
UNIWERSYTET GDAŃSKI
WYDZIAŁ CHEMII
Katedra Analizy Środowiska
ATOMOWA SPEKTROSKOPIA ABSORPCYJNA (AAS)
Gdańsk, 2007

AAS – atomowa spektroskopia absorpcyjna
2
1. Wprowadzenie do atomowej spektrometrii absorpcyjnej (AAS)
Para atomowa czyli medium w stanie gazowym składające się z wolnych atomów
pierwiastka może absorbować promieniowanie elektromagnetyczne. Atom pierwiastka w stanie
podstawowym (o energii E
p
) absorbuje foton promieniowania (energia hν ), co powoduje zmianę
rozkładu elektronów w atomie, przeprowadzając go w stan o wyższej energii (Ep + hν), czyli stan
wzbudzony. Atomy mogą istnieć tylko w określonych stanach energetycznych opisywanych
funkcjami falowymi stanowiącymi rozwiązania odpowiedniego równania Schrödingera. Funkcje
falowe mogą przyjmować tylko pewne określone wartości stałych, zwanych liczbami
kwantowymi: główną, poboczną, magnetyczną i spinową. Zgodnie z zakazem Pauliego, opisy
stanów energetycznych poszczególnych elektronów muszą się różnić wartością przynajmniej
jednej liczby kwantowej.
W stanie podstawowym atomu (w temp. ok. 20ºC) elektrony zapełniają kolejno poziomy
energetyczne wg wzrastającej energii. Konfigurację elektronową np. atomu magnezu można
przedstawić w następujący sposób:
12
Mg: 1s
2
, 2s
2
, 2p
6
, 3s
2
.
Elektron walencyjny może zostać
przeniesiony z poziomu podstawowego (dla magnezu to poziom 3s) na poziom wzbudzony (np. 3p).
Różnica energii między poziomem podstawowym a wzbudzonym (∆E) równa się:
∆E = ( E
p
+ hν ) - E
p
= hν = h c/
λ
(1)
gdzie: E
p
- energia atomu w stanie podstawowym (lub niższym stanie wzbudzonym),
h - stała Plancka (6,626
.
10
-34
J
.
s), ν - częstość promieniowania elektromagnetycz-
nego [s
-1
], c - prędkość rozchodzenia się światła w próżni (3,00
.
10
8
m
.
s
-1
),
λ
- długość fali promieniowania elektromagnetycznego [m].
Ponieważ poziomy energetyczne mogą przyjmować tylko pewne ściśle określone wartości, więc i
różnice energii między nimi nie są dowolne – oznacza to, że tylko promieniowanie o określonej
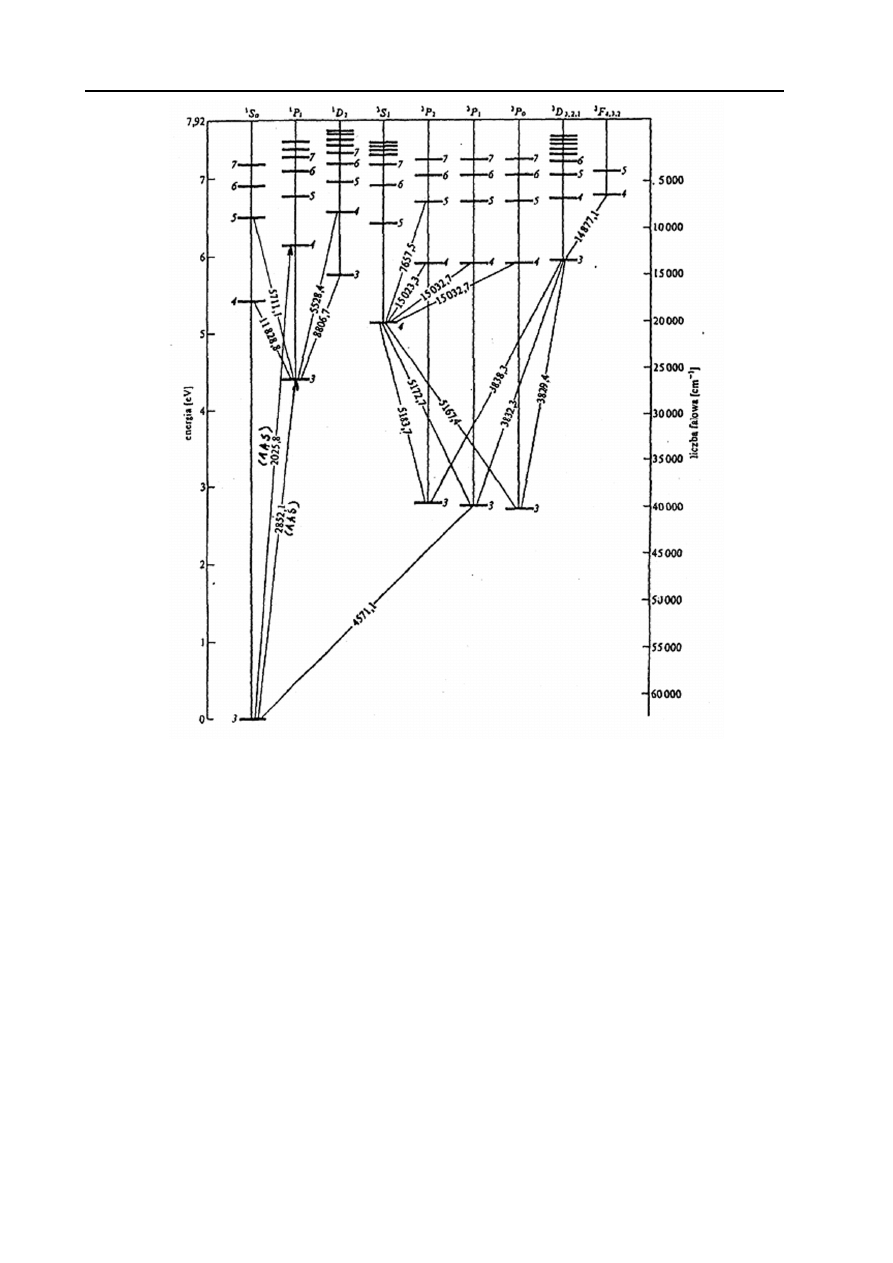
energii czyli o określonej długości fali może być zaabsorbowane. Na Rys.1 przedstawiono schemat
poziomów energetycznych atomu magnezu.
AAS – atomowa spektroskopia absorpcyjna
3
Rys. 1. Schemat poziomów energetycznych atomu magnezu. W metodzie AAS wykorzystuje się linie Mg
związane z przejściami 2852,1 Å i 2025,8 Å.
W atomie jest wiele poziomów energetycznych, na które mogą zostać przeniesione elektrony
wzbudzone. Oznacza to, że atom magnezu może absorbować wiele charakterystycznych długości
fal. Dla przejść elektronów walencyjnych jest to energia promieniowania w zakresie UV-Vis.
Średni czas trwania atomu w stanie wzbudzonym jest bardzo krótki, rzędu 10
-8
s. Po tym
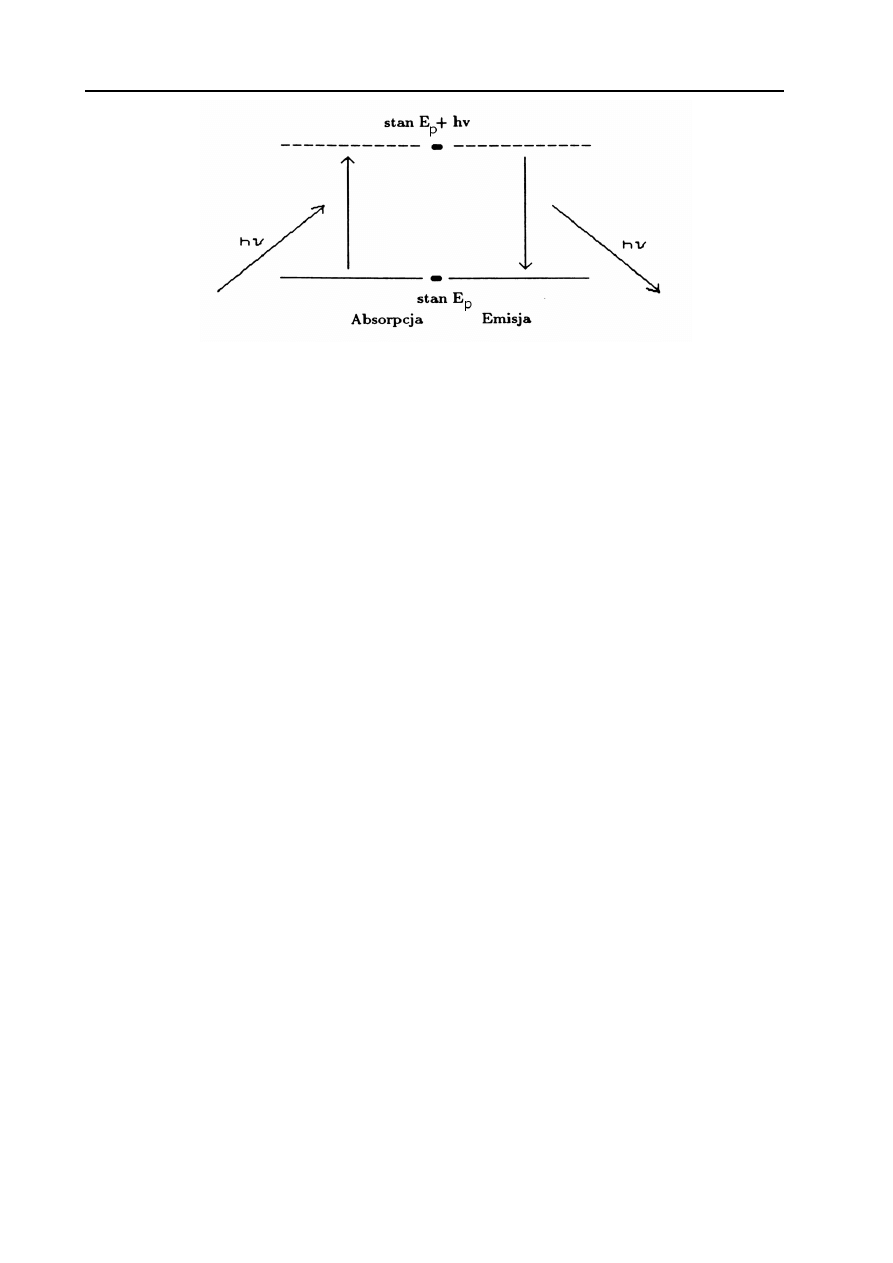
czasie elektron, wracając do stanu podstawowego, emituje energię dokładnie taką, jaka była
potrzebna do przejścia w stan wzbudzony (Rys. 2)
AAS – atomowa spektroskopia absorpcyjna
4
Rys. 2. Przejścia elektronu pomiędzy dwoma poziomami energetycznymi. .
Porcje energii czyli promieniowanie o określonej częstotliwości lub określonej długości
fali, które jest absorbowane przez dany atom jest emitowane podczas powrotu do stanu
podstawowego. Oznacza to, że atom może absorbować promieniowanie elektromagnetyczne tylko o
takiej długości fali, przy której może je emitować i jest ono charakterystyczne dla danego
pierwiastka. Zjawisko to jest podstawą analizy jakościowej metodą atomowej spektrometrii
absorpcyjnej. Dzięki temu możliwe jest oznaczanie wielu pierwiastków zawartych w próbce w
sposób niezależny od siebie (metoda jest bardzo selektywna).
Przejściom elektronów pomiędzy różnymi poziomami energetycznymi odpowiadają różne
częstotliwości promieniowania, których zbiór stanowi charakterystyczne dla danego pierwiastka
widmo atomowe (widmo liniowe). Do celów analitycznych należy dokonać wyboru jednej z wielu
różnych linii absorpcyjnych. W metodzie ASA wykorzystuje się zwykle linię związaną z przejściem
elektronu walencyjnego ze stanu podstawowego na pierwszy (najniższy poziom wzbudzony) i
nazywa się ją linią rezonansową, zaś najniższy stan wzbudzony – stanem rezonansowym.
Miarą intensywności zjawiska absorpcji promieniowania elektromagnetycznego przez wolne
atomy jest absorbancja (A) określana jako
A = lg I
0
/I
(2)
gdzie: I
0
– natężenie wiązki promieniowania padającego, I – natężenie wiązki
promieniowania po przejściu przez ośrodek zawierający wolne, oznaczane atomy
(niezaabsorbowanego przez atomy).
Prawo Lamberta – Beera. Podstawą analizy ilościowej metodą atomowej spektrometrii
absorpcyjnej jest proporcjonalność absorbancji do ilości absorbujących atomów.

AAS – atomowa spektroskopia absorpcyjna
5
Zależność tą opisuje Prawo Lamberta-Beera definiowane następującym wzorem:
A =
ε
λ
· b · N
(3)
gdzie:
ε
λ
- molowy współczynnik absorpcji (wielkość charakterystyczna dla danego rodzaju
atomów i określonej długości fali), b - długości drogi optycznej (długość drogi
promieniowania w ośrodku absorbującym), N - ilość wolnych atomów na drodze
promieniowania.
Ilość wolnych atomów N można zamienić na proporcjonalnie z nią związane stężenie
atomów (c) w próbce, co w stałych warunkach pomiaru dla określonej długości fali daje liniową
zależność:
A = a · c
(4)
gdzie: a – współczynnik proporcjonalności.
Prawo Lamberta-Beera jest spełnione dla małych stężeń, przy których nie mają jeszcze
znaczącego wpływu efekty związane z obecnością zbyt dużej ilości wolnych atomów na drodze
optycznej promieniowania (np. samoabsorpcja).
Drugim warunkiem granicznym spełnienia prawa Lamberta-Beera jest stosowanie
promieniowania monochromatycznego (zależność współczynnika absorpcji zależy od długości fali).
W przypadku absorpcyjnej spektrometrii atomowej konieczna jest, ze względu na wąskie linie
absorpcyjne, znacznie większa monochromatyczność niż w przypadku spektrofotometrii cząsteczek.
Pierwiastki metaliczne występują z reguły w postaci związków organicznych lub
nieorganicznych, zatem do wywołania zjawiska absorpcji należy przeprowadzić je w stan atomowy
- stan pary zdolny do absorpcji promieniowania (poziomy energetyczne w atomach pierwiastka
mają określoną wartość tylko w stanie gazowym).
Ze względu na czułość i selektywność metody korzystne jest, aby wszystkie lub
przynajmniej zdecydowana większość atomów znajdowała się w swoim stanie podstawowym. Gdy
stosuje się plazmy niskotemperaturowe (temperatura od 1000K do 4000K uzyskiwana w płomieniu
i kuwetach grafitowych), większość atomów znajduje się w stanie podstawowym, niezależnie od
tego czy pierwiastek wzbudza się łatwo (np. sód), czy trudno (np. cynk).
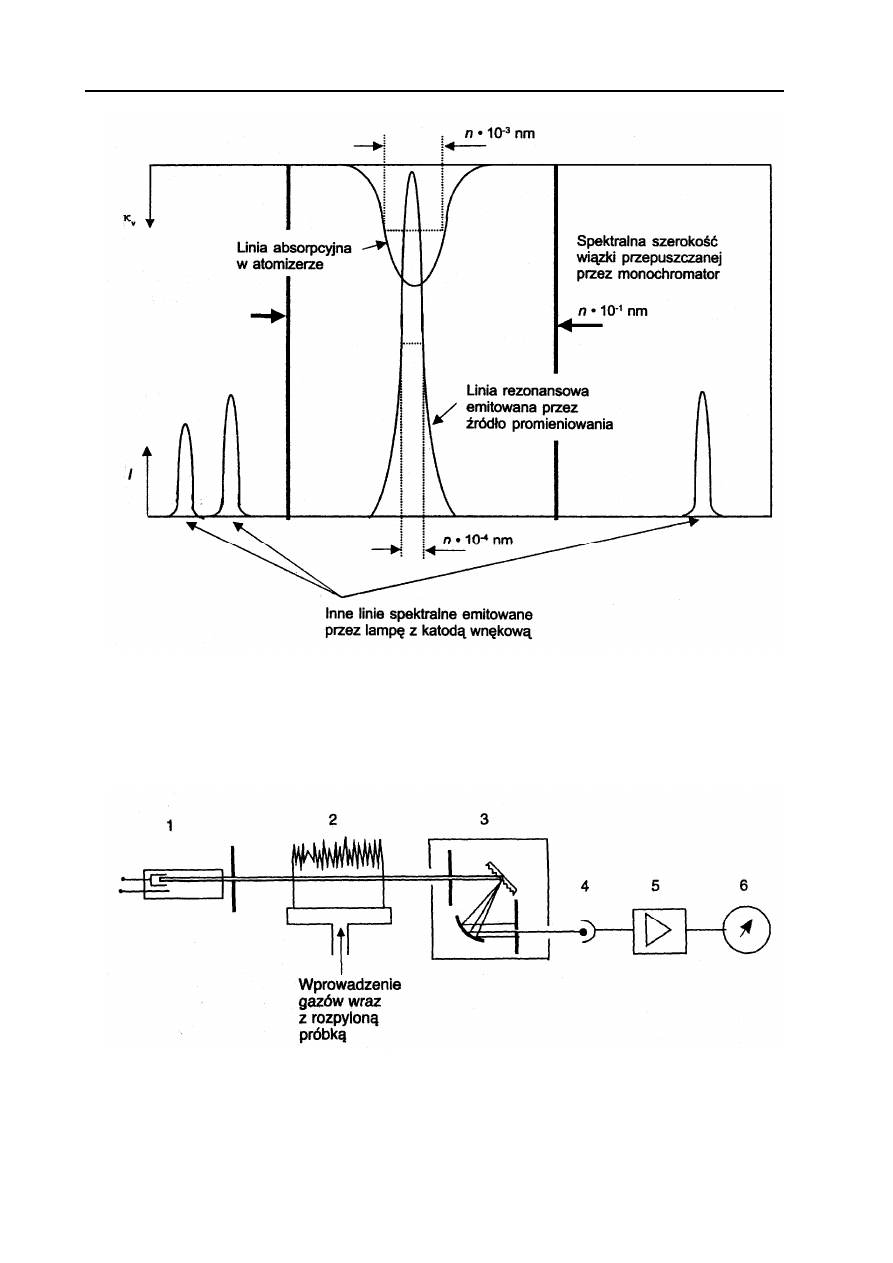
Zasada pomiarów metodą AAS polega na tym, że linia rezonansowa oznaczanego
pierwiastka o natężeniu I
0
, emitowana ze źródła promieniowania przechodzi przez atomizer, w
którym jest absorbowana przez obecne tam wolne atomy. Ta część promieniowania (linii
rezonansowej), która nie została pochłonięta przez wolne atomy, dociera poprzez monochromator
do detektora, który mierzy jej natężenie (I). Porównanie I i I
0
daje absorbancję (wzór 2)
proporcjonalną do stężenia oznaczanego pierwiastka (wzory 3 i 4).
AAS – atomowa spektroskopia absorpcyjna
6
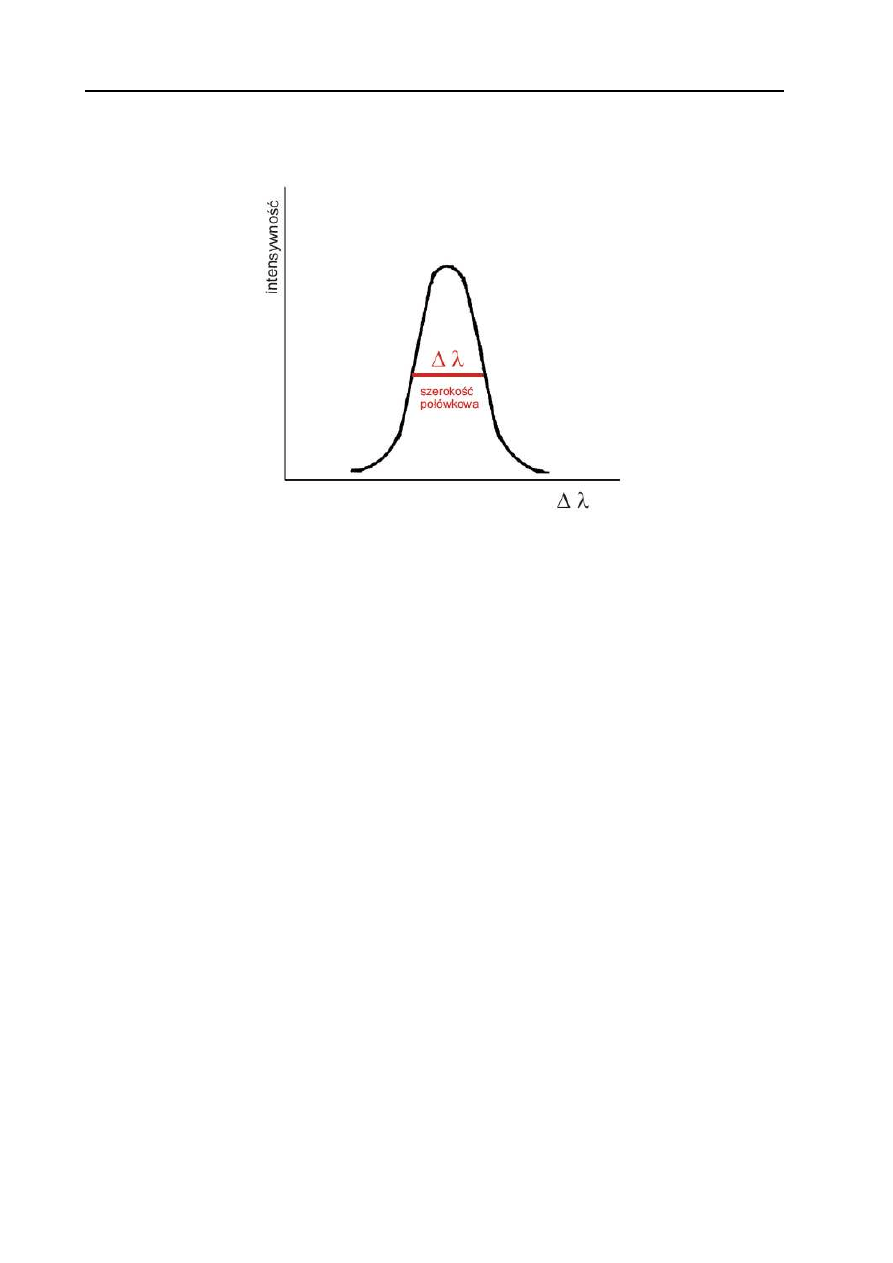
Linie atomowe mają kształt krzywych Gaussa i charakteryzują się intensywnością oraz
szerokością określaną przez szerokość połówkową mierzoną w połowie wysokości piku (Rys. 3).
Rys. 3. Schemat struktury linii atomowej.
Naturalna szerokość linii w zakresie promieniowania UV-Vis stosowanych w AAS wynosi
ok. 10
-6
– 10
-4
nm. W atomizerze szerokość linii absorpcyjnej będzie większa z powodu poszerzenia
temperaturowego (tzw. poszerzenie dopplerowskie) oraz poszerzenia ciśnieniowego (zjawisko
Lorentza). W obu przypadkach wartość tego poszerzenia wynosi ok. 10
-3
nm i jest o dwa rzędy
większa od szerokości naturalnej linii absorpcyjnej. Dlatego szerokość połówkowa linii emitowanej
ze źródła promieniowania powinna być zdecydowanie mniejsza niż szerokość linii absorpcyjnej ze
względu na czułość (im mniejsza szerokość linii emitowanej tym większy jej zakres będzie objęty
absorpcją) i jak najmniejsza ze względu na specyficzność metody (możliwość nakładania się linii
spektralnych innych pierwiastków). Uzyskuje się to przez zastosowanie wyższej temperatury w
atomizerze od temperatury w lampie emitującej.
Zasadę pomiarów metodą AAS ilustruje Rys. 4.
AAS – atomowa spektroskopia absorpcyjna
7
Rys. 4. Zasada pomiarów metodą AAS. (R. Kocjan (red.) Chemia analityczna. Podręcznik dla studentów. Tom
2. PZWL, W-wa, 2000).
2. Aparatura do atomowej spektrometrii absorpcyjnej
Schemat blokowy spektrometru absorpcji atomowej przedstawiono na Rys.5.
Rys. 5. Schemat blokowy spektrometru absorpcji atomowej z atomizerem płomieniowym: 1 - źródło
promieniowania liniowego (lampa z katodą wnękową), 2 – atomizer, 3 – monochromator, 45 – detektor
(fotopowielacz), 56 – wzmacniacz, 67 – rejestrator (komputer). (R. Kocjan, Chemia analityczna. Podręcznik dla
studentów. Tom 2. PZWL, W-wa, 2000).

AAS – atomowa spektroskopia absorpcyjna
8
Aparaty AAS mogą być jedno- lub dwuwiązkowe. W spektrometrach dwuwiązkowych
promieniowanie emitowane ze źródła jest dzielone na dwie wiązki, wiązkę przechodzącą przez
atomizer i wiązkę odniesienia omijającą atomizer. Obie wiązki przechodzą przez ten sam
monochromator a następnie są naprzemiennie rejestrowane przez ten sam detektor. Eliminuje się w
ten sposób błąd pomiaru wynikający ze zmian intensywności promieniowania źródła w czasie
trwania pomiaru lub zmian czułości detektora.
2.1. Źródła promieniowania
Źródła promieniowania stosowane w metodzie AAS muszą się charakteryzować dużą
monochromatycznością promieniowania o częstotliwości zgodnej z częstotliwością rezonansową
oznaczanego pierwiastka. Promieniowanie emitowane przez źródło powinno odznaczać się dużym
natężeniem i stabilnością. W praktyce, w metodzie AAS stosuje się lampy z katodą wnękową lub
wzbudzane wysoką częstotliwością (bądź mikrofalami) lampy bezelektrodowe.
Lampy z katodą wnękową (Hollow Cathode Lamp - HCL) (Rys. 6) są rurkami szklanymi z
okienkami kwarcowymi. Wewnątrz zamkniętej rurki znajduje się gaz szlachetny (Ne lub Ar) pod
niskim ciśnieniem (2-8 hPa). Lampy te zawierają dwie elektrody. Anodą jest drut wolframowy,
katodę stanowi wydrążony cylinder wykonany z metalu, który ma być oznaczany i którego linię
rezonansową lampa ma emitować. Oś cylindra katody odpowiada osi optycznej przyrządu. Gdy
między anodę i katodę zostanie przyłożone dostatecznie duże napięcie (rzędu kilkuset wolt), gaz
wypełniający lampę zostanie zjonizowany. Dodatnie jony gazu, bombardując katodę wybijają, z niej
atomy metalu. Atomy metalu w stanie gazowym ulegają wzbudzeniu i emitują promieniowanie,
które składa się z linii charakterystycznych dla atomów metalu, jonów metalu i gazu szlachetnego.
Natężenie promieniowania można zmieniać regulując natężenie prądu płynącego w lampie.
Odpowiednią linię można wyodrębnić z niezbyt skomplikowanego widma za pomocą prostych
monochromatorów. Lampą z katodą wnękową można oznaczać tylko jeden pierwiastek, ten z
którego została wykonana katoda. Produkuje się też lampy kilkupierwiastkowe, ale nie znajdują one
szerszego zastosowania, głównie z powodu małego natężenia wycinanej wiązki promieniowania.
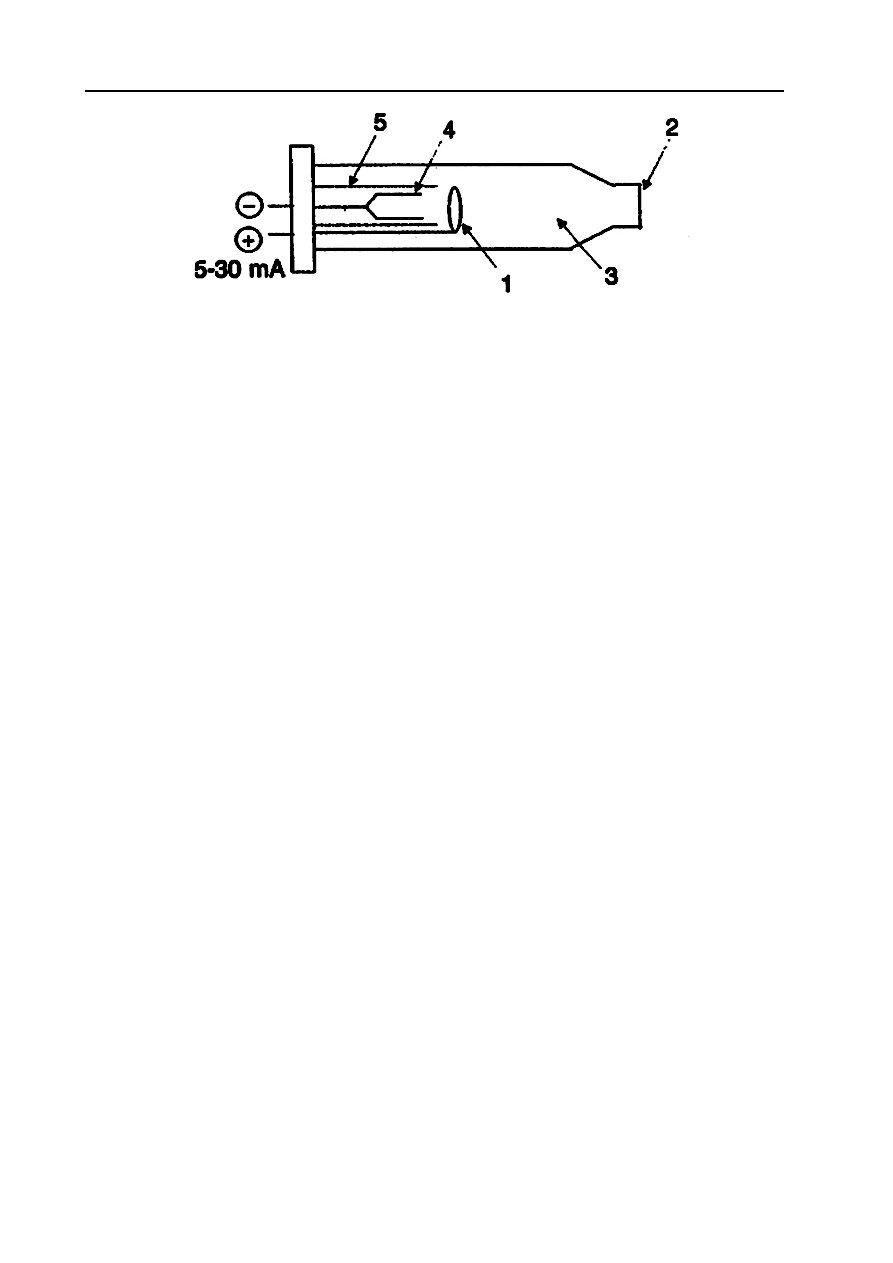
AAS – atomowa spektroskopia absorpcyjna
9
Rys. 6. Lampa z osłoniętą katodą wnękową, 1 – anoda; 2 – okienko kwarcowe; 3 – gaz Ar lub He; 4 – katoda
wnękowa; 5 – ekrany z kwarcu lub miki. (R. Kocjan, Chemia analityczna. Podręcznik dla studentów. Tom 2.
PZWL, W-wa, 2000).
Lampy bezelektrodowe (Electrodeless Discharge Lamp - EDL) ze wzbudzeniem wysoką
częstotliwością są to wąskie, zamknięte rurki kwarcowe zawierające wewnątrz warstwę metalu,
który ma być oznaczony lub/i warstwę soli tego pierwiastka (1 - 2 mg). Rurka wypełniona jest
gazem szlachetnym (Ar, Ne) pod zmniejszonym ciśnieniem (0,2 - 0,8 hPa). Atomizację i
wzbudzenie uzyskuje się przez działanie pola elektromagnetycznego o wysokiej częstotliwości.
Lampy bezelektrodowe charakteryzują się dobrymi parametrami (natężenie linii i szerokość
połówkowa) i są bardzo trwałe. Produkuje się je głównie dla pierwiastków, dla których nie można
zbudować lamp HCL - Sb, As, Se, Te, P, Hg, Bi, Cs, Ge, K, Rb, Tl.
2.2. Atomizery
Zadaniem atomizerów jest otrzymywanie z dużą, powtarzalną wydajnością wolnych atomów
z próbek analitycznych. Im większa wydajność wolnych atomów w stanie podstawowym,
odniesiona do badanej próbki, tym większa czułość metody analitycznej. W procesie atomizacji
musi występować prosta proporcjonalność między stężeniem oznaczanej substancji w próbce a
stężeniem atomów w plazmie absorpcyjnej. Wytworzone atomy powinny w jak najmniejszym
stopniu ulegać wzbudzeniu i jonizacji.
Atomizacja próbki wymaga doprowadzenia energii, co realizowane jest różnymi metodami.
Najczęściej stosuje się:
•
atomizację płomieniową (F - AAS),
•
atomizację bezpłomieniową (ET - AAS) obejmującą takie techniki jak: elektryczne
ogrzewanie oporowe rurki grafitowej (piec Massmanna), atomizacja w łuku prądu
zmiennego (kuwety grafitowe Lwowa), bombardowanie powierzchni metalicznej
elektronami, odparowywanie laserowe,
•
atomizery wykorzystujące zimne pary rtęci (CV - AAS),
•
atomizery wodorkowe (HG - AAS).
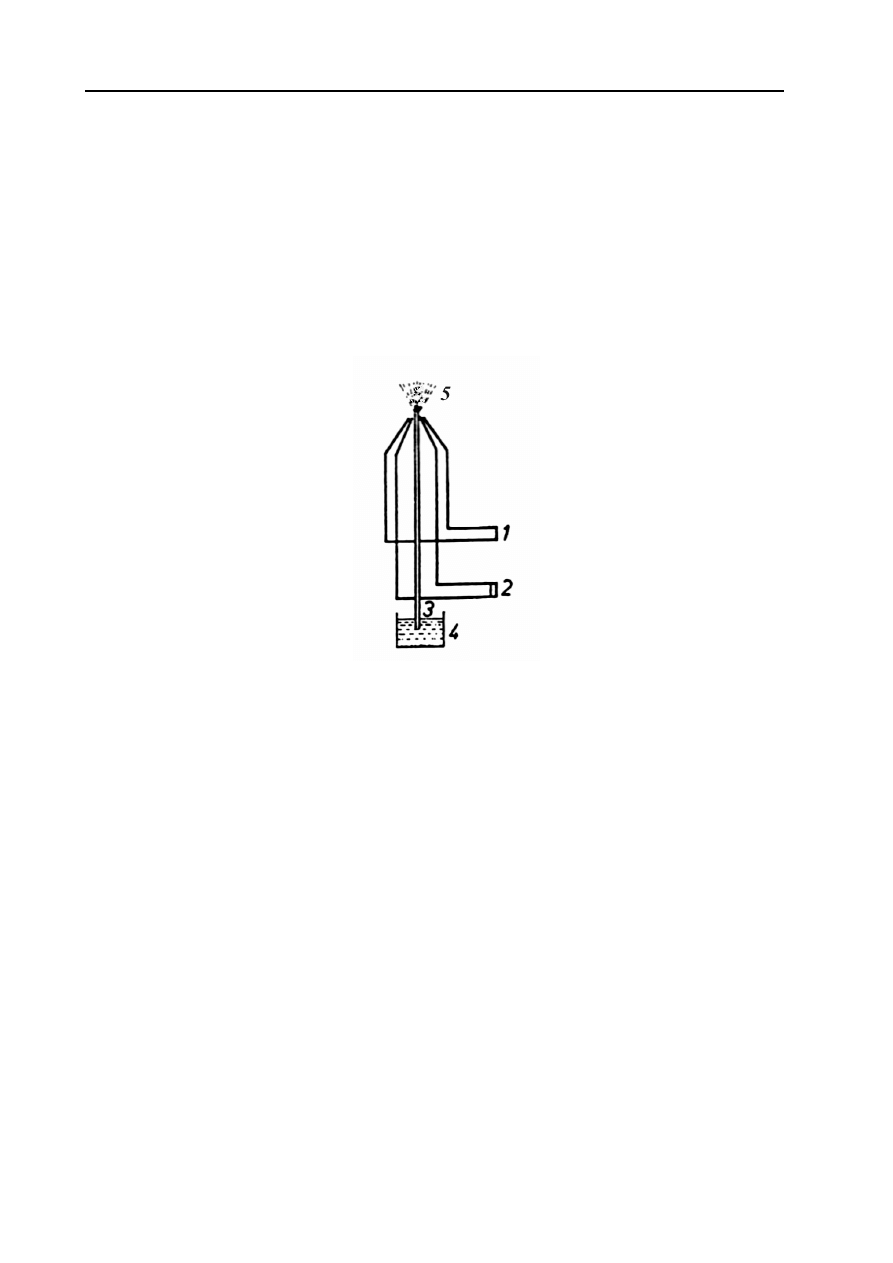
AAS – atomowa spektroskopia absorpcyjna
10
Atomizer płomieniowy. Atomizacja płomieniowa wymaga przeprowadzenia ciekłej próbki
analitycznej w aerozol. Aerozol uzyskuje się najczęściej w nebulizerze pneumatycznym (Rys. 7).
W komorze nebulizera analizowany roztwór przeprowadza się w delikatną mgłę (aerozol),
następnie miesza aerozol z gazem palnym i wprowadza jednorodnie do palnika z zastosowaniem
powierzchni rozpryskowych lub sit dla odrzucenia lub rozbicia większych kropel. Gazem
zasysającym próbkę jest zawsze gaz utleniający. Mieszanina rozpuszczalnika, próbki, gazów
utleniającego i palnego wprowadzana jest do palnika szczelinowego o długości 5 – 10 cm i
szerokości 0,5 – 1,5 mm.
Rys. 7. Nebulizer pneumatyczny.
Płomienie palnika muszą dostarczać energii wystarczającej do przeprowadzenia roztworu w wolne
atomy. Sam płomień powinien absorbować tylko niewielką część promieniowania emitowanego
przez źródło. Stosowane w metodzie AAS mieszaniny gazów to gaz miejski-powietrze (T = 1980
K), propan-butan-powietrze (T = 2200 K), acetylen-powietrze (T = 2600 K), acetylen-tlen (T =
3300 K), acetylen-tlenek azotu (I) (T 3220 K), wodór-powietrze (T= 2275 K) oraz wodór-tlen (T =
2825 K).
Najczęściej stosuje się płomień acetylen-powietrze. Ma on wysoką temperaturę i dopiero
poniżej 230 nm występuje wzrastająca absorpcja własna płomienia. Płomień acetylen-powietrze jest
zalecany do oznaczania następujących pierwiastków: Mg, Ca, Sr, Cr, Mb, Mn, Tc, Fe, Ru, Os, Co,
Rh, Ni, Pd, Pt, Cu, Ag, Au, Zn, Hg, Ga, In, Tl, Pb, Sb, Bi. Dla pierwiastków, które tworzą w
płomieniu trwałe tlenki (np. Ba, Al., B, Be, Si) konieczne jest stosowanie płomienia redukującego z
użyciem gazu utleniającego tlenku azotu (I).
Analizowana próbka może być roztworem prostej soli MA (M
+
i A
-
) lub roztworem
zawierającym inne składniki. Po wprowadzeniu prostej soli do płomienia zachodzą w niej
następujące przemiany fizykochemiczne i reakcje chemiczne (Rys. 8.):
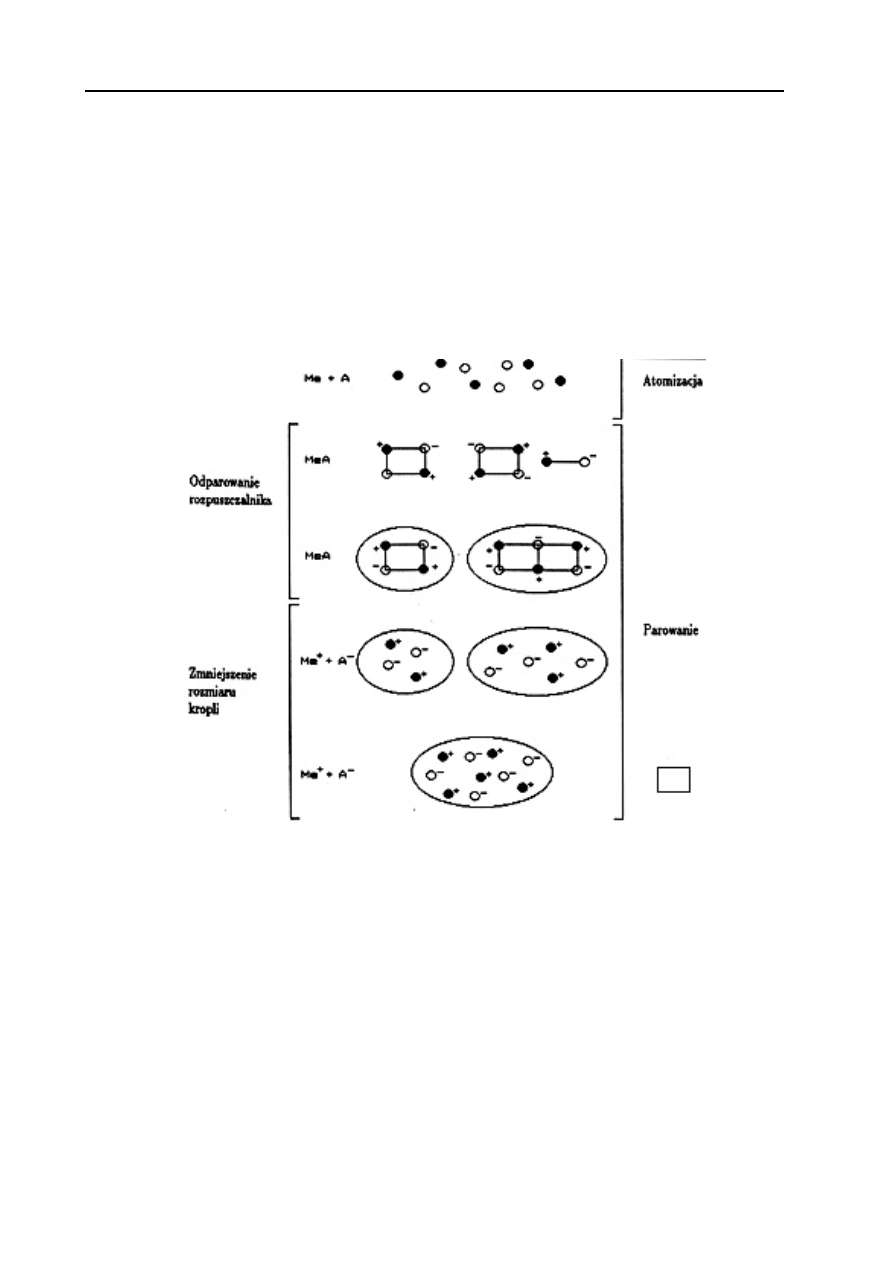
AAS – atomowa spektroskopia absorpcyjna
11
•
Odparowanie rozpuszczalnika
M
+
+ A
-
(mgła)
↔
MA (ciało stałe)
•
Stopienie soli i przeprowadzenie jej w stan pary
MA (ciało stałe)
↔
MA (ciecz)
↔
MA (gaz)
•
Reakcja dysocjacji termicznej
MA (gaz)
↔
M(gaz) + A(gaz)
Rys. 8. Przemiany fizykochemiczne soli w atomizerze płomieniowym.
Równowaga tej reakcji uzależniona jest od temperatury płomienia. Ilość wolnych atomów
rośnie wykładniczo ze wzrostem temperatury.
Inne reakcje, takie jak jonizacja, wzbudzenie i synteza są procesami niekorzystnymi,
ponieważ zmniejszają ilość wolnych atomów w stanie podstawowym a zatem i czułość metody.
•
Reakcja jonizacji M
↔
M
+
+ e
•
Rekcje syntezy M + O
↔
MO
M + H
2
O
↔
MO + H
2
M + OH
↔
MOH
MO + CO
2
↔
MCO
3

AAS – atomowa spektroskopia absorpcyjna
12
•
Reakcje wzbudzenia M ↔ M
*
MA
↔
(MA)
*
MO ↔ (MO)
*
MOH ↔ (MOH)
*
MCO
3
↔ (MCO
3
)
*
W układach złożonych zachodzą w płomieniu dodatkowe reakcje syntezy dające trwałe sole np. z
halogenkami (AlF
3
), z kwasami tlenowymi (CaSO
4
, Ca
3
(PO
4
)
2
lub reakcje tworzenia podwójnych
tlenków metali (MgAl
2
O
4
, CaTiO
3
). Poznanie mechanizmów tych reakcji w płomieniu i znajomość
ewentualnych oddziaływań zakłócających pozwala wyeliminować przyczyny błędów w metodzie
absorpcji atomowej.
Atomizery bezpłomieniowe. Atomizery bezpłomieniowe stosuje się dla ominięcia rozcieńczania
próbek oraz uniknięcia wpływu matrycy. Najczęściej stosowanym sposobem atomizacji
bezpłomieniowej jest atomizacja elektrotermiczna w kuwecie (rurce) grafitowej (piec
Massmanna). Kuwety są to rurki grafitowe o dł. 20 – 50 mm i średnicy wewnętrznej 4 – 6 mm.
Powierzchnia rurki pokryta jest warstwą grafitu pirolitycznego, co zapobiega dyfuzji atomów w
głąb ścianek. Próbkę stałą lub ciekłą wprowadza się bezpośrednio do rurki grafitowej lub na
specjalną płytkę grafitową (platforma Lwowa), która znajduje się w atmosferze bardzo czystego
gazu obojętnego, najczęściej argonu. Ogrzewanie elektryczne, oporowe lub indukcyjne, odbywa się
w sposób programowany, sterowany za pomocą komputera. Cykl pomiarowy składa się
odparowania rozpuszczalnika, mineralizacji próbki (piroliza) i atomizacji, czyli przeprowadzenia
oznaczanej substancji do plazmy termicznej w postaci wolnych atomów.
Zaletami atomizacji elektrotermicznej są m.in. możliwość oddzielenia pierwiastka od
składników matrycy, warunki sprzyjające atomizacji trwałych termicznie tlenków oraz całkowita,
jednorazowa atomizacja wprowadzonej próbki z dużą wydajnością (w płomieniowej AAS – tylko
kilka procent).
Atomizery wodorkowe. Zdolność tworzenia łatwo lotnych wodorków przez niektóre pierwiastki
(Se, Te, As, Sb, Bi, Ge, Sn, Pb) wykorzystano do uwolnienia ich od matrycy. Wodorki tworzy się w
reakcji z silnymi substancjami redukującymi, np. z borowodorkiem sodu w środowisku kwaśnym,
następnie czyste wodorki wypłukuje się wodorem z mieszaniny reakcyjnej i wprowadza do
kwarcowej kuwety pomiarowej, ogrzewanej płomieniem lub elektrycznie do temperatury ok.
1000ºC. W tej temperaturze wodorki ulegają rozpadowi na wolne atomy i gazowy wodór
(atomizacja).
Atomizery wykorzystujące zimne pary rtęci. Stężenie par rtęci powietrzu w temperaturze 300 K
może wynosić ok. 20 ng/cm
3
i jest to wystarczające stężenie do oznaczenia rtęci metodą AAS. Rtęć

AAS – atomowa spektroskopia absorpcyjna
13
w postaci jonów Hg
2+
w roztworach można zredukować za pomocą Sn
2+
i wolną rtęć wypłukać z
mieszaniny reakcyjnej argonem. Rtęć w gazach można zatężyć na wacie złotej; z podgrzanego do
700 -800 K amalgamatu rtęć ulega desorpcji i może być przeniesiona do kuwety pomiarowej w
strumieniu argonu. Kuweta pomiarowa (absorpcyjna) to ogrzewana rurka szklana z okienkami
kwarcowymi znajdująca się w osi optycznej spektrometru absorpcji atomowej.
2.3. Monochromatory
Zadaniem monochromatora jest eliminacja promieniowania własnego płomienia i wycięcie
linii rezonansowej z promieniowania emitowanego przez lampę z katodą wnękową (źródła
promieniowania liniowego). Monochromatory działają na zasadzie siatki dyfrakcyjnej naciętej na
powierzchni zwierciadła, które jest umieszczone na obrotowym uchwycie, umożliwiającym
kierowanie na szczelinę przepuszczającą do detektora różne długości fal (monochromatory typu
Littrowa, Eberta i Czernego – Turnera). Spektrometry AA działają w zakresie od 193,7 do 852,1 nm.
2.4. Detektory
Detektorem w spektrometrze absorpcji atomowej jest fotopowielacz. Jest to układ składający
się z fotokatody, szeregu dynod i anody. Zasada działania fotopowielacza polega na tym, że foton
pada na katodę, wybija z niej elektrony, które trafiają na dynodę. Każdy elektron wybija kilka
nowych elektronów z dynody. Proces ten jest powtarzany na kolejnych dynodach i w ten sposób
otrzymuje się wielokrotne wzmocnienie prądu, który jest proporcjonalny do liczby
zaabsorbowanych fotonów. Prąd przekazywany jest do miernika lub innego urządzenia
pomiarowego wyskalowanego w jednostkach absorbancji lub transmitancji. Jako rejestratory
stosowane są komputery umożliwiające jednocześnie opracowanie statystyczne wyników.
3. Zakłócenia podczas pomiarów i ich eliminacja
Metoda absorpcyjnej spektrometrii atomowej, podobnie jak inne metody instrumentalne,
ograniczana jest zakłóceniami spowodowanymi obecnością w analizowanym roztworze substancji
towarzyszących. Mogą one być przyczyną wielu błędów. Zakłócenia te (zwane interferencjami)
można podzielić na trzy grupy:
•
zakłócenia wynikające z nakładania się linii emisyjnych i absorpcyjnych analizowanych
pierwiastków,
•
zakłócenia wynikające z fizycznych właściwości roztworów i mające wpływ na wydajność
nebulizacji,
•
zakłócenia chemiczne powodowane zakłóceniami chemicznymi zachodzącymi w
atomizerze.

AAS – atomowa spektroskopia absorpcyjna
14
W próbkach złożonych linia rezonansowa oznaczanego pierwiastka może nakładać się z
liniami spektralnymi innych pierwiastków. Przykłady nakładania się linii spektralnych wybranych
pierwiastków przedstawiono w Tab. 1. Interferencje wywołane nakładaniem się linii można
eliminować przez wykonanie pomiarów przy innej długości fali odpowiadającej innej linii
spektralnej oznaczanego pierwiastka lub przez selektywne wyizolowanie pierwiastka oznaczanego
lub zakłócającego.
Tab. 1. Nakładanie się linii spektralnych w AAS
Analizowany
pierwiastek
Długość fali
λ [nm]
Pierwiastek
przeszkadzający
Długość fali
λ [nm]
Cd
228,802
As
228,812
Al.
308,215
V
308,211
Sb
217,023
Pb
216,999
Zn
213,856
Fe
213,859
Ca
422,673
Ge
422,657
Co
252,136
In
252,137
Cu
324,754
Eu
324,753
Fe
271,903
Pt
271,904
Hg
253,652
Co
253,649
Absorpcja linii spektralnej oznaczanego pierwiastka może być pozornie zmniejszona przez
emisję promieniowania przez wzbudzone w atomizerze atomy, cząsteczki czy cząstki ciał stałych.
Duże cząstki plazmy termicznej mogą rozpraszać promieniowanie, przez co pozornie zwiększać
absorpcję. Można temu zapobiec, zwiększając efektywność nebulizacji poprzez zmniejszenie
rozmiaru kropel. Procesy emisji, absorpcji i rozpraszania promieniowania przez składniki plazmy
nie będące agalitem, można wyeliminować aparaturowo poprzez tzw. korektę tła.
Innym typem zakłóceń są interferencje chemiczne, przeważnie specyficzne dla
poszczególnych pierwiastków. Nazywa się je często efektami matrycowymi, gdyż powodowane są
składnikami matrycy. Składniki matrycy mogą powodować inną lepkość i napięcie powierzchniowe
roztworu próbki niż roztworów wzorcowych a tym samym różną wydajność nebulizacji. Problemy
takie występują zwłaszcza przy badaniu płynów fizjologicznych i olejów mineralnych. Lepkość i
napięcie powierzchniowe można zmniejszyć przez dodatek substancji powierzchniowo-czynnych i
rozpuszczalników organicznych.
Innym problemem jest wpływ składu matrycy na tworzenie związków analizowanego
pierwiastka różniących się lotnością i trwałością termiczną, wpływ na stopień dysocjacji termicznej
lub możliwość jonizacji.

AAS – atomowa spektroskopia absorpcyjna
15
Opracowano kilka sposobów eliminacji zakłóceń chemicznych:
•
zastosowanie płomienia redukującego nie dopuszcza do powstania tlenków lub powoduje
ich redukcję: MO + C = M + CO,
•
zastosowanie płomienia o wyższej temperaturze (tlenek azotu (I)-acetylen) umożliwiającego
dysocjację termiczną, która nie zachodzi w płomieniu powietrze-acetylen, np. wapń w
obecności glinu daje trwały związek CaAl
2
O
4
w płomieniu powietrze-acetylen, natomiast
dysocjuje on w płomieniu tlenku azotu (I),
•
dodanie do roztworu analizowanego odczynnika korygującego, powodującego uwolnienie
pierwiastka z trudno dysocjującego związku,
•
dodanie do roztworu analizowanego odczynnika dejonizującego (buforu), zmniejszającego
jonizację oznaczanych atomów,
•
dodatnie do roztworu analizowanego buforu nasycającego, tj. roztworu pierwiastka
zakłócającego o takim stężeniu, przy którym jego wpływ na absorbancję pierwiastka
oznaczanego jest stały.
4. Możliwości zastosowań AAS
•
Metodą AAS można oznaczać około 70 pierwiastków. Problematyczne jest oznaczanie niemetali.
•
AAS jest typową metodą oznaczania pojedynczego pierwiastka. Zastosowanie
spektrometrów wielokanałowych nie dało istotnego postępu w eliminacji tego ograniczenia.
•
AAS jest metodą oznaczania pierwiastków śladowych i składników ubocznych (bardzo
rzadko stosuje się ją do oznaczania składników głównych).
•
Określany zakres stężeń odpowiada w przybliżeniu jednemu rzędowi wielkości. W przypadku
możliwości pomiaru bardzo małych absorbancji zakres ten może objąć 2 – 3 rzędy wielkości.
•
AAS jest metodą względną. Do wyznaczenia stężenia wykorzystuje się krzywe wzorcowe
(wyniki dokładniejsze) lub metodę dodatków (metoda szybsza, ale mniej dokładna).
•
Metoda AAS jest podatna na wszelkiego rodzaju zakłócenia – stąd konieczność obsługi
przez personel o wysokich kwalifikacjach.
•
AAS jest techniką stosowaną w rutynowych oznaczeniach w laboratoriach metalurgicznych,
rolniczych, medycznych, biologicznych, geologicznych, ochrony środowiska i wszędzie tam,
gdzie zachodzi konieczność oznaczeń śladowych ilości pierwiastków.
Granice detekcji dla określonych długości fal wybranych pierwiastków przedstawiono w Tab. 2.

AAS – atomowa spektroskopia absorpcyjna
16
Tab. 2. Granice wykrywalności niektórych pierwiastków w metodach F-AAS i ET-AAS.
Pierwiastek Długość fali
λ [nm]
Granica detekcji F-AAS
(etyn–powietrze)
ppb [µg/dm
3
]
Granica detekcji
ET–AAS
ppb [µg/dm
3
]
Al
309,2710
500
0,01
As
193,759
14
0,12
Cd
228,8072
1
0.0002
Ca
422,673
0,5
0,01
Cu
324,754
1
0,005
Au
242,795
6
0,01
Pb
217,000
9
0.007
Hg
253,652
140
0.2
Ag
328,068
1
0,001
Fe
248,327
5
0,01
Zn
213,856
1
0,001
5. Metodyka pomiarów
Jak wcześniej wspomniano podstawą analizy ilościowej metodą AAS jest prostoliniowa
zależność absorbancji od stężenia analizowanego pierwiastka w próbce. Oznaczenie prowadzi się
dwiema metodami: metodą krzywej wzorcowej (krzywej kalibracyjnej) i metodą dodatku wzorca.
5.1. Metoda krzywej kalibracyjnej
W procesie pomiaru próbka poddawana jest różnym przemianom lub oddziaływaniom, w
wyniku których uzyskiwany jest sygnał analityczny. W metodach instrumentalnych, które w
większości są metodami porównawczymi, mierzony jest parametr fizyczny, będący funkcją stężenia
substancji analizowanej. Aby uzyskać dokładne wyniki ilościowe, wymagana jest kalibracja
względem znanych wzorców. Porównywanie z wzorcami można przeprowadzić najczęściej
następującymi metodami:
•
metodą krzywej kalibracyjnej,
•
metodą dodawania wzorca,
•
metodą wzorca wewnętrznego.
(Dwie ostatnie metody przedstawione są w instrukcjach do ćwiczeń 1 i 3.)
W przeważającej części metod detekcji mierzony parametr jest funkcją liniową stężenia
analitu;
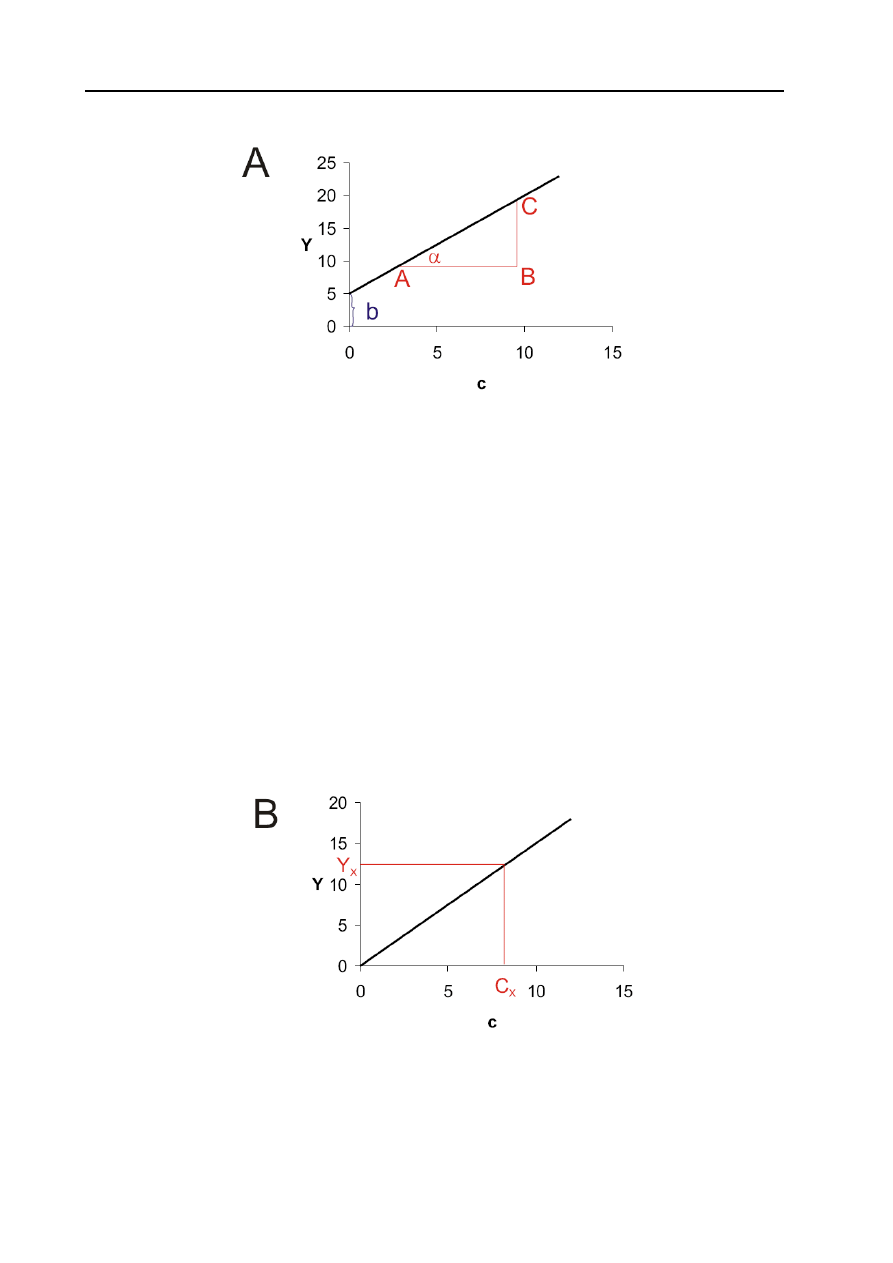
Y = ac + b,
gdzie: Y - wielkość mierzona, c – stężenie analitu, a – współczynnik proporcjonalności,
a = BC/AB = tg α, b – wartość stała, jest często wartością eksperymentalną ślepej
próby.
AAS – atomowa spektroskopia absorpcyjna
17
Taką funkcję liniową można przedstawić graficznie (Rys. 9a)
Rys. 9a. Krzywa kalibracyjna Y = ac + b
Współczynnik proporcjonalności a określa czułość metody; im większe zmiany wartości mierzonej
Y na jednostkę stężenia c, tym większa jego wartość i tym wyższa czułość metody. Metody o małym
kącie nachylenia krzywych kalibracyjnych nie są przydatne do celów analitycznych. Parametr b
może przyjmować wartości dodatnie, ujemne lub zero.
W metodzie krzywej kalibracyjnej przygotowuje się szereg roztworów o znanych stężeniach
substancji analizowanych oraz tzw. ślepą próbę – roztwór, w którym są wszystkie składniki
roztworów wzorcowych z wyjątkiem analitu i dla każdego roztworu mierzy się wartość Y.
Zależność Y od c wzorców wykreśla się (Rys. 9b) lub wylicza równanie prostej. Wartość Y mierzy
się również dla próbki badanej, nanosi na krzywą kalibracyjną i odczytuje stężenie lub oblicza z
równania prostej.
Rys. 9b. Krzywa kalibracyjna Y = a c
Przed przystąpieniem do oznaczeń metodą krzywej kalibracyjnej należy zbadać zakres
prostoliniowej zależności wartości mierzonej od stężenia analitu (Rys. 9c).
D
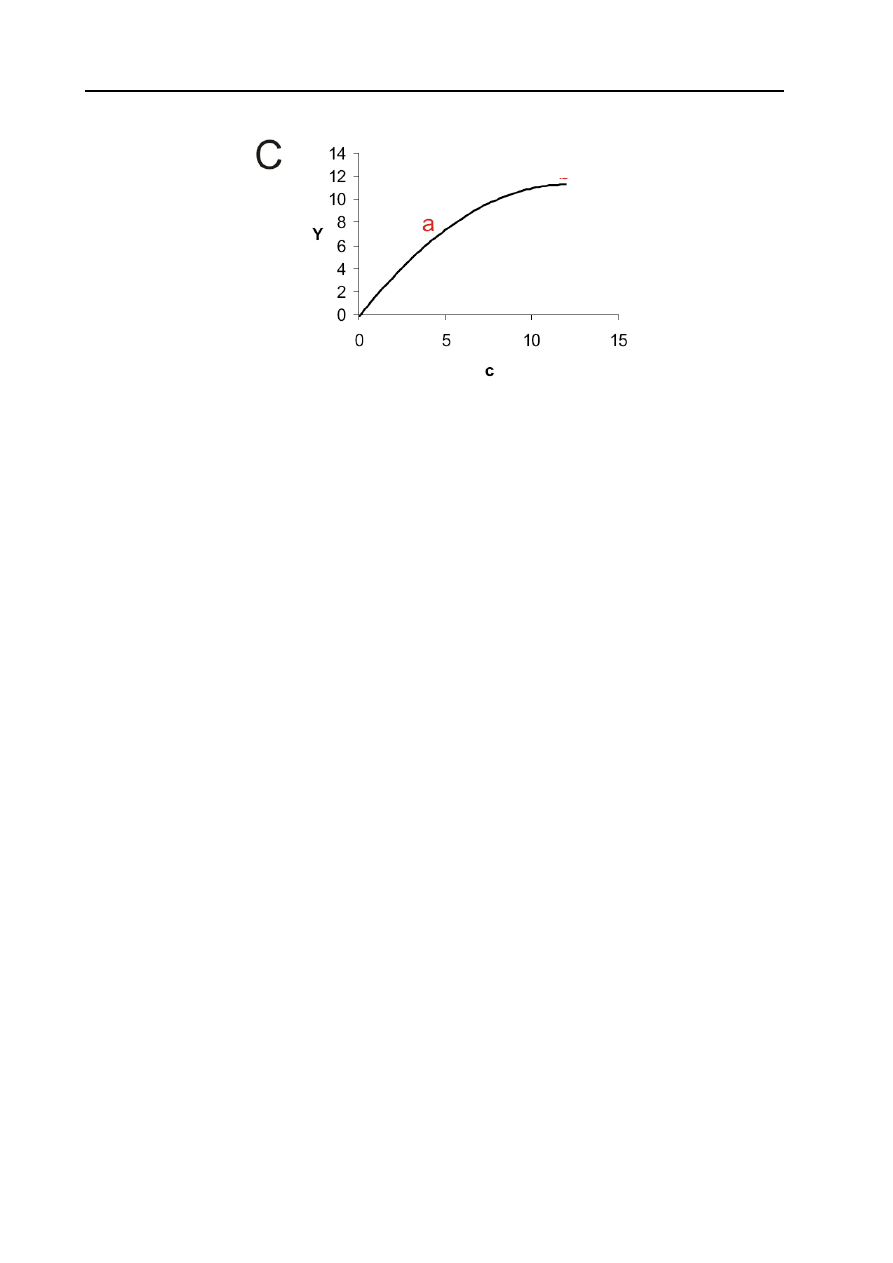
AAS – atomowa spektroskopia absorpcyjna
18
Rys. 9c. Krzywa kalibracyjna o ograniczonym zakresie stosowania
Na przedstawionej na Rys. 9c krzywej kalibracyjnej do celów analitycznych nadaje się zakres stężeń
od 0 do 5 (odcinek krzywej 0D). Krzywą kalibracyjną należy wykonywać w dniu pomiarów, zmiana
warunków pomiarów (np. temperatury) czy użycie innej partii odczynników może powodować
przesunięcie krzywej kalibracyjnej na osi Y lub zmianę nachylenia prostej Y = f(c).
Na wartość wielkości mierzonej może mieć duży wpływ matryca, czyli to wszystko, co
wprowadzamy do układu pomiarowego poza substancją oznaczaną. Udział matrycy należy
uwzględnić przy sporządzaniu roztworów wzorcowych, dbając by roztwory wzorcowe miały skład i
właściwości fizyko-chemiczne jak najbardziej zbliżone do właściwości roztworu analitu.
Zmniejszenie wpływu matrycy na wartość wielkości mierzonej można niekiedy uzyskać przez
wprowadzanie do próbki substancji maskujących. Ograniczenie wpływu matrycy może być trudne
lub niemożliwe, jak często bywa w przypadku próbek środowiskowych, należy wtedy zastosować
metodę dodawania wzorca lub zmienić sposób przygotowania próbki do pomiarów.
Literatura
1.
Dittrich K, Absorpcyjna spektrometria atomowa, PWN Warszawa 1988.
2.
Pinta M, Absorpcyjna spektrometria atomowa. Zastosowanie w analizie chemicznej, PWN, Warszawa
1977.
3.
Ewing GW, Metody instrumentalne w analizie chemicznej, PWN, Warszawa 1980.
4.
Minczewski J, Marczenko Z, Chemia Analityczna, Tom 3, PWN, Warszawa 1985.
5.
Szczepaniak W, Metody instrumentalne w analizie chemicznej, PWN, Warszawa 1996.
6.
Kocjan R, Chemia analityczna, PZWL, Warszawa, 2000.
7.
Namieśnik J, Metody instrumentalne w kontroli zanieczyszczeń środowiska, Politechnika Gdańska,
Gdańsk, 1992.
8.
Reczyński W, Bochnia T, Metody atomowej spektroskopii absorpcyjnej i jej zastosowanie w fizjologii
roślin, Wiadomości Botaniczne, 34, 37, 1990
Wyszukiwarka
Podobne podstrony:
AAS piatek 14 30 id 50013 Nieznany
Abolicja podatkowa id 50334 Nieznany (2)
4 LIDER MENEDZER id 37733 Nieznany (2)
katechezy MB id 233498 Nieznany
metro sciaga id 296943 Nieznany
perf id 354744 Nieznany
interbase id 92028 Nieznany
Mbaku id 289860 Nieznany
Probiotyki antybiotyki id 66316 Nieznany
miedziowanie cz 2 id 113259 Nieznany
LTC1729 id 273494 Nieznany
D11B7AOver0400 id 130434 Nieznany
analiza ryzyka bio id 61320 Nieznany
pedagogika ogolna id 353595 Nieznany
Misc3 id 302777 Nieznany
cw med 5 id 122239 Nieznany
D20031152Lj id 130579 Nieznany
więcej podobnych podstron