Wyjaśni pojęcia, podac przykłady
Proces - w znaczeniu termodyn. polega na zmianie stanu termodyn. układu.
Droga procesu - określona jest przez rodzaj i kolejnośc stanów termodynamicznych układu przy przejściu od stanu początkowego do stanu końcowego. Przez drogę rozumiemy stany termodynamiczne o parametrach np. p1, v1-stan początkowy, a stan końcowy o parametrach p2, v2.
Funkcja stanu - naz. taką, funkcję, której wartośc liczbowa jest określona jednoznacznie stanem termodyn. układu. Przyrost f-cji stanu w dowolnym procesie NIE ZALEŻY OD DROGI PROCESU, A ZALEŻY TYLKO OD STANU POCZĄTKOWEGO I KOŃCOWEGO. Funkcja stanu jest więc wielkością fiz. zależną tylko od stanu układu a nie od sposobu, w jaki został on osiągnięty.
Parametry stanu dzielimy na : ekstensywne i intensywne
Parametry ekstensywne - są proporcjonalne do wielkości układu i służą zasadniczo do charakteryzowania układu jako całości.
Wartość parametru ekstensywnego układu złożonego z podukładów jest sumą wartości tego parametru w poszczególnych podukładach. Do najważniejszych parametrów ekstensywnych należą: objętość układu V, liczba moli określonego składnika chemicznego układzie Nj.
Parametry intensywne - są niezależne od wielkości układu i zasadniczo charakteryzują układ lokalnie (w danym punkcie). Przykładami parametrów intensywnych są temperatura i ciśnienie.
Do grupy parametrów intensywnych należą również parametry właściwe powstające w wyniku odniesienia wielkości ekstensywnych do wielkości układu. W zależności od obranej charakterystyki wielkości układu mogą to by wielkości molowe, masowe, itd. jako przykłady wielkości właściwych możemy podać: objętość molową v = V/N,; gętość liczbowa składnika chemicznego układu nk = Nk / N.
Jeżeli układ termodynamiczny znajduje się w równowadze termodyn. , albo w stanie bliskim równowagi termodynamicznej, staramy się wyrazić energię przez parametry makroskopowe układu, takie jak masa, ciśnienie, temperatura, objętość czy skład chemiczny. Jeżeli jest to możliwe, podanie stanu układu określa jednoznacznie jego energię wewnętrzną U.
3. Omówić wykres zmian energii wew. w przemianie cyklicznej.
Stopień utlenienia danego janu przechodzi np. od kationu +3 do kationu +7
Zależność zmiany f-cji stanu od drogi przemiany
Zmiana f-cji stanu wywołana przejściem ze stanu A do stanu B jest taka sama na drodze A→P1→B jak na drodze A→P2→B. Zmiana f-cji stanu w przemianie cyklicznej A→P1→B→P2→A jest równa zero.
5. Ciepło przemian chemicznych.
Efekty reakcji chemicznych poza p, T, V zależą od ilości moli reagujących . Efekty cielne odnosi się do jednego mola. Ciepło tworzenia lub entalpia tworzenia jest efekt cieplny towarzyszący powstaniu 1 mola związku z pierwiastkiem tych samych warunkach ciśnienia i temperatury. Standardowe ciepło tworzenia jest efekt cieplny towarzyszący powstawaniu 1 mola zw. chem. z pierwszym w warunkach standardowych T = 298K, p = 101325Pa. Ciepło tworzenia ma znak przeciwny do ciepła reakcji roztworu.
7. Pojemność cieplna, ciepło właściwe, wyjaśnić związki pomiędzy Cp i Cv
Pojemność cieplna def. pod stałym cisn. oraz w stałej objętości. Jeżeli Cp [J/k] lub Cv [J/k] podzielimy przez masę, otrzymamy pojemność cieplna właściwą (ciepło właściwe), Cv,p [J/(g*k)], Cp,v [J/(g*k)].
Jeżeli zaś Cp lub Cv podzielimy przez liczbę moli, otrzymamy molową pojemność cieplna (ciepło molowe), Cp [J/(mol*K)] lub Cv [J/(mol*K)].
Ciepło właściwe - Cw danej substancji jest to ilośc ciepła potrzebna do ogrzania 1 grama substancji o 1 K.
Związki między Cp i Cv :
- W przypadku ogrzewania lub oziębienia 1 mola danej substancji w war. Stałej objętości
Cv = (∆U / ∆T)v.
- jeżeli ogrzewanie lub oziębienie następuje pod stałym cisn. to Cp = (∆H / ∆T)p
- jak stwierdzono dośw. Cp > Cv przyczyna tkwi w tym, że wymieniona w procesie izobarycznym energia jest zużywana nie tylko na podniesienie temp. danego układu ale i na wykonanie pewnej pracy objętościowej
- Cp - Cv = R = 8,31 J/mol*K dla gazu doskonałego
- Zależność Cp od temp. Dla gazów Cp = a +bT +cT2
- Dla cieczy i ciał stałych Cp = Cv , a ich wartość zależy od temp.
11. Wykazac na przykładzie, że dana reakcja jest reakcją endo- lub egzotermiczną.
endotermiczna - reakcja z pobraniem ciepła
egzotermiczna - reakcja z oddaniem ciepła.
Praca wykonana na układzie (sprężenie gazów) ozn. „+” . Pracę wykonaną przez układ (rozprężenie gazu) ozn. „-„.
Ciepło wydzielane przez układ np. reakcji egzotermicznej ozn. znakiem „-„. Ciepło pochłonięte przez układ np. w reakcji endotermicznej ozn. znakiem „+”
Zależnie od war. , w jakich dokonuje się dany proces - w zależności od parametrów ustalonych w czasie zachodzenie procesu rozróżniamy:
Proces izotermiczny ( T - const )
Proces izobaryczny ( p = const )
Proces izochoryczny ( V = const )
Proces adiabatyczny ( Q = 0 układ nie wymienia z otoczeniem energii w postaci ciepła)
Proces izoterm. - izobaryczny ( T,p = const )
Proces izoterm. - izochoryczny ( T, V = const )
Układy mogą wymieszać ze sobą energię. Jeżeli układ A przekazał np. 340J energii do układu B, to energie układu A zmalała.
Ani praca ani ciepło nie są rodzajem energii lecz sposobem jej przekazywania.
Przykład
Po wrzuceniu do wody małego kawałka sodu przebiega gwałtowna reakcja chemiczna. Powstaje gazowy wodór i wodorotlenek sodu, a wydzielone ciepło powoduje stopienie sodu, który następnie przybiera kształt kuli. 2Na + 2H2O --> 2NaOH + H2 + ciepło. Równanie pokazuje, że reakcja polega na przegrupowaniu atomów, nie zaś na ich tworzeniu.
13. Reguła Le Chateriera i jej praktyczne zastosowanie
Tzw. reguła przekory. Reguła ta mówi, że jeżeli w danym układzie znajdującym się w stanie równowagi zmieni się tylko jeden z parametrów ( stęż., temp. ciśn. ), wtedy zachodzą wew. układu przemiany zapobiegające zmianom tego parametru. ( substraty ↔ produkty).
Przykłady:
-Układ gazowy w zamkniętym pojemniku z tłokiem: gdy naciskamy na tłok, układ odpowiada zwiększaniem ciśnienia, aż do uzyskania równowagi sił.
-Woda nasycona ditlenkiem węgla pod ciśnieniem (np. woda mineralna): po otwarciu naczynia ciśnienie p ditlenku węgla nad roztworem gwałtownie spada - układ przeciwdziała zmianie wydzielając z roztworu gazowy dwutlenek węgla (po ponownym zamknięciu naczynia ustala się nowy stan równowagi).
15. Rzędowość reakcji
REAKCJE CHEMICZNE RÓŻNYCH RZĘDÓW
Reakcje I rzędu
Reakcja II rzędu
17. Reguła faz Gibasa dla układów 1 składnikowych
REGUŁA FAZ GIBBSA, PUNKT POTRÓJNY
Niektóre wielkości intensywne charakteryzujące układ fazowy jak: temperatura, gęstość, ciśnienie, współczynnik załamania światła są niezależne od objętości.
Wielkości, które zależą od objętości układu to wielkości ekstensywne np. masa.
W celu scharakteryzowania danego układu nie potrzeba podawać wszystkich wielkości.
Najmniejszą liczbę wielkości intensywnych konieczną do opisu stanu układu nazywa się liczba stopni swobody
Liczba stopni swobody z określa nam liczbę parametrów intensywnych, które można zmieniać w pewnych granicach, niezależnie od siebie, tak aby nie uległa zmianie liczba faz będących w równowadze w dany układzie.
REGUŁA FAZ GIBBSA
z = s - f + 2
s - liczba składników znajdujących się w układzie,
f - liczba faz
UKŁAD JEDNOFAZOWY z = s + 1
UKŁAD DWUFAZOWY z = s o jednym składniku np. ciecz - para -woda, lub układ dwu składnikowy - roztwór etanolu w wodzie
UKŁAD TRÓJFAZOWY jednoskładnikowy - woda
Reguła faz lub reguła faz Gibbsa to ważna zależność obowiązująca dla każdego układu w równowadze termodynamicznej łącząca liczbę faz w układzie, liczbę składników niezależnych oraz liczbę stopni swobody:
s = α − β + 2
gdzie:
s - liczba stopni swobody, czyli liczba zmiennych intensywnych, które można zmieniać bez jakościowej zmiany układu (bez zmiany liczby faz w równowadze)
α - liczba niezależnych składników, a więc takich, które nie dają się określić za pomocą zależności chemicznych poprzez stężenia innych składników (niezależnych).
β - liczba faz, a więc postaci materii jednorodnej chemicznie i fizycznie (np. roztwór, faza gazowa, kryształy o określonym składzie)
Punkty inwariantne
Ważnym pojęciem jest tzw. punkt niezmienniczy albo inwariantny dla którego mamy:
s = 0
W takim punkcie nie można zmienić żadnej zmiennej intensywnej bez zmiany ilości faz w układzie - przykładem takiego punktu jest punkt potrójny dla układu jednofazowego (np. punkt potrójny wody) lub punkt poczwórny dla układu dwuskładnikowego.
Układ jednoskładnikowy
Liczba składników, α = 1, stąd liczba stopni swobody s = 3 - β gdzie β to ilość faz w stanie równowagi.
Punkt potrójny
W punkcie potrójnym liczba faz w stanie równowagi β = 3, stąd liczba stopni swobody s = 1 - 3 + 2 = 0. Nie można wówczas zmienić ani żadnej ze zmiennych intensywnych (temperatury ani ciśnienia) bez opuszczenia równowagi 3 faz.
Krzywe równowagi
Krzywe równowagi, linie równowagi albo krzywe współistnienia określają współrzędne ciśnienia i temperatury (p,T) punktów na wykresie fazowym oznaczających β = 2 fazy w równowadze termodynamicznej. Dla krzywych równowagi w układzie jednoskładnikowym otrzymujemy s = 1 - 2 + 3 = 1 stopień swobody, a więc możliwość zmiany ciśnienia albo temperatury (ale nie obu naraz).
Równowaga ciecz-para (parowanie - skraplanie)
W równowadze są 2 fazy (β = 2), stąd liczba stopni swobody s = 1 - 2 + 2 = 1. Można wówczas zmienić temperaturę albo ciśnienie - jeżeli zmienimy temperaturę, ciśnienie musi zmienić się samo, jeżeli zmienimy ciśnienie, wówczas temperatura układu musi się odpowiednio dostosować - nie można zmienić dowolnie (nawet o niewielkie wartości) naraz obu parametrów intensywnych bez opuszczenia krzywej równowagi ciecz-para.
Układ powyżej punktu krytycznego (gaz)
Po przekroczeniu punktu krytycznego (końcowy punkt krzywej ciecz-para od strony wysokich ciśnień i temperatur), mamy do czynienia z 1 fazą (gazową - nie mogą istnieć ani ciecz ani ciało stałe), a więc liczba stopni swobody s = 1 - 1 + 2 = 2, czyli można zmieniać równocześnie 2 zmienne intensywne: ciśnienie i temperaturę.
Równowaga ciecz-ciało (topnienie - krzepnięcie) stałe
W równowadze są 2 fazy, liczba stopni swobody j.w. = 1. Można zmieniać temperaturę albo ciśnienie, ale nie obie zmienne jednocześnie.
Równowaga para-ciało stałe (sublimacja - kondensacja)
W równowadze są 2 fazy, liczba stopni swobody j.w. = 1. Można zmieniać temperaturę albo ciśnienie, ale nie obie zmienne równocześnie.
Czysta pojedyncza faza
Obszar (powierzchnia na wykresie fazowym) poza krzywymi współistnienia i punktem potrójnym oznacza czystą pojedynczą fazę:
powierzchnia ograniczona krzywą ciało stałe-gaz i krzywą ciecz-gaz (niskie ciśnienia i wysokie temperatury) określa obszar występowania pary (a powyżej punktu krytycznego - gazu)
powierzchnia ograniczona krzywą ciało stałe-ciecz i krzywą ciecz-gaz (wysokie ciśnienia i wysokie temperatury) określa obszar występowania cieczy (tylko poniżej punktu krytycznego - powyżej jest tylko 1 faza gazowa)
powierzchnia ograniczona krzywą ciało stałe-gaz i krzywą ciało stałe-ciecz (wysokie ciśnienia i niskie temperatury) określa obszar występowania ciała stałego
Punkt poczwórny
Z reguły faz Gibbsa wynika, że dla czystej chemicznie substancji nie może istnieć punkt poczwórny, czyli punkt gdzie w równowadze znajdują się cztery fazy (punkt poczwórny może istnieć w układach zawierających więcej niż jeden czysty składnik, np. woda + NaCl).
19. Równanie Gibbsa - Duhema
Obniżenie powierzchniowej entalpii swobodnej cieczy może nastąpić nie tylko przez zmniejszenie stosunku swobodnej powierzchni cieczy do jej objętości, ale również - w przypadku roztworów - przez odpowiednią zmianę składu warstwy powierzchniowej cieczy.
W związku z dążeniem układu ( przy T,p = const ) do min. Entalpii swobodnej, której część składową stanowi powierzchniowa entalpia swobodna, występować będzie tendencja do gromadzenia się na powierzchni cieczy cząsteczek substancji rozpuszczanych, obniżających napięcie powierzchniowe roztworu ( tzw. Substancji powierzchniowo aktywnych ). W roztworach wodnych substancjami takimi są Np. alkohol, mydło.
Adsorpcję na powierzchni cieczy określa ilościowo równanie Gibasa
Gdzie:
Г - różnica pomiędzy stężeniem danej substancji w warstwie powierzchniowej a jej stężeniem wewnątrz cieczy, liczona na 1m2 powierzchni, (mol * m-2)
c - stężenie tej samej substancji w całej masie roztworu, (mol * m-3)
R - stała gazowa (J*mol-1*K-1)
T - temperatura w K
dσ - zmiana napięcia powierzchniowego roztworu, odpowiadająca zmianie stężenia całkowitego (tj. w całej masie roztworu) o wartość dc ( σ w N*m-1 = J*m-2).
Z równania wynikają poniższe stwierdzenia.
- Jeśli ![]()
< 0, to Г > 0, czyli dana substancja gromadzi się na powierzchni cieczy. Substancja ta ma charakter powierzchniowo aktywny i powoduje obniżenie napięcia powierzchniowego. Takimi substancjami powierzchniowo aktywnymi są Np. mydła, kwasy tłuszczowe o długich łańcuchach.
- Jeśli ![]()
> 0, to Г < 0, następuje wówczas zubożenie warstwy powierzchniowej cieczy w rozpuszczoną substancję ( substancja jest wypychana z powierzchni). Takimi substancjami są Np. mocne elektrolity.
20. Szybkość reakcji - równanie kinetyczne i stała szybkości, podać 3 przykłady
Szybkość reakcji chemicznej określa się zmianą stężenia reagujących substancji
w jednostce czasu.
Dla substratów - średnia szybkość reakcji
![]()
Dla produktów - średnia szybkość reakcji
![]()
Szybkość rzeczywista reakcji chemicznej w danej chwili gdy

określa się jako:
![]()
Równanie kinetyczne reakcji jest to doświadczalnie wyznaczona krzywa zależności szybkości reakcji od stężenia reagentów uczestniczących w reakcji.
Stałe szybkości reakcji są to współczynniki proporcjonalności w równaniu kinetycznym, łączące szybkość reakcji ze stężeniami reagentów.
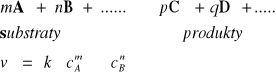
Szybkość reakcji jest proporcjonalna do iloczynu stężeń substratów.
Wykładniki potęgowe, oznaczone symbolami n i m noszą nazwę rzędów reakcji względem składnika A lub B a ich suma nazywana jest ogólnym rzędem danej reakcji.
Przykłady reakcji szybkich (np. wybuch bomby, spalanie wodoru) i wolnych (np. powstawanie węgla kamiennego, rdzewienie żelaza).
21. Równowagi zachodzące w roztworach kwasów i zasad.
Wpływ stężenia na stan równowagi chemicznej:
FeCl3 + KCNS Fe(CNS)3 + KCl
Jeżeli do kolby z Fe( CNS )3 dodamy FeCl3 lub KCNS to następuje przesuniecie równowagi tej reakcji i roztwór zabarwia się intensywniej na czerwono.
Jeżeli do kolby z Fe( CNS )3 dodamy KCl to stężenie rodanku żelaza w roztworze maleje i roztwór się odbarwia.
Wpływ ciśnienie i temperatury na stałą równowagi chemicznej:
Zgodnie z def. wydajności reakcji chem. B to stosunek liczby przereagowanych moli substratu n2 do początkowej ilości moli substratu n0.
B= n2/n0
Jak wpływa temperatura na stała równowagi chemicznej?
N2 + 3 H2 2 NH3
Jeżeli do układu doprowadzimy z zewn. Energie to zostanie zakłócony stan równowagi i w ukł. zacznie przebiegać w takim kierunku, aby zmniejszyć zaistniały wzrost temp.
Proces ten będzie endotermiczny rozkład amoniaku na wodór i azot
STALA ROWNOWAGI CHEMICZNEJ
W celu ilościowego scharakteryzowania stanu równowagi chem. wprowadzono pojecie stałej równania chem. Kc.
Kc= CCp * CDg/ CAn * CBm
W stanie równowagi chemicznej stosunek iloczynu produktu do iloczynu stężen substratu, podniesionych do odpowiednich potęg odpowiadających współczynnikom stechiometrycznym. Jest wielka stała w danych warunkach p i T.
Reguła La Chateriera i Brauna- reguła przekory
Jeżeli w danym układzie znajdującym się w stanie równowagi, zmieni się tylko jeden z parametrów( steż, temp, ciśn. ) wtedy zachodzą wewnątrz układów przemiany zapobiegające zmianom tego parametru.
substraty produkty
23 - Teoria kompleksu aktywnego
Drugą teorią dotyczącą kinetyki reakcji jest teoria kompleksu aktywnego lub inaczej teoria stanów przejściowych.
W teorii tej zakłada się, że reagujące ze sobą cząsteczki lub atomy w czasie reakcji chemicznej tworzą aktywny kompleks, który może się rozpaść, wytwarzając produkty lub odtwarzając substraty.
Podczas zbliżania się do siebie cząstek substratów AB i CD ulegają stopniowemu osłabieniu ich wiązania chemiczne i jednocześnie zaczynają tworzyć się nowe wiązania.
Powstaje przejściowy układ cząsteczek - kompleks aktywny o duż Kompleks aktywny jest w stanie równowagi z substratami
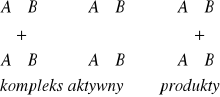
Przebieg reakcji z wytworzeniem aktywnego kompleksu - reakcja egzotermiczna
Przebieg reakcji z wytworzeniem aktywnego kompleksu - reakcja endotermiczn
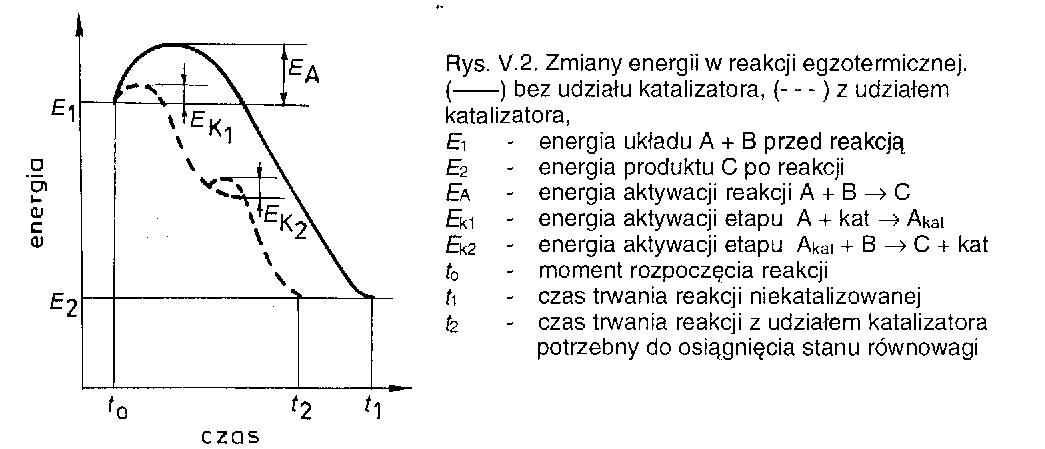
24. Kataliza
Katalizator powoduje jedynie zmianę mechanizmu reakcji, a więc nie jest w reakcji zużywany.
Co więcej, katalizator nie wpływa na równowagowe względne stężenie produktów i substratów, które to są określane wyłącznie przez termodynamikę, natomiast zwiększa szybkość osiągnięcia stanu równowagi.
W przypadku, gdy substrat wiąże się z katalizatorem bardzo mocno, to reakcja z katalizatorem może mieć wyższą barierę energetyczną - mamy wtedy do czynienia z katalizą ujemną a katalizator opóźniający reakcję nazywamy inhibitorem. Odpowiednie inhibitory opóźniają reakcję utleniania lub korozję metali i stopów.
Rozróżniamy katalizę zachodzącą w układach jednorodnych - homogenicznych
w układach niejednorodnych - heterogeniczną
W przypadku katalizy heterogenicznej istotną rolę odgrywają procesy adsorpcji zachodzące na powierzchni stałego katalizatora tworzącego w układzie reagującym odrębną fazę.
Katalizator taki nazywa się często kontaktem.
Zastosowanie tego typu katalizy umożliwia otrzymywanie H2SO4 metodą kontaktową, syntezę amoniaku, otrzymywanie HNO3 przez utlenienie amoniaku
W przypadku katalizy heterogenicznej istotną rolę odgrywają procesy adsorpcji zachodzące na powierzchni stałego katalizatora tworzącego w układzie reagującym odrębną fazę.
Katalizator taki nazywa się często kontaktem.
Zastosowanie tego typu katalizy umożliwia otrzymywanie H2SO4 metodą kontaktową, syntezę amoniaku, otrzymywanie HNO3 przez utlenienie amoniaku.
25. Układy dyspersyjne
Układy koloidalne lub krótko - koloidy, są to układy dyspersyjne, najczęściej dwuskładnikowe, o wyglądzie układów fizycznie jednorodnych, chociaż w rzeczywistości oba składniki nie są ze sobą zmieszane cząsteczkowo. Składnik tworzący fazę ciągłą układu nazywamy ośrodkiem dyspersyjnym lub rozpraszającym, drugi zaś fazą rozproszoną lub składnikiem rozproszonym. Faza rozproszona składa się z cząstek koloidalnych o wymiarach od 1 do 100 nm, a nawet do 500 nm. Należy tu jednak zaznaczyć, że do układów koloidalnych zaliczamy nie tylko te, które mają wszystkie trzy wymiary "koloidalne", lecz także i te, w których dwa a tylko nawet jeden wymiar jest koloidalny, czyli ma wartość od 1 do 500nm. W związku z tym układy koloidalne można podzielić na układy z cząstkami trójwymiarowymi, układy z cząstkami blaszkowatymi i układy z cząstkami nitkowatymi.
Najbardziej rozpowszechnione są układy koloidalne o ciekłym ośrodku dyspersyjnym, zwane roztworami koloidalnymi, liozolami lub zolami. Jeżeli ośrodek dyspersyjny jest wodą, zwane są hydrozolami, jeżeli alkoholem alkozolami, jeżeli benzenem - benzenozolami itd.
26. Właściwości koloidów
• Układami koloidalnymi lub koloidami nazywamy układy dyspersyjne, najczęściej 2 skład-nikowe o wyglądzie układów fizycznie jednorodnych, chociaż w rzeczywistości oba składniki nie są ze sobą zmieszane cząsteczkowo.
•
•Układy dyspersyjne o wymiarach od 2 m do 250 m nazywamy zawiesinami.
•Układy dyspersyjne o wymiarach poniżej 2 m nazywamy koloidami.
•Zole to układy koloidalne, w których fazą ciągłą jest ciecz.
•Jeżeli ośrodkiem dyspersyjnym jest woda, to mówimy o hydrozolach.
•Pirozole to układy koloidalne trwałe jedynie w wysokich temperaturach.
•Kriozole - lód w chloroformie (-200C)
•Koloidy wielkocząsteczkowe - białka - zol znajduje się w równowadze charakterystycznej dla roztworów rzeczywistych (albumina w wodzie)
•Koloidy micelarne (asocjacyjne) - składają się z cząstek powstałych przez asocjację cząstek substancji rozproszonej, ładunki + lub - powierzchni cząstek przyciągają dipole lub jony rozpuszczalnika. Może dojść do wytłumienia ładunku
+- + lub jego zmniejszenia - + -.
•Koloidy liofilowe - Jeżeli cząsteczki fazy rozproszonej łączą się z cząsteczkami ośrodka dyspersyjnego to mówimy o koloidach liofilowych. Ogólnie zjawisko to nazywamy solwatacją a dla wody hydratacją. W wyniku solwatacji następuje stabilizacja cząstek fazy rozproszonej.
Właściwości elektryczne
Ładunek elektryczny cząstek koloidalnych powstaje w wyniku adsorpcji czyli powierzchniowej adsorpcji jonów elektrolitu z roztworu. Tworzy się podwójna warstwa elektryczna złożona z powłoki wewnętrznej czyli adsorpcyjnej- przylegającej mocno do powierzchni i zewnętrznej czyli dywersyjnej. W zależności od tego jakie jony są adsorbowane na powierzchni cząstki koloidalnej to granula może być `'-„ lub `'+ „
29. Adsorpcja
Adsorpcja- proces polegający na pochłanianiu substancji ( tzw. absorbat) przez inną substancje (tzw. absorbent) i równomiernym rozprowadzeniu jej w całej masie absorbentu, najczęściej występuje absorpcja gazów przez ciecz.
Adsorpcja fizyczna- pomiędzy adsorbentem i adsorbatem występuję słabe oddziaływania międzycząsteczkowe zwane siłami Van der Waalsa. Ciepło adsorpcji fizycznej zbliżone jest do ciepła kondensacji adsorbatu i nie przekracza zwykle 40-50 kJ/mol. Adsorpcja fiz. jest na ogół procesem odwracalnym i przebiegającym stosunkowo dość szybko. Na powierzchni adsorbentu może powstać więcej niż jedna warstwa zaadsorbowanych cząstek.
Adsorpcja chemiczna- przy adsorpcji chemicznej jest tylko 1 warstwa zaadsorbowanych cząstek. Adsorpcja chemiczna wymaga zazwyczaj dość wysokiej energii aktywacji i jest procesem stosunkowo powolnym, szybkość jej może być zwiększona przez podwyższenie temperatury. Ciepło adsorpcji jest tego samego rzędu co ciepło reakcji chemicznych i zazwyczaj przyjmuje wartości kilkudziesięciu kJ/mol. Maleje ono w miarę wzrostu stopnia pokrycia powierzchni adsobenta.
30. Lepkość (tarcie wewnętrzne) właściwość płynów i plastycznych ciał stałych charakteryzująca ich opór wewnętrzny przeciw płynięciu. Lepkością nie jest opór przeciw płynięciu powstający na granicy płynu i ścianek naczynia.Zgodnie z laminarnym modelem przepływu lepkość wynika ze zdolności płynu do przekazywania pędu pomiędzy warstwami poruszającymi się z różnymi prędkościami.
Różnice w prędkościach warstw są charakteryzowane w modelu laminarnym przez szybkość ścinania. Przekazywanie pędu zachodzi dzięki pojawieniu się na granicy tych warstw naprężeń ścinających. Wspomniane warstwy są pojęciem hipotetycznym, w rzeczywistości zmiana prędkości zachodzi w sposób ciągły (zobacz: gradient), a naprężenia można określić w każdym punkcie płynu. Model laminarny lepkości zawodzi też przy przepływie turbulentnym, powstający np. na granicy płynu i ścianek naczynia. Dla przepływu turbulentnego jak dotąd nie istnieją dobre modele teoretyczne.
Lepkość dynamiczna wyrażająca stosunek naprężeń ścinających do szybkości ścinania:
Jednostką lepkości dynamicznej w układzie SI jest: kilogram·metr-1·sekunda-1Lepkość kinematyczna czasami nazywana też kinetyczną jest stosunkiem lepkości dynamicznej do gęstości płynu:
31 - Wyprowadzenie wzoru na prędkość opadania kuli w wiskozymetrze Hoeplera
Kulka o gęstości ρk opada z prędkością ν w cieczy o ρc poddana jest działaniu sił:
ciężkości
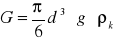
(1)wyporu
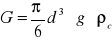
(2)oporu ośrodka
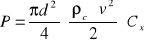
(3)
gdy wypadkowa tych sił = 0, kulka opada ze stałą prędkością ν = ν∞
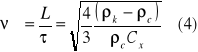
L = droga kulki
τ = czas opadania
d - średnica kulki
Cx - współczynnik oporu
Przy liczbach Reynoldsa Re < 0,2 siła oporu (wzór Stokesa) wynosi:
![]()
z porównania wzorów (3) i (5) dla ruchu laminarnego
![]()
zatem
![]()
32 - Prawo podziału Nernsta
Jeżeli do układu dwóch nie mieszających się ze sobą cieczy wprowadzimy niewielką ilość substancji A, rozpuszczalnej w obu cieczach, to po określonym czasie, w warunkach stałej temperatury i stałego ciśnienia, ustali się stan równowagi, przy którym stosunek stężeń substancji rozpuszczonej w obu cieczach będzie wielkością stałą.
Współczynnik podziału L jest wielkością zależną od temperatury rodzaju cieczy I i II oraz substancji rozpuszczonej A, niezależną natomiast od bezwzględnych wartości stężeń substancji A w obu cieczach.
Prawo podziału Nernsta stosuje się w powyższej formie tylko:
- do roztworów rozcieńczonych (składnik A spełnia w nich prawo Henryjego)
- wówczas gdy stan cząsteczkowy substancji rozpuszczonej jest w obu fazach ten sam.
Matematyczny zapis jednej z wersji prawa podziału Nernsta:
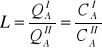
![]()
- aktywność wg Henryjego składnika A I fazy (aktywność przeliczalna na stężenia molowe)
![]()
- stężenie molowe składnika A w cieczy I.
33 - Korozja na styku metali
Korozja - jest to stopniowe niszczenie tworzyw metalowych i niemetalowych pod wpływem chemicznego i elektrochemicznego oddziaływania środowiska w wyniku którego zmieniają się stan i własności niszczonego tworzywa.
W przypadku metali rozróżnia się korozję chemiczną i elektrochemiczną
Korozja chemiczna - metali następuje w wyniku działania suchych gazów lub cieczy nie przewodzących prądu elektrycznego.
Przykładem korozji chemicznej może być działanie tlenu na metale w podwyższonej temperaturze w rezultacie którego na powierzchni metalu powstaje warstwa tlenku. Utlenianie metalu do jego tlenków nie zawsze jest procesem szkodliwym. Jeżeli warstwa tlenku jest dostatecznie zwarta i mocno związana z powierzchnią metalu, chroni ona (pasywuje) metal przed dalszym utlenianiem.
Glin jak wynika z jego położenia w szeregu napięciowym metali jest metalem nieszlachetnym. Jednak ten metal i jego stopy odznaczają się w środowisku obojętnym dużą odpornością na korozję, wynikającą z utworzenia się pasywnej, trudnorozpuszczalnej warstwy tlenku AlOOH. Podobne tlenkowe warstewki ochronne tworzy cynk, chrom i nikiel.
Do korozji chemicznej zalicza się także czernienie przedmiotów srebrnych na powietrzu. Głównym składnikiem ciemnego nalotu jest siarczek srebra powstający w reakcji srebra ze związkami siarki zawartymi w powietrzu.
Korozja elektrochemiczna - Korozja elektrochemiczna jest najbardziej powszechnym rodzajem korozji. Należy do niej powszechnie spotykane zjawisko korozji atmosferycznej, która spowodowana jest działaniem na metale wilgotnego powietrza i zawartych w nim zanieczyszczeń.
Korozja elektrochemiczna powstaje wskutek działania krótkozwartych ogniw na styku metalu z elektrolitem. Ogniwa te powstają w rezultacie niejednorodności chemicznej (lub fizycznej) metalu np. na styku różnych metali, bądź wskutek niejednorodności krystalicznej w strukturze metalu.
Korozja metali najczęściej objawia się poprzez powierzchniowe ubytki metalu (plamy i wżery), bądź przez obniżenie wytrzymałości metali. Najczęściej szybkość korozji określa się przez ubytek masy próbki metalu pod wpływem działania czynnika korodującego na jednostkę powierzchni i czasu.
Pojęcie półogniwa - Półogniwo stanowi układ składający się z metalu zanurzonego do roztworu jego soli. Potencjału półogniwa nie można zmierzyć bezpośrednio, można natomiast zmierzyć różnicę potencjału między półogniwem a innym układem o znanym lub umownie przyjętym potencjale. Takim układem jest elektroda wodorowa, której potencjał umownie przyjęto za równy zero. Elektroda wodorowa składa się z platyny pokrytej czernią platynową (bardzo subtelnie rozdrobniona platyna), która zanurzona jest w roztworze zawierającym jony wodorowe H+ o stężeniu 1 mol/dm3, oraz omywana jest strumieniem gazowego wodoru pod ciśnieniem 1013 hPa w temperaturze 25oC. Elektroda wodorowa stanowi układ odniesienia dla określenia potencjału każdego innego półogniwa
Powłoka anodowa - powłoka z metalu która w określonym środowisku korozyjnym jest mniej szlachetny niż metal chroniony a wiec jego potencjał elektrochemiczny jest bardziej ujemny niż potencjał chronionego metalu.
Potencjał normalny, szereg napięciowy metali - Potencjał normalny stanowi różnicę potencjałów między elektrodą wodorową a półogniwem składającym się z metalu zanurzonego w roztworze jego soli o stężeniu 1 mol/dcm3 w temperaturze 25oC. Wartości normalnych potencjałów półogniw dla różnych metali są różne.
Jeżeli normalne potencjały różnych metali uszeregujemy w kolejności wzrastających (lub malejących) wartości to otrzymamy szereg napięciowy metali. Szereg napięciowy wybranych metal i wodoru wygląda następująco:
Na< Mg< Al< Zn< Fe<Cd<Co<Ni<Pb<H<Cu<Hg<Ag<Au
Każdy metal tego szeregu wypiera następne metale z roztworu ich soli. Wszystkie metale występujące w szeregu przed wodorem posiadają ujemne potencjały normalne. Są to metale “nieszlachetne”, które wypierają wodór z kwasów czyli rozpuszczają się w kwasach z wydzieleniem wodoru. Wszystkie metale występujące w szeregu napięciowym za wodorem posiadają dodatnie potencjały normalne. Są to metale “szlachetne”, które nie wypierają wodoru z kwasów.
Położenie metalu w szeregu napięciowym a więc wartość jego potencjału normalnego posiada bardzo istotne znaczenie dla podatności metalu na korozję elektrochemiczną. Im bardziej ujemna jest wartość potencjału normalnego metalu tym większą posiada on tendencję do przechodzenia do roztworu.
RDZA
Szczególnym przypadkiem korozji elektrochemicznej są zjawiska korozji obserwowane na styku dwóch różnych metali. W obecności wilgoci na styku metali powstaje lokalne ogniwo składające się z dwóch półogniw. W półogniwie o mniejszym potencjale elektrochemicznym będzie dominowała reakcja powodująca przejście metalu w formie jonowej do roztworu:
Me1 ® Men+ + n e-
W półogniwie metalu o większym potencjale elektrochemicznym będzie dominowała reakcja odwrotna, w rezultacie której jony metalu z roztworu będą przechodziły do powierzchni metalu: Me2n+ + n e- ® Me
Zgodnie z tą zasadą metal mniej szlachetny zanurzony w roztworze soli metalu bardziej szlachetnego będzie wypierał jony metalu bardziej szlachetnego, zgodnie z reakcją:
Me1 + Me2n+ ® Me1n+ + Me2
Ochrona metali przed korozją: Typowym przykładem takiego oddziaływania jest zanurzenie blaszki cynkowej do roztworu zawierającego jony miedzi Cu2+. W tym przypadku obserwuje się wydzielanie miedzi z roztworu, która osadza się na blaszce cynkowej zgodnie z reakcją: Cu2+ + Zn ® Cu + Zn2+
Zabezpieczenie przed korozją elektrochemiczną stanowi tak zwana ochrona katodowa. Ochrona katodowa polega na połączeniu chronionej konstrukcji z metalem mniej szlachetnym, tworzącym anodę (protektor) ogniwa, natomiast katodą jest obiekt chroniony. Połączenie takiej anody z konstrukcją chronioną wykonuje się przez bezpośredni styk ( tzw. powłoki anodowe) lub za pomocą przewodnika. Za pomocą protektorów chroni się przed korozją duże obiekty stalowe, takie jak kadłuby statków, rurociągi i podziemne zbiorniki. Protektorami są blachy lub sztaby wykonane z metali aktywnych jak: cynk, magnez lub glin, połączone przewodami z obiektem chronionym. W utworzonym w ten sposób ogniwie anodą jest protektor, który ulega korozji. Po zużyciu protektory wymienia się na nowe. Identyczny efekt daje zastąpienie cynku złomem stalowym połączonym z dodatnim biegunem prądu stałego, podczas gdy chroniona konstrukcja połączona jest z biegunem ujemnym.
Ochrona przed korozją za pomocą powłok ochronnych:
- powłoki nieorganiczne: metalowe i niemetalowe
- powłoki organiczne: farby, lakiery, tworzywa sztuczne, smoła i smary
Niemetaliczne powłoki ochronne wywoływane są na powierzchni metali przez wytworzenie na niej związku chemicznego w wyniku zabiegów chemicznych jak:
- utlenianie (oksydowanie) mające na celu wytworzenie na chronionym metalu pasywnych warstewek tlenkowych
- fosforanowanie za pomocą kwasu fosforowego (tworzą się trudno rozpuszczalne fosforany metali)
- chromianowanie za pomocą mieszaniny kwasu chromowego i siarkowego w wyniku którego tworzą się powłoki chromianowe
34 - Korozja betonu
KOROZJA - korozja betonu
Ługująca
Kwasowa
Karbonizacja betonu
Siarczanowa
Magnezowa
Biologiczna
Wywołana przez substancje nieorganiczne
Wywołana przez substancje organiczne
Ługująca polega na wymywaniu związków rozpuszczalnych przez wody miękkie. Rozpuszczaniu ulega Ca(OH)2
Na płaszczu chłodni kominowej pojawiają się wykwity i nacieki białego osadu CaCO3 na wewnętrznej stronie płaszcza chłodni
Ca(OH)2 + CO2 =CaCO3 + H2O
Kwasowa polega na reagowaniu składników betonu ze środowiskiem kwaśnym - kwaśne deszcze, kwasy.
Eliminacja z betonu Ca(OH)2 węglanów, uwodnionych krzemianów i glinianów wapniowych
Zachodzi bardzo szybko
Efektem reakcji są rozpuszczalne w wodzie związki wapnia i tym samym zniszczona jest struktura betonu
Ca(OH)2 +2H+ =Ca+2 +2H2O
CaCO3 ↓ + 2H+ =Ca+2 +CO2↑ + H2O
CaO x SiO2 x nH2O + 2H+ =Ca+2 +H2SiO3 + nH2O
Karbonizacja betonu - polega na wiązaniu ditlenku węgla z powietrza przez obecny w betonie Ca(OH)2 w CaCO3, co wywołuje obniżenie odczynu betonu
Ca(OH)2 +CO2 =CaCO3 +H2O
CaCO3 +H2O + CO2 = Ca(HCO3)2
Dodatkowo przy zmienionym odczynie pH betonu stal zbrojeniowa nie jest pasywna i w obecności wilgoci, przy dostępie tlenu, ulega silnej korozji.
Skutkiem jest:
- zmniejszenie czynnego przekroju zbrojenia
- przede wszystkim spękanie betonu
- osadzanie otuliny
4. Powstałe produkty korozji mają dużą objętość i narstając warstwowa wokół prętów zbrojeniowych wywołują silne naprężenia w betonie
i powstawanie pęknięć rozchodzących się od zbrojenia w głąb betonu.
5. Trwałość żelbetu zależy od grubości otuliny zbrojenia.
KOROZJA SIARCZANOWA
Powoduje:
- utworzenie się w betonie soli pęczniejących (gipsu i etryngipsu), nierozpuszczalnych w wodzie
Ca(OH)2 + SO4-2 +2H2O = CaSo4 x 2H2O +2OH-
3CaO x Al2O3 x 6H2O +3CaSO4 + 25 H2) = 3CaO x Al2O3 x 3CaSO4 x 31H2O) - pękanie ścian w blokach
- tworzenie się doli Dedala i Candlota i innych jest bardzo niebezpieczne dla betonu
- duża ilość cząsteczek wody sprawie ze sole powiększają swoja objętość
- występują naprężenia wewnętrzne, których nawet najmocniejszy związany beton nie jest w stanie wytrzymać
- w warunkach zimowych powstały lód dodatkowo zwiększa objętość,
a na wiosnę z odpadami rozrastają się pleśnie - grzyby, przerastając całą ścianę!!!
PRZEMARZANIE BUDYNKÓW - DLACZEGO?
- To jest typowy przykład korozji wżernej betonu
- PN 80/B-01800 ograniczenia zawartości jonów siarczanowych w wodzie do 250g SO4-2/m3
KOROZJA MAGNEZOWA
- polega na oddziaływaniu soli magnezowych (skąd pochodzą jony Mg+2) na Ca(OH)2 w betonie
- tworzeniu się Mg(OH)2
- Mg(OH)2 - to osad galaretowaty nie posiadający żadnych właściwości wiążących.
- Ca(OH)2 +Mg+2 = Mg(OH)2 + Ca+2
KOROZJA BIOLOGICZNA
- to głównie korodujące działanie siarkowodoru na beton H2S pochodzi
z gnicia resztek organicznych)
- w reakcji powstaje rozpuszczalny w wodzie CaS
- Ca(OH)2 +H2S =CaS + 2H2O
- NH4+ +3/2 O2 + bakterie =NO- +2H+ +H2O
- syntetyzują metan CH4
Korozja rur kanalizacyjnych betonowych i żeliwnych - siarczany białka
w warunkach beztlenowych w ściekach ulegają redukcji do H2S.
- w przypadku rur żeliwnych na korozję mają wpływ także bakterie żelaziste
- Fe+2= Fe+3
- niszczenie powłok korozyjnych
- zużywanie produktów korozji Fe+2, przez co wzrasta transport elektronów a zmniejsza się wytrzymałość
- powstawanie mikrosfer kwasowych
KOROZJA WYWOŁANA PRZEZ SOLE NIEORGANICZNE
- polega na oddziaływaniu zawartych w wodach np. soli amonowych NH4Cl, NH4NO3, (NH4)2CO3 oraz przez działanie soli krystalizujących np. Na2CO3 x 10H2O i Na2SO4 x 10H2O działają one podobnie jak H2S i siarczany
KOROZJA WYWOŁANA PRZEZ RÓZNE SUBSTANCJE ORGANICZNE
Związki organiczne: kwasy organiczne, fenole, tłuszcze, oleje i cukry wywołują specyficzną korozję z utworzeniem związków organicznych ze składnikami betonu, przez co zmienia się jego wytrzymałość.
KOROZYJNOŚĆ WÓD
Wywołana nadmiarową nie stechiometryczną ilością CO2 w wodzie
(CO2 ogólny = wolny + agresywny + związany lub przynależny CO3-2, HCO3)
Część wolnego CO2 jest konieczna do utrzymania w roztworze równowagi węglanowo wapniowej i to jest przynależny CO2
- obecność w wodzie jonów chlorkowych i siarczanowych musi być normowana ponieważ wywołują one zwiększoną rozpuszczalność CaCO3
- same jony SO4-2 powodują tzw agresywność siarczanową betonu
Jony Cl- w stosunku do betonu i żelaza są agresywne, jeżeli przekroczona zostanie bariera 50 lub 100gCl-/m3
- obecność w wodzie znacznych ilości Na+, K+, Mg+2 przyspiesza procesy korozji.
35 - Ogniwa bimetaliczne i geologiczne - wyjaśnić zasadę działania
Ogniwo bimetaliczne - powstają wtedy kiedy dwa różne metale umieszczone są w środowisku agresywnym pozostają ze sobą w kontakcie elektrycznym.
- metal bardzo szlachetny pełni role katody
- metal bardzo aktywny jest anodą, jego korozja wzmaga się na skutek przepływu prądu
galwanicznego.
Ogniwo geologiczne - wstępnie podczas korozji gruntowej. Przyczyną powstania tego typu ogniw jest niejednorodność elektrolitu glebowego. Nierównomierne nawilżanie gleb różnice ich składu chemicznego różne napowietrzanie sprzyjają trwałemu rozmieszczenie ładunków elektrolitycznych np. metalowych konstrukcjach podziemnych
gleba lekko - spoista
miejscowa niejednorodność gleby
Wpływ głębokości ułożenia np. rozmieszczeń anodowych i katodowych obszarów ogniwa.
36 - Elektroliza
Elektroliza jako układ utleniająco - redukcyjny.
Elektroliza jest procesem wymuszonym, który przebiega w przewodniku elektrolitycznym, gdzie nośnikami ładunków elektrycznych są zarówno jony dodatnie (kationy) jak i ujemne (aniony). Ruch ładunków elektrycznych jest wymuszony poprzez podłączenie zewnętrznego źródła prądu stałego do elektrod zanurzonych w ciekłym przewodniku jonowym.
Proces elektrolizy zachodzi w stopionych solach i roztworach wodnych kwasów, zasad oraz soli i przeprowadzany jest w urządzeniach nazywanych elektrolizerami. Elektrolizery mają inną budowę jak ogniwa galwaniczne. W elektrolizerach w odróżnieniu od ogniwa, elektrody znajdują się w jednym naczyniu zawierającym roztwór jednego elektrolitu.
Przykłady elektrolizy:
Elektroliza stopionego chlorku sodowego NaCl
Elektroliza stężonego wodnego roztworu chlorku sodowego
Elektroliza rozcieńczonego roztworu NaCl
Elektroliza wodnego roztworu kwasu
Elektroliza zasad
37 - Związki powierzchniowo czynne: podział, wyjaśnić w oparciu o zjawisko adsorpcji proces prania
Tensydy i surfaktanty SA to substancje gromadzące się na powierzchni rozdziały faz gaz, ciecz, ciało stałe ciecz, gaz i modyfikujące jej właściwości
Typowa cząsteczka surfaktantu składa się z długiego ogona węglowodorowego rozpuszczalnego w węglowodorach, tłuszczach
i innych niepolarnych związkach oraz z grupy hydrofilowej tworzącej głowę np. z grupy CO2 penetrującej polarny rozpuszczalnik - wodę(CH2)17 - polarny lipofilowy, hydrofobowy łańcuch, nie lubi wody
COONa - polarna, hydrofilowa głowa lubi wodę
Tensydy:
Jonowe: kationowe, anionowe i amfolityczne
Niejonowe
1. grupa hydrofilowa (zwilżana przez wodę) - w tensydach to”
- grupa karboksylowa - COOH, sulfonowa - SO3H, aminowa - NH2, alkoholowa - CH2OH
2. grupa hydrofobowa (nie zwilżana przez wodę) to: reszta alkilowa - kwasy tłuszczowe, alkilobenzeny, tolueny i fenole
Anionowo czynne związki powierzchniowo czynne
R-COO-Me, sole estrów kwasu siarkowego R-O-SO3-Me i sole kwasów sulfonowych R-SO3-Me dysocjują na powierzchni czynny łańcuch hydrofobowy występujący w anionie
R-COO-Na=R-COO- +Na+
(mydła Na, K, oleje, tłuszcze, produkty kondensacji tłuszczów, estry pochodne kwasu fosforowego, polifosforany - detergenty domowe
i zastosowanie w przemyśle)
Kationowo czynne związki powierzchniowo czynne to: sole I, II, III oraz IV rzędowych amin alifatycznych najczęściej pirydyny
Łańcuch hydrofobowy jest w postaci kationowej takie mydła nazywa się często mydłami inwertowanymi
Tensydy amfolityczne - zwierają jednocześnie 2 grupy hydrofilowe - kwasową i zasadową, które nadają cząsteczce charakter amfoteryczny
Tensydy niejonowe nie tworzą jonów w roztworze, natomiast w wodzie ulegają hydratacji (estry kwasów tłuszczowych, alkilo-fenole)
W zależności od zastosowania tensydy dzieli się na: detergenty, zwilżacze, emulgatory, dyspergatory, środki pieniące
MYDŁA - są solami Na, K - kwasów karboksylowych o długich łańuchach
DETERGENTY - substancją powierzchniowo czynna jest z reguły kwas benzenosulfonowy podstawiony grupą alkilową o długim łańcuchu (R-C6H4SO3H).
Mydło jest detergentem wrażliwym na działanie twardej wody, natomiast detergenty nie są wrażliwe na twardość wody i bardzo łatwo wytwarzają pianę, co przy praniu zapewnia znaczną oszczędność środka piorącego.
TENSYDY detergenty syntetyczne:
Twarde ABS niszczą środowisko i jest zakaz ich używania - alkilobenzenosulfoniany
Miękkie LAS - aliloarylosylfoniany
Proszki zawierają około 20-25% substancji powierzchniowo czynnych i innych składników
Niektóre proszki do prania zawierają enzymy proteolityczne, na przykład protezę, które pomagają usunąć zabrudzenia pochodzenia białkowego - takie jak plamy z krwi czy jajek.
Tego typu proszki SA określane mianem proszków „biologicznych”
Płynne detergenty syntetyczne produkuje się dodając do roztworu składników substancji zwanej hydrotropem.
Zwiększa ona rozpuszczalność substancji w wodzie nie pozwalając tym samym składnikom płynu wytrącą się z roztworu.
Działanie substancji powierzchniowo czynnej w detergencie lub mydle polega na rozpuszczeniu się jej w obu fazach: wodnej i węglowodorowej na granicy - styku faz, czego wynikiem jest solubilizacja fazy węglowodorowej, umożliwiająca usunięcie jej z oczyszczonego przedmiotu
Działanie piorące polega na zanurzeniu hydrofobowych ogonów grup węglowodorowych w cząsteczkach tłuszczu i otoczeniu ich przez grupy hydrofilowe aktywnie oddziałujące z wodą
Detergenty syntetyczne używane się także w innych środkach czyszczących takich jak proszki czy mleczka do szorowania. Taki typowy proszek zawiera detergent anionowy, drobno sproszkowany związek mineralny (np. skalen czy marmur) oraz wybielacz chlorowy.
Detergenty, oprócz powszechnie znanego zastosowania w gospodarstwie domowym stosowane są np. w przemyśle włókienniczym do wstępnego oczyszczania włókien naturalnych, na przykład wełny, oraz nadawania włóknom odpowiednich cech użytkowych (np. środki antystatyczne zapobiegają elektryzowaniu się tkanin).
WYPEŁNIACZE
Są dodawane w celu usunięcia z wody jonów Ca+2 i Mg+2 powodujących twardość wody
Tworzą kompleksy chylatowe lub są wymieniaczami jonowymi zamieniając te jony na jony sodu
Podnoszą pH środowiska i wspomagają emulsję olejów
Tripolifosforan sodu(Na5P3O10) - powoduje eutrofizację środowiska wodnego - rozrost i zakwity glonów. Zastępowany jest przez cytrynian sodu, węglan sodu i krzemian sodu.
Wybielacze zawierają chlor (podchloryny). Ze względu na pozostawiony przez chlor zapach zastępowane są przez borany aBrO3 (nadboran(
W reakcji hydrolizy otrzymujemy H2O2, który działa wybielająco
ZMIĘKCZACZE - to kationowe zw powierzchniowo czynne - nadające tkaninie miękkość, puszystość
ENZYMY - proteazy hydrolizują w plamach białka a amylazy by usunąć skrobię, dając łatwo odpłukiwale produkty
CZYNNIKI USZTYWNIAJĄCE - które zmniejszają przyczepność kurzu i piasku do odzieży, są to pochodne estrowe lub estrowe pochodne celulozy
ROZJAŚNIACZE optyczne są to barwniki absorbujące promieniowanie nadfioletowe o niebieskiej fluorescencji (dawniej farbka niebieska do białych tkanin), obecnie są to aminy aromatyczne lub heteroaromatyczne
ŚRODKI ANTYSTATYCZNE - czyli kationowe środki powierzchniowo czynne, by nie gromadził się ładunek na powierzchni tkaniny
ŚRODKI ZAPACHOWE - i inne składniki które powodują że proszek nie kamienieje a jest sproszkowany, bierne wypełniacze.
SKAZENIE DETERGENTAMI
Wzrost zużycia sulfonianów - pochodnych propylenu, spowodował powstawanie pian długo utrzymujących się na powierzchni rzek
Tradycyjne mydła ulegały biologicznemu rozkładowi (biodegradacji), syntetyczne -
z rozgałęzionymi łańcuchami sulfonianów nie ulegają działaniom bakteriiKwas nitrylotioctowy - który zastąpił polifosforany jest też dla środowiska groźny, bo zawiera metale ciężkie ołów kadm. Najlepszym mogą okazać się polimery glinokrzemianów sodu.
W ściekach tworzą się emulsje tłuszczy i olejów oraz wspomagają rozpuszczanie rakotwórczych WWA, działają toksycznie na organizmy wodne i ludzi (zmiany napięcia powierzchniowego ścian komórek)
Powodują zakłócenia zjawisk dyfuzyjnych i zmieniając ciśnienie osmotyczne
w komórkachW procesach uzdatniania wody jedynie przez filtry węglowe są wyłapywane
7
Stan początkowy o energii wewnętrznej Uo
Stan końcowy o energii wewnętrznej Uk
∆U = Uk - Uo
∆ U1 = Up - Uo
∆ U2 = Uk - Up
Stan pośredni o energii pośredniej Up
∆ U = Uk - Uo = ∆ U2 + Up - ( Up - ∆ U1) = ∆ U = ∆ U1 + ∆ U2
P1
A
B
P2
objetośc
Ciśnienie
![]()
![]()
![]()
![]()
Wyszukiwarka
Podobne podstrony:
opracowanie pytań z chemii fiz.calosc, wykłady
Chemia fizyczna -WARUNEK, opracowanie pytań z chemii fiz.calosc
byt-opracowanie-pytan, byt-notatki-wyklad, Dobre oprogramowanie powinno być:
opracowanie pytan na kolokwium wykladowe analiza instrumentalna
Wykład Socjologia opracowanie pytań
nakolosa Kolos wyklad opracowanie pytan z zeszlego ro
Opracowania pytań do wykladu fizyka 2
Opracowanie pytań do wykładu o warstwach, PWR, Chemia Materiałów Inżynieria Materiałowa
wykłady, TUW opracowane pytan na egz, 1
geografia opracowanie pytań, Wykłady rachunkowość bankowość
opracowanie pytań wykłady
Demografia Opracowanie pytań egzaminacyjnych, Studia II stopnia, Demografia - wykład
opracowania pytań na andrago na podstawie wykładów(1), Pedagogika, Andragogika
Opracowanie pytań ściąga, Semestr VII, Semestr VII od Grzesia, Eksploatacja układów technicznych. Wy
opracowanie pytań - wykładyy, Budownictwo UTP, semestr 4, Ekonomika
opracowanie pytań na wykład ze statystyki, STUDIA, SEMESTR IV, Statystyka matematyczna i planowanie
opracowania pytań z prawoznastwa wykłady
więcej podobnych podstron