FARMAKOKINETYKA, FARMAKODYNAMIKA, INTERAKCJE LEKOWE
FARMAKOLOGIA
(gr. pharmacon - lek, logos - słowo, nauka)
nauka o działaniu na organizm różnych związków chemicznych i substancji biologicznie czynnych
zajmuje się właściwościami fizyczno-chemicznymi leku, wpływem leku i mechanizmem działania na organizm, działaniami niepożądanymi i toksycznymi leku
KIERUNKI: farmakodynamika, farmakoterapia, farmakologia kliniczna, farmakologia doświadczalna, toksykologia, farmakokinetyka
DZIAŁY: psychofarmakologia, farmakogenetyka, immunofarmakologia, chronofarmakologia, farmakoekonomia
LEK - (ang. drug) każda substancja wywierającą określone działanie, mogące być wykorzystane w leczeniu
RODZAJE DZIAŁAŃ LEKU:
Przyczynowe (patogenetyczne)
hamuje proces chorobowy.
Działanie objawowe
(lek nie wpływa na proces chorobowy, eliminuje towarzyszące objawy; leki p/gorączkowe, hipotensyjne).
Substytucyjne (zastępcze)
(hormony w stanach ich niedoboru).
Działanie ośrodkowe
na ośrodkowy układ nerwowy.
Działanie obwodowe
na tkanki i narządy poza OUN.
Działanie ogólne
na cały organizm.
Działanie miejscowe
tylko w miejscu podania.
Działanie wybiórcze
wyłącznie na czynność jednego narządu, tkanki lub typu receptora.
LECZENIE: ALLOPATYCZNE, HOMEOPATYCZNE
Allopatia - leki wywołują zmiany czynności przeciwne do chorobowych.
Homeopatia - małe dawki leku wywołują objawy takie same jak chorobowe.
Mechanizmy działania leków:
Mechanizm działania
wpływ leku na żywy organizm, w wyniku którego dochodzi do zmian czynności komórek i tkanek organizmu i w efekcie do powstania określonego działania farmakologicznego.
Mechanizm fizykochemiczny
zmiana przepuszczalności błon komórkowych (leki znieczulające ogólnie), obniżenie napięcia powierzchniowego (kwasy żółciowe, saponiny), leki neutralizujące kwas solny żołądka, osmotyczne środki przeczyszczające.
Mechanizm biochemiczny
lek może reagować chemicznie z enzymem, koenzymem lub substratem reakcji. W efekcie dochodzi hamowania lub nasilania czynności enzymów, a wynikiem tej reakcji jest zmiana czynności komórki, tkanki lub narządu.
Mechanizm receptorowy
lek łączy się z receptorem i powoduje zmianę jego czynności, prowadząc do zmiany procesów metabolicznych zachodzących w komórce
Czynniki wpływające na działanie leków:
Budowa chemiczna
Różna wrażliwość rasowa i gatunkowa
Wiek
Płeć
Stany fizjologiczne
Stany chorobowe
Uwarunkowania genetyczne działania leku
Działania niepożądane i toksyczne:
Działanie niepożądane
Szkodliwy, niezamierzony skutek działania leku obserwowany w dawkach, które służą zapobieganiu, diagnozie, leczeniu lub modyfikacji czynności fizjologicznych. Działania te są charakterystyczne dla danego leku i mogą, ale nie muszą wystąpić przy jego stosowaniu w dawkach leczniczych, zgodnie ze wskazaniami. Nie dotyczą one skutków przedawkowania lub błędnego zażycia leku.
Działania te można podzielić na dwie grupy:
Dające się przewidzieć, związane z mechanizmem działania leku, ustępujące po zaprzestaniu podawania leku
Nie dające się przewidzieć, niezależne od dawki, o podłożu immunologicznym lub genetycznym
Działanie toksyczne
Powstaje w wyniku przedawkowania leku (przekroczenia maksymalnej D leczniczej)
Działania związane z efektem farmakologicznym:
Tachyfilaksja (odwrażliwienie)
zachodzi gdy kolejne podania leku wywołują coraz słabszy efekt aż do całkowitego zaniku działania.
Tolerancja
gdy dla uzyskania tej samej reakcji konieczne są coraz wyższe dawki leku. Zwykle jest używane na określenie zjawiska rozwijającego się wolniej niż tachyfilaksja.
Oba zjawiska mogą być spowodowane zmianami w receptorach, zmniejszeniem ilości receptorów, wyczerpaniem mediatorów, zwiększeniem szybkości metabolizmu, adaptacją fizjologiczną.
Kumulacja
gromadzenie się leku w organizmie; może wystąpić, gdy lek jest podawany w zbyt krótkich odstępach czasu i ustrój nie nadąża z eliminacja leku. Skutkiem może być przedłużenie działania leku, zwiększenie siły działania a nawet przedawkowanie.
Lekozależność
przymus przyjmowania leku w celu osiągnięcia określonych doznań.
DEFINICJE:
FARMAKOKINETYKA
What the body does to the drug
Oddziaływanie organizmu na lek
FARMAKODYNAMIKA
What the drug does to the body
Oddziaływanie leku na organizm
FARMAKOKINETYKA
Schemat losów leku w ustroju:
TABLETKA
Losy leku w organizmie:
L - liberation (uwolnienie)
A - absorbtion (wchłanianie)
D - distribution (rozmieszczenie)
M - metabolism (metabolizm)
E - excretion (wydalanie)
Uwalnianie substancji leczniczych (L) :
Polega na rozpadzie postaci leku (tabletki, kapsułki itp.), rozpuszczeniu substancji czynnej i dyfuzji z miejsca podania do miejsca wchłaniania. Szybkość uwalniania zależy od postaci leku.
Występują postacie leków o przedłużonym działaniu
tabletki wielowarstwowe do podania doustnego,
olejowe roztwory estrów niektórych leków,
leki do wstrzyknięć w postaci zawiesin.
Wchłanianie leku (A):
We wszystkich drogach podania z wyjątkiem dożylnej
Podanie pozanaczyniowe nie zapewnia całkowitego wchłonięcia leku z krążenia
Przenikanie leku
Transport leków przez błony biologiczne:
Dyfuzja bierna
zgodnie z gradientem stężeń, bez zużycia energii; o szybkości decyduje wielkość cząsteczki (im mniejsza tym łatwiej lek dyfunduje), rozpuszczalność w lipidach (im lepiej rozpuszczalna, tym lepiej dyfunduje), stopień jonizacji (niezdysocjowane cząsteczki przechodzą łatwiej niż zdysocjowane).
dotyczy większości leków
słabe wchłanianie (transport bierny) silnych elektrolitów - wysoki stopień jonizacji w jelitach
np. wekuronium, suksametonium - N 4-rz. silne zasady podawane IV
Dyfuzja ułatwiona
zgodnie z gradientem stężeń, bez zużycia energii, przy pomocy nośników białkowych.
aminokwasy, cukry, wit. B12
Transport czynny (aktywny)
wbrew gradientowi stężeń, przy udziale nośników i nakładzie energii (ATP), np. lewodopa (parkinsonizm), fluorouracyl (przeciwnowotw.), Fe2+, Ca2+
Pinocytoza
przenikanie kuleczek cieczy w postaci wodniczki przez błony do komórek.
DROGI PODANIA LEKU:
Droga doustna:
wchłanianie w znacznej większości w jelitach
czynniki wpływające na wchłanianie leków
Szybkość pasażu jelitowego
perystaltyki (migrena, neuropatia cukrz., blokery muskaryn.) wchł.
perystaltyki wchł. (metoklopramid w migrenie) wchł p-bólowych)
perystaltyki wchł.
posiłek szybk. wchł. (wyjątki np. propranolol)
Przepływ krwi przez naczynia trzewne
przepł. (np. posiłek) wchł.
hipowolemia przepł. wchł.
Rozmiar cząstek leku / postać preparatu
kapsułki / tabletki opłaszczone szybk. uwalniania i wchł.
minipompy osmotyczne
Czynniki fizykochemiczne
wiązanie się leków z innymi lekami lub innymi związkami (tetracykliny + Ca 2+, warfaryna/tyroksyna + cholestyramina) wchł.
Niektóre leki specjalnie nie ulegają wchłonięciu, bo służą do leczenia chorób przewodu pokarmowego:
wankomycyna, mesalazyna, olsalazyna
Biodostępność - procent ilości leku, która dostaje się do krwi z miejsca podania; może być różna w różnych preparatach tej samej substancji aktywnej!
nie uwzględnia szybkości wchłaniania (ważne! szybkość osiągane początkowe stężenie we krwi)
Biorównoważność - ta sama substancja + te same własności biofarmaceutyczne i farmakokinetyczne
Droga podjęzykowa
wtedy, gdy lek jest nietrwały w kwaśnym środowisku żołądka/ulega szybkiemu metabolizmowi w wątrobie (pominięcie układu wrotnego - bezpośrednie przejście do krążenia obwodowego)
Nitrogliceryna
Droga doodbytnicza
efekt miejscowy (np. przeciwzapalny, leki na hemoroidy)
efekt ogólny u pacjentów nie mogących przyjmować leków doustnie (operacje, wymioty) lub dożylnie (dzieci w stanie padaczkowym)
duże różnice międzyosobnicze
wchłanianie niepewne (zazwyczaj niewielki stopień wchłaniania)
Na skórę
efekt miejscowy (np. przeciwzapalny)
efekt ogólny - leki transdermalne, toksyny (np. insektycydy fosforoorganiczne)
leki w plastrach (preparaty nikotyny, Evra)
brak efektu pierwszego przejścia (ominięcie wątroby, stała szybkość uwalniania)
aerozole donosowe (leki peptydowe)
Droga inhalacyjna
wziewne narkotyki chirurgiczne (płuca - droga wchłaniania i wydalania) możliwość szybkiej korekty stężenia leków w osoczu
efekt miejscowy na oskrzela (glikokortykosteroidy, rozszerzające oskrzela) powinny słabo wchłaniać się na śluzówce (np. ipratropium - zawiera N 4-rz)
Iniekcja dożylna
najszybsza i najbardziej pewna droga podania
bolus bardzo wysokie stężenie leku
podawanie w kroplówkach uniknięcie wysokich dawek w osoczu
lidokaina, anestetyki (propofol), przeciwpadaczkowe (diazepam)
Iniekcja podskórna/domięśniowa
szybszy efekt niż podanie doustne
szybkość zależy od miejsca podania
dyfuzja przez tkanki
szybkość przepływu krwi i unaczynienie
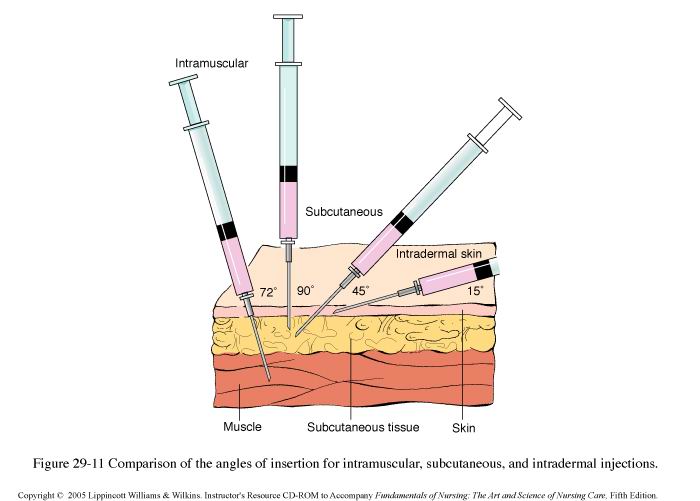
Droga podoponowa
niektóre leki (metotreksat - białaczki u dzieci)
bupiwakaina (znieczulenie całego obszaru ciała lekiem znieczulającym miejscowo)
opioidowe leki przeciwbólowe
aminoglikozydy w leczeniu lekoopornych infekcji OUN
Dystrybucja leków w ustroju (D
Kompartment
zespół tkanek lub narządów, których wspólną cecha jest zdolność do równoczesnej dystrybucji leku.
Komparment centralny (1)
krążenie ogólne.
Kompartment obwodowy (tkankowy, 2)
tkanki i narządy organizmu.
Model jednokompartmentowy
równomierne rozmieszczenie leku w całym organizmie, szybka dystrybucja leku.
Model dwukompartmentowy
gdy lek ma określone powinowactwo do tkanek i narządów i jego rozmieszczenie w organizmie jest nierównomierna.
Substancja lecznicza ulega dystrybucji z kompartmentu 1 do kompartmentu 2. Proces dystrybucji polega na przenikaniu leków i ich metabolitów przez błony biologiczne na zasadzie transportu biernego i czynnego. O szybkości dystrybucji decyduje szybkość przepływu krwi przez tkanki, szybkość transportu przez błony biologiczne, wiązanie leku z białkami krwi i tkanek.
Szybkość przepływu krwi
ponieważ jest różna w równych tkankach i narządach ze względu na ich zróżnicowane ukrwienie, rozmieszczenie leku jest niejednakowe, lek jest szybciej transportowany do tkanek dobrze ukrwionych.
w wodzie równowaga między związanymi z białkami a wolnymi, w tłuszczach tylko formy wolne (mogą przenikać przez błony)
Bariera krew-mózg, bariera naczyniowo-mózgowa
Składa się z kilku barier (krew-mózg, krew-PMR, PMR-mózg).
Z punktu widzenia fizjologicznego zachowuje się jak błona komórkowa i leki przenikają przez nią według ogólnych zasad - cząsteczki małe niezjonizowane, niezwiązane z białkiem i rozpuszczalne w tłuszczach przechodzą najłatwiej.
Szczelniejsza niż inne bariery, ponieważ w OUN praktycznie nie ma przestrzeni pozakomórkowej, a komórki glejowe mogą utrudniać docieranie leku do neuronów
przechodzą tylko leki lipofilowe (penicylina)
w niektórych miejscach nieszczelna
rozszczelnia się pod wpływem stresu
Jej przepuszczalność zwiększa się znacznie dla wielu leków i innych związków w stanach zapalnych opon mózgowych
Bariera łożyskowa
Każdy lek działający na ciężarną działa również na płód (opóźnienie 40-60 min.)
Objętość dystrybucji (Vd)
całkowita wchłonięta dawka leku podzielona przez jego stężenie we krwi. Jeżeli lek jest równomiernie rozmieszczony w organizmie, to wielkość ta odpowiada objętości, w której rozpuszczony jest lek, a więc rzeczywistej objętości dystrybucji. Pozorna objętość dystrybucji - objętość płynu, w jakiej należałoby rozpuścić wchłoniętą dawkę leku, aby uzyskać takie stężenie jak we krwi. Wskazuje w jakim stopniu lek rozprzestrzenia się w organizmie. Mała wartość wskazuje na duże stężenie leku we krwi, duża świadczy o rozprzestrzenieniu leku w tkankach i narządach.
Metabolizm leków (M):
biochemiczny proces przemian leków w organizmie. Zachodzi głównie w wątrobie przy udziale enzymów mikrosomalnych.
Wiele leków może działać nie tylko jako forma wyjściowa, ale ulega przemianie do czynnych metabolitów, mających pożądane działanie farmakologiczne.
Metabolit może wykazywać działanie silniejsze i dłuższe niż macierzysty związek.
Możliwa jest też taka sytuacja, że podawany lek jest nieczynny (prolek, prekursor, pro-drug), a czynny jest dopiero metabolit.
Niekiedy metabolity posiadają nieco inne działanie niż pierwotna postać (kwas salicylowy, feksofenadyna).
Metabolizm zachodzi w 2 fazach:
Reakcje I fazy - procesy rozkładu, do których zalicza się utlenianie, redukcję, hydrolizę. Mogą one:
Zmienić charakter leku - kierunek i siłę działania leku
Stanowić przygotowanie do II fazy
Spowodować unieczynnienie leku
W wyniku reakcji I fazy mogą powstać metabolity aktywne lub nieaktywne.
Reakcje II fazy - procesy syntezy, do których zalicza się acetylację, sprzęganie z aminokwasami (glicyna, kwas glukuronowy), węglowodanami, lub kwasem siarkowym. W wyniku tych reakcji powstają związki nieaktywne, niespolaryzowane, ulegające szybkiemu wydaleniu z organizmu.
Reakcje I fazy
Układ cytochromu P-450 (max absorbcji 450 nm) - enzymy mikrosomalne wątroby
enzym polimorficzny - liczne formy o określonej specyfice substratowej, polimorfizm w obrębie jednej formy
oksydacja (zamiana -H na -OH) przy udziale O2, jonów Fe zawartego w hemoproteinie P-450 oraz reduktazy NADPH-P450
różne formy enzymu u różnych gatunków - znaczenie przy badaniach przedklinicznych leku
niektóre leki współzawodniczą o te same izoformy cytochromu P-450, a niektóre leki nie są substratami, ale hamują te enzymy (np. chinidyna, ketokonazol) - ma to istotne znaczenie przy interakcjach
niektóre leki indukują enzymy mikrosomalne (rifampicyna, etanol, karbamazepina) - zwiększenie toksyczności paracetamolu
zwiększanie metabolizmu leków - zwiększenie lub zmniejszenie ich toksyczności
Inne enzymy
dehydrogenaza alkoholowa metabolizm etanolu i metanolu
oksydaza ksantynowa 6-merkaptopuryna
monoaminooksydaza aminy katecholowe
warfaryna - redukcja przy pomocy P-450
hydroliza wiązań estrowych (szczególnie podatne) i amidowych w osoczu i tkankach
Reakcje II fazy
Głównie - sprzęganie z kwasem glukuronowym
α-glukuronian + związek zwiazek-β-glukuronian (katalizowane przez transferazę glukuronianową)
mała specyfika enzymu - wiele leków i substancji endogennych (bilirubina, kortykosteroidy nadnerczowe)
Acetylacja przy udziale acetylo-CoA
Metylacja przy udziale S-adenozylo-metioniny
najczęściej w wątrobie, częściowo również w płucach i nerkach
Efekt pierwszego przejścia (EPP, first pass effect)
Metabolizm leków zanim dotrą do krążenia obwodowego (wątroba po przejściu przez układ wrotny lub ściana jelita)
Zjawisko zmniejszania się ilości substancji leczniczej w czasie przenikania przez błony przewodu pokarmowego i przejścia przez wątrobę. Jest wynikiem metabolizmu SL między miejscem wchłaniania a krążeniem ogólnym
przy podaniu doustnym - wyższe dawki niż przy podaniu dożylnym (nawet 10-krotnie!)
duże zróżnicowanie osobnicze - trudno przewidzieć efekt określonej dawki leku u konkretnej osoby, ludzie bardzo różnią się między sobą szybkością metabolizmu wątrobowego, jest jedną z przyczyn indywidualnej wrażliwości na leki
U osób z dużym uszkodzeniem wątroby leki podlegające EPP działają znacznie silniej niż u osób zdrowych
nitrogliceryna, lewodopa, lidokaina, salbutamol
Aktywne farmakologicznie metabolity leków
niektóre leki - proleki prekursory, których metabolity dopiero uzyskują aktywność biologiczną
niekiedy metabolity posiadają nieco inne działanie niż pierwotna postać
kw. salicylowy jest pozbawiony działania antyagregacyjnego aspiryny
feksofenadyna nie posiada właściwości proarytmicznych terfenadyny
toksyczne metabolity paracetamolu, cyklofosfamidu, metanolu, glikolu etylenowego
Czynniki wpływające na M leków:
Czynniki genetyczne
głównie genetycznie uwarunkowana aktywność enzymów
Płeć
mężczyźni maja większą aktywność enzymów mikrosomalnych wątroby, stąd szybciej metabolizują leki
Wiek
aktywność enzymów mikrosomalnych wątroby jest mniejsza u małych dzieci i w wieku podeszłym
Stany patologiczne
dotyczące głównie wątroby, mogą wydłużać czas działania lub upośledzać powstawanie aktywnych metabolitów
Droga podania
efekt pierwszego przejścia
Interakcje
z lekami wpływającymi na aktywność enzymów mikrosomalnych wątroby
Eliminacja przez nerki (E):
Filtracja kłębkowa
wszystkie leki z wyjątkiem wysokocząsteczkowych (np. heparyny)
w formie niezwiązanej z białkami osocza - leki silnie wiążące się z białkami (np. warfaryna) nie jest filtrowana
Wydzielanie kanalikowe
ok. 80% leków
transport aktywny (wbrew gradientowi stężeń)
niezależny od wiązania do białek osocza
nieselektywne mechanizmy nośnikowe
dla słabych kwasów (np. furosemid, indometacyna, penicyliny)
dla słabych zasad (np. amylorid, morfina, chinina)
konkurencja pomiędzy lekami o układ nośnika - probenecyd obniża aktywne wydalanie penicyliny
Reabsorpcja
wraz z wodą z moczu pierwotnego
głównie dotyczy leków lipofilowych (np. digoksyna nie jest reabsorbowana i osiąga znacznie wyższe stężenia w moczu niż w osoczu - ok. 100x)
może być regulowana poprzez zmianę pH moczu - leki kwaśne lepiej wydalają się w moczu zasadowym i odwrotnie bo wtedy są zjonizowane (alkalizacja moczu przy przedawkowaniu aspiryny)
Eliminacja z żółcią i krążenie jelitowo-wątrobowe
W postaci sprzężonej z glukuronianami
w jelicie następuje hydroliza wiązania z glukuronianem i reabsorpcja lek przechodzi z powrotem do wątroby i do krążenia obwodowego (morfina, estradiol)
PODSTAWOWE POJĘCIA FARMAKOKINETYCZNE
Stała eliminacji (K)
oznacza szybkość znikania leków z ustroju.
Biologiczny okres półtrwania leku (t1/2)
określa czas, po upływie którego stężenie leku we krwi zmniejszy się o połowę; t1/2 = 0,693/K.
Stan stacjonarny stężenia leku
występuje gdy po wielokrotnym podawaniu leku ustali się równowaga między szybkością wchłaniania i wydalania leku. Zwykle pojawia się po czasie równym 5 okresom biologicznego półtrwania leku.
Powierzchnia pod krzywą (area under the curve; AUC)
pole pod krzywą opisującą przebieg w czasie stężenia leku we krwi. Jest ono miarą biologicznej dostępności leku.
FARMAKODYNAMIKA
Receptory
wysoko wyspecjalizowane białka, które odbierają informacje ze środowiska zewnątrzkomórkowego i przekazują je do elementów wykonawczych komórki (efektorów). Leki naśladują działanie endogennych przekaźników lub hamują działanie receptorów.
Powinowactwo
zdolność wiązania leku z receptorem; określa stopień łączenia się cząsteczek leku z receptorem.
Aktywność wewnętrzna
zdolność do aktywacji receptora.
Ligand
związek łączący się z receptorem, czyli mający do niego powinowactwo.
Agonista
ligand aktywujący receptor, czyli mający powinowactwo i aktywność wewnętrzną; w wyniku jego związania z receptorem dochodzi do uruchomienia układów efektorowych (zmiana potencjału błony komórkowej, zmiana metabolizmu komórki lub ekspresji genów). Pełny agonista - może wywołać maksymalną reakcję, czyli największe działanie, jakie może być osiągnięte w danej tkance w danych warunkach; częściowy agonista - może wywołać tylko mniejsze działanie.
Antagonista - ligand blokujący receptor, posiadający powinowactwo, ale pozbawiony aktywności wewnętrznej; blokując dostęp substancji aktywujących do receptora wywiera określone działanie farmakologiczne.
Aktywność konstytutywna პ brak liganda faworyzuje stan spoczynkowy; pełny agonista - ma silne preferencje do stanu pobudzenia i przesuwa równowagę w tym kierunku; częściowy agonista - ma słabsza preferencję do stanu pobudzenia i nieznacznie przesuwa równowagę w tym kierunku; antagonista - nie ma preferencji, nie przesuwa stanu równowagi, ale uniemożliwia wiązanie agonistów z receptorami; odwrotny agonista - ma silną preferencję do stanu spoczynkowego i przesuwa równowagę w tym kierunku.
Interakcje leków
Zjawisko polegające na wzajemnym oddziaływaniu podanych równocześnie kilku leków, w wyniku czego zmienia się końcowy wynik działania niektórych z nich
Fazy wzajemnego oddziaływania leków:
Faza farmaceutyczna
niezgodności leków występują poza organizmem, najczęściej podczas mieszania kilku leków do iniekcji lub wlewu
Faza farmakokinetyczna
Na etapie jego wchłaniania, wiązania z białkami krwi, transportu przez błony biologiczne, dystrybucji, biotransformacji, wydalania
Faza farmakodynamiczna
Interakcje farmakokinetyczne
Leki oddziałujące na wchłanianie innych leków
szybkość wchłaniania
ilość wchłoniętego leku - niektóre leki mogą wiązać inne leki i zapobiegać ich wchłanianiu np. cholestyramina, inne mogą wpływać ogólnie na wchłanianie wszystkich składników pokarmowych
Leki oddziałujące na dystrybucję innych leków
wiązanie z białkami osocza (warfaryna z tolbutamidem)
Leki oddziałujące na metabolizm innych leków
indukcja enzymów mikrosomalnych wątroby - barbiturany, gryzeofulwina, leki przeciwpadaczkowe, ryfampicyna - zwiększają metabolizm warfaryny i doustnych środków antykoncepcyjnych
obniżenie aktywności enzymów mikrosomalnych wątroby - cymetydyna, ciprofloksacyna, erytromycyna, ketokonazol.
Leki oddziałujące na wydalanie innych leków
np. NLPZ mogą obniżać kanalikowe wydalanie metotreksatu
Interakcje farmakodynamiczne
Leki działające na ten sam receptor/biochemiczny układ efektorowy (np. β-blokery i leki antyastmatyczne oddziałujące β-agonistycznie)
Leki działające na ten sam układ fizjologiczny (synergistyczne działanie grup leków) np. warfaryna i aspiryna
występują u wszystkich lub wielu leków z danej grupy
Leki stosowane równocześnie mogą działać niezależnie od siebie, lub efekt działania jednego z nich może być modyfikowany przez drugi zastosowany w tym samym czasie.
Antagonizm farmakologiczny - występuje gdy leki działają przeciwnie, dając w efekcie zmniejszenie lub całkowite zniesienie działań farmakologicznych.
Antagonizm konkurencyjny (kompetycyjny) - gdy dwa leki (agonista i antagonista) współzawodniczą o ten sam punkt uchwytu na receptorze. Można wówczas znieść działanie agonisty przez podanie antagonisty w odpowiednim nadmiarze i odwrotnie.
Antagonizm niekonkurencyjny (niekompetycyjny) - gdy dwa leki działają na ten sam receptor, ale maja na nim różne punkty uchwytu, czyli nie konkurują o miejsce wiązania. Nie można wówczas całkowicie znieść działania agonisty podając antagonistę.
Antagonizm funkcjonalny (czynnościowy) - gdy dwa leki o różnym punkcie uchwytu wywołują przeciwny efekt.
Antagonizm chemiczny - gdy dwa leki reagują ze sobą tworząc związek o słabszym działaniu lub nieczynny biologicznie.
Synergizm farmakologiczny - jest to jednokierunkowe działanie leków, efekt działania jest wynikiem sumowania lub potęgowania działania równocześnie podanych leków.
Synergizm addycyjny - gdy efekt działania dwóch lub więcej leków podanych razem jest równy sumie działania poszczególnych leków.
Synergizm hiperaddycyjny (potencjalizacja) - gdy efekt działania leków podanych razem jest większy niż suma działania poszczególnych składników.
Rodzaje następstw interakcji leków:
Osłabienie działania farmakologicznego i związana z tym utrata skuteczności leczniczej
Farmakologiczny antagonizm
Utrudnienie wchłaniania
Nasilenie procesów biotransformacji
Zwiększenie wydalania
Zwiększenie siły działania farmakologicznego lub działań niepożądanych i związana z tym zwiększona toksyczność leku
Farmakodynamiczny synergizm
Wypieranie leków z połączeń z białkami
Zahamowanie procesów biotransformacji
Zmniejszenie wydalania
Prawdopodobieństwo wystąpienia interakcji:
Polifarmakoterapia
Ambulatoryjne leczenie chorych i związany z tym brak możliwości konsultacji postępowania terapeutycznego z innymi lekarzami
Niebezpieczne zjawisko samoleczenia się chorych
Starszy lub bardzo młody wiek chorych i związane z tym fizjologiczne odmienności czynności układu krążenia, OUN, wątroby i nerek
Stosowanie leków działających, o niskim (wąskim) wskaźniku terapeutycznym oraz leków określonych jako substancje potencjalnie stwarzające największe niebezpieczeństwo wystąpienia interakcji
Ciężkość schorzenia, które współistnieje zwłaszcza z chorobami (niewydolnością) narządów eliminujących leki, jak wątroba i/lub nerki
Leki stwarzające największe prawdopodobieństwo wystąpienia interakcji:
Stosowane w anestezjologii
Wpływające na układ krzepnięcia krwi
p/cukrzycowe
NLPZ
Stosowane w chorobach układu krążenia
Hipolipemiczne
Teofilina
Antybiotyki
Psychotropowe
p/padaczkowe
p/nowotworowe
Cyklosporyna/takrolimus
Lek i jego metabolity w moczu, kale i żółci
Tkanki
Działanie leku
Dystrybucja
Lek w płynach
przewodu pokarmowego
Wchłanianie
Metabolizm
i wydalanie
Uwalnianie
Lek w ustroju
Lek we krwi
Wyszukiwarka
Podobne podstrony:
Zagadnienia do I kolokwium - poprawka, medycyna UMed Łódź, 3 rok, farmakologia, kolokwium 1
FARMAKOKINETYCZNE INTERAKCJE LEKÓW I ŻYWNOŚCI, KOSMETOLOGIA, ZDROWIE USTAWY I MEDYCYNA
farma kliniczna- pytania, Medycyna, Pobr materiały, V rok UMB-2015-09-30, V rok UMB, Farmakologia Kl
Farmakologia 2010 cd, Medycyna, Farmakologia, farma-testy
Leki objęte specjalnymi wykazami, Medycyna, Farmakodynamika i farmakostatyka
farmakologia kliniczna 2010, MEDYCYNA - ŚUM Katowice, V ROK, Farmakologia kliniczna
farma wstrzas, Medycyna, Pobr materiały, V rok UMB-2015-09-30, V rok UMB, Farmakologia Kliniczna
Farmakoterapia nadciśnienia tętniczeg1, Pomoce naukowe, studia, medycyna
lista lekow ANS CVS, medycyna UMed Łódź, 3 rok, farmakologia, kolokwium 2
Farmakologia Kolokwium nr 2 (IVrok lekarski), MEDYCYNA, III ROK, FARMAKOLOGIA
EGZAMIN FARMAKOLOGIA 2011, MEDYCYNA - ŚUM Katowice, V ROK, Farmakologia kliniczna
dawkowanie Zatrucia, Medycyna, Farmakologia, 24. Toksykologia
osteoporoza, medycyna zabrze SUM lekarski, FARMAKOLOGIA - PRZYDATNE MATERIAŁY, FARMAKOLOGIA KLINICZ
FK112, MEDYCYNA - ŚUM Katowice, V ROK, Farmakologia kliniczna
Farmakoterapia nadciśnienia tętniczeg3, Pomoce naukowe, studia, medycyna
więcej podobnych podstron