b) membranę stanowią duże kationy np. kationy czwartorzędowej soli amoniowej, sole kompleksowe metali przejściowych; są to elektrody czułe na aniony (ClO![]()
, BF![]()
, NO![]()
, Br-, Cl- itp.),
ELEKTRODY JONOSELEKTYWNE. WYZNACZANIE STĘŻENIA JONÓW CHLORKOWYCH METODAMI DODATKU WZORCA.
Elektrody jonoselektywne są to elektrochemiczne półogniwa, które selektywnie reagują na dany jon lub molekułę w obecności innych jonów zawartych w roztworze. Zasadniczą częścią elektrody jonoselektywnej jest membrana (np. trudno rozpuszczalna sól, szkło, półprzewodnik, wymieniacz jonowy itp.), która z jednej strony kontaktuje się z roztworem wewnętrznym membrany lub bezpośrednio z przewodnikiem wyprowadzającym, natomiast z drugiej strony z roztworem badanym. Na granicy faz membrana-roztwór badany wytwarza się różnica potencjałów zależna od aktywności określonego jonu, który występuje w fazie roztworu i w fazie membrany, mogąc łatwo przechodzić z jednej fazy do drugiej.
Elektrody jonoselektywne, ze względu na typ membrany, można podzielić na trzy grupy:
1) elektrody jonoselektywne z membranami stałymi:
a) elektrody z membranami homogenicznymi; elektrody szklane, monokrystaliczne (LaF3, Ag2S, CuS, CuSe), elektrody z membranami wykonanymi ze stopionych lub sproszkowanych, a następnie sprasowanych soli (CuS, Cu2S, AgCl, AgBr, AgI),
b) elektrody z membranami heterogenicznymi; polikrystaliczne (Ag2S w mieszaninie z CuSe, CuS, CdS, PbS, AgCl, AgBr, AgI, AgSCN, AgCN), substancje krystaliczne wbudowane w polimerową matrycę np. polichlorku winylu,
2) elektrody jonoselektywne z membranami ciekłymi:
a) membranoaktywną warstwę stanowi duży anion, np. kwasu alkilofosforowego(V) lub anion tetra-n-chlorofenyloboranowy, rozpuszczony w rozpuszczalniku organicznym, niemieszającym się z wodą. Elektrody takie nadają się do oznaczeń kationów (Ca2+ i Ba2+),
b) membranę stanowią duże kationy np. kationy czwartorzędowej soli amoniowej, sole kompleksowe metali przejściowych; są to elektrody czułe na aniony (ClO![]()
, BF![]()
, NO![]()
, Br-, Cl- itp.),
c) membrany zbudowane na bazie neutralnych ligandów (antybiotyki, cykliczne i acykliczne molekuły), np. elektroda do oznaczania jonów ![]()
zbudowana jest na bazie walinomycyny,
3) trzecia grupa to elektrody, których specyficzna budowa i zasada działania nie pozwala na zaliczenie ich do wcześniej wymienionych grup. Należą tu:
a) elektrody czułe na gazy, w których wykorzystuje się przepuszczającą gaz membranę i odpowiednią elektrodę wskaźnikową (elektrody do oznaczania CO2, SO2 i NH3),
b) elektrody enzymatyczne, reagujące na jony powstałe w wyniku rozkładu substancji organicznych (np. mocznika, L- i D- aminokwasów, amygdaliny itp.) przez określone enzymy.
Potencjał elektrody jonoselektywnej opisuje równanie Nikolskiego
![]()
(1)
gdzie: ![]()
- potencjał standardowy elektrody jonoselektywnej, zależny od rodzaju membrany i konstrukcji elektrody,
![]()
- aktywność jonu oznaczanego,
![]()
- aktywność jonu zakłócającego (interferującego),
R - stała gazowa,
T - temperatura w K,
z - liczba elektronów biorących udział w reakcji elektrodowej,
F - stała Faradaya,
![]()
, ![]()
- wartościowość jonu oznaczanego i zakłócającego,
![]()
- współczynnik selektywności: jon oznaczany - jon zakłócający.
Przez potencjał elektrody rozumiemy siłę elektromotoryczną ogniwa zbudowanego z danej elektrody i elektrody wodorowej o jednostkowej aktywności jonów wodorowych i prężności parcjalnej wodoru równej 1.013 . 105 Pa. W pomiarach potencjałów elektrod, zamiast niewygodnej w użyciu i czułej na różne czynniki elektrody wodorowej, stosuje się inne elektrody odniesienia, charakteryzujące się stałym potencjałem, niezależnym od składu roztworu badanego. Obecnie najczęściej stosowanymi elektrodami odniesienia są elektroda chlorosrebrowa (Ag | AgCl | Cl-) i kalomelowa (Hg | Hg2Cl2 | Cl-) (patrz ćwiczenie: „Wyznaczanie entalpii swobodnej (ΔG), Entalpii (H) i entropii (S) reakcji zachodzącej w ogniwie Clarka”).
Jak wynika z równania Nikolskiego, jednym z istotniejszych parametrów charakteryzujących praktyczną przydatność elektrody jonoselektywnej jest współczynnik selektywności. Im mniejsza jest od jedności wartość współczynnika selektywności tym większa jest specyficzność elektrody w stosunku do jonu oznaczanego w obecności jonu zakłócającego.
W przypadku elektrod z membranami krystalicznymi współczynnik selektywności związany jest z reakcjami tworzenia nowych trudno rozpuszczalnych związków, np. dla elektrody chlorkowej, której membranę stanowi sprasowany pod wysokim ciśnieniem trudno rozpuszczalny AgCl, współczynnik selektywności tej elektrody w stosunku do jonów bromkowych jest równy stosunkowi iloczynów rozpuszczalności chlorku i bromku srebra
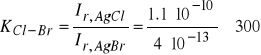
(2)
Taka wartość współczynnika selektywności oznacza, że trzystukrotnie mniejsze stężenie jonów bromkowych w porównaniu z jonami chlorkowymi będzie wnosić taki sam udział w potencjał elektrody jak jony chlorkowe.
W przypadku elektrod z membranami ciekłymi współczynnik selektywności zależy od ruchliwości jonów badanych i zakłócających lub ich asocjatów z substancją jonowymienną w fazie organicznej membrany oraz od współczynników podziału obu jonów pomiędzy fazę wodną i organiczną i stałych asocjacji w fazie membrany .
Innym parametrem charakteryzującym elektrody jonoselektywne jest nachylenie krzywej kalibracji. Krzywą kalibracji elektrody nazywamy wykres zależności potencjału tej elektrody od logarytmu z aktywności (krzywa aktywnościowa) lub stężenia (krzywa stężeniowa) danego jonu. Idealna elektroda powinna mieć tzw. "nernstowskie" nachylenie, równe RT/zAF. Często jednak zdarzają się odchylenia od tej wartości (najczęściej w kierunku wartości niższych), wynikające głównie z właściwości membrany i sposobu jej przygotowania.
Każda elektroda jonoselektywna charakteryzuje się ściśle określoną granicą oznaczalności, związaną z najniższym stężeniem jonu jakie można oznaczać w danym układzie. Jako granicę oznaczalności przyjmuje się stężenie oznaczanych jonów, przy którym odchylenie punktów pomiarowych od prostoliniowego przebiegu krzywej kalibracji wynosi 18/zA mV. Granice oznaczalności dla danej elektrody w znacznym stopniu zależą od typu jej membrany i obecności jonów zakłócających.
Należy również pamiętać, że elektrody jonoselektywne różnią się tzw. czasem odpowiedzi. Jest to czas potrzebny do osiągnięcia potencjału równowagowego przy przeniesieniu elektrody z jednego roztworu do drugiego, różniących się aktywnościami jonów potencjałotwórczych.
Podstawą wszystkich metod analitycznych z zastosowaniem elektrod jonoselektywnych jest empiryczna postać równania Nernsta
![]()
(3)
gdzie: E - SEM pomiarowego ogniwa elektrodowego,
Eo - SEM pomiarowego ogniwa elektrodowego w temperaturze 298 K, przy prężności parcjalnej substancji gazowych równej 1013 hPa i aktywności jonów mierzonych w roztworze równej jedeności,
±S - nachylenie charakterystyki elektrody; (+) dla kationów, (-) dla anionów (dla roztworów wodnych w temperaturze 25oC teoretyczna wartość S jest równa 59.16 mV/zA).
Można wyróżnić kilka metod analitycznych z zastosowaniem elektrod jonoselektywnych takich jak: metoda krzywych wzorcowych, metoda miareczkowania potencjometrycznego i metoda dodatku wzorca.
Metoda krzywych wzorcowych
Sporządza się serię roztworów oznaczanego jonu o różnych stężeniach lub aktywnościach, dla których mierzy się potencjał elektrody jonoselektywnej względem elektrody odniesienia (elektroda chlorosrebrowa, kalomelowa itp.). Nanosząc na wykresie zmierzone wartości potencjałów w zależności od logarytmu ze stężenia lub aktywności otrzymamy krzywą wzorcową (rys. 1). Stężeniowa krzywa wzorcowa w zakresie wysokich stężeń wykazuje odchylenie od liniowogo przebiegu związane ze zmniejszaniem się współczynnika aktywności badanego jonu ze wzrostem siły jonowej roztworu (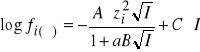
gdzie: ![]()
- współczynnik aktywności i-tego kationu lub anionu, A,B i C stałe, zi - ładunek jonu, a - efektywny promień jonu, I - siła jonowa roztworu: ![]()
).
Mierząc SEM ogniwa pomiarowego w roztworze badanym i porównując uzyskaną wartość z krzywą wzorcową znajdujemy stężenie lub aktywność jonu oznaczanego w roztworze. W metodzie tej należy szczególną uwagę zwrócić na to, aby skład roztworów wzorcowych był zbliżony (najlepiej taki sam) jak matryca roztworu badanego.
Do pomiaru SEM można używać każdego miernika potencjału, którego opór wejściowy jest co najmniej trzy rzędy wielkości większy od oporu wewnętrznego układu pomiarowego (multimetr, pH-metr, jonometr). Obecnie produkowane są jonometry, które w sposób bezpośredni pozwalają na oznaczenie aktywności jonów przy pomocy elektrod jonoselektywnych. Pomiar jest analogiczny do pomiarów pH.
Metoda miareczkowania potencjometrycznego
Dokładność metody miareczkowej zależy od: a) dokładnego ustalenia miana titranta, b) stałej i dokładnie znanej stechiometrii reakcji, c) od dokładności ustalenia punktu końcowego miareczkowania.
Elektrody jonoselektywne dają możliwość najbardziej obiektywnego rozpoznania punktu końcowego miareczkowania. Na krzywej miareczkowania tj. zależności SEM ogniwa od objętości dodanego titranta, występuje skok potencjału związany z punktem końcowym miareczkowania (punkt końcowy leży w punkcie przegięcia krzywej). Dokładną wartość punktu końcowego można uzyskać z pierwszej lub drugiej pochodnej krzywej miareczkowania.
W metodach miareczkowania potencjometrycznego z zastosowaniem elektrod jonoselektywnych można wyróżnić trzy typy miareczkowań: miareczkowanie kwasowo-zasadowe, strąceniowe i kompleksometryczne.
Metoda wykorzystująca ogniwa stężeniowe
W technice tej konstruuje się ogniwo stężeniowe z dwóch jednakowych elektrod jonoselektywnych. Zasada metody polega na tym, że w ogniwie stężeniowym SEM ogniwa równa się zeru, gdy aktywność oznaczanych jonów przy obu elektrodach jest jednakowa. Jeżeli będziemy dodawać do jednego z półogniw znane objętości roztworu wzorcowego lub rozcieńczać jedno z półogniw wodą destylowaną, możemy doprowadzić SEM ogniwa stężeniowego do zera i ze znajomości dodanej objętości obliczyć aktywność jonów w roztworze mierzonym.
Metody dodatku wzorca
Należą do nich metody jednokrotnego, dwukrotnego i wielokrotnego dodatku wzorca oraz metoda dodatku wzorca i rozcieńczenia.
Metodę jednokrotnego dodatku wzorca można stosować wtedy, gdy znamy nachylenie charakterystyki elektrody S. Podstawą tej metody jest wnioskowanie o początkowym stężeniu oznaczanego jonu w odmierzonej ilości roztworu próbki na podstawie zmian SEM po dodaniu dokładnie odmierzonej ilości roztworu wzorcowego tego jonu. Zmiana SEM ogniwa (![]()
E) związana jest ze stężeniem próbki (![]()
) i wzorca (![]()
) następującym równaniem:
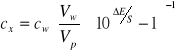
(4)
a uwzględniając efekt rozcieńczenia otrzymujemy:
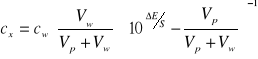
(5)
gdzie: Vp - objętość próbki,
Vw - objętość dodanego wzorca.
Można zastosować metodę odwrotną do opisanej poprzednio, tzn. do znanej objętości roztworu wzorcowego dodaje się określoną objętość próbki. Wówczas
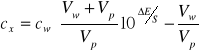
(6)
Metodę dwukrotnego dodatku wzorca stosuje się wtedy, gdy nie znamy nachylenia charakterystyki elektrody (S). Istotą tej metody jest pomiar SEM ogniwa złożonego z elektrody jonoselektywnej i elektrody odniesienia w roztworze badanym i po dwukrotnym dodaniu takiej samej objętości roztworu wzorcowego. Zmiany potencjału elektrody po każdym dodaniu wzorca będą wynosiły odpowiednio:
![]()
(7)
![]()
(8)
Dzieląc powyższe równania stronami, otrzymujemy
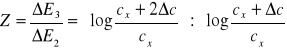
(9)
Ponieważ rozwiązanie tego równania względem cx jest skomplikowane, w załączonej tabeli podano niektóre wartości Z z odpowiadającymi im stosunkami stężeń cx/Δc.
Do wyznaczania stężenia oznaczanego jonu metodą wielokrotnego dodatku wzorca stosuje się metodę Grana, która polega na linearyzacji krzywej dodatku wzorca. W tym celu przekształcamy równanie:
![]()
(10)
do postaci:
![]()
(11)
Z powyższego równania wynika liniowa zależność wyrażenia ![]()
od stężenia wzorca ![]()
. Ekstrapolacja prostej do zerowej wartości ![]()
daje nam stężenie badanej substancji w próbce.
Jeżeli liczba moli substancji badanej w próbce wynosi ![]()
a dodanego wzorca ![]()
to stężenie całkowite w roztworze po dodaniu wzorca będzie równe 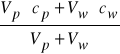
i wówczas równanie (10) przyjmie postać:
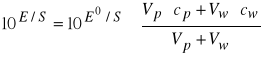
(12)
a dalej
![]()
(13)
Kreśląc zależność (Vp + Vw) 10E/S od objętości dodanego roztworu wzorcowego, otrzymamy linię prostą, która ekstrapolowana do zerowych wartości (Vp +Vw) 10E/S da nam wartość objętości wzorca, odpowiadającej objętości próbki o takim samym stężeniu jak wzorzec.
Metodę Grana stosuje się także do wyznaczania punktu końcowego miareczkowania potencjometrycznego.
Zagadnienia do opracowania
1. Potencjał elektrody, metody pomiaru.
2. Elektrody jonoselektywne - podział, budowa, potencjał.
3. Zastosowanie elektrod jonoselektywnych.
4. Metody dodatku wzorca.
5. Metoda ekstrapolacyjna Grana.
Literatura
1. Koryta J., Dvořák J., Boháčková V., Elektrochemia, PWN, Warszawa 1980, str. 225 - 233.
2. Cammann K., Zastosowanie elektrod jonoselektywnych, WN-T, Warszawa 1977, str. 74-142 i 184-209.
Aparatura
Jonometr lub inny miernik potencjału o dużym oporze wejściowym, chlorkowa elektroda jonoselektywna, chlorosrebrowa lub kalomelowa elektroda odniesienia z kluczem elektrolitycznym wypełnionym 1 M roztworem KNO3, szkło laboratoryjne.
Odczynniki
0,1 M KCl, 1 M KNO3.
Wykonanie ćwiczenia
1. Przygotować serię roztworów o następujących stężeniach jonów ![]()
:10-1, 3.10-2, 10-2, 3.10-3, 10-3, 3.10-4, 10-4, 3.10-5, 10-5, 3.10-6, 10-6 M.
2. Zaczynając od roztworu najbardziej rozcieńczonego, zmierzyć potencjał chlorkowej elektrody jonoselektywnej względem elektrody odniesienia (elektroda chlorosrebrowa lub kalomelowa mogą być wprowadzane do badanego roztworu tylko przez klucz elektrolityczny wypełniony roztworem KNO3 !!!).
3. Zmierzyć potencjał chlorkowej elektrody jonoselektywnej względem elektrody odniesienia w 50 cm3 (odmierzonych dokładnie pipetą) roztworu KCl o stężeniu 3.10-3 M, a następnie po każdorazowym dodatku 0.5 cm3 roztworu KCl o stężeniu 10-1 M (dodać 8 porcji wzorca).
Opracowanie wyników
1. Narysować stężeniową krzywą wzorcową chlorkowej elektrody jonoselektywnej ![]()
.
Obliczyć nachylenie S krzywej kalibracji, ![]()
oraz podać zakres prostoliniowości i granicę oznaczalności.
2. Znając nachylenie krzywej kalibracji S obliczyć stężenie jonów ![]()
w roztworze wyjściowym, korzystając ze zmierzonych wartości potencjałów E:
po dodaniu 0.5 cm3 roztworu wzorcowego,
po dodaniu 3.0 cm3 roztworu wzorcowego.
Obliczenia wykonać według wzorów z pominięciem i uwzględnieniem efektu rozcieńczenia.
3. Obliczyć stężenie jonów chlorkowych w roztworze wyjściowym według metody dwukrotnego dodatku wzorca:
dla ΔV = 0.5 cm3,
dla ΔV = 2.0 cm3.
Wartości Z i odpowiadające im wartości stosunku ![]()
podano w tabeli 1.
Za ![]()
przyjąć następującą wartość 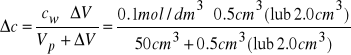
4. Narysować zależność (Vp+ Vw) 10E/S = f(Vw) i z przecięcia prostej z osią Vw (prosta przecina oś po stronie ujemnych wartości Vw), obliczyć stężenie jonów Cl- według równania:
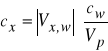
gdzie: ![]()
jest bezwzględną wartością objętości odczytanej z wykresu, ![]()
- stężeniem wzorca, a ![]()
- objętością początkową próbki.
5. Obliczyć procentowy błąd względny pomiaru dla każdej zastosowanej metody (zakładamy, że stężenie rzeczywiste jonów chlorkowych jest równe stężeniu analitycznemu 3.10-3 mola/dm3) i przeanalizować uzyskane wyniki.
Tabela 1.
Wartości Z i odpowiadające im wartości stosunku ![]()
Z |
cx/Δc |
Z |
cx/Δc |
Z |
cx/Δc |
Z |
cx/Δc |
1.270 |
0.100 |
1.480 |
0.548 |
1.590 |
1.029 |
1.700 |
1.894 |
1.280 |
0.113 |
1.485 |
0.565 |
1.595 |
1.056 |
1.705 |
1.948 |
1.290 |
0.126 |
1.490 |
0.582 |
1.600 |
1.086 |
1.710 |
2.006 |
1.300 |
0.140 |
1.495 |
0.600 |
1.605 |
1.116 |
1.715 |
2.066 |
1.310 |
0.154 |
1.500 |
0.618 |
1.610 |
1.147 |
1.720 |
2.126 |
1.320 |
0.170 |
1.505 |
0.637 |
1.615 |
1.179 |
1.725 |
2.190 |
1.330 |
0.186 |
1.510 |
0.655 |
1.620 |
1.213 |
1.730 |
2.256 |
1.340 |
0.203 |
1.515 |
0.675 |
1.625 |
1.245 |
1.735 |
2.326 |
1.350 |
0.221 |
1.520 |
0.694 |
1.630 |
1.280 |
1.740 |
2.397 |
1.360 |
0.240 |
1.525 |
0.714 |
1.635 |
1.315 |
1.745 |
2.470 |
1.370 |
0.260 |
1.530 |
0.735 |
1.640 |
1.353 |
1.750 |
2.549 |
1.380 |
0.280 |
1.535 |
0.756 |
1.645 |
1.391 |
1.755 |
2.629 |
1.390 |
0.302 |
1.540 |
0.778 |
1.650 |
1.430 |
1.760 |
2.711 |
1.400 |
0.325 |
1.545 |
0.801 |
1.655 |
1.469 |
1.765 |
2.801 |
1.410 |
0.349 |
1.550 |
0.823 |
1.660 |
1.510 |
1.770 |
2.892 |
1.420 |
0.375 |
1.555 |
0.847 |
1.665 |
1.554 |
1.775 |
2.985 |
1.430 |
0.399 |
1.560 |
0.870 |
1.670 |
1.598 |
1.780 |
3.088 |
1.440 |
0.427 |
1.565 |
0.896 |
1.675 |
1.643 |
1.785 |
3.193 |
1.450 |
0.455 |
1.570 |
0.920 |
1.680 |
1.691 |
1.790 |
3.301 |
1.460 |
0.485 |
1.575 |
0.946 |
1.685 |
1.738 |
1.795 |
3.416 |
1.470 |
0.516 |
1.580 |
0.973 |
1.690 |
1.787 |
1.800 |
3.536 |
1.475 |
0.532 |
1.585 |
1.000 |
1.695 |
1.840 |
1.805 |
3.664 |
Dyskusja wyników
1. Wytłumaczyć różnicę pomiędzy aktywnościową i stężeniową krzywą kalibracji. W jaki sposób można doświadczalnie sporządzić aktywnościową krzywą kalibracji.
2. Na podstawie wykresu stężeniowej krzywej kalibracji wytłumaczyć dlaczego tylko w pewnych zakresach stężeń można badanej elektrody używać do celów analitycznych. Z czym związany jest obszar, w którym potencjał elektrody nie zmienia się ze zmianami stężenia. Biorąc pod uwagę iloczyn rozpuszczalności AgCl (membrana elektrody chlorkowej) równy 1.1 . 10-10 podać dolną granicę stężenia jonów chlorkowych w stanie równowagi w rozcieńczonych roztworach, nie zawierających jonów interferujących.
3. Ocenić na podstawie obliczonych błędów i szybkości oznaczeń dokładność i zalety przebadanych metod.
4. Zaproponować sposób przeprowadzenia oznaczenia stężenia jonów chlorkowych metodą miareczkowania potencjometrycznego z zastosowaniem chlorkowej elektrody jonoselektywnej.
3
pM (-log cM)
SEM
[mV]
0
1
2
3
4
5
![]()
![]()
![]()
18/z [mV]
SEM0
(Vp+Vw)10E/S
10E/S
Vw
cw
- cp
Vx,w= - Vp . cp / cw
Rys 2. Metoda Grana
Rys. 1. Stężeniowa krzywa kalibracji elektrody