1
1
Termodynamika chemiczna
Funkcja stanu zale
ż
y wył
ą
cznie od stanu układu, czyli od
aktualnych warto
ś
ci takich parametrów jak: masa, licz-
no
ść
materii, st
ęż
enie, temperatura, ci
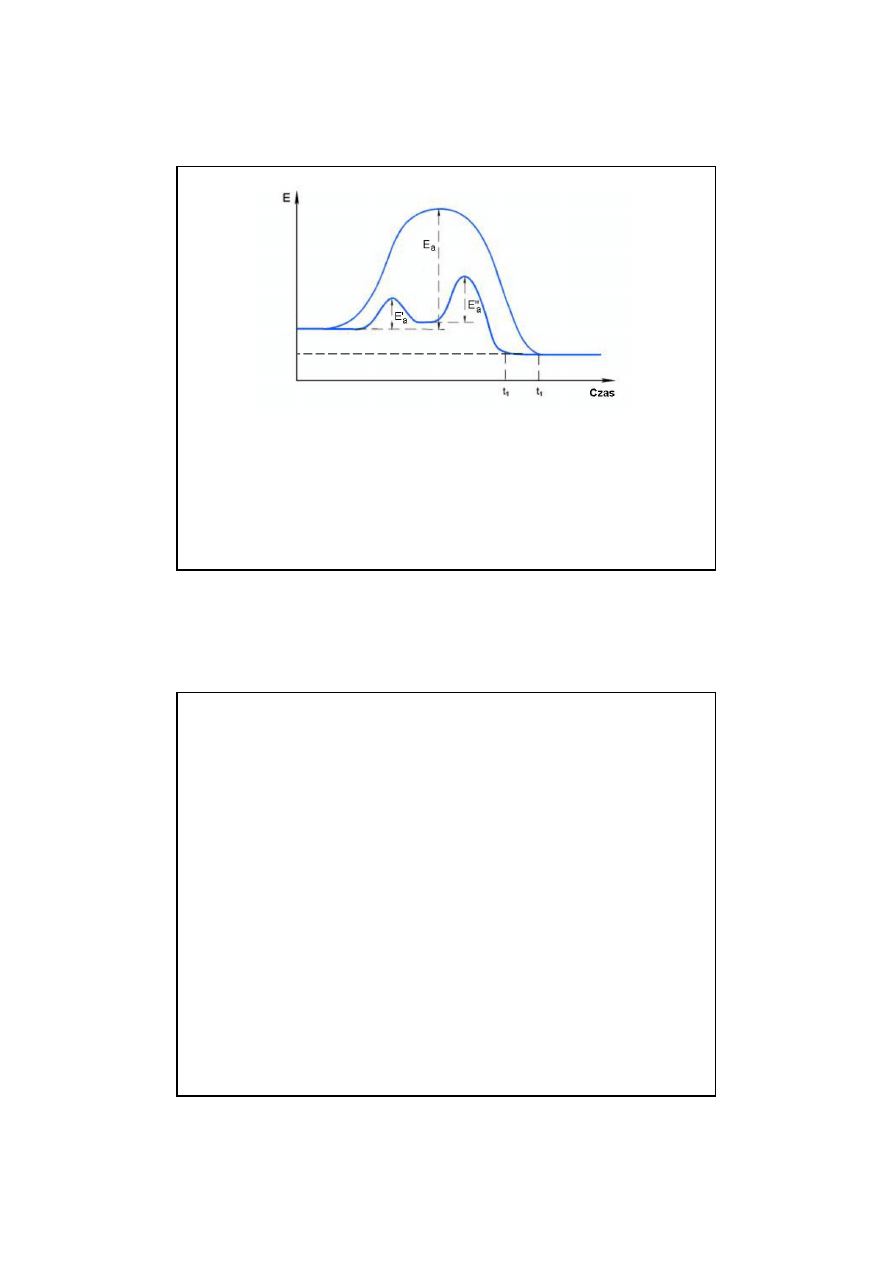
ś
nienie i inne.
Podstawowym poj
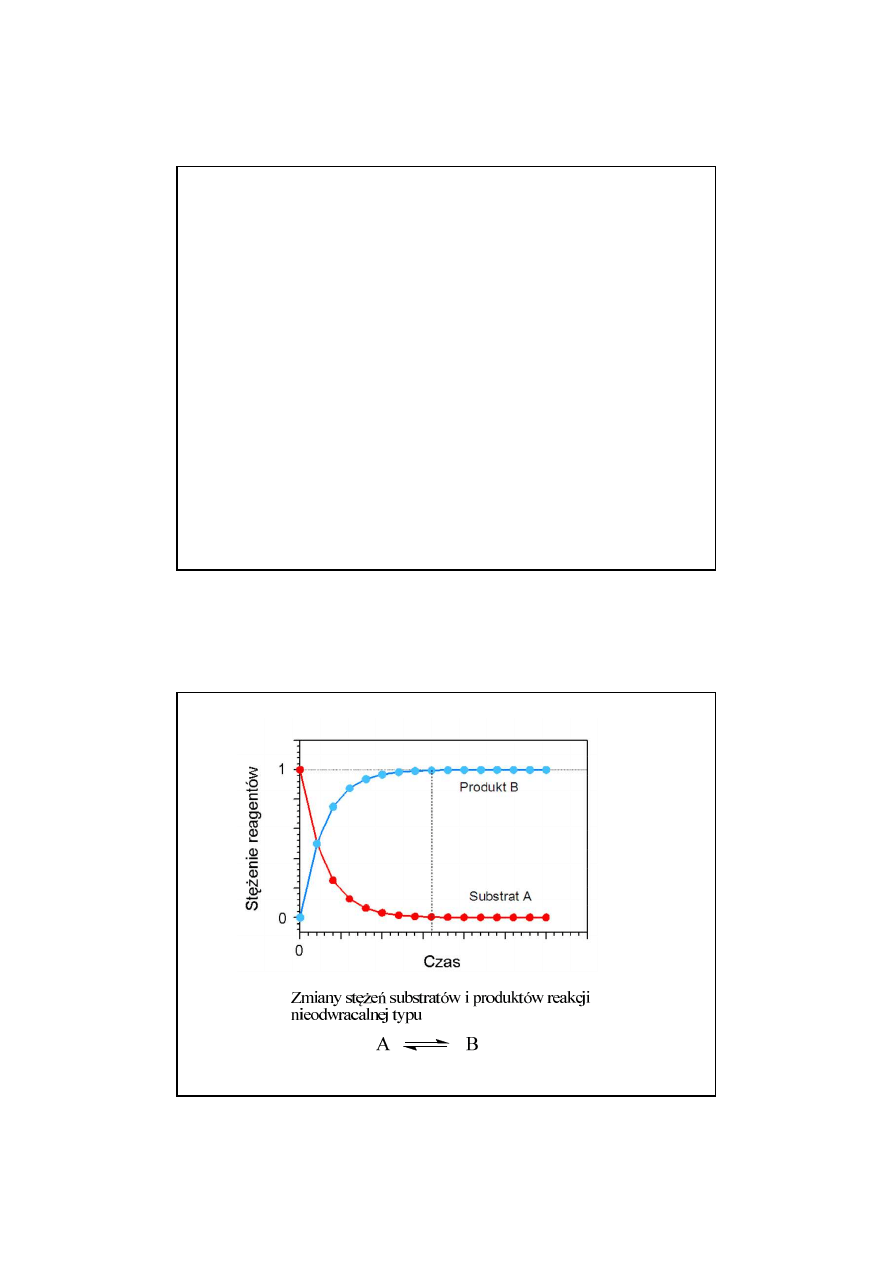
ę
ciem termodynamiki jest energia
wewn
ę
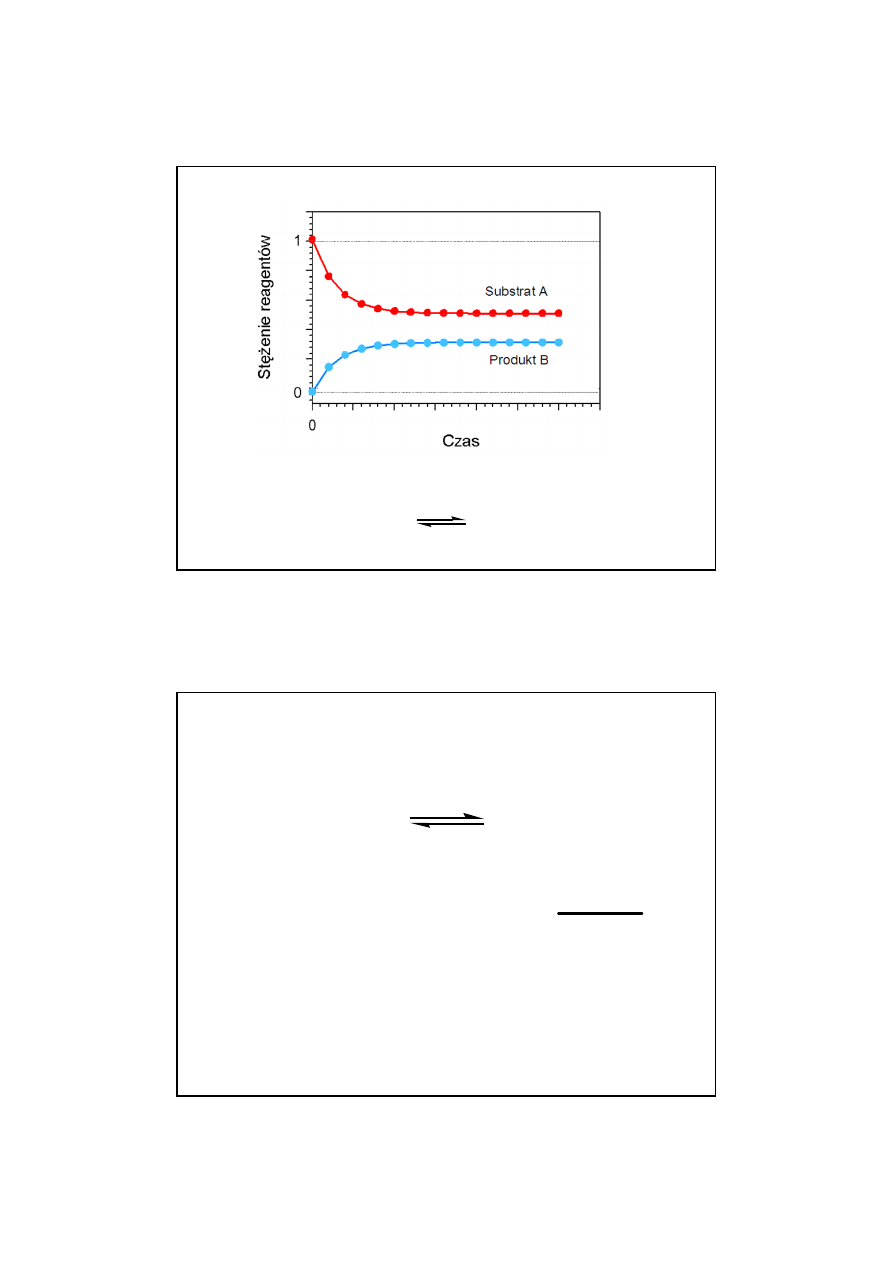
trzna
Energia wewn
ę
trzna (U) jest to cz
ęść
energii układu
zale
ż
na tylko od jego stanu wewn
ę
trznego, stanowi ona
sum
ę
energii oddziaływa
ń
mi
ę
dzycz
ą
steczkowych i
wewn
ą
trz cz
ą
steczkowych układu oraz energii ruchu
cieplnego cz
ą
steczek.
Nie jest mo
ż
liwe zmierzenie energii wewn
ę
trznej danego
układu.
2
Entalpia (H) jest sum
ą
energii wewn
ę
trznej i ciepła
układu.
H = U + pV
Czasem entalpia jest nazywana pojemno
ś
ci
ą
ciepln
ą
układu, czyli ilo
ś
ci
ą
energii cieplnej, któr
ą
nale
ż
y
doprowadzi
ć
do układu aby go ogrza
ć
od temperatury O
bezwzgl
ę
dnego do danej temperatury
.
Nie jest mo
ż
liwe zmierzenie entalpii danego układu,
mo
ż
na mierzy
ć
jej zmiany.
2
3
Entropia (S) ma kilka definicji. B
ę
dziemy stosowa
ć
definicj
ę
wg termodynamiki statystycznej omawiaj
ą
cej
populacje cz
ą
steczek:
S = k ln (W)
k – stała Boltzmana,
W - liczba sposobów na jakie makroskopowy stan
termodynamiczny układu mo
ż
e by
ć
zrealizowany poprzez
mikrostany.
Mikrostan jest stanem układu w danej, bardzo krótkiej
chwili.
4
Kryształ doskonały ma w temperaturze 0 bezwzgl
ę
dnego
entropi
ę
równ
ą
0, gdy
ż
jego stan mo
ż
e by
ć
zrealizowany
tylko na jeden sposób.
Ż
adna z cz
ą
steczek nie mo
ż
e si
ę
porusza
ć
i zamieni
ć
miejscem z inn
ą
).
Oznacza to,
ż
e ka
ż
de rzeczywiste ciało ma w tempera-
turze wi
ę
kszej od zera bezwzgl
ę
dnego entropi
ę
wi
ę
ksz
ą
od zera.
Entropia zwana jest tak
ż
e miar
ą
prawdopodobie
ń
stwa
istnienia układu. Im S wy
ż
sze, tym bardziej jest prawdo-
podobne istnienie układu.
3
5
Energia swobodna (F lub A) jest to cz
ęść
energii
wewn
ę
trznej układu, która mo
ż
e by
ć
w danym procesie
uwolniona na zewn
ą
trz układu.
W odró
ż
nieniu od U mo
ż
na j
ą
bezpo
ś
rednio mierzy
ć
.
Funkcji tej u
ż
ywa si
ę
do opisu reakcji chemicznych.
F = U – TS
Z równania wida
ć
,
ż
e układy o wysokiej entropii maj
ą
nisk
ą
energi
ę
swobodn
ą
.
6
Entalpia swobodna, jest cz
ęś
ci
ą
entalpii, która mo
ż
e by
ć
w danym procesie uwolniona na zewn
ą
trz układu.
Dotyczy to procesów biegn
ą
cych pod stałym ci
ś
nieniem i
bez zmiany obj
ę
to
ś
ci.
G = U + pV – TS
Czyli
G = H – TS
4
7
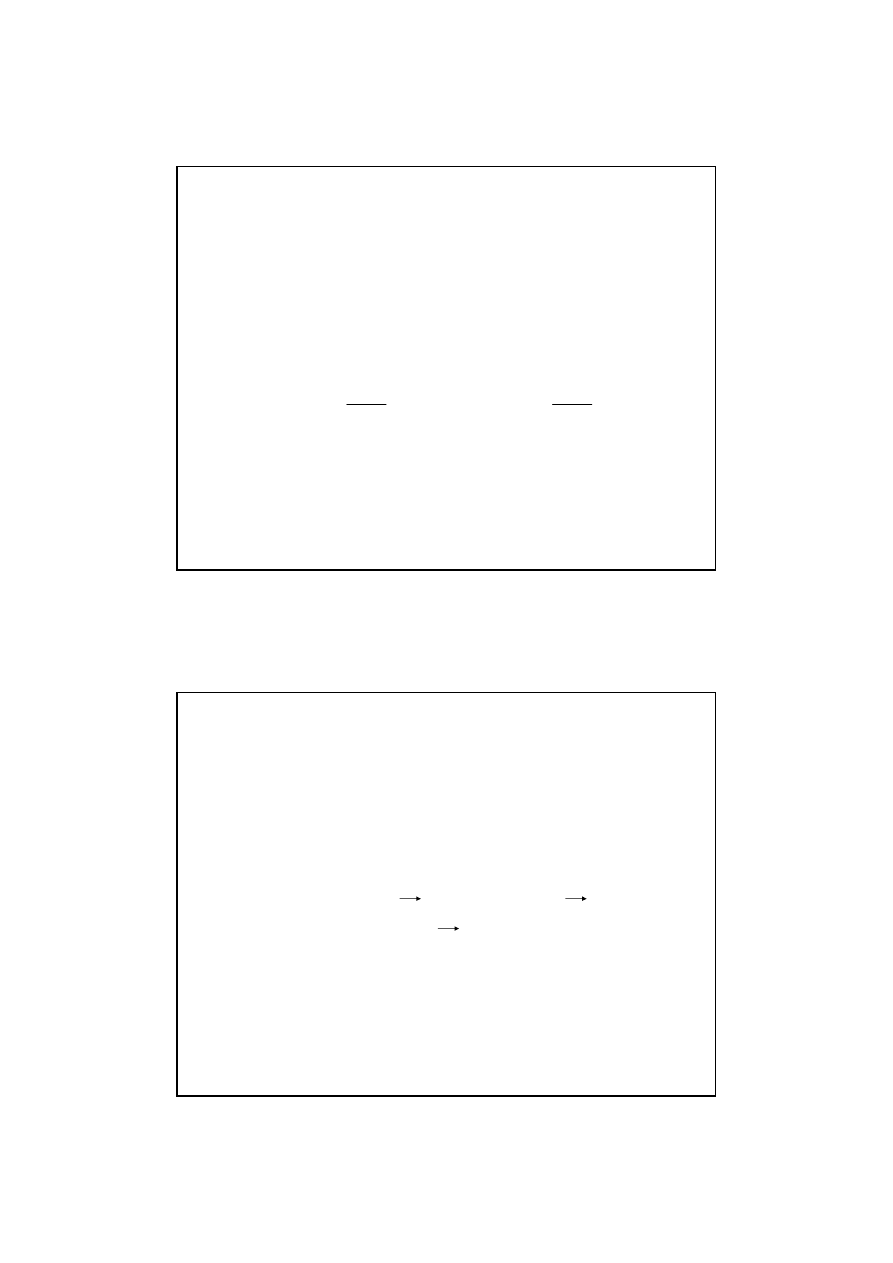
Kinetyka reakcji chemicznych
Szybko
ś
ci
ą
reakcji nazywamy stosunek zmiany st
ęż
enia
substratów do czasu w którym doszło do tej zmiany.
Poniewa
ż
w trakcie przebiegu reakcji szybko
ść
(V) ulega
ci
ą
głym zmianom najwła
ś
ciwszym równaniem odpowia-
daj
ą
cym tej definicji jest:
v = -
dc
s
dt
v =
dc
p
dt
+
lub
Jednostk
ą
szybko
ś
ci reakcji jest mol/dm
3 x
s.
W niektórych badaniach, szczególnie enzymatycznych
stosowane s
ą
inne umowne jednostki.
8
Cz
ą
steczkowo
ść
reakcji
Pojecie cz
ą
steczkowo
ś
ci stosujemy wył
ą
cznie do reakcji
elementarnych, które s
ą
proste i jednoetapowe.
Cz
ą
steczkowo
ś
ci
ą
reakcji nazywamy sum
ę
cz
ą
steczek
substratów w reakcji elementarnej.
Jednocz
ą
steczkowa A B np. glukoza galaktoza
Dwucz
ą
steczkowa A + B C + D
Reakcje dwucz
ą
steczkowe s
ą
najpospolitsze i
najwa
ż
niejsze z punktu widzenia chemii organicznej i
biochemii.
5
9
Szybko
ść
reakcji zale
ż
y od wielu czynników. Niektóre z
nich maj
ą
charakter uniwersalny i wpływaj
ą
na szybko
ść
ka
ż
dej reakcji. S
ą
te
ż
czynniki, które wpływaj
ą
tylko na
okre
ś
lony typ reakcji.
Czynnikami uniwersalnymi s
ą
: temperatura, katalizatory,
st
ęż
enie substratów.
Wzrost temperatury
ś
rodowiska zwi
ę
ksza szybko
ść
wszystkich reakcji, lecz dla reakcji endoergicznych szyb-
ko
ść
ro
ś
nie w wi
ę
kszym stopniu.
Reguła van’t Hoffa – w reakcjach jednofazowych podwy
ż
-
szenie temperatury o 10˚C powoduje wzrost szybko
ś
ci
reakcji 2 - 4 razy.
10
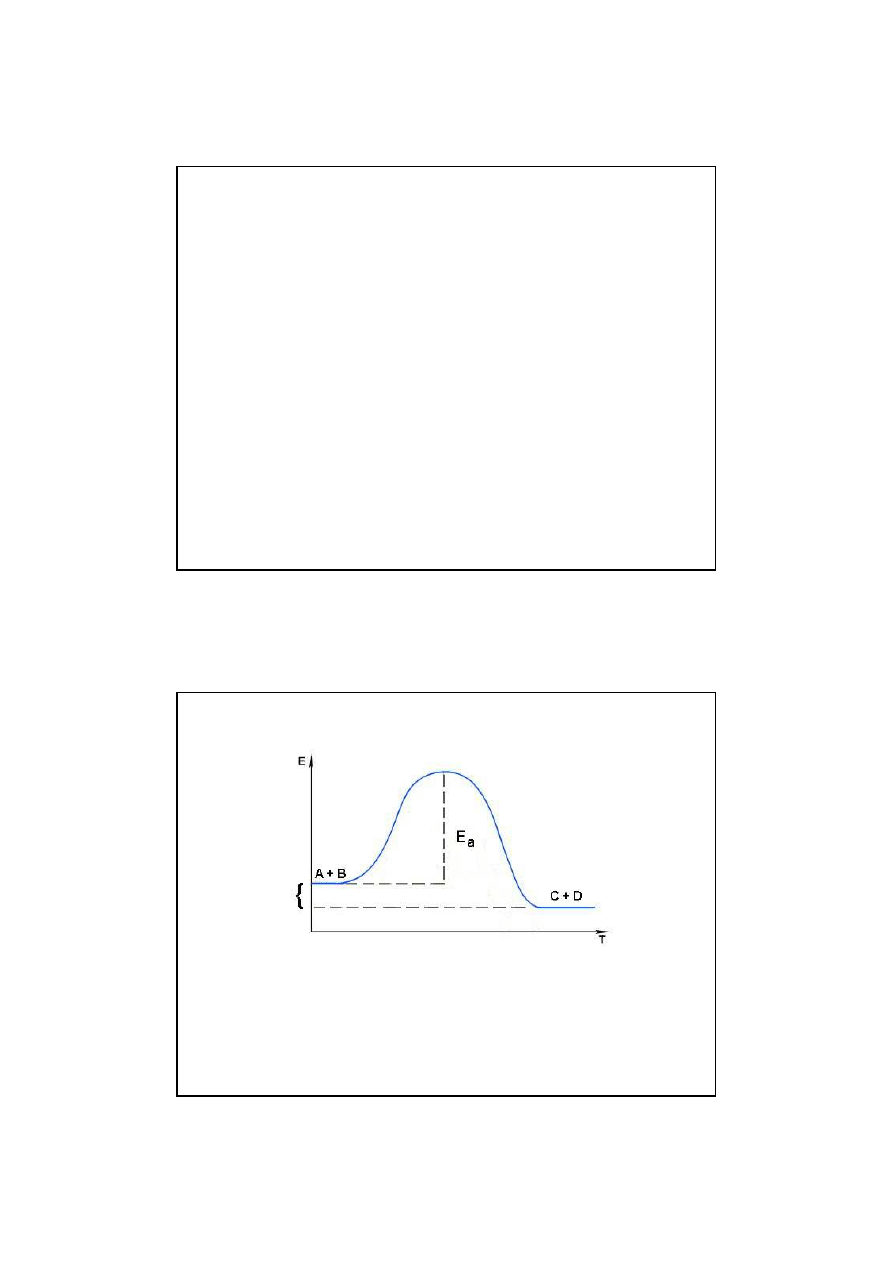
∆
E
Zmiany energii potencjalnej w trakcie przebiegu
reakcji egzoergicznej
Energia reakcji
∆
E = E
p
- E
s
jest ujemna
6
11
Zmiany energii potencjalnej w trakcie przebiegu
reakcji endoergicznej.
Energia reakcji
∆
E = E
p
- E
s
jest dodatnia.
E
r
12
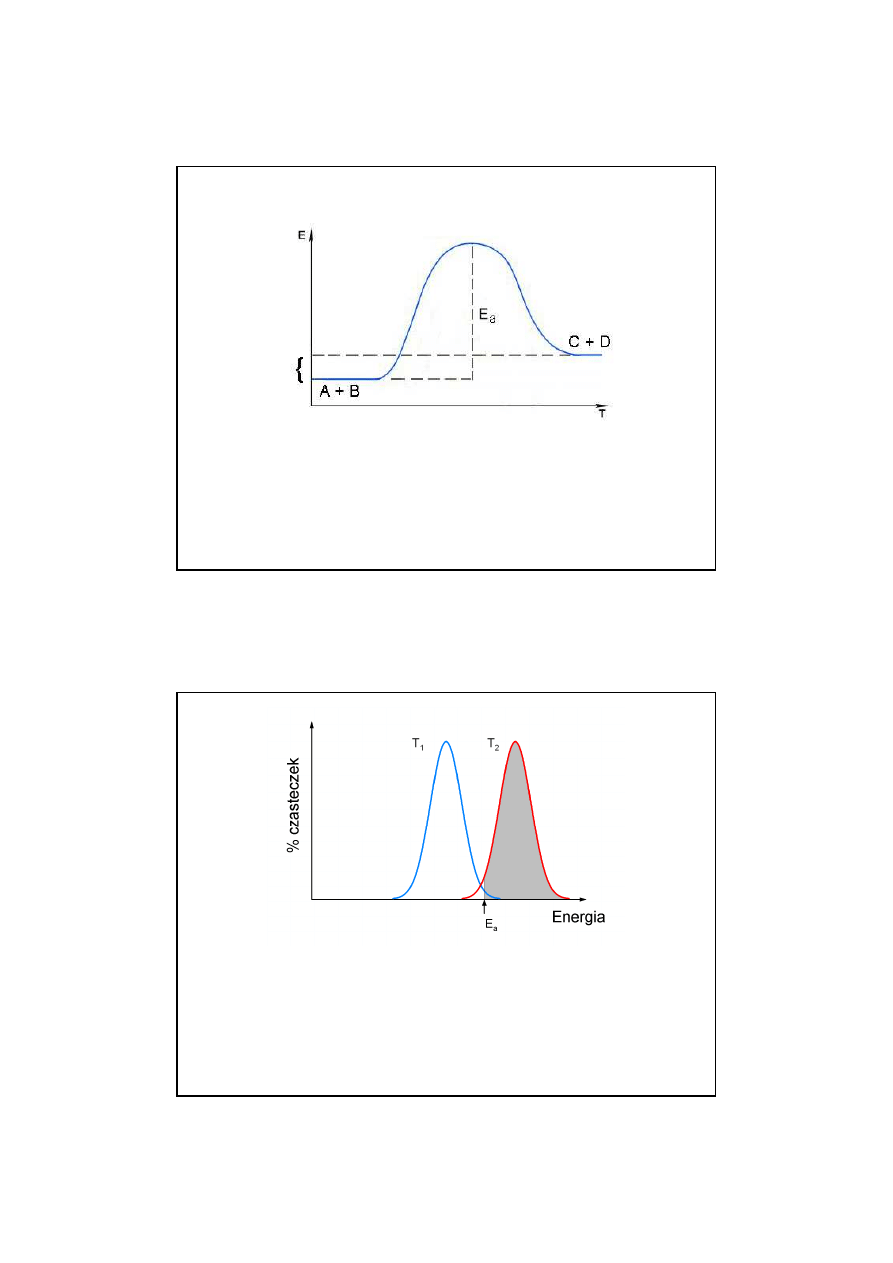
Wzrost temperatury zwi
ę
ksza procentowy udział cz
ą
steczek
w populacji, które maj
ą
energi
ę
równ
ą
lub wy
ż
sz
ą
od E
a
.
Ponadto wzrost temperatury zwi
ę
ksza cz
ę
sto
ść
zderze
ń
mi
ę
dzy cz
ą
steczkami.
7
13
Szybko
ść
reakcji zale
ż
y od st
ęż
enia substratów.
Zale
ż
no
ść
t
ę
przedstawia tzw. równanie kinetyczne reakcji.
V = k [A]
dla reakcji jednocz
ą
steczkowej
V = k [A]
m
[B]
n
dla reakcji dwucz
ą
steczkowej
Gdzie k jest stał
ą
szybko
ś
ci reakcji, m i n s
ą
współczynnika-
mi wyznaczanymi eksperymentalnie. Dla reakcji jednoetapo-
wych m i n mog
ą
by
ć
równe współczynnikom stechiomet-
rycznym.
Powodem wzrostu szybko
ś
ci reakcji spowodowanego wzros-
tem st
ęż
enia jest wi
ę
ksza cz
ę
sto
ść
zderze
ń
, skuteczno
ść
zderze
ń
nie ro
ś
nie.
14
Stała szybko
ś
ci reakcji k, zale
ż
y od energii aktywacji,
temperatury
ś
rodowiska i od katalizatorów.
Reakcje z nisk
ą
E
a
maj
ą
wy
ż
sze warto
ś
ci k.
Katalizatory obni
ż
aj
ą
E
a
i podwy
ż
szaj
ą
stał
ą
szybko
ś
ci, która
w obecno
ś
ci katalizatora oznaczamy jako k’.
V = k [A]
m
[B]
n
Suma wykładników pot
ę
gowych m + n to rz
ą
d reakcji.
Rz
ą
d reakcji jest zwykle zgodny z cz
ą
steczkowo
ś
ci
ą
ale
zdarza si
ę
,
ż
e rz
ę
dowo
ść
jest ni
ż
sza od cz
ą
steczkowo
ś
ci.
8
15
ester + woda kwas + alkohol
Hydroliza estru jest reakcj
ą
dwucz
ą
steczkow
ą
. Szybko
ść
tej
reakcji zale
ż
y od st
ęż
enia estru, a nie zale
ż
y od st
ęż
enia
wody.
V = k [ester] [woda]
0
V = k [ester]
Taka sytuacja zdarza si
ę
w przypadku reakcji, gdzie jeden z
substratów wyst
ę
puje w du
ż
ym nadmiarze.
16
Katalizatory s
ą
substancjami, które zwi
ę
kszaj
ą
szybko
ść
reakcji poprzez obni
ż
enie E
a
.
Mechanizmy działania katalizatorów s
ą
bardzo ró
ż
ne.
Mog
ą
ułatwia
ć
wła
ś
ciw
ą
orientacj
ę
przestrzenn
ą
(katalizato-
ry kontaktowe), mog
ą
zmienia
ć
mechanizm reakcji dzi
ę
ki
czemu powstaj
ą
produkty po
ś
rednie o ni
ż
szej energii.
Najbardziej efektywnymi katalizatorami s
ą
enzymy, które
mog
ą
zwi
ę
ksza
ć
szybko
ść
reakcji o wiele rz
ę
dów wielko
ś
ci
(tysi
ą
ce i miliony razy).
9
17
Katalizatory nie zmieniaj
ą
ko
ń
cowego efektu energetycz-
nego. Przyspieszaj
ą
osi
ą
gni
ę
cie stanu równowagi
E
r
{
18
Katalizatory mog
ą
by
ć
w tej samej fazie, co reagenty (katali-
za homogeniczna) lub w innej fazie (kataliza
heterogeniczna).
Przykładem katalizy homogenicznej jest reakcja estryfikacji z
udziałem H
2
SO
4
jako katalizatora.
Przykład katalizy heterogenicznej – utlenianie SO
2
do SO
3
w
obecno
ś
ci płyt niklowych lub uwodornienie benzenu w
obecno
ś
ci proszku platynowego.
10
19
Równowaga reakcji chemicznych
Reakcje odwracalne i nieodwracalne
Reakcja odwracalna charakteryzuje si
ę
tym,
ż
e w
stanie równowagi obecne s
ą
zarówno substraty jak i
produkty reakcji.
Reakcja nieodwracalna – w stanie równowagi
st
ęż
enia substratów reakcji s
ą
tak niskie,
ż
e nie mo
ż
na
ich wykry
ć
. Dlatego mówi si
ę
,
ż
e w takich reakcjach
dochodzi do całkowitego zu
ż
ycia substratów.
20
Istniej
ą
opinie,
ż
e wszystkie reakcje s
ą
odwracalne,
tylko niedoskonało
ść
metod analitycznych powoduje,
ż
e nie mo
ż
na w stanie równowagi wykry
ć
bardzo
niskich st
ęż
e
ń
substratów.
Termin reakcja nieodwracalna ma wi
ę
c znaczenie
praktyczne. Np. za pomoc
ą
odpowiednio czułej metody
mo
ż
na oznaczy
ć
st
ęż
enia jonów Hg
2+
i S
2-
w nasyco-
nym roztworze HgS.
Hg
2+
+ S
2-
HgS
11
21
St
ęż
enie jonów nad osadem w roztworze nasyconym
wynosi 10
-24
mol/dm
3
.
Istnieje wi
ę
c dowód,
ż
e jest to reakcja odwracalna.
Łatwo mo
ż
na wytr
ą
ci
ć
1 mol osadu HgS z roztworu
Hg(NO
3
)
2
za pomoc
ą
jonów S
2-
.
Ale nie mo
ż
na odwróci
ć
tej reakcji i rozpu
ś
ci
ć
w wodzie
1 mola HgS, poniewa
ż
nie ma odpowiedniej ilo
ś
ci wody
na Ziemi. W tym sensie jest to reakcja nieodwracalna.
22
12
23
Zmiany stężeń substratów i produktów reakcji
odwracalnej typu
A B
24
aA +
b
B
c
C +
d
D
K
eq
=
[A]
a
[B]
b
[C]
c
[D]
d
v
1
v
2
stan równowagi gdy v
1
= v
2
Stała równowagi reakcji
Wykładniki pot
ę
gowe a, b, c i d nie musz
ą
by
ć
zawsze zgodne ze
współczynnikami stechiometrycznymi w równaniu reakcji, poniewa
ż
wyznacza si
ę
je do
ś
wiadczalnie.
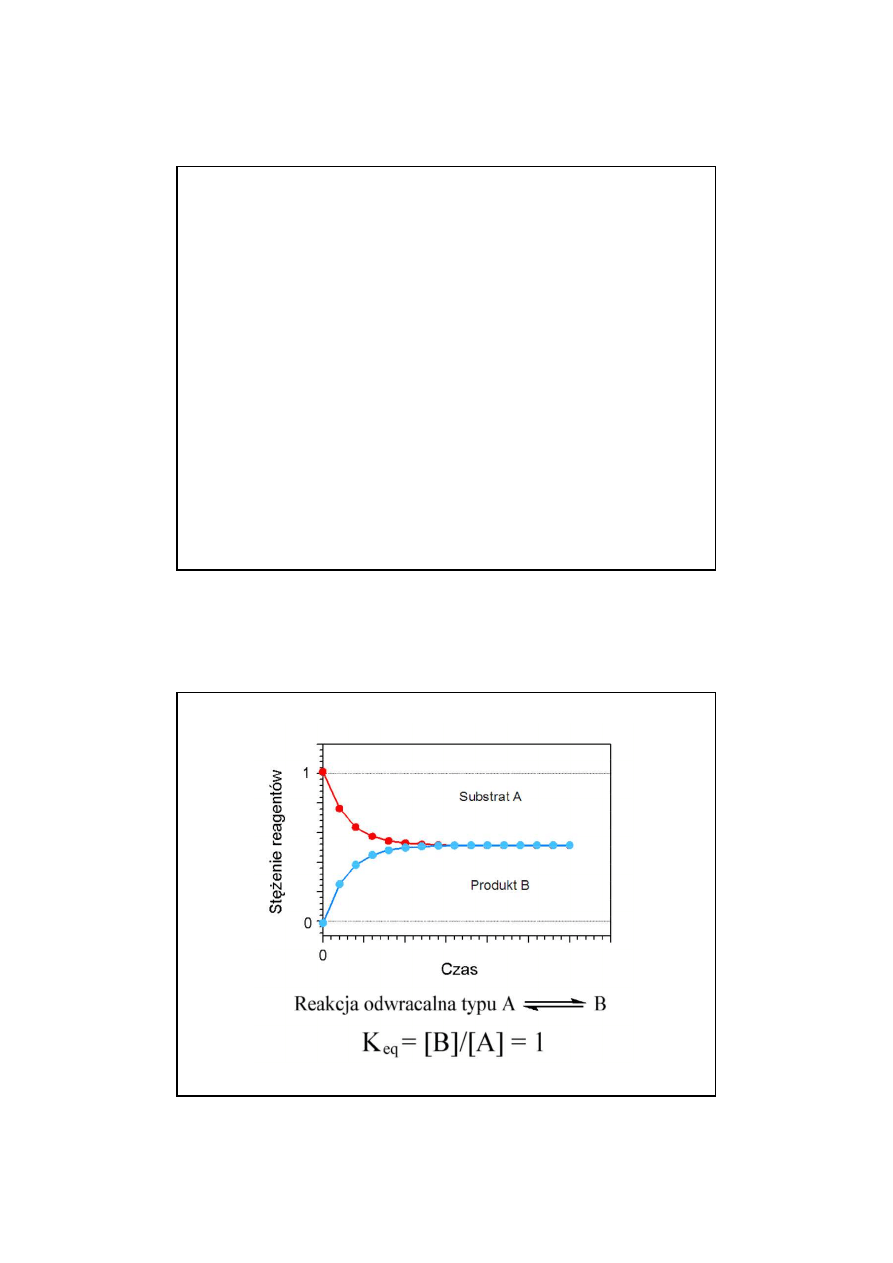
13
25
Stała równowagi w danej reakcji zale
ż
y wył
ą
cznie od temperatury.
Zmiana st
ęż
e
ń
reagentów lub ci
ś
nienia mo
ż
e spowodowa
ć
wytr
ą
cenie
reakcji ze stanu równowagi lecz po ponownym ustaleniu si
ę
równowagi
warto
ść
K
eq
b
ę
dzie nie ulegnie zmianie.
Dodanie katalizatora równie
ż
nie ma wpływu na K
eq
. Mo
ż
e jedynie
przyspieszy
ć
osi
ą
gni
ę
cie stanu równowagi.
Wysoka warto
ść
stałej równowagi mówi nam,
ż
e wydajno
ść
reakcji jest
du
ż
a (znaczna przewaga st
ęż
enia produktów nad st
ęż
eniem
substratów).
Poj
ę
cie stałej równowagi stosujemy wył
ą
cznie dla reakcji odwracalnych.
26
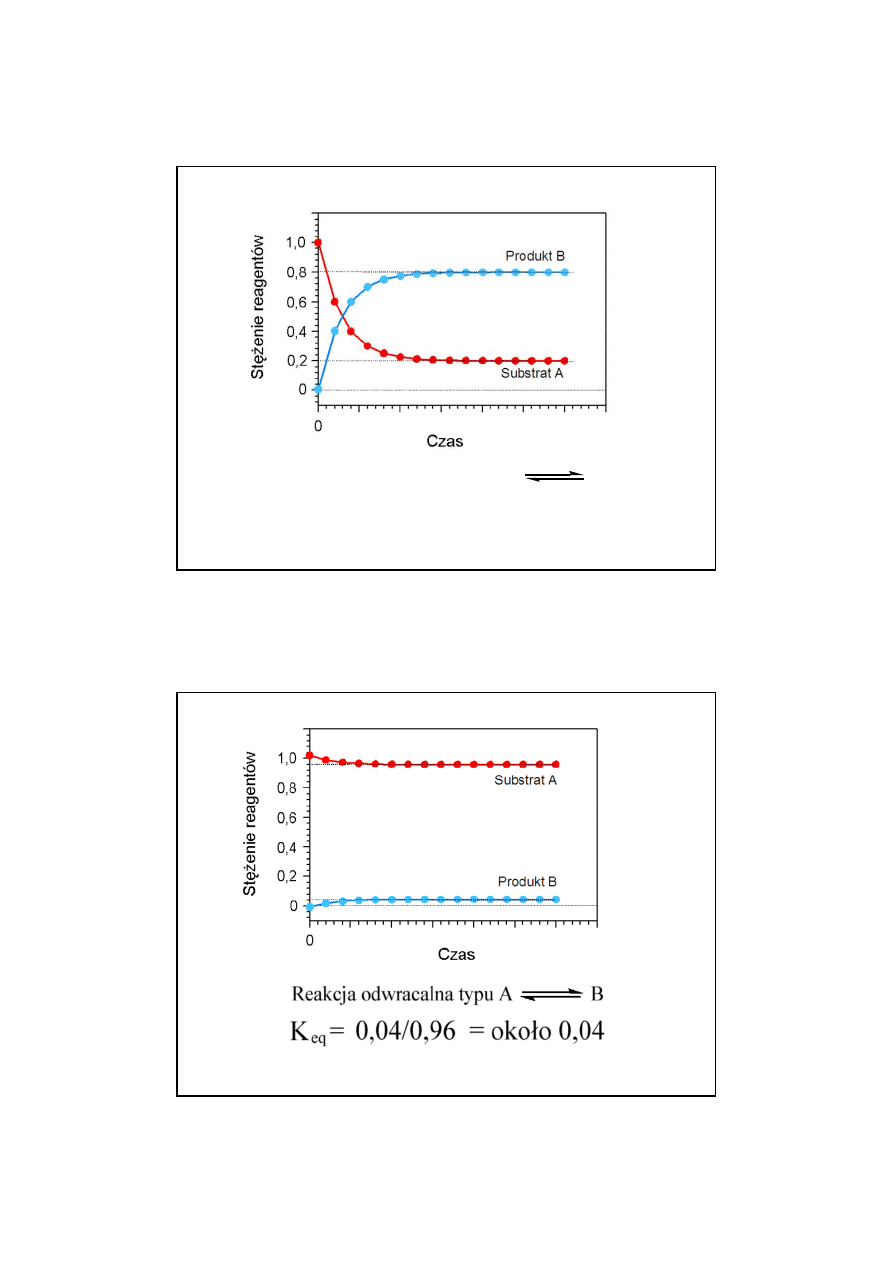
14
27
Reakcja odwracalna typu
A B
K
eq
= 0,8/0,2 = 4
28
15
29
Wydajno
ść
reakcji
W =
Ilość produktów otrzymanych w reakcji
Teoretyczna ilość produktów obliczonych na podstawie równania
Reakcje o du
ż
ej K
eq
posiadaj
ą
te
ż
du
żą
wydajno
ść
.
Równie
ż
w przypadku reakcji o małej warto
ś
ci K
eq
mo
ż
-
liwe jest uzyskanie du
ż
ej wydajno
ś
ci. W tym celu stosuje
si
ę
takie warunki reakcji, aby nie dopu
ś
ci
ć
do osi
ą
gni
ę
cia
stanu równowagi.
Np. usuwa si
ę
produkt(y) lub stosuje bardzo znaczny
nadmiar substratów.
30
W odró
ż
nieniu od stałej równowagi wydajno
ść
reakcji nie
jest wielko
ś
ci
ą
charakteryzuj
ą
c
ą
układ i zale
ż
y nie tylko
od temperatury lecz równie
ż
od innych czynników.
Zmian
ę
wydajno
ś
ci reakcji mo
ż
na przewidzie
ć
w oparciu
o reguł
ę
przekory Le Chateliera-Brauna.
Je
ż
eli na układ b
ę
d
ą
cy w stanie równowagi zadziała jaki
ś
czynnik zewn
ę
trzny, który zaburzy równowag
ę
, to w
układzie zajd
ą
zmiany przeciwdziałaj
ą
ce temu czynnikowi
i układ ponownie uzyska stan równowagi.
16
31
Zale
ż
nie od działaj
ą
cego czynnika stała równowagi mo
ż
e
zosta
ć
zmieniona lub nie. Wydajno
ść
zmienia si
ę
zawsze.
Tylko zmiana temperatury powoduje,
ż
e warto
ść
K
eq
oraz
wydajno
ść
malej
ą
lub rosn
ą
.
Wzrost temperatury podwy
ż
sza K
eq
oraz wydajno
ść
dla
reakcji endotermicznych. Odwrotny skutek wywołuje
podwy
ż
szenie temperatury dla reakcji egzotermicznych.
32
Zmiany ci
ś
nienia maj
ą
wpływ na wydajno
ść
reakcji
biegn
ą
cych całkowicie lub cz
ęś
ciowo w fazie gazowej i
zale
żą
od ilo
ś
ci cz
ą
steczek substratów i produktów w
równaniu reakcji.
C
2
H
4
+ H
2
C
2
H
6
∆
H < 0
Wszystkie reagenty: eten, wodór i etan s
ą
gazami.
Wzrost ci
ś
nienia spowoduje d
ąż
enie układu do jego
obni
ż
enia, co jest osi
ą
gni
ę
te przez zmniejszenie liczby
cz
ą
steczek w układzie. Czyli przez pewien czas ro
ś
nie
szybko
ść
reakcji w prawo do momentu uzyskania
nowego stanu równowagi. K
eq
nie ulega zmianie,
Wydajno
ść
ro
ś
nie.
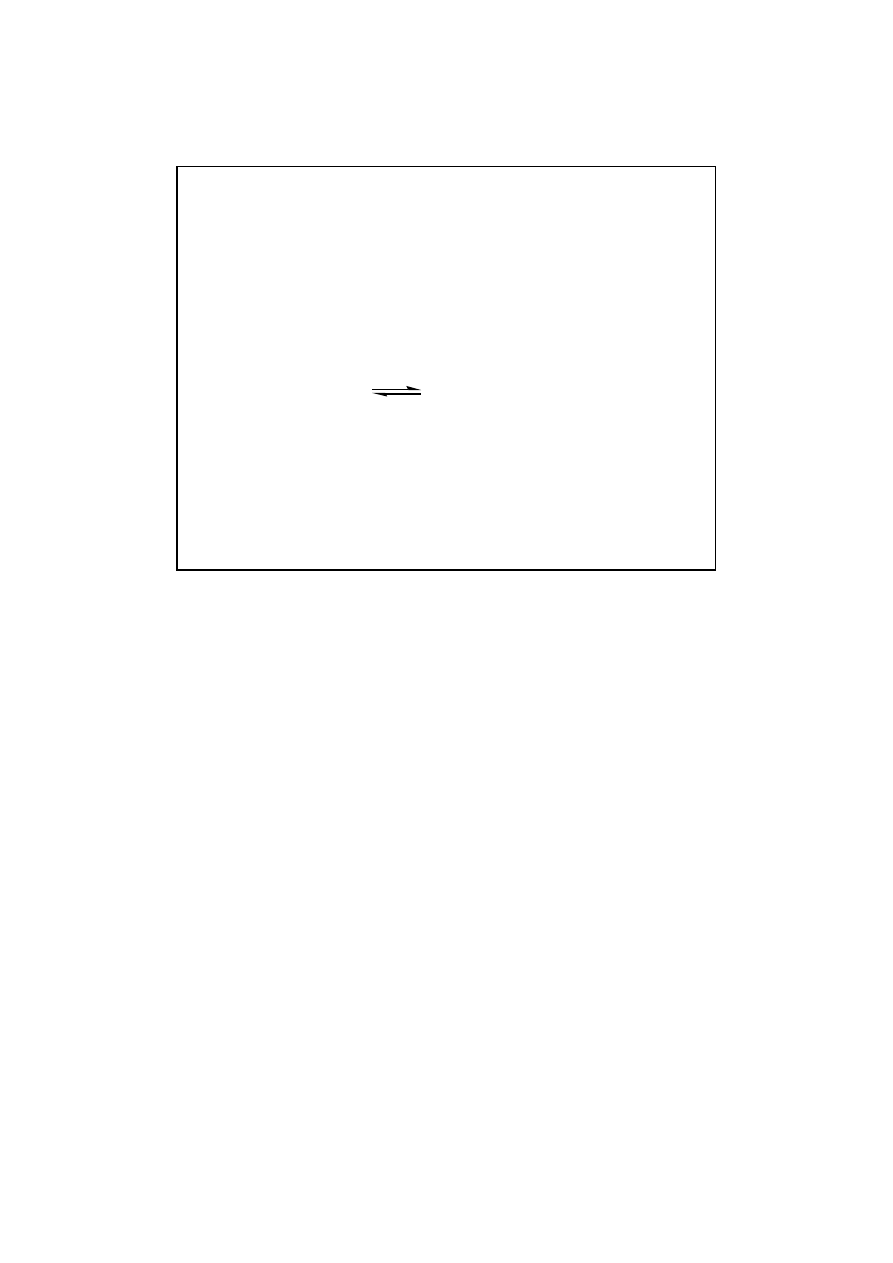
17
33
Uwodorowanie etenu jest reakcj
ą
egzotermiczn
ą
. Wzrost
temperatury spowoduje spadek wydajno
ś
ci tej reakcji
oraz obni
ż
enie Keq.
Czasami stosowane s
ą
warunki reakcji w których dwa
czynniki działaj
ą
w przeciwnych kierunkach, jeden
podwy
ż
sza wydajno
ść
, a drugi obni
ż
a.
N
2
+ 3H
2
2NH
3
∆
H < 0
Wyszukiwarka
Podobne podstrony:
Chemia ogolna wyklady 5 6 2012 Nieznany
Chemia ogolna wyklady 5 6 2012 Nieznany
Chemia ogólna wykład 2 2012
Chemia ogólna wykład 4 2012
Chemia ogólna wykład 7 2012
Chemia ogólna wykład 4-2012
Chemia ogólna wykład 1 2012
chemia 3 etap gim 2012 id 11187 Nieznany
chemia 3 etap gim 2012 id 11187 Nieznany
Cząsteczka (VB), CHEMIA, semestr 1, chemia ogólna, wykłady
MATERIALY DO WYKLADU CZ V id 2 Nieznany
chemia kliniczna cw 1 2011 id Nieznany
więcej podobnych podstron