ZABURZENIA HOMEOSTAZY KOMÓRKOWEJ. APOPTOZA
Wykład prof. zw. dra hab. n. med. Marcina Kamińskiego z kilkoma informacjami dodatkowymi i wyjaśnieniami.
Poniższy tekst to moje notatki z wykładu, który miał miejsce 14 XII 2006 uzupełnione informacjami z dostępnego mi piśmiennictwa, które wydają mi się istotne lub fascynujące. Zapraszam do lektury
HOMEOSTAZA KOMÓRKOWA
Czym jest homeostaza komórkowa i co ma na celu? Jest to pewna równowaga pomiędzy namnażaniem i umieraniem komórek, której celem jest utrzymanie prawidłowej liczebności komórek (objętości tkanek).
Fazy życia komórki:
Proliferacja - Różnicowanie - Śmierć komórki
W życiu człowieka możemy wyróżnić 3 fazy rozwojowe:
1 - faza wzrostu - wszystkie tkanki zwiększają swe rozmiary - do 21 roku życia
2 - faza plateau
3 - faza starzenia się - „zjazd w dół”; koło 35 r. ż. liczba komórek systematycznie maleje.
W komórce istnieje więc system kontrolujący:
a) system proliferacji
b) system umierania.
Nasze organy różnią się pod względem zdolności regeneracyjnych!, np.
- jelita - dziesiątki milionów komórek umierają i rodzą się każdego dnia
- wątroba - dojrzały narząd jest w zasadzie postmitotyczny - 1 kom. na 1 mln. kom. regeneruje w ciągu 24 h
Nasze komórki „chorują” tak jak my. Wpływ na to mają m. in. czynniki środowiskowe. Pod ich wpływem może dojść do przejścia protoonkogenu → onkogen
30 - 60 tys. kom nowotworowych powstaje w ciągu jednej minuty w organizmie każdego z nas! Dlaczego więc nie wszyscy z nas (na szczęście!) zapadają na chorobę nowotworową? Ano dlatego, że większość kom. z błędem genetycznym jest likwidowana.
Spójrzmy na limfocyty B powstające w szpiku lub limfocyty T grasiczo-zależne. Po nabyciu kompetencji immunologicznej przechodzą w fazę G0 cyklu komórkowego. Dopiero po spotkaniu ze swoistym antygenem uaktywniają się i proliferują. Zresztą, np. limfocyty T przechodzą „twardą szkołę” w grasicy - 95 % umiera właśnie na drodze apoptozy. Dlaczego? - są to limfocyty albo posiadające źle wytworzone receptory, albo limfocyty autoreaktywne, czyli stanowiące zagrożenia dla komórek własnych organizmu.
Cykl komórkowy jest nieustannie, w każdym momencie kontrolowany. W każdej z jego faz istnieją odpowiednie punkty kontrolne - tzw. `checkpoints', w których kontrolowana jest jakość genów i prawidłowość zachodzących procesów. Jednym z najważniejszych regulatorów cyklu jest białko P53 („strażnik genomu”). Kontroluje ono przejście do fazy S, ale także prawdopodobnie działa w fazie G2 . Jeśli wszystko jest w porządku - przepuszcza, jeśli coś jest źle, to białko P53 może działać w 2 kierunkach:
1 - zatrzymanie cyklu i w ten sposób umożliwienie próby naprawy genów,
2 - jeśli naprawa jest niemożliwa, skierowanie komórki na drogę apoptozy.
Kiedy checkpointy są sprawne - jesteśmy zdrowi, kiedy nie są - zaczynają się problemy.
Komórki, tak jak ludzie, umierają. Szacuje się, że dziennie umiera 1000000000000 komórek! Jak komórka może umierać?
1 - Apoptoza (o tym za chwilę).
2 - Nekroza (martwica), śmierć cytotoksyczna, brutalna - świadectwo nieporadności komórki, wysoka presja, niestosunek siły negatywnego bodźca (siła znacznie przekraczająca możliwości adaptacyjne komórek). Śmierci tej ulega większość komórek w miejscu zadziałania bodźca. Występuje natychmiast.
Mechanizm:
Zablokowanie oddychania w mitochondriach → siadają pompy → napływają jony sodu → w kom. gromadzi się woda → obrzęk → pęknięcie
Do lat 50-tych był to jedyny znany mechanizm śmierci.
APOPTOZA
W latach 70-tych Kerr, Willie oraz Curie opublikowali doniesienia komunikujące o nowej, dotąd nieznanej i zasadniczo różnej procedurze umierania komórek, zarówno prawidłowych jak i uszkodzonych, która nazwali apoptozą, programowaną śmiercią komórki - PCD (programmed cell death), śmiercią aktywną, śmiercią altruistyczną. Posłużenie się greckim terminem - apoptosis oznaczającym opadanie lub więdnięcie płatków kwiatów lub liści, wyrażało głębokie przekonanie autorów, iż opisany proces ma wymiar fizjologiczny. I nie mylili się
Bodźce negatywne działające na kom. są w tym procesie bardzo subtelne i mogą działać na:
- pole receptorowe błony,
- białka mitochondriów lub siateczki śródplazmatycznej.
Szlaki:
receptorowy
mitochondrialny
mikrosomalny - uszkodzenie białek enzymatycznych w siateczce; ważny w chorobach neurodegeneracyjnych (ch. Alzheimera, ch. Huntingtona)
Odpowiedź komórki jest „rozumna” - śmierć regulowana genetycznie. Jak wygląda morfologia komórki apoptotycznej? Wyróżniamy 3 stadia:
1 - uwolnienia,
- tworzy się swoiste „halo” - świadectwo utraty kontaktu z innymi kom. (brak „partnerów do rozmowy”),
- komórka przyjmuje kształt kulisty w związku ze zmianami w cytoszkielecie,
- jednocześnie dochodzi do fragmentacji DNA (szczegóły w innym miejscu).
2 - uwypuklenia,
- kom. nie pęcznieją jak w nekrozie (nadal produkowany jest ATP, pompy są sprawne, organizacja cytoplazmy jest zachowana),
- nic nie wycieka z komórki; enzymy - transglutaminazy odpowiadają za tworzenie wiązań izopeptydowych między białkami a błoną i w ten sposób „uszczelniają” komórkę,
- integralność błony zostaje zachowana, choć przy udziale floppazy dochodzi do wyeksponowania fosfatydyloseryny na zewnętrznej stronie błony, jej obecność można wykryć za pomocą aneksyny V,
- kom. staje się zdecydowanie mniejsza - obkurcza się (pozbywa się ok. 30% wody), staje się kwasochłonna,
3 - kondensacji
- od. kom. odpączkowują „odpryski” i w efekcie powstają „ciałka apoptotyczne” otoczone błoną komórkową, zawierające organelle, fragmenty cytoplazmy, skondensowanej chromatyny jądrowej.
Programowana śmierć komórki to proces wymagający aktywacji odpowiednich czynników oraz syntezy wielu z nich de novo.(etap uzbrojenia). Wiąże się z tym oczywiście nakład energii i to właśnie dlatego mitochondria prawie przez cały czas trwania procesu funkcjonują normalnie (muszą zapewnić energię!).
Fazy apoptozy:
Faza indukcji albo wzbudzenia (decyzja o śmierci komórki).
zadziałanie czynników spustowych,
przekazanie sygnału z układu receptorowego,
modulowanie sygnału.
Faza wykonawcza (śmierć komórki).
Faza zniszczenia (degradacji) i fagocytozy (pochłonięcia komórki przez fagocyty lub komórki sąsiadujące). Faza III kończy się fagocytozą
Organizmem modelowym, który miał kapitalne znaczenie dla poznania mechanizmów apoptozy jest mały, saprofitujący w glebie nicień - Caenorhabditis elegans. 131 komórek nicienia z 1090 umiera poprzez apoptozę. Geny uczestniczące w PCD są dość konserwatywne w procesie ewolucji i wykazują homologię u różnych grup zwierząt. U człowieka istnieją analogi „genów śmierci” (ced-3, ced-4), czy „genu przeżycia” (ced-9) C. e. Pomiędzy tymi genami toczy się nieustanna konkurencja.
Sygnały apoptozy (wybrane!):
- brak czynników wzrostowych i troficznych,
- promieniowanie UV, jonizujące,
- szok termiczny,
- leki antynowotworowe - wykorzystuje się gotowość tych leków do indukcji śmierci programowanej,
- wybuch tlenowy - produkcja wolnych rodników i innych RFT (reaktywne formy tlenu),
- czynniki zaburzające cykl komórkowy.
Dlaczego apoptozę odkryto tak późno?
1 - Apoptoza dotyczy ograniczonej liczby komórek w narządzie i to wybitnie rozproszonych,
2 - Proces biegnie niezwykle szybko i przykładowo w hepatocytach zamyka się w 169 - 220 minutach.
3 - Apoptozie nie towarzyszy reakcja zapalna, a umierające „altruistyczną” śmiercią komórki są błyskawicznie usuwane (fagocytowane) przez tożsame komórki, komórki sąsiadujące lub makrofagi posiadające odpowiednie receptory.
Mechanizmy regulacji:
rejestracja przez specjalne receptory,
mitochondria,
mikrosomy.
Ad. a) śmierć receptorowa - `extrinsic'
Istnieje szereg receptorów śmierci zaangażowanych w indukcję apoptozy. Jednym z najlepiej poznanych jest receptor CD95 (Apo-1/FAS) obecny, np. na limfocytach. Rozpoznaje cytokinę - ligand CD95-L (FAS-L). Po połączeniu następuje przekazanie sygnału do części wewnątrzkomórkowej receptora. Nie ma tu enzymu roboczego (domeny katalitycznej) i dlatego niezbędne jest połączenie z cytoplazmatycznym białkiem adaptorowym - FADD (fas-associating death domain protein) - inaczej MORT1. Połączenie to jest możliwe dzięki obecności w obu elementach specjalnych „domen śmierci” (DD - death domain). Są to specyficzne sekwencje aminokwasowe. Można powiedzieć, że domena poszukuje partnera, który posiada również tę sekwencję. Adaptor po fuzji z receptorem śmierci tworzy dimer. Oprócz domeny śmierci FADD posiada domenę DED (death efector domain) - efektorową domenę śmierci. Następuje poszukiwanie kolejnego partnera, którym są odpowiednie kaspazy inicjatorowe - m. in. prokaspaza 8 (FLICE - fadd like interleukin-1 converting enzyme). Powstała więc struktura trimera. W jego skład wchodzą:
1 - kompleks ligand-receptor śmierci, który ulega trimeryzacji,
2 - białko adaptorowe,
3 - prokaspaza.
czyli w naszym przykładzie:
1 - FAS-L/FAS
2 - FADD
3 - prokaspaza 8
Kompleks ten nosi nazwę DISC (death-inducing signaling complex). - kompleks śmierci. Po co on jest? Ano jest po to, aby:
1 - przekazać sygnał,
2 - uaktywnić kaspazy inicjatorowe.
Po aktywowaniu kaspazy inicjatorowej następuje kaskadowa aktywacja reszty kaspaz - jest to tzw. kaskada degradacji. Podstawowe znaczenie w procesie ma kondensacja chromatyny i późniejsza fragmentacja DNA. Jak następuje dobranie się do jądra? Kapitalne znaczenie ma tutaj kaspaza 3, która jest aktywowana zarówno w szlaku receptorowym jak i mitochondrialnym (patrz schemat). Degraduje ona ICAD, czyli inhibitor rybonukleazy zależnej od kaspaz, co prowadzi do uaktywnienia CAD - rybonukleazy (DNA-azy) zależnej od kaspaz uprzednio związanej w kompleksie z inhibitorem. To właśnie ona (lecz nie tylko) jest odpowiedzialna za „rozwalanie” DNA. Sam proces fragmentacji DNA jest dwuetapowy, przy czym kluczowy jest etap pierwszy.
1 - tworzenie większych „kawałków” - polinukleosomów,
2 - tworzenie mniejszych fragmentów - oligonukleosomów.
Silnym sygnałem do apoptozy jest TNF-alfa (kachektyna), której głównym receptorem jest TNFR1, a adaptorem TRADD.
Powiedzmy trochę o kaspazach. Są to proteinazy cysteinowe odgrywające podstawową rolę w dokonaniu fazy efektorowej apoptozy. Wszystkie one stanowią grupę białek cechującą się podobieństwem do proteazy CED-3 nicienia. Aktywacja kaspaz odbywa się na różnych, nie w pełni poznanych drogach. Lista ich substratów jest długa, zróżnicowana i imponująca. Wszystkie kaspazy są magazynowane w komórkach w formie nieaktywnej i składają się z dwóch podjednostek, większej - 20 kDa, oraz mniejszej - 10 kDa, powiązanych krótkim odcinkiem łączącym. Ponadto zawierają prodomenę odpowiedzialną za dimeryzację i utrzymywanie nieaktywnych proenzymów. Centrum aktywne enzymu (pentapeptyd zawierający cysteinę) znajduje się w podjednostce większej. Formę aktywną kaspazy stanowi tetramer 2(p20) + 2(p10).
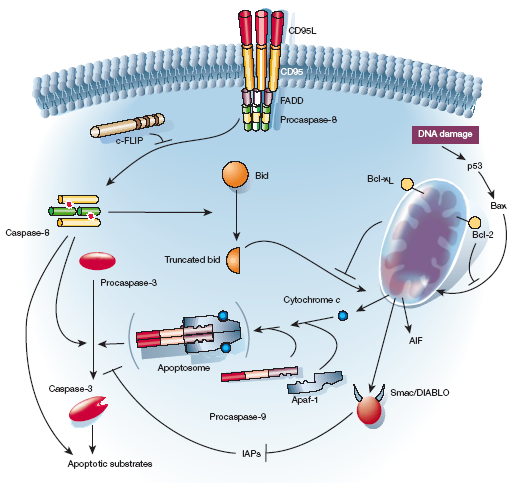
Ad. b) szlak mitochondrialny (obiektem ataku jest mitochondrium) - `intrinsic'
W jednym z artykułów natknąłem się na pewne zdanie, które mnie urzekło: „Mitochondrium jest swoistą elektrownią komórki, ale także jej arsenałem”. Jak wiemy w mitochondrium istnieje mechanizm wytwarzania energii. Opiera się na wytworzeniu gradientu elektrochemicznego, w co zaangażowana jest wewnętrzna błona mitochondrialna. Specjalny enzym - syntaza ATP produkuje AT niezbędne także w procesie apoptozy.
W przestrzeni międzybłonowej znajduje się cytochrom c, który jest niezbędny do zapoczątkowania szlaku wewnątrzkomórkowego PCD. „Wycieka” z mitochondrium gdy dochodzi do „rozszczelnienia” błony. Jakie czynniki sprzyjają takiemu stanowi rzeczy?
- środowisko kancerogenne,
- leki,
- promieniowanie i inne czynniki.
Rodzina białek BCL-2 to liczna rodzina, w której możemy odnaleźć:
białka proapoptotyczne - 9 genów, np. BAX,
białka antyapoptotyczne - 7 genów, np. BCL-2
Wspólną cechą tych białek jest obecność domen BH (BH-2, BH-3,…). Umożliwiają one fuzowanie i w konsekwencji tworzenie rozmaitych homo- i heterodimerów. Białko BAX bierze prawdopodobnie udział w tworzeniu kanałów błonowych („rozszczelnianie”). Homodimer BAX/BAX stymuluje apoptozę, homodimer BCL-2/BCL-2 stymuluje przeżycie komórki, natomiast miareczkowanie BCL-2 przez BAX i tworzenie heterodimeru BCL-2/BAX przechyla szalę w stronę apoptozy. Wzajemna gra pro- i antyapoptotyczna jest o wiele bardziej złożona i nie do końca poznana. Mitochondrium jest więc miejscem, gdzie ważą się losy komórki. Wróćmy jednak do naszego cytochromu c, który przez megakanał opuścił mitochondrium. Przyłącza się do niego Apaf-1 (apoptosis activating factor-1). Następnie prokaspaza 9. Razem z dATP i prawdopodobnie innymi czynnikami tworzy się tzw. apoptosom, którego struktura nie jest do końca poznana. Znamy natomiast główną funkcję kompleksu - jest nią aktywacja prokaspazy 9 do kaspazy 9, która prowadzi do powstawania aktywnej kaspazy 3 (patrz schemat). Oprócz cytochromu c, z mitochondrium wyciekają też inne białka proapoptotyczne. Warto wspomnieć o flawoproteinie AIF (apoptosis inducing factor), czy o białku Smac/DIABLO (second mitochondria-derived activator of caspases/ direct IAP- binding protein with low pI). Drugie z tych białek jest niezwykle intrygujące i to nie dlatego, że ma iście diabelskie rogi na schemacie Jest to inhibitor inhibitorów ( „-” i „-” daje „+”) apoptozy z rodziny IAPs (inhibitors of apoptosis proteins) wykrytych najpierw u bakulowirusów . IAPs hamują aktywację prokaspaz i kaspaz (obrona komórki przed apoptozą). Cechuje je obecność homologicznej domeny BIR (baculoviral IAP repeats). Aby nie przeszkadzały w procesie apoptozy rozprawia się z nimi sam DIABLO.
Czy istnieje jakiś łącznik pomiędzy szlakiem mitochondrialnym a receptorowym? Owszem, istnieje. Jest to białko proapoptotyczne Bid/P15 z rodziny BCL-2. Umożliwia tzw. `cross-talk' i integrację obu szlaków. Należy jednak pamiętać, że `cross-talk' jest minimalna i de facto obie drogi są niezależne. Wyjaśnijmy jeszcze mechanizm działania. Bid jest aktywowany przez kaspazę 8 szlaku receptorowego. Skutkuje to jego translokacją do mitochondrium, gdzie promuje „wyjście” cytochromu c. Następuje więc amplifikacja sygnału proapoptotycznego.
Ad. c) Szlak mikrosomalny.
Związany jest z rolą siateczki śródplazmatycznej (ER) w apoptozie (istotna w neuronach, kom. glejowych).Białka BAX mogą tworzyć kanały nie tylko w mitochondrium, ale także w siateczce, która jest wewnątrzkomórkowym magazynem jonów Ca 2+. Jony wapnia są niezbędne w procesie apoptozy. Stwierdzono, że apoptozę poprzedza wzrost stężenia wolnego wapnia we wnętrzu komórki. W przypadku niższego stężenia wapnia początek apoptozy jest zwykle opóźniony. Jony wapniowe odgrywają kluczową rolę w aktywacji endonukleaz, DNA-azy I oraz kalpain. Białko BCL-2 zlokalizowane w siateczce prawdopodobnie ma wpływ na uwalnianie jonów Ca 2+. Prokaspaza 12 „robi porządek” z nieprawidłowymi białkami („wyciekanie” z przestrzeni lumenalnej). Przeciwne działanie niż wapń wykazują jony cynku, które blokują aktywność endonukleaz fragmentujących DNA.
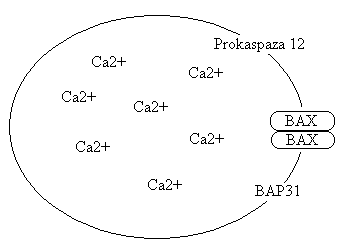
Skoro istnieje `cross-talk' pomiędzy dwoma głównymi szlakami apoptozy opisanymi powyżej, to czy siateczka śródplazmatyczna porozumiewa się w jakiś sposób z mitochondrium? Wyniki ostatnich badań sugerują, że tak - istnieje „ER-mitochondria cross-talk”. Białko BAP31 to integralne białko błonowe ER. Jest ono substratem aktywnej kaspazy 8 aktywowanej przez DISC. Produkt aktywacji BAP31 może kierować sygnał proapoptotyczny między ER a mitochondrium. Sygnałem tym jest uwalnianie jonów Ca 2+ z ER. Pobierane przez mitochondrium są niezbędne dla aktywności Drp1 (dynamin-related protein 1), która pośredniczy w rozszczelnianiu zewnętrznej błony mitochondrialnej i tym samym uwalnianiu cytochromu c.
Kolejnym istotnym zagadnieniem jest aktywacja kaspaz bez pośrednictwa receptorów, czy organelli. Mowa tu o szlaku śmierci limfocytów cytotoksycznych (CTL) oraz naturalnych killerów (NK). W trakcie egzocytozy komórki te wydzielają granule zwierające protezy serynowe, zwane granzymami. Granzymy (np. granzym B) wprowadzają komórkę na tor samounicestwienia przez bezpośrednią aktywację kaspaz (3, 7, 10,…). Aby jednak mogły wniknąć do komórki musza zadziałać perforyny tworzące kanał w błonie komórkowej. Proces ten prawdopodobnie nie zależy od inhibitorów kaspaz, co pozwala limfocytom działać bardzo efektywnie.
Krótko o wolnych rodnikach w regulacji apoptozy. Skrócony zapis przedstawia się następująco:
Wolne rodniki tlenowe → Polimeraza poli-ADP-rybozy (obniżenie) → Wewnątrzkomórkowa pula NAD/NADH i ATP (obniżenie) → Energia (obniżenie) → APOPTOZA
Wolne rodniki mogą być pochodzenia egzogennego i endogennego. BCL-2 działa w komórce także jako zmiatacz wolnych rodników, hamując w ten sposób apoptozę.
Jak już zostało powiedziane apoptoza jest procesem fizjologicznym istotnym przede wszystkim w embriogenezie, ale także w każdym etapie życia. Może jednak wiązać się z niektórymi patologiami na 2 sposoby:
- zbyt intensywna apoptoza - spadek liczby komórek,
- zahamowana apoptoza - wzrost liczby komórek.
Na koniec kilka słów na temat interesującego wykorzystania zjawiska apoptozy w medycynie sądowej. Wydaje mi się, że nie muszę uzasadniać dlaczego poruszam tą tematykę W praktyce sądowo-lekarskiej diagnostyka nagłych zgonów w następstwie wczesnego zawału mięśnia sercowego nadal stanowi poważny problem. Wiele prac wykazało możliwość indukcji apoptozy w wyniku hipoksji oraz reperfuzji. Co ciekawe, apoptoza stanowi podstawową formę obumierania komórek w pierwszych godzinach (2-4h) zawału. Apoptotyczne kardiomiocyty lokalizują się głównie w obszarze granicznym strefy zawału. W centrum dominuje nekroza. Specjalne techniki wykrywające fragmentację DNA dają pozytywne wyniki badania wczesnych etapów zawału wtedy, gdy klasyczne badania dają wyniki negatywne. Ma to duże znaczenia dla potwierdzenia nagłej śmierci sercowej.
Tym „optymistycznym” akcentem kończę ten krótki zarys tematu i życzę owocnej nauki!
autor: Rafał Skowronek
gr 7 II rok WL
e-mail: rafal-skowronek@wp.pl
1
Wyszukiwarka
Podobne podstrony:
No to dalej o tych kościach, materiały medycyna SUM, histologia, wykład
Histologia semestralka 2006, materiały medycyna SUM, histologia, test
Patomorfologia wykład 1 03.10.2007 Angiogeneza, materiały medycyna SUM, patomorfologia, wykłady
Wykład nr 3 2009 - enzymologia kliniczna, materiały medycyna SUM, biochemia, wykłady
Niedokrwistoÿci, materiały medycyna SUM, patofizjologia, wykłady
Niedokrwistoÿci, materiały medycyna SUM, patofizjologia, wykłady
HISTOLOGIA -test, materiały medycyna SUM, histologia, test
Niedokrwistoÿci, materiały medycyna SUM, patofizjologia, wykłady
Niedokrwistoÿci, materiały medycyna SUM, patofizjologia, wykłady
ETIOLOGIA OGÓLNA, materiały medycyna SUM, patofizjologia, wykłady
Niedokrwistoÿci, materiały medycyna SUM, patofizjologia, wykłady
Niedokrwistoÿci, materiały medycyna SUM, patofizjologia, wykłady
BIOCHEMIA - VII - 13.11.2000, materiały medycyna SUM, biochemia, Kolokwium III, wykłady do II
więcej podobnych podstron