Pytania na egzamin z chemii fizycznej
Biologia lic. I rok (pytania z 2004) i dużo się powtórzyło w 2005
Uwagi:
1. Pytania zostały podzielone na działy, numeracja pozostała wg tekstu pierwotnego, z tym że, nie można się nią sugerować, podobnie jak literkami (kolejnością odpowiedzi),
gdyż są różne grupy.
2. Materiał został uzupełniony po konsultacjach 9.IX, pochodzące stamtąd komentarze Wykładowcy zaznaczone są literką G.
3. Na niebiesko zakreślone są odpowiedzi, w których w stosunku do poprzedniej wersji zaszły istotne zmiany - poprawiono błędy lub dostosowano odp. do interpretacji Wykładowcy (G.). Jeśli ktoś wydrukował poprzednie wersje - zakreślone miejsca należy koniecznie poprawić.
4. Jeżeli przy równaniach nie wyświetla się znak (delta) oznacza brak w systemie czcionki Sybmol (powinna ona standartowo zainstalowania), tekst taki można otworzyć i wydrukować na innym kompie.
Działy tematyczne: (hipertekst)
|
I. Błony biologiczne, dyfuzja, osmoza
1. Spontaniczne formowanie się dwuwarstwy fosfolipidowej możliwe jest dzięki:
A. amfifilowości cząsteczek wody;
B. oddziaływaniom hydrofobowym pomiędzy łańcuchami węglowodorowymi;
C. amfifilowości cząsteczek fosfolipidów;
D. wiązaniom wodorowym pomiędzy łańcuchami węglowodorowymi a wodą.
Odp. C amfifilowość - to cecha cząsteczki (fosfolipidu), że jest ona jednocześnie hydrofobowa i hydrofilowa.
2. Której z cech nie można przypisać błonom komórkowym:
A. symetria poprzeczna i izotropowość;
B. przepuszczalność dla jonów;
C. asymetria poprzeczna;
D. obecność dwuwarstwy fosfolipidowej;
Odp. A - błony są półpłynne, przepuszczalne i zawsze asymetryczne, przepuszczalność dla jonów nie jest cechą błony jako takiej (wymaga specjalnych kanałów jonowych).
3. "Płynność" dwuwarstwy lipidowej i błon to określenie związane bezpośrednio z: (chyba):
A. wartościami współczynników przepuszczalności dla wody;
B. przemieszczaniem się cząsteczek lipidów w poprzek dwuwarstwy;
C. uporządkowaniem i ruchliwością łańcuchów węglowodorowych;
D. brakiem lub obecnością domen lipidowych i białkowych;
Odp. C - Łańcuchy węglowodorowe lipidów mogą występować w uporządkowanym stanie stałym lub w słabo uporządkowanym stanie płynnym, co zwiększa ich ruchliwość. Zależy to od temperatury oraz struktury łańcuchów. Ze strukturą warstwy nie ma związku przepuszczalność wody (nie A); Lipidy w błonie przemieszczają się co do zasady wzdłuż błony, a nie w poprzek (nie B).
4. Dynamikę czyli zdolność do względnie szybkiej wymiany swoich składników błony zawdzięczają:
A. oddziaływaniem z otaczającymi cząsteczkami wody;
B. oddziaływaniem z jonami soli obecnymi w ich otoczeniu;
C. względne małą przewaga energii wiązań międzycząsteczkowych w stosunku do energii ruchów cieplnych (lub sformułowanie podobne);
D. konkurencji cząsteczek lipidów i białek o wiązanie wodorowe.
Odp. C. Żeby coś z błony „wyrwać” musi być mała energia wiązań - jako przykład przeciwny G. podał kryształ krzemu, który nie wymienia składników bo występują w nim mocne wiązania.
5. Potencjał spoczynkowy na błonie komórkowej jest wynikiem:
A. ....coś z prądami jonowymi
B. stanu (nie) równowagi, w którym poszczególne strumienie jonów równe są zero
C. zahamowanego (?) działania pomp sodowo-potasowych
D. stanem nierównowagi, w którym suma strumieni wszystkich substancji jest równa zero.
Odp. D - powstanie potencjałów wymaga powstania stanu stacjonarnej nierównowagi ładunków, przy czym przepływ jonów nie musi być całkiem zablokowany, wystarczy żeby wielkość ładunków w jedną i druga stronę była jednakowa; dopiero otwarcie kanałów jonowych spowoduje wyzerowanie ładunków (potencjał czynnościowy, pobudzony, aktywny); C jest źle, bo sód i potas są dodatnie, stąd też „pompowanie” ich nie ma wpływu na potencjał.
13. Zjawisko charakterystyczne i samorzutne dla dwuwarstwy lipidowej:
A. dyfuzja jonów w poprzek dwuwarstwy
B. ?
C. ?
D .?
Odp. Na pewno nie A - bo dwuwarstwa lipidowa jest z zasady nieprzepuszczalna dla jonów, oraz cząstek polarnych dużych (jak glukoza) i małych naładowanych - dla ich transportu wymagane są specjalne białka (transport ułatwiony); stosunkowo łatwo przepuszcza natomiast cząsteczki O2, CO2, N2, NO, i małych, nie naładowanych cząsteczek polarnych - np. H20, etanol, mocznik (transport bierny). Samorzutnie m.in. zachodzi przemieszczanie się lipidów wzdłuż dwuwarstwy (taka lub podobna odpowiedź będzie poprawna).
22. Błona półprzepuszczalna. Jak się doda chyba białko nieprzepuszczalne przez błonę to jak będzie z H2O? Czy zostanie wydalona z kom, czy pobrana z otoczenia czy nic się nie będzie działo. Coś takiego.
G. nie doprecyzował tego pytania, ale zwrócił uwagę, że skoro aktywność osmotyczna zależy od stężania molowego, to dla substancji wielkocząsteczkowych (np. białek, nawet dobrze rozpuszczalnych ) będzie ona znikoma - czyli dodanie takiego białka nie spowoduje przepływu wody.
23. Wzór Ficka, czego nie uwzględnia?
Wzór Ficka określa prędkość dyfuzji: d n / d t = - DS * d c / d x
gdzie: d μ - przyrost moli cząsteczek dyfunujących; d t - przyrost czasu; D - współczynnik dyfuzji = ruchliwość elektroforetyczna); S - powierzchnia dyfuzji; x - droga dyfuzji (grubość błony); d c - różnica stężeń po obu stronach (czyli bodziec termodynamiczny). Po podzieleniu przez S, wzór ten przyjmie postać: d n / S *d t = - D * d c / d x = J (Strumień dyfuzji), gdzie D / d x oznacza współczynnik przepuszczalności błony.
J zależy zatem od: gradientu stężeń, odległości (grubości błony) i od współczynnika D. Z kolei D zależy od rodzaju substancji, temperatury, częściowo ciśnienia, ale zdaniem G, współczynnik D jest specyficzny dla danego procesu, to tych parametrów cząstkowych (wpływających na jego wielkość) mamy nie brać pod uwagę. G zaznaczył także, że Fick w swoim wzorze nie uwzględnia strumieni innych składników roztworu.
24. wzór Nernsta - jakie parametry nie wpływają na strumień dyfuzji?
Pomiędzy dwoma substancjami o różnych stężeniach zachodzi różnica potencjałów chemicznych μ = μo + RT ln c ten wzór wymaga uwzględnienia ładunku i ma postać:
μ = μo + RT ln cz/cw + ZFф,
gdzie Z - to ładunek jonu, F - stała Faradaya, ф - potencjał równowagowy elektryczny, cz / w - ciśnienie zewnętrzne / wewnętrzne, μ - potencjał chemiczny. Patrz też pyt. 37.
33. Jeśli po jednej stronie jest cukier a po drugiej woda - czy potencjał wody będzie ten sam?
Odp. Nie, bo liczy się go z uwzględnieniem wszystkich składników układu.
37. Od czego zależy potencjał elektrodyfuzyjny?
- jak w pyt. 24 - od ładunku jonów, temperatury, różnicy (stosunku) stężeń po obu stronach.
38. Co się dzieje gdy pompy sodowo- potasowe zostaną zatrzymane?
Odp. zgon (albo inna podobna) - bo sód będzie gromadził się wewnątrz komórek, a potas w przestrzeni zewnątrzkomórkowej. W tkankach żywych jest odwrotnie, dzięki tzw. transportowi aktywnemu, czyli przerzucania przez błonę K+ do wewnatrz, a Na+ na zewnątrz wbrew gradientowi stężeń. Pamiętamy, że potas ma być w komórce, a nie odwrotnie, bo w USA podaje się go dożylnie skazanym na śmierć.
39. Różnica między dyfuzją ułatwioną a prostą?
Odp. Oba rodzaje zachodzą pod wpływem potencjału chemicznego (gradientu stężeń), ułatwiona dodatkowo wymaga nośników (białek transportowych), jest jeszcze dyfuzja aktywna, która odbywa się wbrew gradientowi stężeń np. przy pomocy ATP (np. Na+, K+).
50. Czym jest spowodowana równowaga przy osmozie?
Odp. Wyrównaniem potencjału chemicznego po obu stronach błony
51. Plazmoliza, deplazmoliza.
Odp. Plazmoliza - to „liza” czyli zmniejszanie się objętości plazmy komórki poprzez osmotyczny wypływ wody - np. poprzez zanurzenie w roztworze hipertonicznym (o większym stężeniu). Dlatego soląc potrawy zabijamy bakterie. Przy niezbyt długim czasie ekspozycji plazma może „odżyć”, jeżeli otoczymy ją roztworem hipotonicznym (o mniejszym stężeniu). Taki proces odwrotny nazywany deplazmolizą.
52. Jaki proces sprawia, że liposomy mogą wnosić lekarstwa do wnętrza komórki?
Odp. fuzja - dwie struktury dwuwarstwowe mogą zlewać się w jedną.
53. Od czego zależy potencjał spoczynkowy ?
Odp. od stężenia jonów na zewnątrz i wewnątrz, od kanałów jonowych, przepuszczalności, aktywności pomp jonowych.
59. Równowaga, a stan stacjonarny - różnice
Równowaga jest wtedy, gdy nic się nie dzieje (objawem zewnętrznym jest stałość parametrów termodynamicznych). Z kolei stan stacjonarny oznacza zerowanie się procesów - są przepływy ale entropia się nie zmienia, stężenia są zachowane.
II. Oddziaływania międzycząsteczkowe
6. Wskaż zestaw, w którym we wszystkich podanych przykładach występuje wiązanie wodorowe:
A. gazowy wodór, skroplony wodór, woda
B. białko, benzen, aceton
C. lód, DNA, skroplony tlen
D. etanol, kwas octowy, etanal
Odp. D - wszystkie wymienione tu związki (alkohol, kwas organ., aldehyd) mają grupy o właściwościach polarnych -OH lub -O więc mogą tworzyć wiązanie. Szczególnie charakterystyczne jest ono dla kwasów, których grupy -OH łączą się parami tworząc tzw. dimery. Wiązanie wodorowe natomiast nie występuje: (a) w wodorze H2 (jest kowalencyjne), (b) w benzenie - ze względu na brak odpowiedniego akceptora (F,O,N,Cl, sam węgiel nie wystarczy), (c) w tlenie - nie ma tam wodoru, bez którego nie może powstać wiązanie wodorowe.
7. Wartość energii wiązania wodorowego w kJ/ mol leżą w zakresie:
A. kilka
B. kilkanaście
C. kilkadziesiąt
D. kilkaset
Odp. C. - Energia wynosi od 12 do 35 kJ/ mol ale uwaga! zdaniem G jest to już na poziomie kilkadziesiąt, a nie kilkanaście !!!
W starszych podręcznikach jako jednostkę energii stosuje się kilokalorię, którą przelicza się: 1 kcal = 4,19 kJ. Wtedy wartość ta będzie wynosić na poziomie kilku kcal.
8. Wskaż poprawne zdanie o wiązaniu wodorowym:
A. brak odczytu - ... końcowe nie mogą być zaangażowane w wiązania...... (nie mam
pojęcia o co tam chodziło)
B. hydratacja jon - dipol polega na tworzeniu wiązań wodorowych
C. wiązania wodorowe między cząsteczkami wody
D. ?
Odp. Warianty nie są odtworzone dokładnie - nie powinno być A , gdyż wiązanie wodorowe raczej nie zależy od lokalizacji. Też nie B - bo hydratacja nie polega na wiązaniach wodorowych. Może być C (są tam wiązania krótkotrwałe), albo coś zgodnie z definicją wiązania (komentarz do pyt. 6)
9. Jakim rodzajem oddziaływań jest hydratacja w cząsteczce soli?
Odp. jon- dipol (a właściwie to nawet dipol - jon gdyż to dipole wody atakują cząsteczki soli i „rozbijają” ją na jony, ale z punktu testu to jest to samo).
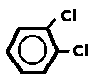
11. Która cząsteczka ma trwały charakter dipolowy:
A. benzen
B. etanu
C. metan
D. orto- di-chlorobenzen
Odp. D - (wzór na rysunku) rozmieszczenie „orto” oznacza, że oba podstawniki są osadzone na sąsiadujących atomach węgla - Cl jest elektroujemny więc we wiązaniu kowalencyjnym z -C ściąga parę elektronowa w swoją stronę, co oznacza, że cząsteczka D jest nie tylko polarna (taka byłaby również przy naprzeciwległym rozmieszczeniu Cl „para”), ale i polarna, ponieważ ładunek ujemny jest wyraźnie po jednej stronie; (pozostałe odpowiedzi - to wszystko węglowodory, które nie są polarne, jak też mają symetryczne rozmieszczenie C i H). Gdyby zamiast tego pojawił się kwas chlorowy (w poprzedniej wersji), to on też jest dipolowy.
12. Czy między cząsteczkami węglowodorów nienasyconych możliwe jest wiązanie wodorowe:
A. tak, tylko w stanie ciekłym
B. tak, coś tam
C. nie występuje
D. tak, coś tam.
Odp. C. Wszystkie węglowodory zawierają tylko C i H, a nie mają atomów elektroujemnych (N, F, O, Cl) niezbędnych do „ściągnięcia” wodoru. Nasycenie nie ma tu nic do rzeczy.
15. Jaka jest jednostka momentu dipolowego - wg układu SI ?
A. kulombometr
B. debaj
C, D ???
Odp. A (1 culomb * 1 metr) - moment dipolowy jest wprost proporcjonalny do wielkości ładunków cząsteczki oraz odległości pomiędzy nimi (nie debaj !, bo to ma być w jednostkach SI)!
47. Gdzie występuje chwilowe wiązanie wodorowe?
Odp. w wodzie (w ciekłym benzenie nie z powodów j.w.)
III. Fale elektromagnetyczne
14. Jednostka natężenia strumienia światła
Odp. W/ m2
30. Stosunek fali emitowanej do absorbowanej - która jest dłuższa?
Odp. Zgodnie z prawem Stockesa - fala emitowana („odbita”) jest dłuższa, gdyż ma mniejszą energię - np. w znanym z ekologii efekcie cieplarnianym - pada widzialne - emitowane jest ciepło (podczerwień).
35. Jaka jest multipletowość 1 niesparowanego elektronu?
Odp. M =2 (2*S + 1 = 2 * ½ +1)
S - to łączna liczba spinowa elektronów (wynosi +½ lub -½ w zależności od kierunku)
przy dwóch elektronach byłoby to: M=1 dla tripletu (oba spiny przeciwne: S= ½ - ½ = 0), a M= 3 dla singletu (oba spiny jednakowe: S = ½+ ½ = 1)
36. Zależność między energią wiązania a jego długością
Odp. czym krótsze wiązanie, tym większa energia (bo trudniej było zbliżyć atomy). Jeśli w odpowiedziach byłoby tu coś o wiązaniu wodorowym - to należy pamiętać, że mając energię znacznie niższą od chemicznego - będzie dłuższe.
45. Co nie może służyć, jako monochromator w spektrofotometrze?
A. pryzmat
B. siatka dyfrakcyjna
C. filtr
D. układ pryzmatów
Odp. na pewno będzie pryzmat i siatka (dalej mam pewne wątpliwości - muszę już puścić to co jest, najwyżej uzupełnimy jutro)
48. Prawo Lamberta Beera - do czego proporcjonalna jest absorbancja?
Odp. Do grubości warstwy x i stężenia roztworu c.
Absorpcja to stosunek początkowego natężenia promieniowania Io do natężenia Ix po przejściu przez próbkę: A = Io * 100% / Ix . Jej zależność od grubości próbki i stężenia ma charakter wykładniczy: Ix = Io e-k c x, (co oznacza, że w przypadku absorpcji proporcjonalny do x i c będzie jej log10). Równanie to po zlogarytmowaniu daje absorbancję: E = log Io / Ix = € c x, gdzie: k - współczynnik absorpcji, € współczynnik absorbancji.
49. Jakie rodzaje widma absorbcyjnego odpowiadają emisji podczerwieni?
Odp. Widmo oscylacyjne i rotacyjne (a nie widmo oscylacyjne lub rotacyjne, gdyż występują one łącznie).
51. Uporządkować fale wg rosnącej długości (podane 4 warianty):
Odp. Właściwa kolejność to: fale radiowe, telewizyjne, radarowe (mikrofale), IR (podczerwień), VIS (widzialne), UV (ultrafiolet), RTG (rentgenowskie), promieniowanie gamma, promieniowanie kosmiczne. Kolejność wg rosnącej częstotliwości (a tym samym energii) będzie odwrotna.
54. Fluorescencja i fosforescencja
Są to zjawiska emisji promieniowania - pierwsze w wyniku przejść, zachodzących bez zmiany multipletowości (czyli triplet / triplet lub singlet/ singlet), a drugie - triplet/ singlet lub singlet / triplet.
IV. Termodynamika
10. Jaka jest zmiana energii układów, pomiędzy którymi (nie mogłam odczytać)
A. wzrost energii
B. spadek energii
C. możliwy jest brak zmiany
D.?
Trudno skojarzyć o co tu konkretnie chodzi, ale gdyby był przepływ ciepła - to układ, do którego ciepło dopłynie zwiększy jego energię, jeśli przepływ masy - to też (każda dodana masa nawet zimniejsza - ma jakąś energię wewnętrzną, stąd tez powiększy stan dotychczasowy). Gdyby dopłynęła entropia - to energia może wzrosnąć lub zmaleć. P. też. pyt. 43
40. Reakcja egzotermiczna, samorzutna, nieodwracalna.
Przy wyborze właściwego wariantu trzeba uwzględnić następujące zasady:
proces zajdzie samorzutnie, gdy entalpia swobodna G zmaleje (G < 0);
zgodnie z wzorem Gibbsa Helmholtza G = H - Q = H - T S - czyli G zmaleje tym bardziej, im bardziej obniży się entalpia i im bardziej wzrośnie entropia;
reakcja samorzutna nie musi być egzotermiczna (wtedy H > 0), ale wtedy przyrost entalpii układu musi być zrekompensowany odpowiednio większym wzrostem entropii, (T S > 0, przy czym T S > H); mamy tak np. przy rozpuszczaniu niektórych soli w wodzie, co powoduje obniżenie się jej temperatury;
wzrost samej entropii S (S > 0) przesądza o nieodwracalności procesu.
Patrz też pyt. 43
41. Entropia w układzie izolowanym.
Odp. S = 0 (dla procesów odwracalnych) lub S > 0 (dla nieodwracalnych). Zmniejszenie entropii wymagałoby dostarczenia energii z zewnątrz. W układzie izolowanym jest to niemożliwe, gdyż nie tam ma wymiany energii ani masy z otoczeniem.
42. Entalpia = Q w jakiej reakcji?
A. Izobarycznej
B. Izochorycznej
C. Izotermicznej
Nie wiem, czy dobrze rozumiem intencje pytania, ale zasady są takie:
Zgodnie z wzorem Gibbsa Helmholtza H = Q +G, czyli przyrost entalpii będzie równy ciepłu w przemianach, gdzie nie zachodzi zmiana entalpii swobodnej i wykonywanie pracy objętościowej.
Również z wzoru na en. wewnętrzną U = Q + pV, po przyrównaniu do wzoru na entalpię: U = H+ pV i skróceniu pV wychodzi, że H = Q.
Zasada ta obowiązywałaby w układzie izobarycznym i izochorycznym, tam gdzie nie zachodzi praca objętościowa.
W układzie spełniającym kryteria A + B + C (czyli o stałej objętości, temperaturze i ciśnieniu), który nie wymienia z otoczeniem energii na sposób pracy mechanicznej (objętościowej), cała wymiana energii wewnętrznej odbywa się na sposób ciepła (Q = U).
43. I zasada termodynamiki- zależności
Prawo zachowania energii - w układzie izolowanym (który nie wymienia energii z otoczeniem) energia wewnętrzna nie zmienia się. W nieizolowanym (zamkniętym lub otwartym) możliwe są przepływy energii na sposób ciepła lub pracy (objętościowej lub nieobjętościowej - w obie strony dodatkowo w u. otwartym część energii wewnętrznej można wynieść/dostarczyć razem z masą).
44. II zasada termodynamiki - zależności
Procesy nieodwracalne i samorzutne przebiegają przy wzroście entropii: Δ S > Q / T, jej obniżenie wymaga dostarczenia energii z zewnątrz.
Dla procesów odwracalnych Δ S = Q / T = 0 (odwracalność w tym kontekście rozumiemy inaczej niż w kinetyce chemicznej)
59. Poprawne określenie ciepła to miara:
Odp. przepływu energii związanej z nieuporządkowanymi ruchami cząsteczek; ciepło nie jest funkcją stanu.
V. Kinetyka chemiczna
16 Prawa Arrheniusa - jak się zmieni szybkość reakcji jeśli temperatura
wzrośnie.
Wzrost temperatury wpływa na przyspieszenie reakcji. Szybkość reakcji jest to ubytek substratu w jednostce czasu. Najczęściej zależy ona od stężenia substratów, oraz warunków przebiegu reakcji.
17. Od czego nie jest zależna stała równowagi?
Odp. Od stężenia.
Dla reakcji odwracalnej mA + nB <----> pC + qD, stała równowagi jest stosunkiem współczynników proporcjonalności k1 i k2 wziętych z równań szybkości reakcji w obie strony i wynosi: ![]()
gdzie m,n,p,q, to współczynniki stechiometryczne. Stała ta jest charakterystyczna dla określonej temperatury i innych warunków, w jakich reakcja przebiega.
18. Od czego nie jest zależna stała szybkości?
Odp. j.w. Stała szybkości k określa zależność pomiędzy ubytkiem stężenia substratu A w jednostce czasu, a stężeniem tego substratu: v = dc/ dt = k [A]. Zależy ona m.in. od temperatury i in. warunków przebiegu reakcji (np. dla reakcji w stanie gazowym od ciśnienia), a nie zależy od stężenia substratów.
29. Czas połówkowy reakcji. Mamy Ao, które mamy doprowadzić do 3t1/2, wiedząc, że po czasie 2t jest równe:
Odp. Ma być 1/8.
Czas połówkowy t1/2 stanowi okres, w jakim początkowa ilość (stężenie) substratu obniży się o połowę. Jeżeli więc na początku był 1 g substratu, to po upływie jednego t1/2 zostanie go ½ g, po upływie 2 t1/2 będzie ¼ g, a po 3 t1/2 już 1/8 g.
32. Co robi katalizator?
Obniża energię aktywacji i przyspiesza reakcję, natomiast sam się przy tym nie zużywa.
34. A+B C Jaki jest rząd reakcji?
Odp. Drugorzędowa, bo v = k * [A] * [B] [mol/ l] * [mol/ l] = mol 2/ l2] - stężenia będą w jednostkach podniesionych do potęgi drugiej. Podobnie reakcja: 2 A C też jest drugorzędowa bo v = k * [A] * [A] = [A] 2. Rząd reakcji oznacza stopień równania kinetycznego (sumę wykładników potęg do jakiej należy podnieść stężenia poszczególnych substratów, aby były proporcjonalne do prędkości. Wyznaczamy go analizując mechanizm konkretnej reakcji na poszczególnych etapach jej przebiegu, a najczęściej eksperymentalnie.
VI. Gazy, ciecze, stany fazowe, roztwory
19. Co oznacza xi w prawie Routa?
Prawo Routa stanowi, że prężność pary danego składnika nad roztworem jest równa iloczynowi prężności pary tego roztworu w stanie czystym pomnożonej przez jego ułamek molowy w roztworze (xi): pa= poa xa; pb = pob xb. Sumaryczna prężność pary zatem wynosi: p= poa xa; pb + pob xb. Od tego prawa występują odchylenia w przypadku, gdy składniki oddziałują między sobą (azeotropia).
Ułamek molowy xi to wielkość charakterystyczna dla danego składnika i roztworu i oznacza stosunek moli tego składnika do łącznej sumy moli wszystkich składników roztworu. Suma ułamków molowych wszystkich składników roztworu równa się jedności.
25. Ile jest stanów swobody wrzącej wody?
Odp. jeden - bo z dwóch wchodzących tu w grę parametrów termodynamicznych - możemy swobodnie zmieniać tylko jeden - np. jak zmienimy temperaturę, to musimy natychmiast odpowiednio dostosować ciśnienie - bo „zniknie” nam jedna z faz (będzie sama woda, albo tylko para). Określa to reguła faz Gibbsa.
31. Co wrze w wyższej temperaturze?
A. czysta woda;
B. 1 molowy roztwór NaCl;
C. 0,5 molowy roztwór glukozy;
D. 1 molowy roztwór glukozy.
Odp. B. - Dlatego, że wzrost temperatury wrzenia (i spadek krzepnięcia) jest tym większy, im więcej cząstek jest rozpuszczonych w roztworze (gdyż zgodnie z prawem Routa ułamek molowy wody jest wówczas odpowiednio niższy i tak samo obniży się prężność pary). Dla 1m NaCl jest najwięcej ze względu na dysocjację (1 mol Na+ i 1 mol Cl-) - czyli razem 2 mole cząstek.
VII. Elektrochemia, dysocjacja
20. Mając Ir=10-8 jakie jest stężenie jonów [ Ca+2] w CaSO4 ?
Odp. [ Ca+2] = 10-4 Iloczyn rozpuszczalności liczymy mnożąc stężenia molowe poszczególnych obecnych w roztworze jonów: Ir = [Ca+2] + [SO4-2]. Ponieważ CaSO4 --> Ca+2 + SO4-2 to na 1 mol Ca+2 przypada 1 mol SO4-2, czyli w tym przypadku Ir = [Ca+2]2. Uwaga: W przypadku dysocjacji w innych proporcjach np. dla AlCl3 byłyby inne wzory.
21. Jakie jest pH gdy mamy [OH]= 10-4?
Odp. pH = 10. Liczymy to wiedząc, że iloczyn jonowy wody tj. [H+] * [OH-] = 10-14, a pH jest ujemnym logarytmem ze stężenia [H+] - stąd: [H+] = 10-14 / 10-4 = 10-10. log 10-10 = -10, a -log 10-10 = 10.
26. Mamy ogniwa wodorowe, czy jest tam utlenianie czy redukcja, które będzie anodą, a które katodą?
Odp. Ogniwo wodorowe składa się z blaszki platynowej, zanurzonej w 1M roztworze H+ i obmywanej wodorem gazowym o ciśnieniu 1 atm. Jest to tzw. ogniwo stężeniowe. Przyjmuje się, ze takie ogniwo wytwarza potencjał standardowy - czyli 0.
Mając więc zadaną różnicę potencjałów - trzeba szybko zorientować się w którym kierunku przepływają elektrony - np. potencjał wyższy w stosunku do ogniwa wodorowego oznaczałby, że na tym drugim ogniwie jest niedobór elektronów, czyli że zachodzi tam redukcja (będzie to katoda). W takim razie, dla zachowania równowagi, na ogniwie wodorowym będzie zachodziło utlenianie (wtedy będzie ono anodą).
27. Jak się zmieni stopień dysocjacji roztworu gdy jego rozcieńczenie zwiększy się dwukrotnie? Odp. Stopień się zwiększy o 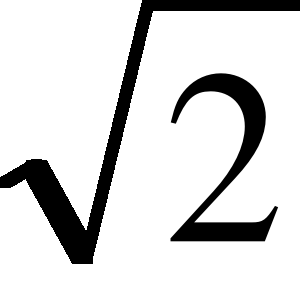
- krotnie.
Zależności określa wzór: 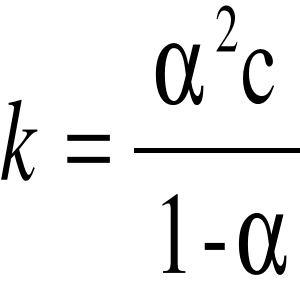
; lub po uproszczeniu: k = 2 c ; Stąd: 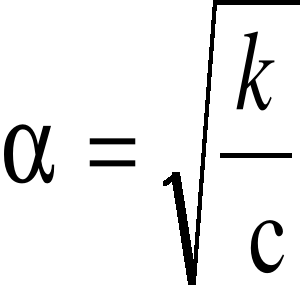
gdzie: to stopień dysocjacji, c - stężenie roztworu, k - stała dysocjacji (przy danej temperaturze jest niezmienna bez względu na stężenie). Jeśli więc stężenie zmaleje:
2 - krotnie - to wzrośnie
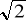
- krotnie;4 - krotnie - to wzrośnie 2 - krotnie;
n - krotnie - to wzrośnie
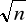
- krotnie. Odpowiednio jeżeli c wzrośnie:
2 - krotnie - to zmaleje
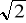
- krotnie (bo wzrośnie * 1 /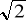
)n - krotnie - to zmaleje
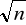
- krotnie.
28. Jak się zmieni stała dysocjacji 1 dm3 elektrolitu, gdy dodamy do niego 3 dm3 wody?
Stała dysocjacji się nie zmienia, bo nie zależy od stężeń.
46. Jaką substancję dodamy do buforowania pH?
Odp. Słaby kwas i jego sól z mocną zasadą lub słaba zasada i jej sól z mocnym kwasem. Np. NH4Cl/ / NH4OH (bufor amoniakalny) lub CH3COOH/ CH3COONa (bufor octanowy). Właściwości takich nie ma np. NaCl (jeden z wariantów w teście).
48. Co jest reduktorem w reakcji siarczanu miedzi i drutu żelaznego Cu+2 SO4 + Fe0 ---> Fe+2 SO4 + Cu0 ?
Odp. żelazo to reduktor = donor elektronów = samo się przy tym utlenia;
miedź to utleniacz = akceptor elektronów = sam się przy tym redukuje.
2
Wyszukiwarka
Podobne podstrony:
Chem fiz egz grupa A, Studia, Chemia fizyczna, Od Anki, egzaminy
chf wykład 6, Studia, Chemia, fizyczna, wykłady
ćw7 - Refrakcja i wyznaczanie momentu dipolowego, studia, chemia fizyczna
pytania z examinu, Studia, Chemia, ogólna, examin
chf wykład 3, Studia, Chemia, fizyczna, wykłady
ćwiczeniee 43, materiały naukowe do szkół i na studia, chemia fizyczna moja, Chemia fizyczna, Opraco
ch.fiz.12, Chemia fizyczna
chf wykład 8, Studia, Chemia, fizyczna, wykłady
chf wykład 1, Studia, Chemia, fizyczna, wykłady
fiz 36, Chemia fizyczna
chf wykład 5, Studia, Chemia, fizyczna, wykłady
7, Studia, Chemia fizyczna, Laborki
Sprawozdanie 12 do druku, Studia, Chemia fizyczna, Laborki, 12 - Równowaga fazowa ciecz-para
2, Studia, Chemia fizyczna, Laborki
ćw2 - Napięcie powierzchniowe, studia, chemia fizyczna
chf wykład 4, Studia, Chemia, fizyczna, wykłady
ćw9 - Elektrody jonoselektywne, studia, chemia fizyczna
Fizyczna I termin 22 VI 209, Studia, Chemia fizyczna, Od Anki
więcej podobnych podstron