodują wstępną degradację substratów białkowych. Produkty proteolizy pozakomórkowej wnikają do komórki drogą endocytozy i dalej rozkładane są przez proteazy wewnątrzkomórkowe.
Trawienie kolagenu (kolagenoliza): kolagenazy trawią kolagen w jego naturalnej postaci, czyli potrójnej helisy. Struktura potrójnej helisy czyni kolagen opornym na działanie większości enzymów proteolitycznych. Pod wpływem kolagenazy tkankowej cząsteczka kolagenu: tropokolagen rozpada się na dwie wielkocząsteczkowe produkty: tropokolagen A i tropokolagen B, które charakteryzują się niską temperatura denaturacji. Tracą strukturę potrójnej helisy i wnikają do komórki droga endocytozy i są trawione dalej przez proteazy wewnątrzkomórkowe do produktów drobnocząsteczkowych.
Biosynteza aminokwasów. Organizm człowieka potrafi syntetyzować niektóre aminokwasy, zwane aminokwasami endogennymi. Są nimi: Ala, Arg, Asp, Asn, Cys, Gly, Glu, Gln, Pro, Ser, Tyr. Głównym producentem aminokwasów są mięśnie. Znaczna część aminokwasów powstaje z ketokwasów, przez przyłączenie grupy aminowej w miejsce grupy ketonowej (proces transaminacji). Niektóre aminokwasy powstają drogą przekształcania innych aminokwasów np. endogenna Tyr powstaje poprzez hydroksylację egzogennej Phe; endogenna Cys dzięki siarce przenoszonej z egzogennej Met, Gly jest produktem rozpadu Ser i Thr. Pomiędzy narządami dochodzi do wymiany aminokwasów. Ala i Glu produkowane w dużej ilości przez mięśnie, przechodzą do krwi i są wychwytywane przez wątrobę. Nerki produkują i uwalniają Ser i Ala, a pobierają z krążenia Glu, Pro, Gly. Mózg pobiera duże ilości Val.
Metabolizm grup aminowych aminokwasów.
Głównym miejscem rozkładu aminokwasów jest wątroba. Grupa α-aminowa aminokwasu może być odłączona w dwojaki sposób: poprzez przekazanie na inny akceptor (ketokwas) lub poprzez bezpośrednie odłączenie w postaci amoniaku. Obydwa procesy prowadzą do przekształcenia azotu aminokwasowego w mocznik. Pozostający szkielet węglowodorowy zostaje przekształcony w ciała ketonowe (aminokwasy ketogenne) lub glukozę (aminokwasy glukogenne).
Transaminacja: proces przemiany aminokwasów rozpoczyna się od przeniesienia grupy α-aminowej na jeden z trzech ketokwasów: pirogronian, szczawiooctan, α-ketoglutaran. Reakcje te są katalizowane przez enzymy zwane aminotransferazami. Dawcami grup aminowych są wszystkie aminokwasy, z wyjątkiem Lys i Thr oraz Pro i hydroksyproliny. Aminokwas pozbawiony grupy aminowej staje się ketokwasem. Ketokwas, który przyłączył grupę aminową staje się aminokwasem. Przenośnikiem grup aminowych jest fosforan pirydoksalu.
Ryc. 1. Transaminacja aminokwasów.
α- aminokwas + pirogronian → α-ketokwas + alanina
(AlAT - aminotransferaza alaninowa)
α- aminokwas + szczawiooctan → α-ketokwas + asparaginian
(AspAT - aminotransferaza asparaginowa)
α- aminokwas + α-ketoglutaran → α-ketokwas + glutaminian
(aminotransferaza glutaminianowa)
Deaminacja: odłączenie grupy aminowej od aminokwasu w postaci amoniaku. Niektóre reakcje deaminacji zachodzą drogą oksydoredukcji. Noszą one nazwę oksydacyjnej deaminacji. Głównie glutaminian ulega deaminacji tym szlakiem.
Oksydacyjna deaminacja glutaminianu polega na odłączeniu grupy aminowej i utlenieniu węgla α do grupy ketonowej. Powstaje α-ketoglutaran i NH3. Reakcję tę katalizuje enzym dehydrogenaza glutaminianowa.
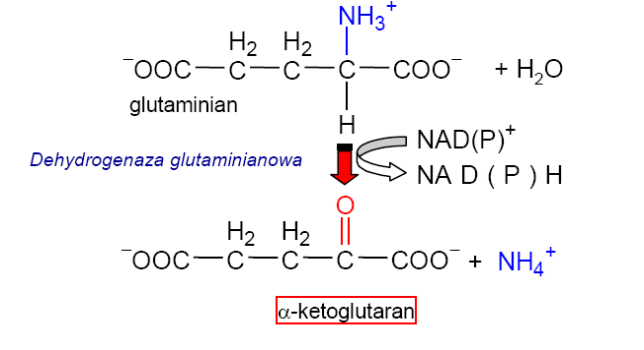
Ryc. 2. Oksydacyjna deaminacja glutaminianu.
Pewne znaczenie w procesie deaminacji przypisuje się oksydazom aminokwasowym, współdziałającym z nukleotydami flawinowymi: FMN i FAD. Pod działaniem oksydazy aminokwas utlenia się do ketokwasu, a FMN redukuje się do FMN2 lub FAD do FADH2. Grupa aminowa odłącza się w postaci amoniaku.
Detoksykacja amoniaku.
Amoniak jest związkiem toksycznym. Powstaje nie tylko w procesie deaminacji aminokwasów, ale także w następstwie daeminacji zasad purynowych i pirymidynowych, hydrolizy amidów kwasowych.
3.1. Proces przekształcania toksycznego amoniaku w nietoksyczny mocznik nosi nazwę cyklu mocznikowego (ornitynowego).
Ryc. 3 . Cykl mocznikowy.
Amoniak wchodzi w reakcję z CO2 i dwiema cząsteczkami ATP, tworząc karbamoilofosforan. Reakcja ta jest katalizowana przez syntetazę karbamoilofosforanową. Jedna cząsteczka ATP rozpada się do ADP i nieorganicznego fosforanu, druga natomiast przekształca się w ADP, przekazując grupę fosforanową do produktu reakcji: karbamoilofosforanu.
Przeniesienie grupy karbamoilowej z karamoilofosforanu na ornitynę w obecności karbamoilotransferazy ornitynowej. Produktem reakcji jest cytrulina.
W wyniku kondensacji cytruliny z asparaginianem, zachodzącej przy udziale syntetazy argininobursztynianowej, powstaje argininobursztynian. Podczas reakcji zużywana jest cząsteczka ATP, która rozpada się do AMP i pirofosforanu.
Argininobursztynian pod działaniem liazy argininobursztynianowej rozpada się na argininę i fumaran.
Hydroliza argininy zachodząca pod działaniem arginazy uwalnia mocznik i odtwarza ornitynę.
Uwolniona ornityna wchodzi w reakcję z kolejną cząsteczką karamoilofosforanu i cykl się powtarza.
Bilans cyklu mocznikowego:
2 NH3 + CO2 + asparaginian + 3 ATP
→ mocznik + fumaran + 2 ADT + 2 Pi + AMP +PPi
Reakcje cyklu mocznikowego zachodzą w cytosolu wątrobowym i mitochondriach. Uwalnianie amoniaku z glutaminianu, tworzenie karamoilofosforanu i jego wiązanie z ornityną zachodzą w macierzy mitochondrialnej, a pozostałe reakcje w cytosolu.
Stężenie mocznika w surowicy krwi jest ważnym parametrem biochemicznym, przydatnym w diagnostyce nerek. Osocze zdrowego człowieka zawiera od 20 do 40 mg mocznika na decylitr. Wzrost jego stężenia wskazuje na upośledzenie funkcji nerek.
Niesprawne funkcjonowanie cyklu mocznikowego prowadzi do akumulacji amoniaku w tkankach i płynach ustrojowych. Stan ten jest określany nazwą hiperamonemii. Przyczyną może być wrodzony niedobór jednego z enzymów uczestniczących w syntezie mocznika, a także ciężkie uszkodzenie wątroby przez czynnik toksyczny lub zakaźny.
Wiązanie amoniaku w glutaminian.
Część amoniaku wiązana jest przez grupę γ-karboksylową kwasu glutaminowego z wytworzeniem glutaminy. Reakcja ta katalizowana jest przez syntetazę glutaminy przy udziale ATP, który rozpada się do ADP i fosforanu.
Metabolizm energetyczny szkieletów węglowodorowych aminokwasów.
Przemiany aminokwasów glukogennych. Szkielety węglowodorowe aminokwasów glukogennych przekształcają się bezpośrednio lub pośrednio w szczawiooctan, kluczowy metabolit w procesie glukoneogenezy. Typowymi przedstawicielami tej grupy aminokwasów są: Ala, Ser, Cys, Asn, Gln. W wyniku transaminacji lub deaminacji przechodzą w odpowiednie ketokwasy: pirogronian, szczwiooctan, α-ketoglutaran i ulegają przemianie do szczawiooctanu.
Przemiana aminokwasów ketogennych. Typowymi aminokwasami ketogennymi są leucyna, która przekształca się w acetooctan (ciało ketonowe) i lizyna, która przekształca się w acetoacetylo∼S-CoA.
Przemiana aminokwasów glukoketogennych. Aminokwasami glukoketogennymi są: Phe, Tyr, Ile, Thr i Trp. Dwa pierwsze z nich są typowymi przedstawicielami tej grupy. Przekształcają się one do dwóch produktów końcowych: acetooctanu oraz fumaranu. Fumaran po przekształceniu w szczwiooctan drogą cyklu Krebsa, może być substratem zużywanym w procesie glukoneogenezy. Wrodzony niedobór enzymów biorących udział w przemianie tych aminokwasów jest przyczyną chorób metabolicznych:
Fenyloketonuria - niedobór enzymu hydroksylazy fenyloalaninowej, odpowiedzialnego za przekształcenie Phe w Tyr; alternatywny szlak przemiany fenyloalaniny prowadzi do powstania fenyloketokwasu, od którego pochodzi nazwa choroby; nadmiar Phe upośledza transport innych aminokwasów do wnętrza komórki, konkurując o białka przenośnikowe. Brak hydroksylacji Phe ogranicza syntezę innych związków powstających z Phe: dopaminy, noradrenaliny, adrenaliny. W związku z tym fenyloketonuria objawia się głównie pod postacią różnych zaburzeń funkcji centralnego układu nerwowego.
Alkaptonuria - efekt wrodzonego niedoboru dioksygenazy homogentynizowanej, biorącej udziale przemianie aminokwasu aromatycznego tyrozyny. Związana jest z wydalaniem z moczem dużych ilości kwasu homogentyzynowego (ciemniejącego na powietrzu), niebieskawo-czarnym zabarwieniem tkanki łącznej (ochronoza) oraz zmianami zwyrodnieniowymi stawów i kręgosłupa.
Tyrozynemia - jest spowodowana wrodzonym niedoborem aminotranferazy tyrozynowej i hydrolazy fumaryloacetooctanowej. W moczu chorego pojawia się nadmiar tyrozyny i jej metabolitów. Pojawiają się uszkodzenia wielu narządów, głównie wątroby, nerek i narządu wzroku.
Aminokwasy źródłem związków biologicznie czynnych.
Aminokwasy są substratami wielu związków biologicznie czynnych. Są to: aktywne fragmenty jednowęglowe, barwniki porfirytowe, zasady purynowe i pirymidynowe, niektóre hormony i neurotransmitery, melaniny, aminy i poliamidy, karnityna, koenzymy.
Z aminokwasów powstają aktywne fragmenty jednowęglowe: metylo-THF i formylo-THF
Z tyrozyny powstają hormony i neuroprzekaźniki: dopomina, adrenalina, noradrenalina, tyroksyna, trijodotyronina
Z tyrozyny powtają melaniny, następstwem nieprawidłowości w ich syntezie jest albinizm (bielactwo), spowodowany wrodzonym brakiem tyrozynanzy
Z tryptofanu powstaje kwas nikotynowy (niacyna), substratu do biosyntezy koenzymów: NAD+ i NADP+.
Cysteina przekształca się w taurynę, składnika soli kwasów żółciowych oraz pełniącej funkcję neuroprzekaźnika w centralnym układzie nerwowym
Metionina uczestniczy w transmetylacji
Z aminokwasów powstaje kreatyna, będąca akceptorem reszt fosforanowych z ATP
Z lizyny i metioniny powstaje karnityna, będąca nośnikiem reszt długołańcuchowych kwasów tłuszczowych poprzez wewnętrzna błonę mitochondrialną
Dekarboksylacja aminokwasów dostarcza amin biogennych: histamina, serotonina, etanoloamina
Z argininy powstaje tlenek azotu, odpowiedzialny za obniżenie ciśnienia tętniczego krwi, regulację krzepliwości krwi, przekazywanie sygnałów wewnątrz komórki i pomiędzy nimi.
Wyszukiwarka
Podobne podstrony:
AMINOKWASY, Ratownicto Medyczne, BIOCHEMIA
Transportery glukozy, Ratownicto Medyczne, BIOCHEMIA
Izomeria monosacharydów, Ratownicto Medyczne, BIOCHEMIA
PEPTYDY, Ratownicto Medyczne, BIOCHEMIA
biochemia wyklad 2, Ratownicto Medyczne, BIOCHEMIA
Kod genetyczny, Ratownicto Medyczne, BIOCHEMIA
biochemia test!!!!!!!!!!!!!!!!!!!, Ratownicto Medyczne, BIOCHEMIA
Harmonogram zajęć z Biochemii, Ratownicto Medyczne, BIOCHEMIA
biochemia 1 wykład, Ratownicto Medyczne, BIOCHEMIA
metabolizm aminokwasów i białek, Biologia, Biochemia
Biochemiczne markery chorób stawów, Ratownictwo medyczne, Ortopedia
BIOCHEMIA Metabolizm aminokwasów i białek
Badanie biochemiczne krwi, Studia - ratownictwo medyczne, 3 rok, Zawansowane procedury ratunkowe
Biochemia 11 Metabolizm aminokwasówK
ZAKRES MATERIAŁU Z BIOCHEMII WYMAGANY, Ratownictwo Medyczne Studia, Giełda, 1. rok, Chemia ogólna, B
Zasady zaliczania przedmiotu do zawieszenia dla ratownictwa, Ratownictwo Medyczne Studia, Giełda, 1.
więcej podobnych podstron