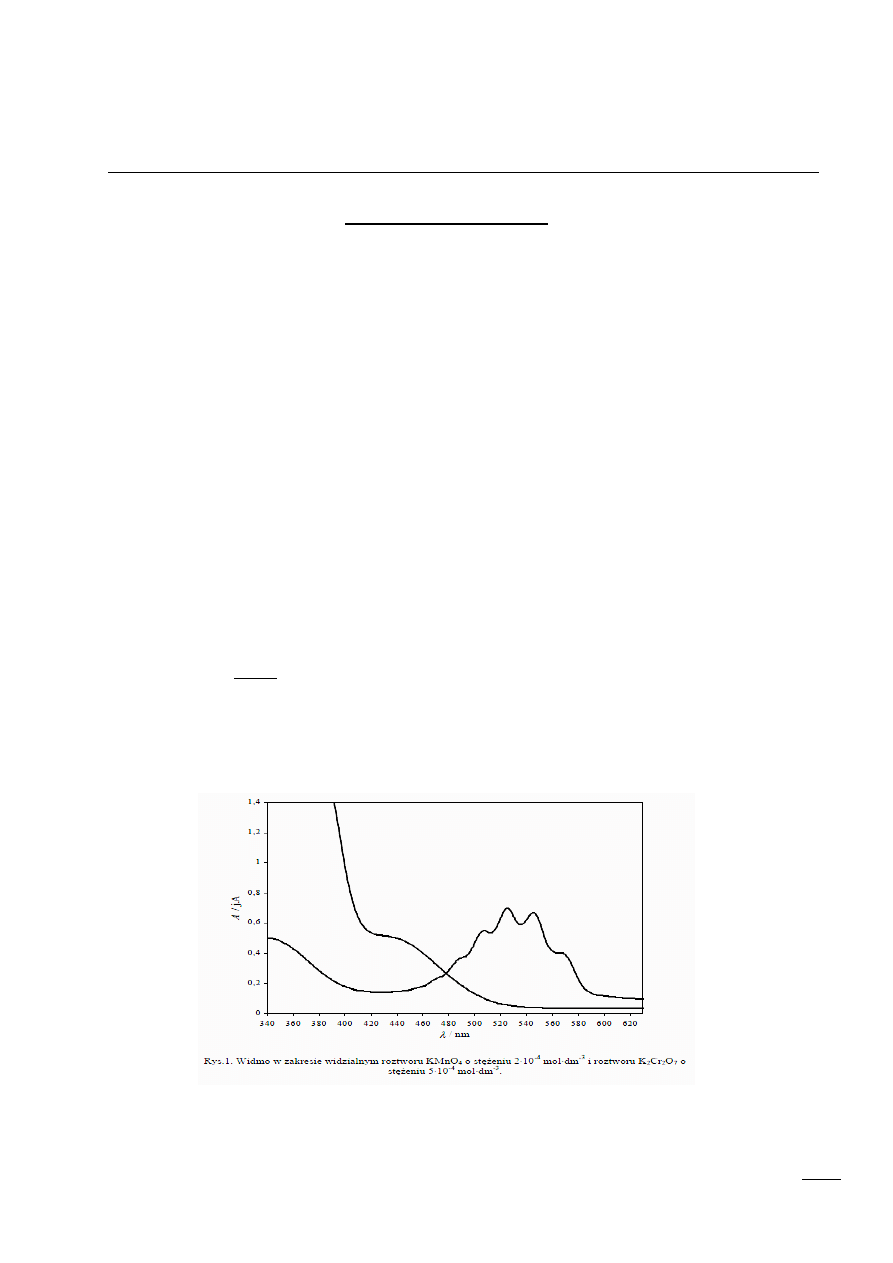
A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
1
1. Absorpcjometria w świetle widzialnym
Absorbcja, absorbancja
Absorpcja – w optyce proces pochłaniania energii fali elektromagnetycznej przez substancję.
Absorbancja – jest miarą absorpcji promieniowania.
Sposób przedstawiania widma absorpcji – oś x: długość fali [nm], oś y: absorpcja a
Prawo addytywności absorbancji
dotyczy roztworów i mieszanin wieloskładnikowych. Wyraża ono absorbancje całkowitą środowiska,
A, jako sume niezależnych absorbancji poszczególnych składników (A
1
, A
2
, .....A
n
)
n
i
i
n
A
A
A
A
A
1
2
1
....
Molowy współczynnik absorbancji
oznaczany przez ε - stała proporcjonalności związana z pochłanianiem promieniowania przez
częściowo absorbujący i rozpraszający ośrodek.
bezpośrednio zależy od długości fali promieniowania elektromagnetycznego
jednostka:
𝑑𝑚
3
𝑚𝑜𝑙∙𝑐𝑚
Jeżeli w pomiarach występują ujemne wartości A, to znaczy że kuweta z odnośnikiem jest zabrudzona
i absorbuje więcej promieniowania niż kuweta z próbką. Należy dokładnie umyć kuwetę i osuszyć ją
bez pozostawiania odcisków palców. Przyjmujemy A=0.

A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
2
Transmitancja
Transmitancja wskazuje, jaka część promieniowania padającego została przepuszczona przez
substancję.
Prawo Lamberta-Beera
absorbancja jest wprost proporcjonalna do stężenia c i grubości warstwy l roztworu, przez który
przechodzi promieniowanie.
gdzie:
– absorbancja
– stała proporcjonalności (współczynnik pochłaniania promieniowania, często nazywany
współczynnikiem absorpcji)
– stężenie substancji w roztworze
– grubość warstwy absorbującej (droga jaką pokonuje promieniowanie przechodząc przez
roztwór)
Odstępstwa
Prawo Lamberta-Beera jest spełnione dla wiązki promieniowania
skolimowanego (promienie są równoległe, wiązka płaska).
Gdy warunki te nie są spełnione obserwujemy odstępstwa od tego prawa. Odstępstwa mogą mieć
charakter zarówno chemiczny jak i fizyczny.
Odstępstwa chemiczne
zachodzą gdy cząsteczki substancji lub rozpuszczalnika bezpośrednio ze sobą oddziaływują w
wyniku na przykład solwatacji lub dysocjacji.
Odstępstwa fizyczne:
niemonochromatyczność przechodzącego promieniowania.
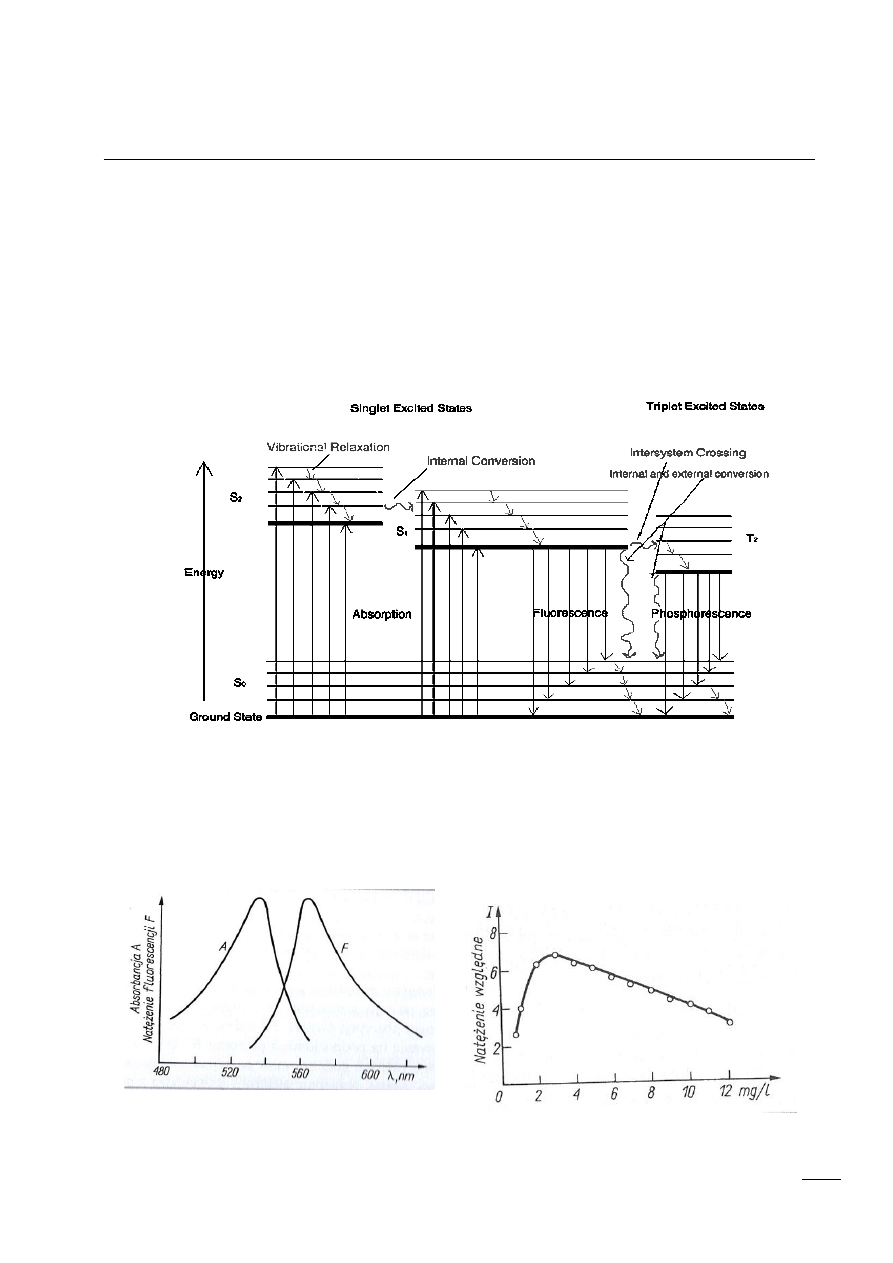
A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
3
2. Luminescencja
Luminescencja- wszelkie rodzaje emisji światła z wyjątkiem promieniowania cieplnego.
- promieniowania samorzutna - świecenie występujące natychmiast po naświetleniu,
znikające po zaniku naświetlania; zjawisku podlegają niektóre gazy, pary, substancje organiczne i ich
roztwory; średni czas wzbudzenia
- promieniowanie wymuszone – fosforescencja ; po wzbudzeniu część elektronów powraca
na poziom podstawowy, natomiast cześć przechodzi samorzutnie na poziom metastabilny, pozostaje
tam do momentu wzbudzenia, kosztem np. energii termicznej ukłsadu; krótki czas wzbudzenia;
występuje w substancjach stałych i bardzo lepkich roztworach ciekłych.
Diagram Jabłońskiego
Cecha charakterystyczna fotoluminescencji jest widmo promieniowania absorbowanego i
emitowanego. Widmo fal jest przesunięte w kierunku fal o większej długości, długość fali emitowanej
jest większa od długości fali absorbowanej.
1 Krzywa zależności natężenia
promieniowania fluorescencyjnego od
stężenia
2 Porównanie widma absorpcji [A] i fluorescencji
[F] tego samego związku

A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
4
Pomiar
- pomiar natężenia promieniowania fluorescencyjnego emitowanego po odpowiednim wzbudzeniu
substancji
𝑐
𝑥
=
𝐼
𝑥
𝐼
𝑤𝑧
𝑐
𝑤𝑧
c
x
- stężenie próbki
I
x
- natężenie fluorescencji próbki
I
wz
- natężenie fluorescencji wzorca
c
wz
-stęzenie wzorca
NIEDOKŁADNOŚCI pomiaru powodowane są wpływem:
obcych substancji potęgujących zdolność f
temperatury ( wzrost powoduje spadek f)
rozpuszczalnika
pH
nieodpowiedniej długości fali
ZALETY:
najbardziej czuła metoda analityczna
wykrywalność związków nietrwałych
możliwość analizy substancji nieprzeźroczystych
WADY:
tylko do substancji wykazujących zdolność fluorescencji
wygaszenie stężeniowe
(W rozcieńczonych roztworach intensywność fluorescencji jest proporcjonalna do stężenia
luminoforu. Jednak gdy stężenie substancji fluoryzującej przekroczy pewną granicę,
intensywność fluorescencji zaczyna maleć)
małe stężenie substancji
nie nadaje się do oznakowania podstawowych składników próbek
Schemat spektrofotometru
1. Źródło promieniowania
2. Monochromator układu wzbudzającego (siatka dyfrakcyjna, filtr)
3. Naczynie z próbką
4. Monochromator układu emisyjnego
5. Detektor

A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
5
5. Interferometria
Zjawisko interferencji
Interferencja – zjawisko powstawania nowego, przestrzennego rozkładu amplitudy fali
(wzmocnienia i wygaszenia) w wyniku nakładania się (superpozycji fal) dwóch lub więcej fal.
Warunkiem trwałej interferencji fal jest ich spójność, czyli korelacja faz i częstotliwości.
Interferencja konstruktywna - prowadzi do dodawania się i wzmocnienia fal. Kiedy
nakładające się fale mają ten sam kierunek i długość fali λ oraz są zgodne w fazie wtedy amplituda fali
wypadkowej jest dwa razy większa niż amplituda A obu fal oddzielnie.
Interferencja destruktywna – prowadzi do odejmowania się i wzajemnego wygaszania fal.
Jeżeli obie spotykające się fale są przesunięte względem siebie o połówkę długości fali, czyli drgają w
przeciwfazie to rezultatem ich nałożenia się będzie ich całkowite wygaszenie.
Zjawisko załamania światła
Współczynnik załamania ośrodka jest miarą zmiany prędkości rozchodzenia się fali w danym ośrodku
w stosunku do prędkości w innym ośrodku (pewnym ośrodku odniesienia).
gdzie
– prędkość fali w ośrodku, w którym fala rozchodzi się na początku,
– prędkość fali w ośrodku, w którym rozchodzi się po załamaniu.
Ogólnie mówiąc współczynnik załamania światła jednego ośrodka względem drugiego zależy od
długości fali (dyspersja światła). Współczynnik załamania światła zależy również od stanu ośrodka -
np. jego temperatury i ciśnienia.
PRAWO SNELLIUSA
Współczynnik załamania wiąże się bezpośrednio z kątem padania i kątem załamania. Związek ten
wyraża prawo Snelliusa:
gdzie
α – kąt padania promienia fali na granicę ośrodków
β – kąt załamania.
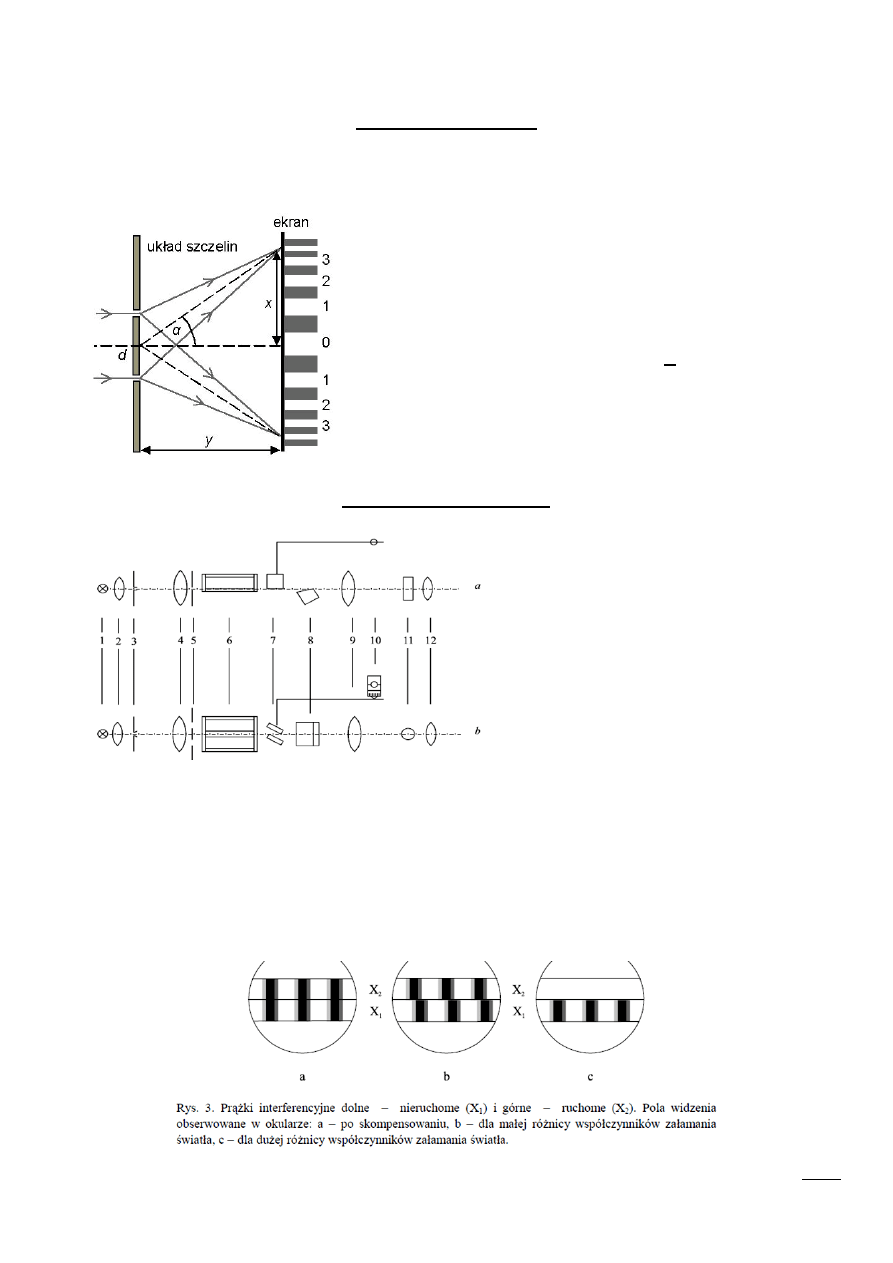
A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
6
Prążki jasne i ciemne
W doświadczeniu Younga wskutek interferencji na ekranie powstają jasne i ciemne prążki w
obszarach, w których światło jest wygaszane lub wzmacniane.
Jasne prążki (maksima) powstają w miejscach, dla ktorych
spełniony jest warunek:
𝑑
・
𝑠𝑖𝑛𝛼 = 𝑛𝜆
Ciemne prążki (minima) powstają w miejscach, dla ktorych
spełniony jest warunek:
𝑑
・
𝑠𝑖𝑛𝛼 = (2𝑛 − 1)
𝜆
2
gdzie:
d – stała siatki (odległość między dwoma sąsiednimi szczelinami)
n = 0, 1, 2... – rząd widma
Budowa interferometru
Wiązka światła wychodząca ze źródła (1)
pada na soczewkę (2), która skupia ją
dokładnie na szczelinie (3) kolimatora (4).
Wiązka światła po przejściu przez soczewkę
zamykającą kolimator i dalej szczelinę staje
się równoległa i ma kształt świetlnego
walca. Trafiając z kolei na przesłonę z
dwiema
równoległymi szczelinami (5)
rozszczepia się na dwie równolegle biegnące
wiązki
przechodzące
następnie
przez
termostat, w którym znajdują się dwie
identyczne kuwety interferometryczne (6).
Termostat ten może być wypełniony
powietrzem,
wodą
destylowaną
lub
odpowiednim
rozpuszczalnikiem.
Część
promieni przechodzi przez naczyńka wypełnione roztworem badanym i odnośnikiem, a część pod
naczyńkami. Promienie górne, po przejściu przez kuwety interferometryczne, spotykają na swojej drodze
dwie płytki kompensacyjne (7). Jedna z nich jest nieruchoma, ale druga może być obracana za pomocą
śruby mikrometrycznej (10). Obydwie wiązki promieni górnych po przejściu przez następną soczewkę
skupiającą (11), tworzą w okularze (12) obraz prążków interferencyjnych. Pomiar przy użyciu opisanego
wyżej interferometru, polega na znalezieniu różnicy przesunięć prążków, która odpowiada różnicy
współczynników załamania światła substancji badanej i ośrodka odniesienia.
3 Schemat przekroju pionowego (a) i poziomego (b) interferometru
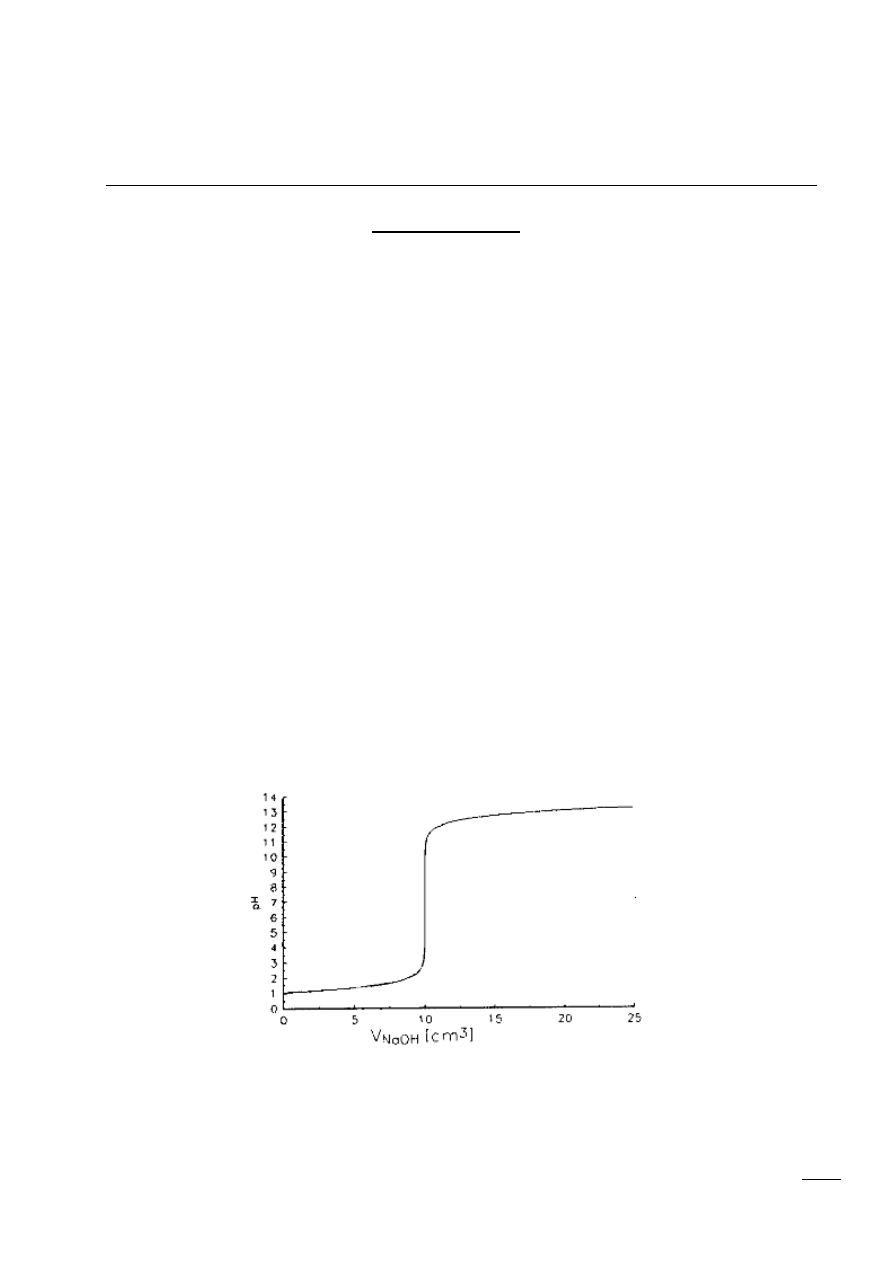
A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
7
7. Miareczkowanie do martwego punktu
Miareczkowanie
Miareczkowanie - chemiczna technika analizy ilościowej polegająca na dodawaniu roztworu -
tzw. titranta w postaci kropel do roztworu zwanego analitem.
Roztwory odczynników o znanym stężeniu (mianie) używane do miareczkowania nazywa się
roztworami mianowanymi. Stężenia roztworów mianowanych wyraża się molowością (mol/l).
Punkt równoważnikowy (punkt równoważności, punkt nasycenia równoważnikowego, PR, martwy
punkt) - jest to moment miareczkowania, w którym oznaczany składnik (analit) przereagował
ilościowo z dodanym z biurety odczynnikiem (titrantem).
W praktyce zamiast punktu równoważnikowego obserwuje się punkt końcowy miareczkowania (PK),
który jest zwykle przesunięty w stosunku do PR o 0,05%~1%, a różnicę między PR i PK określa się jako
błąd miareczkowania.
REAKCJE CHEMICZNE STOSOWANE W MIARECZKOWANIU:
przebiegające stechiometrycznie (ilościowo)
przebiegające szybko
których punkt równoważnikowy można dokładnie wyznaczyć
w których biorą udział związki chemiczne tworzące roztwory trwałe w warunkach
miareczkowania.
Inne nazwy miareczkowania do martwego punktu: amperometryczne
Krzywa miareczkowania:

A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
8
8. Fotometria płomieniowa
Fotometria
płomieniowa
jest metodą
analityczną
opartą
na pomiarze promieniowania
emitowanego przez odpowiednio wzbudzoną próbkę. Jako źródło wzbudzenia stosuje się
płomień palnika, do którego wprowadza się badaną substancję, zwykle w postaci
rozpylonego roztworu.
Atomy spalanej substancji emitują charakterystyczne widmo. Światło płomienia przechodzi przez
układ optyczny z filtrem i pada na fotoogniwo.
Filtr – przepuszcza jedynie widmo badanego pierwiastka,
Fotoogniwo – powstaje w nim prąd elektryczny będący miarą ilości badanej substancji.
Fotometrię płomieniową stosuje się do naturalnych materiałów ciekłych:
Wody różnego pochodzenia, ścieki
Badanie moczu, surowicy krwi/tkanek
Analiza rud i minerałów
Badanie gleb, nawozów itp.
WPŁYW TEMPERATURY
Najważniejszym czynnikiem, od którego zależy wynik analizy, jest temperatura płomienia.
W zbyt niskiej temperaturze:
- wzbudzeniu ulega tylko mała część atomów
- intensywność promieniowania jest za mała.
W zbyt wysokiej temperaturze:
- może w płomieniu nastąpić jonizacja atomów oznaczanego pierwiastka,
- jony powodują zmniejszenie stężenia i natężenia promieniowania atomów (widmo jonów
jest inne niż widmo atomów).
Od temperatury płomienia zależą również procesy zachodzące w płomieniu po wprowadzeniu do
niego badanego roztworu w postaci aerozolu.
WPŁYW SAMOABSORPCJI
Wzrost stężenia pierwiastka w roztworze powoduje wzrost ilości niewzbudzonych atomów w
chłodniejszej części płomienia. Wskutek tego promieniowanie wychodzące ze strefy wzbudzenia jest
w większym stopniu absorbowane.
REAKCJE ZACHODZĄCE W PŁOMIENIU
Odparowanie/spalenie rozpuszczalnika
Dysocjacja i redukcja cząsteczek
Wzbudzenie atomów
Jonizacja
A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
9
9. Miareczkowanie fotometryczne
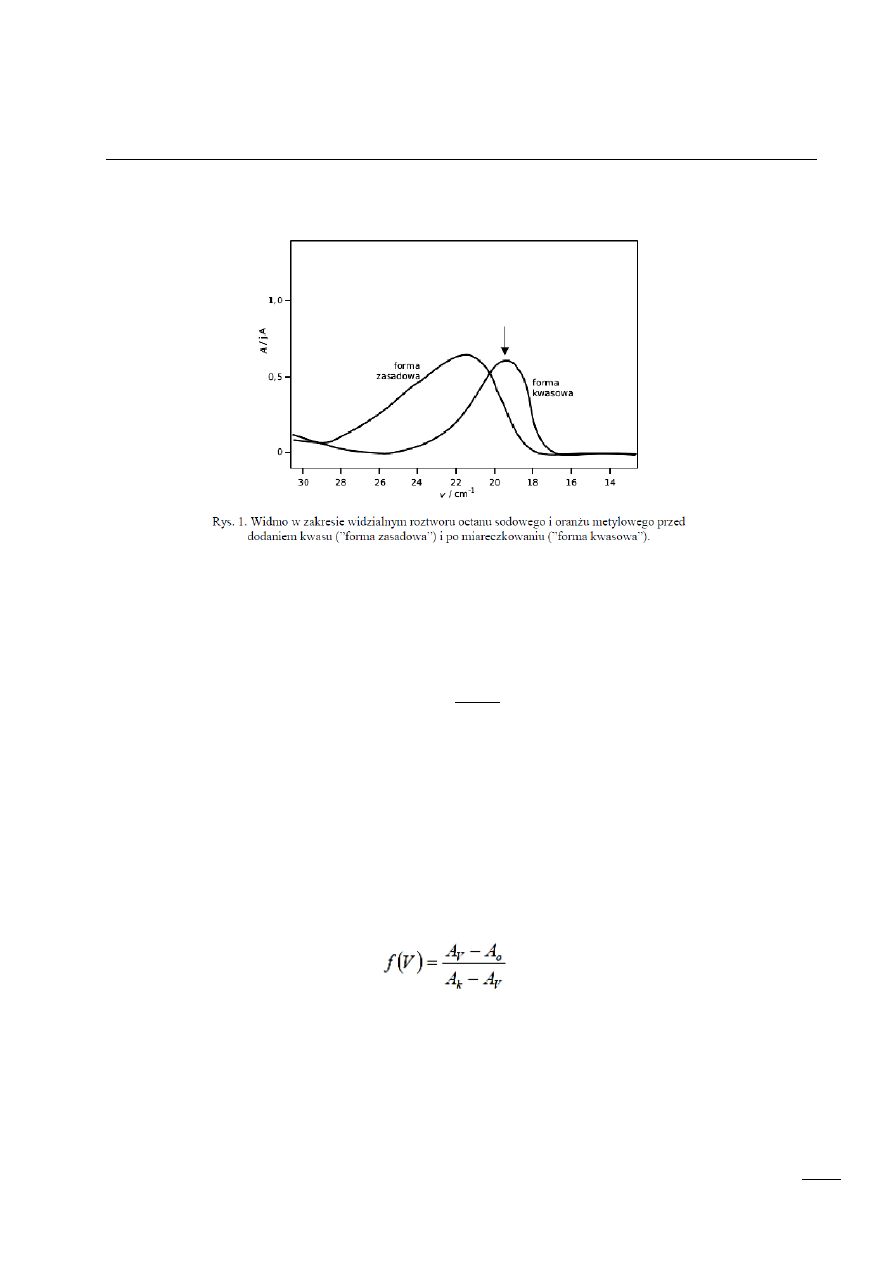
Indykator – substancja, która poprzez zmianę zabarwienia wskazuje jaki jest odczyn roztworu.
POPRAWKA NA ROZCIEŃCZENIE
Zanim użyjemy „surowych” danych, trzeba je skorygować korzystając z tzw. poprawki na
rozcieńczenie roztworu podczas miareczkowania.
Poprawkę obliczamy korzystając z poniższego wzoru:
𝐴
′
= 𝐴 ∙
𝑉 + 𝑉
𝑡
𝑉
K – odczytana wartość absorbancji,
V – początkowa objętość roztworu, rowna V
w
+V
pr
,
V
w
– objętość wody destylowanej,
V
pr
– objętość próbki
V
t
– objętość titranta
FUNKCJA LINEARYZUJĄCA:
A
0
- początkowa wartość absorbancji (wartość absorbancji formy zasadowej
indykatora) w jednostkach j.A;
A
k
- końcowa wartość absorbancji z uwzględnieniem poprawki na rozcieńczenie (wartość absorbancji formy kwasowej
indykatora) w jednostkach j.A;
V - objętość dodanego odczynnika miareczkującego w jednostkach cm3.
A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
10
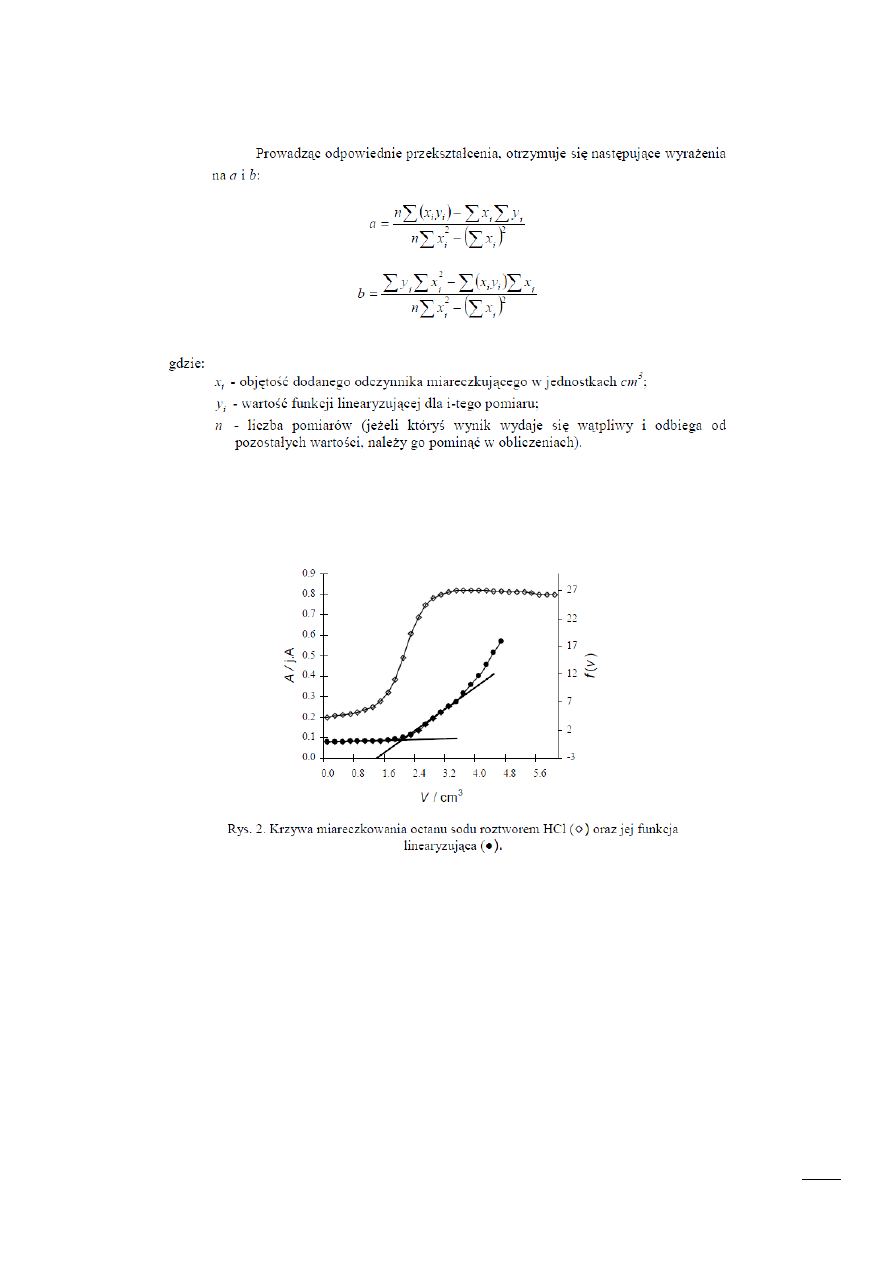
METODA NAJMNIEJSZYCH KWADRATÓW:
KRZYWA MIARECZKOWANIA WRAZ Z FUNKCJĄ LINEARYZUJĄCĄ
REAKCJE STOSOWANE DO MONITOROWANIA:
zobojętnianie
resold
strąceniowe
kompleksowanie

A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
11
10. Miareczkowanie konduktometryczne
Definicja
Miareczkowanie konduktometryczne - w chemii analitycznej chemiczna technika miareczkowa
polegająca na pomiarze zmian przewodnictwa elektrycznego analizowanego roztworu w trakcie
stopniowego dodawania do niego odczynnika miareczkującego. Znajduje zastosowanie w przypadku
roztworów silnie zabarwionych lub mętnych, a także w reakcjach, w których tworzą się kompleksy.
Miareczkowanie konduktometryczne przeprowadzane jest zwykle w układzie kwas-zasada. O
przewodnictwie układu kwas-zasada decydują głównie bardzo ruchliwe jony hydroniowe, a zatem
jest ono funkcją pH układu.
Ruchliwość jonów
Ruchliwość definiowana jest jako prędkość dryfu nadawana przez jednostkowe pole elektryczne:
μ jest ruchliwością.
Najczęściej wyraża się ją w m
2
/Vs.
Pomiędzy ruchliwością jonów w roztworze nieskończenie rozcieńczonym a granicznym
przewodnictwem molowym zachodzi następująca zależność
:
)
(
o
o
o
u
u
F
Λ
,
F – oznacza stałą Faradaya, 96500 C.
W rozcieńczeniu nieskończenie dużym, gdy przewodnictwo równoważnikowe osiąga wartość
maksymalną
o
Λ
, elektrolit rozpada się całkowicie na jony (
=1).
W mniejszych rozcieńczeniach, tzw. stężeniach skończonych:
słabe elektrolity nie są całkowicie zdysocjowane
mocne elektrolity są zdysocjowane, ale występują oddziaływania międzyjonowe
zmniejszające przewodnictwo.
Przy założeniu, że przewodnictwa jonowe nie zależą od stężenia, wyraził Arrhenius przewodnictwo
molowe w skończonych stężeniach za pomocą następującej zależności uwzględniającej stopień
dysocjacji
.
)
(
o
B
o
A
AB
Λ
Λ
α
Λ
Stopień dysocjacji można zatem obliczyć jako stosunek przewodnictwa równoważnikowego w danym
stężeniu do przewodnictwa równoważnikowego w rozcieńczeniu nieskończenie dużym:
o
Λ
Λ
α
A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
12
12. Miareczkowanie potencjometryczne
Definicja
Miareczkowanie potencjometryczne -
technika miareczkowa polegająca na pomiarze zmian
napięcia elektrycznego generowanego przez ogniwo złożone z elektrody wskaźnikowej i elektrody
odniesienia w funkcji objętości dodanego titranta. Pomiar prowadzi się wobec elektrody pH-czułej
(np. elektrody szklanej)
jako elektrody wskaźnikowej. Uzyskuje się wynik w postaci zależności
napięcia od objętości dodanego titranta. Taka zależność może zostać łatwo przekształcona w
zależność pH od objętości dodanego titranta.
Elektrody
SEM ogniwa
– parametr charakteryzujący źródło energii elektrycznej, jest to praca wykonana przez
źródło podzielona przez wartość przenoszonego ładunku.
𝜀 =
𝑊
𝑞
Półogniwo kalomelowe – II rodzaju, odwracalne z dwoma granicami faz
Schemat: Hg | HgCl
2(s)
| KCl
(nas)
(Cl
-
)
Reakcja: H
2
Cl
2
+ 2e
-
↔ 2Hg
(c)
+ 2Cl
-
Półogniwo srebrowe – I rodzaju, odwracalne z jedną granicą faz
Schemat: Ag | Ag
+
Reakcja: Ag
↔ Ag
+
+ e
-
Półogniwo platynowe – wrażliwa na zmiany potencjału redoks
Schemat: Pt | X
+
/ X
-
Składa się z platyny i roztworu w którym zachodzi jedna lub więcej reakcji redoks.
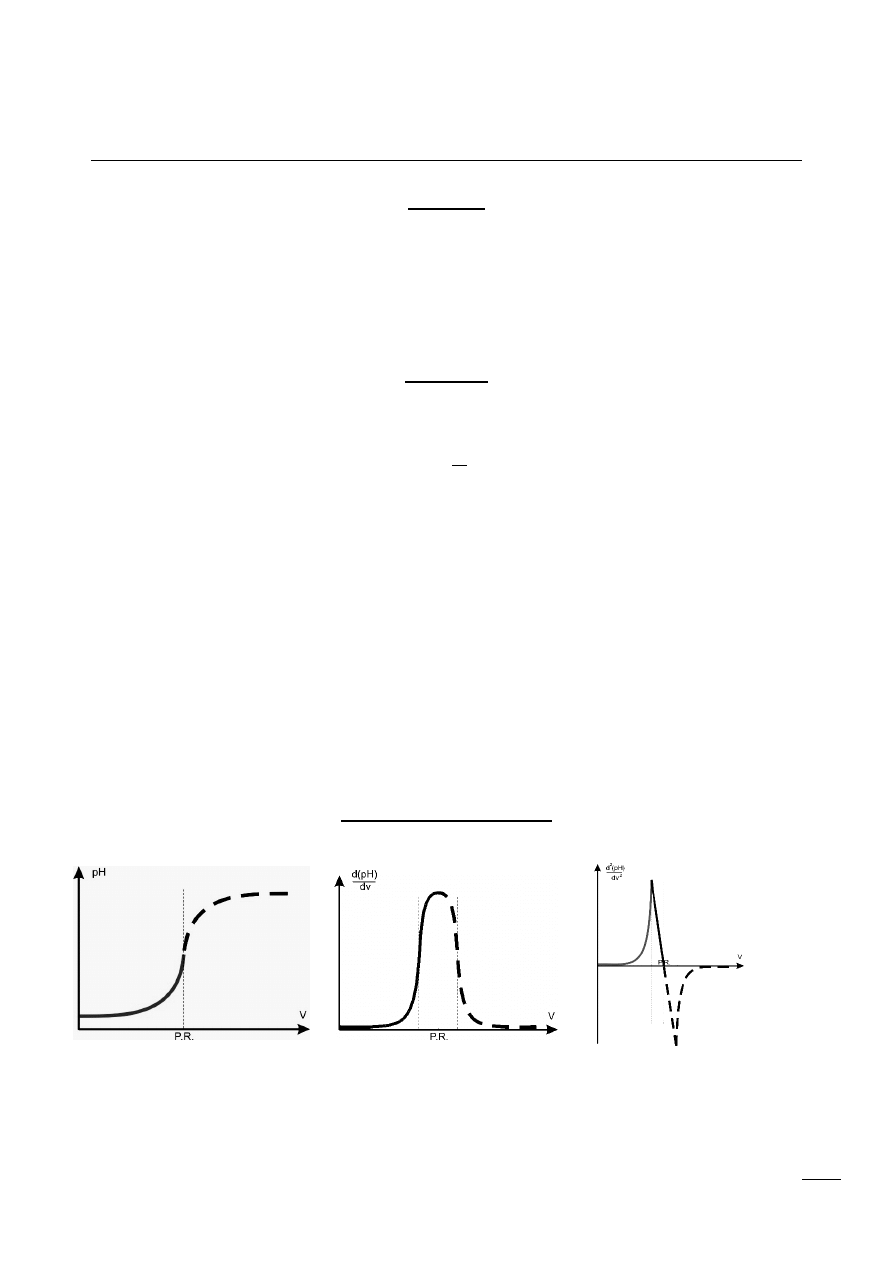
Krzywe miareczkowania
6 Zwykła
5 Pierwsza pochodna
4 Druga pochodna
A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
13
14. Przewodnictwo elektrolitów
Przewodnictwo
Wielkością charakteryzującą przewodnictwo elektryczne materiału jest przewodność elektryczna
właściwa (konduktywność) σ.
Przewodność elektryczna zależy od koncentracji nośników prądu i ich ruchliwości:
q - ładunek nośników, n - koncentracja nośników,
- ruchliwość nośników.
Czynnikiem wpływającym na przewodność elektryczną wszystkich ciał jest temperatura. Zmiana
temperatury może spowodować zmianę zarówno koncentracji, jak i ruchliwości nośników ładunku
elektrycznego.
Jony H+ i OH- mają większą przewodność niż pozostałe jony.
Metoda wielokrotnego dodatku wzorca
Procedura oznaczeń w metodzie dodawania wzorca jest następująca:
przygotowujemy próbkę analizowaną i przeprowadzamy dla niej pomiar,
do tej samej próbki dodajemy znaną ilość substancji oznaczanej (wzorca), roztwór
mieszamy i mierzymy ponownie.
Wartość parametru mierzonego wzrasta, a wzrost jest proporcjonalny do ilości dodanego wzorca.
Wynikiem dodania wzorca jest zmiana objętości roztworu próbki badanej, stąd należy skorygować
stężenia.
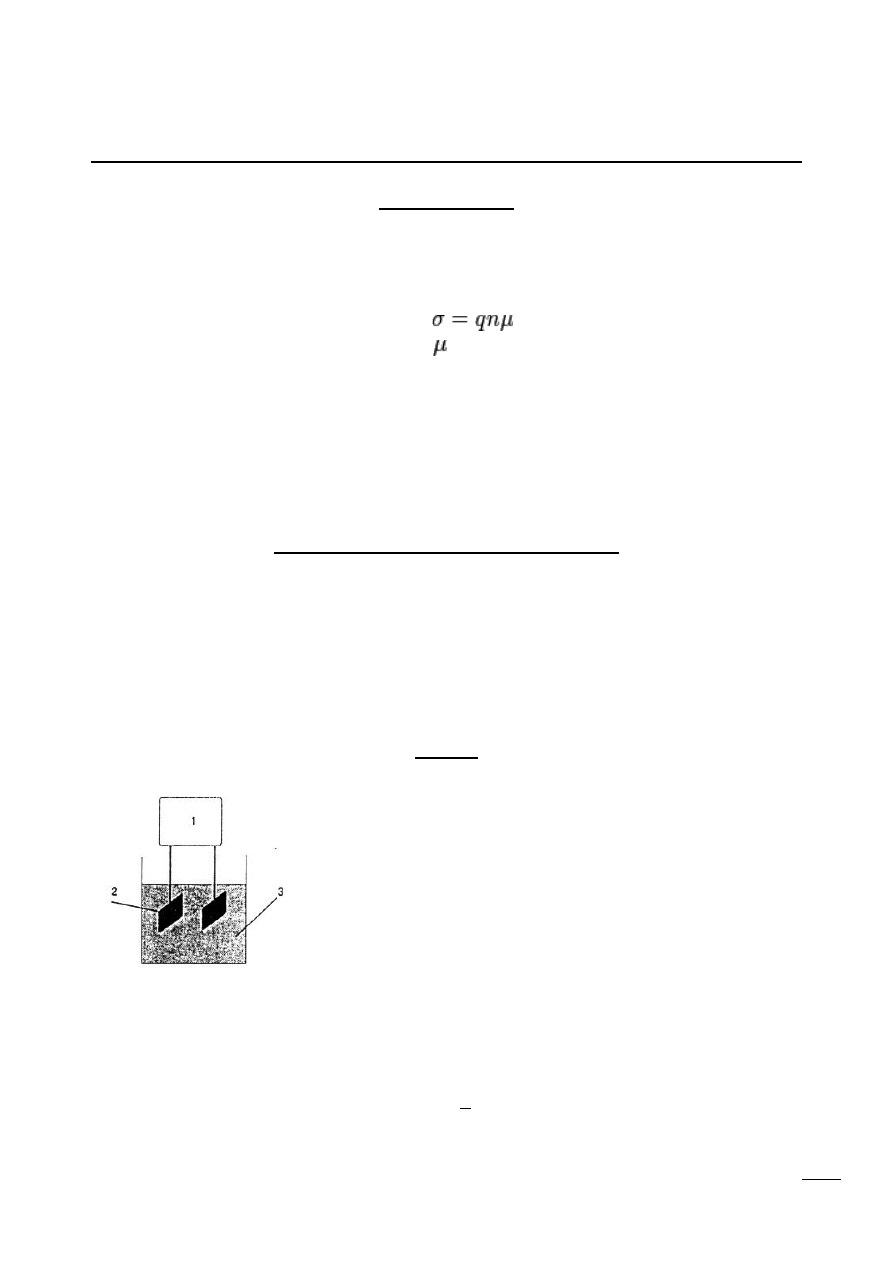
Pomiar
Schemat układu konduktometrycznego.
1 – konduktometr,
2 – elektrody,
3 – badany elektrolit
W pomiarach przewodności roztworu nie można stosować prądu
stałego, powodującego niekorzystne zjawiska na powierzchni elektrod
i zmiany składu elektrolitu.
Pomiary przewodnictwa elektrolitów wykonywane są w naczyńkach konduktometrycznych, które
zawierają dwie elektrody platynowe. Istotne jest zachowanie stałej odległości między elektrodami i
możliwie małej pojemności naczyńka. Zakładając, że powierzchnie elektrod są równe i wynoszą A,
odległość miedzy elektrodami l, stała naczyńka konduktometrycznego – k wynosi:
𝑘 =
𝑙
𝐴

A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
14
15. Polarymetria
Fala świetlna to poprzeczna fala elektromagnetyczna w której wektory pola elektrycznego (E)
i magnetycznego (H) sa jednocześnie prostopadłe do siebie i do kierunku rozchodzenia się fali.
Drgania fali świetlnej wychodzącej z źródła odbywaj się we wszystkich możliwych kierunkach –
światło naturalne (niespolaryzowane).
Światło spolaryzowane – drgania poprzeczne fali światła rozchodzą się na jednej płaszczyźnie,
wektor E i kierunek promienia (r) wyznaczaj płaszczyznę polaryzacji
Skręcalność
Wiele substancji przeźroczystych charakteryzuje się niesymetryczną budową cząstkowa, posiadają
one zdolność skręcania płaszczyzny światła spolaryzowanego (kwarc, cukry). Zależnie od budowy kąt
skręcania polaryzacji zmienia się, w prawo lub w lewo. Skręcalność płaszczyzny w prawo oznacza się
znakiem (+) – zgodnie z ruchem wskazówki zegara - substancja prawoskrętna. Skręcalność
płaszczyzny w lewo (-) - substancja lewoskrętna.
Związki optycznie czynne charakteryzuje:
brak elementów symetrii cząsteczki ( kwas mlekowy)
brak węgla asymetrycznego
brak płaszczyzny symetrii w cząsteczce (spiryny - zw. organiczne o budowie pierścieniowej)
brak osi symetrii
UWAGA! Równocząsteczkowa mieszanina obu izomerów optycznie czynnych jest roztworem
optycznie nieczynnym.
Wielkość kata skręcenia zależy od: ( dla danego związku)
liczby cząsteczek znajdujących się na drodze promieni / stężenia
długości fali
temperatury
Skręcalność właściwa- [∝)
D
20
– kąt o jaki płaszczyzna polaryzacji ulegnie skręceniu w świetle linii D
sodu λ=589,3nm i w temp= 20℃
[𝛼]
𝐷
20
=
𝛼1000
𝑙𝜌
l- grubość warstwy (dm)
α- kat skręcania płaszczyzny polaryzacji (
0
)
ρ- stężenie roztworu (g/100ml)
[𝛼]
𝐷
20
=
𝛼1000
𝑙𝑝𝑑
d-gęstość roztworu (g/ml)
p-stęzenie roztworu (%)
A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
15
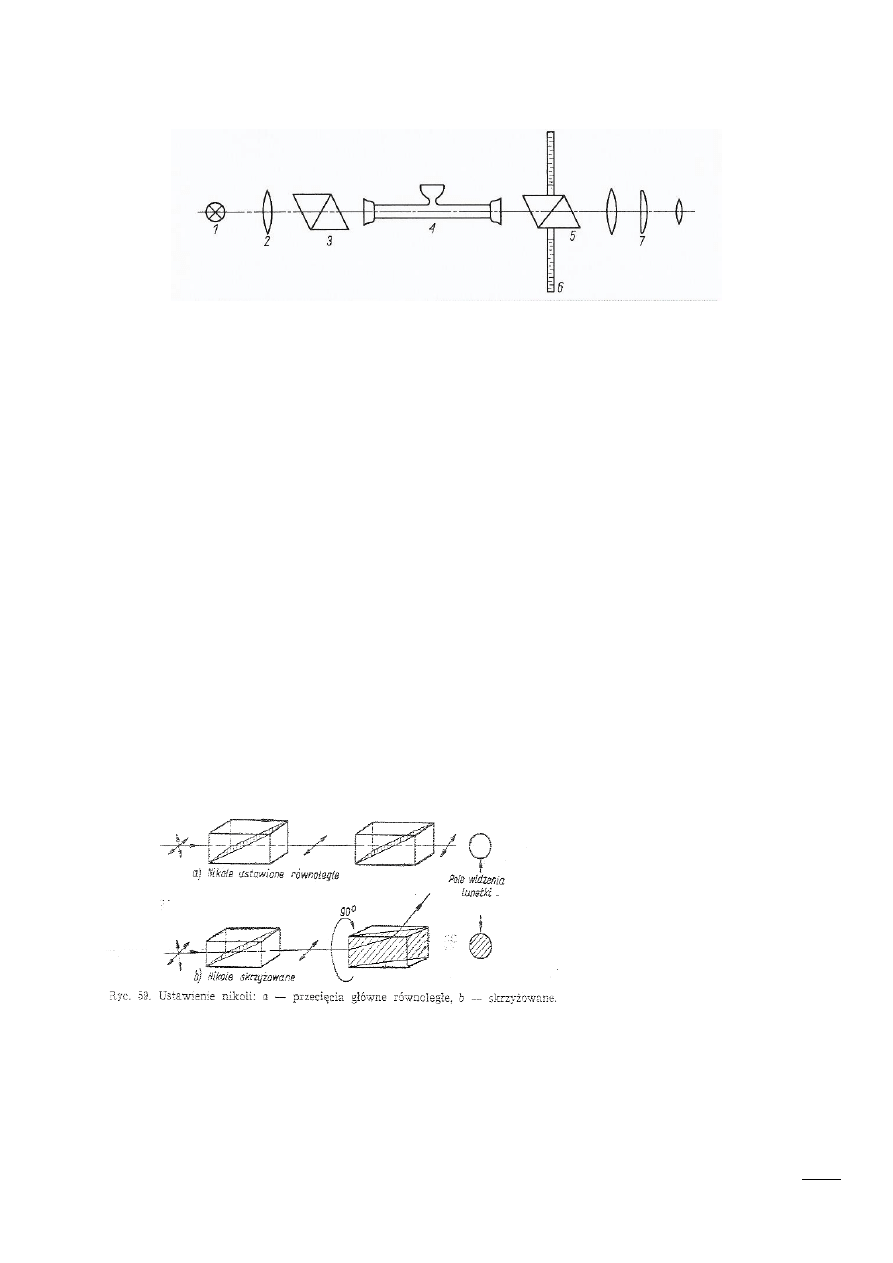
Polarymetr- urządzenie do pomiaru kata skręcenia płaszczyzny
Budowa polarymetru
1) Lampa sodowa
2) Kolimator – przyrząd przetwarzający padające światło lub strumień cząstek w równoległą
wiązkę
3) Polaryzator- urządzenie optyczne przepuszczające światło o określonej polaryzacji liniowej.
4) Szklana rurka z badana substancja
5) Analizator- ruchomy pryzmat służący do pomiaru kata skręcalności.
6) Podziałka kątowa
7) Okular
Działanie pryzmatu Nicola - Promień Z pada na wchodzi do pryzmatu w punkcie Pi dzieli się na
promień zwyczajny Z i nadzwyczajny N. Promień Z pada pod kątem większym od kata granicznego i
zostaje całkowicie odbity, następnie pochłonięty. Promień N, przechodzi przez pryzmat.
W polarymetrze światło przechodzi przez dwa nikole. Jeżeli przecięcia obu ustawione s
równolegle to światło wykazuje maksymalne natężenie. Natomiast minimum przypada gdy
przecięcia Nicoli są do siebie prostopadłe.
Światło przechodzi przez dwa nikole. Jeżeli przecięcia obu ustawione s równolegle to światło
wykazuje maksymalne natężenie. Natomiast minimum przypada gdy przecięcia Nicoli są do siebie
prostopadłe.
W
naszych
pomiarach
odczytujemy kąt kiedy w
okularze całe pole jest jasne.
Możemy
odczytać
dwa
położenia różniące się o 90
0
.
Poprawny wynik jest kątem
mniejszym od 90
0
. Pomiary
polarymetryczne pozwalają na
identyfikacje
i
określenie
stężenia badanej substancji.
Zalety tej metody:
prostota
niski koszt
mała pracochłonność

A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
16
17. Refraktometria
Podstawowe pojęcia
Refraktometria - metoda badania własności fizykochemicznych substancji na podstawie pomiarów
ich współczynników załamania, których dokonuje się za pomocą refraktometrów.
Refrakcja – zmiana kierunku rozchodzenia się fali elektromagnetycznej lub akustycznej, załamanie
fali związane ze zmianą jej prędkości, gdy przechodzi do innego ośrodka. Zmiana prędkości wiąże się
ze zmianą długości fali, podczas gdy częstotliwość pozostaje stała.
Prawo Snelliusa
Promienie padający i załamany oraz prostopadła padania (normalna) leżą w jednej płaszczyźnie,
a kąty spełniają zależność:
n
1
– współczynnik załamania światła ośrodka pierwszego,
n
2
– współczynnik załamania światła ośrodka drugiego,
n
21
– względny współczynnik załamania światła ośrodka drugiego względem pierwszego,
θ
1
– kąt padania, kąt między promieniem padającym a normalną do powierzchni granicznej ośrodków,
θ
2
– kąt załamania, kąt między promieniem załamanym a normalną.
- - -
Jeżeli światło przechodzi z ośrodka o mniejszym współczynniku załamania światła do ośrodka o współczynniku większym (np.
powietrze-woda), tak jak na rysunku, to kąt załamania jest mniejszy od kąta padania. Jeżeli na odwrót (szkło-powietrze) –
kąt załamania jest większy.
Współczynnik załamania dla danego ośrodka rośnie wraz z gęstością, np. w atmosferze maleje wraz z wysokością. Dla
różnych ośrodków tendencja ta jest na ogół również zachowana, ale nie jest regułą. Przykładem może być etanol, który ma
mniejszą gęstość niż woda, ale większy współczynnik załamania.
- - -
Kąt graniczny - maksymalny kąt, pod jakim promień świetlny może padać na granicę ośrodków,
ulegając przy tym załamaniu. Występuje tylko w sytuacji, gdy światło rozchodzące się w ośrodku
o współczynniku załamania n
1
pada na granicę z ośrodkiem o współczynniku załamania n
2
, takim
że n
2
< n
1
.
Przy wzroście kąta padania promienia powyżej wartości kąta granicznego, promień nie załamuje się i
pojawia się efekt całkowitego wewnętrznego odbicia.

A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
17
21. Fotometryczna analiza śladowa
Benzen absorbuje w nadfiolecie (zakres: 10-400nm).
Nitrobenzen absorbuje w promieniach widzialnych (zakres: 380-780nm)
Wzbudzeniu promieniowaniem UV-VIS ulegają elektrony walencyjne, ponieważ ...
W celu zwiększenia selektywności reakcji analitycznych (selektywności odczynników) stosuje się
często reakcje kompleksowania do maskowania jonów przeszkadzających w analizie. Jest to jedno z
ważniejszych zastosowań reakcji tworzenia się kompleksów w chemii analitycznej.
Pod pojęciem maskowania jonu przeszkadzającego rozumiemy przeprowadzenie go w wyniku reakcji
kompleksowania z odpowiednim ligandem w związek kompleksowy o odpowiedniej trwałości. Związany w tej
postaci jon jest niezdolny do reakcji zakłócającej prawidłowy przebieg reakcji wykrywania łub oznaczania
innego jonu.
Opracowanie ogólne
1. Metody pomiarowe
a) Porównawcze (względne)
- wymagają kalibracji względem znanych wzorców
- w procesie kalibracji wyznacza się współczynnik proporcjonalności m celem skorzystania z
zależności Y = mc (Y – wartość mierzona, c – stężenie).
b) Krzywej wzorcowej (kalibracyjnej)
- przedstawienie zależności w postaci Y = mc + b (b – stała eksperymentalna)
- współczynnik proporcjonalności jest nachyleniem krzywej (m=tgα), określa czułość metody
- przygotowuje się serię wzorców zewnętrznych i mierzy Y dla rosnących wartości stężeń by
wykreślić krzywą wzorcową i odczytać z niej szukaną wartość
- należy minimalizować wpływ matrycy (maskowanie jonów, regulacja siły jonowej)
c) Dodawania wzorca
- wykorzystuje się dwa pomiary: dla próbki bez dodatku wzorca i dla próbki z dodatkiem
wzorca
- stężenie obliczamy na podstawie dwóch równań: Y
0
=mc oraz Y
i
=m(c+c
wz
)
- wymagana jest poprawka na stężenie
d) Wielokrotnego dodawania wzorca
- polega na pomiarze wielkości wielokrotnie, po każdym dodaniu wzorca, by otrzymać wykres
funkcji ∆𝒀 =
𝒀
𝟎
𝑪
∙ 𝑪
𝒘𝒛
(stężenie wynosi zatem 𝒄 =
𝒀
𝟎
𝒕𝒈 𝜶
)
- brak wpływu matrycy na efekt wzorcowania

A
n
ali
za in
strum
entaln
a
–
S.
Bartk
iew
icz
18
e) Wzorca wewnętrznego
- stosowana, gdy współczynnik m zmienia się w kolejnych pomiarach Y
- mierzy się dwa sygnały: dla analitu oraz dla wzorca nie będącego analitem
(Y
1
=m
1
c
1
, Y
2
=m
2
c
2
)
- współczynnik odpowiedzi wynosi: 𝑅 =
𝑚
1
𝑚
2
, po wielokrotnym dodawaniu wzorca można
wykreślić krzywą kalibracyjną w krórej R jest nachyleniem prostej
- WZORZEC: nie może być obecny w analizie, jego sygnał musi być oddzielony od sygnału
analitu, nie może reagować ze składnikami próbki
2. Statystyczna ocena wyników
a) Odchylenie standardowe
b) Średnie odchylenie standardowe
c) Wariancja
d) Przedział ufności
e) Test t-studenta
f) Krzywa Gaussa
Wyszukiwarka
Podobne podstrony:
Analiza matematycza opracowanie pytań
analiza sensoryczna opracowane pytania testowe poprawione
diagnozowanie - analiza bezrobocia, Opracowanie bezrobocia - ćwiczenia
analiza istrumentalna, spr fotom płom
Analiza matematyczna 2 - opracowane zagadnienia na egzamin, Wykłady - Studia matematyczno-informatyc
analiza istrumentalna kolo 2 id Nieznany (2)
opracowanie zad dobre!
analiza istrumentalna, spr interferencja
Analiza matematycza opracowanie pytań
Analiza matematyczna 2 i 3 opracowanie Michał Musielak
Analiza matematyczna 1 opracowanie Michał Musielak
JHP, Informacja naukowa i bibliotekoznastwo 2 semestr, Analiza i opracowaniw dokumentów, Analiza i o
Opracowania pytań na analizę instrumentalną
Nowe Grocholice3, MAGAZYN DO 2015, Nowe Grocholice - wersje maj 2014, opracowanie ng1, NG1 - ANALIZA
Analiza błędów Statystyczne opracowanie wyników pomiarów
ANALIZA bakoma, Ekonomia- wykłady, opracowania
Ikolo-opracowanie, biotechnologia Sem 5 Olsztyn, III rok, III rok BARDZO DOBRE !!!!
więcej podobnych podstron