
Wydział Inżynierii Procesowej i Ochrony Środowiska
Katedra Systemów Inżynierii Środowiska
LABORATORIUM
CHEMII ANALITYCZNEJ
ANALIZA ILOŚCIOWA
INŻYNIERIA ŚRODOWISKA

2
HARMONOGRAM ZAJĘĆ NA PRACOWNI CHEMII ANALITYCZNEJ
Kierunek: Inżynieria Ochrony Środowiska , IV semestr Wymiar:
45godz..11 pracowni po 4godz.+ 1godz.
1. Wstępne informacje. Przepisy BHP. Odebranie szafek laboratoryjnych (1godz.).
2. Sprawdzian. Przygotowanie roztworów: kwasu solnego i tiosiarczanu sodu.
3. Kolokwium 1- Wiadomości wstępne.
Otrzymywanie osadu jodku ołowiu(II) metodą wagową
4. Kolokwium 2- Alkacymetria.
Oznaczanie wodorotlenku sodu.
5. Kolokwium 3- Redoksymetria.
Oznaczanie miedzi.
6. Kolokwium 4- Argentometria.
Oznaczanie chlorków.
7. Kolokwium 5- Kompleksometria.
Oznaczanie cynku.
8. Kolokwium 6- Twardość wody.
Oznaczanie wapnia i magnezu.
9. Kolokwium 7- Spektrofotometria. Statystyczna interpretacja wyników.
Spektrofotometryczne oznaczanie bizmutu(III) metodą jodkową.
10. Kolokwium 8- Metody rozdziału.
Spektrofotometryczne oznaczanie żelaza(III) metodą tiocyjanianową
11. Termin rezerwowy-1.
12. Termin rezerwowy-2.
Zdanie szafek. Zaliczenie ćwiczeń.

3
KSIĄŻKI W BIBLIOTECE WYDZIAŁOWEJ
1. Francik Renata „Chemia w pigułce" - 1 egz
2. Arni Arnold „Repetytorium z chemii: chemia ogólna i nieorganiczna" - 1 egz. (1995)
3. Badzińska Jadwiga „Testy z chemii: chemia ogólna i nieorganiczna" - I egz. (1996)
4. Materiały przygotowawcze dla kandydatów na akademie medyczne -CHEMIA- testy
z rozwiązaniami. - 1 egz. (1996)
5. Cygański Andrzej, Krystek Jacek, Ptaszyński Bogdan „Obliczenia z chemicznych i
instrumentalnych metod analizy" - 5 egz. (1996)
6. Hulawicki Adam „Reakcje kwasów i zasad w chemii analitycznej" - I egz. (1992)
7. Cygański Andrzej „Metody spektroskopowe w chemii analitycznej" - 3 egz. (1993)
8. Cygański Andrzej „Chemiczne metody analizy ilościowej" - 5 egz. (1987), - 1 2 egz.
(1994)
9. Sołoniewicz Rajmund „Obliczenia z chemii ogólnej i nieorganicznej" - 1 egz. (1993)
10. Cygański A., Sołoniewicz R. „Laboratorium analizy ilościowej" zeszyt 1: 12 egz.,
zeszyt 2 : 9 egz., zeszyt 3:12 egz.
11. Minczewski Jerzy, Marczenko Zygmunt „Chemia analityczna" tom 1: 7 egz., tom 2 :
10 egz., tom 3: 11 egz.
12. Drapała T. „Chemia ogólna nieorganiczna" - 2 egz. (1986)
13. Bielański Adam „Podstawy chemii nieorganicznej" tom 1: 5egz.,tom 2: 5 egz., tom 3
: 5 egz.
14. „Poradnik chemika analityka" 1 i 2 tom po 2 egz. (1989)
15. Szczepaniak Walenty „Metody instrumentalne w analizie chemicznej" - 4 egz. (1995)
16. Witekowa S., Witek T. „Ćwiczenia z analizy jakościowej i ilościowej" cz.2 - 1 egz.
(1970)
17. Lipiec - Szmal „Chemia analityczna" - 3 egz. (1976)
18. Szyszko Edmund „Instrumentalne metody analityczne" - 3 egz. (1975)
19. Cygański Andrzej „Metody elektroanalityczne" - 3 egz. (1991)
20. Williams A.F. „Chemia nieorganiczna. Podstawy teoretyczne." - 1 egz. (1986)
21. Kryściak Jan „Chemiczna analiza instrumentalna" - 1 egz. (1989)
22. Całus Henryk „Obliczenia chemiczne" - 12 egz. (1987)
23. Korczyński A., Sołoniewicz R, „Obliczenia chemiczne" - 10 egz. (1974)
24. Śliwa Alfred (red.) „Zbiór zadań z chemii ogólnej i analitycznej nieorganicznej" -
6 egz. (1987)
25. Witekowa S. „Ćwiczenia z chemii ogólnej" - 5 egz. (1974) wyd.3
26. Bielański Adam „Chemia ogólna i nieorganiczna" - 16 egz. (1973, 1976)
27. Pazdro Krzysztof M. „Zbiór zadań z chemii dla szkół średnich" (1992) wyd. 5
28. Wesołowski M. i inni „Zbiór zadań z analizy chemicznej" - 3 egz. (1997)

4
SZCZEGÓŁOWY ZAKRES MATERIAŁU DO KOLOKWIÓW
KOLOKWIUM I - WIADOMOŚCI WSTĘPNE
1. Wiadomości podstawowe: Układ okresowy pierwiastków-liczba atomowa, liczba
masowa, jednostka masa atomowa, masa atomowa, przynależność pierwiastków do grup
i okresów (konsekwencje).
Właściwości pierwiastków: metale, niemetale.
Podstawowe kwasy (tlenowe, beztlenowe): nazwy, wzory i nazwy ich soli.
Wodorotlenki: nazwy i wzory.
Sole: nazwy i wzory.
2. Podstawowe pojęcia w obliczeniach analitycznych: definicja mola i masy molowej,
sposoby wyrażania stężeń roztworów. Cyfry znaczące.
3. Rodzaje naczyń. Naczynia miarowe. Mycie naczyń (różne roztwory myjące).
4. Wagi - ich charakterystyka i podział wag. Technika ważenia na wagach analitycznych.
Przygotowywanie odważek.
5. Rodzaje wody w związkach nieorganicznych. Metody oznaczania wody.
6. ZADANIA - obliczenia na podstawie stechiometrii reakcji i stężenia molowego.
KOLOKWIUM 2 - ALKACYMETRIA
1. Analiza miareczkowa.
Podział metod miareczkowych według rodzaju titrantu oraz sposobu prowadzenia
miareczkowania. Błędy w analizie miareczkowej.
Obliczenia w analizie miareczkowej - obliczenia na podstawie stechiometrii reakcji
i stężenia molowego.
2. Alkacymetria
Reakcje kwas-zasada, reakcje zobojętniania i hydrolizy. Teoria Arrheniusa, stała
dysocjacji, stopień dysocjacji, stała i stopień hydrolizy, prawo rozcieńczeń Ostwalda.
Teoria kwasów i zasad wg Bronsteda. Iloczyn jonowy wody, definicja i obliczanie pH.
Wskaźniki kwasowo-zasadowe według teorii Ostwalda. Krzywe miareczkowania:
mocnego kwasu mocną zasadą, mocnej zasady mocnym kwasem, słabego kwasu mocną
zasadą, słabej zasady mocnym kwasem, słabego kwasu słabą zasadą, wieloprotonowych
kwasów i zasad. Punkt równoważnikowy i punkt końcowy. Roztwory buforowe,
pojemność buforowa, obliczanie pH roztworów buforowych. Roztwory mianowane,
substancje wzorcowe do mianowania kwasów i zasad.
3. Oznaczanie wodorotlenku sodowego.
Wyprowadzenie wzorów na: miano kwasu solnego ustawiane na węglan sodowy oraz na
masę NaOH w próbie.
4. ZADANIA
KOLOKWIUM 3 - REDOKSYMETRIA
1. Reakcje utleniania-redukcji, umiejętność dobierania współczynników oraz umiejętność
przewidywania kierunku reakcji na podstawie znajomości potencjałów redoks
półogniw.
2. Charakterystyka układów redoks, potencjał redoks, równanie Nernsta, potencjał
normalny, normalna elektroda wodorowa, stała równowagi układów redoks i jej związek z
równaniem Nernsta. Wpływ pH na przebieg reakcji redoks. Wskaźniki redoks. Przebieg i
krzywe miareczkowania redoks. Substancje wzorcowe stosowane do mianowania
roztworów.
3. Jodometria: roztwory mianowane jodu i tiosiarczanu sodu. Substancje wzorcowe

5
stosowane do mianowania roztworów.
4. Oznaczanie jodometryczne miedzi (II).
Wyprowadzenie wzorów na: miano tiosiarczanu ustawiane na dwuchromian potasu oraz
na masę miedzi (II) w próbie.
5. ZADANIA
KOLOKWIUM 4 - ARGENTOMETRIA
1. Miareczkowe metody wytrąceniowe (charakterystyka, podział).
2. Iloczyn rozpuszczalności i rozpuszczalność.
3. Substancje wzorcowe i wskaźniki. Przygotowanie mianowanego roztworu azotanu
srebra.
4. Metody oznaczania chlorków według Mohra i Volharda (zalety, wady, warunki
oznaczenia, jakie halogenki można oznaczać).
5. Wyprowadzenie wzoru na masę chlorków w badanej próbie w metodzie Mohra.
6. ZADANIA
KOLOKWIUM 5 - KOMPLEKSOMETRIA
1. Charakterystyka metody: ogólny schemat reakcji, metale oznaczane tą metodą.
Kompleksometria, chelatometria, kompleksonometria.
2. Pojęcia podstawowe w kompleksometrii: atom centralny, liczba koordynacyjna Ligandy
jedno i wielofunkcyjne, sól
kompleksowa, sfera koordynacji, kompleksy
homoligandowe.
3. Trwałość kompleksów. Stała trwałości. Bierność i labilność kompleksów. Czynniki
wpływające na trwałość kompleksów.
4. Kompleksy chelatowe. Kompleksony. Kwas etylenodiaminotetraoctowy i jego sól
disodowa. Czynniki wpływające na trwałość kompleksu z EDTA (pH, stopień
utlenienia, rodzaj atomów donorowych, konfiguracja elektronowa ).
5. Wskaźniki kompleksometryczne i ich podział (czerń eriochromowa T, mureksyd)
6. Metody miareczkowania roztworem EDTA (bezpośrednie, odwrotne, podstawieniowe).
Przykłady oznaczeń.
7. Kompleksometryczne oznaczanie cynku. Wyprowadzenie wzoru na masę cynku w badanej
próbie.
8. ZADANIA
KOLOKWIUM 6 - TWARDOŚĆ WODY
1. Oznaczanie wapnia w wodzie wobec mureksydu.
2. Oznaczanie wapnia w wodzie wobec czerni eriochromowej T.
3. Oznaczanie magnezu w wodzie wobec czerni eriochromowej T.
4. Jednoczesne oznaczanie wapnia i magnezu w wodzie wobec czerni eriochromowej T.
5. Twardość wody - rodzaje i jednostki.
Przy wszystkich oznaczeniach należy znać: równania reakcji, rodzaje i barwy kompleksów.
6. ZADANIA
KOLOKWIUM 7 - SPEKRTOFOTOMETRIA ORAZ STATYSTYCZNA
INTERPRETACJA WYNIKÓW
1. Co to jest światło białe i monochromatyczne?
2. Jaka jest zależność między absorpcją promieniowania a barwą substancji?
3. Długość fali promieniowania. Zakres UV i widzialny widma
4. Na czym polega zjawisko absorpcji światła w roztworach związków chemicznych?

6
5. Co się dzieje w cząsteczkach związku chemicznego, gdy absorbują światło z zakresu
widzialnego i ultrafioletu?
6. Widmo elektronowe związku (zależność absorbancji od długości fali).
7. Co to są chromofory i auksochromy?
8. Absorbancja i transmitancja.
9. Prawo Lamberta-Beera: warunki stosowalności i odchylenia.
10. Metoda krzywej wzorcowej.
11. Budowa i działanie spektrofotometru Specol.
12. Oznaczanie bizmutu.
13. Podstawy statystycznego opracowania wyników: krzywa Gaussa, precyzja oznaczeń,
przedział ufności oznaczenia
14. ZADANIA
KOLOKWIUM 8 - METODY ROZDZIAŁU
1. Metody rozdzielania i zagęszczania: wytrącanie osadów, współstrącenie, elektroliza,
metody chromatograficzne, ekstrakcja.
2. Ekstrakcja. Prawo podziału Nernsta. Współczynnik ekstrakcji. Krotność ekstrakcji.
3. Oddzielanie ekstrakcyjne żelaza od manganu.
4. Oznaczanie żelaza metodą tiocyjanianową (rodankową).
5. Wyprowadzenie wzoru na masę żelaza(III) w badanej próbie.
6. ZADANIA
Literatura do kolokwium:
1. Instrukcja: „Laboratorium chemii analitycznej”
2. A. Cygański „Metody spektroskopowe w chemii analitycznej" WNT(1993)
3. A. Cygański „Chemiczne metody analizy ilościowej” WNT(1987), (1992),(1994)
4. A. Cygański, R. Sołoniewicz, „Laboratorium analizy ilościowej" zeszyt 1, 2 i 3
Skrypt Politechniki Łódzkiej.
5. J. Minczewski, Z. Marczenko „Chemia analityczna” tom 1-3, PWN
6. D. A. Skoog, D. M. West, F. James Holler, S. R. Grouch, “Podstawy chemii analitycznej” , t.1,PWN
2006.
7. A. Persona, „Chemia analityczna. „Podstawy klasycznej analizy ilościowej” Medyk 2007.
8. T. Lipiec, Z. S. Szmal - Chemia analityczna z elementami analizy instrumentalnej. PZWL,
Warszawa 1972.
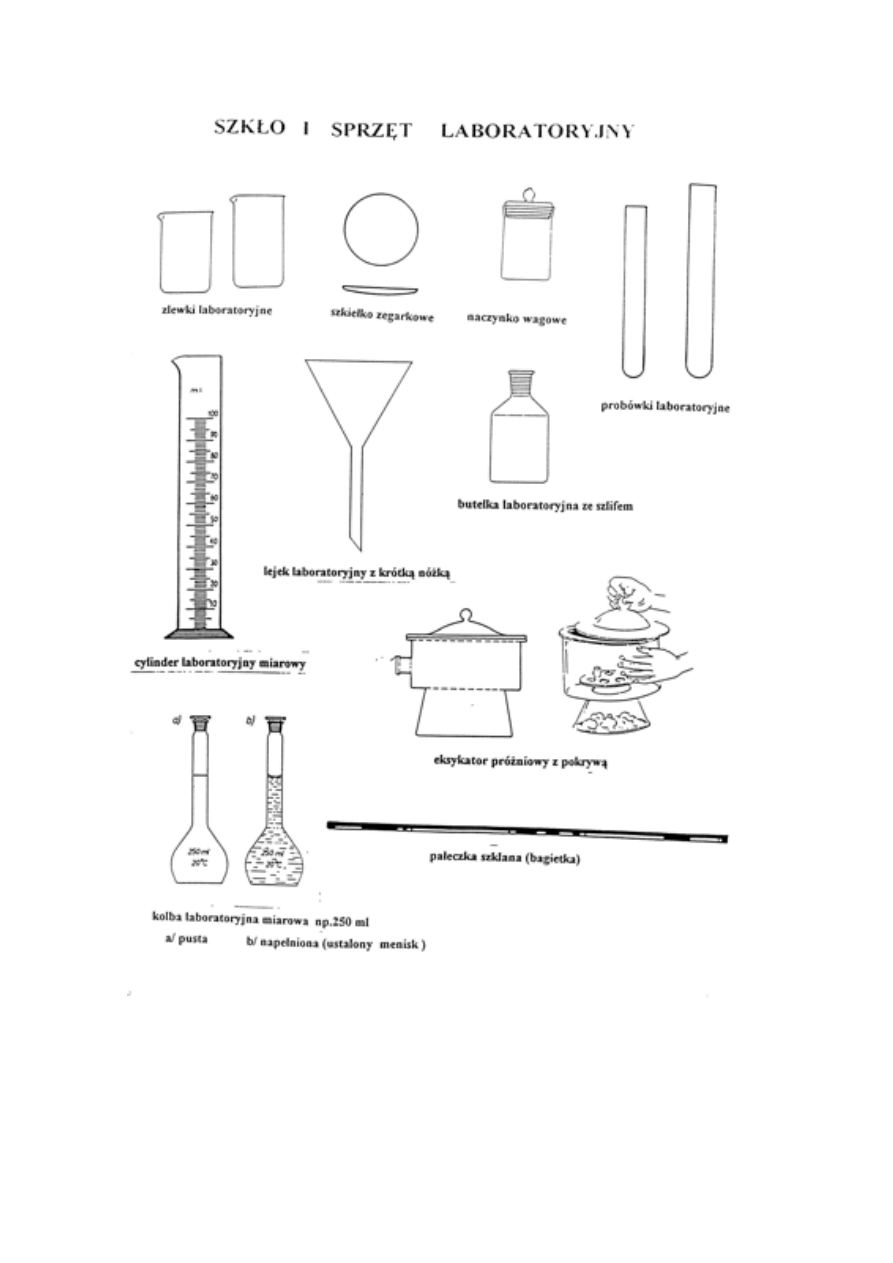
7
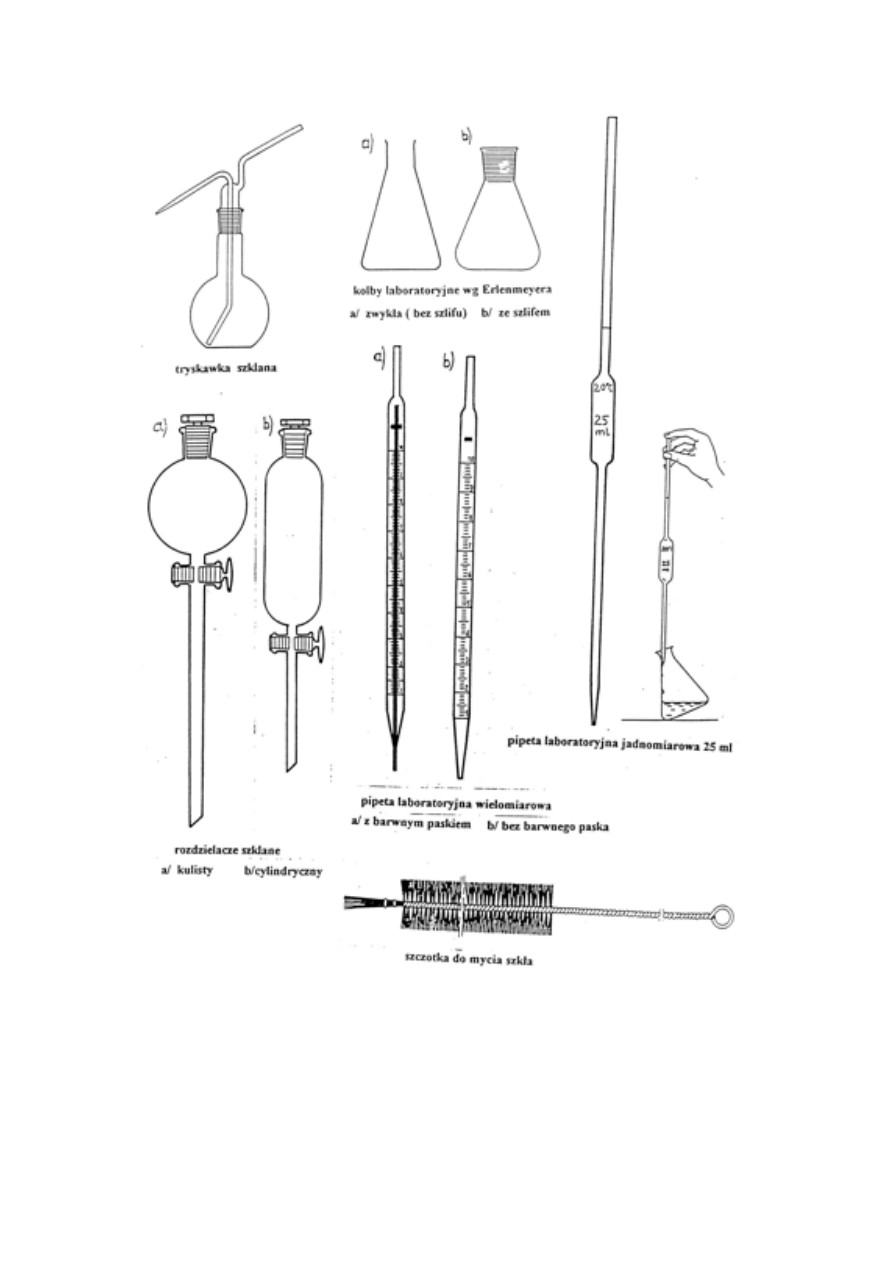
8
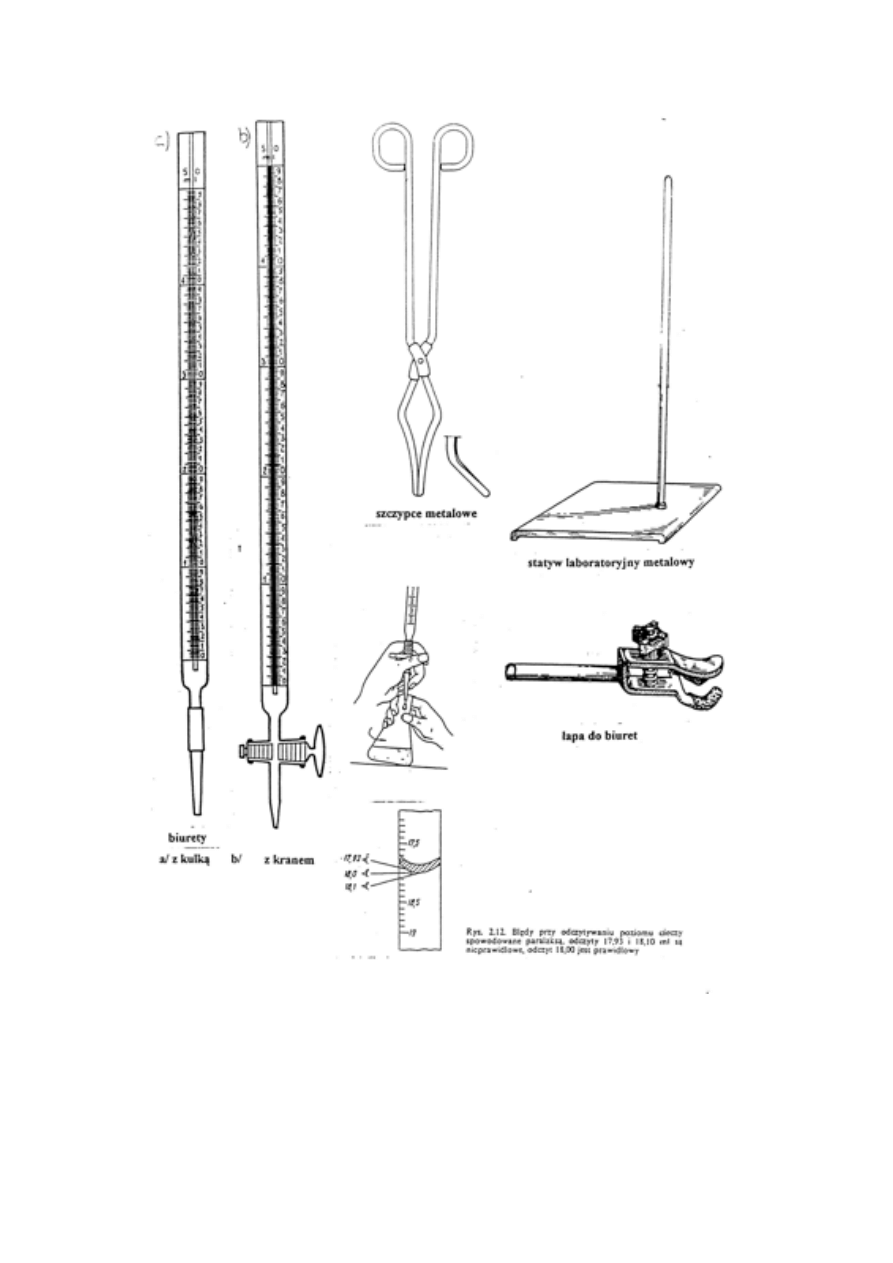
9

10
ĆWICZENIE NR 1
Otrzymywanie jodku ołowiu(II) metodą wagową
W reakcji azotanu(V) ołowiu(II) z jodkiem potasu wytrąca się jodek ołowiu(II) w postaci
żółtych kryształów. Związek ten topi się w temperaturze 405 C. Nie rozpuszcza się w
alkoholu i trudno rozpuszcza się w wodzie (w 20 C ok.0.069%).
Pb(NO
3
)
2
+ 2 KJ
PbJ
2
+ 2 KNO
3
Wykonanie oznaczenia
Do zlewki o pojemności 250cm
3
dodać odmierzone za pomocą pipety 25cm
3
roztworu jodku
potasu o stężeniu 0.12 mol/dm
3
oraz 50 cm
3
roztworu azotanu ołowiu stężeniu 0.03 mol/dm
3
.
Roztwory wymieszać, a wytrącony osad przesączyć przez sączek ułożony i dopasowany do
szklanego lejka. Sączek przed ułożeniem na lejku ważymy wraz ze szkiełkiem zegarkowym
na wadze analitycznej. Osad na sączku przemywamy wodą destylowaną z tryskawki. Sączek
wraz z osadem umieszczamy na szkiełku zegarkowym i przenosimy do suszarki
i suszmy w temperaturze120 C. Po upływie 1 godziny wysuszony osad wraz ze szkiełkiem
zegarkowym przenosimy do eksykatora i po 15 min. ważymy. Obliczamy ile osadu
otrzymano. Obliczamy teoretyczną i rzeczywistą wydajność reakcji.

11
ĆWICZENIE NR 2
Oznaczanie wodorotlenku sodowego NaOH
1. Przygotowanie mianowanego roztworu kwasu solnego
Kwas solny o stężeniu 0.1mol/dm
3
sporządza się przez rozcieńczenie stężonego roztworu HCl
ok. 35% masowych o gęstości 1.18 g/cm
3
. Sposób obliczenia objętości stężonego kwasu
solnego, którą należy wziąć do przygotowania 1l roztworu o stężeniu 0,1 mol/dm
3
HCl jest
następujący: Z definicji stężenia wyrażonego w procentach masowych wynika, że 35 g HCl
znajduje się w 100 g roztworu stężonego, tj. w
3
84,7cm
1,18
100
.
Masa 0.1 mol roztworu HCl wynosi 0.1 M
HCl
= 0.1·36,5 = 3.65 g HCl. Ta masa znajduje się w
następującej objętości stężonego kwasu solnego:
35 g HCl - 84,7 cm
3
3.65 g HCl - V
x
V
x
=
3
8,83cm
35
84,7
3,65
Należy, więc cylinderkiem odmierzyć 9 cm
3
stężonego roztworu HCl i rozcieńczyć do
objętości 1 dm
3
lub odpowiednio mniejszą objętość stężonego kwasu, jeśli przygotowujemy
0.5 lub 0.3 dm
3
rozcieńczonego kwasu. Otrzymuje się w ten sposób roztwór o stężeniu
przybliżonym, którego dokładne miano ustala się przez miareczkowanie odważek sody
otrzymanej z NaHCO
3
. Podczas takiego miareczkowania wobec oranżu metylowego
przebiega reakcja:
Na
2
CO
3
+ 2 HCl → 2 NaCl + H
2
O + CO
2
UWAGA! Przygotować 0.5 dm
3
0.1 mol/dm
3
kwasu solnego w butelce z korkiem na szlif.

12
2. Nastawianie miana 0.1 mol/dm
3
roztworu HCl przy użyciu węglanu sodowego.
Bezwodny węglan sodowy zawiera zwykle nieco wilgoci oraz kwaśnego węglanu sodowego.
Ogrzewanie w ciągu ok. godziny w temp. 270-300
o
C pozbawia sodę wilgoci oraz domieszek
NaHCO
3
w wyniku termicznego rozkładu:
2 NaHCO
3
→ Na
2
CO
3
+ CO
2
+ H
2
O
Ogrzewanie sody przeprowadza się w tyglu porcelanowym w piecu elektrycznym
z regulowana temperaturą lub w łaźni piaskowej ogrzewanej palnikiem gazowym.
Temperaturę sody w tyglu sprawdza się termometrem, który jednocześnie służy do mieszania,
co pewien czas zawartości tygla. Tygiel z wysuszoną sodą umieszcza się w eksykatorze, a po
ostudzeniu przesypuje się sodę do szczelnego naczynka wagowego, które także przechowuje
się w eksykatorze.
Odważki sody powinny być takiej wielkości, aby podczas miareczkowania zużywano
z biurety 30-40 ml 0.1 mol/dm
3
HCl. Miareczkowanie prowadzi się wobec oranżu
metylowego w sposób podany poniżej.
W naczynku wagowym odważa się dokładnie (przez odsypywanie) 0.15-0.20 g Na
2
CO
3
i przenosi ilościowo do kolbki stożkowej o pojemności 250 cm
3
. Odważony węglan
rozpuszcza się w ok. 60 cm
3
wody destylowanej, dodaje 1-2 krople oranżu metylowego
i roztwór miareczkuje z biurety roztworem przygotowanego kwasu solnego. Miareczkowanie
prowadzi się aż do pojawienia się barwy cebulkowej, przejściowej miedzy żółtą i czerwoną.
Wykonuje się, co najmniej 3 oznaczenia.
Pod koniec miareczkowania roztwór jest nasycony CO
2
, który obniża nieco pH roztworu
NaCl. W przypadku trudności w ustaleniu końca miareczkowania należy przygotować
roztwór porównawczy, tzw. wzorzec (świadek) miareczkowania, którym jest roztwór
o objętości i stężeniu równych objętości i stężeniu roztworu miareczkowanego w końcu
miareczkowania.

13
Stężenie kwasu oblicza się wg wzoru:
106
V
m
1000
1
2
c
3
2
CO
Na
HCl
gdzie: c
HCl
- stężenie molowe roztworu HCl;
3
2
CO
Na
m
-
odważka Na
2
CO
3
,
V - objętość roztworu HCl zużyta na miareczkowanie odważki sody o masie
3
2
CO
Na
m
.
3. Oznaczenie NaOH
Otrzymane zadanie (próbkę) rozcieńcza się wodą destylowaną do kreski w kolbie miarowej
o objętości 250 cm
3
i dokładnie miesza. Następnie odmierza się pipetą 25 cm
3
tego roztworu
do kolbki stożkowej, dodaje 1-2 krople oranżu metylowego i miareczkuje przygotowanym
roztworem kwasu solnego (ok. 0.1 mol/dm
3
) do zmiany barwy z żółtej na cebulkową.
Wynik oblicza się z wzoru:
40
1000
V
c
M
1000
V
c
m
HCl
HCl
NaOH
HCl
HCl
NaOH
gdzie: c
HCl
- stężenie molowe HCl;
V
HCl
- objętość roztworu HCl zużyta podczas miareczkowania.

14
ĆWICZENIE NR 3
Jodometryczne oznaczanie miedzi
/. Roztwór mianowany tiosiarczanu sodu
Mianowany roztwór tiosiarczanu sodowego jest titrantem stosowanym w jodometrii obok
mianowanego roztworu jodu. Utlenianie jonu tiosiarczanowego (ditionianowego) do
tetrationianowego przebiega zgodnie z reakcją:
2 S
2
O
3
2-
→ S
4
O
6
2-
+ 2 e ( w reakcji utleniania oddawane są 2 elektrony)
Pod działaniem silnych utleniaczy jon tiosiarczanowy utlenia się do innych kwasów
wielotionowych, a nawet do jonu siarczanowego(VI) SO
4
2-
. Z tego względu zastosowanie
tiosiarczanu ogranicza się w analizie do reakcji z jodem, która stanowi podstawę metod
jodometrycznych
2 S
2
O
3
2-
+ I
2
—> S
4
O
6
2-
+ 2 e
Reakcja tiosiarczanu z jodem przebiega szybko, co ma znaczenie ze względu na środowisko
reakcji. Miareczkowanie tiosiarczanem prowadzi się w środowisku kwaśnym, ponieważ
w alkalicznym S
2
O
3
2-
utlenia się częściowo do siarczanu. Jednak w środowisku bardzo
kwaśnym jon S
2
O
3
2-
jest nietrwały i rozkłada się z wydzieleniem siarki. Rozkład
tiosiarczanu pod działaniem kwasów odbywa się tak wolno, że można miareczkować jod
tiosiarczanem w środowisku kwaśnym. Miareczkuje się roztwory zawierające nawet do
1 mol/l HC1 pod warunkiem, że są one w czasie miareczkowania intensywnie mieszane.
Stały krystaliczny tiosiarczan sodu Na
2
S
2
O
3
·5 H
2
O można otrzymać w stanie czystym, jednak
ten hydrat nie jest trwały i dlatego nie można związku traktować jako substancji
wzorcowej. Świeżo przygotowany roztwór tiosiarczanu też nie jest trwały i zmienia
nieco swoje stężenie w ciągu 8 - 1 4 dni. Główną przyczyną nietrwałości roztworów
tiosiarczanu jest obecność w wodzie CO
2
(i ewentualnie innych kwasów) oraz bakterii.
Kwas węglowy reaguje z tiosiarczanem powodując jego rozkład
S
2
O
3
2-
+ H
+
→ HSO
3
-
+ S
Powstanie wodorosiarczanów(IV) zwiększa miano roztworu. Dlatego zaleca się
przygotowanie roztworów tiosiarczanu przy użyciu wody destylowanej pozbawionej CO
2
przez gotowanie i dodanie niewielkiej ilości Na
2
CO
3
(0,1 g/l) w celu zalkalizowania
roztworu. Aby uniknąć działania bakterii, które również powodują wydzielenie siarki, stosuje
się dodatek środków aseptycznych, np. chloroformu (0,5 g/l) lub alkoholu amylowego (l g/l).

15
Roztwór tiosiarczanu o stężeniu c=0,1 mol/l przygotowuje się przez rozpuszczenie 25 g
Na
2
S
2
O
3
·5 H
2
O
w wodzie i rozcieńczenie do 1 litra oraz dodanie 0.1 g Na
2
CO
3
i 0.5 g
chloroformu. Miano tiosiarczanu nastawia się po upływie 10 dni, stosując jako substancje
wzorcowe K
2
Cr
2
O
7
, bromian potasu, jod lub wodorojodan potasu KH(IO
3
)
3
.
2. Nastawianie miana tiosiarczanu na K
2
Cr
2
O
7
Dwuchromian potasu można otrzymać w bardzo czystej postaci, toteż jest on dobrą
pierwotną substancją wzorcową. Roztwory wodne K
2
Cr
2
O
7
odznaczają się nieograniczoną
trwałością. Związek ten suszy się w suszarce w temp. 150°C. Nastawianie miana odbywa się
drogą pośrednią przez wydzielenie jodu, ponieważ dwuchromian ma zbyt silne właściwości
utleniające i reakcja utleniania tiosiarczanu do czterotionianu nie przebiegałaby ilościowo,
na skutek częściowego tworzenia się siarczanów. Natomiast reakcja utlenienia jodków
dwuchromianem przebiega w środowisku kwaśnym stechiometrycznie i ilość wydzielonego
elementarnego jodu jest równoważna ilości dwuchromianu. Reakcję prowadzi się wobec
kilkukrotnego nadmiaru jodku.
Zachodzące przy nastawianiu miana reakcje przebiegają wg następujących równań;
Cr
2
O
7
2-
+ 14 H
+
+ 6 e → 2 Cr
3+
+ 7 H
2
O
·1
E° = 1,36 V
2 I
-
→ I
2
+ 2 e
·3
Cr
2
O
7
2-
+ 6 I
-
+ 14 H
+
→ 2 Cr
3+
+ 3 I
2
+ 7 H
2
O
Z równania pierwszego wynika, że redukcja jonu Cr
2
O
7
2-
zachodzi z pobraniem sześciu
elektronów. Reakcja utleniania jonu S
2
O
3
2-
jodem zachodzi wg omówionego poprzednio
równania
2 S
2
O
3
2-
+ I
2
—> S
4
O
6
2-
+ 2 e
Nastawienie miana tiosiarczanu prowadzi się w sposób następujący: do kolby stożkowej
z dopasowanym korkiem wsypuje się odważkę ok. 0,2 g K
2
Cr
2
O
7
wysuszonego w temp.
150°C do stałej masy i rozpuszcza w 70 ml wody, dodaje 25 ml roztworu 1 mol/l H
2
SO
4
i 2 g KI. Kolbę zamyka się korkiem i odstawia w ciemne miejsce na 15 min. Spłukać korek
wodą destylowaną z tryskawki. Następnie wydzielony jod miareczkuje się roztworem Na
2
S
2
O
3
.
Pod koniec miareczkowania, gdy roztwór ma słabe pomarańczowo-żółte zabarwienie,
dodaje się 2 ml roztworu skrobi i dalej miareczkuje do zmiany barwy
z ciemnogranatowej na zieloną. Zabarwienie zielone pochodzi od obecności jonów Cr
3+

16
Zmiana barwy w punkcie końcowym jest wyraźna. Miareczkowanie należy powtórzyć min
trzykrotnie.
3. Obliczanie stężenia molowego tiosiarczanu
Stosunek współczynników stechiometrycznych tiosiarczanu i dwuchromianu wynosi 6:1,
ponieważ 1 mol jonów Cr
2
O
7
2-
odpowiada 3 molom I
2
, które odpowiadają 6 molom jonów
S
2
O
3
2-
co można zapisać schematycznie:
Cr
2
O
7
2-
→ 3 I
2
→ 6 S
2
O
3
2-
c
Na
2
S
2
O
3
=
m
K
2
Cr
2
O
7
1000
294.2 V
Na
2
S
2
O
3
6
1
gdzie
K
2
Cr
2
O
7
m
masa odważki K
2
Cr
2
O
7
V
Na
2
S
2
O
3
objętość zużytego roztworu tiosiarczanu
4. Oznaczanie miedzi
Miedz oznacza się jodometrycznie metodą pośrednią. Do lekko kwaśnego roztworu o pH 4-5
dodaje się nadmiar KI. Miedź redukuje się do miedzi (I), utleniając jony jodkowe do jodu,
który miareczkuje się mianowanym roztworem Na
2
S
2
O
3
2 Cu
2+
+ 4 I
-
→ 2 CuI + I
2
Potencjał normalny układu I
2
/2I
-
jest wyższy niż potencjał układu Cu
2+
/Cu
+
, ale wskutek
wytrącenia się trudnorozpuszczalnego osadu Cul, potencjał układu Cu
2+
/Cu
+
ulega
znacznemu podwyższeniu do ok.0,76 V i dlatego zachodzi reakcja ilościowego utleniania
jodków. Reakcja ta jest odwracalna i dlatego potrzebny jest duży nadmiar jodku potasu
(ok. 40-60-krotny). Miareczkowanie należy prowadzić dość szybko, aby zapobiec utlenieniu
jodków tlenem powietrza (jony Cu
2+
katalizują tę reakcję). W roztworze nie powinno być
chlorków tworzących kompleksy z jonami miedzi.
Wykonanie oznaczenia.
Do około 25 ml roztworu zawierającego ok. 150 mg Cu(II) w postaci siarczanu dodaje się
50 ml wody i 5 ml roztworu H
2
SO
4
o stężeniu 1 mol/l, a następnie dodaje się 20 ml

17
10%-owego roztworu KI (świeżo przygotowanego). Roztwór natychmiast miareczkuje
się mianowanym roztworem Na
2
S
2
O
3
do uzyskania cielistego (jasnożółtego) zabarwienia.
Następnie dodaje się 4 ml roztworu skrobi i miareczkuje aż do zaniku niebieskiego zabarwienia
roztworu.
W celu zaoszczędzenia KI można jego ilość zmniejszyć do 0,3 g i po wprowadzeniu H
2
SO
4
dodać 15 ml 10%-owego roztworu KSCN. Tworzący się CuSCN jest trudniej rozpuszczalny
niż CuI, co dodatkowo zmniejsza stężenie jonów Cu
2+
w roztworze, powodując jeszcze
większy wzrost potencjału utleniającego układu Cu
2+
/Cu
+
.
Obliczanie wyników
Stosunek współczynników stechiometrycznych wynosi 1, ponieważ
2 Cu
2+
→ I
2
→ 2 S
2
O
3
2-
m
V
Na
2
S
2
O
3
c
Na
2
S
2
O
3
Cu
=
1000
1
1
63.54

18
Ć
WICZENIE NR
4
Oznaczanie chlorków metodą Mohra.
/. Mianowany roztwór AgNO
3
;
Argentometria to dział analizy miareczkowej wytrąceniowej, w której wykorzystuje się
oznaczanie substancji w wyniku tworzenia podczas miareczkowania trudnorozpuszczalnego
osadu. W metodzie oznaczania chlorków metodą Mohra jako roztwór miareczkujący stosuje
roztwór azotanu srebrowego AgNO
3
, który można przygotować następującymi sposobami:
przez rozpuszczenie w wodzie odważki azotanu srebra o wysokim stopniu czystości lub
przez
rozpuszczenie odważki chemicznie czystego srebra (w postaci druciku) w 10
cm
3
ok. 30%-owego HNO
3
. Po rozpuszczeniu srebra roztwór odparowuje się w celu usunięcia
tlenków azotu (odbarwienie roztworu). Miano roztworu AgNO
3
wyznacza się używając NaCl
lub KC1jako substancję wzorcową.
2. Nastawianie miana AgNO
3
na chlorek sodowy.
Miano roztworu AgNO
3
o stężeniu 0,05 mol/dm
3
ustala się na naważki wysuszonego (ok.2
godz) w temp. 110°C chlorku sodowego. Odważkę NaCl (70-80 mg dokładnie odważoną
przez odsypywanie) rozpuszcza się w ok. 60 ml wody destylowanej (nie zanieczyszczonej
chlorkami), dodaje kilka kropli roztworu K
2
CrO
4
i miareczkuje roztworem AgNO
3
aż do
powstania wyraźnego czerwonobrunatnego zabarwienia nie znikającego w ciągu 20 sekund
mieszania. Roztwór azotanu srebra rozkłada się powoli pod wpływem światła i dlatego
roztwory AgNO
3
należy przechowywać w ciemnych butelkach.
3.
Oznaczanie chlorków metodą Mohra
Metoda Mohra polega na bezpośrednim miareczkowaniu obojętnego roztworu chlorku
mianowanym roztworem AgNO
3
w obecności K
2
CrO
4
jako wskaźnika. Podczas
miareczkowania wytrąca się najpierw trudnorozpuszczalny osad AgCl
Ag
+
+ Cl
-
→ AgCl
↓
Gdy praktycznie cała ilość jonów chlorkowych zostanie wytrącona, nadmiar roztworu
jonów srebrowych Ag
+
reaguje z jonami chromianowymi CrO
4
2-
wytrącając
brunatnoczerwony
osad
chromianu
srebrowego.
Powstanie
brunatnoczerwonego
zabarwienia roztworu wskazuje na koniec miareczkowania.

19
2 Ag
+
+ CrO
4
2-
→ Ag
2
CrO
4
↓
Odczyn roztworu powinien być obojętny, ponieważ w roztworze kwaśnym jony wodorowe
reagują jonami CrO
4
2-
, tworząc jony wodorochromianowe HCrO
4
-
i dichromianowe Cr
2
O
7
2-
2 CrO
4
2 -
+ 2 H
+
↔ Cr
2
O
7
2 -
+ H
2
O
Powoduje to zmniejszenie stężenia jonów CrO
4
2-
, a w bardziej kwaśnych roztworach osad
może się wcale nie wytrącić.
Ag
2
CrO
4
, jako sól słabego kwasu, ulega rozpuszczeniu w kwaśnych roztworach.
W roztworach silnie zasadowych pH>10,5 następuje wytrącanie Ag
2
O
2 Ag
+
+ 2 OH
-
-→ Ag
2
O + H
2
O
Metody Mohra nie można stosować do oznaczania chlorków w obecności anionów
tworzących w roztworach obojętnych trudnorozpuszczalne sole srebrowe (Br
-
, I
-
, AsO
4
3-
,
PO
4
3-
, CO
3
2-
), kationów tworzących trudnorozpuszczalne chromiany (Ba
2+
, Pb
2+
) oraz
substancji redukujących AgNO
3
do srebra metalicznego ( np. jony Fe
2+
).
Metodą Mohra można oznaczać bromki. Nie można jednak stosować tej metody
do oznaczania jodków i tiocyjanianów, ponieważ jodek i tiocyjanian srebra silnie adsorbują
jony chromianowe, przez co punkt równoważności nie jest wyraźny.
Wykonanie oznaczenia
Po rozcieńczeniu próbki w kolbie miarowej odmierza się pipetą 25 cm
3
(lub 20 cm
3
)
roztworu i przenosi do kolby stożkowej. Roztwór rozcieńcza się do objętości ok. 60 cm
3
,
dodaje 5-6 kropli roztworu K
2
CrO
4
o stężeniu 1 mol/dm
3
i miareczkuje mianowanym
roztworem AgNO
3
aż do powstania zabarwienia beżowego, nie znikającego w ciągu 20 s
mieszania.

20
Ć
WICZENIA NR
5.
Kompleksometryczne oznaczanie cynku.
1. Mianowany roztwór EDTA (sól disodowa kwasu etylenodiaminotetraoctowego, wersenian
disodowy)
Jeżeli dysponuje się odpowiednio czystym wersenianem disodowym to można roztwór EDTA
przygotować przez rozpuszczenie w wodzie odpowiedniej odważki dwuwodnej soli. Nie
należy jednak wersenianu suszyć, ponieważ bezwodna sól jest higroskopijna i musi być
przechowywana w eksykatorze nad P
2
O
5
, ale sól uwodniona jest trwała w dużym zakresie
wilgotności powietrza.
Masa molowa wersenianu disodowego C
10
H
14
N
2
Na
2
O
8
·2H
2
O wynosi 372.24 g/mol. Do
przygotowania roztworu o stężeniu 0.05 mol/dm
3
należy rozpuścić 18.6050 g wersenianu
disodowego w wodzie i rozcieńczyć wodą do objętości 1 dm
3
.
W przypadku związku o mniejszej lub niepewnej czystości należy nastawić jego miano
stosując odpowiednie substancje wzorcowe. Substancje wzorcowe, które zostały
z pozytywnymi wynikami zastosowane do nastawiania miana roztworów EDTA, można
podzielić na dwie grupy:
1/ metale i tlenki organiczne Mg, Cu, Ni, Zn, Cd, MgO, PbO, ZnO;
2/ nieorganiczne sole bezwodne MgSO
4
, CaCO
3
, PbCl
2
, Pb(NO
3
)
2
Najlepiej ustalać miano roztworu EDTA na roztwór wzorcowy oznaczanego metalu
w warunkach określonej metody. Nie jest to jednak regułą w kompleksometrii. Z substancji
wzorcowych często stosowany jest węglan wapnia.
W miareczkowaniach kompleksometrycznych stosuje się najczęściej roztwory o stężeniach
0.1-0.01 mol/dm
3
. Duża czułość wskaźników kompleksometrycznych umożliwia stosowanie
roztworów nawet o stężeniu 0.001 mol/dm
3
.
2. Oznaczanie cynku
Jony cynkowe miareczkuje się roztworem EDTA w środowisku buforu (NH
4
Cl-NH
3
)
wobec czerni eriochromowej jako wskaźnika.
Oznaczaniu cynku przeszkadzają przede wszystkim jony miedzi, niklu i kobaltu. Metale te
oraz żelazo (po zredukowaniu do Fe(II) kwasem askorbinowym) maskuje się
przeprowadzając je w trwałe kompleksy cyjankowe. Dodany cyjanek potasowy przeprowadza
także cynk w kompleks cyjankowy. Cynk demaskuje się, (czyli uwalnia z kompleksu

21
cyjankowego) za pomocą aldehydu mrówkowego. Aldehyd wiąże cyjanki w trwałą
cyjanohydrynę uwalniając jony cynkowe, które można teraz odmiareczkować roztworem
EDTA. Podobnie jak cynk zachowuje się kadm.
Glin oraz żelazo(III) można maskować
fluorkami. Fluorki wiążą jony wapnia i magnezu w trudno rozpuszczalne związki.
Odczynniki:
1. EDTA, 0,05 mol/dm
3
2. Roztwór buforowy o pH ok. 10.
Rozpuścić 70 g chlorku amonowego cz.d.a. w wodzie, dodać 570 dm
3
stężonego
amoniaku i rozcieńczyć roztwór wodą do 1 dm
3
.
3.
Czerń eriochromowa T
Zmieszać dokładnie przez roztarcie w moździerzu porcelanowym 0.10 g czerni
eriochromowej T z 20 g chlorku sodowego cz.d.a. Przechowywać mieszaninę
w szklanym słoiku z doszlifowanym korkiem.
Wykonanie oznaczenia
Otrzymane zadanie (badany roztwór) rozcieńczyć wodą w kolbie miarowej do objętości
250 cm
3
. Pobrać 25 cm
3
do kolby stożkowej o poj. 250 cm
3
i rozcieńczyć wodą destylowaną
do ok. 60 cm
3
. Do prawie
o
bojętnego roztworu dodać 5 cm
3
roztworu buforowego, szczyptę
mieszaniny czerni eriochromowej T z NaCl i miareczkować roztworem EDTA do zmiany
zabarwienia
roztworu
miareczkowanego
z
różowofiołkowego na niebieskie.
Miareczkować w obecności świadka (niebieskiego roztworu przemiareczkowanego).
Obliczenia
Zawartość cynku w g obliczyć ze wzoru:
m
Zn
=
c
EDTA
V
EDTA
1000
65.37
Reakcje zachodzące w roztworze podczas oznaczania cynku
W roztworach wodnych metale znajdują się nie w postaci wodnych jonów, lecz w postaci
akwakompleksów, tj. kompleksów, w których ligandami są cząsteczki wody Reakcje
tworzenia się kompleksu można przedstawić równaniem ogólnym
[M(H
2
O)
x
]
n+
+ L
p-
→ ML
n-p
+ xH
2
O
Reakcja tworzenia kompleksów z EDTA jest zgodna z tym równaniem. Sól disodowa kwasu
etylenodiaminotetraoctowego w roztworze wodnym dysocjuje na jony

22
Na
2
H
2
Y → H
2
Y
2-
+ 2Na
+
Jon H
2
Y
2-
ma silne właściwości kompleksotwórcze i reaguje z kationami M
n+
zgodnie
z następującym równaniem:
[M(H
2
O)
x
]
n+
+ H
2
Y
2-
↔ MY
n -4
+ 2H
3
O
+
+ (x-2)H
2
O
Trwałość kompleksów EDTA zależy od czynników wewnętrznych tj. właściwości danego
kationu, ale również od czynników zewnętrznych jak pH roztworu. Wzrost stężenia jonów H
+
powoduje przesuwa równowagę reakcji w kierunku substratów, co powoduje zmniejszenie
trwałości kompleksu:
M
n +
+ H
2
Y
2-
↔ MY
n -4
+ 2 H
+
Oznaczanie metali (Ca, Mg, Zn) odbywa się w obecności czerni eriochromowej T,
zachowującej się jak wskaźnik alkacymetryczny mający dwa przejścia barwne
odpowiadające dwustopniowej dysocjacji protonów:
Oznaczenie wykonuje się najczęściej w roztworach o pH 10, przy którym następuje
wyraźna zmiana barwy.
Reakcje zachodzące w roztworze podczas oznaczania cynku:
a) reakcja ze wskaźnikiem
b) reakcja podczas miareczkowania EDTA
W czasie miareczkowania następuje zmiana barwy z czerwonej (barwa kompleksu cynku ze
wskaźnikiem) na niebieską, charakterystyczną dla jonów czerni eriochromowej.
Wydzielające się w tej reakcji jony H
+
zostają związane przez bufor.
niebieski
Zn
2+
+ HF
2-
ZnF
-
+ H
+
winnoczerwony
ZnF
-
+ H
2
Y
2-
ZnY
2-
+ HF
2-
+ H
+
winnoczerwony
bezbarwny
niebieski
H
2
F
-
HF
2-
F
3-
pH 6.3
pH 11.5
różowy
niebieski
pomarańczowy

23
ĆWICZENIE NR 6
Oznaczanie wapnia i magnezu. Twardość ogólna wody.
Miareczkowanie metali za pomocą EDTA może być prowadzone w sposób bezpośredni,
odwrotny lub podstawieniowy.
Wapń można oznaczać zarówno poprzez miareczkowanie
bezpośrednie roztworem mianowanym EDTA wobec mureksydu jak i poprzez
miareczkowanie podstawieniowe.
Bezpośrednie miareczkowanie wapnia roztworem EDTA wymaga stosowania jako
wskaźnika mureksydu lub kalcesu, ponieważ z czernią erichromowej kompleks zbyt mało
trwały, aby można było miareczkować go bezpośrednio wobec tego wskaźnika. Mureksyd
(HInd) jest to sól amonowa kwasu purpurowego - w skrócie. Jego zabarwienie zależy od
pH roztworu:
Zmiana zabarwienia jest spowodowana dysocjacją protonów grup imidowych. Z czterech
grup imidowych tylko dwie dysocjują ze wzrostem pH.
Mureksyd jest najczęściej stosowany
do oznaczeń Ni, Co i Cu w roztworach amoniakalnych i Ca w roztworze silnie alkalicznym
pH>13
Podczas oznaczania wapń najpierw reaguje ze wskaźnikiem (mureksydem) tworząc kompleks
o różowej barwie:
a następnie podczas miareczkowania roztworem EDTA następuje wypieranie wapnia z jego
kompleksu ze wskaźnikiem i zmiana zabarwienia roztworu pod koniec miareczkowania na
niebiesko-fioletowe (barwa wolnego wskaźnika):
Należy podkreślić, że Ca
2
* nie tworzy tylko jednego kompleksu ze wskaźnikiem, ale istnieją w
równowadze trzy różniące się zabarwieniem kompleksy:
H
4
Ind
-
H
3
Ind
2-
H
2
Ind
3-
pH 9
pH 11
czerwonofioletowy
fioletowy
niebieskofioletowy
Ca
2+
+
H
2
Ind
3-
CaH
2
Ind
-
różowoczewony
CaH
2
Ind
-
+ H
2
Y
3-
+ 2 OH
-
CaY
2+
+ H
2
Ind
3-
+ 2 H
2
O
niebieskofioletowy

24
Oznaczanie ilości wapnia w wodzie.
Do kolby stożkowej o pojemności 200-300 ml odmierza się 200 ml wody, dodaje z biurety
mianowanego roztworu HC1 w ilości odpowiadającej uprzednio wyznaczonej twardości
węglanowej, następnie dodaje się 10 ml roztworu NaOH o stężeniu 1 mol/l, 0.1 mg
mureksydu i miareczkuje mianowanym roztworem EDTA o stężeniu 0.05 mol/l aż do
zmiany barwy z różowej na niebiesko-fioletową.
Zawartość wapnia w gramach w 1l wody oblicza się wg wzoru
gdzie: a - ilość ml roztworu EDTA zużyta do miareczkowania badanej wody (200 ml);
C
EDTA
- stężenie molowe roztworu EDTA ,
V
H2O
- ilość ml wody pobranej do oznaczenia (200 ml).
Uwaga 1:
Jeśli analizowana próba nie zawiera wodorowęglanów dodatek HCl jest niepotrzebny.
Uwaga 2:
W warunkach oznaczenia roztwór zalkalizowany zasadą sodową wykazuje pH ok. 11.
W tych warunkach wytrąca się z roztworu biały osad wodorotlenku magnezu, który nie bierze
udziału w reakcji (I
Mg(OH)2
= 1.1·10
-11
, I
Ca(OH)2
= 5.5·10
-6
w 25
o
C)
Oznaczanie wapnia i magnezu w wodzie.
Oznaczanie sumy wapnia i magnezu wykonuje się wobec czerni eriochromowej T.
(Równania reakcji magnezu z EDTA i z czernią eriochromowa T są identyczne jak
w przypadku oznaczania cynku - patrz ćwiczenie „Oznaczanie cynku"). W wodzie musi być
obecny magnez, ponieważ sam wapń tworzy z czernią nietrwały kompleks i zmiana
zabarwienia nie byłaby wyraźna. Kompleks wapnia z EDTA jest trwalszy niż kompleks
magnezu.
Wynik oznaczenia (tzw. twardość całkowita lub ogólna ) podaje się w stopniach niemieckich:
CaH
4
Ind
+
CaH
3
Ind
CaH
2
Ind
-
żółtopomarańczowy
czerwonopomarañczowy
czerwony
m
=
Ca
40.08 a
c
EDTA
V
H
2
O

25
1°d=10 mg CaO w 1 l wody
Twardość wody jest to właściwość polegająca na zużywaniu pewnych ilości mydła bez
wytworzenia piany podczas wytrząsania próby wody. Właściwość tę nadają naturalnej wodzie
głównie obecne w niej jony wapnia i magnezu (jony innych metali występują w niewielkich
ilościach), które tworzą z mydłem dodawanym do wody nierozpuszczalne mydlą wapniowe
i magnezowe nie tworzące piany podczas wytrącania.
Twardość wody surowej nazywa się twardością ogólną (T
wo
) i oznacza m.in. w stopniach
niemieckich. Twardość może być wywołana przez wodorowęglany, węglany i wodorotlenki
wapnia i magnezu i nazywa się wtedy twardością węglanową (T
ww
). Twardość wywołana
przez inne związki wapnia i magnezu nazywa się twardością niewęglanową (T
wn
). Twardość
węglanowa i niewęglanowa stanowią w sumie twardość ogólną wody. T
w o
= T
w w
+ T
w n
Wykonanie oznaczenia.
Do kolby stożkowej o pojemności 200-300 ml odmierza się 100 ml badanej wody, dodaje się
z biurety mianowanego roztworu HC1 w ilości odpowiadającej uprzednio wyznaczonej
twardości węglanowej. Następnie dodaje się 10 ml buforu amonowego o pH 10, 0.1 mg czerni
eriochromowej T i miareczkuje roztworem EDTA o stężeniu 0.05 mol/l aż do zmiany barwy
z różowej na niebieską.
Uwaga:
Jeżeli badana próbka nie zawiera wodorowęglanów dodatek HC1 jest niepotrzebny.
Obliczenie wyników:
1.
Obliczanie zawartości magnezu w gramach zawartego w 1 l wody:
gdzie:
b- ilość ml roztworu EDTA zażyta na zmiareczkowanie sumy wapnia i magnezu
w 100 ml wody;
a-ilość ml roztworu EDTA zużyta na zmiareczkowanie wapnia w 200 ml wody;
V
H2O
- ilość ml wody pobrana do oznaczenia sumy wapnia i magnezu (tu 100 ml);
C
EDTA
- stężenie molowe roztworu EDTA.
2. Obliczanie twardości ogólnej wody w stopniach niemieckich korzystając ze wzoru:
m
Mg
24.30 (b -
c
EDTA
V
H
2
O
a
2
)
=

26
T
wog
=56.08·c
EDTA
·b
gdzie: T
wog
- twardość całkowita w stopniach niemieckich (°d);
C
EDTA
stężenie molowe roztworu EDTA;
b - ilość ml roztworu EDTA w ml zużyta do miareczkowania sumy wapnia i magnezu w
100 ml wody.
Literatura
1. A.Cygański „Chemiczne metody analizy" WNT 1994.
2. B. E. Gomółka „Ćwiczenia laboratoryjne z chemii wody" Wrocław 1992.
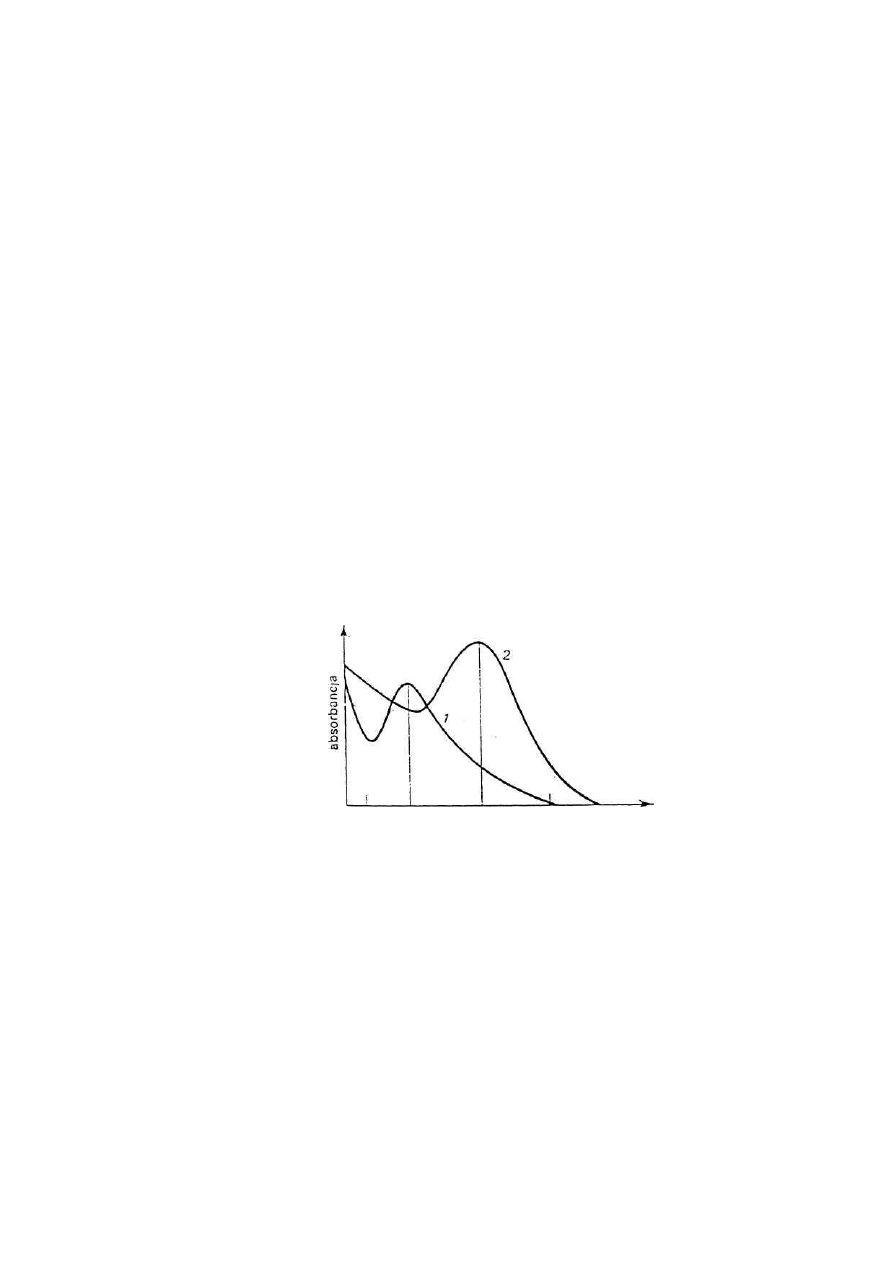
27
ĆWICZENIE NR 7
Spektrofotometryczne oznaczanie bizmutu(III) metodą jodkową.
W środowisku 0.2-2 mol/l kwasu siarkowego bizmut tworzy z jonami jodkowymi (wobec ich
nadmiaru
pomarańczowożółty
kompleks
BiI
4
)
stanowiący
podstawę
spektrofotometrycznego oznaczania bizmutu. Do stężenia 3% KI w roztworze zabarwienie
rośnie; przy dalszym wzroście stężenia jodku zabarwienie roztworu pozostaje
stałe. Wydzielaniu się wolnego jodu, w wyniku utleniania jodku tlenem powietrza lub przez
substancje utleniające obecne w roztworze badanym, zapobiega dodatek środków
redukujących. Stosuje się w tym celu kwas askorbinowy, siarczyny, podfosforyny
lub mieszaniny wymienionych odczynników. Jako reduktor może być stosowany także
tiomocznik. Jodkowy kompleks bizmutu wykazuje wyższe maksimum absorbcji przy 337 nm
i niższe w widzialnej części widma przy 465 nm. Molowy współczynnik absorbcji kompleksu
przy długości fali 465 nm wynosi 9.1·10
3
400 425
465
500
d
ługość fali, nm
Rys. Krzywe absorbcji jodkowych kompleksów antymonu(III) (1) i bizmutu(III) (2)
w roztworach wodnych.
Oznaczaniu bizmutu metodą jodkową przeszkadza antymon. W nieco zmienionych
warunkach antymon nie przeszkadza oznaczaniu bizmutu. Przy 1.5%-owym stężeniu KI
antymon nie daje już zabarwienia, zabarwienia pochodzące zaś od bizmutu jest przy tym,
obniżonym w stosunku do optymalnego, stężeniu jodku słabsze o ok.10%.
Poza antymonem barwne kompleksy jodkowe tworzą: platyna(IV), pallad(IV) i cyna(IV)
Srebro, tal, miedź i ołów wydzielają się w postaci trudnorozpuszczlnych jodków. Chlorki

28
i fluorki osłabiają zabarwienie jodkowego kompleksu bizmutu.
Wykonanie oznaczenia
Odczynniki, roztwory i aparatura
1. Jodek potasowy, 1% roztwór.
2. Roztwór podstawowy bizmutu o stężeniu l mg Bi/ml.
Rozpuścić 2.3210 g azotanu bizmutu Bi(NO
3
)
2
·5 H
2
O w 100 ml HNO
3
(1+3) i rozcieńczyć
roztwór wodą w kolbie miarowej do 1 l. Roztwory robocze otrzymuje się przez
odpowiednie rozcieńczenie roztworu podstawowego.0.01 M kwasem azotowym.
3. Kwas askorbinowy, 2% roztwór.
4. Kwas siarkowy, (1+1) roztwór
5. Kwas azotowy, (1+3) roztwór
6. Kwas azotowy, ok.0.01 M roztwór
7. Spektrofotometr
1 Przygotoanie roztorów wzorcowych.
Do 5 kolb miarowych o pojemności 50 ml przenieść kolejno:0.2;0.3;0.4;0.5;0.7 ml
roztworu podstawowego o stężeniu 1 mg Bi/ml. Następnie dodać do każdej kolby 5 ml kwasu
siarkowego /l+l/, 2 ml 2% kwasu askorbinowego i 10 ml 1% roztworu jodku potasowego.
Dopełnić wodą roztwór do kreski i wymieszać. Po upływie 5 min można mierzyć absorbancję.
a) Wykreślanie widma absorbcyjnego kompleksu BiI
4
-
Krzywą absorbcji jodkowego kompleksu bizmutu BiI
4
-
należy zmierzyć dla roztworu
bizmutu o największym stężeniu. Jako odnośnik stosuje się roztwór o identycznym składzie jak
roztwór badany nie zawierający bizmutu. (Ze względu na jego niewielką absorbancję można
także w tym przypadku stosować wodę jako odnośnik.)
Absorbancję roztworu bizmutu mierzymy dla różnych długości fali w zakresie 400-500 nm
co 10 nm. Na podstawie zmierzonych wartości należy wykreślić wykres zależności
absorbancji od długości fali i wybrać długość fali do oznaczeń.
b) Wykreślenie krzywej wzorcowej
Ustawić długość fali w na wyznaczoną w punkcie a/ i zmierzyć absorbancję przygotowanych
roztworów wzorcowych bizmutu. Jako odnośnik stosować identyczny roztwór

29
nie zawierający bizmutu. Na podstawie zmierzonych wartości absorbancji wykreślić wykres
zależności absorbancji od stężenia bizmutu.
c) Oznaczenie ilości bizmutu w próbie badanej.
Do otrzymanego roztworu /próby badanej/ umieszczonego w kolbie miarowej o pojemności
50 ml dodać 5 ml kwasu siarkowego (l+l), 2 ml 2% roztworu kwasu askorbinowego i 10 ml
roztworu jodku potasowego. Dopełnić roztwór wodą do kreski i wymieszać. Po upływie 5 min
zmierzyć absorbancję roztworu w identycznych warunkach w jakich wykonywano krzywą
wzorcową.
Nanieść na wykres krzywej wzorcowej absorbancję badanej próbki A
x
i odczytać
odpowiadające jej stężenie bizmutu w roztworze c
x
. Znając stężenie c
x
i objętość roztworu
badanego V można obliczyć ilość bizmutu w próbie:
m
Bi
= c
x
·V
Literarura:
1. Z. Marczenko „Spektrofotometryczne oznaczanie pierwiastków", PWN, Warszawa 1979.

30
Ć
WICZENIE NR
8.
Spektrofotometryczne oznaczanie żelaza(III) metodą tiocyjanianową
Zasada oznaczenia żelaza metodą tiocyjanianową (rodankową)
Jon żelazowy tworzy z jonami tiocyjanianowymi (rodankowymi) zabarwienie
krwistoczerwone. Przy małej ilości jonów tiocyjanianowych powstaje początkowo FeSCN
2-
, a
zwiększenie ich stężenia prowadzi do powstania jonu [Fe(SCN)
6
]
3-
. Intensywność barwy
roztworu zależy od stężenia jonów SCN
-
i od innych czynników. W obecności dużego
nadmiaru jonów tiocyjanianowych roztwór podlega prawu Beera w szerokim zakresie stężeń.
Reakcję należy wykonywać w środowisku kwaśnym, najlepiej od kwasu azotowego, w celu
cofnięcia hydrolizy jonu żelazowego
Fe
3 +
+ 3H
2
O → Fe(OH)
3
↓ + 3H
+
W wykonaniu oznaczenia przeszkadza obecność wielu anionów i kationów. Szereg
anionów tworzy z jonami żelazowymi jony zespolone, jak np. fluorki, fosforany, chlorki,
winiany, arseniany, octany. Z tego względu w razie nieobecności dużego nadmiaru tych
anionów najlepiej jest strącić najpierw żelazo amoniakiem w postaci Fe(OH)
3
, następnie osad
rozpuścić w kwasie azotowym i w otrzymanym roztworze kolorymetrycznie oznaczyć żelazo.
W wykonaniu oznaczenia przeszkadza obecność kationów tworzących osady i kompleksy z
jonami tiocyjanianowymi. Należą tu: Hg
2+
(tworzą Hg(SCN)
2
i [Hg(SCN)
4
]
2-
, dopuszczalna
zawartość 1 mg/ cm
3
), Cd
2+
, Zn
2+
, Mn
2+
, Ag
+
,Cu
2+
', Bi
3+
, Co
2+
, Ni
2+
,Cr
3+
i inne.
Pod nieobecność podanych wyżej jonów można oznaczyć metodą tiocyjanianową nawet
2.5 µg Fe w 50 cm
3
roztworu. Do oznaczenia najodpowiedniejsze są stężenia żelaza od
0.04 do 0.1 mg w 50 cm
3
.
Barwa roztworu po pewnym czasie słabnie wskutek redukcji jonów Fe
3+
przez jony SCN
-
.
W eterze, acetonie i innych rozpuszczalnikach organicznych kompleks tiocyjaniano-żelazowy
jest znacznie trwalszy. Z tego względu podczas oznaczania bardzo małych ilości żelaza,
prowadzi się oznaczania w roztworze zawierającym 50% acetonu. Kompleks tiocyjaniano-
żelazowy można również ekstrahować eterem (lub alkoholem izoamylowym)
i prowadzić oznaczenie w tym rozpuszczalniku. Czułość metody tiocyjanianowej (rodankowej)
wzrasta 2-3 krotnie przez zastosowanie rozpuszczalnika organicznego zamiast wody.
Natężenie barwy porównuje się z roztworem wzorcowym, zawierającym znaną ilość żelaza
31
Roztwór wzorcowy sporządza się zwykle z naważki Fe
2
(SO
4
)
3
·(NH
4
)
2
SO
4
·24H
2
O lub
FeNH
4
(SO
4
)
2
·12H
2
O.
Wykonanie oznaczenia.
1. Sporządzenie roztworu wzorcowego żelaza..
J eden mol, tj. 481 .98 g FeNH
4
(SO
4
)
2
·12 H
2
O uwodnionego siarczanu żelazowo-
amonowego zawiera 55.85 g Fe. Zwykle sporządza się roztwór podstawowy zawierający
0.1 mg Fe w 1 cm
3
. W tym celu rozpuszcza się w wodzie destylowanej zakwaszonej 10 cm
3
stężonego HNO
3
odważkę 0.4317 g soli FeNH
4
(SO
4
)
2
·12 H
2
O i rozcieńcza wodą do
objętości 1 dm
3
.
Jeżeli potrzebny jest roztwór wzorcowy o stężeniu 0.01 mg Fe/cm
3
to wówczas 25 cm
3
roztworu podstawowego przenosi się do kolby miarowej o pojemności 250 cm
3
, dodaje
się 5 cm
3
stężonego HNO
3
i rozcieńcza wodą destylowaną do kreski.
2. Przygotowanie krzywej wzorcowej
Do pięciu kolb miarowych o pojemności 50 cm
3
zawierających 25 cm
3
acetonu dodaje się
kolejno od 0.125 do 1.5 cm
3
roztworu Fe(III) o stężeniu 0.1 mg Fe/ cm
3
, następnie dodaje się
po 10 cm
3
10%-owego roztworu NH
4
SCN (do każdej kolby). Kolbę należy uzupełnić wodą
destylowaną do kreski i dokładnie wymieszać.
Mierzymy absorbancję otrzymanych roztworów wzorcowych na kolorymetrze wobec wody,
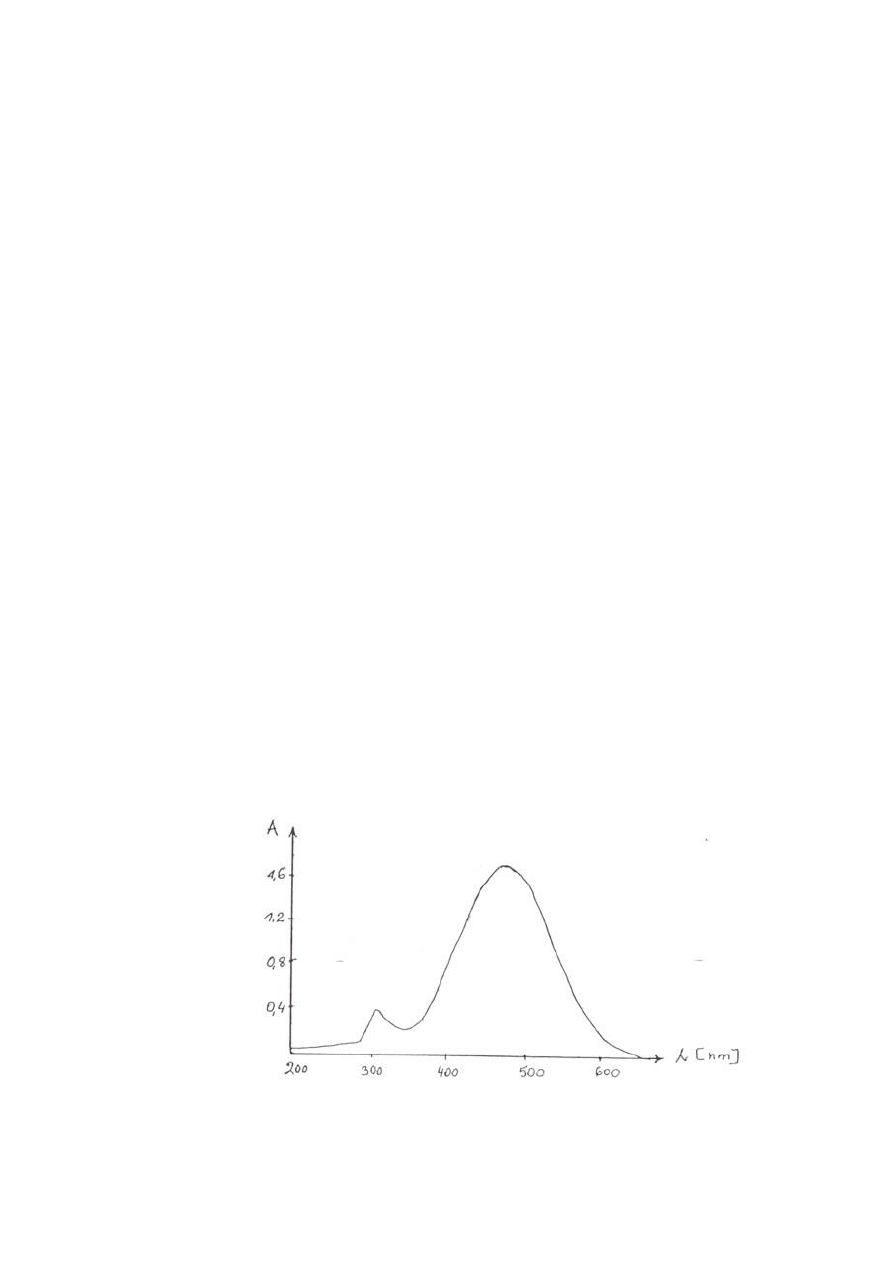
jako odnośnika przy długości fali λ.= 495 nm (Rys. 1). Należy uważać, aby roztwory nie były
poddawane bezpośredniemu działaniu promieni słonecznych.
Rys. 1. Widmo spektralne kompleksu tiocyjanianożelazowego(III).
Na podstawie otrzymanych wyników wykreśla się krzywą wzorcową (kalibracji) w jednym z
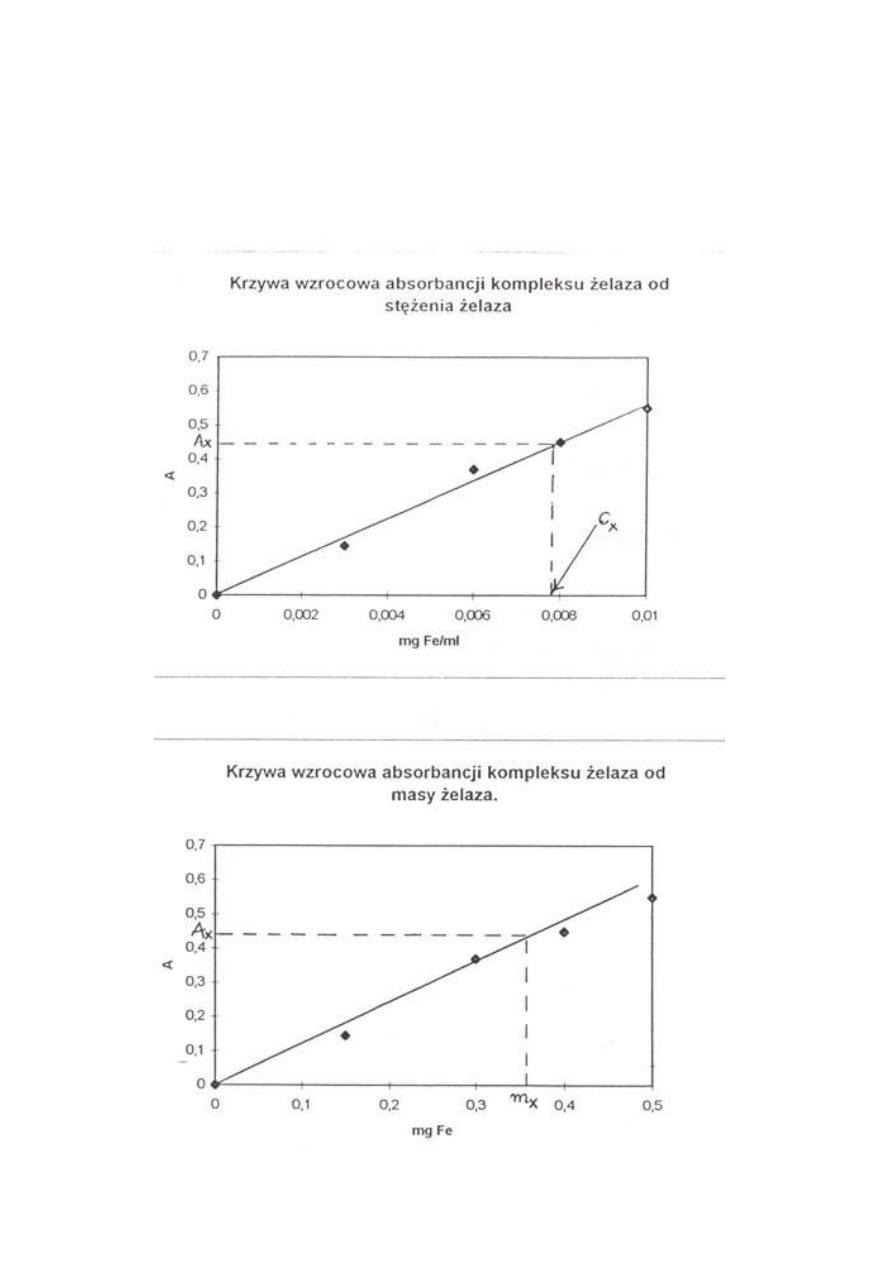
32
układów:
absorbancja vs. stężenie Fe w roztworze lub absorbancja vs masa Fe w
roztworze (przy stałej objętości roztworu V=50 cm
3
).

33
3. Oznaczanie żelaza w badanej próbie
Do otrzymanego w kolbie miarowej na 50 cm
3
zadania zawierającego nieznaną ilość żelaza
należy dodać: 25 cm
3
acetonu, 10 cm
3
roztworu 10%-owego roztworu NH
4
SCN, uzupełnić
roztwór wodą destylowaną do kreski i dokładnie wymieszać.
Zmierzyć absorpcję otrzymanego roztworu na spektrofotometrze wobec wody, jako odnośnika
Należy uważać, aby roztwory nie były poddawane bezpośredniemu działaniu promieni
słonecznych.
Na podstawie przygotowanej krzywej wzorcowej (kalibracji) obliczyć masę
żelaza w otrzymanym od asystenta roztworze do analizy ( w badanej próbie).
m
Fe
[mg] = ( c
x
)·50
gdzie
c
x
- stężenie Fe odczytane z wykresu A = f(c
Fe
)
lub odczytanej z wykresu A = f(m
Fe
)
Wyszukiwarka
Podobne podstrony:
M Deka,M Turowska Laboratorium Analizy Ilościowej
laboratorium analizy ilosciowej
instrukcja - ANALIZA ILOŚCIOWA-OBJĘTOŚCIOWA (miareczkowa), Inżynieria środowiska, inż, Semestr II, C
instrukcja - ANALIZA ILOŚCIOWA-WAGOWA, Inżynieria środowiska, inż, Semestr II, Chemia ogólna, labora
Cz VII Analiza ilosciowa
analiza ilosciowa 6 id 60541 Nieznany (2)
analiza ilosciowa 2 id 60539 Nieznany
Analiza ilosciowa substancji farmakopealnych metoda bromianometryczna
Laboratorium Analizametalograficznastalikonstr
Projekt I Analiza ilościowa i jakościowa rynku
Test sprawdzający Z. Hak, VII, VII Analizy ilościowe i graficzne przedstawienie wyników
lab, MetNum2 lab, Laboratorium: ANALIZA I PROJEKTOWANIE KOMPUTEROWE UKŁADÓW ELEKTRONICZNYCH
analiza ilościowa 3
Cwiczenie nr 10 Analiza ilościowa Alkacymetria Oznacznie weglanow i wodoroweglanow
BS Laboratorium 7 analiza ruchu sieciowego id 934 (2)
Analiza ilościowa
więcej podobnych podstron