
p r a k t y k a m e d y c z n a
przewodnik
l e k a r z a
36
Heterogenność lipoprotein
osocza jest czynnikiem od-
zwierciedlającym różnice w ich
biosyntezie, budowie i meta-
bolizmie. Mogą być one uwa-
runkowane genetycznie, lecz
częściej są pochodną diety,
stylu życia oraz wpływów me-
tabolicznych. Przedmiotem
licznych badań jest ustalenie,
czy analiza heterogenności
w obrębie frakcji lipoprotein
osocza ma wyższą wartość
predykcyjną w ocenie zagro-
żenia incydentami sercowo-
-naczyniowymi w stosunku do
tradycyjnych lipidowych czyn-
ników ryzyka, oraz czy jest
przydatna do monitorowania
terapii hipolipemizującej. Spo-
śród lipoprotein osocza naj-
większe zainteresowanie ba-
daczy wzbudza jedna z frakcji
lipoprotein o małej gęstości
(LDL) – tzw. małe gęste LDL.
S
Sttrru
uk
kttu
urra
a ii ffu
un
nk
kc
cjja
a
lliip
po
op
prro
otte
eiin
n o
os
so
oc
cz
za
a
Lipoproteiny osocza s¹ sfe-
rycznymi cz¹stkami zbudowany-
mi z hydrofobowego rdzenia, bo-
gatego w cholesterol i triglicery-
dy oraz z p³aszcza utworzonego
z apolipoprotein (bia³ka), fosfoli-
pidów i wolnego (niezestryfiko-
wanego) cholesterolu. Gêstoœæ
wzglêdna oraz œrednica cz¹stki
zale¿¹ od sk³adu chemicznego.
Im wiêksza zawartoœæ apolipo-
protein, tym mniejsza œrednica
cz¹stki (bia³ko wykazuje du¿y
stopieñ upakowania) oraz wy¿sza
gêstoœæ wzglêdna (bia³ko posiada
du¿¹ masê cz¹steczkow¹ w sto-
sunku do lipidów). Najwy¿sz¹ za-
wartoœci¹ apolipoprotein cechuj¹
siê lipoproteiny o du¿ej gêstoœci
(HDL), a najni¿sz¹ chylomikrony
(ryc. 1.).
Apolipoproteiny warunkuj¹ bio-
syntezê cz¹stek lipoprotein, umo¿-
liwiaj¹ transport lipidów w wod-
nym œrodowisku osocza, s¹ akty-
watorami i inhibitorami enzymów
zaanga¿owanych w metabolizm li-
poprotein osocza, a czêœæ z nich
pe³ni funkcje ligandów receptoro-
wych, dziêki czemu mo¿liwe jest
przedostawanie siê cholesterolu do
wnêtrza komórek. Spoœród apoli-
poprotein osocza kluczowe znacze-
nie odgrywa apoB-100 oraz apoA-
I. Podwy¿szone stê¿enie lipoprote-
in zawieraj¹cych apoB-100 oraz
obni¿one posiadaj¹cych apoA-I,
sprzyja progresji mia¿d¿ycy têtnic
oraz rozwojowi choroby niedo-
krwiennej serca.
Lipoproteiny
zawieraj¹ce
apoB-100 obejmuj¹ lipoproteiny
o bardzo ma³ej gêstoœci (VLDL)
i ich remnanty, lipoproteiny o po-
œredniej gêstoœci (IDL), LDL oraz
lipoproteinê (a). VLDL – syntety-
zowane w w¹trobie s¹ g³ównym
transporterem triglicerydów endo-
gennych w uk³adzie kr¹¿enia. Pod
wp³ywem ektoenzymu œródb³on-
kowego – lipazy lipoproteinowej
(LPL) dochodzi do hydrolizy tri-
glicerydów zawartych w VLDL
z uwolnieniem kwasów t³uszczo-
wych, które s¹ najistotniejszym
paliwem energetycznym w warun-
kach tlenowych, a ponadto s³u¿¹
do budowy t³uszczu zapasowego.
Prowadzi to do powstania rem-
nantów VLDL – cz¹stek o wy¿-
szej gêstoœci, znacznie ubo¿szych
w triglicerydy, a nastêpnie IDL,
bêd¹cych bezpoœrednim prekurso-
rem LDL. Wiêkszoœæ IDL jest wy-
chwytywana przez receptor dla
apoB/E obecny na powierzchni
hepatocytu, a ok. 30 proc. podle-
ga dzia³aniu lipazy w¹trobowej
M
Ma
ałłe
e g
gę
ęs
stte
e L
LD
DL
L
–
– w
wyyzzw
wa
an
niie
e tte
erra
ap
pe
eu
uttyyc
czzn
ne
e d
dlla
a s
stta
attyyn
n
Jan Gmiñski, Joanna Kopeæ
RRyycc.. 11..
HHeetteerrooggeennnnooœœææ
lliippoopprrootteeiinn
oossoocczzaa
p r a k t y k a m e d y c z n a
przewodnik
l e k a r z a
38
triglicerydowej (HL) zlokalizowa-
nej na œródb³onku zatok w¹trobo-
wych, przekszta³caj¹c siê do
LDL. G³ównym sk³adnikiem lipi-
dowym LDL jest cholesterol
w formie zestryfikowanej i wol-
nej, który za ich poœrednictwem
przedostaje siê do tkanek obwo-
dowych, s³u¿¹c tam m.in. do syn-
tezy b³on, prowitaminy D oraz
hormonów steroidowych. Apoli-
poproteina B-100 pokonuje drogê
od w¹troby, gdzie zostaje wbudo-
wana do VLDL, poprzez IDL
i LDL do tkanek obwodowych,
w których jej interakcja z recep-
torem dla apoB/E (receptor wyso-
kiego powinowactwa) warunkuje
prawid³owy klirens LDL. Ka¿da
cz¹stka lipoprotein wymienio-
nych frakcji zawiera jedn¹ cz¹-
steczkê apo-B100.
Podwy¿szone stê¿enie frakcji
LDL sprzyja infiltracji œciany na-
czyniowej przez te cz¹stki. Lipo-
proteiny LDL ulegaj¹ licznym mo-
dyfikacjom, m.in. oksydacji, glika-
cji, tiolacji oraz angiotensynizacji,
staj¹c siê tym samym ligandem dla
receptorów typu scavenger, zloka-
lizowanych na makrofagach. Gro-
madzenie cholesterolu w tych ko-
mórkach prowadzi do powstania
komórek piankowatych, sta³ego
elementu blaszki mia¿d¿ycowej.
H
He
ette
erro
og
ge
en
nn
no
oś
ść
ć ffrra
ak
kc
cjjii L
LD
DL
L
Rutynowe metody analitycz-
ne, obejmuj¹ce oznaczenia cho-
lesterolu ca³kowitego oraz we
frakcjach LDL i HDL, oznacze-
nia triglicerydów, czy nawet po-
szczególnych apolipoprotein nie
pozwalaj¹ na wgl¹d w hetero-
gennoœæ lipoprotein w obrêbie
poszczególnych frakcji. Wyma-
ga to zastosowania technik bar-
dzo wyrafinowanych, a przez to
kosztownych i niedostêpnych
nawet w wielu specjalistycz-
nych oœrodkach. Obejmuj¹ one,
m.in. ultrawirowanie analitycz-
ne, gradientow¹ elektroforezê
¿elow¹, immunopowinowactwo
oraz spektroskopiê j¹drowego re-
zonansu magnetycznego (NMR).
Metody te pozwalaj¹ na rozró¿-
nienie w obrêbie g³ównych klas
lipoprotein osocza podfrakcji,
ró¿ni¹cych siê w zale¿noœci od
zastosowanej techniki badawczej
– wielkoœci¹ lub gêstoœci¹ cz¹-
stek. Mimo niejednorodnej no-
menklatury zazwyczaj stosuje siê
podzia³ na podfrakcje o du¿ej,
poœredniej i ma³ej gêstoœci.
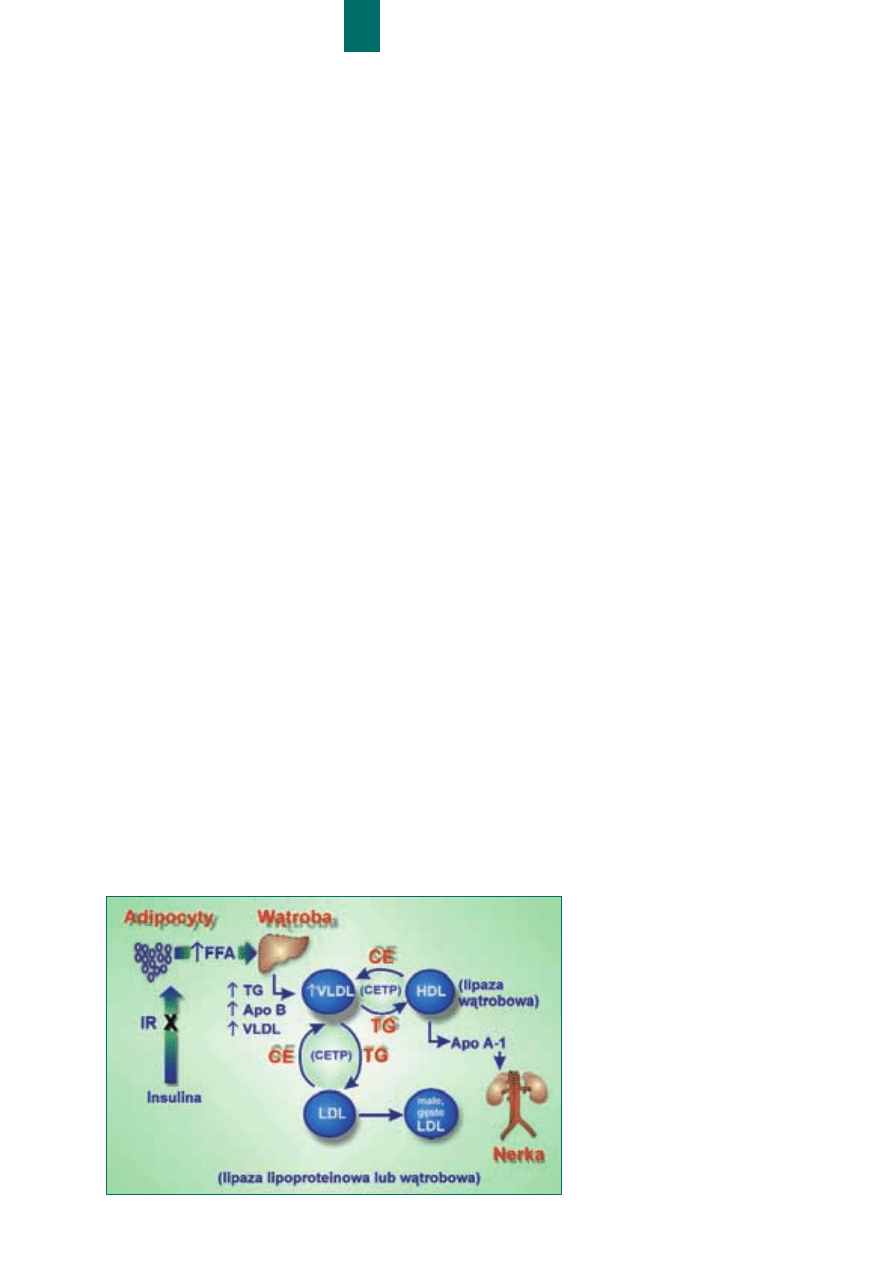
Kluczowe znaczenie dla pozna-
nia przyczyn heterogennoœci frak-
cji LDL ma zrozumienie wymia-
ny sk³adników lipidowych pomiê-
dzy
lipoproteinami
osocza.
Enzymem odpowiedzialnym za
wspomniany proces jest bia³ko
przenosz¹ce estry cholesterolu
(CETP). CETP przenosi estry cho-
lesterolu z cz¹stek HDL na
VLDL, z których poprzez IDL do-
staj¹ siê one do w¹troby, a cz¹stki
HDL zostaj¹ wzbogacone w trigli-
cerydy pochodz¹ce z VLDL.
Wzbogacone w triglicerydy HDL
staj¹ siê substratem dla lipazy w¹-
trobowej, która degraduj¹c je pro-
wadzi do obni¿enia stê¿enia HDL
w osoczu. Podobna wymiana ma
miejsce pomiêdzy LDL a VLDL.
Wzbogacone w estry cholesterolu
cz¹stki VLDL staj¹ siê bardziej
aterogenne, a wzrost zawartoœci
triglicerydów w LDL sprzyja ich
hydrolizie przez HL lub LPL
z wytworzeniem podfrakcji ma-
³ych gêstych LDL (ryc. 2.).
Stanem, w którym przedsta-
wiona kaskada zdarzeñ przyjmu-
je szczególnie niebezpieczne na-
silenie jest zespó³ metaboliczny,
a zw³aszcza jeden z jego sta³ych
elementów – insulinoopornoœæ.
Wskutek braku skutecznego ha-
mowania lipolizy wewn¹trz adi-
pocytów przez insulinê, dochodzi
do nasilonego uwalniania wol-
nych kwasów t³uszczowych
(FFA) do krwiobiegu. W przypad-
ku, gdy dotyczy to tkanki t³usz-
czowej zlokalizowanej w dorze-
czu ¿y³y wrotnej powódŸ wol-
nych kwasów t³uszczowych
zalewa hepatocyty, nasilaj¹c syn-
tezê triglicerydów endogennych,
wbudowywanych nastêpnie do
VLDL. Sprzyja temu anaboliczne
dzia³anie insuliny, która wzmaga
syntezê apo-B100. Nadprodukcja
cz¹stek VLDL, wykrywana w ru-
tynowym badaniu laboratoryjnym
jako hipertriglicerydemia, prowa-
dzi do wzrostu aktywnoœci CETP
z nastêpow¹ degradacj¹ HDL (ob-
ni¿enie stê¿enia cholesterolu
HDL) i wzrostem aterogennoœci
remnantów VLDL. Wzmo¿ona
wymiana sk³adników lipidowych
miêdzy VLDL i LDL, po³¹czona
z typowym dla zespo³u metabo-
licznego i cukrzycy typu 2 wzro-
stem aktywnoœci HL, warunkuje
wzrost tworzenia cz¹stek ma³ych
gêstych LDL.
Przedstawione w ogólnym za-
rysie mechanizmy odpowiedzial-
ne s¹ za wystêpowanie triady li-
pidowej – wzrostu stê¿enia trigli-
RRyycc.. 22
PPoow
wssttaaw
waanniiee
m
maa³³yycchh
ggêêssttyycchh LLDDLL

p r a k t y k a m e d y c z n a
przewodnik
l e k a r z a
39
cerydów, obni¿enia stê¿enia cho-
lesterolu frakcji HDL, wzrostu
zawartoœci ma³ych gêstych LDL
– charakteryzuj¹cej dyslipidemiê
cukrzycow¹ [1].
M
Ma
ałłe
e g
gę
ęs
stte
e L
LD
DL
L a
a c
ch
ho
orro
ob
ba
a
n
niie
ed
do
ok
krrw
wiie
en
nn
na
a s
se
errc
ca
a
Proaterogenne w³aœciwoœci ma-
³ych gêstych LDL by³y przedmio-
tem szeregu badañ eksperymental-
nych i klinicznych (ryc. 3.).
W badaniu AMORIS (Apoli-
poprotein-Related Mortality Risk
Study), obejmuj¹cym ponad 170
tys. Szwedów przez 5,5 roku wy-
kazano wy¿szoœæ oznaczeñ apoB
nad stê¿eniem cholesterolu LDL,
w stosunku do ryzyka wyst¹pie-
nia zawa³u miêœnia sercowego,
u obu p³ci i we wszystkich kate-
goriach wiekowych [2].
Szereg du¿ych badañ prospek-
tywnych wskazuje na istotn¹ ko-
relacjê dodatni¹ miêdzy wystê-
powaniem ma³ych gêstych LDL
a zapadalnoœci¹ na chorobê nie-
dokrwienn¹ serca. Obecnoœæ ma-
³ych gêstych LDL zwiêksza ry-
zyko choroby niedokrwiennej
serca nawet 7-krotnie, chocia¿
w wielu z tych badañ zale¿noœæ
ta nie by³a niezale¿na od innych
tradycyjnych czynników ryzyka
[3]. W dwóch spoœród wykona-
nych badañ ma³e gêste LDL po-
zostawa³y czynnikiem predyk-
cyjnym w stosunku do wystêpo-
wania choroby wieñcowej po
korekcji w stosunku do stê¿enia
triglicerydów i cholesterolu frak-
cji HDL. W innych badaniach
wykazano znamienne korzyœci
angiograficzne, zwi¹zane z re-
dukcj¹ stê¿enia ma³ych gêstych
LDL w osoczu krwi.
Niezale¿nie od zmian w profi-
lu lipidowym osocza podwy¿szo-
ne stê¿enie ma³ych gêstych LDL
zaburza funkcje œródb³onka na-
czyniowego, co zgodnie z teori¹
jednolitej odpowiedzi na uraz
mo¿e prowadziæ do inicjacji
i progresji aterogenezy [4].
Wg raportu National Choleste-
rol Education Program ATP III
obecnoœæ ma³ych gêstych LDL
w osoczu sprawia, ¿e ryzyko
wieñcowe wynikaj¹ce ze stê¿enia
cholesterolu frakcji LDL staje siê
niedoszacowane. Tak wiêc iloœæ
cz¹stek LDL mo¿e byæ lepszym
wskaŸnikiem potencja³u atero-
gennego w porównaniu ze stê¿e-
niem cholesterolu w tej frakcji.
Wobec braku dostêpnych i ta-
nich metod oceniaj¹cych stê¿enie
cz¹stek LDL (a nie cholesterolu
LDL) na uwagê zas³uguje przy-
datnoœæ oznaczania stê¿enia apo-
lipoproteiny B. Oznaczenie stê¿e-
nia apoB metod¹ immunoturbidy-
metryczn¹ jest relatywnie tanie,
dok³adne i powtarzalne. Jest to
lepszy wskaŸnik iloœci cz¹stek
LDL, ni¿ stê¿enie cholesterolu
frakcji LDL. Mo¿e byæ to rów-
nie¿ wartoœciowy wskaŸnik od-
powiedzi na terapiê statynami
u pacjentów z prawid³owym lub
granicznym stê¿eniem choleste-
rolu LDL, lecz du¿ym globalnym
ryzykiem wieñcowym.
NCEP ATP III rekomenduje
oznaczanie cholesterolu nie-
-HDL, którego stê¿enie jest œci-
œle skorelowane z zawartoœci¹
apoB. Cholesterol nie-HDL wy-
licza siê poprzez odjêcie stê¿enia
cholesterolu HDL od cholesterolu
ca³kowitego. Obejmuje on chole-
sterol zawarty we frakcjach
VLDL oraz LDL i jest istotnym
celem terapeutycznym u pacjen-
tów z hipertriglicerydemi¹ [5].
D
Dy
ys
slliip
piid
de
em
miia
a c
cu
uk
krrz
zy
yc
co
ow
wa
a
Przez dziesiêciolecia cukrzyca
uwa¿ana by³a za klasyczny czyn-
nik ryzyka choroby niedokrwien-
nej serca. Haffner i wsp. w wie-
loletnim badaniu prospektywnym
wykazali, ¿e ryzyko zgonu z po-
wodu choroby niedokrwiennej
serca w cukrzycy jest równowa¿-
ne temu, które wystêpuje po prze-
byciu zawa³u miêœnia sercowego
[6]. Raport National Cholesterol
Education Program ATP III kla-
syfikuje cukrzycê jako równo-
wa¿nik choroby niedokrwiennej
serca, a nie czynnik ryzyka. Cho-
cia¿ priorytetem terapii hipolipo-
mizuj¹cej jest uzyskanie stê¿enia
cholesterolu frakcji LDL <100
mg%, na wzrost ryzyka wieñco-
wego wp³ywaj¹ równie¿ inne
czynniki ryzyka okreœlonych po-
wy¿ej jako triada lipidowa. Obok
zmian w stylu ¿ycia – wzrost ak-
tywnoœci fizycznej, modyfikacje
dietetyczne, obni¿enie masy cia³a
– korekcja zaburzeñ lipidowych
wymaga leczenia farmakologicz-
nego. U osób z hipertrigliceryde-
mi¹ dodatkowym celem leczenia
hipolipemizuj¹cego jest, obok re-
dukcji cholesterolu LDL, obni¿e-
nie stê¿enia cholesterolu nie-
-HDL <130 mg%, które jak
wspomniano powy¿ej koreluje
z zawartoœci¹ apoB [5].
Wczeœniejsze rekomendacje
ogniskowa³y siê na redukcji stê-
¿enia triglicerydów oraz wzro-
œcie stê¿enia cholesterolu frakcji
HDL jako celach terapii hipoli-
pemizuj¹cej. Odzwierciedleniem
tego by³y zalecenia dotycz¹ce
stosowania fibratów i kwasu ni-
kotynowego w dyslipidemii cu-
krzycowej, które jednak nie by³y
RRyycc.. 33..
PPrrooaatteerrooggeennnnee
w
w³³aaœœcciiw
wooœœccii
m
maa³³yycchh
ggêêssttyycchh LLDDLL
â
â ma³e powinowactwo do receptora
â
â d³ugi okres pó³trwania
â
â intensywne naciekanie œciany naczyniowej
â
â silne wi¹zanie z proteoglikanami
â
â przyspieszona oksydacja
â
â intensywny wychwyt przez makrofagi

p r a k t y k a m e d y c z n a
przewodnik
l e k a r z a
40
poparte dowodami p³yn¹cymi
z badañ randomizowanych [7].
Zalecenia American Heart As-
sociation pozycjonuj¹ fibraty do
stosowania w ciê¿kiej hipertrigli-
cerydemii (TG >500 mg%), ce-
lem prewencji ostrego zapalenia
trzustki oraz w dysbetalipoprote-
inemii [8].
Wg aktualnych wytycznych
NCEP ATP III (5) oraz American
Diabetes Association (ADA) [9]
terapi¹ I linii dla redukcji stê¿e-
nia cholesterolu LDL s¹ inhibito-
ry reduktazy HMG-CoA, czyli
statyny. W szeregu wieloletnich
badañ randomizowanych wyka-
zano obni¿enie umieralnoœci
ogólnej oraz z przyczyn sercowo-
-naczyniowych, zarówno w pre-
wencji pierwotnej, jak i wtórnej
choroby niedokrwiennej serca.
Chocia¿ jak dotychczas nie wy-
konano badania dedykowanego
wp³ywowi terapii hipolipemizu-
j¹cej na twarde punkty koñcowe
u pacjentów z cukrzyc¹ typu 2, to
w poszczególnych badaniach (4S
– simwastatyna, CARE – prawa-
statyna, HPS – simwastatyna,
LIPS – fluwastatyna) chorzy na
cukrzycê byli reprezentowani.
Wykazano podobn¹ lub wy¿sz¹
skutecznoœæ kliniczn¹ statyn
u diabetyków w porównaniu
z populacj¹ generaln¹ [10].
Statyny poprzez wzrost eks-
presji receptora dla apoB/E w ko-
mórce mi¹¿szowej w¹troby obni-
¿aj¹ nie tylko stê¿enie choleste-
rolu LDL, ale tak¿e triglicerydów
zawartych w aterogennych rem-
nantach VLDL i IDL. Powoduj¹
tak¿e wzrost stê¿enia cholestero-
lu frakcji HDL, zw³aszcza u osób
z niskim wyjœciowym stê¿eniem
tego parametru oraz z hipertrigli-
cerydemi¹.
Przeciwmia¿d¿ycowe dzia³a-
nie statyn wykracza znacznie po-
za wp³ywy lipidowe. Efekty te
okreœlane jako dzia³ania plejotro-
powe obejmuj¹, m.in. ochronny
wp³yw na œródb³onek naczynio-
wy, dzia³anie antyproliferacyjne
w stosunku do komórek miêœni
g³adkich, zmniejszenie nacieku
monocytów w œcianie naczynio-
wej, dzia³anie antyoksydacyjne,
dzia³anie przeciwzapalne, zmniej-
szenie ekspresji receptora AT1
dla angiotensyny II [11].
Jeœli pocz¹tkowe leczenie sta-
tyn¹ nie prowadzi do osi¹gniêcia
w ci¹gu 6 tyg. docelowego stê¿e-
nia cholesterolu frakcji LDL, ko-
nieczne jest zwiêkszenie jej daw-
ki. Innym podejœciem rekomen-
dowanym przez ADA
jest
zastosowanie terapii skojarzonej,
polegaj¹cej na do³¹czeniu fibratu
do statyny. Terapia ta oparta jest
na przes³ankach patofizjologicz-
nych. Jak dotychczas bowiem
brak jest badañ pokazuj¹cych
skutecznoœæ kliniczn¹ (twarde
punkty koñcowe) takiego postê-
powania. OdpowiedŸ dadz¹ byæ
mo¿e wyniki trwaj¹cego obecnie
badania ACCORD, poœwiêcone-
go wp³ywowi dodania fibratu do
wczeœniej zastosowanej statyny
u chorych na cukrzycê [10].
Terapia skojarzona statyn¹
z fibratem wzbudza czêsto pyta-
nia o ryzyko wyst¹pienia miopa-
tii. Szczególnie rekomendowa-
nym po³¹czeniem ze wzglêdu na
skutecznoœæ hipolipemizuj¹c¹
i bezpieczeñstwo jest do³¹czenie
fibratu do fluwastatyny. Kombi-
nacja taka nie powoduje wzrostu
aktywnoœci CPK, nawet u osób
w wieku podesz³ym, u których
czêstoœæ dyslipidemii, zw³aszcza
dyslipidemii cukrzycowej jest
wysoka.
S
Stta
atty
yn
ny
y a
a m
ma
ałłe
e g
gę
ęs
stte
e L
LD
DL
L
Spoœród leków hipolipemizu-
j¹cych dobrze znany jest wp³yw
fibratów i kwasu nikotynowego
na zmiany dystrybucji w obrêbie
podfrakcji LDL.
W
œwietle uznania przez
NCEP ATP III i ADA statyn, ja-
ko leków hipolipemizuj¹cych I li-
nii w dyslipidemii cukrzycowej,
zrozumia³e zainteresowanie mu-
si wzbudzaæ ich wp³yw na stê¿e-
nie ma³ych gêstych LDL, które
bêd¹c elementem triady lipidowej
wi¹¿¹ siê ze zwiêkszonym ryzy-
kiem incydentów sercowo-naczy-
niowych.
W przeprowadzonych bada-
niach u pacjentów leczonych sta-
tynami, podfrakcje LDL ocenia-
ne by³y technik¹ gradientowej
elektroforezy ¿elowej (GGE) lub
ultrawirowania. GGE daje jedy-
nie przybli¿ony wgl¹d w wiel-
koœæ cz¹stek LDL. W ¿adnym
z dotychczas wykonanych t¹
technik¹ badañ nie stwierdzono
zmian w stê¿eniu podfrakcji LDL
w przebiegu leczenia statynami.
W czêœci badañ, gdzie do oceny
dystrybucji podfrakcji LDL wy-
korzystywano technikê ultrawiro-
wania, wykazano korzystny
wp³yw statyn na obni¿enie stê¿e-
nia ma³ych gêstych LDL, pod-
czas gdy inni badacze takich
efektów nie uzyskali [12].
Podstawowymi mankamenta-
mi dotychczasowych badañ by³o
stosowanie techniki obarczonej
du¿ym b³êdem (GGE), brak po-
dzia³u grupy badanej wg wyjœcio-
wego stê¿enia ma³ych gêstych
LDL oraz brak grupy otrzymuj¹-
cej placebo w czêœci z nich. Pod-
sumowuj¹c, wp³yw statyn na stê-
¿enie ma³ych gêstych LDL pozo-
staje kwesti¹ otwart¹.
Pojawienie siê na rynku far-
maceutycznym pierwszej statyny
o przed³u¿onym dzia³aniu – flu-
wastatyny XL – ponownie zogni-
skowa³o uwagê badaczy na kwe-
stii wp³ywu statyn na stê¿enie
ma³ych gêstych LDL. Fluwasta-
tyna XL dziêki powolnemu
wch³anianiu z przewodu pokar-
mowego, z jednej strony zapew-
nia wielogodzinn¹ kontrolê nad
komórk¹ w¹trobow¹, a z drugiej
osi¹ga bardzo niskie stê¿enie sys-
temowe, redukuj¹c do absolutne-
go minimum ryzyko wyst¹pienia
miopatii.
Efektem d³ugofalowego wp³y-
wu na hepatocyt jest trójkierun-
kowe dzia³anie fluwastatyny XL,
prowadz¹ce do redukcji choleste-
rolu frakcji LDL o 38 proc., tri-
glicerydów o 31 proc. oraz wzro-

p r a k t y k a m e d y c z n a
przewodnik
l e k a r z a
41
stu stê¿enia cholesterolu frakcji
HDL o 21 proc. [13]. Istotnym
czynnikiem decyduj¹cym o po-
wstawaniu ma³ych gêstych LDL
jest wzmo¿ona synteza VLDL
w w¹trobie, znajduj¹ca odbicie
w rutynowym badaniu laborato-
ryjnym w postaci hipertriglicery-
demii. Fluwastatyna XL istotnie
obni¿aj¹c stê¿enie triglicerydów
mo¿e prowadziæ wiêc do reduk-
cji stê¿enia aterogennych ma³ych
gêstych LDL.
Wychodz¹c z
powy¿szych
przes³anek, Winkler i wsp. [14]
zbadali wp³yw 8-tygodniowego
leczenia fluwastatyn¹ XL metod¹
podwójnie œlepej próby kontrolo-
wanej placebo na stê¿enie pod-
frakcji LDL u pacjentów z dysli-
pidemi¹ cukrzycow¹. Badaniem
objêto 89 pacjentów obu p³ci
z cukrzyc¹ typu 2 i dyslipidemi¹.
Œrednie stê¿enie HbA1c wynosi-
³o 7,1 proc., cholesterolu LDL
3,37 mM, a triglicerydów 2,41
mM. Lipoproteiny osocza izolo-
wane by³y technik¹ sekwencyjne-
go preparatywnego ultrawirowa-
nia, a w obrêbie frakcji LDL me-
tod¹ ultrawirowania w gradiencie
gêstoœci wyodrêbniono 6 podfrak-
cji. Po 4-tygodniowym stosowa-
niu diety wg re¿imu NCEP, pa-
cjentów randomizowano do stoso-
wania fluwastatyny XL w dawce
80 mg przed snem lub placebo.
U 79 proc. pacjentów z cu-
krzyc¹ typu 2 stwierdzono pod-
wy¿szone wartoœci stê¿enia ma-
³ych gêstych LDL. W ci¹gu 8 tyg.
leczenia fluwastatyn¹ XL dosz³o
do 29-procentowej redukcji cho-
lesterolu LDL oraz 26-procento-
wego obni¿enia stê¿enia apoB.
Wœród pacjentów z wyjœciowym
stê¿eniem TG >300 mg% zaob-
serwowano ponadwudziestoczte-
roprocentow¹ redukcjê stê¿enia
triglicerydów.
Interesuj¹ce wyniki uzyskano
w grupie pacjentów z wyjœciowo
podwy¿szonym stê¿eniem ma-
³ych gêstych LDL w grupie otrzy-
muj¹cej fluwastatynê XL. Stwier-
dzono nie tylko 28-procentow¹
redukcjê stê¿enia ma³ych gêstych
LDL, lecz równie¿ statystycznie
znamienne obni¿enie frakcji
o poœredniej gêstoœci. Mo¿na
przypuszczaæ,
¿e
korzystny
wp³yw fluwastatyny XL w tej
grupie wi¹¿e siê ze zmniejsze-
niem stê¿enia triglicerydów, któ-
rych rola patogenetyczna w two-
rzeniu ma³ych gêstych LDL jest
dobrze poznana.
W grupie pacjentów z wyj-
œciowo niskim stê¿eniem ma³ych
gêstych LDL fluwastatyna XL
spowodowa³a znamienne obni¿e-
nie flotuj¹cych lipoprotein zawie-
raj¹cych apoB (VLDL, IDL oraz
du¿ych lekkich LDL), pozostaj¹c
bez wp³ywu na ma³e gêste LDL.
Wydaje siê, ¿e g³ówny mecha-
nizm dzia³ania fluwastatyny w tej
grupie pacjentów polega na wzro-
œcie wychwytu lipoprotein po-
przez receptor dla apoB/E na po-
wierzchni hepatocytu.
Reasumuj¹c, fluwastatyna XL
wywiera efekt na dystrybucjê
frakcji LDL w dwojaki sposób.
U pacjentów z dyslipidemi¹ cu-
krzycow¹ i podwy¿szonym stê¿e-
niem ma³ych gêstych LDL, re-
dukcja ich zawartoœci w osoczu
zachodzi poprzez obni¿enie stê-
¿enia triglicerydów, podczas gdy
w grupie z lekkimi i poœrednimi
LDL ich stê¿enie ulega redukcji
poprzez wzrost klirensu za po-
œrednictwem
receptora
dla
apoB/E. Przeciwmia¿d¿ycowe
w³aœciwoœci fluwastatyny u cho-
rych na cukrzycê typu 2 mog¹
wiêc znacznie wykraczaæ poza
wyliczone z redukcji stê¿enia
cholesterolu LDL i nie-HDL.
Piœmiennictwo
1. Taskinen M-R. Diabetic dyslipidaemia:
from basic research to clinical practi-
ce. Diabetologia 2003, 46, 733-49.
2. Walldius G, Jungner I, Holme I,
Aastveit AH, Kolar W, Steiner E.
High apolipoprotein B, low apolipo-
protein A-I, and improvement in the
prediction of fatal myocardial infarc-
tion (AMORIS study): a prospective
study. Lancet 2001, 358, 2026-33.
3. Gardner CD, Fortmann SP, Krauss
RM. Association of small low-density
lipoprotein particles with the incidence
of coronary artery disease in men and
women. JAMA 1996, 276, 875-81.
4. Vakkilainen J, Makimattila S, Seppa-
la-Lindroos A, Vehkavaara S, Lah-
denpera S, Froop PH, Taskinen MR,
Yki-Jarvinen H. Endothelial dysfunc-
tion in men with small LDL particles.
Circulation 2000, 102, 716-21.
5. Third Report of the National Chole-
sterol Education Program (NCEP)
Expert Panel on Detection, Evalu-
ation, and Treatment of High Blood
RRyycc.. 44..
CCeecchhyy
ffaarrm
maakkoollooggiicczznnee
ii kklliinniicczznnee
fflluuw
waassttaattyynnyy
â
â poœrednia rozpuszczalnoœæ
â
â w³aœciwoœci antyoksydacyjne
â
â odmienny metabolizm w¹trobowy
â
â skutecznie redukuje ma³e gêste LDL
â
â bezpieczna w chorobach nerek
â
â bezpieczna w skojarzeniu z fibratami
â
â spowalnia progresjê mia¿d¿ycy (LCAS)
â
â skuteczna u osób z niskim HDL (LCAS)
â
â skuteczna w zaawansowanej mia¿d¿ycy têtnic
(LiSA)
â
â skuteczna po PTCA (FLARE, LIPS)

p r a k t y k a m e d y c z n a
przewodnik
l e k a r z a
42
Cholesterol in Adults (Adult Treat-
ment Panel III) NIH Publication No.
02-5215.
6. Haffner SM, Lehto S, Ronnemaa T.
Mortality from coronary heart diseases
in subjects with type 2 diabetes and in
nondiabetic subjects with and without
prior myocardial infarction. N Engl J
Med 1998, 339, 229-34.
7. American Diabetes Association: Detec-
tion and management of lipid disorders
in diabetes (Consensus Statement).
Diabetes Care 1993, 16, 828-34.
8. ATP III Final Report. Circulation
2002, 106, 3303.
9. American Diabetes Association. Ma-
nagement of Dyslipidemia in Adults
with Diabetes. Diabetes Care 2003,
26 Suppl. 1, S83-S86.
10. Haffner SM, Goldberg RB. New stra-
tegies for the treatment of diabetic
dyslipidemia. Diabetes Care 2002,
25, 1237-9.
11. Takemoto M, Liao JK. Pleiotropic ef-
fects of 3-hydroxy-3-methylglutaryl
coenzyme A reductase inhibitors. Ar-
teriosclerosis Thrombosis and Vascu-
lar Biology 2001, 21, 1712-8.
12. Marz W, Scharnagl H, Abletshauser
C, Hoffmann MM, Berg A, Keul J,
Wieland H, Baumstark MW. Fluva-
statin lowers atherogenic dense low-
-density lipoproteins in postmeno-
pausal women with the atherogenic
lipoprotein phenotype. Circulation
2001, 103, 1942-8.
13. Ballantyne CM, Pazzucconi F, Pinto
X, Reckless JP, Stein E, McKenny J,
Bortolini M, Chiang YT. Efficacy
and tolerability of fluvastatin exten-
ded-release delivery system: a pooled
analysis. Clin Ther 2001, 23, 177-92.
14. Winkler K, Abletshauser C, Hoff-
mann MM, Friedrich I, Baumstark
MW, Wieland H, Marz W. Effect of
fluvastatin slow-release on low den-
sity lipoprotein (LDL) subfractions in
patients with type 2 diabetes melli-
tus: baseline LDL profile determines
specific mode of action. J Clin Endo-
crinol Metab 2002, 87, 5485-90.
prof. dr hab. med. Jan Gmiñski
mgr Joanna Kopeæ
Katedra i Zak³ad Biochemii
Doœwiadczalnej i Klinicznej
Wydzia³u Lekarskiego w Katowicach
Œl¹skiej Akademii Medycznej
Wyszukiwarka
Podobne podstrony:
Abolicja podatkowa id 50334 Nieznany (2)
4 LIDER MENEDZER id 37733 Nieznany (2)
katechezy MB id 233498 Nieznany
metro sciaga id 296943 Nieznany
perf id 354744 Nieznany
interbase id 92028 Nieznany
Mbaku id 289860 Nieznany
Probiotyki antybiotyki id 66316 Nieznany
miedziowanie cz 2 id 113259 Nieznany
LTC1729 id 273494 Nieznany
D11B7AOver0400 id 130434 Nieznany
analiza ryzyka bio id 61320 Nieznany
pedagogika ogolna id 353595 Nieznany
Misc3 id 302777 Nieznany
cw med 5 id 122239 Nieznany
D20031152Lj id 130579 Nieznany
mechanika 3 id 290735 Nieznany
więcej podobnych podstron