1
1. E
STRYFIKACJA I ETERYFIKACJA
1.1. M
ECHANIZM REAKCJI ESTRYFIKACJI KWASAMI ORGANICZNYMI ALKOHOLI
I-
I
III-
RZĘDOWYCH
1.1.1. M
ECHANIZM ESTRYFIKACJI Z ALKOHOLAMI
I-
RZĘDOWYMI
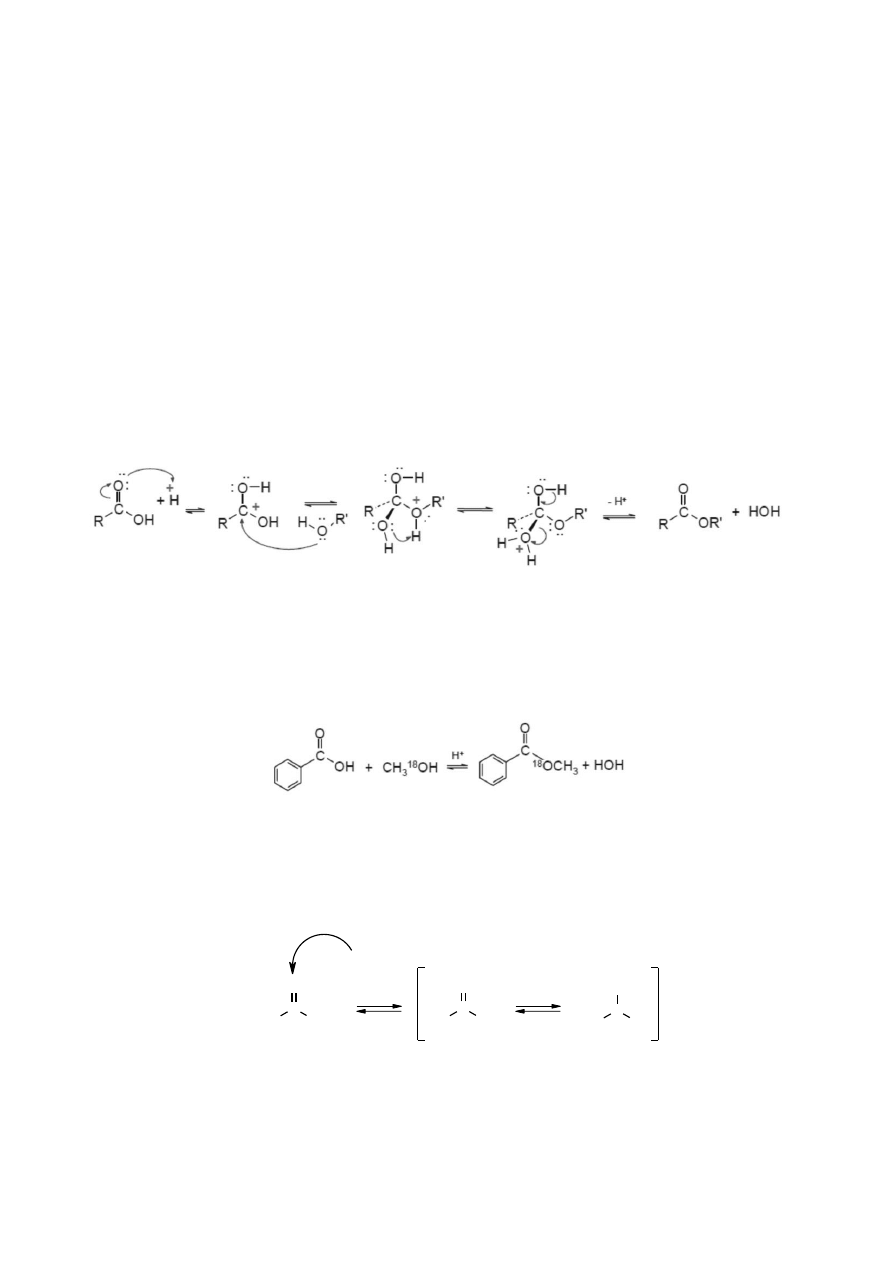
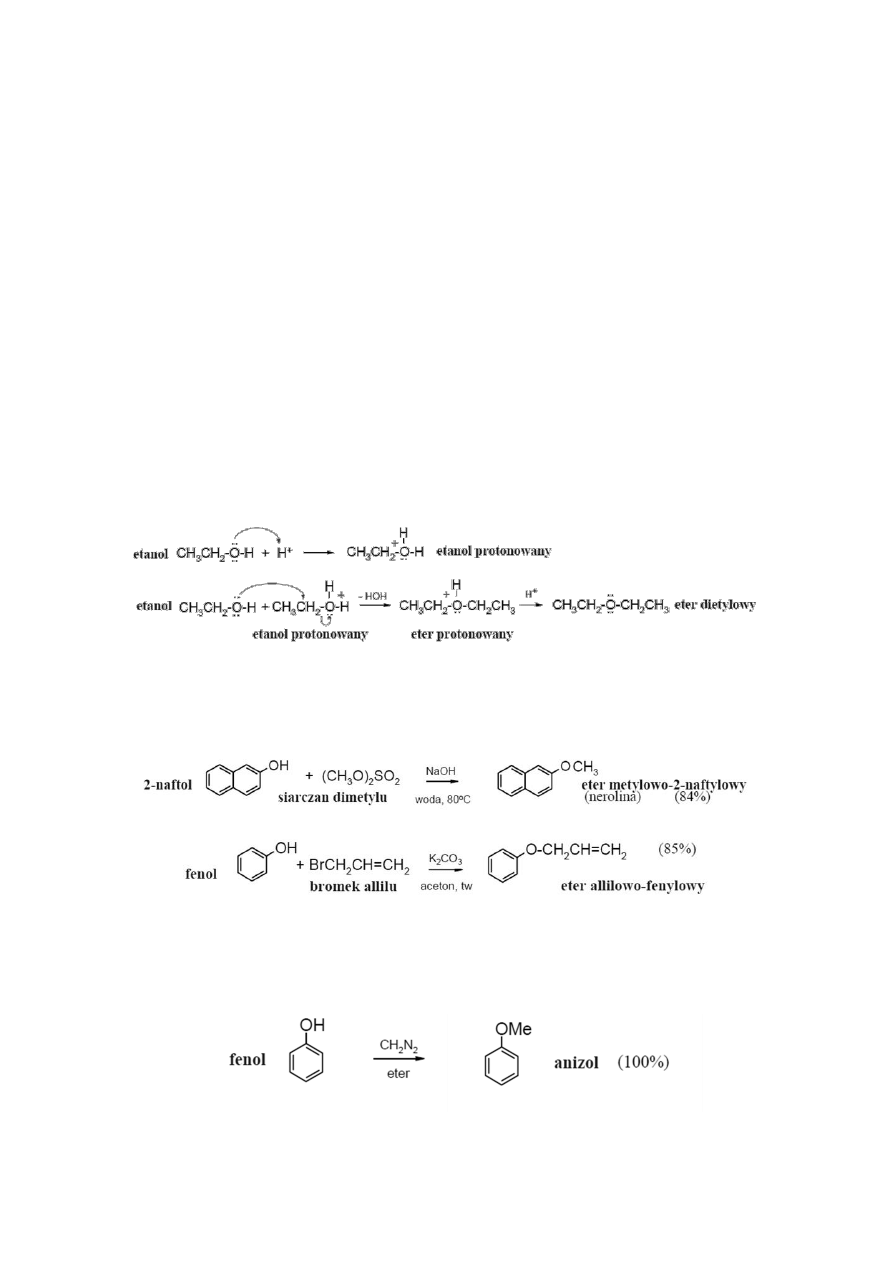
Mechanizm reakcji estryfikacji jest zależny od rzędowości reagującego alkoholu.
Pierwszym etapem reakcji z udziałem alkoholi
i
jest protonowanie atomu tlenu grupy
karbonylowej, przez co zostają zwiększone właściwości elektrofilowe karbonylowego atomu węgla.
Następnie dochodzi do ataku cząsteczki alkoholu (nukleofila) na tak uaktywniony atom węgla.
Utworzony czterowiązalny addukt stabilizuje się poprzez protonowanie atomu tlenu grupy -OH,
eliminację cząsteczki wody i protonu.
Atom tlenu łączący resztę alkilową R’ z karbonylowym atomem węgla w estrze otrzymanym
w reakcji estryfikacji z udziałem 1
o
lub 2
o
alkoholu pochodzi z cząsteczki alkoholu. Fakt ten został
udowodniony za pomocą reakcji kwasu z alkoholem znaczonym izotopem tlenu
18
O. Izotop
18
O
został wbudowany w cząsteczkę estru.
Mechanizm reakcji estryfikacji obejmuje więc następujące etapy:
a) Przyłączenie protonu do karbonylowego atomu tlenu grupy karboksylowej – zwiększenie
elektrofilowości acylowanego atomu węgla:
C
OH
O
C
H
3
H
+
OH
C
C
H
3
OH
+
OH
C
+
C
H
3
OH
+
-
b) Przyłączenie alkoholu do arylowanego atomu węgla:

2
OH
C
+
C
H
3
OH
C
H
3
OH
C
H
3
C
OH
OH
O
+
CH
3
H
+
c) Migracja protonu – powstanie grupy odchodzącej
:
C
H
3
C
OH
OH
O
+
CH
3
H
C
H
3
C
OH
OH
2
+
O
CH
3
d) Eliminacja wody z utworzeniem karbokationu przy acylowym atomie węgla:
C
H
3
C
OH
OH
2
+
O
CH
3
C
H
3
C
+
OH
O
CH
3
-H
2
O
e) Eliminacja protonu.
C
H
3
C
+
OH
O
CH
3
C
H
3
C
O
CH
3
O
-H
2
1.1.2. M
ECHANIZM ESTRYFIKACJI Z ALKOHOLAMI
III-
RZĘDOWYMI
Pierwszym etapem reakcji z udziałem alkoholi
jest protonowanie atomu tlenu alkoholu
i eliminacja wody – powstaje III-rzędowy karbokation.
H
+
C
C
H
3
CH
3
H
3
OH
C
C
H
3
CH
3
H
3
OH
2
+
C
+
C
H
3
CH
3
H
3
Następnie dochodzi do ataku karbokationu na atom tlenu cząsteczki kwasu. Utworzony
związek stabilizuje się poprzez eliminacje protonu.
C
OH
O
C
H
3
C
+
C
H
3
CH
3
H
3
C
C
H
3
CH
3
H
3
O C
+
OH
CH
3
C
C
H
3
CH
3
H
3
O C
O
CH
3
+
-
+
-H
2
Atom tlenu łączący resztę alkilową z karbonylowym atomem węgla w estrze
otrzymanym w reakcji estryfikacji z udziałem
z cząsteczki kwasu.
3
1.2. M
ETODY OTRZYMYWANIA ESTRÓW ALKOHOLI I FENOLI
1.2.1. A
LKOHOLIZA HALOGENKÓW KWASOWYCH
Alkoholizę halogenków kwasowych prowadzi się w środowisku zasadowym, w celu
pochłaniania wydzielającego się chlorowodoru. Rolę zasady pełni pirydyna, aminy 3
o
lub NaOH.
Reakcja chlorków kwasowych z alkoholami (fenolami) w wodzie, w obecności NaOH nazywa
się reakcją Schottena – Baumanna. Wymaga ona silnego mieszania lub wytrząsania, ponieważ
chlorki kwasowe, podobnie jak bezwodniki są nierozpuszczalne w wodzie.
1.2.2. A
LKOHOLIZA BEZWODNIKÓW KWASOWYCH
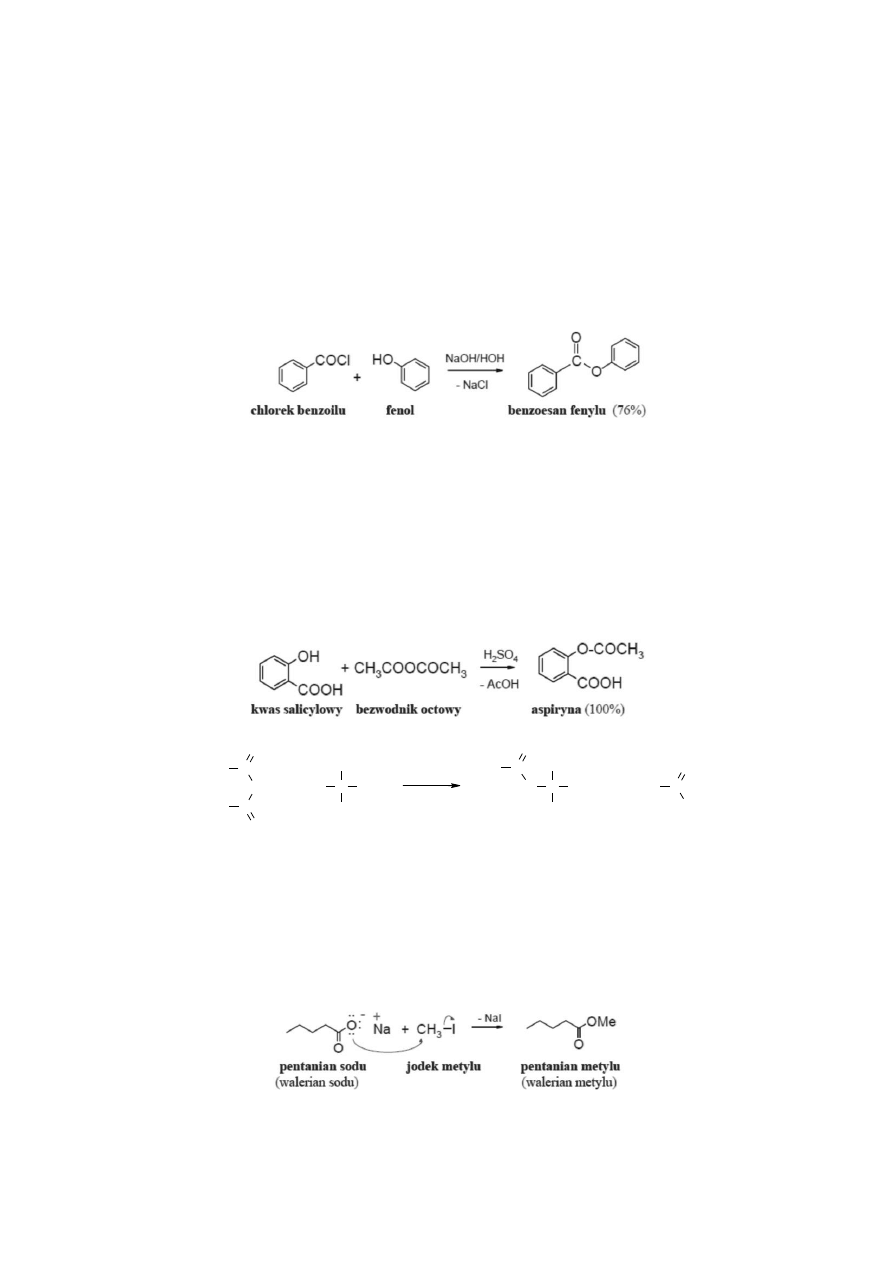
Alkoholizę bezwodników stosuje się jako metodę otrzymywania estrów najczęściej
wówczas, kiedy bezwodnik jest łatwo dostępny i tani. Te warunki spełniają bezwodnik octowy
i benzoesowy. Podczas acylowania bezwodnikiem jego połowa jest niewykorzystywana,
co zmniejsza wydajnośd atomową reakcji. Bezwodnik octowy jest używany, np. do otrzymywania
aspiryny, czyli kwasu acetylosalicylowego.
C
CH
3
CH
3
CH
3
O
H
C
C
H
3
O
O
C
O
C
H
3
C
CH
3
CH
3
CH
3
O
C
C
H
3
O
C
C
H
3
OH
O
alkohol t-butylowy
+
bezwodnik octowy
octan t-butylu (57%)
+
kwas octowy
ZnCl
2
1.2.3. A
LKILOWANIE SOLI KWASÓW KARBOKSYLOWYCH
Na sole kwasów karboksylowych działa się odczynnikami alkilującymi (halogenkami
alkilowymi lub estrami kwasów sulfonowych). Metoda ta jest ograniczona do pochodnych
zawierających 1° reszty alkilowe.
4
1.2.4. A
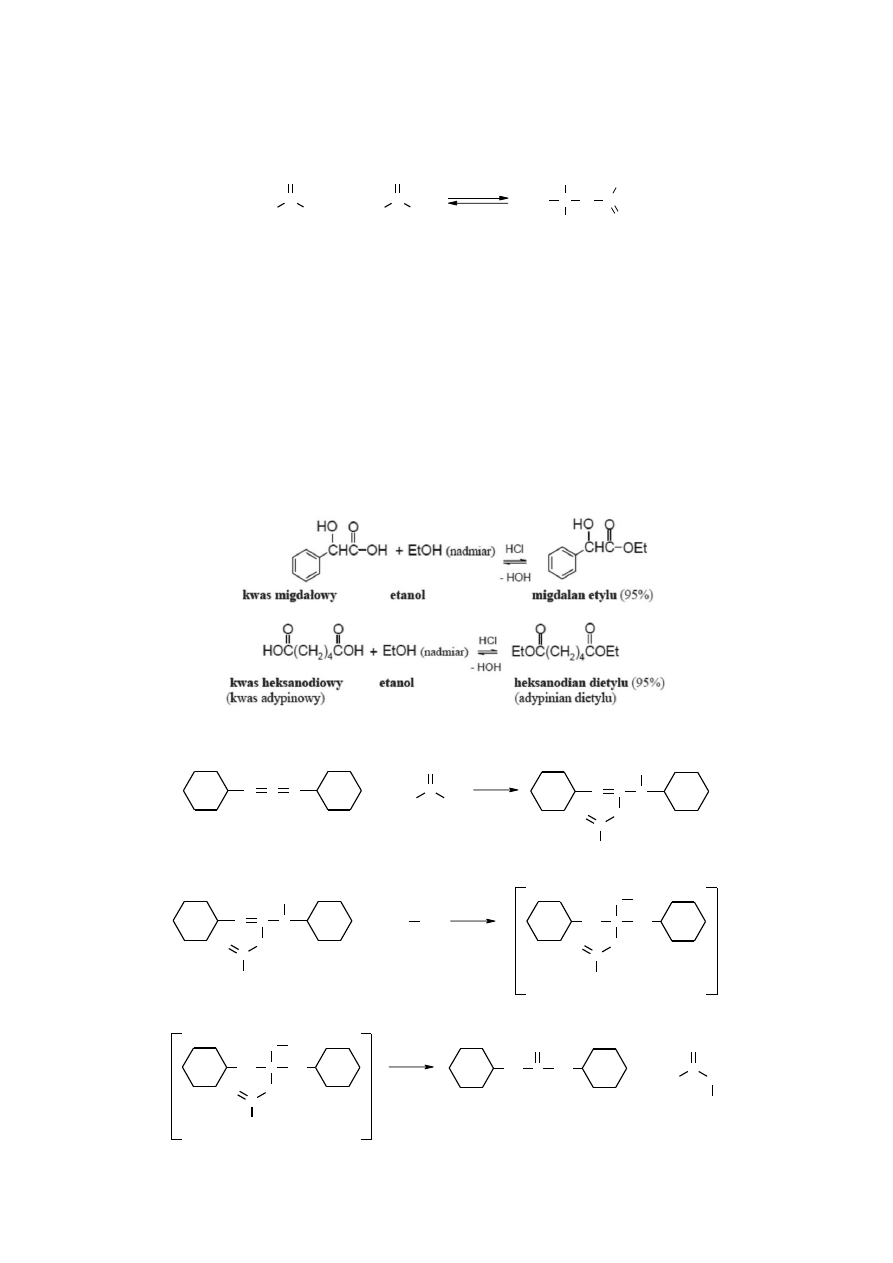
DDYCJA KWASÓW KARBOKSYLOWYCH DO OLEFIN
W wyniku przyłączenia kwasów karboksylowych do podwójnego wiązania alkenów tworzą
się estry. W ten sposób otrzymuje się, np. estry t-butylowe znacznie łatwiej niż innymi metodami.
C
OH
O
C
H
3
C
C
H
3
CH
3
H
3
O C
O
CH
3
H
2
SO
4
C
CH
3
CH
2
C
H
3
+
octan t-butylu (95%)
kwas octowy
izobuten
1.2.5. R
EAKCJA ESTRYFIKACJI
Reakcja estryfikacji to synteza estrów w wyniku działania kwasu karboksylowego
na alkohol. Wymaga ona obecności silnego kwasu jako katalizatora.
Reakcja estryfikacji jest reakcją odwracalną, wobec czego do osiągnięcia wysokiej
wydajności potrzebne jest stosowanie nadmiaru jednego z reagentów, oczywiście tego taoszego.
Najczęściej są to niższe alkohole, tj. metylowy, etylowy czy propylowy. Nadmiar alkoholu pełni
równocześnie rolę rozpuszczalnika.
1.2.6. R
EAKCJA ESTRÓW AKTYWOWANYCH
O
C
N
N
H
C
O
CH
3
C
H
3
C
O
OH
N
C
N
H
H
H
H
+
OH
C
H
3
O
C
N
N
H
C
O
CH
3
O
C
NH
NH
C
O
CH
3
O
CH
3
H
H
+
H
H
O
C
NH
NH
C
O
CH
3
O
CH
3
C
NH
NH
O
C
H
3
C
O
O
CH
3
T
H
H
H
H
+
5
1.2.7. R
EAKCJA TRANSESTRYFIKACJI
C
O
O
R'
R
R''
OH
OH
R'
R
C
O
R''
O
+
+
R''
C
O
R'
O
R
C
O
R'
O
C
O
OH
R''
O
C
R
OH
+
+
1.3. K
INETYKA REAKCJI ESTRYFIKACJI
O
C
R
OH
2
+
R OH
R C
+
O
OH
R
O
C
OH
R
R
+
R OH
2
+
OH
C
R
OH
2
+
O R
C
O
O
R
R'
H
+
O
C
R
OH
+
R'
O
C
+
R
OH
R'
O
C
R
O
R'
H
O
C
R
OH
2
+
+H
2
O
-H
+
alken
ALKOHOLE 1
o
v=k[ester][H
2
O]
karbokation stabilizowany
przez rezonans
H
2
O
-H
2
O
v=k[ester]
v=k[ester]
ALKOHOLE 2
o
ALKOHOLE 3
o
szybko
wolno
+
+
+
+
wolno
wolno
+
1.4. M
ECHANIZM REAKCJI HYDROLIZY ESTRÓW W ZALEŻNOŚCI OD ŚRODOWISKA REAKCJI
Hydrolizę estrów katalizują zarówno kwasy, jak i zasady. W środowisku obojętnym estry
reagują z wodą bardzo wolno. W wielu wypadkach potrzeba dni, żeby zauważyd ich wyraźny
rozkład. Wydzielający się jednak w reakcji kwas karboksylowy zwiększa szybkośd hydrolizy.
To zjawisko autokatalizy polega na katalitycznym działania powstającego kwasu. Hydroliza estrów
w środowisku kwaśnym jest reakcją odwracalną i stanowi odwrócenie procesu syntezy estrów
w reakcji Fischera.
Zasadowa hydroliza estrów jest reakcją nieodwracalną i wymaga stechiometrycznej ilości
zasady, ponieważ jest ona zużywana w miarę postępu reakcji – z powstającym kwasem tworzy
sole, zwane mydłami. Z tego powodu zasadowa hydroliza estrów nazywana jest zmydlaniem
lub z łaciny saponifikacją.
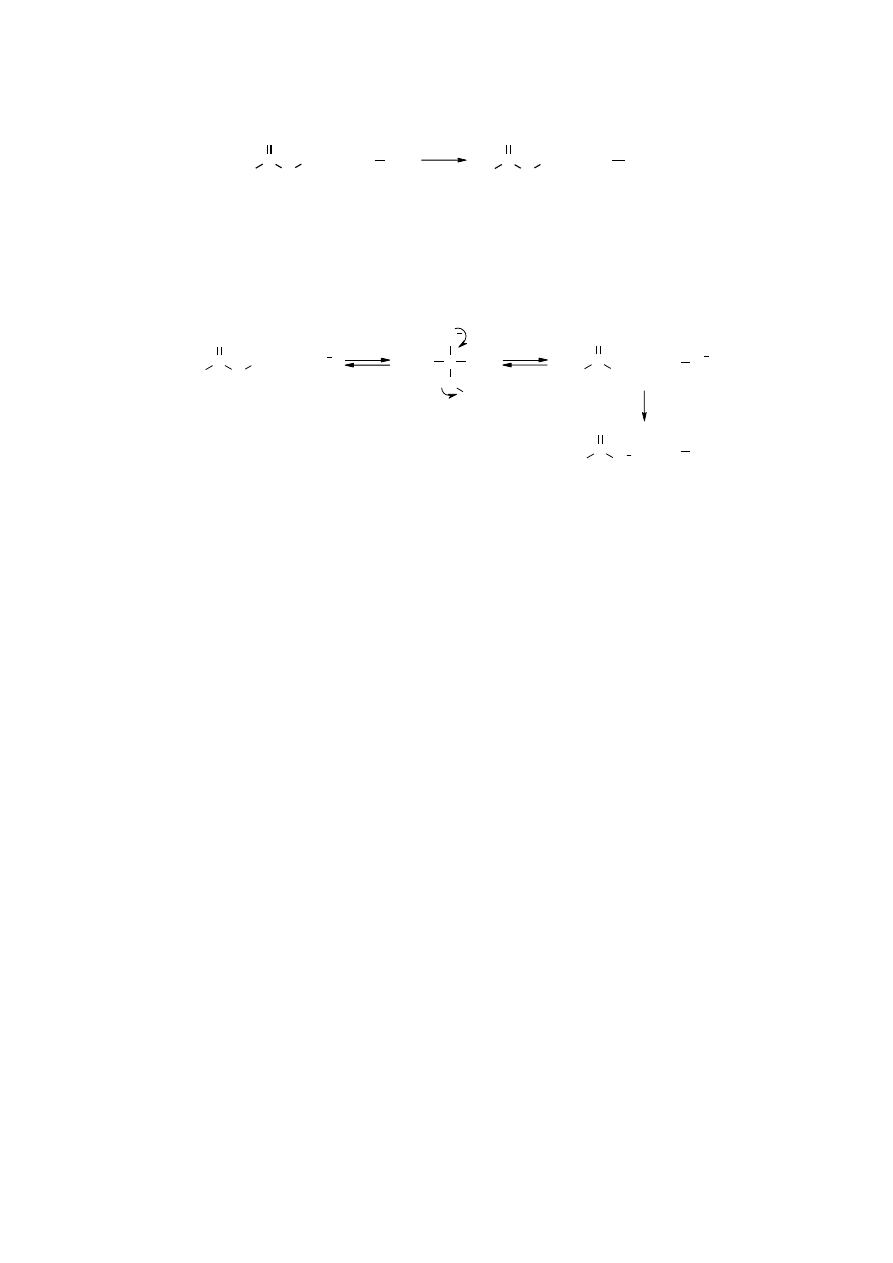
6
1.4.1. Z
ASADOWA HYDROLIZA ESTRÓW
Zasadą najczęściej używaną do reakcji z estrami jest wodorotlenek sodu.
C
O
O
R
R'
Na
OH
C
O
O
R
Na
R'
OH
+
+
Hydroliza estru w zasadowym środowisku może byd również prowadzona w bezwodnym
otoczeniu, np. w obecności alkoholu.
Zasadowa hydroliza estrów jest przykładem nukleofilowego podstawienia przy acylowym
atomie węgla. Mechanizm reakcji (na przykładzie octanu metylu):
C
O
O
CH
3
C
H
3
O
C
H
3
OH
C
H
3
C
O
O
OH
CH
3
C
O
OH
C
H
3
OH
C
H
3
C
O
O
C
H
3
+
+
+
Dwie pierwsze reakcje powyższego mechanizmu są odwracalne, a położenia ich równowagi
są przesunięte w kierunku substratów. Dopiero przeniesienie protonu od kwasu karboksylowego
do jon alkoholanowy w ostatnim etapie jest nieodwracalne i przesuwa równowagę w prawą stronę
aż do całkowitego przereagowania substratów. Nieodwracalnośd przeniesienia protonu
jest wywołana różnicą zasadowości anionów karboksylanowych i alkoholanowych.
1.4.2. K
WASOWA HYDROLIZA ESTRÓW
Kwasowa hydroliza estrów (jednocząsteczkowa) przebiegająca wg kinetyki reakcji
pierwszego rzędu.
Pierwszym etapem tej reakcji jest przyłączenie protonu do cząsteczki estru
i z wytworzeniem tautomeryzującego kationu. W drugim etapie następuje rozpad powstałego
kationu, przy czym tworzy się cząsteczka alkoholu i kation acylowy. Ta ostatnia reakcje jest
procesem jednocząsteczkowym. Ponieważ dalszy etap polegający na reakcji kationu acylowego
determinuje wypadkowy charakter reakcji hydrolizy.
Kwasowa hydroliza estrów (dwucząsteczkowa) przebiegająca wg kinetyki reakcji drugiego
rzędu.
Omawiany mechanizm polega na przyłączeniu do cząsteczki estru protonu. Następnie
do
tak
powstałego
karbokationu
acylowego
przyłącza
się
woda
/nukleofilowo/,
która za pośrednictwem swojej wolnej pary elektronowej włącza się do luki elektronowej
karbonylowego atomu węgla. Kompleks aktywny stabilizuje się przez odczepienie protonu
i cząsteczki alkoholu.
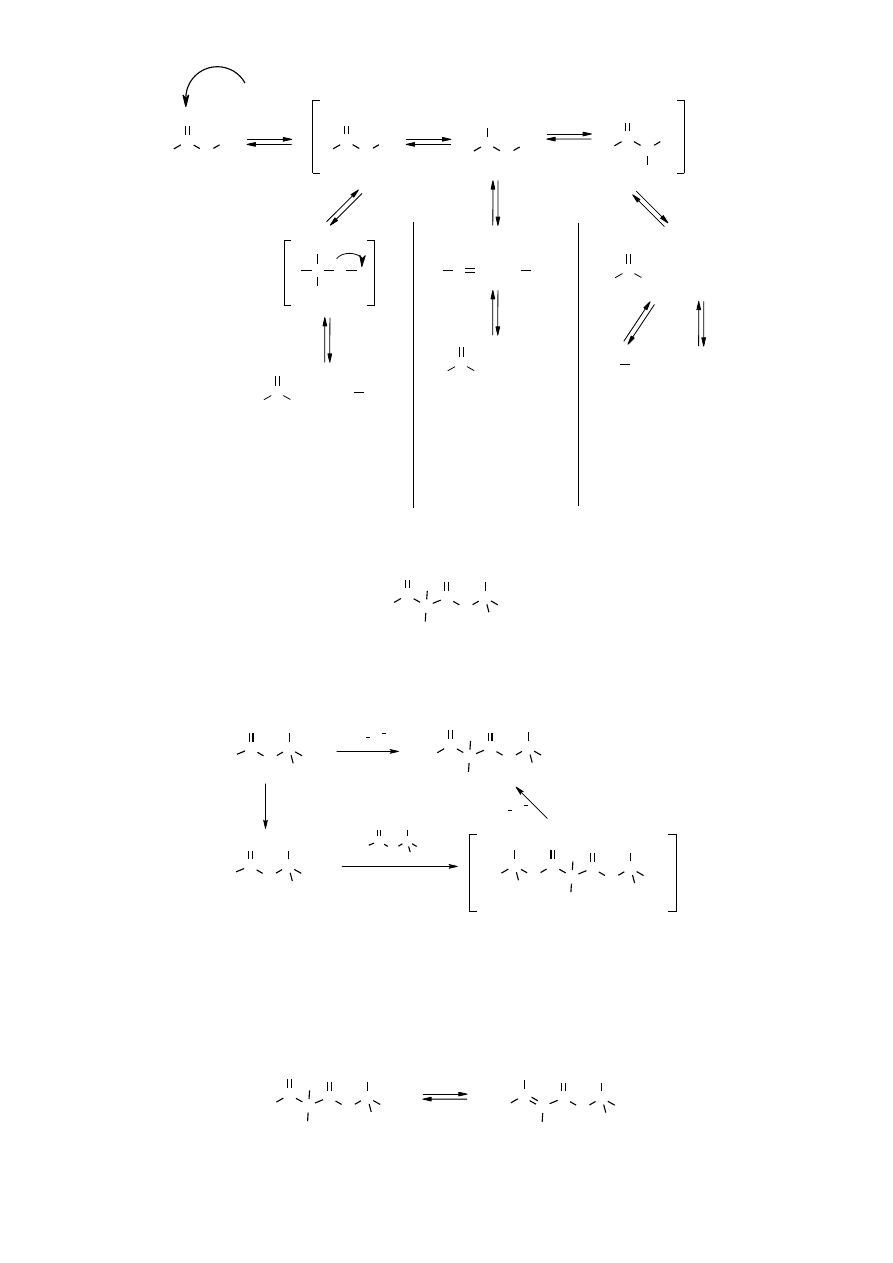
7
O
C
R
OH
2
+
R OH
R C
+
O
OH
R
O
C
OH
R
R
+
R OH
2
+
OH
C
R
OH
2
+
O R
C
O
O
R
R'
H
+
O
C
R
OH
+
R'
O
C
+
R
OH
R'
O
C
R
O
R'
H
O
C
R
OH
2
+
+H
2
O
-H
+
alken
ALKOHOLE 1
o
v=k[ester][H
2
O]
karbokation stabilizowany
przez rezonans
H
2
O
-H
2
O
v=k[ester]
v=k[ester]
ALKOHOLE 2
o
ALKOHOLE 3
o
szybko
wolno
+
+
+
+
wolno
wolno
+
1.5. A
CETYLOOCTAN ETYLU
–
SYNTEZA
,
WŁAŚCIWOŚCI I ZASTOSOWANIE
C
C
O
C
H
3
C
O
O
C
CH
3
H
H
H
H
Acetylooctan etylu w warunkach laboratoryjnych otrzymuje się w wyniku kondensacji
Claisena z octanu etylu katalizowanej etanolanem sodu.
C
C
O
C
H
3
C
O
O
C
CH
3
H
H
H
H
C
H
3
C
O
O
C
CH
3
H
H
C
2
H
5
O
C
H
3
C
O
O
C
CH
3
H
H
C
H
3
C
O
O
C
CH
3
H
H
C
C
O
O
C
O
O
C
CH
3
H
H
H
H
C
C
H
3
H
H
- C
2
H
5
O
Acetylooctan etylu jest bezbarwną, łatwopalną cieczą działającą drażniąco na błony
śluzowe. Przenika do organizmu przez drogi oddechowe i skórę.
Związek ten istnieje w dwóch formach tautomerycznych, enolowej i ketonowej, dzięki temu
może reagowad jak alkohol i jak kwas.
C
C
O
C
H
3
C
O
O
C
CH
3
H
H
H
H
C
C
OH
C
H
3
C
O
O
C
CH
3
H
H
H
forma ketonowa
forma enolowa

8
Protony w pozycji α względem grupy estrowej łatwo odczepiają się z wytworzeniem
karboanionów. Powstający ambidentny karboanion jest trwały ze względu na delokalizację pary
elektronowej w obrębie dwóch grup karbonylowych. Obecnośd zasady warunkuje
zapoczątkowanie reakcji.
C
C
C
O
O
H
O
C
2
H
5
CH
3
Zastosowanie: rozpuszczalnik organiczny, oraz substrat lub półprodukt w otrzymywaniu
aminokwasów, antybiotyków itd. W postaci wolnej stosowany jako związek zapachowy
posiadający zależnie od stężenia woo owocową i rumową.
Acetylooctan etylu służy do otrzymywania cząsteczek o bardziej rozbudowanym łaocuchu
węglowym. W reakcji alkilowania lub dialkilowania estru acetylooctowego i następczych,
czyli hydrolizy oraz dekarboksylacji powstają ketony.
Mechanizm:
Reakcja zaczyna się od oderwania kwaśnego protonu z grupy CH
2
znajdującej pomiędzy
dwiema karbonylowymi grupami funkcyjnymi acetylooctanu etylu i alkilowaniu tak otrzymanego
karboanionu za pomocą bromku alkilu. Po hydrolizie i dekarboksylacji monoalkilowej pochodnej
powstaje keton.
Monoalkilowa pochodna poddana ponownemu działaniu zasady również zostaje
przekształcona w karboanion, który także można alkilowad (np. za pomocą R’-X) do pochodnej
dialkilowej, a po hydrolizie i dekarboksylacji otrzymuje się keton, tym razem rozgałęziony.
R X
O
C
O
C
H
C
O
C
2
H
5
CH
3
R
Na
+
X
Na
O
C
O
C
H
C
O
C
2
H
5
CH
3
-
+
+
O
C
O
C
H
C
O
C
2
H
5
CH
3
R
OH
C
O
C
H
C
O
CH
3
R
H
C
H
C
O
CH
3
R
CO
2
H
+
H
2
O
Mechanizm dekarboksylacji
Dwie silnie elektroujemne grupy przyłączone do jednego atomu węgla destabilizują
cząsteczkę. Taka sytuacja występuje w anionie karboksylanowym acetylooctanu – z atomem
C
2
związana jest grupa karbonylowa (C
3
) i anion karboksylanowy (C
1
). W podwyższonej
temperaturze, w pobliżu temperatury wrzenia wody tego typu związki rozkładają się
z wydzieleniem cząsteczki CO
2
. Przemiana polega na tym, że miejsce grupy COOH zajmuje atom H.
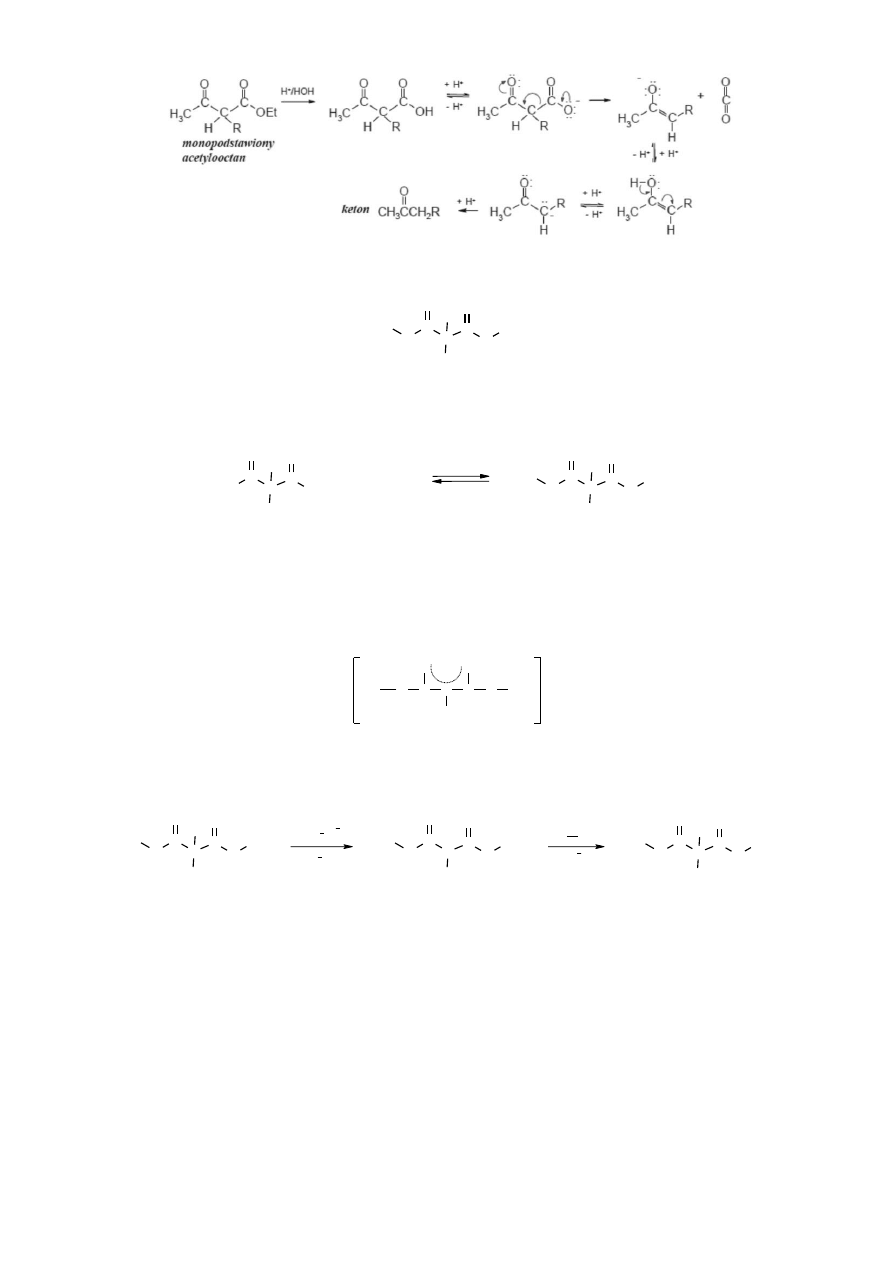
9
1.6. M
ALONIAN DIETYLU
–
SYNTEZA
,
WŁAŚCIWOŚCI I ZASTOSOWANIE
C
C
O
O
C
O
O
C
2
H
5
H
H
C
2
H
5
Malonian dietylu można otrzymywad w reakcji estryfikacji kwasu 1,3
–
propanodikarboksylowego i alkoholu etylowego:
C
C
O
O
C
O
O
C
2
H
5
H
H
C
2
H
5
C
C
O
O
H
C
OH
O
H
H
H
5
C
2
-OH
H
2
SO
4
+ 2
H
2
O
2
W cząsteczce tego związku grupa metylowa znajduje się pomiędzy dwiema silnie
elektroujemnymi grupami estrowymi, w związku z czym atomy wodoru tej grupy są stosunkowo
ruchliwe i odszczepiają się w postaci protonów pod wpływem mocnych zasad z utworzeniem
ambidentnego karboanionu utrwalany poprzez delokalizację pary elektronowej.
C
C
C
O
O
H
O
C
2
H
5
O
H
5
C
2
Nukleofilowy karboanion malonianu dietylu reaguje z halogenkami alkilów, tworząc
odpowiednie alkilopochodne estru malonowego:
C
2
H
5
O
C
C
O
O
C
O
O
C
2
H
5
H
H
C
2
H
5
C
C
O
O
C
O
O
C
2
H
5
H
C
2
H
5
R
X
X
C
C
O
O
C
O
O
C
2
H
5
H
R
C
2
H
5
C
2
H
5
OH
-
Dwukrotne alkilowanie prowadzi do kwasów rozgałęzionych w pozycji 2 (α).
Hydroliza uzyskanego alkilodiestru prowadzi do alkilopochodnej kwasu malonowego. Kwas
alkilomalonowy łatwo ulega dekarboksylacji przekształcając się w kwas jednokarboksylowy.
Opisana synteza jest wygodną metodą otrzymywania kwasów monokarboksylowych
o dowolnej długości łaocucha, z takich substratów jak: halogenki alkilowe i malonian dietylu.

10
1.7. T
ŁUSZCZE I WOSKI ORAZ ICH ZASTOSOWANIE
Tłuszcze to estry wyższych kwasów tłuszczowych i glicerolu. Tłuszcze wydzielane
z materiałów roślinnych i zwierzęcych zawierają od kilku do kilkunastu różnych kwasów
tłuszczowych. Rozdział indywidualnych glicerydów z mieszaniny tłuszczów jest niemożliwy.
Stan skupienie tłuszczów zależy od zawartości nienasyconych kwasów tłuszczowych.
Tristearynian glicerolu jest ciałem stałym o temperaturze topnienia wynoszącej 71 stopni
Celsjusza, natomiast trioleinian glicerolu jest cieczą krzepnącą w -17 stopniach.
Obniżenie temperatury topnienia tłuszczów przez nienasycone kwasy jest konsekwencją
kształtu łaocuchów węglowych. Podwójne wiązania w nienasyconych kwasach tłuszczowych
posiadają najczęściej konfigurację Z przez co są zgięte w miejscu podwójnego wiązania. Zgięte
łaocuchy węglowe nie wypełniają przestrzeni tak szczelnie, jak łaocuchy proste, a to pociąga
za sobą zmniejszczenie oddziaływao międzycząsteczkowych i mniejszą trwałośd kryształów.
Reakcje charakterystyczne tłuszczy to saponifikacja – zasadowe zmydlanie tłuszczy
oraz katalityczne uwodornienie tłuszczy płynnych (zastosowanie: produkcja stałych tłuszczy
roślinnych).
Woski są estrami wyższych kwasów tłuszczowych monokarboksylowych i wyższych alkoholi
jednowodorotlenowych o parzystych liczbach atomów węgla od C
16
do C
36
. Ponadto w woskach
występują domieszki węglowodorów szeregu parafinowego, wolne alkohole jak i kwasy
tłuszczowe.
Wśród alkoholi jakie otrzymuje się w wyniku hydrolizy wosków znajdują się następujące:
alkohol cetylowy, alkohol cerylowy, alkohol mirycylowy i inne.
Z kwasów spotykamy w woskach związane estrowo kwasy palmitynowy, stearynowy,
oleinowy, a także nie występujące w tłuszczach kwasy u większych masach cząsteczkowych,
między innymi kwas ligpocerynowy, kwas cerotynowy, kwas melicynowy i inne.
Estry występujące w woskach odznaczają się większą trwałością, trudniej ulegają hydrolizie
enzymatycznej i jełczeniu niż tłuszcze. Na przykład wosk pszczeli może byd przechowywany bez
dostrzegalnych zmian w ciągu wielu lat. Również temperatury topnienia wosków są wyższe od
tłuszczów i wahają się w granicach 50-90°C.
Wosk pszczeli stosowany jest w przemyśle włókienniczym, w produkcji powłok
wodoodpornych i różnego rodzaju past, a wosk olbrotowy stosuje się w przemyśle
farmaceutycznym, kosmetycznym i tekstylnym.
Woski spełniają u roślin i zwierząt rolę ochronną. Powlekają one cienką warstwą ochronną
liście, owoce, pióra, skórę uodparniając je na działanie szkodliwych mechanicznych bodźców

11
zewnętrznych oraz niekorzystnych czynników chemicznych i biologicznych. Woski niektórych roślin
chroniąc je przed zbytnią utratą wody drogą parowania.
W związku ze wzrastającym zapotrzebowaniem, ostatnio coraz większą uwagę zwraca się
na woski syntetyczne i pochodzenia mineralnego.
1.8. W
AŻNIEJSZE ESTRY KWASÓW NIEORGANICZNYCH
Estry kwasów nieorganicznych to związki powstające w reakcji alkoholi i kwasów
nieorganicznych, takich jak: kwas siarkowy(VI), kwas azotowy(V), kwas fosforowy(V), kwas borowy
i innych. Kwasy, które posiadają w swojej cząsteczce tylko jeden atom wodoru, mogą reagowad
tylko z jedną cząsteczką alkoholu, podczas gdy kwasy dwuprotonowe czy trzyprotonowe mogą
reagowad odpowiednio z dwiema lub trzema cząsteczkami alkoholi. Poniżej przedstawiono reakcje
otrzymywania estrów kwasów nieorganicznych wraz z nazwami uczestniczących w nich związków
i wzorami ogólnymi grup estrów pochodzących od tych samych kwasów nieorganicznych.
ESTRY KWASU AZOTOWEGO (V)
Estry kwasu azotowego tworzą się z dużą łatwością w reakcji HNO
3
z alkoholami:
Estry kwasu azotowego rzadko są otrzymywane w laboratorium, ponieważ nie znajdują w
chemii organicznej godnych uwagi zastosowao. Najbardziej znanym estrem jest tzw. nitrogliceryna
– stosowana do produkcji dynamitu i jako lek nasercowy. Triazotan(V) gliceryny jest cieczą bardzo
wrażliwą na wstrząsy i łatwo wybuchającą. W dużej skali przemysłowej produkuje się
nitroglicerynę i tetraazotan pentaerytrytu. Są to bardzo silne środki wybuchowe.
Nazwa “nitrogliceryna” nie jest poprawna, bo przedrostek nitro- zarezerwowany jest dla
związków, w których azot grupy nitrowej NO
2
jest bezpośrednio połączony z atomem węgla.
Estryfikacja alkoholi kwasem azotowym jest zabiegiem bardzo niebezpiecznym. Nawet w ściśle
kontrolowanych warunkach przemysłowych, ciągle zdarzają się tragiczne w skutkach wybuchy
instalacji do produkcji nitrogliceryny.
R O NO
2
R O H
HNO
3
R O NO
2
+
+
H
2
O
azotan alkilu
CH
OH
CH
2
CH
2
OH
OH
HNO
3
CH
CH
2
CH
2
NO
2
NO
2
NO
2
+
+
H
2
O
triazotan gliceryny
(nitrogliceryna)
3
3
C
H
3
O H
HNO
3
C
H
3
O NO
2
+
+
H
2
O
azotan (V) metylu
kwas
azotowy (V)
metanol
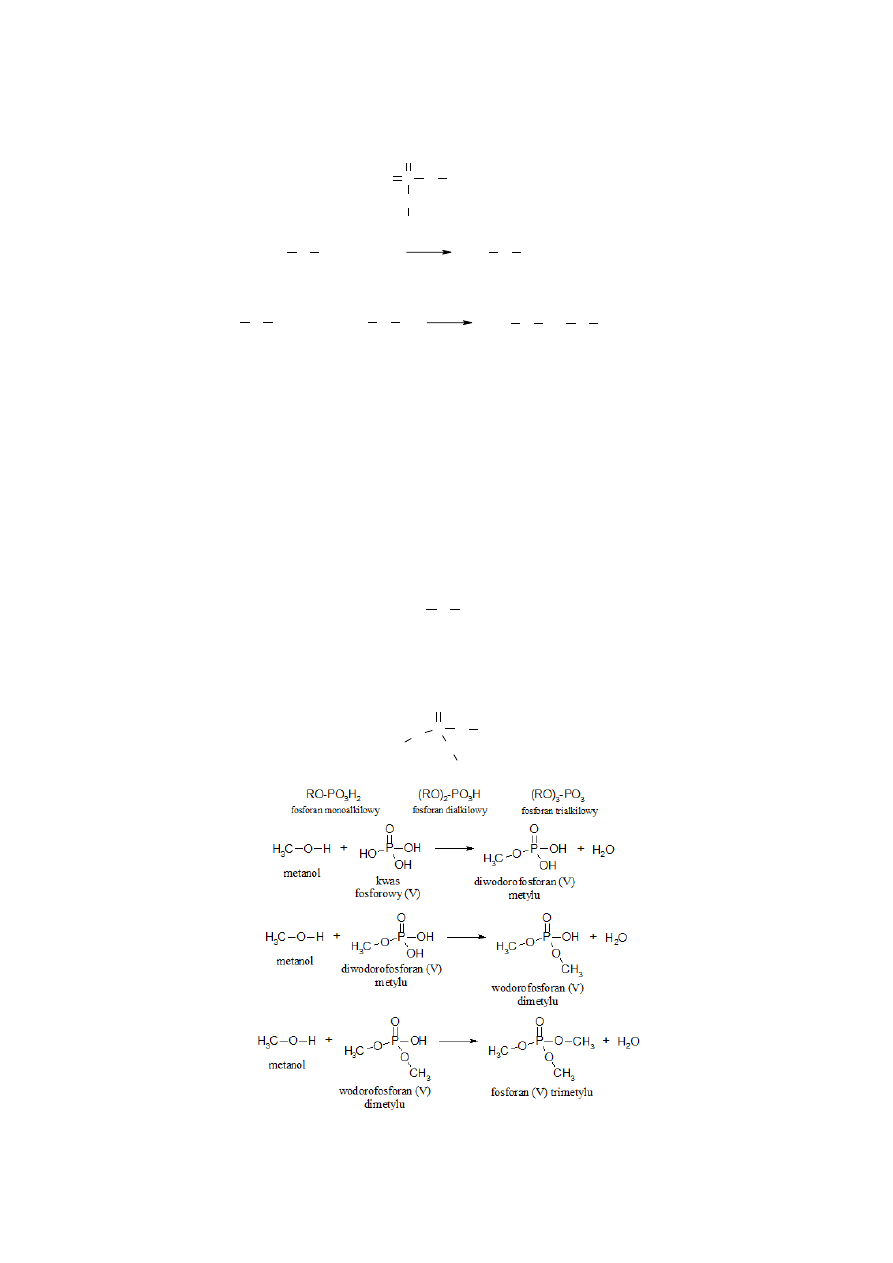
12
ESTRY KWASU SIARKOWEGO (VI)
Kwas siarkowy tworzy dwa szeregi estrów. Są to wodorosiarczany alkilowe i siarczany dialkilowe:
Duże znaczenie praktyczne mają wodorosiarczany otrzymane z alkoholi o długich
łaocuchach węglowych. Niektóre z nich są powszechnie stosowanymi detergentami.
Detergentami nazywamy substancje powierzchniowo czynne, czyli zmniejszające napięcie
powierzchniowe wody. Najdawniej używanym detergentem jest mydło.
Nazwa detergentów pochodzi od ich zdolności do usuwania brudu (łac. detergere – usuwad).
Do szeroko stosowanych detergentów należy dodecylosiarczan sodu:
ESTRY KWASU FOSFOROWEGO (V)
S
O
O
O
O
R1
R2
C
H
3
O H
H
2
SO
4
C
H
3
O SO
3
H
+
+
H
2
O
wodorosiarczan (VI)
metylu
kwas
siarkowy (VI)
metanol
C
H
3
O H
C
H
3
O SO
3
H
C
H
3
O SO
2
O CH
3
+
wodorosiarczan (VI)
metylu
metanol
siarczan (VI) dimetylu
+
H
2
O
CH
3
-(CH
2
)
11
O SO
3
Na
dodecylosiarczan sodu
O P
O
O
O
R1
R2
R3
13
1.9. E
TERY ŁAŃCUCHOWE
–
OTRZYMYWANIE
,
WŁAŚCIWOŚCI I ZASTOSOWANIE
Etery są związkami o wzorze ogólnym R-O-R
‘
, gdzie symbole R i R
‘
oznaczad mogą zarówno
podstawnik alifatyczny jak i aromatyczny. Etery mogą byd traktowane jako pochodne
hydroksyzwiązków organicznych, w których wodór grupy hydroksylowej został zastąpiony
podstawnikiem węglowodorowym R’.
W eterach obydwa podstawniki związane z tlenem mogą byd jednakowe (R=R’), mamy
wówczas do czynienia z eterami symetrycznymi, lub różne (R ≠ R’), jak to jest w przypadku
Otrzymywanie eterów:
a) Katalizowana kwasem siarkowym dehydratacja alkoholi
Przemysłowa metoda wytwarzania eteru etylowego polega na dehydratacji etanolu,
reakcji katalizowanej stężonym kwasem siarkowym. Do przekształcania etanolu w eter używa się
większych ilości kwasu, a alkohol dodaje się do reaktora w sposób ciągły w miarę postępu reakcji.
Przy użyciu tej metody można otrzymywad odpowiednie łaocuchowe etery symetryczne
z innych alkoholi 1
o
. Reakcja przebiega wg mechanizmu S
N
2.
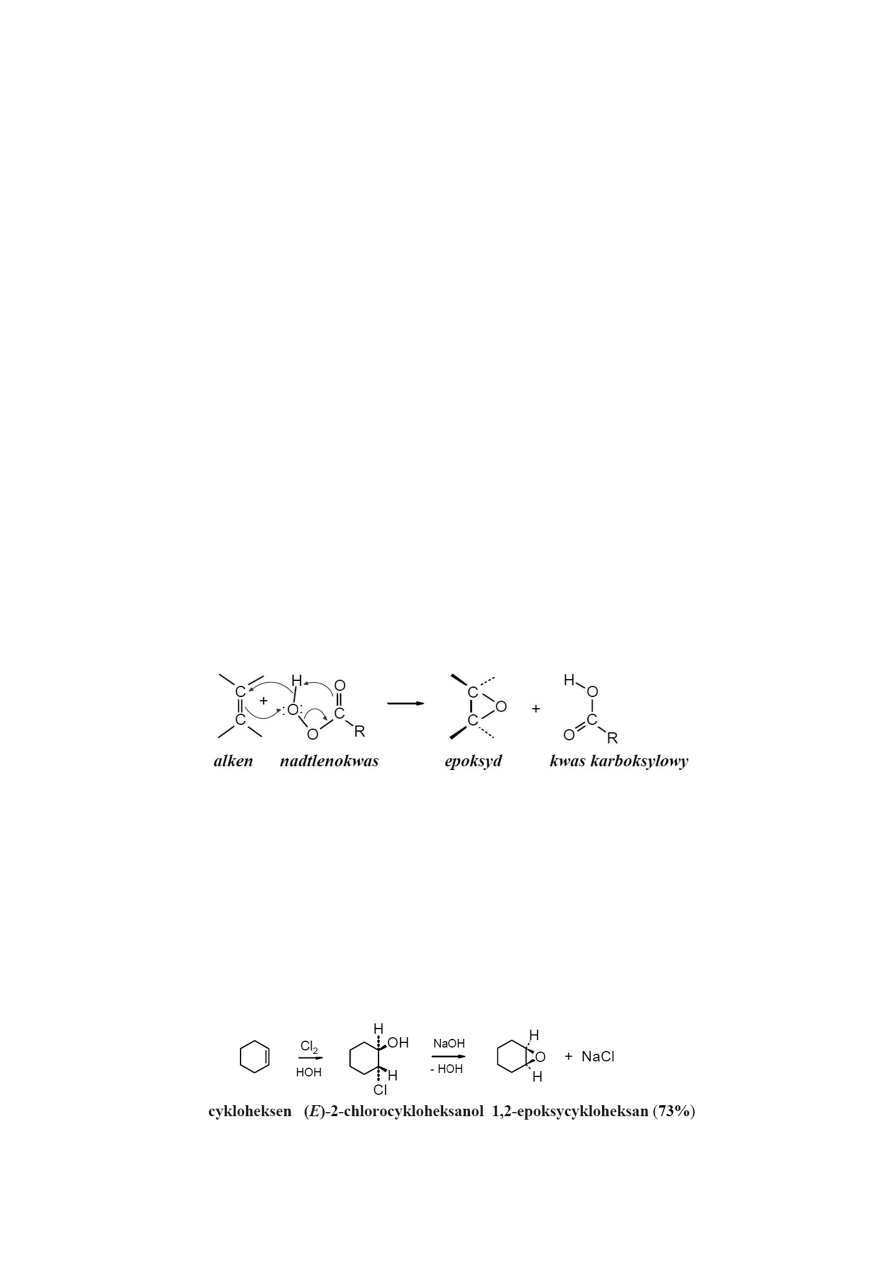
b) Metoda Williamsona
Jest to ogólna metoda otrzymywania eterów poprzez alkilowanie alkoholanów
halogenkami albo siarczanami alkilowymi. Reakcja katalizowana zasadami.
Fenole są bardziej reaktywne od alkoholi, dlatego reakcja przebiega w wodzie w obecności
słabej zasady, jaką jest węglan potasu. Atom wodoru grupy fenolowej jest wystarczająco kwaśny
by podstawienie grupą metylową mogło nastąpid przy użyciu diazometanu.
14
Właściwości chemiczne:
Ogólnie biorąc etery odznaczają się małą reaktywnością chemiczną, ponieważ wiązanie
węgiel – tlen nie ulega łatwo rozerwaniu. Z tego powodu często stosuje się etery w syntezie
organicznej w charakterze rozpuszczalników. Szczególne zastosowanie wykazują: eter dietylowy,
eter diizopropylowy.
W przeciwieostwie do alkoholi, etery nie mają kwasowego charakteru i zwykle nie reagują
z zasadami. Odczynniki wyjątkowo silnie zasadowe, w szczególności związki metaloorganiczne
zawierające metale alkaliczne, działają na etery powodując ich rozkład.
Etery reagują z tlenem z powietrza i tworzą, silnie wybuchowe nadtlenki.
1.10. E
TERY CYKLICZNE
–
OTRZYMYWANIE
,
WŁAŚCIWOŚCI I ZASTOSOWANIE
Związki cykliczne zawierające w pierścieniu atom tlenu związany z dwoma atomami węgla
nazywane są eterami cyklicznymi. W zależności od wielkości pierścienia występują etery cykliczne
trójpierścieniowe, czteropierścieniowe, pięciopierścieniowe, itd.
Otrzymywanie:
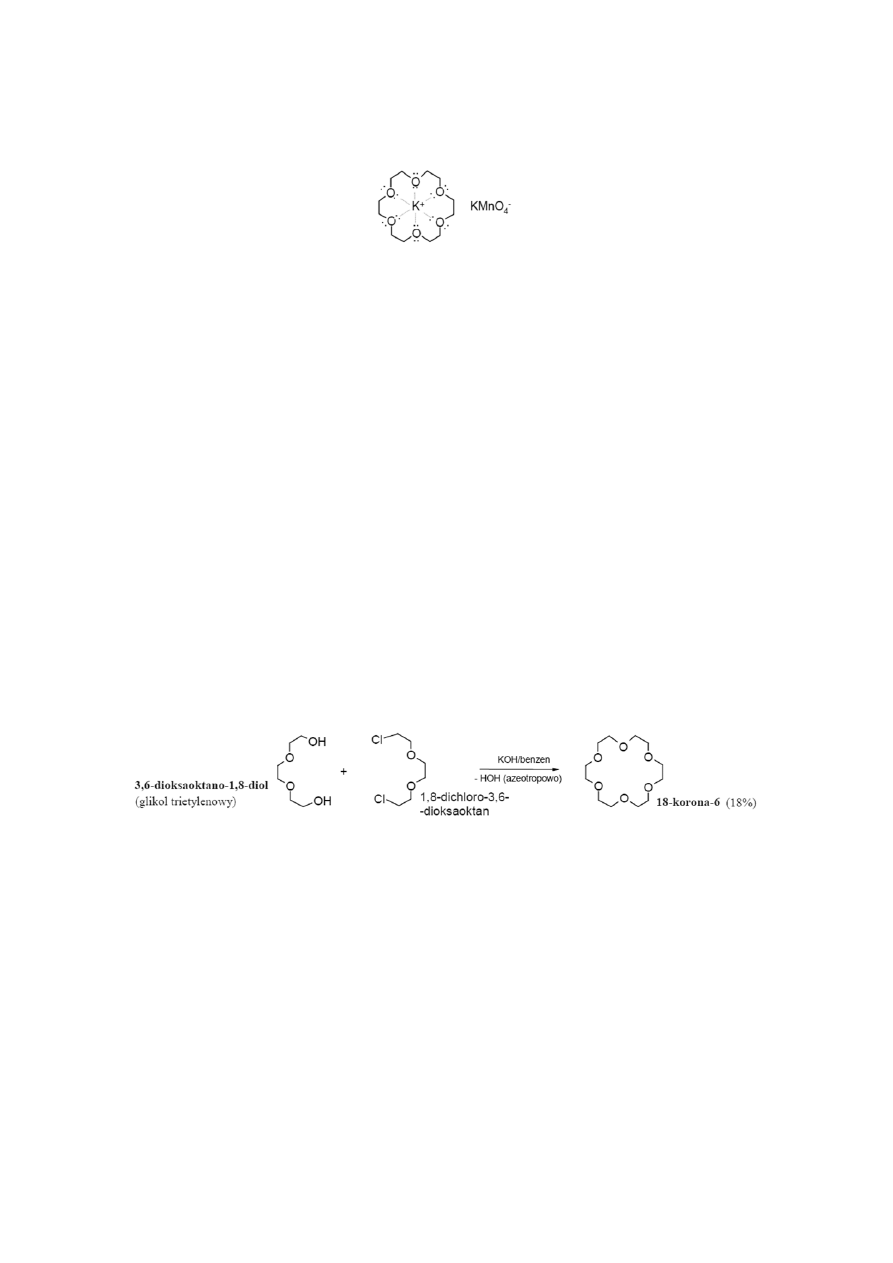
a) Utlenianie alkenów kwasami peroksykarboksylowymi:
Laboratoryjne metody otrzymywania epoksydów polegają na utlenieniu alkenów peroksykwasami
– RCOOOH:
b) Eliminacja halogenowodoru z halogenohydryn w środowisku zasadowym
Innym
sposobem
otrzymywania
epoksydów
jest
eliminacja
halogenowodoru
z 1,2-hydroksyhalogenozwiązków (halohydryn) w środowisku zasadowym.
Traktowanie 2-chloroetanolu zasadą – Ca(OH)
2
– było przez lata przemysłową metodą
otrzymywania tlenku etylenu. Jednak ze względów ekologicznych, tj. z powodu powstawania
ogromnych ilości ścieków zawierających zanieczyszczony chlorek wapnia, technologia ta została
zaniechana.

15
c) Utlenianie alkenów
Ugrupowanie epoksydowe powstaje podczas utleniania alkenów tlenem z powietrza
w obecności srebra jako katalizatora. Proces jest tak prowadzony, żeby konwersja etenu nie była
wysoka. Zapobiega to dalszemu utlenieniu epoksydu.
Właściwości chemiczne:
Niższe cykliczne etery wykazują wysoką reaktywnośd. W wodnych roztworach kwasów
ulegają przekształceniu 1,2-diole.
Rozszczepianie epoksydów za pomocą odczynników nukleofilowych, charakterystyczna
reakcja tych związków, daje 1,2-difunkcyjne pochodne. Nukleofil zajmuje pozycje po przeciwnej
stronie pierścienia niż grupa –OH pochodząca z rozszczepionego pierścienia epoksydowego.
W reakcji z alkoholami powstają alkoksyalkohole, z amoniakiem lub aminami – aminoalkohole,
z halogenowodorami – halogenohydryny.
Wyższe etery cykliczne.
Wyższe etery cykliczne, szczególnie te o pierścieniach od pięcioczłonowego wzwyż,
właściwościami chemicznymi przypominają etery alifatyczne. Na uwagę zasługuje tetrahydrofuran,
który jest szeroko stosowany w laboratoriach chemicznych jako rozpuszczalnik.
Otrzymuje się go z furanu poprzez uwodornienie w obecności niklu jako katalizatora.
Reaktywnośd eterów cyklicznych w zależności od wielkości pierścienia przypomina
właściwości chemiczne cykloalkanów – najbardziej reaktywny jest cyklopropan, potem
reaktywnośd spada; cykloheksan jest podobny właściwościami chemicznymi do alkanów. Podobnie
jest z eterami cyklicznymi.
Do popularnych rozpuszczalników należy dioksan. Jego rozpuszczalnośd w wodzie wynosi
8g/100 ml w 20
o
C. Tetrahydrofuran miesza się z wodą w każdym stosunku.
Etery koronowe
Etery koronowe:
Odkrycie eterów koronowych w latach 60. XX w. było nie lada sensacją naukową, ponieważ
okazało się, że mają one niezwykłe właściwości. Potrafią, np. spowodowad rozpuszczenie soli
nieorganicznych, w tak niepolarnych rozpuszczalnikach jak, np. benzen.
Umieszczone w rozdzielaczu dwie nie mieszające się z sobą ciecze – bezbarwny benzen
i fioletowy wodny roztwór KMnO
4
nawet po wielokrotnym wytrząsaniu rozdzielą się i górna
16
warstwa organiczna pozostanie bezbarwna, a dolna wodna – fioletowa. Co oznacza,
że nadmanganian potasu jest nierozpuszczalny w benzenie. Jednak po dodaniu do rozdzielacza
szczypty 18-korony-6 (eteru koronowego) – warstwa benzenowa zabarwi się na fioletowo.
Jony nadmanganianu potasu w obecności eteru 18-korony-6 zostają rozdzielone – kation K
+
skoordynowany we wnętrzu korony traci wysoką polarnośd, stając podatny na solwatowanie przez
cząsteczki benzenu. Anion MnO
4
-
, który także ulega rozpuszczeniu w benzenie jest w stanie
reagowad z niepolarnymi substancjami znajdującymi się w niepolarnym rozpuszczalniku,
np. utleniad je.
Możliwośd
rozpuszczenia
polarnych
odczynników,
np.
utleniaczy
w
niepolarnych
rozpuszczalnikach, to nie jedyna korzyśd, jaką daje stosowanie eterów koronowych w tego typu
reakcjach. Uwięzienie kationu we wnęce eteru koronowego wywołuje podobny wpływ
na reaktywnośd anionu jak działanie polarnych rozpuszczalników aprotycznych, typu DMSO, DMF
czy HMPA. Uwolniony od kationu anion staje się bardziej reaktywny.
a) Otrzymywanie – metoda Williamsona
Etery koronowe można otrzymad poprzez alkilowanie alkoholanów, zgodnie z reakcją
Williamsona. Konkurencyjnie tworzą się etery liniowe. Dodatek do mieszaniny reakcyjnej
odpowiednich kationów katalizuje cyklizację, np. jony K
+
sprzyjają tworzeniu się 18-korony-6.
1.11. C
HARAKTER CHEMICZNY I HYDROLIZA ETERÓW
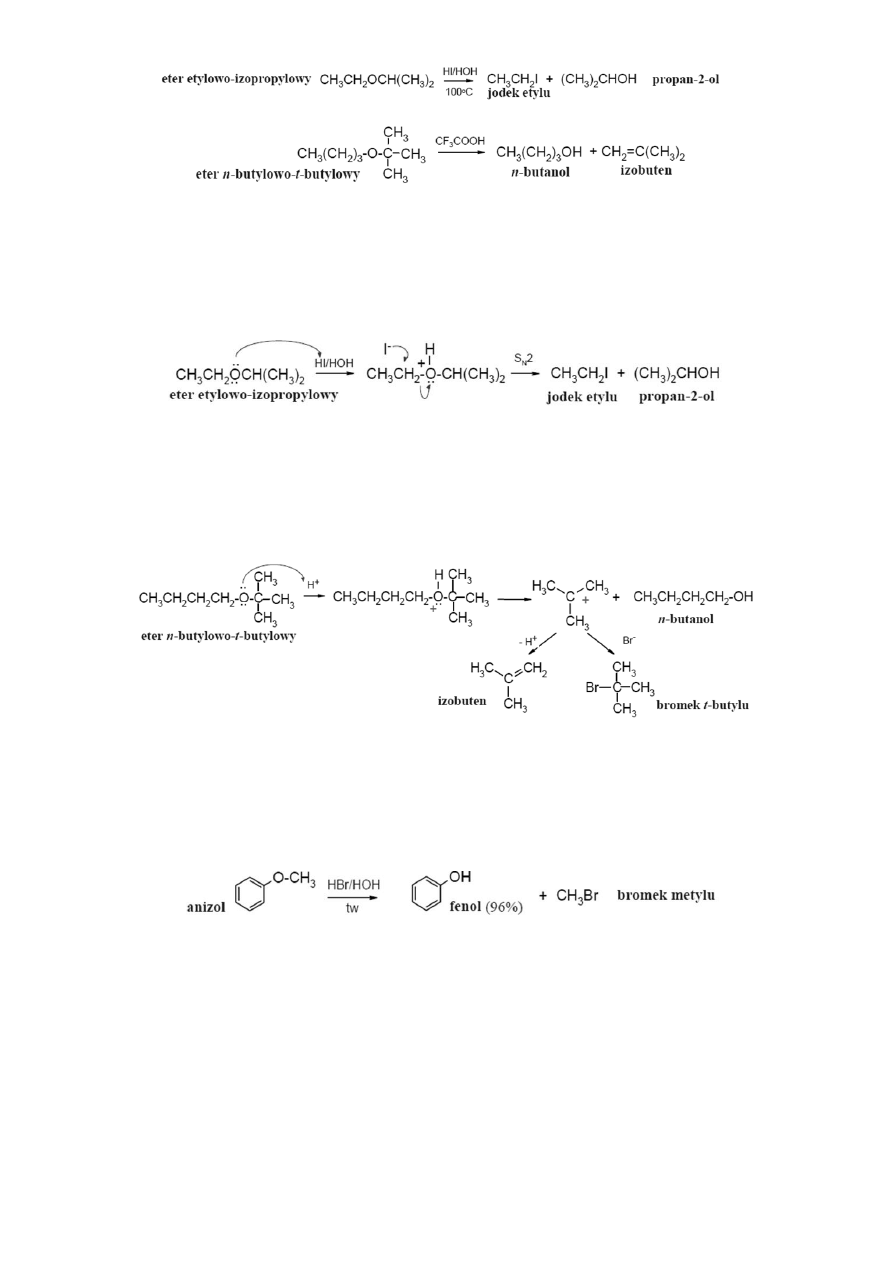
Pod wpływem silnych kwasów (halogenowodorów) w eterach dochodzi do rozerwania
wiązania C-O, czyli reakcji zwanej rozczepieniem eterów na halogenek i alkohol. Szereg eterów,
wg podatności na rozszczepienie kwasowe:
R-O-R’< R-O-Ar < Ar-O-Ar’ < R-O-t-Bu
Etery alifatyczne rozkłada się za pomocą najsilniejszego kwasu halogenowodorowego,
jakim jest jodowodór i to jest możliwe dopiero w podwyższonej temperaturze, etery alkilowo-
aromatyczne ulegają tej reakcji już pod wpływem kwasu bromowodorowego (w temperaturze
wrzenia), a odszczepieniu ulega bromek alkilu, zaś etery t-butylowe rozkładają się już w 0
o
C
w obecności kwasu trifluorooctowego, przy czym wydziela się izobuten.
17
Mechanizm:
Reakcję rozpoczyna protonowanie atomu tlenu z utworzeniem soli oksoniowej, która pod
wpływem anionu halogenkowego (nukleofila) ulega przekształceniu w halogenek alkilowy
i alkohol, wg mechanizmu S
N
2 (ścieżka A):
Sól oksoniowa może ulegad również rozpadowi na alkohol i karbokation, którego
stabilizacja następuje poprzez przyłączenie nukleofila prowadzące do powstania halogenku
alkilowego tudzież odczepienia protonu z wytworzeniem olefiny. Drugi mechanizm dotyczy eterów
zawierających 3
o
resztę alkilową (ścieżka B):
Rozszczepianie eterów metylowo – arylowych zachodzi ilościowo, dlatego wprowadzanie
osłon metylowych na grupę fenylową jest sposobem na ochronę wrażliwych grup funkcyjnych
przed niepożądanymi reakcjami chemicznymi.
W wyniku rozszczepienia eterów alkilowo – arylowych powstaje fenol i halogenek alkilu
Wyszukiwarka
Podobne podstrony:
5 estryfikacja
kwasy i pochodne Synteza?nzoesanu?nylu reakcja estryfikacji
reakcje estryfikacji i estry
Estryfikacja
,chemia, WPŁYW KATALIZATORÓW PRZENIESIENIA?ZOWEGO NA ETERYFIKACJĘ SORBITU CHLORKIEM ALLILU
12 ESTRYFIKACJA
estryfikacja
estryfikacja
estryfikacja
Kinetyka estryfikacji
Kinetyka estryfikacji
REAKCJA ESTRYFIKACJI ćwiczenie laboratoryjne otrzymywanie kwasu?etylosalicylowego
Sprawozdanie estryfikacja
więcej podobnych podstron