
1. Uszkodzenie komórki, procesy adaptacyjne, śmierć komórki
2. Zapalenie ostre i przewlekłe
3. Naprawa tkanek: regeneracja i włóknienie
4. Zaburzenia hemodynamiczne, zakrzepica, wstrząs
5. Choroby układu odpornościowego
6. Nowotwory
7. Choroby uwarunkowane genetycznie i choroby wieku dziecięcego
8. Choroby środowiskowe
9. Patologia ogólne chorób zakaźnych
10. Naczynia
11. Serce
12. Układ krwionośny i limfatyczny
13. Płuca i górne drogi oddechowe
14. Nerka i drogi wyprowadzające mocz
15. Jama ustna i przewód pokarmowy
16. Wątroba i drogi żółciowe
17. Trzustka
18. Narządy płciowe męskie
19. Narządy płciowe żeńskie i pierś
20. Układ dokrewny
21. Układ mięśniowo-szkieletowy
22. Skóra
23. Układ nerwowy
USZKODZENIE KOMÓRKI, PROCESY ADAPTACYJNE I ŚMIERĆ KOMÓRKI
1. Rodzaje uszkodzeń komórki
Komórki mogą adaptować się do fizjologicznych lub patologicznych bodźców, dążąc do zachowania
stanu równowagi, co umożliwia im przeżycie. Podstawowe odpowiedzi adaptacyjne to :
•
Zanik (atrophia)
•
Przerost (hypertrophia)
•
Rozrost ( hyperplasia)
•
Metaplazja
Po przekroczeniu zdolności adaptacyjnych następuje uszkodzenie komórki ( jest uszkodzenie
odwracalne oraz nieodwracalne prowadzące do śmierci komórki)
Schematy śmierci komórki:
- MARTWICA (najczęściej skrzepowa) następuje w efekcie niedokrwienia lub zadziałania
substancji toksycznych. Charakterystyczne- obrzęk komórek, denaturacja białek, rozpad
organelli. Śmierć dużej liczby komórek może prowadzić do niewydolności narządu.
- APOPTOZA- efekt uruchomienia programu „komórkowego samobójstwa”. Obumarłe w ten
sposób komórki usuwane są z minimalnym uszkodzeniem sąsiadujących tkanek. Występuje gdy
konieczne jest usunięcie zbędnych tkanek (np. w rozwoju zarodkowym) i w stanach
patologicznych ( np. zmiany powstałe w przebiegu mutacji).
2. Przyczyny uszkodzeń komórki
- NIEDOBÓR TLENU- hipoksja- nie mylić z niedokrwieniem, które wprawdzie jest najczęstszą
przyczyną niedotlenienia, ale nie jedyną. Poza tym hipoksja występuje w:
Stanach niedostatecznego utlenowania krwi (zapalenie płuc)
Zmniejszeniu zdolności krwi do przenoszenia tlenu (niedokrwistość, zatrucie CO)
- CZYNNIKI CHEMICZNE- Inie tylko znane subst.chem. (azbest, środki owadobójcze etc) W dużych
stężeniach nawet glukoza i sól mogą być szkodliwe (bo zmiana osmotyczności środowiska).
Również wysokie ciśnienie parcjalne tlenu może być toksyczne. Leki też.
- CZYNNIKI ZAKAŹNE- wszystko- pasożyty, bakterie, wirusy, grzyby
- REAKCJE IMMUNOLOGICZNE
- DEFEKTY GENETYCZNE
- ZABURZENIA ODŻYWIANIA- niskobiałkowa i n niskoenergetyczna dieta ludzi Trzeciego Świata,
niedobory witamin, nadmierne spożycie pokarmów (otyłość otyłość cukrzycy), dieta
miażdżycorodna.
- CZYNNIKI FIZYCZNE- uraz, wysoka temperatura promieniowanie, porażenie prądem, nagłe zmiany
ciśnienia atm.
1

- STARZENIE SIĘ- powtarzające się urazy prowadzą do zwyrodnienia tkanek. Starzenie prowadzi do
upośledzenia zdolności replikacji i naprawy komórek i tkanek. Powoduje to zmniejszenie
odpowiedzi na bodźce i urazy zewnętrzne, prowadząc do śmierci.
3. Mechanizm uszkodzenia komórki
- odpowiedź komórki na uszkodzenie zależy od rodzaju uszkodzenia, czasu trwania i nasilenia
bodźca.
- skutki zadziałania bodźca zależą od rodzaju, stanu, zdolności do adaptacji i genetycznie
uwarunkowanych właściwości uszkodzonej komórki, np.
mięsień poprz.prążkowany wytrzymuje niedokrwienie przez 2- 3 godz, a martwica mięśnia
sercowego następuje po 20- 3- minutach.
lepiej radzą sobie dobrze odżywione komórki np. bogate w glikogen hepatocyty
lepiej tez mają te, które mają genetycznie uwarunkowaną zdolność wytwarzania wielu enzymów
rozkładających szkodliwe czynniki powstałe w niedotlenieniu.
- CZTERY WRAŻLIWE UKŁADY WEWNĄTRZKOMÓRKOWE:
1) integralność błony kom. (homeostaza osmotyczna i jonowa)
2) produkcja ATP
3) synteza białek
4) integralność aparatu genetycznego
- ustanie czynności komórki następuje dużo wcześniej niż jej śmierć,
np. komórki serca tracą zdolność kurczenia po 1-2 min od niedokrwienia, a obumierają po 20- 30
minutach. Wewnętrzne zmiany w komórkach są widoczne jeszcze później, np. w sercu pod
mikroskopem świetlnym po 6-12 godz a w elektr.po 2- 3 godz
•
Ogólne mechanizmy biochemiczne
Wyczerpanie ATP- zatrzymanie szlaków metabolicznych, utrata osmolarności, transportu,
syntezy białek
Brak tlenu/ powstanie reaktywnych związków tlenu
utrata równowagi wapniowej- normalnie stęż Ca w komórce jest 10 000 razy mniejsze niż na
zewnątrz. Niedokrwienie lub toksyny prowadzą do napływu wapnia do cytozolu lub
uwolnienia go z mitochondriów itp. W ten sposób uczynniane są fosfolipazy (uszkadzają
błonę), proteaz (białka), ATP-az i ednonukleaz.
zaburzenia przepuszczalności błony- bezpośrednio przez toksyny bakt., białka wirusowe,
dopełniacz, limfocyty Tc itp. Mogą mieć też charakter wtórny (brak ATP, uczynnienie Ca-
zależnych fosfolipaz)
uszkodzenie mitochondriów- wzrost stęż. Wapnia, stres oksydacu=yjny, produkty rozpadu
tłuszczów prowadzą do powstania kanałow w wewn. Błonie mitochondrialnej, oraz
wyciekania cytochromu c (który aktywuje apoptozę komórki).
•
Uszkodzenie wywołane niedokrwieniem i niedotlenieniem
niedokrwienie uszkadza tkanki szybciej niż niedotlenienie, ponieważ w niedotlenieniu mimo iż
nie ma tlenu to komórka może wytwarzać energię w mechanizmie beztlenowym, a w
niedokrwieniu nie ma dostarczania zarówno tlenu jak i substancji potrzebnych do metabolizmu
beztlenowego. Nie ma też wypłukiwania szkodliwych metabolitów przez krążącą krew.
Podstawowym procesem, na jaki wpływa niedotlenienie jest fosforyzacja oksydacyjna (spadek
produkcji ATP). Skutki:
- spadek aktywności pompy sodowej- nagromadzenie sodu, utrata potasu- ostry obrzęk
komórki (gromadzenie innych metabolitów wzmaga obrzęk)
- wzrost glikolizy beztlenowej- spadek ATP, wzrost AMP który aktywuje fosfofruktokinazę. F.
prowadzi do wytwarzania ATP z glikogenu. To prowadzi do gromadzenia się kwasu mlekowego
mlekowego fosforanów nieorganiczny, przez co SPADA pH.
Jeśli niedokrwienie będzie się utrzymywać, dojdzie do coraz większych zaburzeń ( a w
konsekwencji do nieodwracalnego uszkodzenia), jeśli dopływ tlenu powróci to wszystkie
zaburzenia się cofną.
•
Uszkodzenia związane z niedokrwieniem i reperfuzją
nie zawsze przywrócenie krążenia jest korzystne i powoduje cofnięcie zaburzeń. Czasem może
spowodować pogłębienie uszkodzenia. Mechanizmy tego procesu:
- w wyniku porotu krążenia następuje wzrost stężenia wapnia w uszkodzonych omórkach, gdy
nie są one jeszcze zdolne do kontroli jonowej.
- reperfuzja uszkodzonych komórek prowadzi do napływu kom órek zapalnych, wydzielających
wolne rodzniki
2

-nie w pełni zdrowe mitochondria nie potrafią całkowicie redukować tlenu, wytwarzają więc
wolne rodniki. Uszkodzone komórki nie mają wystarczających mechanizmów obrony przed
nimi.
•
Uszkodzenie przez wolne rodniki
-peroksydacja lipidów błonowych. Prowadzi do powstania reaktywnych, niestabilnych
nadtlenków, co zapoczątkowuje reakcją autokatalityczną
-fragmentacja DNA. Wolne rodniki łączą się z wyminą prowadząc albo do śmierci albo do
przemiany złośliwej.
-powstanie wiązań krzyżowych krzyżowych białkach. Przyspiesza to ich degradację i utratę
zdolności enzymatycznej.
ANTYOKSYDANTY: katalaza, glutation, dysmutaza, witaminy A,C,E, beta karoten
•
uszkodzenie chemiczne
- działanie bezpośrednie- wiązanie ze składnikiem molekularnym lub organellą komórkową (np.
chlorek rtęci)
- nieaktywne subst chemiczne zostają przetworzone do aktywnych metabolitów. Najczęściej
odpowiedzialny jest cytP450 (np. czterochlorek węgla CCl4- środek czyszczący)
4.
Adaptacja komórki do uszkodzenia
a) adaptacja fizjologiczna- odp komórki na stymulację hormonalną lub mediatorami chemicznymi
(np. powiększenie gruczołow piersiowych i laktacja w ciąży)
b) adaptacja patologiczna- opiera się na tym samym, tylko że tu komórka dopasowuje się do
niekorzystnych warunków żeby przetrwać.
Mechanizmy adaptacyjne dzielimy na „w górę” lub „w dół” (zwiększona lub zmniejszona ilość
receptorów)
Adaptacja komórki może się wiązać z produkcją białek (np. białka szoku termicznego, synteza kolagenu
w przewlekłych zapaleniach, włóknienie itp.)
•
Zmiany adaptacyjne dotyczące wzrostu i różnicowania komórek:
1) ZANIK- atrofia
- Jest to zmniejszenie wymiarów komórki, spowodowane utratą substancji komórkowej.
- jeśli dotyczy wielu komórek może prowadzić do zmniejszenia całego narządu
- przyczyny: zmniejszenie obciążenia, odnerwieni, zmniejszony dopływ krwi, nieodpowiednie
odżywienie, utrata stymulacji hormonalnej, starzenie
- na poziomie komórkowym jest to zmniejszenie komórki do wymiaru który umożliwia
jeszcze przeżycie
-następuje redukcja elementów strukturalnych komórki
-kluczowa rola należy do regulacji rozpadu białek (lizosomy, szlak ubikwityno-
proteasomowy, wakuole autofagocytarne)
2) PRZEROST- hipertrofia
- Jest to zwiększenie wymiarów komórek, a co za tym idzie- całych narządów.
- fizjologiczny i patologiczny
- w wyniku zapotrzebowania funkcjonalnego lub stymulacji hormonalnej
- przerost i rozrost mogą wystąpić jednocześnie (macica w ciąży)
- patologia- serce w nadciśnieniu, przerost kardiomiocytów po zawale (kompensacja
obumarłych komórek)
- komórki poprzecznie prążkowane mogą reagować tylko przerostem (bo są dojrzałe i nie
mają zdolności podziału)
3) ROZROST- hiperplazja
- jest to zwiększenie liczby komórek narządu lub tkanki.
- rozrost fizjologiczny dzieli się na R.HORMONALNY- sutek w ciąży i R.KOMPENSACYJNY po
usunięciu lub uszkodzeniu narządu (np po resekcji wątroby dochodzi do odbudowania
brakującej części narządu), lub gojenie ran.
- rozrost patologiczny jest w większości spowodowany nadmierną stymulacją przez hormony
lub czynniki wzrostu.
4) METAPLAZJA
- jest to odwracalna zmiana jednego typu dojrzałych komórek (nabłonkowych lub
mezenchymalnych) w inny dojrzały typ, który lepiej toleruje niekorzystne warunki.
- np. pojawienie się nabłonka płaskiego w miejsce oddechowego oddechowego palaczy
(takąmetaplazję może też powodować niedobór wit A)
3

- nowy nabłonek jest trwalszy ale oznacza utratę ważnych elementów obronnych (np.
wydzielanie śluzu)
- utrzymujące się czynniki, które wywołały metaplazję mogą prowadzić do powstania
nowotworu w nabłonku zmienionym metaplastycznie.
•
Rodzaje subkomórkowej odpowiedzi na uszkodzenie:
a)katabolizm lizosomalny (hetero- i autofagocytoza)
-heterofagocytoza- fago i pinocytoza (granulocyty obojętnochłonne i makrofagi)
-autofagocytoza (usuwanie starych organelli i remodeling komórki)
b) przerost SER
c) zmiany w mitochondriach (wzrost ilości w przeroście, zmniejszenie w zaniku, mogą rosnąć
do dużych rozmiarów rozmiarów hepatocytach u osób niedożywionych i alkoholików,
miopatie mitochondrialne- duża liczba dużych mitoochondriów z nieprawidłowymi
grzebieniami)
d) nieprawidłowa budowa cytoszkieletu (zaburzenia organizacji mikrotubul mogą być
przyczyną niepłodności, oraz przewlekłych zapaleń dróg oddechowych- zesp. Kartagenera.
Mikrotubule są potrzebe do migracji leukocytów leukocytów fagocytozy. Tworzą też
wrzeciono podziałowe.
d) białka wstrząsu termicznego (HSP)- aktywowane są przez bodziec uszkadzający i
przywracają białkom prawidłowy kształt, dzięki czemu mogą one odzyskać swoją funkcję.
Jeśli białku nie zostanie przywrócony prawidłowy kształt to jest ono niszczone w
proteasomie.
•
Złogi wewnątrzkomórkowe
1) stłuszczenie (steatosis)- każde nagromadzenie TAG w obrębie komórek podścieliska
- jest wyznacznikiem odwracalnego uszkodzenia
- może pojawić się w sąsiedztwie komórek, które uległy martwicy
- najczęściej dotyczy wątroby (też w sercu, mięśniach szkieletowych, nerce innych)
- może być pod wpływem toksyn, w niedożywieniu, niedoborze białek, otyłości, niedotlenieniu,
ALKOHOL!
- morfologicznie- obecność wakuol w komórkach podścieliska; występuje głównie w wątrobie
wątrobie i w sercu.
W wątrobie we wczesnym stłuszczeniu małe okołojądrowe wakuole (makro niewidoczne); w
późnym wakuole zlewają się spychając jądro na brzeg komórki (makro widoczne żółte
zabarwienie wątroby)
W sercu stłuszczenie jest wynikiem niedotlenienia (wtedy „serce tygrysie”- szkliste pasma
słuszczonych komórek na przemian z pasmami prawidłowych kardiomiocytów) lub pewnych
rodzajów zapaleń (np. błonicy).
2) cholesterol i jego estry
-komórki fagocytarne często w procesach patologicznych przeładowane są lipidami (Tag,
cholesterol)
- makrofagi przeładowane lipidami wypełniają siędrobnymi wakuolami otoczonymi błoną=
komórki piankowate
- w miażdżycy komórki te przyczyniają się do żółtego zabarwienia blaszek miażdżycowych
miażdżycowych uczestniczą w patogenezie uszkodzenia
- w hiperlipidemiach powodują powstanie xanthomata na skórze
3) białka
-gromadzenie się białek może być efektm ich nadmiernej syntezy lub nadmiernego
przedostawania się
-np. gdy uszkodzona jest błona filtracyjna kłębuszka (zesp.nerczycowy) to wychwyt białek drogą
pinocytozy wzmaga się i prowadzi do powstania różowych hialinowych kropelek kropelek
cytoplazmie (proces odwracalny)
-ciałka Russela- nowo zsyntetyzowane immunoglobuliny w RER niektórych plazmocytów
- ciałko Mallory’ego (hialina alkoholowa)- kwasochłonny cytoplazmatyczny wtręt w hepatocytach
alkoholików
-sploty neurofibrylarne w mózgu chorych na Alzheimera
4) glikogen
-efekt zaburzenia metabolizmu glikogenu lub glukozy
- w źle kontrolowanej cukrzycy odkłada się w nabłonku cewek nerkowych, kardiomiocyach i
kom. Beta wysp Langerhansa
4

- w glikogenozach defekty enzymatyczne powodują nadmierne spichrzanie glikogenu i śmierć
komórek
5) barwniki
-mogą być egzo lub endogenne
-najczęstszym egzogennym jest pył węglowy, który zaczernia miąższ płuca(anthracosis) i
okoliczne węzły chłonne. Masywne złogi mogą prowadzić do pylicy węglowej.
-barwniki endogenne to lipofuscyna, melanina i pochodne hemoglobiny
-lipofuscyna (barwnik zużycia)- brunatna, gromadzi się głównie w sercu, wątrobie mózgu, jako
wyraz zużycia lub starzenia. Jest kompleksem tłuszczowo-białkowym powst.przez peroksydacje
kwasów tłuszczowych błon kom. Jest nieszkodliwa. Może być wyznacznikiem uszkodzenia
wolnymi rodnikami. Jej obfite występowanie w tkance to ZANIK BRUNATNY.
-melanina- brązowo-czarna. Powst.przez utlenianie tyrozyny. Syntetyzowana tylko w
melanocytach, w naskórku. Chroni przed promieniami UV. Może występować w warstwie
podstawnej naskórka (piegi) lub w makrofagach skóry.
-hemosyderyna- złotożółta lub brązowa, pochodzi z hemoglobiny. Gromadzi si…ę w tkankach,
w których jest nadmiar żelaza( Fe w tkankach związane jest z apoferrytyną, która tworzy micele
ferrytyny). Hemosyderyna to duże złogi mieli ferrytyny— H. pojawia się w miejscu krwawienia
np. siniak.
-
Hemosyderoza- przeładowanie organizmu żelazem. Odkłada się ono w różnych narządach i
tkankach. Najpierw pojawia się w wątrobie, szpiku, śledzionie i w węzłach chłonnych (wszędzie
żelazo obecne jest w makrofagach), makrofagach czasem obejmując inne narządy.
Hemosyderoza występuje w przebiegu: 1.zwiększonej absorpcji żelaza z pożywieniem
2.upośledzenia utylizacji żelaza 3.niedokrwistości hemolitycznych 4.przetoczeń krwi. W
większości przypadków układowych hemosyderoz żelazo nie prowadzi do uszkodzenia funkcji
ani struktury. Z kolei masywne odkładanie żelaza prowadzi do HEMOCHROMATOZY, w której
uszkodzenie tkanek powoduje włóknienie wątroby, niewydolność serca, cukrzycę.
•
Zwapnienia patologiczne
morfologicznie- złogi wapnia mają wygląd białych grudek, są twarde. Histologicznie złogi
powstają wewnątrz- i zewnątrzkomórkowo. Z czasem może pojawić się heterotopowa tkanka
kostna.
wapnienie dystroficzne- gdy zwapnienia powstają w tkankach martwych lub obumierających.
Stężenie wapnia w surowicy jest prawidłowe.
- występuje w większości martwic (np. blaszka miażdżycowa)
- często jest przyczyną zaburzenia czynności jakiejś struktury (np. wapnienie płatków
zastawek w wyniku starzenia)
- patogeneza obejmuje inicjację (w pęcherzykach macierzy powstają fosforany wapnia) oraz
przyrastanie (wzrost kryształu). Proces zależy od stężenia wapnia, fosforanów, obecności
inhibitorów i kolagenu (kol.przyspiesza)
wapnienie przerzutowe- odkładanie soli wapnia w prawidłowych tkankach. Niemal zawsze
oznacza jakieś zaburzenie metabolizmu wapnia (hiperkalcemia).
-przyczyny hiperkalcemii : zwiększone wydzielanie PTH; niszczenie kości (np.choroba Pageta,
unieruchomienie, lub nowotwór- szpicak, białaczka, lub przerzuty do kości); zaburzenia
związane z wit. D (zatrucie witaminą D, lub sarkoidoza); niewydolność nerek (retencja
fosforanów prowadzi do wtórnej nadczynności przytarczyc).
5) Odwracalne i nieodwracalne uszkodzenie komórki
a) mechanizmy ogólne
jak wyżej- istnieją cztery podatne systemy:
1) integralność błony kom. (homeostaza osmotyczna i jonowa)
2) produkcja ATP
3) synteza białek
4) integralność aparatu genetycznego
W określonym przedziale komórka potrafi równoważyć zaburzenia w tych systemach. Jednak bardzo
silny lub długo trwający bodziec prowadzi do „progu nieodwracalnego uszkodzenia” . nieodwracalne
uszkodzenie zaburza fosforyzację oksydacyjną, a co za tym idzie- syntezę ATP. Przerwanie ciągłości
błony kom. Jest czynnikiem krytycznym prowadzącym do uszkodzenia komórki, a wapń jest
mediatorem zmian morfologicznych podczas śmierci komórki.
b) Mechanizmy uszkodzenia nieodwracalnego
Nieodwracalność procesu charakteryzują dwa zjawiska:
5

1. niemożność odwrócenia zaburzeń funkcjonowania mitochondriów (zahamowanie fosforyzacji
oksydacyjnej i produkcji ATP) nawet po ustąpieniu czynnika wywołującego (np. przywrócenie
ukrwienia)
2. powstanie głębokich zaburzeń funkcji błony komórkowej (utrata regulacji objętości,
zwiększona przepuszczalność, defekty ultrastrukturalne). Czynniki powodujące zniszczenie
błony:
- utrata fosfolipidów błonowych
- zaburzenia budowy cytoszkieletu
- wolne rodniki
- produkty rozpadu lipidów (powodują obniżenie napięcia powierzchniowego)
Końcowym efektem uszkodzenia błony jest masywny wyciek substancji wewnątrzkomórkowych i
masywny napływ wapnia.
c) Mechanizmy uszkodzenia odwracalnego oraz śmierć komórki- martwica
1. uszkodzenie odwracalne
Zmiany ultrastrukturalne:
- zmiany w obrębie błony (uwypuklenia-blebs, zaburzenia wyglądu mikrokosmków, rozluźnienie
połączeń międzykomórkowych)
- zmiany w mitochondriach (obrzęk i bezpostaciowe zagęszczenia bogate w fosfolipidy)
- zmiany w retikulum (odłączanie się rybosomów, rozpad polisomów)
- zmiany w jądrze (rozpad elementów ziarnistych ziarnistych włókienkowych)
Zmiany morfologiczne w mikroskopie świetlnym:
- obrzęk komórki- zwyrodnienie wodniczkowe (degeneratio hyropica)
- słuszczenie- pojawienie się wakuol lipidowych (pod wpływem np.niedotlenienia)- zachodzi głównie
w hepatocytach i kardiomiocytach.
2. Martwica czyli zmiany nieodwracalne
Jest to sekwencja zmian morfologicznych następującyh w martwej komórce w obrębie żywej tkanki.
Najczęstsza jest martwica skrzepowa, charakteryzująca się obrzękiem komórek, denaturacją białek
i rozpadem organelli.
enzymatyczne trawienie białek- autoliza (jeśli enzymy pochodzą z martwych komórek) lub
heteroliza (jeśli pochodzą z komórek zapalnych).
denaturacja białek
Do pełnego rozwoju procesów potrzeba kilku godzin (pod mikroskopem po 4- 12 godz)
Morfologicznie:
- martwe komórki są kwasochłonne (barwienie eozyną), bo brak RNA odpowiedzialnego za
zasadochłonność.
- cytoplazma „zjedzona przez mole” (wakuole)
- zmiana w jądrze może przebiegać na trzy sposoby:
1.rozpad (karyolisis) spopod. DNA-zą;
2.obkurczenie (pyknosis) ze zwiększoną zasadochłonnością
3.fragmentacja (karryorhexis)
po 1- 2 dniach jądro zanika całkowicie
W zależności od tego który proces wystąpił najpierw (denaturacja czy auto(hetero)liza), martwicę
dzielimy na:
a) skrzepową
-pierwotny proces- denaturacja (zarówno białek strukturalnych jak i enzymatycznych- dlatego
zahamowana jest liza enzymat.)
-np. zawał serca
- śmierć z powodu niedokrwienia we wszystkich tkankach z wyjątkiem mózgowej.
b) rozpływną
- charakterystyczna dla miejscowych zakażeń bakteryjnych i grzybiczych (silny bodziec dla białych
krwinek)
- także śmierć OUN
c) martwica serowata
- najczęściej w gruźlicy
-centralny obszar martwicy przypomina biały ser
-mikro: bezpostaciowa ziarnista masa otoczona rąbkiem ziarniniakowatej reakcji zapalnej
i teraz martwice nie stanowiące oddzielnego rodzaju ale omówione oddzielnie:
- zgorzelinowa- niedokrwienna martwica skrzepowa (często w obrębie kończyn) z komponentą
rozpływną.
- m. tkanki tłuszczowej- ogniskowe obszary destrukcji tkanki tłuszczowej (typowo w uszkodzeniu
trzustki- OZT; aktywowane enzymy trzustkowe wydostające się z przewodów trzustkowych, upłynniają
6

lipidy błon komórkowych i hydrolizują estry TAG. Uwolnione kwasy tłuszczowe wiążą się z wapniem,
tworząc widoczne makroskopowo kredowobiałe obszary (zmydlone tłuszcze).
Ostatecznie większość komórek martwiczych zostaje strawiona enzymatycznie pozakomórkowo lub
przez fagocyty. Jeśli komórki martwicze lub ich pozostałości nie zostaną zupełnie wyeliminowane to
mogą przyciągać sole wapniowe i prowadząc do wapnienia dystroficznego.
6) Apoptoza- zaprogramowana śmierć komórki
Zachodzi podczas następujących procesów:
- zaprogramowanej destrukcji komórek podczas embriogenezy w czasie implantacji,
organogenezy i inwolucji towarzyszącej rozwojowi zarodka
- inwolucji fizjologicznej zależnej od hormonów (endometrium w czasie menstruacji, lub prostata
po kastracji)
- śmierć komórek w tkankach szybko proliferujących, jak nabłonek krypt jelitowych lub komórki
nowotworowe
- eliminacja autoreaktywnych limfocytów T w grasicy, śmierć limfocytów bez cytokin lub śmierć
komórek indukowana przez limfocyty Tc
- po zadziałaniu czynników uszkadzających DNA (np. temperatura, leki przeciwnowotworowe)-
uruchamiają one autodestrukcję komórki (np. przez białko TP35)
Mechanizmy apoptozy:
1. bodźce wywołujące:
- zaprogramowana odśrodkowo (podczas rozwoju)
- brak czynnika wzrostowego
- interakcje receptora z liganiem i uwolnienie limf. Tc
- czynniki uszkadzające (promieniowanie)
- sygnały przezbłonowe wzmagające apoptozę: TNFR (np. cząstka FAS), zawierają one
wewnątrzkomórkową „domenę śmierci”, która aktywuje kaspazy i enzymy, prowadząc do
śmierci komórki.
- istnieją też sygnały osłabiające apoptozę
2. kontrola i integracja
- istnieją dwa alternatywne szlaki:
a) bezpośrednie przekazanie bodźców śmierci przez białka adaptorowe do mechanizmu
wykonawczego
b) regulacja przepuszczalności mitochondriów przez białka z rodziny BCL-2 ( hamują one
apoptozę, zapobiegając zwiększeniu przepuszczalności błony mitochondrialnej i stabilizują
białka). Białka BAX i BAD przyspieszają apoptozę.
Wykonanie:
- przecinanie białek przez kaspazy (caspas ma aktywną cysteinę, białko przecina tam gdzie
jest kwas asparaginowy= c jak cysteina, aspas jak kwas asparaginowy)
-masywne tworzenie wiązań krzyżowych wywołane aktywacją transglutaminazy
- rozpad DNA pod wpływem nukleaz zależnych od Ca i Mg
Usuwanie martwych komórek
- kom. apoptotyczne i ich fragmenty mają swoiste receptory, które pozwalają na ich wykrycie i
wyeliminowanie
7) Starzenie się komórki
- Są dwie teorie- wewnętrznego starzenia się komórki (proces ten zapisany jest w materiale
genetycznym) i zużycia komórki (mimo procesów naprawczych, długotrwałe skutki wpływów
zewnętrznych w końcu przeważają i komórka się starzeje)
1. teoria wewnętrznego starzenia ma dwa mechanizmy:
•
Niekompletna replikacja końciwych odcinków chromosomów (skracanie telomerów)
•
Geny zegarowe
2.teoria zużycia
•
Polega na kumulacji błędów w DNA na skutek niewydolnych procesó naprawczych- szybkość
kumulacji tych błędów decyduje o szybkości starzenia (jak w progerii)
•
Wadliwa naprawa DNA występuje u ludzi z zespołem Cockayne’a i ataksja- teleangiektazja-
pacjenci z tymi chorobami szybciej się starzeją
•
Uszkodzenia następują pod wpływem wolnych rodników- promieniowanie X, brak glutationu
i innych przeciwutleniaczy
•
Drugi mechanizm zużycia komórek polega na posttranslacyjnej modyfikacji białek-
oksydacja przez wolne rodniki lub glikozylacja nieenzymatyczna prowadząca do powstania
7

AGE (tworzy wiązania krzyżowe w białkach), np. W soczewce glikozylacja prowadzi do
powstania zaćmy starczej
ZAPALENIE OSTRE I PRZEWLEKLE
(w tym rozdziale koniecznie obejrzyjcie rysunki!!!)
Patologia ogólna zapaleń
Zapalenie jest reakcją obronna organizmu, której celem jest usuniecie lub zniszczenie przyczyny
uszkodzenia, a także tkanek objętych martwicą a wyniku pierwotnego uszkodzenia.
W procesie zapalenia bierze udział wiele różnych Komorek- krwi, osocza, ścian naczyń oraz komórki
macierzy pozakomórkowej otaczającej tkankę łączną
Do Komorek krążących w krwiobiegu należą granulocyty obojętnochłonne, kwasochłonne i
zasadochłonne, limfocyty, monocyty i płytki
Wśród białek osocza znajdują się czynniki krzepnięcia, kinino geny i składniki dopełniacza
Komórki ścian naczyń: komórki śródbłonka i komórki mięśni gładkich
W obrębie tkanki łącznej znajdują się makrofagi, komórki tuczne i fibroblasty.
- schemat reakcji zapalnej:
Czynnik wywołujacy→uwolnienie mediatorów chemicznych zapalenia z osocza lub tkanki
lacznej→odpowiedź komorkowa/ naczyniowa. Odczyn zapalny jest wygaszany gdy czynnik
uszkadzający oraz mediatory zapalenia zostaną usunięte.
ZAPALENIE OSTRE
- stosunkowo krotki czas trwania (kilka godzin do kilku dni)
- wysiek zawiera plyny i bialka osocza
- naciek z neutrofili
1. Zmiany naczyniowe
•
zmiany średnicy naczynia i przepływu naczyniowego zależą od siły i czasu trwania bodźca.
Po przejściowym (kilka sek) skurczu naczyń dochodzi do rozszerzenia tętniczek→miejscowy
wzrost przepływu krwi i przekrwienie łożyska naczyniowego. Jest powodem wystąpienia
rumienia (erytema) i wzmożonego ucieplenia
↓
Nastepnie- wzrost przepuszczalności naczyn mikrokrazenia prowadzi do wydostawania się
bogatobialkowego plynu do przestrzeni zewnatrznaczyniowej→ zwiekszenie stężenia RBC,
wzrost lepkości krwi i zwolnienie przepływu (mikro dużo krwinek czerw. W naczyniach= stasis
czyli zastój)
↓
W warunkach zastoju leukocyty (głównie neutrofile) gromadzą się na powierzchni śródbłonka=
marginalizacja. Następnie przylegają do niego i przechodzą do tk.śródmiąższowej
•
wzrost przepuszczalności ścian naczyń
W najwcześniejszej fazie zapalenia rozszerzenie się tętniczek i wzmożony przepływ prowadzą do
wrostu ciśnienia hydrostatycznego i przechodzenia płynu do przestrzeni zewnątrzkomórkowej
(jest to przesięk). Płyn ten zawiera niewielkie ilości białka.
Natomiast wskutek wzrostu przepuszczalności dochodzi do wysięku, który zawiera dużo białka,
powodując spadek ciśnienia osmotycznego w naczyniach, a wzrost w przestrzeni
zewnątrzkomórkowej→ gromadzenie się wody i jonów w przestrzeni pozanaczyniowej (obrzęk)
przyczyny wzrostu przepuszczalności:
-w wyniku skurczu komórek śródbłonka powstają szczeliny międzykomórkowe w ścianie żyłek.
Jest to proces odwracalny indukowany przez histaminę, bradykininę, LTE i in.
Skurcz trwa 10-15 minut – jest to odpowiedź
natychmiastowa przemijająca.
Dotyczy tylko żyłek pozawłośniczkowych
-Rozsuwanie się komórek śródbłonka
Biorą tu udział cytokiny (TNF i IL-1) powodując reorganizację białek cytoszkieletu →
rozerwanie połączeń międzykomórkowych → odrywanie się komórek od siebie (po 4-6
godzin od zadziałania bodźca)
Też jest przemijająca, ale kończy się 24h później niż to wyżej
-Bezpośrednią przyczyną uszkodzenia śródbłonka jest martwica komórek prowadząca do
masywnego przecieku płynów i białek
8
Występuje przy silnych bodźcach (oparzenia, zakażenia)
Często towarzyszy temu gromadzenie płytek i powstanie zakrzepów
Przeciek może utrzymywać się kilka dni, aż do naprawienia uszkodzenia lub powstania
zakrzepicy
Jest to odpowiedź
natychmiastowa utrwalona
Zachodzi zarówno w żyłkach, kapilarach jak i tętniczkach
Bezpośrednie uszkodzenie może spowodować opóźniony (2-12 h po zadziałaniu czynnika)
przewlekły wzrost przepuszczalności który trwa wiele godzin (dni) i dotyczy żyłek oraz
naczyń kapilarnych. Np. urazy termiczne o średnim/niewielkim nasileniu , promieniowanie
X lub UV
-Uszkodzenie komórek śródbłonka zależne od leukocytów
Następstwo gromadzenia się LEU, które uwalniają toksyczne związki tlenu i enzymy
proteolityczne
W żyłkach i kapilarach płucnych
-Wzmożona transcytoza
Odbywa się drogą pęcherzyków śródkomórkowych
Zwiększa przepuszczalność żyłek (zwłaszcza pod wpływem mediatorów jak VEGF)
Trans cytoza zachodzi przez kanały, które powstają ze zlewających się pęcherzyków
-Wyciek z nowo utworzonych naczyń
Bo są słabo wykształcone
Mają dużą ekspresję receptorów (np. dla VEGF)
2
Procesy komórkowe
Proces przechodzenia leukocytów z naczynia do tkanki można podzielić na (1) marginalizacja i toczenie
się. (2) adhezja i przechodzenie między komórkami śródbłonka i (3) migracja LEU z przestrzeni
śródmiąższowej w kierunku sygnału chemotaktycznego
funkcja
śródbłonek
leukocyt
Toczenie się (neutrofile,
monocyty, limfocyty)
P- selektyna
Białka połączone ze sjalo-
Lewis X
Toczenie się (neutrofile,
monocyty)
GlyCam-1, CD34
L-selektyna
Toczenie się i adhezja
(neutrofile, monocyty,
limfocyty T)
E-selektyna
Białka połączone ze sjalo-
Lewis X
Adhezja (eozyno file,
monocyty, limfocyty)
VCAM-1
Integryna VLA-4
Adhezja, zatrzymanie i
przejście przez ścianę
naczynia (neutrofile,
monocyty, limfocyty)
ICAM-1
Integryny CD11/CD18
(LFA-1, Mac-1)
Zatrzymanie, przechodzenie
przez ścianę naczynia
(neutrofile, monocyty,
limfocyty)
CD31 (PECAM-1)
CD31 (PECAM-1)
1. Marginalizacja i toczenie się
Słabe przejściowe toczenie się LEU związane jest z selekty nami (znajdują się na LEU i na
śródbłonku)
E- selektyna (na śródbłonku), P- selektyna (śródbłonek i płytki) i L-selektyna (LEU) wiążą się z
oligocukrami + kwasem sjalowym (sjalo-Lewis X na LEU), wchodzącymi w skład komórek
docelowych
W niepobudzonych komórkach śródbłonka P- selektyna występuje jedynie w
wewnątrzkomórkowych ciałkach Weibel- Palade’a. pod wpływm zadziałania np.histaminy/
trombiny, cząsteczki przedostają się na powierzchnię komórki i wiążą z leukocytami.
Podobnie E-selektyna, lub ICAM-1 pojawia się na komórce dopiero po zadziałaniu TNF i IL-1
2. Adhezja i przechodzenie komórek
Zachodzi za pośrednictwem immunoglobulin śródbłonka, które łączą się z integrynami na LEU
Śródbłonkowe cząstki adhezyjne: ICAM-1, VCAM-1
9

Ekspresja tych cząstek wzbudzana jest przez IL-1 i TNF
Integryny (przezbłonowe glikoproteiny-heterodimery łańcuchów α i β- będące też receptorami
dla macierzy pozakomórkowej). Np. dla ICAM-1 są LFA1 i Mac-1, a dla VCAM-1 jest VLA-4.
Normalnie integryny nie mogą wiązać się z CAMami. Dopiero pod wpływem pewnych czynników
(chemokiny, C5a dopełniacza, PAF) zwiększa się ich powinowactwo do cząstek adhezyjnych i
wtedy mogą się z nimi łączyć.
LEU przechodzą przez ściany naczyń głównie w żyłkach pozawłośniczkowych (rzadziej kapilary
płucne)
W procesie przeciskania się między komórkami bierze udział płytkowo-śródbł.cząstka adhezyjna
PECAM-1 (lub CD31- immunoglobulina)
Po przekroczeniu wiązań międzykomórkowych leukocyty miejscowo trawią błonę podstawną za
pomocą kolagenaz.
Pierwsze 6- 24 godziny przeważają neutrofile, potem 24-48 monocyty (ponieważ istnieje
sekwencja ekspresji cząstek adhezyjnych różnego typu w różnych fazach odczynu zapalnego;
poza tym neutrofile żyją tylko 24-48h)
3. Chemotaksja i aktywacja
Działanie chemotaktyczne na leukocyty mają: rozpuszczalne produkty komórek bakteryjnych,
składniki dopełniacza (zwłaszcza C5a), metabolity kwasu arachidonowego (zwłaszcza LTB4) i
cytokiny (zwłaszcza hemokiny, np. IL-8)
Cząsteczki chemotaktyczne wiążą się ze swoistymi receptorami na powierzchni komórek,
powodując aktywację fosfolipazy C zależnej od białka G.
fosfolipazaC
PIP2 →→→ DAG + IP3
↓
Wzrost stężenia Ca
↓
Powstanie kurczliwych elementów
cytoszkieletu, potrzebnych do ruchu
komórki
Leukocyty poruszają się za pomocą pseudopodiów, kotwiczących się w macierzy
pozakomórkowej
Kierunek ruchu określa gęstość receptorów, reagujących z ligandami chemotaktycznymi
Powstanie DAG prowadzi do de granulacji i wydzielania enzymów lizosomalnych i generację
przełomu oksydacyjnego
DAG i Ca aktywują fosfolipazę A2, która uczestniczy w przemianie kwasu arachidonowego
Wapń moduluje liczbę cząstek adhezyjnych i ich powinowactwo do ligandów
4. Fagocytoza i de granulacja
- fagocytoza zachodzi w trzech etapach:
(1)rozpoznanie i związanie cząstki docelowej przez LEU, (2) wchłonięcie cząstki z utworzeniem
wakuoli, (3) neutralizacja i rozpad pochłoniętego materiału.
Rozpoznanie odbywa się za pośrednictwem opsonin, np. IgG (zwłaszcza fragment Fc), składnik
C3b dopełniacza, lektyny osoczowe (wiążą one węglowodany zwane kolektynami które mają
powinowactwo do grup cukrowych na powierzchni mikroorganizmów)
Receptory na LEU: rec dla Fc IgG, rec dla skł. Dopełniacza (CR1, CR2, CR3) oraz C1q dla
kolektyn.
Wiązanie cząstek docelowych otoczonych osponinami jest bodźcem do ich pochłaniania
Proces zabijania mikroorganizmów zachodzi dzięki aktywnym związkom tlenu
Następnie mikroorg. Jest trawiony przez kwaśne hydrolazy w obrębie lizosomu
Inne związki w LEU zabijające bakterie i inne takie paskudztwa: bakteriobójcze białko
zwiększające przepuszczalność, lizozym, główne białko zasadowe, defensyny.
5.
Zaburzenia funkcji leukocytów
-zaburzenia adhezji
Niedobór adhezji leukocytów 1 typu (LAD-1)- nieprawidłowa synteza podj. CD18β w LFA1 i Mac-1
LAD-2- brak sjalo- Lewis X
-zaburzenia funkcji bakteriobójczej
Przewlekła choroba ziarniniakowi (CGD)- niedobór elementów oksydazy NADPH- nie da się
wytworzyć nadtlenków
- defekty tworzenia fagolizosomu
10

Zespół Chediaka- Higashiego- choroba genetyczna, w której zachodzi zaburzenie dystrybucji
organelli cytoplazmatycznych- prowadzi to do upośledzonej de granulacji lizosomów do
fagosomów. Zaburzone jet też uwalnianie ziarnistości litycznych przez Tc (ciężkie upośledzenie
odporności)
6. Chemiczne mediatory zapalenia
Mediatory mogą krążyć w osoczu (m.układowe wytwarzane przeważnie w wątrobie) lub
powstawać na miejscu (m.miejscowe)
osoczowe- dopełniacz, kininy, czynniki krzepnięcia- krążą w postaci nieaktywnych prekursorów.
Miejscowe magazynowane są w ziarnistościach w komórce i uwalniane są w momencie
aktywacji komórki (np. histamina w kom.tucznych), lub syntetyzowane de novo pod wpływem
bodźca (np. prostaglandyny)
większość działą przez związanie się z receptorami
mogą pobudzać komórki docelowe do produkcji innych mediatorów o podobym działaniu
(intensyfikacja zjawiska) lub przeciwstawnym (prowadzą do hamowania reakcji)
mogą być swoiste i nieswoiste (zal.od komórki docelowej)
ich funkcja podlega ścisłej regulacji- szybki rozpad (poch.kw.arachidon.), inaktywacja
enzymatyczna (kininaza deaktywuje bradykininę), eliminacja (wymiatające przeciwutleniacze)
lub zahamowanie (białka hamujące dopełniacz)
a) aminy naczynioaktywne
histamina występuje w kom tucznych, płytkach i neutrofilach
bodźce uwalniające: urazy fizyczne, reacje immunologiczne (IgE), fragmenty C3a i C5a
dopełniacza (anafilatoksyny), białka leukocytarne, neuropeptydy (substancja P), cytokiny (IL-1 i
IL-8)
powoduje rozszerzenie tętniczek
główny mediator wzrostu przepuszczalności naczyń (powoduje skurcz śródbłonka i rozsunięcie
komórek)
inaktywowana wkrótce po uwolnieniu przez histaminazę
serotonina działa podobnie, występuje w płytkach i uczestniczy w procesie ich agregacji
b) neuropeptydy
np. substancja P
przewodzą bodźce bólowe, regulują napięcie ścian naczyń i ich przepuszczalność
włókna nerwowe które je uwalniają znajdują się głównie w płucach i przewodzie pokarmowym
c) proteazy osoczowe
czynnik Hagemana (XII)- produkowany przez wątrobę, krąży w osoczu w nieaktywnej postaci
aktywacja po połączeniu z kolagenem, błoną podstawną lub pobudzonymi płytkami
(uszkodzenie śródbłonka)
kofaktor- kininogen ciężki (HMWK) zmienia formę nieaktywną w aktywną (XIIa)
aktywacja układu kinin prowadzi w końcu do powstania bradykininy (z jej prekursora HMWK).
Innym aktywatorem cz. XII jest kalikreina.
bradykinina powoduje wzrost przepuszczalności ścian naczyń, rozszerzenie naczyń i skurcz
mięśniówki gładkiej oskrzeli
podana podskórnie powoduje ból
dezaktywowana szybko przez kininazy
cz. XIIa w układzie krzepnięcia prowadzi do aktywacji trombiny, a w konsekwencji wytworzenia
nierozpuszczalnego skrzepu włóknika
cz. Xa (jeden z El. Pośrednich układu krzepnięcia) powoduje wzrost przepuszczalności ścian
naczyń i migrację leukocytów
trombina wmaga adhezję LEU do śródbłonka i wytwarza fibryno peptydy
fibrynopeptydy zwiększają przepuszczalność naczyń i działają chemotaktycznie na LEU
cz. Hagemana inicjuje proces krzepnięcia i pobudza jednocześnie fibrynolizę (żeby była
równowaga)
aktywator plazminogenu i kalikreina rozcinają plazminogen, powstaje plazmina (prozeaza, która
rozcina fibrynę), rozpuszczając skrzep.
Produkty rozpadu fibryny powodują wzrost przepuszczalności ścian naczyń
Plazmina zmienia C3 na C3a, przez co wpływa na rozszerzenie naczyń i wzrost ich
przepuszczalności (aktywuje też czynnik Hagemana przez co wzmaga cały proces)
11

UKŁAD DOPEŁNIACZA składa się z kaskady białek osoczowych
W ukł.immunologicznym ostatecznym efektem działania tego układu jest powstanie kompleksu
atakującego błonę (MAC)
MAC powoduje perforowanie błon bakterii
Składniki dopełniacza obecne są w osoczu w postaci nieaktywnej
Najważniejszym elementem układu jest aktywacja C3 która może odbyć się na drodze
klasycznej, lub alternatywnej
Droga klasyczna polega na wiązaniu C1 przez kompleks antygen-przeciwciało (kompleks ten
aktywuje C3)
Droga alternatywna wywoływana jest przez wielocukry bakteryjne (np.endotoksyny), wielocukry
złożone lub agregaty IgA. W procesie tym bierze udział wiele białek osoczowych, np.
properdyna, białka B i D
Niezależnie od drogi aktywacji, konwertaza C3 rozcina C3 na C3a i C3b
C3b wiąże się z konwertazą C3, tworząc konwertazę C5, która rozcina C5 z wytworzeniem C5a i
aktywacją końcowych etapów tworzenia MAC
Powstałe czynniki biorą udział w procesach zapalnych:
-zmiany naczyniowe: C3a i C5a (anafilatoksyny) zwiększają przepuszczalność naczyń i powodują
ich rozszerzenie za pośrednictwem histaminy uwolnionej z komórek tucznych. C5a uczestniczy
też w przemianach kwasu arachidonowego
-aktywacja, adhezja i chemotaksja leukocytów: C5a aktywuje leukocyty (wzrost powinowactwa
integryn), wywołuje też działanie chemotaktyczne dla neutrofili, monocytów, eozynofili i bazofili
-fagocytoza- C3b i C3bi są silnymi opsoninami
d) metabolity kwasu arachidonowego: prostaglandyny, leukotrieny, lipoksyny
Pełnią funkcję „hormonów o niewielkim zasięgu”
Działają w miejscu uwolnienia, po czym są szybko trawione enzymatycznie lub tracą aktywność
Kwas arachidonowy jest składnikiem fosfolipidów błonowych
Bodźce mechaniczne, fizyczne, cemiczne, lub mediatory zapalenia (np. C5a) aktywują fosolipazy
które uwalniają ten kwas
Jeśli zadziała na niego cyklooksygenaza to powstaną prostaglandyny i tromboksany
Jeśli zadziała lipooksygenaza to powstaną leukotrieny i lipoksyny
Działanie eikozanoidów w zapaleniu:
Działanie
metabolit
Skurcz naczyń
Tromboksan A, leukotrieny C4, D4, E4
Rozszerzenie naczyń
Prostacyklina
PGI2,
PGE1,
PGE2,PGD2,lipoksyny
Wzrost przepuszczalności
Leukotrieny C4, D4, E4
Chemotaksja i adhezja leukocytów
Leukotrien B4, lipoksyny
Cyklooksygenaza COX-1 obecna jest w żołądku, a produkowane przez nią prostaglandyny
chronią błonę śluzową przed działąniem kwasu. Zahamowanie COX powoduje zmniejszenie
reakcji zapalnej ale jednocześnie sprzyja owrzodzeniom żołądka
e)czynnik aktywujący płytki (PAF)
Działą za pośrednictwem receptora związanego z białkiem G
Poza aktywacją płytek powoduje też skurcz naczyń krwionośnych i oskrzeli
Jego działanie naczyniorozszerzające i zwiększające przepuszczalność jest 100- 10 000 razy
silniejsze niż histaminy
Zwiększa adhezję LEU, chemotaksję, degranulację i przełom oksydacyjny
Stymuluje syntezę eikozanoidów i in.mediatorów
f) cytokiny
produkowane głównie przez pobudzone limfocyty i makrofagi
modulują aktywność komórkową
należą tu: czynniki stymulujące tworzenie kolonii (CSF), czynniki wzrostu, interleukiny i
chemokiny powodujące adhezję i chemotaksję LEU
można podzielić je na 5 klas:
1. cytokiny regulujące czynność limfocytów, np. IL2 (pobudza proliferację), lub TGFβ (hamuje
wzrost limfocytów)
2. związane z odpornością wrodzoną (TNF i IL-1)
3. aktywujące komórki zapalne (zwłaszcza makrofagi), np. IFNγ i IL-12
4. wykazujące działanie chemotaktyczne dla różnych leukocytów
5. pobudzające krwi otworzenie, w tym pobudzające tworzenie kolonii GM-CSF i IL-3
IL1 i TNF
12

Produkowane przez makrofagi
Ich wydzielanie stymulowane jest przez endotoksynę, kompleksy immunologiczne, toksyny,
uszkodzenie mechaniczne, różne mediatory zapalne
Mają zdolność aktywacji komórek śródbłonka
Aktywują fibroblasty powodując nasilenie ich proliferacji i wytwarzania macierzy
pozakomórkowej
Wywołują odpowiedź układową ostrej fazy, związaną z uszkodzeniem lub zakażeniem
( gorączka, senność, synteza białek w wątrobie, wyniszczenie, uwolnienie do krążenia
neutrofilów i ACTH)
TNF obniża ciśnienie we wstrząsie septycznym i powoduje zmniejszenie kurczliwości serca i
zwiotczenie mięsni gładkich
Chemokiny
Określone hemokiny powodują rekrutację poszczególnych populacji komórkowych w miejsce
zakażenia
Mogą pobudzać komórki prekursorowe szpiku
Mogą przyciągać i pobudzać fibroblasty i miocyty gładkie
Utrzymują gradient chemotaktyczny w macierzy pozakomórkowej, niezbędny do ruchu
komórek zapalnych
Chemokiny CKC (np. IL-8) działają głównie na neutrofile, w odpowiedzi na IL-1 i TNF
Chemokiny CC (np. białko chemotaktyczne monocytów- MCP-1 i białko zapalne
makrofagówMIP-1α, RANTES- czynnik chemotaktyczny dla limfocytów TCD4+ oraz
monocytów, eotaksyna działająca na eozyno file
g) tlenek azotu i wolne rodniki tlenowe
NO syntetyzowany jest z argininy, O2 i NADPH. NOS- synteza tlenku azotu katalizuje reakcję (jej
aktywność zależna jest od stężenia Ca)
Funkcja NO w zapaleniu:
Rozszerzenie naczyń
Przeciwdziała aktywacji płytek na wszystkich etapach
Hamuje rekrutację LEU w ogniskach zapalnych
Działa zabójczo na mikroorganizmy w pobudzonych makrofagach
Wolne rodniki
Uwalniane są z neutrofilów i makrofagów po zadziałaniu czynników chemotaktycznych,
kompleksów immunologicznych lub fagocytozy
Jon nadtlenkowy zamieniany jest na aktywne związki tlenowe
W małych stężeniach związki te powodują zwiększoną ekspresję mediatoró zapalnych
potęgując samo zapalenie
W dużych stężeniach uszkadzają tkanki (inicjują krzepnięcie i zwiększają przepuszczalność
naczyń uszkadzając śródbłonek,; aktywują proteazy; inaktywują antyproteazy, trawiąc
macierz pozakomórkową; bezpośrednio uszkadzają komórki)
Mechanizmy obronne organizmu: katalaza, dysmutaza, glutation
h) składniki lizosomów
Proteazy kwaśne (tylko w lizosomach)
Proteazy obojętne- elastaza, katepsyna, kolagenaza (zachowują aktywność w macierzy, poza
lizosomom) rozcinają C3 i C5, prowadząc do powstania bradykininy
Mechanizmy obronne przed nadmiernym naciekiem- antyproteazy:
α2- makroglobulina
α1- antytrypsyna
tabelka podsumowująca ten przydługi wywód o chemokinach których mam już mega dość!!!!
Zjawiska towarzyszące zapaleniu
Odpowiedzialne mediatory
Rozszerzenie naczyń
Prostaglandyny, NO
Wzrost przepuszczalności naczyń
Histamina, serotonina
C3a, C5a(uwalniają ww aminy biogenne)
Bradykinina
Leukotrieny C4, D4, E4
PAF
Chemotaksja i aktywacja leukocytów
C5a
Leukotrien B4
Produkty bakteryjne
13

Chemokiny (np. IL-8)
gorączka
IL-1, IL-6, TNF
Prostaglandyny
ból
Prostaglandyny
Bradykinina
Uszkodzenie tkanek
Enzymy lizosomalne neutrofilów i
makrofagów
Metabolity tlenu
NO
6. Następstwa ostrego zapalenia
•
Rozejście
Gdy zapalenie trw krótko i jest słabo nasilone
Gdy uszkodzenie jest minimalne a tkanka jest zdolna do odtworzenia komórek
Tkanka wraca do stanu wyjściowego pod względem histologicznym i czynnościowym
Współdziałające siły drenażu limfatycznego i zdolności makrofagów prowadzą do ustąpienia
obrzęku , usunięcia komórek zapalnych i pochłonięcia pozostałości zniszczonych komórek.
•
Bliznowacenie lub włóknienie
Gdy uszkodzenie jest bardzo rozległe lub tkanka niezdolna do regeneracji
Masywny wysięk włóknikowy nie zostaje całkowicie wchłonięty. Ulega on organizacji z
tworzeniem tkanki łącznej, prowadząc do włóknienia
W przypadku masywnych nacieków może dojść do powstania ropni
•
Progresja do zapalenia przewlekłego
ZAPALENIE PRZEWLEKŁE
Cechy charakterystyczne:
Naciek z komórek jednojądrowych- makrofagi, limfocyty, komórki plazmatyczne
Niszczenie tkanek
Naprawa z angiogenezą i włóknieniem
Zapalenie przewlekłe jest wtedy, gdy odczyn zapalny nie może się rozejść ze względu na utrzymujący
się bodziec zapalny lub z powodu zaburzeń gojenia
Np. owrzodzenie trawienne dwunastnicy
•
Zapalenie przewlekłe rozwija się w przypadkach:
Zakażeń wirusowych
Zakażeń wywołanych przez określone mikroorganizmy- prątki krętka bladego i niektóre grzyby
(wywołują odpowiedź typu późnego- delayed hipersensivity), która może kończyć się reakcją
ziarniniakowi
Przedłużającej się ekspozycji na związki toksyczne- cząstki krzemu prowadzące do pylicy-
silicosis, przewlekle podwyższone stężenie lipidów prowadzące do miażdżycy
Chorób autoimmunologicznych, np. RZS, lub stwardnienie rozsiane
•
Mediatory zapalenia przewlekłego:
Makrofagi
Mogą być rozproszone w tkance łącznej lub tworzyć skupiska (np. komórki Kupffera w
wątrobie)- wtedy pełnią rolę filtrów wychwytujących materiał cząsteczkowy i informują o
tym limfocyty T i B
T1/2 wynosi 1 dzień
Pod wpływem czynników chemotaktycznych monocyty opuszczają krew i zamieniają się w
większe makrofagi
Mają zdolność fagocytozy
Proces aktywacji makrofaga polega na zwiększeniu jego wymiarów, wzroście enzymów
lizosomalnych i aktywności metabolicznej
Aktywacja następuje pod wpływem IFNγ, endotoksyn bakteryjnych, różnych mediatorów
zapalenia ostrego, oraz białek macierzy (np. fibronektyny)
Substancje uwalniane przez makrofag:
- kwaśne i obojętne proteazy
- składniki dopełniacza C1- C5
- properdyna
- czynniki krzepnięcia V i VIII
- czynnik tkankowy
14

- reaktywne związki tlenu
- NO
- eikozanoidy
- IL-1, TNF
- czynniki wzrostu, powodujące wzrost mm. Gładkich, fibroblastów i produkcję macierzy
IL-4 i TNF mogą powodować zlewanie się makrofagów prowadząc do powstania komórek
olbrzymich wielojądrowych
Limfocyty, komórki plazmatyczne, eozynofile i komórki tuczne
Limfocyty T i B używają tych samych cząstek adhezyjnych co makrofagi (mają też te same
hemokiny powodujące ich migrację)
Limfocyty produkują IFNγ który pobudza makrofagi, a makrofag IL-1 i TNF które pobudzają
limfocyt
Komórki plazmatyczne to zróżnicowane limfocyty B, produkujące przeciwciała przeciw
antygenom w ognisku zapalnym
Eozynofile- ich ziarnistości zawierają główne białko zasadowe MBP działające toksycznie na
pasożyty i powodujące lizę komórek nabłonkowych
Komórki tuczne = strażnicy. Zaopatrzeni są w IgE przeciw niektórym antygenom. Po
kontakcie z antygenem uwalniają histaminę i eikozanoidy. Odgrywają kluczową rolę w
reakcji anafilaktycznej. Wytwarzają też TNF
•
Zapalenie ziarniniakowe (inflammatio granulomatosa)
-zapalenie przewlekłe, charakteryzujące się skupianiem się pobudzonych makrofagów
przypominających swoim wyglądem komórki nabłonka płaskiego (komórki nabłonkowate)
- formowanie ziarniniaków nie zawsze prowadzi do eradykacji czynnika sprawczego który
zazwyczaj jest bardzo oporny, jednak może „odgrodzić” czynnik ten od reszty tkanek
- przykłady
1. Bakteryjne:
Gruźlica
Trąd
Kiła
Choroba kociego pazura
2. Pasożytnicze
schistosomiaza
3. grzybicze
histoplazmoza
blastomykoza
kryptokokoza
kokcydioidomykoza
4. związane z metalami i pyłami
pylica krzemowa
pylica berylowa
5. związane z ciałem obcym
szwy chirurgiczne, implanty piersi, protezy naczyniowe
6. idiopatyczne
sarkoidoza
-zapalenie ziarniniakowe morfologicznie:
Centralnie położony jest obszar martwicy (nie zawsze)
Otoczony jest skupiskiem makrofagów nabłonkowatych, naokoło których przebiega pasmo
limfocytów stale pobudzająych makrofagi.
Pod wpływem cytokin uwalnianych przez makrofagi, dookoła tego wszystkiego pojawiają się
fibroblasty i komórki tkanki łącznej.
Często w obrębie ziarniniaków znajdują się komórki olbrzymie wielojądrowe, powstałe w
wyniku zlania się z sobą wielu makrofagów
Udział naczyń limfatycznych i węzłów chłonnych w procesie zapalnym
Połączenia międzykomórkowe w naczyniach limfatycznych mają luźną formę, co umożliwia
pozostanie płynu pozakomórkowego i chłonki w równowadze
W przypadku zapalenia, przepływ chłonki zostaje przyspieszony, co umożliwia usunięcie obrzęków,
leukocytów i fragmentów zniszczonych komórek
W przypadku nasilonego zapalenia, naczynia chłonne mogą też transportować mediatory zapalenia.
Prowadzi to do zapalenia naczyń chłonnych (lymphangitis) oraz węzłów chłonnych (lymphadenitis)
Powiększanie węzłów chłonnych jest zazwyczaj związane z prolfiferacją limfocytów i makrofagów
oraz przerostu fagocytów (jest to odczynowe zapalenie ww chłonnych- lymphadenitis reactiva)
15

Zazwyczaj zapora z węzłów chłonnych wystarcza do zatrzymania szerzenia się zapalenia
Jeśli zapalenie złamie tą zaporę i bakterie dostaną się do naczyń krwionośnych to wystąpi
bakteriemia
Następną barierą obronną są komó®ki żerne wątroby, śledziony i szpiku kostnego
W masywnych zakażeniach mikroorganizmy rozsiewają się dalej, zajmując najczęściej zastawki
serca, opony mózgowo- rdzeniowe, nerki (ropne zapalenie nerek) i stawy (septyczne zapalenie
stawów)
Typy morfologiczne zapaleń ostrych i przewlekłych
1.
zapalenie surowicze
obfita, wodnista, ubogobiałkowa wydzielina (wysięk)
pochodzi z osocza, lub międzybłonka wyściełającego otrzewną, opłucną i osierdzie
np. Pęcherz skórny, w następstwie oparzenia lub zakażenia wirusowego
2. zapalenie włóknkowe
•
następstwo poważnych uszkodzeń, w których dochodzi do znacznego wzrostu
przepuszczalności naczyń
•
barierę śródbłonka może pokonać nawet fibrynogen
•
histologicznie widoczne są skupiska włóknika, stanowiące kwasochłonną sieć nitek
•
zlep ten może ulec organizacji z całkowitym powrotem pierwotnej struktury lub może
powtać blizna
3. zapalenie ropne
•
towarzyszy mu powstanie ropy (pus), składającej się z neutrofilów, obumarłych komórek i
płynu obrzękowego
•
szczególnie ropotwórcze są gronkowce
•
ropień jest ograniczonym skupiskiem wysięku ropnego, powstającym w wyniku rosiewu
drobnoustrojów do głębokich warstw tkankowych lub nadkażenia obszarów martwiczych
•
ropnie składają się z centralnej martwicy, otoczonej pasmem neutrofilów. Obecne są też
poszerzone naczynia i proliferujące fibroblasty
•
z czasem ropień może zostać zastąpiony tkanką łączną
4. Owrzodzenie
•
Zapalenie prowadzące do przerwania ciągłości nabłonka
•
Może być następstwem urazowego lub toksycznego uszkodzenia nabłonka
•
W wartswie podnabłonkowej zazwyczaj toczy się już proces zapalny
•
Np. Owrzodzenie trawienne żołądka i dwunastnicy
•
Początkowo naciek z neutrofilów i poszerzone naczynia
•
W zmianach przewlekłych komórki zapalenia przewlekłego
Ogólnoustrojowe następstwa zapaleń
•
Może być reakcja ostrej fazy- czyli objawy ostrego zakażenia wirusowego- gorączka, senność,
złe samopoczucie, brak łaknienia, przyspieszony rozpad białek mięśni, spadek ciśnienia
tętniczego, synteza białek wątrobowych- dopełniacz itp. I przesunięcia w puli leukocytów
krążących
•
Najważniejsze mediatory ostrej fazy: IL-1, IL-6 i TNF- cytokiny uwalniane kaskadowo przez
limfocyty i in. W odpowiedzi na jakiś bodziec uszkadzający.
•
TNF pobudza IL1 która pobudza IL6
•
Leukocytoza- liczba LEU wzrasta do 15- 20 tys/µl. Moze osiągać wartość 40 – 100 tys (odczyn
białaczkowy)
•
W leukocytozie występuje przesunięcie w lewo- dużo młodych neutrofilów. Leukocyty wzrastają
po TNF, IL1 i CSF
•
Bakterie powodują neutrofilię
•
Pasożyty powodują eozynofilię
•
Mononukleoza, świnka, różyczka powodują limfocytozę. Jednak w większości chorób wirusowych
limfocyty spadają
•
Leukopenia występuje w nowotworach
ODROST I NAPRAWA
16

1. Odrost = odnowa (regeneratio)
To powszechna cecha organizmów żywych do odtwarzania komórek bądź tkanek. W odroście
następuje zupełny powrót do stanu poprzedniego pod względem anatomicznym i czynnościowym.
Wiąże się ściśle z rozmnażaniem komórek i ich różnicowaniem.
Komórki międzymitotyczne (labilne)-komórki które zachowują zdolność podziału i dzielą się w
ciągu całego życia.
■
komórki szpiku kostnego
■
komórki nabłonka powierzchniowego naskórka
■
komórki nabłonka jamy ustnej, pochwy, szyjki macicy
■
komórki nabłonkowe wyścielające przewody gruczołów wydzielania zewnętrznego
■
komórki jednowarstwowe walcowate pp, macicy ,jajowodu
■
komórki przejściowe dróg moczowych
■
spermatogonie
Komórki pomitotyczne odwracalne (stabilne)- utraciły zdolność podziałową ale przy znacznych
uszkodzeniach mogą ją odzyskać)
■
komórki śródmiąższu większości tkanek gruczołowych (wątroba, nerki ,trzustka)
■
komórki śródbłonka
■
fibroblasty
■
komórki mezenchymalne tkanki łącznej i mięśni gładkich
Komórki pomitotyczne nieodwracalne (ostatecznie zróżnicowane)- nie dzielą się w okresie po
urodzeniu
■
większość neuronów
■
komórki mięśnia sercowego
2. Naprawa ( reperatio )
Występuje gdy zniszczona została tkanka o małej zdolności odnowy, pozbawiona jej zupełnie lub
uszkodzenie było zbyt duże. W tym wypadku ubytek wypełnia tkanka łączna obfitująca w siec
naczyń włosowatych → ziarnina (granulatio).
Proces ten ma 4 zasadnicze etapy:
tworzenie nowych naczyń krwionośnych (angiogeneza)
migracja i proliferacja fibroblastów
okładanie się macierzy pozakomórkowej
dojrzewanie i reorganizacja tkanki włóknistej (remodeling)
Naprawa zaczyna się w ciągu 24 h od wystąpienia urazu pod napływem fibroblastów oraz
pobudzeniem proliferacji fibroblastów i komórek śródbłonka. Po 3-5 dni pojawia się
wyspecjalizowana tkanka typowa dla procesu gojenia, zwana ziarniną. Ziarnina gromadzi coraz
więcej macierzy łącznotkankowych , co może prowadzić ostatecznie do nasilonego zwłóknienia
(bliznowacenie), które z czasem może ulegać przebudowie.
Angiogeneza
Naczynia krwionośne powstają w toku dwóch procesów: waskulogenezy, podczas której pierwotna
siec naczyniowa powstaje z angioblastów w czasie rozwoju zarodkowego lub neowaskularyzacji w
której istniejące naczynia krwionośne wysyłają pączki włosowate dające początek nowym
naczyniom.
Na angiogenezę wpływa wiele czynników z których najważniejsze to:
zasadowy czynnik wzrostu fibroblastów (bFGF)
naczyniowy czynnik wzrostu (VEGF)
Włóknienie (powstawanie blizny)
Odbywa się na osnowie z nowo powstałych naczyń krwionośnych i luźnej macierzy pozakomórkowej
wytworzonej we wczesnych fazach naprawy uszkodzonych tkanek. Proces włóknienia zachodzi w
dwóch etapach:
napływ i proliferacja fibroblastów
odkładanie macierzy zewnątrzkomórkowej
Największą role w rekrutacji fibroblastów odgrywają czynniki wzrostowe wydzielane przez komórki
zapalenia.
Podczas gojenia zmniejsza się liczba proliferujących fibroblastów i nowych naczyń krwionośnych.
Fibroblasty rozpoczynają aktywność nastawioną na syntezę, zwiększa się też okładanie macierzy
pozakomórkowej.
17

Ziarnina przekształca się ostatecznie w bliznę zbudowaną najczęściej z nieaktywnych wrzecionowato
fibroblastów, gęsto upakowanego kolagenu, fragmentów tkanki sprężystej. Podczas dojrzewania
blizny zanikanie naczyń krwionośnych powoduje przemianę dobrze unaczynionej ziarniny w bladą,
niemal pozbawioną naczyń bliznę.
Przebudowa (remodeling) blizny
Przekształcenie ziarniny w bliznę wiąże się ze zmianą składu macierzy pozakomórkowej; nawet po
jej powstaniu i odłożeniu macierz pozakomórkowa blizny jest modyfikowana i przebudowywana.
Każdy z tych etapów jest wypadkową syntezy i degradacji macierzy pozakomórkowej.
(metaloproteineazy – główne enzymy biorące udział w degradacji kolagenu koniecznej do
remodelingu)
3. Gojenie się ran
pobudzenie ostrej reakcji zapalnej przez powstałe uszkodzenie
regeneracja komórek podścieliska
migracja i proliferacja komórek podścieliska oraz tkanki łącznej
synteza białek macierzy pozakomórkowej
remodeling składowych podścieliska w celu odtworzenia funkcji tkanki
remodeling tkanki łącznej w celu wzmocnienia blizny
Gojenie przez rychłozrost
np. gojenie czystej, niezakażonej rany operacyjnej zaopatrzonej szwem (nacięcie powoduje jedynie
przerwanie ciągłości błony podstawnej naskórka i śmierć stosunkowo niewielkiej ilości kom.
naskórka i tk. Łącznej; wynikiem tego jest przewaga regeneracji naskórka nad procesami
włóknienia)
Gojenie przez ziarniowanie
występuje gdy nastąpi masywna utrata komórek lub tkanek (zawał, owrzodzenie zapalne, rozległa
rana itp.) regeneracja komórek śródmiąższu nie wystarcza do przywrócenia pierwotnej architektury.
4. Patologiczne aspekty wpływające na gojenie się ran
zakażenie - najważniejszy czynnik wpływający na opóźnienie gojenia się rany
niedożywienie (niedobór białek, niedobór wit. C)
podawanie glikokortykosteroidów – działają hamująco na włóknienie
obecność ciał obcych w ranie
czynniki mechaniczne – ucisk, skręcenie brzegów rany
zła perfuzja spowodowana np. miażdżycą naczyń
umiejscowienie uszkodzenia- procesy zapalne np. W jamie opłucnowej czy otrzewnowej
wywołują nasilony odczyn wysiękowy wymagający najpierw tzw. Resorpcji
Bliznowce (kleoidy) – rozległe wyniosłe blizny powstałe w wyniku odkładania się zbyt dużych ilości
kolagenu ( skłonność rodzinna, częściej w populacji czarnoskórej)
„dzikie mięso”=wybujała ziarnina – nadmiar ziarniny wystający ponad poziom otaczającej skóry
4
5
6
CHOROBY UWARUNKOWANE GENETYCZNIE
1. STATYSTYKA I DEFINICJE:
*dotykają ponad 20% hospitalizowanych dzieci
*abberacje chromosomowe stwierdza się w ponad 50% poronień samoistnych w I trymestrze ciąży
*dziedziczne zburzenia- przekazywane genetycznie
*plejotropia- mutacja jednego genu może powodować różne efekty fenotypowe
18

*heterogenność genetyczna- mutacje wielu loci mogą powodować podobny efekt
*kodominacja- oba allele ulegają pełnej ekspresji (dominujący i recesywny) np. geny zgodności
tkankowej, antygeny grup krwi
*polimorfizm- istnienie wielu form allelicznych tego samego genu
*wrodzone zaburzenia- stwierdzane w momencie urodzenia
*MUTACJE – utrwalone zmiany w obrębie DNA:
-PUNKTOWE- zastąpienie pojedynczego nukleotydu innym, może powstać:
inny aminokwas (mutacja zmiany sensu) np. w anemii sierpowatej
kodon terminacyjny (stop codon)
-PRZESUNIĘCIA RAMKI ODCZYTU- delecje lub insercje jednej lub dwóch par zasad zmienia ramkę
odczytu (nie zmienia się, gdy wypadają trzy lub wielokrotność trzech par zasad)
-POWTÓRZENIA TRIPLETÓW- amplifikacja sekwencji trzech nukleotydów (we wszystkich
występuje guanina i cytozyna) np. zespół łamliwego chromosomu X (250-4000 powtórzeń CGG
w genie FMR1). Mają charakter dynamiczny (stopień amplifikacji wzrasta w gametogenezie).
2. PODZIAŁ ZABURZEŃ GENETYCZNYCH:
zaburzenia o dziedziczeniu mendlowskim (spowodowane defektami pojedynczych genów):
- są przyczyną hospitalizacji 1% osób dorosłych i 6-8% dzieci
- dzielą się na:
*autosomalne dominujące
np. rodzinna hipercholesterolemia, wielotorbielowatość nerek, choroba Huntingtona,
dziedziczna sferocytoza, zespół Marfana, neurofibromatoza (nerwiakowłókniakowatość),
dystrofia miotoniczna, stwardnienie guzowate, rodzinna polipowatość jelita grubego,
choroba von Willebranda, zespół Ehlersa- Danlosa (niektóre postaci), osteogenesis
imperfecta, achondroplazja, ostra porfiria przerywana
*autosomalne recesywne
np. niedokrwistość sierpowata, CF (mukowiscydoza), choroba Taya-Sachsa,
fenyloketonuria, mukopolisacharydozy, glikogenozy, galaktozemia, homocysteinuria,
deficyt alfa1-antytrypsyny, choroba Wilsona, hemochromatoza, talasemie, wrodzony
przerost kory nadnerczy, zespół Ehlesra-Danlosa (niektóre postaci), alkaptonuria,
neuropochodne atrofie mięśniowe, ataksja Friedreicha, rdzeniowy zanik męśni
*sprzężone z X
np. dystrofia Duchenne’a, hemofilia A i B, przewlekła choroba ziarniniakowi, niedobór
dehydrogenazy glukozo-6-fosforanowej, agammaglobulinemia, zespół Wiskotta-Aldricha,
moczówka prosta, zespół Lesh-Nyhana, zespół łamliwego chromosomu X
3. CHOROBY ZALEŻNE OD MUTACJI WW BIAŁKACH STRUKTURALNYCH:
ZESPÓŁ MARFANA:
*mutacja w obrębie genu FBN1 (15q12)-> defekt fibryny 1
*objawy głównie w:
- układzie kostnym: smukła, szczupła sylwetka, wydłużone kończyny, wydłużone palce
(arachnodaktylia), wysoko wysklepione podniebienie, nadmierna ruchomość w stawach
(hyperextensibility), ciężkie kifozy i inne deformacje kręgosłupa, klatka kurza lub lejkowata
-gałce ocznej: przemieszczenie lub podwichnięcie soczewek (z powodu wiotkości więzadeł
wieszadłowych)
-układzie sercowo-naczyniowym: fragmentacja włókien elastycznych w ścianie aorty
predysponuje do poszerzenia ściany, rozwarstwienia i powstawania tętniaków, poszerzenie
pierścienia zastawkowego i niedomykalność zastawki aortalnej
-skórze: rozstępy
-mięśniach: sprawiają wrażenie miopatycznych (hipotonia, utrata masy)
-płucach: uszkodzenie tkanki łącznej może objawiać się odmą
ZESPOŁY EHLERSA-DANLOSA (EDS):
*nieprawidłowa synteza lub struktura kolagenu, różnie dziedziczone
*objawy:
-skóra nadmiernie rozciągliwa, krucha, podatna na urazy pozbawiona prawidłowej
elastyczności
-możliwe powikłania: pęknięcie jelit i dużych naczyń (EDS typu IV), kruchość gałk ocznych z
pękaniem rogówki i odklejaniem się siatkówki (EDS typu IV), przepukliny przeponowe (EDS
typu I)
*przyczyny EDS:
-niedobór hydroksylazy lizynowej (typ VI)- zakłócenie tworzenia prawidłowych wiązań
krzyżowych, dziedziczony autosomalnie recesywnie,
-mutacja genu pro-alfa III (typ IV) - niedostateczna synteza kolagenu typu III- mała
wytrzymałość tkanek, dziedziczona autosomalnie dominująco
19
-mutacja w genie dla kolageny typu I- nieprawidłowe przekształcanie pro kolagenu do
kolagenu (typ VII)
4. CHOROBY ZALEŻNE OD MUTACJI W BIAŁKACH RECEPTOROWYCH:
RODZINNA HIPERCHOLESTEROLEMIA:
*występowanie 1:500
*mutacja w obrębie genu warunkującego funkcję recepora dla LDL:
-klasa I: brak możliwości syntezy receptora
-klasa II (najczęstsza): zaburzony transport z retikulum endoplazmatycznego do aparatu
Golgiego
-klasa III: receptor niezdolny do prawidłowego przyłączania LDL
-klasa IV: receptor po przyłączeniu LDL nie jest zdolny do internalizacji
-klasa V: nie dochodzi do dysocjacji receptora ze związanym z nim LDL
*zaburzenie transportu i katabolizmu LDL oraz transportu IDL do wątroby (IDL w osoczu
przekształca się w LDL) -> wysokie stężenie cholesterolu we krwi (u heterozygot 2-3 krotne, u
homozygot 5-krotne) z jego zredukowanym katabolizmem i nadmierną biosyntezą, odkładaniem
się w makrofagach i ścianach naczyń
*występowanie żółtaków ścięgien, miażdżycy, choroby wieńcowej
5. CHOROBY ZALEŻNE OD MUTACJI W BIAŁKACH ENZYMATYCZNYCH:
FENYLOKETONURIA (PKU):
*częsta u Skandynawów, rzadka u Żydów i w rasie czarnej
*ciężki niedobór hydroksylazy fenyloalaninowej -> hiperfenyloalaninemia rośnie w ciągu kilku
tygodni -> uszkodzenie mózgu -> ciężkie upośledzenie umysłowe w 6 miesiącu życia; u mniej
niż 4% nieleczonych IQ ok. 50-60;
wymaga ścisłej diety ze znacznym ograniczeniem fenyloalaniny do końca życia
*rzadziej niedobór innych enzymów, np. DHRP (reduktazy dyhydrobiopterynowej) nie mogą być
leczone eliminacją fenyloalaniny z diety
*1/3 nieleczonych nie chodzi, 2/3 nie mówi
*objawy neurologiczne (nasilone wymioty, padaczka)
*brak tyrozyny-> brak melaniny-> obniżona pigmentacja skóry, włosów, rumień skóry
*metabolity fenyloalaniny wydalane z moczem i potem -> mysi zapach
*fenyloketonuria matczyna: dzieci nieleczonych matek z pku rodzą się z wadami rozwojowymi,
upośledzeniem umysłowym, wzrasta ryzyko poronienia
*diagnostyka: test Guthriego
GALAKTOZEMIA:
*70% przypadków w USA zależy od mutacji zmiany sensu
*brak UDP-transferazy galaktozo-1-fosforanu -> gromadzenie m. In. galaktozo-6-fosforanu,
galaktitolu głównie w:
-wątrobie (zmiany stłuszczeniowe, marskość)i, nerkach, śledzionie, soczewce oka (zaćma
rozwija się w ciągu kilku tygodni), korze mózgu
*zmiany nieswoiste: wymioty i biegunka (po kilku dniach od podania mleka), powiększenie
wątroby z żółtaczką (w I tygodniu), utrata aminokwasów z moczem
*posocznica o ciężkim przebiegu wywołana E. coli
*diagnostyka: rozpoznanie w moczu cukrów innych, niż glukoza, wykrywanie niedoboru
transferazy w leukocytach i erytrocytach, hodowla komórek płynu owodniowego (oznaczenia
enzymatyczne lub badanie DNA)
CHOROBY SPICHRZENIA LIZOSOMOWEGO (GANGLIOZYDOZA GM2):
Choroba Taya-Sachsa:
*niedobór alfa-N-acetyloheksozaminidazy, ponad 85 typów mutacji, większość zakłóca
strukturę lub transport białka
*uszkodzenie mózgu, upośledzenie umysłowe, ślepota, gromadzenie GM2 w neuronach,
osłonkach cylindrycznych wypustek nerwowych, komórkach glejowych w OUN, AUN, rdzeniu
kręgowym, nn. obwodowych, siatkówce, zgon w 2-3 roku życia
*rozpowszechniona wśród Żydów
Choroba Niemanna-Picka:
*typ A i B: pierwotny niedobór kwaśnej sfingomielinazy -> spichrzenie sfingomieliny
*typ C: mutacja NPC1 (białko włączone w przetwarzanie i transport LDL)
*dotyka najbardziej: śledzionę, wątrobę, szpik, węzły chłonne, płuca, OUN
*powiększenie narządów, zaburzenia neurologiczne, zgon w pierwszych latach życia
*diagnostyka: oznaczenie aktywności sfingomielinazy w leukocytach lub fibroblastach
Choroba Gauchera:
20

*mutacja w genie kodującym cerebrozydazę, dziedziczona autosomalnie recesywnie,
występuje w trzech wariantach
*niedobór glukocerebrozydazy -> gromadzenie glukocerebrozydów w jednojądrzastych
komórkach (komórki Gauchera- powiększają się do 100um, rozdęte lizosomy w cytoplazmie
dają obraz „pomarszczonego papieru”)
*typ I: 99% przypadków, głównie u Żydów Aszkenzyjskichnieneuropatyczny,
hepatosplenomegalia, zajęcie szpiku -> spadek elementów morfotycznych krwi, zmiany
szkieletowe, daje szanse długiego przeżycia
*typ II: początek w 6-miesiącu życia, dominują zaburzenia OUN, zmiany wątrobie i
śledzionie, duża letalność
*typ III: (młodzieńczy) zmiany w OUN i narządowe, pośredni między I a II
*diagnostyka: ocena aktywności glukocerebrozydazy w leukocytach lub fibroblastach
MPS (mukopolisacharydozy):
*nieprawidłowa degradacja i spichrzenie mukopolisacharydów w tkankach, większość
autosomalne recesywne
*typy od I do VII (w zależności od niedoboru enzymu)
*akumulowane m.in. siarczany (dermatanu, keratanu, heparanu, chondroityny)
*zaburzenia wielonarządowe (wątroby, śledziony, serca, naczyń), pogrubiene rysów twarzy,
zmętnienie rogówki sztywność stawów, dotknięte fagocyty jednojąądrzaste, fibroblasty,
mięśnie gładkie, śródbłonek
-> zespół Hurlera (MPS I H)- niedobór L-iduronidazy, przeżycie chorych 6-10 lat,
„gargoilizm” (deformacje szkieletowe), zgony z powodu niewydolności krążenia (w
wyniku pogrubienia wsierdzia i odkładania MPS w tętnicach wieńcowych i zastawkach)
->zespół Huntera: wyjątek dziedziczony w sprzężeniu z X- łagodniejszy przebieg,
niedobór sulfatazy iduronianu
GLIKOGENOZY:
*zwykle autosomalnie recesywnie dziedziczone zaburzenia syntrzy lub degradacji glikogenu,
gromadzony jest w lizosomach (choroba Pompego), cytoplazmie lub jądrze komórkowym,
podział:
- postaci wątrobowe np. choroba Gierkego (glkogenoza typu I): utrara aktywności glkozo-6-
fosfatazy
- postaci miopatyczne: obnżenie aktywności enzymów glikolizy np. zespół McArdle’a
(glikogenowa typu V- obniżona aktywność fosforylazy mięśniowej)
- inne: choroba Pompego (glikogenoza typu II)- obniżenie kwaśnej maltazy lizosomalnej,
kardiomegalia; glikogenowa typu IV
6. MUTACJE BIAŁEK REGULUJĄCYCH WZROST KOMÓRKI:
NERWIAKOWŁÓKNIAKOWATOŚĆ (NEUROFIBROMATOZY):
*typ I (choroba von Ricklinhausena): ponad 90% chorych:
-liczne guzki uszypułowane wystające ponad powierzchnię skóry, czasem splotowa te,
proliferacja wszystkich elementów nn. Obwodowych
-plamy cafe au lait
-zmiany barwnikowe tęczówki typu hamartoma (guzki Lisha)
-większe ryzyko wystąpienia innych guzów, zwłaszcza glejaków, oponiaków
-gen NF1 (na chromosomie 17) koduje białko supresorowe dla białka RAS
*typ II
-obustronne nerwiaki nerwu słuchowego, liczne oponiaki
-plamy cafe Au lait
-gen NF2 (na chromosomie 22)koduje merlinę (supresor nowotworzenia)
7. DZIEDZICZENIE ZABURZEŃ WIELOCZYNNIKOWYCH:
Cukrzyca, nadciśnienie, schizofrenia, dna, choroby maniakalno depresyjne, wrodzone wady serca,
układu kostnego. Do choroby dochodzi przy połączeniu czynników genetycznych i środowiskowych
(oddzielnie nie wywołują choroby)- efekt progowy.
8. CHOROBY ZALEŻNE OD ABBERACJI CHROMOSOMOWYCH:
*EUPLOIDIE- wielokrotność liczby haploidalnej, najczęściej nierozdzielenie w mejozie
*3n lub 4n- POLIPLOIDIE
*ANEUPLOIDIA- niedokładna wielokrotność n
*ABBERACJE STRUKTURALNE: translokacja, delecja, insercja, chromosom pierścieniowy
9. NIEPRAWIDŁOWOŚCI CYTOGENETYCZNE:
ZESPÓŁ DOWNA (TRISOMIA 21):
*najczęstsza choroba zależna od abberacji
21

*częściej u kobiet < 20 r.ż i > 45 r.ż.
*do nierozdzielenia chromosomów dochodzi podczas oogenezy
*4%- translokacja ramienia długiego 21 na chromosom 14 lub 22, u 1% mozaikowatość
*cechy kliniczne: zmarszczka nakątna, spłaszczenie profilu twarzy, ciężkie upośledzenie
umysłowe IQ: 25-50, małpia bruzda, zwężenia przewodu pokarmowego, wady serca,
pogrubienie fałdu skórnego na karku, przepuklina pępkowa, hipotonia, predyspozycja do
wystąpienia białaczki, objawy choroby Alzheimera
ZESPÓŁ MIKRODELECJI 22q11:
*wrodzona wada stożka tętniczego, nieprawidłowy rozwój podniebienia, dysmorfia twarzy,
opóźnienie rozwoju, hipoplazja grasicy (niepoprawna odpowiedź limfocytów T)hipoplazja
przytarczyc -> hipokalcemia
*zespół DiGeorge’a- dominują objawy T-komórkowego deficytu odporności i hipokaliemia
* VCSF (zespół podniebienno-sercowo-twarzowy)- dominuje dysmorfia i wady serca
*diagnostyka: obserwacja kliniczna i FISH
10.CHOROBY ZALEŻNE OD CHROMOSOMÓW PŁCIOWYCH:
ZESPÓŁ KLINEFELTERA:
*hipogonadyzm męski z minimum dwoma X i minimum jednym Y, większość ma kariotyp 47,
XXY (wynik nierozdzielenia w czasie mejozy)
*ryzyko zachorowania zwiększa starszy wiek matki i promienie jonizujące (na któregokolwiek z
rodziców)
*większy wymiar stopy od kości łonowej, wydłużenie sylwetki, enuchoidalny wygląd, skąpy
zarost i owłosienie, ginekomastia, zmniejszenie jąder, niższe stężenie testosterony, zwiększone
wydalanie gonadotropin z moczem, niepłodność (generalnie) przez zakłócenie spermatogenezy
-> azoospermia
*może występować upośledzenie umysłowe i zaburzenia rozwoju fizycznego
ZESPÓŁ TURNERA:
*pierwotny hipogonadyzm u fenotypowych kobiet, u 57% utrata całego X-> 45,X
*znaczne opóźnienie wzrastania, niedobory wzrostu osobniczego <3 centyla, obrzmienie karku,
poszerzenie naczyń limfatycznych, płetwiasta szyja, niska linia owłosienia, koślawość łokci,
puklerzowa ta klatka piersiowa z szeroko roztawionymi brodawkami, wysoko wysklepione
podniebienie, obrzęk dłoni i stóp, nerka podkowiasta, zwężenie aorty, dwupłatkowa zastawka
aortalna, brak II-rzędowych cech płciowych, często pierwotny brak miesiączki, jajniki
pozbawione pęcherzyków, może współwystępować niedoczynność tarczycy
*43% ma kariotyp mozaikowy
8
PATOLOGIA OGÓLNA CHORÓB ZAKAŹNYCH
Kochani! Tytułem wstępu…
ten rozdział (9) po pierwsze nie jest ujęty w żaden sposób w naszym materiale, raczej nie powinno być
go na egzaminie a przede wszystkim to jakieś pierdu-pierdu na tematy bliskie mikrobiologii – dlatego
też pozwoliłam sobie na jego dosadne skrócenie i wymienienie najważniejszych rzeczy a w ramach
rekompensaty dostaniecie później bardzo ważny wykład z miażdżcy, który na 100% będzie na
egzaminie. PZDR! AM
1. Postulaty Kocha:
•
organizm chorobotwórczy można znaleźć w mianach chorobowych
•
może być wyizolowany jako pojedyncza kolonia na podłożach stałych
•
zaszczepienie koloni zwierzęciu eksperymentalnemu wywołuje chorobę
•
organizm może być odzyskany ze zmian chorobowych zwierzęcia
2.
Nowe i nowopojawiające się choroby zakaźne:
- dopiero teraz mamy techniki do hodowli/izolacji czynników chorobotwórczych np. HBV
- odkrywane są w dalekich zakątkach świata np. Ebola
- pojawiają się nowe wirusy np. HIV
- mogą się ujawnić w wyniku immunosupresji np. CMV u pacjentów z AIDS
- ich ujawnieniu/rozwojowi mogą sprzyjać zmiany w środowisku np. zalesianie (więcej saren
więcej kleszczywięcej boreliozy)
Na mechanizm rozwoju choroby:
22

•
specyficzne właściwości organizmu chorobotwórczego
•
odpowiedź gospodarza na czynniki zakaźne
3. Kategorie chorób zakaźnych:
a) priony
b) wirusy
c) bakteriofagi, plazmidy, transpozony
d) bakterie
e)
riketsje, chlamydie, mikoplazmy
f) grzyby
g) pierwotniaki
h) robaki pasożytnicze
i) pasożyty zewnętrzne
4. Bariery chroniące gospodarza przed zakażeniem:
a) skóra:
•
złuszcza się i odnawia w ten sposób eliminuje bakterie
•
podobnie pH 5,5 i kwasy tłuszczowe
•
wilgotna jest przepuszczalna dla niektórych mikroorganizmów np. krętka bladego czy
Papiloma virus lub sprzyja rozwojowi innych np. S.aureus lub grzybice
•
inne dostają się od organizmu przez ukłucia, głębokie rany, oparzenia, odleżyny i
owrzodzenia, ukąsznia owadów, kleszczy, większych zwierząt etc.
b) układ moczowo- płciowy
•
„normalny mocz” powinien być sterylny
•
u kobiet częściej dochodzi do zapaleń ze względu na krotkość cewki
•
dodatkowo zapaleniom sprzyja refluks i utrudnienie przepływu moczu
•
przekroczenie granicy pęcherza – odmieniczkowe zapalenie nerek
•
patogeny –ostre - najczęściej E.Coli, przewlekłe – Proteusz, Pseudomonas, Kliebsiella,
Enterococus, inne: Gonococus
c) układ oddechowy:
•
im mniejszy organizm tym dalej zajdzie
•
aparat śluzowo-rzęskowy połykanie lub odkrztuszanie mikroorganizmów
•
mniejsze niż 5 mikrometrów – docierają do pęcherzyków
•
palenie tytoniu, mukowiscydoza, intubacja, aspiracja treści żołądkowej uszkodzenie
tegoż aparatu
•
charakterystyczne dla drobnoustrojów dróg oddechowych – hemaglutyniny wiążące się z
węglowodanami komórek nabłonka chroniące je przed wymiataniem
•
neuraminidaza – zmniejsza lepkość śluzu (grypa, paragrypa, zapalenie przyusznic)
•
H. influenza – hamuje ruch rzęsek
•
Saphylo/Stroptococcus – wtórne wrota zakażenia (pierwotnie infekcja wirusowa)
•
M. tuberculosis – hamuje fagocytozę przez makrofagi
•
B. często infekcje oportunistyczne P.carinii – AIDS, Aspergillus – chemioterapia
d) układ pokarmowy
•
kwaśny sok żołądkowy
•
śluz pokrywający jelito
•
enzymy lityczne trzuski, detergenty żółci
•
wytwarzanie IgA w MALT (limfocyty B układu chłonnego błon śluzowych jelita) na
antygeny zaprezentowane im przez komórki M (nabłonka)
•
naturalna flora jelitowa
•
wirusy posiadające otoczkę są unieszkodliwiane przez sok żołądkowy, te nie posiadające
otoczki nie
•
efekty patogenne: enterotoksyny gronkowca, egzotoksyny cholery i E.Coli, Shigelle
Salmonella – owrzodzenia i krwotoki, S.typhi – przechodzi do krwi i szerzy się na całe
ciało
5. Jak czynniki zakażne wywołują chorobę:
•
Bezpośrednia śmierć komórki po wniknięciu mikroorganizmu
•
Endo-egzotoksyny zabijające „na odległość”
•
Uszkodzenia tkanek wyniku odpowiedzi obronnej gospodarza
23

a) wirusy:
•
zahamowanie syntezy DNA, RNA i białek
•
wbudowanie białek wirusowych w błonę komórkową i jej bezpośrednie uszkodzenie
•
intensywna replikacja wirusa – cytoliza komórek gospodarza (za dużo wirusów –
rozerwanie komórki)
•
białka na powierzchni komórki – atakują ją limfocyty gospodarza
•
niszczenie komórek układu odpornościowego przez wirusy
•
zniszczenie jednego rodzaju komórki pociąga za sobą zniszczenie komórek od nich
zależnych np. polio niszczy neurony ruchowe to prowadzi do zaniku mięśni
szkieletowych
b) bakterie:
•
adhezyjny bakteryjne – wiążą bakterie z komórkami gospodarza
gram dodatnie – kwas tejchojowy, białko F
gram ujemne – fimbrie, pilie
•
endotoksyny bakteryjne – liposacharyd powoduje wzrost temperatury, DIC, ARDS,
wstrząs septyczny
•
superantygeny – wiążą się MHC klasy II i pobudzają limfocyty T (Wiele klonów), te
produkują dużo IL-2 a ta powoduje wzrost wydzielania TNF i inych cytokin
•
egzotoksyny – wydzielane przez bakterię białka, które bezpośrednio niszczą komórkę
np. toksyna błonicza (jednosta A katalizuje ADPrybozylację czynnika EF-2
6. Sposoby ucieczki patogenów przed układem immunologicznym
•
pozostanie niedostępnym – np. patogen bytuje w komórkach nabłonka
•
rozkład przeciwciał i oporność na lizę zależną od dopełniacza lub przeżycie w komórkach
fagocytarnych
•
zmienność antygenów
•
wywoływanie immunosupresji
7. Odpowiedź na czynniki zapalne
a) zapalenia ropne z udziałem neutrofili:- gram dodatnie ziarniniaki i pałęczki
b) zapalenie z udziałem komórek jednojądrowych - są wyrazem zapalenia przewlekłego
zapalenia jeżeli towarzyszy im np. włóknienie a jeżeli pojawiają się na ostro – zakażenia wirusowe
c) zapalenie rozplemowi z uszkodzeniem tkanek – zakażenia wirusowe, niewielka odpowiedź ze
strony gospodarza
d) zapalenia z wytwarzaniem martwicy – również małe nacieki zapalne
e) zapalenia przewlekłe z włóknieniem i tworzeniem blizn -
PATOLOGIA NACZYŃ
1. Podział naczyń:
Tętnice:
- typu sprężystego – aorta i jej bezpośrednie odgałęzienia (pień r-g, podobojczykowe, szyjne
wspólne, biodrowe) – ich błona środkowa bogata zawiera dużo włókien sprężystych, powoduje
odrzut krwi na obwód
- typu mięśniowego (wieńcowe, nerkowe) – ze względu i te o małej średnicy (poniżej 2 mm) i
tętniczki (20-100 mikrometrów) leżące w obrębie tkanek i narządów – ze względu na przewagę
komórek mięśniowych mają zdolność do regulowania przepływu krwi poprzez zwężanie-
rozszerzanie naczynia.
Tętniczki są głównym punktem oporu krwi (spadek promienia o ½ powoduje wzrost oporu 16 razy)
Włośniczki: mają średnicę 7-8 mikrometra, wyściółkę ze śródbłonka i nie posiadają blony
środkowej
żyły - stosunku do odpowiednich tętnic mają większe światło, większą średnicę i cieńszą ścianę bo
mają mniej komórek mięśniowych gładkich (WOW!) przez to są bardziej podane na nieregularne
rozszerzenia, ucisk, nacieki nowotworowe i procesy zapalne, w żyłach kończyn – zastawki. Mamy
żyłki pozawłośniczkowe (przechodzenie płynu i leukocytów), żyłki zbiorcze i żyły małego,
średniego i dużego kalibru
24

naczynia chłonne – praktycznie sam śródbłonek, mogą stanowić drogę rozsiewu chorób
nowotworowych i zakaźnych
2. Budowa prawidłowych naczyń – tętnice
intima – błona wewnętrzna – pojedyńcza warstwa śródbłonka, tkanka łączna (mało), włókna
sprężyste (błona sprężysta wewnętrzna)
media – komórki mięśniowe gładkie odżywiana za pomocą małych fenestracji lub vasa vasorum, na
jej „spodzie” błona sprężysta zewnętrzna
adventitia – przydanka – tkanka łączna osłaniająca włókna nerwowe i tętniczki odżywcze
3. Komórki śródbłonka (endothelium):
- charakterystyczne dla niego są ciałka Weibela-Palade’a zawierające czynnik von Willenbranda
oraz czynnik CD31
- właściwości i funkcje śródbłonka tabelka na str 375: ja w skrócie tylko wymienie:
•
Wytwarzanie cząsteczek o działaniu przeciwkrzepliwym i przeciwzakrzepowym
•
Wytwarzanie cząsteczek o działaniu sprzyjającym krzepnięciu
•
Produkcja macierzy pozakomórkowej
•
Modulacja przepływu i reaktywności naczyń
•
Regulacja procesów zapalnych i odporności
•
Regulacja wzrostu komórek
•
Utlenianie LDLi
•
BARIERIA!
- nic więc dziwnego że jak zostanie on uszkodzony (fizycznie – np. nieprawidłowy przepływ krwi)
chemicznie (np. histamina) czy zakaźnie – siada nam cała hemodynamika i hemostaza (tendencje do
zakrzepów, miażdżyca i zmiany naczyniowe)
Dysfunkcja śródbłonka: kilka typów potencjalnie odwracalnych zmian w jego funkcjonowaniu, które
są skutkiem działania czynników środowiskowych, może to być np. zaburzenie w poszerzeniu naczynia,
spadek produkcji NO lub wzrost produkcji endoteliny lub wolnych rodników
Do czynników wywołujących dysfunkcję należą: cytokiony, bakterie (czyli odczyny zakaźne lub sepsa),
stresy hemodynamiczne, produkty lipidowe biorące udział w tworzeniu miażdżycy powodują one
produkcję czyników przylegania i substancji sprzyjających zakrzepicy, czynników wzrostu etc.
4. Komórki mięśniowe gładkie
- regulują światło naczynia skurczem i rozkurczem
- produkcja kolagenu, włókien sprężystych, proteoglikanów, czynników wzrostu,endoteliny mogą
także przechodzić do błony wewnętrznej i ulegać tam proliferacji głównie pod wpływem nagłego
uszkodzenia śródbłonka lub jego przewlekłej dysfunkcji (tworzy się tzw neointima). Dzieje się tak pod
wpływem płytkopodobnego czynnika wzrostu, zasadowego czynnia wzrostu fibroblastów i IL-
1. Inhibitory tego procesu to: siarczan heparanu, NO, INF gamma, TGF beta
Teraz coś co interesuje nas najbardziej czyli choroby naczyń
1. Wrodzone zaburzenia rozwojowe
a) przetoka tętniczo-żylna – oprócz tego,że może być wrodzona może powstać także w wyniku
pęknięcia tętniaka do sąsiadującej żyły, uszkodzenia ścian żyły i tętnicy (uraz penetrujący) lub
wtórnie do zmian martwiczych – mogą powodować hiperkinetyczną niewydolność krążenia lub
krwotoki
b) tętniaki wrodzone
2. Stwardnienie ścian tętnic – arteriosclerosis
a)
atherosclerosis – miażdżyca (ATH) dotyczy błony środkowej
b)
stwardnienie błony środkowej ze zwapnieniami – typu Monckeberga (w tętniczkach
mięśniowych), po 50 r.ż, widoczne radiologicznie i co ważne nie dają zwężenia światła
naczynia
c)
arteriolosclerosis- zmiany w małych tętnicach tętniczkach – szkliwienie (hyalinica) i
rozrost komórek (hyperplastica) – daja pogrubienie ścian naczyń ze zwężeniem światła,
najczęstszy powód – nadciśnienie tętnicze i cukrzyca
3.
Miażdżyca – obecność w błonie wewnętrznej blaszek miażdżycowych (włóknisto
-tłuszczowych lub kaszowatych), które zwężają światło naczynia, osłabiają błonę środkową i dają
25

inne powikłania: zamknięcie światła niedokrwienie, przerwanie – zakrzepy, tętniaki, zatory
w krążeniu dystalnym
- zmiany miażdżycowe zajmują głownie tętnice typu sprężystego i duże/srednie
tt.mięśniowe
- najczęstsze narządy: serce, mózg, nerki, konczyny dolne zawał serca, zawał mózgu
(udar), tętniaki aorty, zgorzel kończyn
Morfologia miażdżycy:
Blaszka miażdżycowa
- jest to uniesiona zmiana w błonie wewnętrznej
- ma 0,3 – 1,5cm średnicy i obejmuje tylko część obwodu naczynia (zmiana ekscentryczna)
- zawiera rdzeń lipidowy (cholesterol i jego estry) – tzw debris:
•
martwiczo zmienione komórki,
•
kryształy cholesterolu,
•
komórki piankowate – czyli makrofagi tkankowe rzadziej kom. mięśniowe obładowane
lipidami
•
wapń,
•
włóknik i skrzepliny w różnych stadiach organizacji
pokryty „czapeczką”:
•
komórki mięśniowe gładkie (które imigrowały z bł. Środkowej),
•
makrofagi,
•
komórki piankowate,
•
limfocyty,
•
kolagen,
•
włókna sprężyste,
•
proteoglikany,
•
nowo powstające naczynka (neowaskularyzacja)
Elementy morfologiczne:
a)
komórki: mięśniowe gładkie, makrofagi i inne WBC
b)
macierz pozakomórkowa: kolagen włókna sprężyste i proteoglikany
c)
lipidy wewnątrzkomórkowe i pozakomórkowe
kolejność lokalizacji blaszek:
najczęściej zajęta jest chyba aorta brzuszna natomiast nie jest to
wyraźnie powiedziane w tekście(przypisek autora AM)
- tętnice wieńcowe
- tętnice podkolanowe
- tętnica szyjna wewnętrzna
- koło tętnicze Willisa
Wtórne procesy w blaszcze miażdżycowej:
- ogniskowe pęknięcie, owrzodzenie, nadżerka zakrzep(ica) lub zator
- krwotok do blaszki po pęknięciu „czapeczki” lub naczynek zaopatrujących blaszkę
- tętniak (poszerzenia światła) w wyniku ucisku błony środkowej przez blaszkę lub jej zaniku w wyniku
niedokrwienia, utraty włókien sprężystych
Stłuszczenie błony zewnętrznej:
- skupiska komórek piankowatych wypełnionych lipidami, nie powodują zaburzeń w przepływie
krwi, występują już we wczesnym wieku czasami poniżej 1 r.ż a w 10 r.ż u wszystkich. W
naczyniach wieńcowych w miejscach stłuszczenia rozwiną się później pełne blaszki miażdżycowe ale
nie każde stłuszczenie jest podłożem miażdżycy
(trochę to pokręcone)
Epidemiologia i czynniki ryzyka:
- częściej w krajach rozwiniętych mniej np. w Afryce,Azji
- czynniki ryzyka których nie można kontrolować:
•
wiek
•
płeć (mężczyźni są bardziej obciążeni) ale po 70-8 r.ż ryzyko jest równe dla M i K
•
wywiad rodzinny – dziedziczenie wielogenowe
•
zaburzenia genetyczne – rodzinna hipercholesterolemia, homocystynuria
- czynniki ryzyka które można kontrolować:
26

•
hiperlipidemia zwłaszcza hipercholesterolemia
•
nadciśnienie
•
palenie tytoniu
•
cukrzyca
- czynniki, w których do końca nie wiadomo o co chodzi
•
otyłość
•
mała aktywność fizyczna
•
osobowość typu a
•
niedobór estrogenów (menopauza) ochronna rola HTZ
•
dużo cukrów w diecie
•
lipoproteina a – zawiera apo B-100 (LDL)związany z apoA
•
Chlamyda pneumoniae
•
homocysteinemia (żeby obniżyć poziom homocyteiny trzeba zażywać wit. B12,B6 i kw. foliowy)
- dodatkowe czynniki mające wpływ na hemostazę i rozwój zakrzepicy (jako powikłania miażdżycy)
•
wzrost stężenia inhibitora aktywatora plazminogenu
•
wzrost białka C reaktywnego
Patogeneza:
hipoteza odpowiedzi na uszkodzenie – ATH jest przewlekłą odpowiedzią ścian naczyń o
charakterze zapalnym zapoczątkowanym uszkodzeniem śródbłonka
Etapy tego procesu:
•
przewlekłe uszkodzenie śródbłonka dysfunkcja
•
przedostawanie się LDLi w obręb ściany naczyń i ich utlenienie
•
przyleganie monocytów, migracja do błony wewnętrznej, transformacja do makrofagów i kom
piankowatych
•
przyleganie płytek
•
uwalnianie przez te wszystkie nadprogramowe komórki czynników chemicznych powodujących
migrację kom. mięśniowych gładkich
•
proliferacja kom mięśniowych gładkich i produkcja macierzy pozakomórkowej
Ad. Uszkodzenia śródbłonka:
- substancje z papierosów
- homocysteina
- wirusy i inne czynniki zakaźne
- hipercholesterolemia
-zaburzenia w przepływie krwi
Ad. Lipidy
- mogą bezpośrednio zaburzać funkcjonowanie komórek śródbłonka przez zwiększoną produkcję
wolnych rodników, które dezaktywują NO
- utlenione LDL są pochłaniane przez makrofagi (receptor dla resztek), zwiększają napływ monocytów,
pobudzają uwalnianie czynników wzrostu i cytokin, są cytotoksyczne dla komórek śródbłonka i mięśni
gładkich żeby zapobiec utlenianiu – witamina E i beta-karoten
Ad. Makrofagi
– utleniają lipidy dzieje się to co powyżej,
- dodatkowo uwalniają IL-1 i TNF i inne chemokiny (MCP1) zwiększające przyleganie i migrację
leukocytów wgłąb a także czynniki wzrostu – proliferacja kom. mięśniowych gładkich
Ad. Zakażenia
- CMV, Herpes, chlamydia (najpewniejszy)– nie wystepują w ścianach nie zmienionych
- wywołują przewlekły proces zapalny dysfunkcja śródbłonka i znowu to samo
Zapobieganie:
Prewencja pierwotna – modyfikacja czynników ryzyka nie palić, ruszać się, pić winko kontrolować
ciśnienie i wagę etc
Prewencja wtórna – statyny, leki p.płytkowe
Mała modyfikacja ważnej tabelki z Robinsa, pojawiła się też na wykładzie jeżeli dobrze pamiętam
27
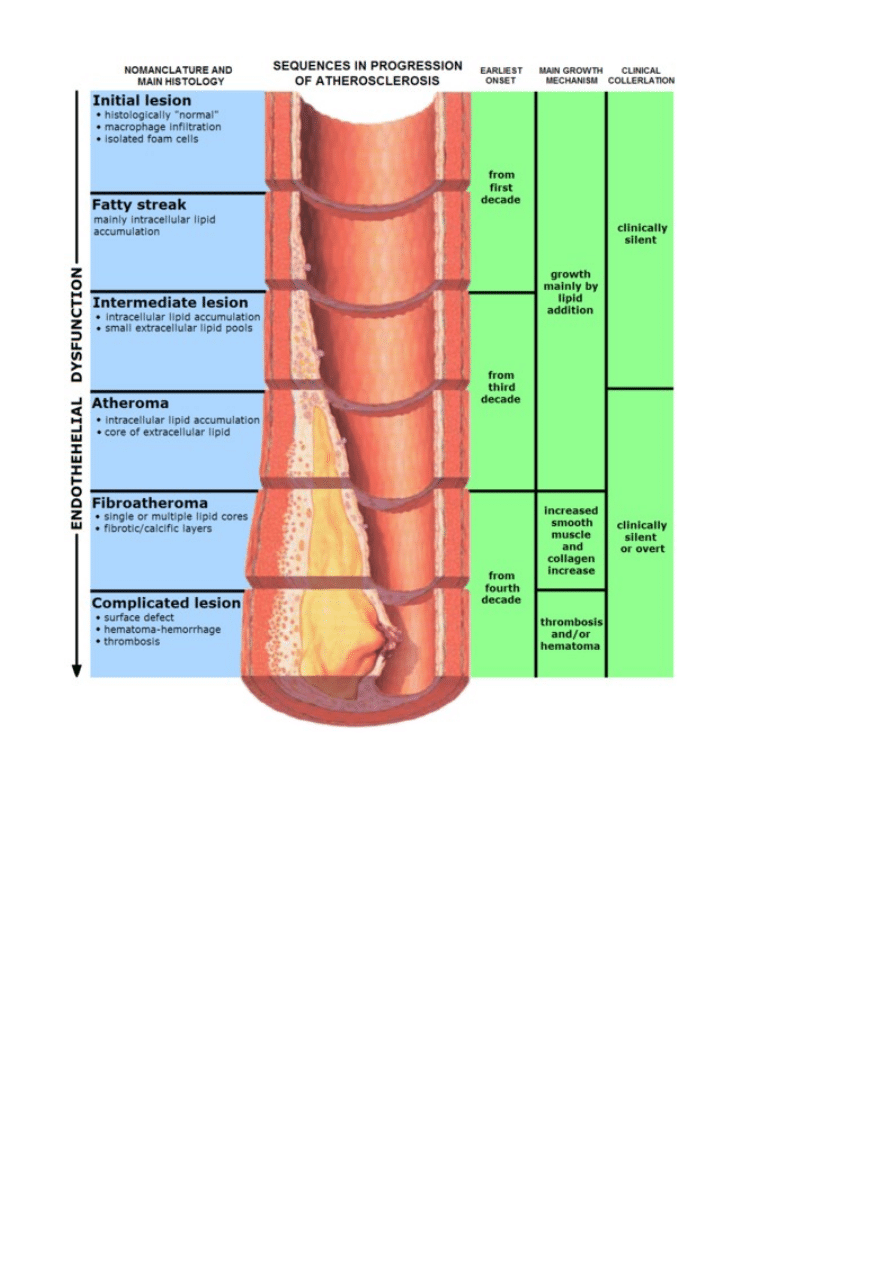
4. Choroba
nadciśnieniowa (hipertonia arterialis):
a) Pierwotne (samoistne) 90-95%
b) Wtórne: nerkopochodne, endokrynne, sercowo-naczyniowe, neurologiczne
HA złośliwe- nagły wzrost ciśnienia, rozkurczowe >120, niewydolność nerek, krwotoki do
siatkówki obrzęk tarczy nerwu wzrokowego, może pojawić się u osób poprzednio zdrowych ale
częściej u tych, które już wcześniej miały HA
regulacja prawidłowego RR
(nie mylić z respiratory rate ku rozpaczy dr Naskręta)
- pojemność minutowa (objętość krwi)
- opór obwodowy
Rola nerek w regulacji RR:
- układ RAA
- produkcja prostaglandyn
- homeostaza sodowa
- ANP działający na cewki dalsze
Mechanizmy nadciśnienia samoistnego:
- obniżenie wydalania sodu przez nerki np. w wyniku mutacji alfa adducyny,
- zwężenie naczyń (zmiany morfologiczne w ścianach lub skurcz mm.gladkich) np. pogrubienie
ściany w wyniku zaburzeń hematologicznych
- czynniki środowiskowe – stres, oyłość, palenie, mała aktywność fizyczna, dużo soli w diecie
Morfologia nadciśnienia:
28

Stwardnienie tętniczek ze szkliwieniem – różowe, szkliste pogrubienie ścian tętniczek z
utratą elementów morfologicznych leżących poniżej oraz ze zwężeniem światła. Występuje
często u osób starszych bez HA. Występuje także w cukrzycy – mikroangiopatia cukrzycowa
a także w łagodnym stwardnieniu naczyń nerkowych (nephrosclerosis). Jest efektem
przechodzenia składników osocza przez śródbłonek naczyń i nadmiernej produkcji macierzy
pozakomórkowej przez komórki mięśniowe gładkie.
Stwardnienie tętniczek z rozplemem – charakterystyczne jest (na ogół) dla HA złośliwego
koncentryczne, warstwowe pogrubienie ścian tętniczek „listki cebuli” ze stopniowym
swężeniem ich świała zbudowanych z kom. mięśniowych gładkich i pogrubiałej błony
podstawnej mogą także być złogi włóknikowate, ostra martwica ścian naczyń (zapalenie
tętniczek z martwicą)
5. Tętniaki i krwiaki rozwarstwiające:
Tętniak (aneurysma) - nieprawidłowe poszerzenie światła naczynia lub serca
•
Prawdziwe (verum) – jeżeli ciągłość ścian jest niezmieniona – tętniaki miażdżycowe,
kiłowe, , wrodzone, tętniaki LK serca po zawale
•
Rzekome (spurium)-pseudotętniaki- powstają w wyniku przerwania ciągłości ścian
naczynia – krew wynaczynia się i tworzy się krwiak pulsujący
•
Rozwarstwienie tętnicy – gdy krew dostaje się pomiędzy warstwy ściany naczynia
•
Przyczyny- ATH i zwyrodnienie torbielowate błony środkowej, inne:urazy, defekty
genetyczne – Marfan, zmiany wrodzone – powstają wtedy małe tętniaki prosowate (berry
aneurysm), zakażenia, kiła, zapalenia naczyń
Podział ze względu na kształt
•
tętniaki workowate - sacciformae
•
tętniaki wrzecionowate - fusiformae
Tętniaki rozwarstwiające podział wg De Bakeya (
taka ciekawostka dla wytrwałych– wiecie, że
DeBakey sam miał tętniaka rozwarstwiającego aorty, kilka lat temu przeszedł operację, obecnie jest
już najdłużej żyjącym po takm zabiegu pacjentem a za kilka m-cy stuknie mu setka i ponoć nadal
udziela się zawodowo – Woźniak ma konkurenta ;))
•
typ I – obejmuje wstępującą i co najmniej łuk i może ciągnąć się dalej
•
typ II –tylko wstępująca
•
typ III – obejmuje zstępującą
podział Stanford
typ A – obejmuje wstępującą bez względu na miejsce powstania
typ B – nie obejmuje wstępującej
Tętniak aorty brzusznej – najczęściej miażdżycowe, zlokalizowane poniżej ujścia tętnic nerkowych,
zmienny kształt i długość, często zawierają owrzodziałe blaszki miażdżycowe pokryte zakrzepami,
często towarzyszom im mniejsze tętniaki naczyń biodrowych
Tętniaki brzuszne zapalne – gęste włóknienie okołoaortalne z nasiloną reakcją zapalną z limfocytów
i komórek plazmatycznych a także licznych makrofagów
Tętniaki brzuszne bakteryjne – na tle miażdżycy + dodatkowe nadważenie mikroorganizmami np.
Salmonella
Patogeneza:
- po 50 r.ż.
- faceci częściej
-defekty genetyczne – np. Marfan i inne
Przebieg kliniczny:
- pęknięcie z masywnym krwotokiem
- zamknięcie światła naczynia
- zator masami kaszowatymi lub zakrzepem przyściennym
- Przemieszczenie sąsiednich struktur (ubytki żeber, ucisk moczowodu)
Tętniaki kiłowe – zrostowe zapalenie tętnic – w kile IIIrzędowej może występować w każdym
miejscu ale najniebezpieczniejsza w tętniczkach odżywczych (vasa vasorum) ściany aorty
zwłaszcza aorty piersiowej (charakterystyczna lokalizacja!)
29

Morfologia:
- zarostowe zapalenie naczyń przydanki niedokrwienie utrata włókien sprężystych i komórek
mięśniowych, zapalenie i bliznowacenie
- może być przyczyną poszerzenia pierścienia zastawki aotralnej niedomykalność
Objawy:
- trudności w oddychaniu (zajęcie płuc) i przewlekły kaszek porażenie nerwu krtaniowego
wstecznego
- zajęcie śródpiersia
- trudności w oddychaniu
- ubytki kostne bóle
- choroby serca (bo zastawka)
- pęknięcie
Rozwarstwienie aorty (krwiak rozwarstwiający) – wnikanie krwi pomiędzy i wzdłuż blaszek, może
pęknąć dając masywny krwotok, nie zawsze towarzyszy mu poszerzenie naczynia.
Pacjenci:
A) mężczyźni między 40-60 r.ż. z HA
B) młodsi z nieprawidłowościami genetycznym jak np. nieśmiertelny zespół
Marfana
C) pacjenci u których doszło do uszkodzenia jatrogennego np. przy kaniulacji
tętnicy
Morfologia – rozdarcie w błonie wewnętrznej, krwiak może dojść do błony środkowej ale jej nie
przekracza.
- Lokalizuje się między środkową i zewnętrzną 1/3 intimy,
- zaczyna się około 10 cm. od zastawki aorty, może rozchodzić się proksymalnie lub dystalnie
-najczęsciej zajmuje aortę wstępującą lub wstępującą i zstępującą (I i II DeBakey
Stanford A)
-czasami może ponownie przebić się do wewnątrz – powstaje zdwojona aorta z kanałem rzekomym,
któ®e mogą po dłuższym czasie zostać wyścielone śródbłonkiem
- zmiana poprzedzająca (występuje w zespole Marfana) - zwyrodnienie torbielowate błony
środkowej (fragmentaryzacja tkanki sprężystej oraz oddzielenie elementów sprężystych i włóknisto-
mięśniowych błony środkowej przez małe torbiele wypełnione macierzą pozakomórkową
Objawy rozwarstwienia:
-nagły rozdzierający ból, w przedniej części klatki piersiowej, promieniujący do grzbietu i przesuwający
się w dół (można pomylić z zawałem)
- pęknięcie do: jany osierdzia, opłucnej, otrzewnej- najczęstsza przyczyna zgonu
- tamponada serca
- niedomykalność zastawki aortalnej
- zawał mięśnia sercowego
-objawy niedokrwienia ważnych narządów np. nerek, rdzenia kręgowego (poprzeczne zapalenie)
11
12
PŁUCA I GÓRNE DROGI ODDECHOWE
Patologia płuc dotyczy dróg oddechowych, miąższu i systemu naczyniowego.
1.
Wady wrodzone. (Nie obowiązują na egzamin.)
2. Zmiany w upowietrznieniu:
Pozapłucne
30

Niedodma (atelectasis) znana również jako zapadniecie, jest utratą objętości płuca spowodowaną
nieodpowiednim rozprężeniem przestrzeni powietrznych. Towarzyszy temu przetaczanie
nieodpowiednio utlenowanej krwi z tętnic płucnych do żył, co prowadzi do zaburzenia równowagi
wentylacja-perpuzja i hipoksji. Uwzględniają mechanizm, który znajduje się u podstaw tej zmiany lub
lokalizację zapadniętych pęcherzyków, niedodmę dzieli się na cztery kategorie:
- resorpcyjna – gdy zaczopowanie uniemożliwia przedostawanie się powietrza do dystlanego odcinka
dróg oddechowych, znajdujące się tam powietrze jest stopniowo wchłaniane z następowym
zapadnięciem się pęcherzyków, może być zajęte całe płuco, cały płat lub jeden czy więcej segmentów.
Najgęstsze przyczyny zaczopowania: czopy śluzowe lub śluzowo-ropne, czasem zaaspirowanie ciał
obcych lub skrzep krwi, nowotwory, powiększone węzły chłonne i tętniaki naczyniowe.
- z ucisku – spowodowana gromadzeniem się płynu, krwi lub powietrza w jamie opłucnej, co powoduje
mechaniczne zapadnięcie się przylegającego płuca (zastoinowa niewydolność serca, pneumothorax,
wysokie ustawienie przepony)
- mikroniedodma – utrata możliwości rozprężania się płuca w następstwie: braku surfaktantu, RDS,
schorzenia płuc związane z zap. śródmiąższowym, pooperacyjna.
- z obkurczenia – w skutek miejscowych lub uogólnionych zmian włókniejących płuca lub opłucnej i
może utrudniać rozprężanie i elastyczne zwiększanie wyrzutu podczas wydechu.
Jako skutek rozlanych chorób płuc:
a)ch. obturacyjne (ch.dróg powietrznych) charakteryzujące się ograniczeniem przypływu
powietrza w wyniku wzrastającego oporu wskutek częściowego lub całkowitego zaczopowania na
dowolny poziomie
dychawica oskrzelowa (astma bronchiale)- odwracalne skurcze oskrzeli spowodowane
nadmierną reakcją oskrzelowo-skurczową na różne czynniki stymulujące.
Dychawica oskrzelowa = Astma oskrzelowa
- najczęściej objawia się chorobami alergicznymi
- napady utrudnionego wydechu
- przyczyną są alergeny, często te zawarte w kurzu(uczulenie na kurz), ale także przyczyną mogą być:
zakażenia wirusowe, niektóre leki (beta-Blokery i Aspiryna), działanie niektórych gazów (SO
2
, NO, O
3
),
stresowa sytuacja, zły stan psychiczny, wyczerpanie fizyczne, zimne powietrze
Charakterystyczne cechy morfologiczne:
- skurcz mm. małych oskrzeli
- obecność gęstego śluzu i treści surowiczej
- granulocyty (przede wszystkim kwasochłonne), limfocyty, kom. plazmatyczne, mastocyty w świetle
oskrzeli
To wszystko utrudnia głównie wydech (ale wdech także może być czasem utrudniony).
Z czasem dochodzi do:
*nacieku tych samych komórek w bł. śluzowej oskrzeli i tk. łącznej okołooskrzelowej
*przerostu gruczołów małych oskrzeli (one najpierw nadmiernie wydzielają, a potem ulegają
przerostowi)
*przerostu mięśniówki oskrzeli
*pogrubienia bł. podstawnej nabłonka oskrzelowego
*rozdęcia tk. płucnej z ogniskami niedodmy (jest to niedodma wtórna)
rozdęcie jest to mechaniczne rozrywanie ścian pęcherzyków
Ogólnie astmę dzielimy na dwie główne kategorie, biorąc pod uwagę obecność lub brak podłoża
immunologicznego:
Astmę zewnątrzpochodną cechuje epizod astmatyczny najczęściej inicjowany przez reakcję
nadwrażliwości typu I, wywoływaną przez ekspozycję na antygen zewnętrzny, gdzie ważna rolę
odgrywają limfocyty Th2 (dają reakcję z przeciwciałem CD 34), które wydzielają przede wszystkim
interleukinę 4 i 5 wzmożone wydzielanie Ig E przez limf.B, zwiększenie liczby mastocytów oraz
31

zwiększenie liczby eozynofilów i ich aktywacja. Mastocyty z kolei od wpływem antygenu uwalniają w bł.
śluzowej różne dalsze chemiczne mediatory zapalenia. Jednocześnie dochodzi do podrażnienia
receptorów n. błędnego skurczu mięśniówki oskrzeli. Wyróżnia się trzy typy astmy zewnątrzpochodnej:
atopową, zawodową (wiele postaci ) i alergiczną aspergilozę płucno-oskrzelową.
Astmę wewnątrzpochodną, której mechanizm wyzwalający nie ma uwarunkowania
immunologicznego. W tej postaci astmy liczne czynniki stymulujące, które u zdrowych osób maja mały
wpływ lub w ogóle go nie mają, wywołują skurcz oskrzeli. Do takich czynników należą aspiryna,
zakażenia płuc, zwłaszcza wirusowe, zimno, stresy i substancje drażniące, takie jak ozon i dwutlenek
siarki. Zwykle w wywiadzie bark danych o osobniczych lub rodzinnych objawach alergicznych, a
stężenie IgE w surowicy jest prawidłowe.
rozedma (emphysema)
Rozedma jest określana jako trwałe powiększenie przestrzeni powietrznej obwodowo w stosunku do
oskrzelików końcowych, czyli jest ograniczona do gronka, któremu towarzyszy uszkodzenie ich ścian.
R. śródmiąższowa(emphysema interstitiale) – powstaje w następstwie rozrywania utkania płuca
np. przy gwałtownym wtłaczaniu powietrza przy stosowaniu respiratorów, jeśli jest to nieumiejętnie
zrobione; czasami występuje u dzieci w następstwie gwałtownie przebiegających zapaleń – dochodzi do
uszkodzenia ścian pęcherzyków i dostawania się powietrza do przegród międzypęcherzykowych.
Możliwym, ale na szczęście bardzo rzadkim powikłaniem jest rozerwanie opłucnej odma
R. pęcherzykowa(e. vesiculare)
– rozwija się powoli i wiąże ze zmianami chorobowymi płuca,
wyróżniamy 3 postaci: pierwotna=samoistna=obturacyjna, wtórna i zastępcza.
R. p. pierwotna stanowi wyosobnioną jednostkę, której podstawą jest zaburzenie równowagi elastyna/
alfa
1
-antytrypsyna czyli proteaza/antyproteaza. Pod wpływem różnorakich czynników może dojść do
wytworzenia nadmiernej ilości elastazy, która uszkadza płuco. Inne znowu czynniki powodują, że nie
ma dostatecznej aktywności antytrypsyny, która broni płuco przed elastazą. Elastaza jest wytwarzana i
uwalniana przez neutrofile, makrofagi, kom. żółtakowe, podobno też przez trzustkę i niektóre bakterie.
Główną rolę w rozedmie najczęściej odgrywa elastaza wytwarzana przez neutrofile. U palaczy zwykle
obserwujemy spadek aktywności alfa
1
-antytrypsyny(oksydanty z dymu tytoniowego i WRT uwalniane
przez neutrofile ja unieczynniają), poza tym mają oni zwiększoną liczbę neutrofilów i makrofagów w
płucach uwalniają one elastazę, która nie jest unieczynniana przez antytrypsynę.
Morfologicznie wyróżnia się rozedmę centralną (środkowej części zrazika), brzeżną (okołoprzegrodowa)
i zrazikową (całego zrazika).
W centralnej poszerzone są środkowe i obwodowe części zrazika utworzone przez oskrzeliki
oddechowe oraz bliższe odcinki przewodów pęcherzykowych, a więc to, co jest bliżej głównych oskrzeli.
W obwodowej=marginalnej=brzeżnej: zmienione są proksymalne części zrazika a więc woreczki
pęcherzykowe i same pęcherzyki. Ściany pęcherzyków pękają i tworzą się takie duże „balony”.
W zrazikowej: wszystkie te anatomiczne struktury są zajęte.
=>zwiększa się objętość nabłonków, ale nie zwiększa się pojemność oddechowa płuca, bo ściany te są
niewydolne. Następuje utrudnienie wymiany gazowej, uciśnięcie oskrzeli. Miąższ jest blady, suchy, a
płuca są duże, puszyste (można powiedzieć, że niemal nie mieszczą się w klatce piersiowej). Klatka
piersiowa jest beczkowata. Prowadzi to do zespołu serca płucnego.
Rozedma starcza odnosi się do nadmiernego rozdęcia płuc u osób starszych w wyniku
uwarunkowanych wiekiem zmian w wewnętrznej geometrii płuca (np. większe przewody pęcherzykowe
i mniejsze pęcherzyki). Nie ma tu wyraźnego niszczenia tk. i lepszym określeniem dla tych zmian
byłoby starcze rozdęcie.
Rozdęcie zatorowe odnosi się do stanu, w którym opluca zostaj rozdęte prze uwięzione w nich
powietrze.
Rozedma kompensacyjna oznacza rozdęcie pęcherzyków w wyniku utraty struktury płucnej, np. w
pozostałym miąższu płuca po chirurgicznym usunięciu chorego płuca lub jego płata.
Rozedma zastępcza – rozdęcie płuca w otoczeniu tkanki zmienionej niedodmowo z różnych przyczyn
( blizny, nacieki, zmiany zapalne, etc.), jako wyrównanie.
Emphysema bullosum – gdy makroskopowo są widoczne pęcherze.
Rozedma występuje często, ale trudno jest oszacować częstość jej występowania, ponieważ ostateczne
rozpoznanie, które opiera się na morfologii, może być ustalone tylko na podstawie sekcyjnego badania
płuca. Genezą dwóch częstych postaci rozedmy – środkowej części zrazika i całego zrazika – jest
zaburzenie równowagi proteaza-antyproteaza oraz oksydacja-antyoksydacja, ich efekty sumują się w
postaci uszkodzenia tkanki. Istnieje jednoznaczny związek pomiędzy intensywnym paleniem
papierosów a rozedmą, wpływ składników dymu tytoniowego, zwłaszcza na rozwidlenie oskrzelików
32

oddechowych powoduje napływ neutrofilów i makrofagów, które wydzielają proteazy. Wzrost
aktywności proteaz zlokalizowany w środkowej części zrazika, łącznie z indukowanym przez palenie
tytoniu uszkodzeniem oksydacyjnym powoduje u palaczy rozedmę centralnej części zrazika. Rozpad
tkanki jest wzmożony w następstwie inaktywacji ochronnej antyproteazy przez aktywne rodniki tlenowe
obecne w dymie tytoniowym. Ten schemat wyjaśnia również nakładanie się wpływu palenia i niedoboru
α1-antytrypsyny na indukowaniu poważnej choroby zamykającej drogi oddechowe. (To zdanie dedykuje
Dr G:p)
Pierwszym objawem jest duszność, która systematycznie narasta, , częsta jest utrata masy ciała,
płucne testy czynnościowe wykazuja obniżenie FEV
1
oraz FEV
1
/FVC, u osób u których nie występuje zap.
oskrzeli jest beczkowata klatka piersiowa i duszność z wyraźnie przedłużonym wydechem,
hiperwentylacja, pacjent siadając w przygarbionej pozycji stara się wycisnąć powietrze z płuc przy
każdym wydechu. Do późnej fazy choroby wymiana gazowa jest odpowiednia , wartośći wysycenia krwi
gazami prawidłowe. Ze względu na znaczna duszność i prawidłowe utlenownai hemoglobiny pacjentow
takich okreśła się jako „różowi dyszący” (pink puffers).
Drugą skrajnością są pacjenci z przewlekłym zap. oskrzeli lub astmatycznym zap. oskrzeli, u których
kaszel i sapanie mogą być pierwszymi dolegliwościami, w wywiadzie nawracające infekcje z ropną
plwociną. Maja oni zwykle mniej nasilona duszność i zmniejszona wydolność oddechową, dlatego
następuje zatrzymanie CO
2
, niedotlenienie i często sinica, zastoinowa niewydolność serca i obrzęki,
określamy ich jako „sini obrzęknięci” (blue bloaters).
U wszystkich tych pacjentów rozwinie się stopniowo wtórne nadciśnienie płucne, wynikające z skurczu
naczyń płucnych indukowanego hipoksją i zmniejszenia powierzchni włośniczek płucnych w wyniku
uszkodzenie pęcherzyków. Śmierć z powodu rozedmy jest spowodowana niewydolnością płuc z kwasicą
oddechową, hipoksją i śpiączką lub niewydolnością prawokomorową serca ( serce płucne).
Przewlekłe zapalenie oskrzeli jest określane na postawie objawów klinicznych, jak obecność
przewlekłego i nawracającego kaszlu z odksztuszaniem (utrzymujący się kolejno przez 3 miesiące
przynajmniej w ciągu 2 lat) z nadmiernym wydzielaniem śluzu, anatomiczna struktura rozmieszczenia
obejmuje zarówno duże, jak i małe drogi oddechowe(przewlekłe zapalenie oskrzelików).
Najważniejsza przyczyna jest palenie papierosów, znaczenie mają też inne zanieczyszczenia powietrza,
takie jak dwutlenek siarki i dwutlenek azotu. Czynniki drażniące indukują nadmierną sekrecję
gruczołów śluzowych oskrzeli, powodują przerost gruczołów śluzowych i prowadzą do
metaplastycznego tworzenia się wydzielniczych komórek kubkowych w powierzchniowym nabłonku
oskrzeli. Ponadto powodują zapalenie z naciekaniem limfocytów T CD8+, makrofagów i neutrofilów. W
przeciwieństwie do astmy nie ma eozynofilów.
Makroskopowo bł. śluzowa dużych dróg oddechowy jest zwykle przekrwiona i obrzęknięta. Często
pokryta jest wartą wydzieliny śluzowej lub śluzoworopnej. Histologiczną cechą diagnostyczną jest
powiększenie śluzowych gruczołów sekrecyjnych. Często widać zwiększającą się liczbę komórek
kubkowych ze współistniejąca utratą komórek urzęsionych. Często powstaje metaplazja
płaskonabłonkowa z następującymi po niej zmianami dysplastycznymi. Dochodzi do włóknienia,
rozplemu tk. łącznej, więc może dojść również do obturacji. Kaszel i wytwarzane plwociny mogą trwać
nieokreślony czas bez zaburzeń wentylacji, jednak u niektórych pacjentów rozwija się wyraźny COPD.
Rozstrzenie oskrzeli jest stałym rozdęciem oskrzeli i oskrzelików spowodowanym zniszczeniem
mięśni i elastycznej tkanki podporowej, wynikającym lub współwystępującym z przewlekłymi
zakażeniami martwiczymi. Zmianami predysponującymi do rozstrzenia oskrzeli są zaczopowanie
oskrzeli oraz stany wrodzone (mukowiscydoza, niewydolność immunologiczna, zespół Kartagenera),
martwicze lub ropne zapalenie płuca.
Występują zwykle obustronnie w dolnych płatach, zwłaszcza w płatach z najbardziej pionowym
przepływem powietrza, jeśli przyczyna są nowotwory lub ciała obce mogą być ograniczone do jednego
segmentu. Nasilone zmiany zwykle w obwodowych oskrzelach i oskrzelikach. W zaawansowanych
przypadkach intensywny wysięk zapalny oraz złuszczanie się wyścielającego nabłonka prowadza do
powstania rozległych owrzodzeń. Włóknienie rozwija się w bardziej przewlekłych przypadkach. W
33

zaawansowanych przypadkach martwica niszczy ściany oskrzeli lub oskrzlików i tworzy się ropień
płuca.
b)ch. restrykcyjne charakteryzujące się zmniejszoną podatnością płuc - ograniczenie rozprężania
miąższu płucnego, któremu towarzyszy ograniczenie całkowitej pojemności płuca:
Zespół błon szklistych - ciężka niedomoga oddechowa noworodków(IRDS) Podstawowym
zaburzeniem jest niezdolność niedojrzałych płuc do tworzenia odpowiedniej ilości surfaktantu, którego
niedobór w płucach powoduje tendencję pęcherzyków do zapadania się, w skutek czego niezbędny jest
odpowiednio większy wysiłek oddechowy, by z każdym wdechem otworzyć pęcherzyki. U noworodka
szybko następuje wyczerpanie oddechowe i rozpoczyna się uogólniona niedodma. Rozwijające się
niedotlenienie wprowadza w ruch łańcuch zdarzeń prowadzących od uszkodzenie nabłonka i
śródbłonka, a wreszcie do tworzenia się błon szklistych.
Ostre uszkodzenie płuc (ALI) uważa się za wczesną fazę ARDS, w której łagodne zaburzeni funkcji
oddechowej mogą stopniowo przekształcać się w bardziej typowy ARDS o ciężkim przebiegu klinicznym
Zespół ostrej niewydolności oddechowej (ARDS). ALI i ARDS charakteryzują się: nagłym
wystąpieniem duszności, zmniejszeniem ciśnienia parcjalnego tlenu we krwi tętniczej (hipoksemia),
rozwojem obustronnych nacieków widocznych w obrazie radiologicznym płuc oraz brakiem klinicznych
cech pierwotnej lewokomorowej niewydolności serca.
Następuje pośledzenie integralności bariery śródbłonkowej, nabłonkowej lub – najczęściej – obydwu.
Ostre następstwa uszkodzenia błony pęcherzykowo-włośniczkowej to zwiększona przepuszczalność
naczyń krwionośnych i gromadzenie płynu w świetle pęcherzyka, utrata zdolności dyfuzji oraz rozsiane
zaburzenia w obrębie surfaktantu, wywołane uszkodzeniem pneumocytów typu II. Najogólniej można
powiedzieć, że uszkodzenie płuca jest wywołane prze zaburzenie równowagi między cytokinami
nasilającymi i hamującymi procesy zapalne, istotną role w patogenezie odgrywają neutrofile.
Dwie fazy rozwoju:
1) zastój krwi
pojawiają się neutrofile
następnie martwica nabłonka pęcherzyków płucnych
wylew krwi do pęcherzyków
zapadanie się ściany pęcherzyków
zakrzepica
=> zaburzenia w krążeniu i martwica nabłonka
2) jeśli chory wyjdzie z ciężkiej fazy pierwszej, następuje ona po 2-3 tyg.
faza proliferacyjna: rozplem pneumocytów typu II i fibroblastów
=> dochodzi do włóknienia śródmiąższowego
Przebieg kliniczny jest bardzo ciężki, objawy u około 85% pacjentów rozwijają się w ciągu 72 godzin od
zadziałania czynnika uszkadzającego. Dawniej śmiertelność sięgała 100%, obecnie 30-40%, ale w
przypadkach wywołanych posocznicą jest zwykle wyższa. Nawet jeżeli pacjent przeżyje może dojść do
rozlanego włóknienia śródmiąższowego.
Przewlekłe restrykcyjne choroby płuc
Idiomatyczne włóknienie płuc (IPF) zwane też zwłókniającym zapaleniem pęcherzyków płucnych
jest schorzeniem płuc histologicznie charakteryzującym się rozlanym włóknieniem śródmiąższowym,
którego następstwami w przypadkach zaawansowanych są ciężka hipoksemia i sinica. Choroba częściej
dotyczy mężczyzn, a około 2/3 pacjentów ma w momencie rozpoznania ponad 60 lat.
Początkiem jest uszkodzenie ściany pęcherzyków płucnych o nieznanym charakterze. Jego
następstwem jest obrzęk śródmiąższowy i gromadzenie się komórek zapalnych. Jeśli proces
uszkadzający ma umiarkowane nasilenie, dochodzi do wycofania się procesu chrobowego i odtworzenia
prawidłowej struktury miąższu płuca. W innych przypadkach interakcje komórkowe pomiędzy
limfocytami, makrofagami, neutrofilami i komórkami nabłonka pęcherzykowego prowadzą do
proliferacji fibroblastów i postępującego włóknienia śródmiąższowego. Podejrzewa się, że ciąg zderzeń
inicjują mechanizmy immunologiczne.
34

Sarkoidoza jest wieloukładową chorobą o nieznanej etiologii, charakteryzującą się obecnością
nieserowaciejących zarniniaków w wielu tkankach i narządach, które mogą jednak występować również
w innych schorzeniach (w pewnych typach gruźlicy, w niektórych zmianach grzybiczych, w kile,
leiszmanozie, brucelozie, turalemii, berylozie, ch. Leśniowskiego-Crohna, a także w zmianach
odczynowych np. w przebiegu niektórych nowotworów) dlatego rozpoznanie sarkoidozy jest diagnozą
przez wykluczenie. Obustronna limfadenopatia lub obustronne zajęcie płuc (lub obie te zmiany)
widoczne w obrazie radiologicznym klatki piersiowej są w większości przypadków głównymi objawami,
mogą być zajęte oskrzeliki, oskrzela, śródmiąższ. Charakterystyczną cechą są guzki sarkoidalne, które
występują nie tylko w płucu (skóra, nerki, inne narządy) Zajęcie oczu i skóry występuje w 25%
przypadków i może być czasami dominującym objawem choroby.
Cechą charakterystyczną wyłącznie dla guzków sarkoidalnych (która jednak nie występuje często) jest
obecność ciałek Schaumanna-Weissnera (koncentryczne warstwy zasadochłonne, zawierają Fe, Ca) i
tzw. asteroidów (zawierają szczawiany wapnia, drobne wł. kolagenu)
- patogeneza niejasna, prawdopodobnie związana z dysfunkcją krążących limfocytów T i z nadmierną
aktywnością limfocytów B
- nierzadka
- doprowadza do włóknienia płuca
- dobrze poddaje się leczeniu kortykosteroidami, ale niektórzy chorzy wykazują na to leczenie oporność
(przyczyna nieznana)
Alergiczne zapalenie płuc jest zapalną chorobą płuc o podłożu immunologicznym, która pierwotnie
dotyczy pęcherzyków i dlatego nazywana jest alergicznym zapaleniem pęcherzyków. Może występować
bądź jako ostra reakcja z gorączką, kaszlem, dusznością i ogólnoustrojowymi dolegliwościami w 4-8
godzin po ekspozycji, bądź jako choroba przewlekła z podstępnym początkiem, kaszlem, dusznością,
złym samopoczuciem i utratą masy ciała. Alergiczne zapalenie płuc jest odpowiedzią immunologiczna
na zewnętrzny antygen, która obejmuje reakcję nadwrażliwości zarówno typu III (kompleks
immunologiczny), jak i typu IV (reakcja opóźniona).
Zespoły rozlanego krwawienia pęcherzykowego - „pierwotna” choroba o podłożu
immunologicznym, która występuje jako triada – krwioplucie, niedokrwistość i rozlane nacieczenie
płucne. Prototypem tego zaburzenie jest zespół Goodpasture’a charakteryzujący się zapaleniem
kłębuszków nerkowych i krwotocznym śródmiąższowym zapaleniem płuc wskutek odkładania się
przeciwciał przeciw antygenom wspólnym dla kłębuszkowych i płucnych błon podstawnych będących
wyrazem nadwrażliwości cytotoksycznej typu II. Inną odmianą tego schorzenia jest idiomatyczna
hemosyderoza płucna, która ma podobny obraz w płucach, bez towarzyszącej choroby nerkowej, ale
nie występują przeciwciała przeciw błonom podstawnym.
Zapalenie naczyń płucnych i zarniniakowatość - ziarniniak Wegenera jest klasyczną chorobą
układową, może jednak ograniczać się do płuc. WG jest prototypem zaburzenia w grupie zapaleń
naczyń, znanego jako zapalenie tętnic płucnych i zarniniakowatość. Zmiany płucne w WG
charakteryzują się połączeniem martwiczego zapalenia naczyń ze śródmiąższowym zapaleniem
martwiczo-ziarniniakowym. W naczyniach płucnych mogą również występować zapalenia
współistniejące z martwicą włóknikowatą. W WG mogą występować objawy zarówno ze strony górnych
dróg oddechowych jaki i objawy płucne.
Płuco w zaburzeniach kolagenowych naczyń (toczeń układowy, reumatoidalne zapalenie stawów,
sklerodermia oraz zapalenie skórno-mięśniowe i wielomięśniowe) Najczęściej stwierdzaną zmianą
płucną jest zapalenie śródmiąższowe, któremu towarzyszy lub się nim kończy rozlane zwłóknienie
śródmiąższowe.
Pylica, azbestoza, patrz choroby środowiskowe….. :p
Patologia przeszczepu dotyczy zarówno dróg oddechowych jak i naczyń. Podobnie jak inne
przeszczepy, również przeszczepy płucne są narażone na niepowodzenie wskutek odrzucenia
immunologicznego. Zostały one podzielone na odrzuty ostre, w których wyróżniamy 4 stopnie
uszeregowane w kolejności jego nasilenia oraz odrzuty przewlekły. Oba rodzaje wymagają oceny
zarówno dróg oddechowych jak i naczyń. Należy pamiętać, że pacjenci z przeszczepem są prawie
zawsze poddani leczeniu immunosupresyjnemu i w związku z tym są podatni na wiele zakażeń
oportunistycznych, które mogą klinicznie i histopatologicznie imitować odrzut.
35

3. Zaburzenie w krążeniu płucnym:
zator (embolia)
zawał (infarctus)
DIC (disseminated intravascular coagulation)
przekrwienie bierne (hyperaemia passiva)
obrzęk (oedema)
nadciśnienie płucne
Patrz odpowiednio inne opracowania.
4. Zapalenia oskrzeli:
ostre
przewlekłe
Patrz odpowiednio inne miejsca opracowania :p
5. Zapalenie płuc
Definiujemy jako każde zakażenie w płucu. Może objawiać się jako ostra, gwałtownie przebiegająca
choroba lub jako przewlekła o przedłużonym działaniu. Histologicznie obraz zapalenia płuc może się
bardzo różnić, od włóknikowo-ropnego wysięku w pęcherzykach płucnych, stwierdzanego w ostrym
bakteryjnym, do śródmiąższowego nacieku komórek jednojądrzastych w wirusowym i innych
nietypowych, wreszcie do ziarniniaków i zmian jamistych stwierdzanych w wielu przewlekłych
zapaleniach płuc. Ostre bakteryjne zapalenie płuc może występować jako jedna z dwóch postaci
określanych anatomicznie i radiologicznie jako odoskrzelowe i płatowe zapalenie płuc. W zapaleniu
odoskrzelowym (bronchopneumonia) ogniska zapalenia są rozproszone, z reguły stwierdza się je w
więcej niż jednym płacie. Wynika to z wstępnego zakażenia oskrzeli i oskrzelików, a następnie
rozszerzania się go na sąsiadujące pęcherzyki. Natomiast w płatowym zapaleniu płuc przylegające
przestrzenie powietrzne w części bądź w całości wypełnione są jednorodnym płynem wysiękowym, co
można uwidocznić na zdjęciach rentgenowskich jako naciek obejmujący płat lub segment. Anatomiczny
obraz pomiędzy nimi może być jednak niekiedy zatarty dlatego też najlepiej dzielić zapalenia płuc na
podstawie swoistego czynnika etiologicznego lub jeżeli nie może być wyizolowany na podstawie
sytuacji klinicznej co w istotny sposób zawęża listę podejrzanych czynników patogennych dla
empirycznego zastosowania leczenia przeciwbakteryjnego.
Środowiskowo nabyte zapalenia płuc - Streptococcus pneumoniae, Haemophilus influenzae,
Moraxella catarrhalis, Staphylocosccus ureus, Legionella pnumophila, Enterobacterie (Klebsiella
pneumoniae) i Pseudomonas.
Nierzadko występują po zakażeniach wirusowych górnego odcinka układu oddechowego, z nagłym
początkiem, wysoką gorączka, dreszczami, bólem opłucnowym i wilgotnym, wytwórczym, śluzowo-
ropnym kaszlem. Najczęstszą przyczyna są infekcje pnumokokowe zarówno jako zakażenia płatowe lub
odoskrzelowe o typowym przebiegu, który obejmuje 4 stadia: przekrwienie, zwątrobienie czerwone,
zwątrobienie szare i rozejście, obecnie ze względu na skuteczną antybiotykoterapie praktycznie nie
spotykane. Coraz częściej stosowane są szczepionki pneumokokowe z polisacharydów ścian
komórkowych.
Haemophilus influenzae – zap. wywołuja postaci z otoczką i bez, zagrożeni szczególnie pacjenci z
przewlekłymi chorobami płuc (przewlekłe zapalenie oskrzeli, mukowiscydoza, rozstrzenie oskrzeli).
Moraxella catharrhalis – coraz częstsza przyczyna, zwłaszcza u osób starszych, u dzieci często prowadzi
do zapalenia ucha środkowego.
36

Staphylococcus ureus – istotny czynnik wtórnych, bakteryjnych zakażeń, w zapaleniach
gronkowcowych zwiększona jest częstość występowania powikłań, takich jak ropień i ropniak opłucnej,
współistniejące z zapaleniem wsierdzia jest powikłaniem u nadużywających leków dożylnych.
Klebsiella pneumoniae – najczęstsza przyczyna Gram-ujemnego zap. płuc, często dotyczy osób
niedożywionych i alkoholików, charakterystyczna jest gęsta, galaretowata plwocina ponieważ Klebsiella
wytwarza obfite, lepkie polisacharydy otoczkowe, które powodują trudności w odksztuszaniu.
Psuedomonas aeruginosa – równie często spotykana w zakażeniach szpitalnych, u pacjentów z
neutropenią, z rozległymi oparzeniami, u sztucznie wentylowanych, ma właściwości wnikania do
naczyń krwionośnych w miejscu zakażenia z następowym rozsiewem wewnątrzpłucnym, posocznica
Pseudomonas jest chorobą o piorunującym przebiegu.
Legionella pnumophila – zapalenia wywołanych przez nią często u osób z predyspozycjami do chorób
serca, nerek oraz chorób immunologicznych i hematologicznych, szczególnie biorczy przeszczepów,
często jest to zapalenie o ciężkim przebiegu, gorączka Pontiac odnosi się do samoustępującego
zakażenia górnych dróg oddechowych.
Środowiskowo uwarunkowane atypowe zapalenia płuc (Mycoplasma pneumoniae, Chlamydia
pneumoniae, C. psittaci, C. trachomatis, Coxiella burnetti, wirusy (RSV, wirus paragrypy(dzieci), grypa
A i B, adenowirus)
Przebieg kliniczny pierwotnego atypowego zapalenie płuc jest bardzo zróżnicowany, może naśladować
ciężkie zakażenie górnych dróg oddechowych, które przemija nierozpoznane lub może objawiać się
jako piorunujące, zagrażające życiu zakażenie u pacjentów z obniżoną odpornością immunologiczna.
Bardziej typowo ma charakter ostrego, nieswoistego schorzenia charakteryzującego się gorączka,
bólem głowy, złym samopoczuciem, a później kaszlem z nieznacznym odksztuszaniem plwociny.
Objawy przedmiotowe są charakterystycznie minimalne i nieodróżnialne od zapalenia odoskrzelowego,
w Mycoplasma można stwierdzić naciek płatowy. Obraz radiologiczny ujawnia zwykle przejściowe,
słabo odgraniczone ogniska, głownie w płatach dolnych. Zdefiniowanie czynnika przyczynowego jest
trudne.
Morfologiczna struktura jest podobna, proces może być jednorodnie rozproszony, obejmować
jednostronnie lub obustronnie całe płaty. Makroskopowo zmienione obszary są czerwono-niebieskie,
przekrwione i trzeszczące. Odczyn zapalny w większości ograniczony do ścian pęcherzyków, w
przeciwieństwie do go bakteryjnych gdzie nacieczone są też przestrzenie pęcherzykowe. W ciężkich w
pełni rozwiniętych przypadkach może rozwinąć się rozlane uszkodzenie pęcherzyków z tworzeniem się
błon szklistych.
Zapalenie grypowe – rozpoczyna się zapaleniem błon śluzowych w obrębie jamy nosowej, gardła,
krtani, tchawicy, oskrzeli. Bł. śluzowa jest obrzęknięta, czerwona, niekiedy z nadżerkami. Może dojść
do dużego wyniszczania nabłonka w ciężkim przebiegu, aż do ogołocenia bł. podstawnych – gromadzi
się wtedy wysięk włóknikowy i nazywamy to
bronchitis fibrinosa profunda
. Wtedy zwykle dołącza się,
w takim ciężkim przebiegu, zapalenie śródmiąższowe płuc.
Bł. szkliste odkładają się przede wszystkim w przewodach pęcherzykowych i w pęcherzykach płucnych.
W odróżnieniu od zespołu bł. szklistych noworodków, gdzie bł. szkliste odkładają się przede wszystkim
w oskrzelikach oddechowych i w przewodach pęcherzykowych, a nie w pęcherzykach.
Zapalenie grypowe jest często powikłane infekcją bakteryjną, przede wszystkim paciorkowcową.
Zapalenie odrowe płuc – występuje o chorych z obniżoną odpornością i zwykle wtedy brak jest
wysypki (ciężko tę odrę rozpoznać). Liczne komórki olbrzymie wielopłatowe wyściełają pęcherzyki
płucne i przewody pęcherzykowe. Te wielojądrzaste formy powstają w wyniku fuzji kom. nabłonka.
Charakterystyczne są też obecne w tych komórkach ciała wtrętowe ( w jądrach i w cytoplazmie).
Inne przyczyny zapaleń śródmiąższowych:
Zapal. śródmiąższ. – wspólne cechy morfologiczne:
37

- nacieki zapalne w przegrodach pęcherzykowych i w tkance okołooskrzelowej
- częsta obecność wysięku białkowego w pęcherzykach ( niekiedy z wytwarzaniem bł. szklistych)
Wewnątrzszpitalne zapalenia płuc określamy zakażenie płucne nabyte podczas hospitalizacji
(powyżej 48h) Wywoływane głównie przez Gram-ujemne pałeczki należące do enterobakterii
(Klebsiella, Serratia marcescens, Escherichia coli) i Pseudomonas oraz penicylinooporny
Staphylococcus ureus. Spektrum zapaleń szpitalnych jest ubocznym efektem skuteczności leczenie. Są
częste u pacjentów z poważnymi chorobami podstawowymi, obniżeniem odporności, przedłużającą się
terapią antybiotykową lub ze stosowanie inwazyjnych narzędzi.
Zachłystowe zapalenia płuc wywoływane przez beztlenową florę ustną, są zwykle zapaleniami
typu martwiejącego z następującym po nich piorunującym przebiegiem kliniczny i często zgonem u
pacjentów ze skłonnościami do zachłyśnięcia.
Przewlekłe zapalenia płuc (Nocardia, Actinomyces, Ziarniniakowe: Mycobacterium tuberculosis i
nietypowe prątki)
Gruźlica jest zakaźna przewlekłą chorobą ziarniniakową powodowana przez Mycobacterium
tuberculosis. Zwykle zajmuje płuca, ale może dotyczyć każdego narządu i tkanki ustroju. Ważne jest
odróżnienie zakażenia od choroby. Zakażenie oznacza zagnieżdżenie ogniska z bakteriami, które
mogą, ale nie muszą powodować klinicznie istotnego uszkodzenia tkanek – choroby. Zakażenie typowo
prowadzi do nadwrażliwości opóźnionej, która może być wykryta testem tuberkulinowym (Mantoux).
Gruźlica pierwotna jest postacią choroby, która pozwija się u osoby uprzednio nie narażonej na
ekspozycje i dlatego nie uczulonej. Głównymi implikacjami są wywołanie nadwrażliwości i zwiększenie
oporności, ogniska bliznowacenia mogące stanowic schronienie dla żywych prątków przez lata, a nawet
prze całe życie i w ten sposób będące siedliskiem reaktywacji, gdy zostanie upośledzona obrona
gospodarza, czasami choroba bez etapu przejściowego może przekształcić się w tzw. postępująca
gruźlicę pierwotną. Wtórna gruźlica pojawia się u uprzednio uczulonego gospodarza, może nastąpić
krótko po gruźlicy pierwotnej, ale znacznie częściej rozwija się po reaktywacji uśpionych ognisk
pierwotnych.
Zapalenia płuc w przebiegu chorób układowych (np. gośćcowe, w przebiegu mocznicy).
Zapalenie płuc u osób o zmniejszonej odporności (Wirus cytomegalii, Pneumocystis carinii,
Mycobacterium avium-intracellulare, inwazyjna aspergiloza, inwazyjna kandydiaza).
Pneumocystis carinii – niemowlęta, zwłaszcza wcześniaki i osoby bardzo stare, chorzy na AIDS. W
przegrodach międzypęcherzykowych są nie tyle limfocyty, co przede wszystkim kom. plazmatyczne
zapalenie plazmatyczno-komórkowe. W świetle pęcherzyków obecna jest „pianka”, a w niej te
pierwotniaki.
Ropień płuca odnosi się do ograniczonego obszaru ropnej martwicy w obrębie miąższu płuca, w
następstwie czego dochodzi do tworzenia się jednej lub więcej dużych jam. Organizmy będące
przyczyna ropnia mogą być wprowadzone do płuc na różnych drogach:
Aspirowanie zakażonego materiału
Aspirowanie zakażonej treści żołądkowej
Jako powikłanie martwiczego bakteryjnego zapalenia płuc
Zaczopowanie oskrzeli
Septyczny zator
Jako wynik hematogennego rozsiewu infekcji bakteryjnej.
6. Nowotwory.
Płuca są bardzo częstym miejscem przerzutów nowotworów wywodzących się z narządów znajdujących
się poza klatką piersiową. Równie częsty jest pierwotny rak płuca. 95% nowotworów płuca wywodzi się
38

z nabłonka oskrzelowego (rak oskrzelopochodny), pozostałe 5% to rakowiaki oskrzela, guzy z
gruczołów oskrzelowych , mezenchymalne nowotwory złośliwe, chłoniaki i kilka zmian łagodnych.
Rak oskrzelopochodny jest główną przyczyną śmierci z powodu nowotworów. Rokowanie w raku
płuca jest bardzo niepomyślne, wskaźnik 5-letniego przeżycia we wszystkich stopniach zaawansowania
wynosi 14%, u pacjentów z miejscową chorobą 45%. Wyróżnia się 4 główne typy histologiczne, ze
względu na leczenie podzielone na 2 duże grupy raki niedrobnokomórkowe i drobnokomórkowe:
1. rak niedrobnokomórkowy płuc (NCLC) (70-75%)
- rak płaskonabłonkowy (epidermoidalny) (25-30%)
- gruczolakorak łacznie z rakiem pęcherzykowo-oskrzelikowym (30-35%)
- rak wielokomórkowy (10-15%)
2. rak drobnokomórkowy płuc (SCLC) (20-25%)
3. Struktury złożone – raki mieszane - (5-10%)
Podstawą rozróżnienia na raki drobno i niedrobnokomórkowe jest fakt, że w chwili rozpoznania
wszystkich przypadków raków drobnokomórkowych stwierdza się przerzuty i w związku z tym nie może
być rozważane leczenie chirurgiczne. W związku z tym najlepszą metodą leczenie jest chemioterapia.
Przeciwnie raki niedrobnokomórkowe zwykle bardzo słabo reagują na chemioterapię i najlepszą
metodą ich leczenia jest leczenie chirurgiczne.
Istnieje liniowa korelacja między intensywnością ekspozycji na palenie papierosów i pojawianiem się
niepokojących zmian nabłonkowych, które rozpoczynają się jako pozornie niewinna hiperplazja
komórek podstawnych i metaplazja płaskonabłonkowa, która przechodzi w dysplazję płaskonabłonkową
i raka in situ, nim zakończy się rakiem inwazyjnym. Wśród głównych podtypów histologicznych raka
płuc raki płaskonabłonkowy i drobnokomórkowy wykazują najważniejszy związek z ekspozycją na
palenie papierosów. Inne czynniki to ekspozycja na azbest, arsen, chrom, uran, nikiel, chlorek winylu i
iperyt.
Rak płaskonabłonkowy występuje częściej u mężczyzn, ma tendencje do powstawanie centralnie w
oskrzelach głównych z rozprzestrzenianiem się do węzłów chłonnych wnęki, ale rozsiew poza klatkę
piersiową stwierdza się później niż w innych typach histologicznych. W dużych zmianach następuje
martwica w części środkowej, tworząc strukturę jamistą. Często przez wiele lat raka poprzedza
metaplazja płaskonabłonkowa i dysplazja nabłonka oskrzelowego, która przekształca się w raka
przedinwazyjnego, a proces ten może trwać wiele lat. W tym czasie atypowe komórki mogą być
stwierdzane w rozmazach cytologicznych. Zmiany nowotworowe osiągają stopień zmiany objawowej
kiedy masa guza zamyka światło głównego oskrzela powodując często obwodową niedodmę i
zakażenia jednocześnie zmiana nacieka obwodową część płuca. Obraz histologiczny od wysoko
dojrzałego raka płaskonabłonkowego z obecnością pereł rogowych do postaci nisko zróżnicowanych.
Gruczolakorak może być postacią zlokalizowaną centralnie, ale zwykle występuje obwodowo i może
wywoływać tzw. bliznowacenie płuc („rak w bliźnie”). Wykazuje najsłabszy związek z paleniem
papierosów. Ogólnie rośnie wolniej, tworzy mniejsze guzy niż inne raki, ale wykazuje tendencje do
dawania odległych przerzutów we wczesnych stadiach. Histologicznie występują różne postacie –
zrazikowa(tworzenie gruczołów), brodawkowata i lita. Prawdopodobnym prekursorem jest atypowa
hiperplazja gruczołowa. Podtypem gruczolakoraka jest rak pęcherzykowo-oskrzelikowy, którego
podstawową właściwością jest rozrost między istniejącymi strukturami i zachowanie architektoniki
pęcherzyków płucnych, może on jednak przejść w inwazyjnego gruczolakoraka.
Rak wielokomórkowy wykazuje brak zróżnicowania. Komórki są duże, anaplastyczne i zwykle mają duże
pęcherzykowate jądro z wyraźnym jąderkiem.
Rak drobnokomórkowy występuje ogólnie jako bladoszare, centralnie zlokalizowane masy z
naciekiem otaczającego miąższu i wczesnym zajęciem węzłów chłonnych wnęki i śródpiersia. Martwica
jest stałym składnikiem i niekiedy jest bardzo rozległa. Charakterystyczne jest formowanie się
(modelowanie) jąder w związku z ciasnym ułożeniem komórek i skąpa cytoplazmą. Nowotwory te
pochodzą z komórek neuroendokrynnych płuca i dlatego wykazują ekspresję różnych markerów
neuroendokrynnych i hormonów polipeptydowych czego wynikiem mogą być zespoły
paraneoplastyczne.
Rakowiak to dosyć częsty nowotwór w drogach oddechowych, głównie w dużych oskrzelach,
wywodzący się z neuroendokrynnych komórek nabłonka oskrzeli. Rozrasta się powoli bądź jako
polipowaty twór zamykający światło oskrzela lub w postaci okrągłej masy wpuklającej się do światła
bądź jako zgrubienie śluzówki penetrujące do ściany oskrzela i tkanki około oskrzelowej. Przeważnie
łagodny. Niekiedy wykazuje wyższy wskaźnik mitotyczny, występuje bardzo zróżnicowany obraz
cytologiczny i ogniskowa martwica – cechy, które zaliczają go do zmian określanych jako krakowiak
atypowy, który cechuje wyższa częstość dawania przerzutów do węzłów chłonnych. Czasem może dać
zespół rakowiaka. Typowy krakowiak, atypowy krakowiak i rak drobnokomórkowy tworzą pewną
ciągłość wzrastającej histologicznej agresywności i potencjalnego złośliwego charakteru w ramach
ogólnego spektrum nowotworów endokrynnych płuca.
Wśród raków anaplastycznych natomiast wyróżniamy:
39

drobnokomórkowego (owsianokom.)
pośredniokom. ta sama złośliwość
olbrzymio-/wielkokomórkowego
Nowotwory nienabłonkowe - wyjątkowo rzadkie.
Nowotwory mieszane - jeszcze rzadsze
Przerzuty - częste
7. Zmiany w opłucnej:
Złośliwy międzybłoniak opłucnej (mezothelioma) jest rzadko występującym nowotworem
wywodzącym się z komórek mezotelialnych, który zwykle powstaje w opłucnej ściennej lub trzewnej,
ale może tez rozwija się, choć znacznie rzadziej, w jamie otrzewnej i worku osierdziowym. Jest związany
z zawodową ekspozycją na azbest. Złośliwe międzybłoniaki są często poprzedzane rozległym
włóknieniem opłucnej, z tworzeniem się zgrubień. W badaniu sekcyjnym zmienione płuco typowo
otoczone jest grupową, białożółtawą, twardą, niekiedy galaretowatą warstwą guza, która zarasta
opłucną. Rzadko występują odległe przerzuty. Nowotwór może bezpośrednio naciekać ścianę klatki
piersiowej lub tkankę podopłucnową. Histologicznie występują 3 postaci: nabłonkowa, mięsakowa,
dwufazowa – zarówno pola utkania mięsakowego i nabłonkowego.
Płyn wysiękowy to obecność płynu w jamie opłucnej, który może być przesiękiem(hydrothorax) –
przyczyny to niewydolność układu krążenia, zastój pochodzenia sercowego(zastoinowa niewydolność
serca) lub wysiękiem charakteryzującym się ciężarem właściwym większym niż 1,020 i często
obecnością komórek zapalnych wskazujących na zapalenie. Istnieją 4 zasadnicze przyczyny wysięku:
inwazja drobnoustrojów (bezpośrednio z zakażenia płuc lub pośrednio przez krew), nowotwór, zawał
płuc i wirusowe zapalenie opłucnej.
Odma patrz wcześniej
Hemothorax to gromadzenie się pełnej krwi (w przeciwieństwie do wysięku krwawego) w jamie
opłucnej, jest powikłaniem pęknięcia tętniaka aorty piersiowej, w hemothorax krew krzepnie w jamie
opłucnej.
Chylothorax jest gromadzeniem się w opłucnej chłonki, ogólna ilość płynu może nie być duża, ale ma
duże znaczenie ponieważ wskazuje na zamknięcie dużych naczyń chłonnych limfatycznych.
8. Choroby górnych dróg oddechowych:
Zakażenia ostre górnych dróg oddechowych należą do najpowszechniejszych przypadłości u ludzi,
powszechnie określane jako przeziębienia.
Najczęstsze czynniki patogenne rhinoviruses oraz coronavirusy, RSV, wirusy grypy i paragrypy,
adenowirusy, enterowirusy, czasem paciorkowce β-hemolizujące grupy A. Czasem przeziębienia mogą
być komplikowane przez rozwój bakteryjnego zapalenia ucha środkowego lub zatok. Objawom
przeziębienia może towarzyszyć ostre zapalenie gardła, którego objawy zwykle nie są nasilone,
trzeba jednak mieć na uwadze postacie z zapaleniem migdałków ze względu na potencjalną
możliwość rozwoju okołomigdałkowych ropni lub popaciorkowcowego zapalenia kłębuszków nerkowych
i ostrego zapalenia reumatycznego.
Ostre bakteryjne zapalenie nagłośni – wywoływane przez H.influenzae, występujące głównie u
młodszych dzieci, u których jest szczególnie niebezpieczne ponieważ w swoim ostrym przebiegu może
prowadzić do zamknięcia dróg oddechowych i śmierci.
Ostre zapalenie krtani powodowane jest wdychanie drażniących czynników, reakcją alergiczną, a
także skutkiem działania czynników prowadzących do przeziębienia. U dzieci wirus paragrypy jest
najczęstszą przyczyną zapalenia krtani, tchawicy i oskrzeli, znanego powszechnie jako krup. Mimo
wyraźnych objawów jak wdechowy świst krtaniowy i charczący, uporczywy kaszel na ogół jest
procesem samoograniczającym. W niektórych przypadkach jednak reakcje zapalne krtani mogą zwężać
drogi oddechowe wystarczająco, aby spowodowac niewydolność oddechową.
Rak nosogardła bardzo rzadki, ale wyraźnie powiązany z EBV i predyspozycjami genetycznymi o
czym świadczy wysoka częstość występowania w Chinach.
40

Nowotwory krtani
Zmiany niezłośliwe to guzki strun głosowych jako skutek ich przewlekłego drażnienia u palaczy i
śpiewaków oraz brodawczaki krtani zwykle pojedyncze u dorosłych, u dzieci często mnogie w postaci
brodawczakowatości młodzieńczej krtani.
Rak krtani stanowi 2% wszystkich nowotworów złośliwych. Najczęściej występuje po 40r.ż., częściej u
mężczyzn, wyraźnie powiązany z paleniem papierosów, piciem alkoholu i ekspozycja na azbest. 95% to
typowe raki płaskonabłonkowe, rzadko stwierdza się gruczolakoraki. Zwykle rozwija się bezpośrednio
na strunach głosowych (guzy głośni) 60-75%, powyżej strun (nagłośniowy) 25-40%, lub poniżej strun
(podgłośniowy) poniżej 5%. Początkowo są one zmianami in situ, które następnie stają się
nieregularnymi zgrubieniami na powierzchni śluzówki, które w efekcie mogą przekształcić się w
owrzodzeni lub zmiany typu grzybiastego. Klinicznie objawia się przewlekła chrypką. 90% raków krtani
w chwili rozpoznania jest ograniczonych, ponieważ rejon głośni ma ubogie unaczynienie limfatyczne
dlatego rzadko rozsiewa poza rejon głośni, przeciwnie część nagłośniowa. Nowotwory części
podgłośniowej mają tendencję do klinicznego utajenia. Możliwe leczenie chirurgiczne, radioterapia i
skojarzone, jednak jedna trzecia chorych umiera.
Carcinoma adenoides cysticum - cylindroma: charakterystyczny dla ślinianek, występuje również
w oskrzelach, daje przerzuty, ale nie bardzo złośliwy np. może naciekać wokół pnia nerwowego
Carcinoma mucoepidermale: charakterystyczny dla ślinianek, utkanie płaskonabłonkowe i
gruczołowe, im więcej śluzu wydziela, tym mniej złośliwy, im mniej śluzu, tym gorzej daleko mniej
złośliwy niż typowe raki odoskrzelowe.
NERKI
Zapalenia nerek:
-> zap. kłębuszkowe nerek- glomerulonephritis (GN)
-> zap. cewkowo-śródmiąższowe- nephritis tubulo-interstitialis
Ogólnie:
Zap. kłębuszkowe na tle immunologicznym
Zap.cewk.-śródmiąższ. związane z substancjami toksycznymi i zakażeniami
ZAPALENIE KŁĘBUSZKOWE NEREK GN:
->pierwotne- gdy choroba zajmuje wyłącznie lub niemal wyłącznie nerki
->wtórne- towarzyszące chor.układowym (gł. SLE, cukrzyca, srobiawica, z.Goodpasture`a, z.Wegenera)
Charakterystyka i podział GN ze względu na mechanizm powstania:
1. Wywołane krążącymi kompleksami immuno.
Kłębuszek nerkowy jest przypadkową ofiarą, w kt. osiadają kompleksy, antygen wywołujący nie
pochodzi ze struktur kłębuszka, może mieć char. endogenny (np. w SLE) lub egzogenny (w następstwie
zak. paciork.,gronko.,wir.-WZW B, C, świnka, odra, ospa wietrzna).
Złogi podśródbłonkowe, rzadko podnabłonkowe. Następuje wiązanie dopełniacza, naciek WBC,
proliferacja kom. śródbłonka, kom. mezangium i kom. nabłonkowych. W mikroskopie
immunofluorescencyjnym ziarnisty układ złogów w kłębuszku
2. Wywołane kompleksami immuno. powstającymi in situ (Ig reagują bezpośrednio z antygenami
związanymi ze strukturą kłębuszka:
a) GN anty-GBM, czyliIg p.błonie podstawnej, w mikroskopii immunoflu.- linijny układ złogów
np. z.Goodpasture`a (Ig p. bł.podst. kłębuszka i płuc)
Ta postać GN może ciężko uszkadzać kłębuszki, dając gwałtownie postępujące GN z obecnością
półksiężyców
b) GN typu Heymanna - reakcja Ig z kompleksami antygenowymi występującymi w sposób nieciągły w
zagłębieniach podocytów, w mikr. immunoflu. zazwyczaj układy ziarniste.
Ogólnie:
-> gdy złogi znajdują się proksymalnie w stosunku do bł. podstawnej (w śródbłonku lub
podśródbłokowo)-> r. zapalna i nacieki WBC
->gdy złogi dystalnie (w nabłonku, podnabłonkowo)-> bez r. zap., zmiany jak w zap. błoniastym
41

Z. NERCZYCOWY
dobowa utrata bialka z moczem > 3,5g
hipoalbuminemia < 3g\dl
uogólnione obrzęki
hiperlipidemia, lipiduria, azotemia (podwyższony poziom kreatyniny i mocznika w surowicy), ew.
krwiomocz, nadciśnienie
Przyczyny z. nerczycowego:
1. pierwotne:
- zmiana minimalna (nerczyca lipidowa)
- glomerulopatia błoniasta
- glomerulopatia błoniasto-rozplemowa
- blomerulopatia rozplemowa
- ogniskowe segmentalne stwardnienie kłębuszków
2. wtórne:
- w przebiegu chor. układ. tj. SLE, cukrzyca, skrobiawica
- związane z lekami np. solami złota; zakażeniami np. WZW B, AIDS, malaria
- towarzyszące now. złoślwym np. czerniakowi
U dzieci < 15 rż. zazwyczaj przyczyna pierwotna, u dorosłych częściej wtórna.
SUBMIKROSKOPOWE ZAP. NEREK = ZMIANA MINIMALNA = NERCZYCA LIPIDOWA
Jest to najczęstsza przyczyna z. nerczycowego u dzieci (szczególnie małych, 2-3 rż.). Białkomocz
selektywny. Zmiana łagodna, odwracalna. W mikroskopie elektronowym - rozległy zanik wypustek
podocytów. Przyczyna - zaburzenia syntezy nefryny (główne białko błony filtracyjnej). Rokowanie
dobre, dobrze reaguje na sterydoterapię.
BŁONIASTE KŁĘBUSZKOWE ZAP. NEREK (NEFROPATIA BŁONIASTA)
Gł. 30-50 rż., białkomocz nieselektywny, w 85% char. idiopat. (pierwotny), pozostałe przypadki to
nefopatia wtórna do :
- zak. (WZW B, kiła, malaria, schistosomiaza)
- now. złośliwe (r. płuc, j. grubego, czerniak)
- SLE
- sole nieorg. (np. złota, rtęci)
- leki (NLPZ, kaptopryl, penicylamina)
Nefropatia błoniasta to przewlekłe zap. nerek, związane z kompleksami immuno. powstającymi in situ
(mechanizm typu Heymanna)
Charakterystyczne podnabłonkowe złogi wzdłuż bł. podst. tworzą ziarnisty układ w mikro. immunoflu.,
w formie zaawansowanej rozlane pogrubienie bł. podst. widoczne w mikro. świetlnym, a w mikro.
elektron. z czasem widoczne inkorporowanie złogów do bł. podst. i zanik wypustek podocytów.
Brak r. na sterydy, u ok. 40% progresja do krańcowej NN
OGNISKOWE SEGMENTALNE STWARDNIENIE KŁĘBUSZKÓW NERKOWYCH (FSG)
Zajęcie części kłębuszków, w poszczególnych kłęb. zmiany dot. tylko niekt. segmentów. Trzeba
różnicować FSG ze zmianą minimalną, ponieważ inny przebieg klin. (w FSG: krwiomocz, nadciśnienie,
białkomocz nieselekt.) i rokowanie (FSG nie reaguje na sterydy, szybko dochodzi do krańcowej NN - ok.
10 lat).
W FSG uszkodzenie i zaburzenie układu podocytów -> odkładanie białek i lipidów w miejscach
zwiększonej przepuszczalności, prolif. kom. mezangium -> sklerotyzacja kłęb.
W mikro. elektr. - zanik wypustek podocytów + oddzielanie się podocytów -> naga bł. podst.
BŁONIASTO-ROZPLEMOWE KŁĘBUSZKOWE ZAP. NEREK
Klin. może dać z. nerczycowy lub z. nerczycowo-nefrytyczny.
Obraz w mikro. świetlnym: duże kłęb. o zrazikowatym kształcie, prolif. kom. mezangium i nacieki WBC,
pogrubienie bł. podst., charakterystyczne rozwarstwienie bł. podst. przez wnikające wypustki kom.
mezangium -> obraz szyn tramwajowych.
Ze względu na lokal. złogów wyróżniamy 3 typy:
1.typ - GN diffusa cum depositis subendothelialibus - złogi podśródbłonkowe z kompleksów immuno. i
dopełniacza - 2\3 przypadków GN diffusa membrano-proliferativa, związane z SLE, WZW B i C (gł. C)
2.typ - GN diffusa cum depositis membranace basalis - złogi wewnątrzbłonowe - tzw. chor. gęstych
złogów, złogi z subst. o nieznanym składzie
3.typ - GN diffusa cum depositis subepitheliale - złogi podnabłonkowe
Ogólnie rokowanie złe, gorzej w typie 2.
42

Z. NEFRYTYCZNY
Nagle występujące: krwiomocz, skąpomocz, nadciśnienie, także pewnego stopnia białkomocz i obrzęki.
Przyczyna - proliferacja kom. w obrębie kłęb. (czyli wszystkie zap. rozplemowe) z obecnością nacieków
z WBC; dochodzi do uszkodzenia ściany kapilary -> przechodzenie krwinek i spadek GFR -> krwiomocz
i skąpomocz; wzrost produkcj reniny przez niedokrwioną nerkę -> nadciśnienie.
Ostry z. nefrytyczny może być następstwem chor. układ. np. SLE albo pierwotny, np. ostre rozlane
rozplemowe GN.
W klasyf. p.prof. Salwy do rozlanych rozplemowych GN zaliczamy:
1. GN diffusa endocapillaris (wewnątrzwłośniczkowe)
2. GN diffusa exocapillaris (zewnątrzwłośniczkowe):
-> subacuta
-> rapid progressiva
3. GN diffusa mesangialis
OSTRE ROZPLEMOWE (POPACIORKOWCOWE, POINFEKCYJNE) GN
Prawdopodobnie jest to odpowiednik GN diffusa endocapillaris.
Przyczyna to najczęściej kompleksy immuno. (patrz mech. powstawania GN wywołany krążącymi
kompleksami immuno.).
Jest to jedna z najcz. występujących glomerulopatii, częsta u dzieci.
W mikro. immunofluor. ziarnisty układ złogów z C3i IgG podśródbłonkowo, podnabłonkowo lub
wewnątrz bł. podst.
W mikro. świetlnym - zwiększona komórkowość, ponieważ prolifer. i obrzęk kom. śródbł., kom.
mezangium + nacieki zap. z neutrofilów i monocytów. Mogą być zakrzepy w świetle kapilar i martwica
ich ścian.
W mikro. elektr. - kompleksy immuno. tworzą garbiki (garby wielbłądzie).
Klin. : w klasycznej postaci 1-4 tyg. od ustąpienia objawów inf. paciorkowc. z gr. A (gardło lub skóra).
Początek nagły, osłabienie, gorączka, nudności + z. nefrytyczny. Niski poziom dopełniacza w surowicy
(w złogach kumulowane składowe dopełniacza), gdy etiologia paciork. to wzrost antystreptolizyny O.
Rokowanie: u dzieci dobre, zazwyczaj pełne wyzdrowienie, u dor. gorsze, może przejść w gwałtownie
postępujące GN lub w PNN.
GWAŁTOWNIE POSTĘPUJĄCE GN ( Z PÓŁKSIĘŻYCAMI )
Odpowiednik GN exocapillaris rapid progressiva.
Charakterystyczne półksiężyce nabłonkowe w większości kłęb. nerk. (tzw. diadem). Półksiężyc to efekt
prolif. kom. nabłonka ściennego torebki Bowmana + nacieki z monocytów i makrofagów. Półksiężyce
mogą z czasem ulegać bliznowaceniu, doprowadzając do zamknięcia przestrzeni Bowmana i uciśnięcia
kłęb.
Podtypy GN z półksiężycami:
1. typ - Ig p. bł. podstawnej (patrz mech. GN wywołany kompleksami immuno. powstającymi in situ)
np. z. Goodpasture`a z charakt. linijnymi złogami IgG i C3 na pow. bł. podst.; lecz.: plazmafereza
2. typ - związany z kompleksami immuno., ziarnisty układ złogów; zazw. jest to powikłanie innych GN
np. popaciorkowcowego GN lub zap. nerek w przebiegu np. SLE, plamicy Schonleina-Henocha.
3. typ - skąpoimunologiczny, związany z ANCA; w przebiegu np. z. Wegenera, mikroskopowego zap.
tętnic.
Klin.: Z. nefrytyczny, może być też intensywny białkomocz; lecz.: dializy, przeszczepienie nerki,
plazmafereza; rokowanie zależy od liczby zajętych nefronów.
NEFROPATIA IgA = CHOR. BERGERA
Odpowienik GN mezangialis
Przyczyna - nadmierna produkcja IgA w odpowiedzi na ekspozycję dróg oddech. czy p. pok. na wir.,
bakt.; IgA odkłada się w mezangium, dochodzi do akt. dopełniacza i uszkodzenia kłębuszka. Chor. dot.
gł. dzieci i młodych dorosłych. Daje objawy z. nefrytycznego. Nefropatia IgA jest jedną z najczęstszych
przyczyn nawrotnego krwiomoczu i najczęściej występującą na świecie glomerulopatią.
NEFROPATIE WRODZONE
1. Z. ALPORTA
Zaburzenie syntezy kolagenu typu 4.
Progresywne uszkodzenie nerek, dodatkowo upośledzenie słuchu, zmiany neurologiczne, oczne,
uszkodzenie PLT, chor. częściej u męż, u męż. też cięższy przebieg.
Objawy ze str. nerek:
- krwiomocz\krwinkomocz
- może być mierny białkomocz, rzadko z. nerczycowy
- prolif. mezangium i ogniskowe pogrubienie ścian -> sklerotyzacja kłębuszków (glomerulosclerosis)
43

- w mikro. elektr. widoczne odcinkowe pogrubienia bł. podst. i ich rozwarstwienie!!!, bł. podst.
ogniskowo może być ścieńczała
- w śródmiąższu włóknienie i charakt. kom. piankowate
2. Z. CIENKICH BŁON PODSTAWNYCH = ŁAGODNY RODZINNY KRWIOMOCZ
Częstszy i łagodniejsz od z. Alporta
Jest rozlane ścieńczenie bł. podst. -> krwio\krwinkomocz; po wielu latach fibrosklerotyzacja
kłębuszków.
PRZEWLEKŁE GN
Gł. młodzi dorośli i osoby w średnim wieku, jest to forma zejściowa innych zap. nerek, gł.: gwałtownie
postępującego GN, zap. błoniastego, błoniasto-rozplemowego i FSG.
Nerki zmniejszone, rozległe wlóknienie kłęb., śródmiąższu, zanik cewek warstwy korowej, pogrubiałe,
zwężone ściany naczyń - jest to obraz nerki zejściowej.
Klin.: nadciśn. i azotemia (progresja do mocznicy) + epizody z. nerczycowego i nefrytycznego
Lecz.: dializa, transplantacja
CHOROBY CEWKOWO-ŚRÓDMIĄŻSZOWE
CEWKOWO-ŚRÓDMIĄŻSZOWE ZAP. NEREK = ODMIEDNICZKOWE ZAP. NEREK - PYELONEPHRITIS:
1. OSTRE
Najcz. E. coli, rzadziej Proteus, Klebsiella, Pseudomonas.Towarzyszy mu zakaż. dolnego odc. dróg
mocz, gdy jest to zak. wstępujące; rzadziej zak. krwiopochodne w następstwie posocznicy czy bakt.
zap. wsierdzia.
Czynn. ryzyka: zabiegi na drogach mocz., ciąża, cukrzyca, zaburzenia odpływu moczu, wady ukł. mocz.
np. niedomykalność ujścia moczowodowo-pęcherzowego powodująca refluks pęcherzowo-
moczowodowy.
Obraz: niewielkie żółtawe ropnie unoszące powierzchnię nerki, odpowiadające histo. zmianom
martwiczo-ropnym w miąższu, kt. z czasem naciekają cewki.
Postacie ostrego odmiedniczkowego zap. nerek:
- roponercze (pyonephrosis), czyli wysięk ropny w moczowodach, miedniczce i kielichach - przyczyną
jest zaburz. odpływu moczu
- martwica brodawek nerkowych - zmiany martwiczo-ropne obejmują 2\3 szczytowe piramidy, może
dot. jednej lub wszystkich brodawek nerkowych, charakter. dla cukrzycy.
2. PRZEWLEKŁE
Bliznowacenie i znieksztalcenie ukł.kielichowo-miedniczkowego, które towarzyszy śródmiąższowemu
zap. i bliznowaceniu
Postacie chor.:
- związana z zaburzeniem odpływu moczu -> nawrotowe zap. nerek i bliznowacenie
- związana z refluksem - nefropatia refluksowa - częściej
Cechy char.: zmiana dot. jednej\obu nerek, ogniskowa\rozproszona; nieregularne bliznowacenie, w
odróżnieniu do typowo symetrycznych zmian bliznowatych w przebiegu miażdżycy. Bliznowacenie
obejmuje miedniczkę nerkową i\lub kielichy -> spłaszczenie brodawek nerkowyh i deformacja
kielichów.
Mikroskop.:
- nieregularne włóknienie śródmiąższu + nacieki zapalne
- poszarzenie\zwężenie cewek + zanik nabł. wyścielającego; poszerzona cewka może zawierać treść
przypominającą tk. tarczycy - tzw. "nerka tarczycowata"
- włóknienie ściany kielichów + nacieki zapalne
- zmiany naczyniowe przypominające arteriolosclerosis
- może być stwardnienie kłęb. - glomerulosclerosis
Klin.: PNN, nadciśnienie
W pielografii b. char. niesymetr. zaciągania + poszerzenie i zniekształcenie kielichów
ŚRÓDMIĄŻSZOWE POLEKOWE ZAP. NEREK:
1. OSTRE
Leki najcz. wywołujące: metycilina, ampicylina, ryfampicyna, tiazydy, NLPZ, cymetydyna
Klin.: ok 2 tyg. po ekspozycji na lek - gor., eozynofilia, wysypka i zaburzenia funkcji nerek - krwiomocz,
białkomocz, WBC w moczu w tym eos, może być ONN z bezmoczem.
W śródmiąższu obrzęki, nacieki zap., mogą powstawać ziarniniaki.
Podejrzewane tło immuno. chor.
2. PRZEWLEKŁE = NEFROPATIA POANALGETYCZNA
Jest to śródmiąższowe zap. nerek z często towarzyszącą, wtórną martwicą brodawek nerkowych(!)
Gł. winowajcami są: aspiryna, kt. hamuje PG -> skurcz naczyń -> niedokrwienie brodawki nerkowej i
acetaminofen (metabolit fenacetyny) ->uszkodzenie oksydacyjne brodawek
44
Klin.: PNN, nadciśnienie, niedokrwistość; wzrost ryzyka r. urotelialnego miedniczki nerkowej lub
pęcherza moczowego
OSTRA MARTWICA CEWEK NERKOWYCH
Jest to najczęstsza postać ostrej, niezapalnej NN u doroslych.
1. TOKSYCZNA - w 95% wyleczalna, wywołana przez: leki -cyklosporyna, gentamycyna, śr. cieniujące,
rozpuszczalniki org., grzyby trujące, pestycydy, czterochlorek węgla, rtęć, alkohol metylowy (tu
dodatkowo ślepota)
2. NIEDOKRWIENNA (ISCHEMICZNA) - w 60-80% prowadzi do zgonu, dochodzi do anoksji i odwodnienia
kom. nabł. cewek w przebieg np.: wstrząsu septycznego, kardiogennego, w masywnym z.
zmiażdzenia, po operacjach, po przetoczeniu krwi niezgodnej grupowo.
Obraz: odcinkowe zniszczenie nabł. cewek (dla postaci niedokrwiennej dot. to gł. cewek 1-rz. i cz.
wstęp. pętli Henlego), subtelne pęknięcia bł. podst. (p.prof. Salwa temu przeczy), mocz przedostaje się
do śródmiąższu - obrzęki i nacieki zap., w świetle cewek białka Tamma-Horsfalla, Hgb, b. osocza
Ewolucja zmian w martwicy cewek:
1. faza - od zadziałania czynn. do wystąpienia ostrych objawów
2. faza - oliguria, narasta mocznica, kw. metab., hiperkaliemia, hemodializa
3. faza - regeneracja nabł. cewek - zdrowienie, poliuria
NACZYNIA NERKOWE W NADCIŚNIENIU
1. STWARDNIENIE NEREK W PRZEBIEGU NADCIŚNIENIA ŁAGODNEGO - NEPHROSCLEROSIS BENIGNA
Stwardnienie tętniczek ze szkliwieniem - arteriolosclerosis hyalinica - pogrubienie ściany + zwężenie
światła, co powoduje zaburzenia w przepływie krwi przez nerki -> utrata nefronów -> obraz
symetrycznie pozaciąganej nerki.
Arteriolosclerosis hyalinica należy także do części składowej mikroangiopatii cukrzycowej.
2. STWARDNIENIE NEREK W PRZEBIEGU NADCIŚNIENIA ZŁOŚLIWEGO - NEPHROSCLEROSIS MALIGNA
Nadciśn. złośl.: ciśn. rozk.>120mmHg, obrzęk tarczy n. wzrokowego, encefalopatia, zaburz. serc.-nacz.,
NN
Stwardnienie tętniczek z rozplemem - arteriolosclerosis hyperplastica - koncentryczne, warstwowe
pogrubienie ścian tętniczek (listki cebuli) + stopniowe zwężenie światła. Zmianom rozplemowym
towarzyszą złogi włóknikowate i ostra martwica ścian naczyń -> zap. tętniczek z martwicą - arteriolitis
necrotisans. Martwicze zap. może też czasem dot. kłęb. - glomerulitis necrotisans.
NOWOTWORY ZŁOŚLIWE NEREK
Ogólna charakt.:
- 2% wszystkich now. złośl. u dor.
- męż. 2-razy częściej, szczyt zachor. to 6-7 dek. życia
- wywodzą się z kom. nabł. cewkowego -> lokalizacja gł. w w-wie korowej
- przerzuty najcz. do: 1. płuc, 2. kości, wątroby, 3. innych tk. i narz.
- możliwa jest obecność utkania sarkomatoidnego (wrzecionowato-komórkowego), kt. znacznie
pogarsza rokowanie
- objawy: początkowo pseudogrypowe, póżniej bezbolesny krwiomocz, rzadko ból w j. brzusznej czy
palpacyjnie wyczuwalny guz, dodatkowe obj. niespecyficzne, związane z naturą guza (z.
paraneoplastyczne)
Czynn. ryzyka r. nerki:
- tytoń
- otyłość, szczególnie u kobiet
- leczenie estrogenami
- nadciśn.
- narażenie na: ropę naft., azbest, kadm
- nabyta torbielowatość w przebiegu długotrwałej dializoterapii
- większość to guzy sporadyczne, tylko 2% guzy rodzinne - np. w chor. VHL (aut. dom.,
wielotorbielowatość nerek, wątroby, śledziony + naczyniaki płodowe w móżdzku i siatkówce)
1. RAK KONWENCJONALNY (JASNOKOMÓRKOWY)
70-80% raków nerki
Zwykle guz pojed., duży o śr. 3-15cm, zlokal. w w-wie korowej, gł. w biegunie górnym; żółtawy (lipidy),
na przekroju zmiany martwicze, krwotoczne, torbielowate.
Może naciekać ż. nerkową, szerząc się w postaci litej masy do ŻGD, sporadycznie osiągając prawy
przedsionek.
Może wywoływać z. paraneoplastyczne:
- EPO -> czerwienica (policytemia)
- rPTH -> hiperkalcemia
- renina -> nadciśnienie
- glikokortykosteroidy -> z. Cushinga
45

- h. płciowe ->feminizacja\maskulinizacja
2. RAK BRODAWKOWATY (CHROMOFILNY) - ADENOCARCINOMA PAPILLARE
ok 10-15%
tendencja do wieloogniskowości, często obustronnie, koloru beżowobrązowego, zazw. struktura
brodawkowata z kom. piankowatymi w rdzeniu, na przekroju zmiany martwicze, krwotoczne i
torbielowate.
Złośliwość mniejsza niż w r. jasnokom.
3. RAK CHROMOFOBNY
ok 5%
Rozrost zrazikowaty, nieotorebkowany, koloru nerki, w 10% wieloogniskowy, na przekroju ogniska
martwicze i krwotoczne.
W mikrosk. elektr. charakt. halo wokół jąder.
Najlepsze rokowanie.
Należy go różnicować z łagodnym guzem onkocytarnym nerki.
NOWOTWORY PĘCHERZA MOCZOWEGO
1. ŁAGODNE - gł. brodawczaki (papilloma) - rdzeń włóknisto-naczyniowy pokryty dobrze zróżnicowanym
nabł. urotelialnym, zwykle pojedyńcze o char. nienaciekającym, usuwane chir.
2. ZŁOŚLIWE
ok 3% wszystkich now zł.
3-razy częściej u męż., gł. 50-70 rż.
Czynn. ryzyka:
- tytoń
- amin aromatyczne
- przewlekłe zap. pęcherza mocz.
- leki, np. cyklofosfamid
- schistosomiaza pęch. mocz. (związek gł. z r. płaskonabł.)
Objawy:
- bezbolesny krwiomocz
- obj. związane z zamknięciem odpływu moczu -> hydronephrosis -> zap. odmiedniczkowe ->
pyonephrosis
Mogą charakteryzować się wzrostem płaskim lub brodawkowatym, naciekającym lub nienaciekającym.
Najcz. r. z nabł. przejściowego (urotelialne) - ok 80%, pozostałe to r. płaskonabłonkowy i gruczołowy.
Trzeba pamiętać, że u dzieci w pęch. mocz. może rozwinąć się rhabdomyosarcoma rosnący w postaci
winogronowych mas.
WĄTROBA I DROGI ŻÓŁCIOWE
I. Uszkodzenie wątroby
1. Zapalenie (hepatitis) – napływ do wątroby komórek ostrego i przewlekłego nacieku zapalnego,
często limfocytów T. Jest ograniczone do przestrzeni wrotnych lub zajmuje miąższ.
2. Zwyrodnienie:
a) balonowate – obrzmienie hepatocytów, zbrylona cytoplazma i duże puste przestrzenie (toksyny,
reakcje immunologiczne),
b) piankowate – rozlany, piankowaty, obrzmiały wygląd (zastój żółci),
c) stłuszczenie (steatosis) – krople lipidów w hepatocytach:
stłuszczenie drobnokomórkowe (steatosis microvacuolaris) – choroba alkoholowa, zespół Reye’a,
ostre ciążowe stłuszczenie wątroby,
stłuszczenie wielkokropelkowe (steatosis macrovacuolaris) – jądro na obwodzie – uszkodzenie
alkoholowe, osoby otyłe, cukrzycy.
3. Śmierć komórki:
a) martwica skrzepowa (necrosis coagulativa) – niedokrwienna,
b) apoptoza (apoptosis) – toksyny, reakcje immunologiczne,
46

c) zwyrodnienie wodniczkowe (hydropic degeneration)/ martwica rozpływna (necrosis colliquativa,lysis)
– obrzmiewanie i pękanie hepatocytów.
Zwykle następuje martwica hepatocytów wokół żyły środkowej (martwica strefy środkowej).
Graniczne zapalenie wątroby (interface hepatitis) – apoptoza na pograniczu między zapalnie
zmienionymi przestrzeniami wrotnymi a miąższem okołowrotnym.
Martwica mostkująca – tworzenie mostków wrotno-wrotnych, wrotno-centralnych lub centralno-
centralnych.
Martwica subtotalna – niszczenie całych zrazików.
Martwica masywna (rozległa) – niszczenie większości miąższu wątroby.
4. Włóknienie (fibrosis).
5. Marskość (cirrhosis hepatis).
Wątroba ma nadzwyczajną rezerwę czynnościową!!!
II. Żółtaczka i zastój żółci
Żółtaczka (icterus) – bilirubina > 2,0mg/dl.
Cholestaza (zastój żółci) – retencja bilirubiny i in. substancji (kwasów żółciowych, cholesterolu).
Kwasy cholowy i chenodezoksycholowy – pierwotne kwasy żółciowe.
Kwasy dezoksycholowy i litocholowy – wtórne kwasy żółciowe.
Bilirubina niesprzężona – ściśle związana z albuminami surowicy, nierozpuszczalna w wodzie, nie
może być wydalona z moczem. Frakcja wolna od albumin może przenikać do tkanek (zwłaszcza
mózgu u noworodków) i powodować ich toksyczne uszkodzenie. Bilirubina niesprzężona może
gromadzić się w mózgu w chorobie hemolitycznej noworodków, powodując poważne uszkodzenia
neurologiczne (kernicterus).
Bilirubina sprzężona – rozpuszczalna w wodzie, nietoksyczna, luźno związana z albuminami, jej
nadmiar może być wydalony z moczem. Frakcja „delta” kowalentnie związana z albuminami.
Hiperbilirubinemia z nadmiarem bilirubiny niesprzężonej:
1) nadmierna produkcja bilirubiny (niedokrwistości hemolityczne, resorpcja krwi z krwotoków
wewn., nieefektywna erytropoeza),
2) zmniejszony wychwyt bilirubiny przez wątrobę (leki, zespół Gilberta),
3) zaburzenia sprzęgania bilirubiny (żółtaczka fizjologiczna noworodków, żółtaczka u noworodków
karmionych piersią, zespół Gilberta, zespoły Criglera-Najjara typu I i II, rozlane uszkodzenie
wątroby – zapalenie wirusowe lub polekowe, marskość).
Hiperbilirubinemia z nadmiarem bilirubiny sprzężonej:
1) zmniejszone wydalanie dwuglukoronianów bilirubiny przez wątrobę (zespół Dubina-Johnsona,
zespół Rotora, leki – doustne środki antykoncepcyjne, cyklosporyna, uszkodzenie wątroby –
zapalenie wirusowe lub polekowe, całkowite odżywianie pozajelitowe, zakażenia
ogólnoustrojowe),
2) zmniejszony wewnątrzwątrobowy przepływ żółci (zaburzenia przepływu żółci przez kanaliki
żółciowe, np. polekowa dysfunkcja mikrofilamentów, uszkodzenie wewnątrzwątrobowych dróg
żółciowych w przebiegu zapaleń),
3) zamknięcie pozawątrobowych dróg żółciowych (kamica dróg żółciowych, rak głowy trzustki, rak
pozawątrobowych przewodów żółciowych, rak brodawki Vatera, zwężenia, torbiele, zarośnięcie,
pierwotne zapalenie ze stwardnieniem, zakażenie wątroby przez przywry).
Żółtaczka fizjologiczna noworodków – niedojrzałość wątroby, niedobór UGT1A1 (transferazy
urydyno-dwufosfo-glukuronylowej).
Żółtaczka u noworodków karmionych piersią – wzmożone działanie β-glukuronidaz, które
powodują rozprzęganie bilirubiny.
Zespół Gilberta – dziedziczony heterogenicznie, dość częsty, obniżona aktywność UGT1A1.
Zespół Criglera-Najjara typu I – dziedziczony autosomalnie recesywnie, rzadki, całkowity niedobór
aktywności UGT1A1, zgon z powodu uszkodzenia mózgu (kernicterus) zwykle w ciągu 18 miesięcy po
urodzeniu.
47

Zespół Criglera-Najjara typu II – dziedziczony autosomalnie recesywnie, częściowy brak aktywności
UGT1A1.
Zespół Dubina-Johnsona – dziedziczony autosomalnie recesywnie, defekt białka transportującego,
ciemne zabarwienie wątroby, hepatomegalia.
Zespół Rotora – odmiana zespołu Dubina-Johnsona. Wątroba pozbawiona ciemnego zabarwienia.
Zastój żółci:
- świąd (wzrost stężenia kwasów żółciowych w surowicy krwi i ich odkładanie się w tkankach
obwodowych, zwłaszcza w skórze),
- kępki żółte (xanthomata) – skutek hiperlipidemii i zaburzonego wydalania cholesterolu,
- ↑ fosfatazy zasadowej w surowicy krwi,
- zaburzenia wchłaniania (np. niedobór rozp. w tłuszczach witamin A, D, K).
Morfologia cholestazy:
- powiększone hepatocyty z kropelkami barwnika żółciowego,
- zwyrodnienie pierzaste lub piankowate,
- poszerzone kanaliki żółciowe z czopami żółci,
- obecność komórek apoptycznych,
- obecność komórek Browicza i Kupffera,
- poszerzenie przewodów żółciowych,
- proliferacja komórek nabłonka przewodzików żółciowych, obrzęk, nacieki z neutrofilów,
- w zaawansowanym stadium włóknienie (marskość żółciowa).
III. Niewydolność wątroby
Przyczyny:
1) masywna (rozległa) martwica wątroby - piorunujące wirusowe zapalenie wątroby, leki
(paracetamol, halotan, ryfampicyna, izoniazyd, inhibitory oksydazy monoaminowej), związki
chemiczne (czterochlorek węgla, toksyny grzyba Amanita phalloides),
2) przewlekła choroba wątroby – najczęstsza przyczyna niewydolności,
3) nieprawidłowa czynność wątroby bez oznak martwicy (ostre ciążowe stłuszczenie wątroby,
zatrucie tetracyklinami, zespół Reye’a).
Objawy kliniczne:
1) żółtaczka,
2) hipoalbuminemia, obrzęki obwodowe,
3) hiperamonemia (↑ amoniaku w surowicy krwi),
4) charakterystyczny zapach ciała (foetor hepaticus),
5) hiperestrogenemia (rumień dłoniowy – erythema palmaris, pajączki naczyniowe, u mężczyzn
hipogonadyzm i ginekomastia),
6) niewydolność wielonarządowa,
7) skaza krwotoczna (diathesis haemorrhagica) – zaburzona synteza czynników krzepnięcia II, VII,
IX, X, krwotoki z przewodu pokarmowego, wybroczyny krwawe (petechiae),
8) encefalopatia wątrobowa,
9) zespół wątrobowo-nerkowy.
Objawy kliniczne ujawniają się, gdy nastąpi wyczerpanie 80-90% rezerwy czynnościowej wątroby.
Encefalopatia wątrobowa:
- zaburzenia świadomości (od niewielkich zmian w zachowaniu poprzez nasilone splątanie i osłupienie
aż do głębokiej śpiączki i zgonu),
- sztywność, wygórowane odruchy, nieswoiste zmiany elektroencefalograficzne, ataki drgawek,
- asterixis – grubofaliste, trzepoczące ruchy rąk,
- nierytmiczne, gwałtowne ruchy zgięcia i wyprostne głowy oraz kończyn,
- skutek zaburzeń metabolicznych w OUN (przepływ krwi omijający uszkodzoną wątrobę →
nieprawidłowe środowisko metaboliczne < ↑amoniaku w surowicy krwi > → zaburzenia działania
neuronów, obrzęk mózgu).
48

Zespół wątrobowo-nerkowy:
- znaczne uszkodzenie wątroby + niewydolność nerek,
- przyczyna – zmniejszenie przepływu krwi przez nerki, obkurczenie naczyń w krążeniu układowym,
- ↑ mocznika i kreatyniny w surowicy krwi,
- mocz – mała ilość, hiperosmolarny, pozbawiony białek, w osadzie mało sodu.
IV. Marskość wątroby
Morfologia:
- mostki włókniste tworzące przegrody,
- guzki w miąższu – wynik regeneracji hepatocytów (mikroguzki <3mm średnicy, makroguzki),
- zaburzenie architektoniki całej wątroby.
Przyczyny:
- choroba alkoholowa wątroby (najczęściej!!!),
- WZW,
- choroby dróg żółciowych,
- wrodzona hemochromatoza,
- choroba Wilsona,
- niedobór α
1
-antytrypsyny,
- galaktozemia lub tyrozynoza u niemowląt i dzieci,
- leki (np. α-metyldopa),
- kiła,
- choroba serca,
- marskość kryptogenna.
Patogeneza:
- we wszystkich strefach zrazika jest odkładany kolagen typu I i III (produkowany przez
okołozatokowe komórki gwiaździste Ito),
- komórki śródbłonka tracą fenestracje → brak możliwości wymiany między osoczem a hepatocytami
(zwłaszcza białek),
- powstają omijające połączenia naczyniowe („przecieki”) między żyłą wrotną a żyłą wątrobową oraz
tętnicą wątrobową a żyłą wrotną,
- stymulacja włóknienia → TNF, limfotoksyny, Il-1.
Objawy kliniczne:
- początkowo jadłowstręt, utrata masy ciała, osłabienie,
- w zaawansowanej chorobie wyniszczenie,
- zgon w wyniku niewydolności wątroby, powikłań nadciśnienia wrotnego, raka
wątrobowokomórkowego.
Nadciśnienie wrotne:
- przedwątrobowe - zakrzepica, zwężenie głównego pnia żyły wrotnej, splenomegalia,
- wątrobowe – marskość wątroby, schistosomiaza, stłuszczenie wątroby, sarkoidoza, gruźlica
prosówkowa, guzkowa hiperplazja regeneracyjna,
- pozawątrobowe – ciężka prawokomorowa niewydolność krążenia, zaciskające zapalenie osierdzia,
zamknięcie przepływu przez żyłę wątrobową.
W marskości nadciśnienie jest wynikiem wzrostu oporu w przepływie wrotnym na poziomie
naczyń zatokowych oraz ucisku żył środkowych przez włóknienie okołożylne i guzki.
Skutki kliniczne:
1)
wodobrzusze (ascites) – głównie płyn surowiczy zawierający do 3g/dl białek (głównie
albumin), glukozę, sód, potas, kom. śródbłonka, leukocyty jednojądrowe; możliwy wysięk
opłucnowy (hydrothorax); spowodowane usuwaniem płynu z przestrzeni Dissego przez
wątrobowe naczynia limfatyczne, przesączaniem chłonki z wątroby do jamy otrzewnowej,
wtórnym hiperaldosteronizmem,
2)
tworzenie omijających zespoleń żylnych („przecieków”) wrotno-układowych - żylaki odbytu,
przełyku, okołopępkowe (caput medusae), ściany brzucha,
3)
splenomegalia – wynik przewlekłego przekrwienia biernego,
4) encefalopatia wątrobowa,
5) inne (niedożywienie, zanik jąder).
49

V. Zapalenia wątroby
Zdecydowaną większość stanowią wirusowe zapalenia wątroby. Inne przyczyny to gruźlica
prosówkowa, malaria, bakteriemia gronkowcowa, salmonellozy, kandydioza, pełzakowica. Zajęcie
wątroby jest prawie wyłącznie krwiopochodne.
Wirusowe zapalenia wątroby:
1) wirusy WZW,
2) mononukleoza zakaźna (wirus Epsteina-Barr),
3) cytomegalia, wirusy herpes,
4) żółta febra,
5) różyczka, adenowirusy, enterowirusy.
Wirus zapalenia wątroby typu A (HAV):
- okres inkubacji – 2-6 tygodni,
- pikornawirus, jednoniciowy bezotoczkowy RNA,
- droga przenoszenia: feralno-oralna,
- zakażenie z reguły w dzieciństwie, rozsiew wirusa za pośrednictwem zanieczyszczonej wody lub
żywności,
- zachorowania endemiczne w krajach, gdzie panują złe warunki higieniczne i sanitarne,
- nie prowadzi do przewlekłego zapalenia wątroby, stanu nosicielstwa ani raka
wątrobowokomórkowego, wyjątkowo zdarza się piorunujące zapalenie wątroby,
- p/ciała anty-HAV klasy IgM – wiarygodny marker ostrego zakażenia,
- p/ciała anty-HAV klasy IgG – utrzymują się przez całe życie.
Wirus zapalenia wątroby typu B (HBV):
- okres inkubacji – 4-26 tygodni,
- hepadnawirus, dwuniciowy otoczkowy DNA,
- droga przenoszenia: pozajelitowa,
- zakażenie podczas przetaczania krwi, dializ, zakłucia igłą, przyjmowania narkotyków, przenoszenie
drogą płciową, z matki na dziecko podczas porodu, poprzez kontakt ze śliną, potem, łzami, mlekiem,
patologicznym płynem wysiękowym chorego,
- klinicznie bezobjawowy (subkliniczny) przebieg choroby lub ostre wirusowe zapalenie wątroby z
wyzdrowieniem, przewlekłe zapalenie bez postępu choroby, przewlekła choroba kończąca się
marskością, piorunujące zapalenie wątroby z rozległą martwicą,
- ryzyko raka wątrobowokomórkowego,
- HBsAg – antygen osłonki – pojawia się przed wystąpieniem objawów, osiąga najwyższy poziom
podczas objawowego okresu choroby, spada do wartości niewykrywalnych,
- HbcAg – białko „części rdzennej” nukleokapsydu,
- HBeAg – transkrypt obejmujący rejon przed częścią rdzenną oraz część rdzenną, wydzielany do krwi,
- polimeraza DNA,
- białko X HBV – aktywator transkrypcji,
- HBeAg, HBV-DNA, polimeraza DNA – świadczą o aktywnej replikacji wirusa,
- anty-Hbc klasy IgM – marker świeżego zakażenia i ustępowania ostrej infekcji,
- anty-HBs i anty-Hbc klasy IgG,
- 2 fazy zakażenia HBV (proliferacyjna – episomalny HBV, aktywacja T CD8+, integracyjna –
wbudowanie DNA do genomu gospodarza).
Wirus zapalenia wątroby typu C (HCV):
- okres inkubacji – 2-26 tygodni,
- flawiwirus, jednoniciowy otoczkowy RNA,
- droga przenoszenia: pozajelitowa,
- zakażenie poprzez wszczepienie, przetaczanie krwi, dożylne zażywanie narkotyków, drogą płciową i
wertykalną (z matki na dziecko podczas porodu),
- przewlekłe zapalenie wątroby > 70%, marskość, rzadko piorunujące zapalenie wątroby,
- ryzyko raka wątrobowokomórkowego,
- istnienie wielu typów i podtypów wirusa – brak szczepionki,
- anty-HCV klasy IgG nie zapewnia efektywnej odporności na kolejne zakażenia HCV,
- epizodyczny wzrost stężenia aminotransferaz w surowicy krwi.
Wirus zapalenia wątroby typu D (HDV):
- okres inkubacji – 4-7 tygodni w czasie superinfekcji z HBV,
50

- jednoniciowy otoczkowy RNA z defektem replikacji (zakażenie tylko przy współistniejącym HBV),
- droga przenoszenia: pozajelitowa,
- 2 drogi zapalenia wątroby typu D:
1) koinfekcja – równoczesne zakażenie HBV i HDV,
2) superinfekcja – nadkażenie HDV u przewlekłego nosiciela HBV,
- w przypadku konfekcji większość pacjentów zdrowieje,
- w przypadku superinfekcji zwykle dochodzi do przyspieszenia przebiegu zapalenia wątroby i bardziej
nasilonego przewlekłego zapalenia wątroby, przewlekłe zapalenie wątroby > 80%,
- HDV RNA, HDVAg,
- anty-HDV IgM – świeże zakażenie.
Wirus zapalenia wątroby typu E:
- okres inkubacji - 2-8 tygodni,
- kalciwirus, jednoniciowy bezotoczkowy RNA,
- droga przenoszenia: jelitowa, zakażenie przez wodę,
- nie powoduje przewlekłego zapalenia wątroby ani raka,
- endemicznie w Indiach, sporadycznie u osób podróżujących,
- wysoka śmiertelność wśród kobiet w ciąży (20%),
- HEVAg w cytoplazmie hepatocytów i stolcu,
- HEV-RNA, anty-HEV w surowicy krwi.
Wirus zapalenia wątroby typu G (HGV):
- flawiwirus, jednoniciowy RNA,
- droga przenoszenia: pozajelitowa,
- może spowodować przewlekłe zapalenie wątroby,
- miejscem replikacji są najprawdopodobniej komórki jednojądrowe, HGV nie jest wirusem
hepatotropowym, nie powoduje wzrostu stężenia aminotransferaz w surowicy krwi,
- zakażenie HGV ma efekt ochronny u pacjentów z konfekcją HIV.
Zespoły objawów klinicznych:
1) stan nosicielstwa – bez jawnych objawów choroby lub z przewlekłym zapaleniem wątroby
przebiegającym subklinicznie; rezerwuar zakażenia; nosicielstwo HBV - głównie u osób
zakażonych podczas porodu, u osób z zaburzeniami odporności,
2) zakażenie bezobjawowe – obecne jedynie serologiczne wykładniki choroby, ↑ ALAT, AspAT,
3) ostre wirusowe zapalenie wątroby – (1) okres wylęgania, (2) faza objawowa przedżółtaczkowa,
(3) faza objawowa żółtaczkowa, (4) rekonwalescencja; w fazie przedżółtaczkowej (2) – złe
samopoczucie, łatwe męczenie się, nudności, utrata apetytu, utrata masy ciała, niewielka
gorączka, bóle głowy, mięśni, stawów, wymioty, biegunka; w 10% objawy przypominające
chorobę posurowiczą (gorączka, wysypka skórna, bóle stawowe); ↑ aminotransferaz w surowicy
krwi, powiększona i tkliwa wątroba; w fazie żółtaczkowej (3) nieswoiste objawy zaczynają
ustępować; zwykle żółtaczka z nadmiarem bilirubiny sprzężonej → ciemne zabarwienie moczu;
faza żółtaczkowa nie występuje u dzieci z HAV; zwykle występuje u dorosłych z HAV, w połowie
przypadków zakażeń HBV, w mniej niż połowie HCV,
4) przewlekłe wirusowe zapalenie wątroby – charakter ciągły lub nawracający, >6 miesięcy;
uczucie zmęczenia, złe samopoczucie, utrata apetytu, rzuty łagodnej żółtaczki; pajączki
naczyniowe, rumień dłoniowy, niewielka hepatomegalia, wrażliwość wątroby na ucisk,
nieznacznego stopnia splenomegalia; ↑aminotransferaz w surowicy krwi, ↑czasu
protrombinowego, hipergammaglobulinemia, hiperbilirubinemia, nieznaczny wzrost poziomu
fosfatazy zasadowej; HBV, HCV – choroba kompleksów immunologicznych – zapalenie naczyń
(vasculitis), zapalenie kłębuszków nerkowych (glomerulonephritis); HCV – krioglobulinemia,
5) piorunujące zapalenie wątroby z submasywną lub rozległą martwicą.
Morfologia ostrego WZW:
- zwyrodnienie balonowate,
- cholestaza (brązowe ziarnistości w hepatocytach, czopy żółci w kanalikach),
- stłuszczenie – HCV,
- hepatocyty „o wyglądzie matowego szkła” – HBV,
- „szlifowane” jądra hepatocytów – HBV,
- cytoliza (pękanie błon kom.), apoptoza (obkurczone, eozynochłonne hepatocyty, fragmentacja jąder),
- martwica okołowrotna,
51

- martwica mostkująca (wrotno-wrotna, centralno-centralna, wrotno-centralna),
- dezorganizacja zrazików,
- zmiany zapalne – skupiska makrofagów (często obładowane lipofuscyną), nacieki zapalne o
charakterze mieszanym głównie w przestrzeniach wrotnych, zmiany reaktywne i proliferacja
przewodów żółciowych.
Morfologia przewlekłego WZW:
- martwica hepatocytów (okołowrotna, mostkująca),
- nacieki zapalne głównie z limfocytów, makrofagów, komórek plazmatycznych,
- włóknienie wrotne, okołowrotne, mostkujące,
- marskość.
VI. Masywna (rozległa) martwica wątroby
Przyczyny:
- ostre zakażenia wirusowe,
- leki, związki chemiczne (paracetamol, izoniazyd, antydepresanty < zwł. inhibitory oksydazy
monoaminowej >, halotan, α-metyldopa, mykotoksyny grzyba Amanita phalloides),
- udar cieplny,
- zamknięcie światła żył wątrobowych,
- choroba Wilsona,
- zespoły drobnokropelkowego stłuszczenia,
- nowotwór złośliwy,
- zapalenie autoimmunizacyjne.
Morfologia:
- utrata masy wątroby,
- zapadnięta sieć włókien retikulinowych, zachowane przestrzenie wrotne,
- prawie zupełny brak reakcji zapalnej,
po kilku dniach masywny napływ komórek nacieku zapalnego i regeneracja.
Objawy kliniczne:
- żółtaczka,
- encefalopatia w ciągu 2-3 tyg. od pierwszych objawów klin.,
- faetor hepaticus,
- brak objawów typowych dla przewlekłej choroby wątroby,
- powikłania: skaza krwotoczna, niestabilność układu krążenia, niewydolność nerek, ARDS, zaburzenia
wodno-elektrolitowe i kwasowo-zasadowe, posocznica.
VII. Zapalenie autoimmunologiczne wątroby
Cechy:
- głównie u kobiet,
- ↑ IgG (>2,5g/dl),
- wysokie stężenie autoprzeciwciał,
-zwiększona częstość występowania antygenów HLA-B8 oraz HLA-DRw3,
- współwystępowanie innych chorób autoimmunizacyjnych,
- ryzyko rozwoju marskości,
- bardzo dobra odpowiedź na leczenie immunosupresyjne.
Typ 1 – najczęstszy – p/ciała p/jądrowe i/lub przeciw kom. mięśniowym gładkim.
Typ 2 – najrzadszy – p/ciała przeciw antygenom mikrosomalnym kom. wątroby i nerek.
Typ 3 – p/ciała przeciwko rozpuszczalnemu antygenowi wątroby.
VIII. Ropnie wątroby
Etiologia:
- pasożytnicza (pełzakowica, tasiemczyca i inne) – kraje rozwijające się,
- bakteryjna, grzybicza – kraje rozwinięte.
Drogi inwazji:
1) wstępujące zapalenie dróg żółciowych (cholangitis ascendens),
2) rozsiew przez naczynia krwionośne,
3) bezpośrednia inwazja ze źródła zakażenia w pobliżu wątroby,
4) zranienie penetrujące do wątroby.
Zakażeniom sprzyja spadek odporności (podeszły wiek, immunosupresja, chemioterapia, niewydolność
szpiku).
52

Ropnie bakteryjne (ropotwórcze):
- pojedyncze lub mnogie,
- bardzo rzadko ropnie okolicy podprzeponowej mogą rozprzestrzeniać się do klatki piersiowej, tworząc
ropniaka opłucnej i ropnie płuc,
- klinicznie: gorączka, ból w prawym górnym kwadrancie jamy brzusznej, wrażliwość wątroby na ucisk,
hepatomegalia, żółtaczka,
- leczenie: antybiotyki, drenaż chirurgiczny.
IX. Uszkodzenia polekowe i toksyczne
Reakcja na lek lub związek chemiczny:
- możliwa do przewidzenia (paracetamol, tetracykliny, leki przeciwnowotworowe, toksyna Amanita
phalloides, czterochlorek węgla, alkohol),
- typu idiosynkrazji (chloropromazyna, halotan, sulfonamidy, α-metyldopa, allopurinol).
Typy uszkodzenia:
- stłuszczenie drobnokropelkowe (tetracykliny),
- stłuszczenie wielkokropelkowe (etanol, metotreksat),
- martwica strefy środkowej zrazików (CCl
4
, paracetamol, halotan),
- rozlana lub masywna martwica (halotan, izoniazyd, paracetamol, α-metyldopa, Amanita phalloides),
- zapalenie wątroby (α-metyldopa, izoniazyd),
- włóknienie (etanol, metotreksat),
- tworzenie ziarniniaków (sulfonamidy, α-metyldopa),
- zastój żółci (chloropromazyna, steroidy anaboliczne, erytromycyna),
- choroba wenookluzyjna (leki cytotoksyczne),
- zakrzepica żył wątrobowych lub żyły wrotnej (estrogeny, leki cytotoksyczne),
- peliosis hepatis (steroidy anaboliczne, doustne środki antykoncepcyjne),
- gruczolak (doustne środki antykoncepcyjne),
- rak wątrobowokomórkowy (polichlorek winylu, aflatoksyny, Thorotrast),
- rak z dróg żółciowych (Thorotrast),
- mięsak naczyniowy (polichlorek winylu, związki arsenu, Thorotrast).
X. Choroba alkoholowa wątroby
1) Stłuszczenie wątroby (steatosis hepatis):
- początkowo drobnokropelkowe, w miarę upływu czasu wielkokropelkowe,
- początkowo zmiany w środkowych częściach zrazików,
- wątroba powiększona,
- po pewnym czasie rozwija się włóknienie,
- hepatomegalia, niewielki wzrost stężenia bilirubiny i fosfatazy zasadowej w surowicy krwi.
2) Zapalenie alkoholowe wątroby (hepatitis alcoholica):
- zwyrodnienie balonowate,
- martwica komórek,
-
ciałka Mallory’ego – konglomeraty cytokeratyn i innych białek,
- odczyn z neutrofilów, nacieki z limfocytów i makrofagów,
- włóknienie (okołozatokowe, okołożylne, okołowrotne),
- czasem zastój żółci i niewielkiego stopnia gromadzenie hemosyderyny (żelaza) w hepatocytach oraz
kom. Browicza i Kupffera,
- złe samopoczucie, jadłowstręt, utrata masy ciała, uczucie dyskomfortu w górnej części jamy
brzusznej, wrażliwość powiększonej wątroby na ucisk, gorączka,
- hiperbilirubinemia, wzrost stężenia fosfatazy zasadowej, leukocytoza, wzrost stężenia
aminotransferaz.
3) Marskość alkoholowa:
- początkowo marskość drobnoguzkowa, z czasem tworzenie większych guzków,
- szerokie pasma łykowatej, bliznowatej tkanki,
53

- typowe objawy marskości, czasem podstępny rozwój choroby (złe samopoczucie, osłabienie, utrata
masy ciała, brak apetytu),
- wzrost stężenia aminotransferaz, hiperbilirubinemia, wzrost stężenia fosfatazy zasadowej,
hipoproteinemia, niedokrwistość,
- bezpośrednie przyczyny zgonu – niewydolność wątroby, masywne krwawienie z żylaków przełyku,
towarzyszące zakażenia, zespół wątrobowo-nerkowy, rak wątrobowokomórkowy.
Patogeneza uszkodzenia alkoholowego:
- stłuszczenie jest wynikiem (1) działania dehydrogenazy alkoholowej i dehydrogenazy aldehydu
octowego → powstaje nadmiar zredukowanego dwunukleotydu nikotynoamidoadeninowego → dochodzi
do biosyntezy lipidów, (2) zaburzeń w tworzeniu i wydzielaniu lipoprotein, (3) zwiększonego rozpadu
tłuszczu w tkankach obwodowych,
- aktywacja cytochromu P-450 → wzmożone przekształcanie leków do toksycznych metabolitów,
- utlenianie alkoholu → tworzenie wolnych rodników,
- zaburzenie czynności mikrotubul i mitochondriów oraz przepuszczalności błon,
- aldehyd octowy – peroksydacja lipidów, łączenie białek z aldehydem octowym → zaburzenie czynności
cytoszkieletu i błon,
- zapalenie alkoholowe → nacieki z neutrofilów → uwalnianie toksycznych metabolitów tlenowych,
- atak immunologiczny przeciwko hepatocytom,
- najbardziej wrażliwe na uszkodzenie przez toksyny są strefy środkowe zrazików.
XI. Stłuszczenie niealkoholowe wątroby
Cechy:
- wzrost stężenia aminotransferaz w surowicy krwi,
- niskie ryzyko rozwoju marskości,
- u części pacjentów obecne są mieszane nacieki zapalne z neutrofilów i komórek jednojądrowych w
miąższu, ciałka Mallory’ego, destrukcja hepatocytów,
- przyczyny – otyłość, cukrzyca typu 2, nietolerancja glukozy, hiperlipidemia,
- przebieg zwykle bezobjawowy, niekiedy uczucie zmęczenia, złe samopoczucie, uczucie dyskomfortu w
prawym górnym kwadrancie.
XII. Hemochromatoza
Patogeneza wrodzonej hemochromatozy:
- dziedziczona autosomalnie recesywnie (gen HFE), związana z układem HLA,
- HFE reaguje z receptorem transferyny w błonie plazmatycznej,
- nadmierne gromadzenie żelaza w organizmie (głównie w wątrobie i trzustce),
- żelazo gromadzi się wskutek nadmiernego wchłaniania w jelitach (0,5-1,0 g rocznie),
- pierwsze objawy w piątej lub szóstej dekadzie życia,
- przeważają mężczyźni.
Objawy kliniczne:
- marskość drobnoguzkowa (wszyscy pacjenci) z hepatomegalią, bóle brzucha,
- cukrzyca (3/4 pacjentów),
- przebarwienie skóry (3/4 pacjentów),
- zaburzenia czynności serca (arytmie, kardiomiopatia),
- atypowe zapalenie stawów,
- hipogonadyzm (np. zanik miesiączki u kobiet, utrata popędu płciowego i impotencja u mężczyzn)
związany z zaburzeniem prawidłowej czynności osi podwzgórze-przysadka,
- ryzyko rozwoju raka wątrobowokomórkowego,
- objawy kliniczne przy ilości żelaza >20g,
- toksyczność związana z peroksydacją lipidów (wolne rodniki), pobudzeniem tworzenia kolagenu,
bezpośredniej interakcji żelaza z DNA,
- leczenie – upusty krwi, związki chelatujące.
Morfologia:
- odkładanie hemosyderyny (wątroba, trzustka, mięsień sercowy, przysadka, nadnercze, tarczyca,
przytarczyce, stawy, skóra),
54
- marskość,
- włóknienie trzustki, nieznaczny zanik miąższu,
- w wątrobie brak wykładników zapalenia (żelazo jest toksyną działającą bezpośrednio),
- serce z ziarnistościami hemosyderyny, delikatne włóknienie śródmiąższowe,
- skóra zabarwiona w wyniku odkładania się hemosyderyny i zwiększonej produkcji melaniny w
naskórku,
- odkładanie się hemosyderyny w wyściółce maziowej stawów, ostre zapalenie stawów, gromadzenie
się nadmiaru pirofosforaów wapnia, niszczenie chrząstek stawowych, czasami zapalenie wielostanowe i
kalectwo (
rzekoma dna moczanowa),
- zanik jąder.
Wtórne gromadzenie żelaza:
1)
pozajelitowe dostarczanie (przetoczenia krwi – hemodializowanie, niedokrwistość aplastyczna,
niedokrwistość sierpowatokrwinkowa, zespoły mielodysplastyczne, białaczki, podawanie
dekstranu zawierającego żelazo),
2) nieefektywna erytropoeza (β-talasemia, niedokrwistość syderoblastyczna, niedobór kinazy
pirogronianowej),
3) nadmierne doustne spożycie żelaza (syderoza Bantu),
4) wrodzony brak transferyny,
5) przewlekłe choroby wątroby (przewlekła choroba alkoholowa wątroby, porfiria skórna późna).
Hemochromatoza noworodków:
- rozwój niewydolności wątroby zapoczątkowany w czasie ciąży,
- czynniki dziedziczne związane z mitochondriami lub narażenie w czasie ciąży na działanie czynników
środowiskowych.
XIII. Choroba Wilsona (zwyrodnienie wątrobowo-soczewkowate)
Patogeneza:
- dziedziczona autosomalnie recesywnie (gen ATP7B),
- gromadzenie miedzi w toksycznych ilościach w wielu tkankach i narządach, głównie w wątrobie,
mózgu i gałce ocznej,
- wchłonięta miedź nie może przedostać się do układu krążenia w formie ceruloplazminy, wydalanie
miedzi z żółcią znacząco zmniejszone,
- uszkodzenie wątroby przez (1) tworzenie wolnych rodników, (2) wiązanie miedzi z grupami
sulfohydrylowymi białek komórkowych, (3) przemieszczanie innych metali z metaloenzymów
wątrobowych,
- zwykle przed 5 r.ż. miedź nie związana z ceruloplazminą zaczyna być obecna w krążeniu, powodując
hemolizę i zmiany patologiczne w mózgu, rogówce, nerkach, kościach, stawach, przytarczycach,
- spadek stężenia ceruloplazminy w surowicy krwi, wzrost zawartości miedzi w wątrobie, wzrost
wydalania miedzi z moczem.
Morfologia:
- wątroba – stłuszczenie, zmienione wodniczkowo jądra (zawierają glikogen lub wodę), ogniskowa
martwica hepatocytów, ostre lub przewlekłe zapalenie,
ciałka Mallory’ego, może rozwinąć się
marskość; rzadko występującym objawem jest masywna, rozległa martwica wątroby,
- mózg – zmiany toksyczne w obrębie jąder podstawy, zwłaszcza w skorupie; zaniki, jamiste
przestrzenie,
- gałka oczna –
pierścienie Kaysera-Fleischera (zielonkawe lub brązowe złogi miedzi w błonie
Descemeta w rąbku rogówki).
Objawy kliniczne:
- objawy ostrej lub przewlekłej choroby wątroby,
- objawy neurologiczne lub psychiatryczne (zmiany w zachowaniu, psychoza, objawy przypominające
chorobę Parkinsona),
- obecność pierścieni Kaysera-Fleischera,
- leczenie – związki chelatujące (D-penicylamina), niekiedy niezbędne jest przeszczepienie wątroby.
XIV. Niedobór α
1
-antytrypsyny
Patogeneza:
- choroba dziedziczona autosomalnie recesywnie,
55

- niedobór α
1
-antytrypsyny (inhibitora proteaz).
Morfologia:
- wątroba – hepatocyty zawierają kuliste wtręty cytoplazmatyczne zatrzymanej AAT,
różny obraz – od nasilonego zastoju żółci z martwicą hepatocytów u noworodków poprzez marskość
ujawniającą się u dzieci aż do przewlekłego zapalenia lub marskości ujawniającej się w późniejszym
okresie życia,
- płuca – rozedma płuc.
Objawy kliniczne:
- zastój żółci,
- objawy zapalenia lub marskości wątroby,
- objawy rozedmy płuc,
- ryzyko rozwoju raka wątrobowokomórkowego,
- leczenie – w rozwiniętej chorobie jedynym sposobem jest przeszczepienie wątroby.
XV. Zapalenie wątroby noworodków
Główne przyczyny cholestazy noworodków:
- zamknięcie przewodów żółciowych (zarośnięcie pozawątrobowych dróg żółciowych),
- zakażenia u noworodków,
- toksyny (leki, odżywianie pozajelitowe),
- choroby metaboliczne (tyrozynemia, choroba Niemanna-Picka, galaktozemia),
- niedobór α
1
-antytrypsyny,
- mukowiscydoza,
- postępująca rodzinna cholestaza wewnątrzwątrobowa,
- wstrząs/hipoperfuzja,
- marskość występująca u dzieci w Indiach,
- zespół Alagille’a (niedobór lub zupełny brak przewodów żółciowych),
- idiopatyczne zapalenie wątroby noworodków.
Cholestaza noworodków
– przedłużona hiperbilirubinemia z nadmiarem bilirubiny sprzężonej. Objawy kliniczne: żółtaczka,
oddawanie ciemnego moczu, jasne stolce, hepatomegalia, dysfunkcja wątroby w zakresie syntez (np.
hipoprotrombinemia). Główną przyczyną jest idiopatyczne zapalenie wątroby noworodków, następnie
zarośnięcie pozawątrobowych dróg żółciowych.
XVI. Zespół Reye’a
Cechy:
- stłuszczenie wątroby i encefalopatia,
- głównie u dzieci przed 4 r.ż. i typowo rozwija się 3-5dni
po zakażeniu wirusowym,
- wymioty, drażliwość lub ospałość, hepatomegalia,
- u 25% rozwija się śpiączka, której towarzyszy wzrost stężenia bilirubiny, aminotransferaz, a zwłaszcza
amoniaku w surowicy krwi,
- przyczyną zgonu są zaburzenia neurologiczne lub niewydolność wątroby,
- leczenie objawowe i podtrzymujące funkcje życiowe,
- patogeneza – prawdopodobnie dysfunkcja mitochondriów, która występuje samoistnie lub towarzyszy
zakażeniu wirusowemu,
- obserwowano przypadki zespołu Reye’a spowodowane przyjmowaniem
salicylanów podczas
zakażenia wirusowego,
- morfologia – stłuszczenie drobnokropelkowe wątroby, obrzęk mózgu (obrzmiałe astrocyty), czasem
stłuszczenie drobnokropelkowe w mięśniach szkieletowych, nerkach oraz sercu.
XVII. Choroby wątroby z blokadą przepływu w drogach żółciowych
Marskość żółciowa wtórna:
- najczęstszą przyczyną jest kamica pozawątrobowych dróg żółciowych, ale także zarośnięcie dróg
żółciowych, nowotwory złośliwe dróg żółciowych oraz głowy trzustki, zwężenia pooperacyjne,
- jeśli blokada przepływu nie jest całkowita, dochodzi do wtórnego zakażenia bakteryjnego dróg
żółciowych (zapalenie wstępujące dróg żółciowych, cholangitis ascendens) → E. coli, enterokoki,
- świąd, żółtaczka, osłabienie, ciemne zabarwienie moczu, odbarwienie stolców, hepatosplenomegalia,
- hiperbilirubinemia z nadmiarem bilirubiny sprzężonej, wzrost stężenia fosfatazy zasadowej, kwasów
żółciowych, cholesterolu w surowicy krwi,
56

- przed rozwojem marskości wyraźny zastój żółci, proliferacja przewodów żółciowych, nacieki z
neutofilów, obrzęki przestrzeni wrotnych.
Marskość żółciowa pierwotna:
- etiologia autoimmunizacyjna,
- niszczenie wewnątrzwątrobowych dróg żółciowych, zapalenie ziarniniakowe,
- głównie u kobiet w średnim wieku,
- początek skryty; podstępny, utajony przebieg,
- objawy kliniczne jak w marskości żółciowej wtórnej (jednak pierwszym objawem zwykle świąd,
żółtaczka pojawia się późno),
- wykładniki laboratoryjne jak w marskości żółciowej wtórnej, hiperbilirubinemia występuje późno,
dodatkowo wzrost stężenia IgM oraz obecność
autoprzeciwciał, zwłaszcza przeciwko
mitochondrialnej dehydrogenazie pirogronianowej,
- związek z innymi chorobami autoimmunizacyjnymi,
- przed rozwojem marskości gęste nacieki limfocytarne w przestrzeniach wrotnych oraz ziarniniaki
niszczące przewody żółciowe.
Pierwotne zapalenie dróg żółciowych ze stwardnieniem:
- etiologia prawdopodobnie autoimmunizacyjna,
- związek z zapaleniami idiopatycznymi jelit (zwłaszcza CU),
- częściej u mężczyzn,
- zapalenie, włóknienie i zwężenie oraz odcinkowe poszerzenie zablokowanych wewnątrz- i
zewnątrzwątrobowych dróg żółciowych,
- objawy kliniczne jak w marskości żółciowej wtórnej; podstępny, utajony przebieg,
- wykładniki laboratoryjne jak w marskości żółciowej wtórnej, dodatkowo wzrost stężenia IgM i IgG w
surowicy krwi,
- ryzyko rozwoju pierwotnego raka dróg żółciowych.
Morfologia:
-
marskość żółciowa wtórna – grube, włókniste przegrody, rozległa proliferacja małych
przewodzików żółciowych, obrzęk; zakażenie bakteryjne → nacieki z neutrofilów wokół przewodów
żółciowych; mogą rozwinąć się zmiany zapalne żył i ropnie dróg żółciowych,
-
marskość żółciowa pierwotna – brak międzyzrazikowych przewodów żółciowych (niszczone przez
zapalenie –
kwitnące zmiany przewodowe), wewnątrznabłonkowe nacieki z limfocytów, zapalenie
ziarniniakowe, w przestrzeniach wrotnych nacieki z limfocytów, makrofagów, kom. plazmatycznych
oraz pojedynczych eozynofilów,
-
pierwotne zapalenie dróg żółciowych ze stwardnieniem – włókniejące zapalenie przewodów
żółciowych, w zajętych przestrzeniach wrotnych włóknienie o układzie „listków cebuli”, zarośnięcie
światła, niewielkie nacieki limfocytarne.
XVIII. Zaburzenia w krążeniu
Zaburzenia w dopływie krwi do wątroby (blok przedwątrobowy):
1) zaburzenia napływu krwi tętnicą wątrobową (zakrzepica, ucisk, zator, nowotwór, posocznica) –
powikłania przeszczepienia wątroby, utrata narządu, ograniczony zawał miąższu,
2) blokada przepływu żyłą wrotną (zakrzepica, guz, posocznica, zespół Bantiego)– ból brzucha,
wodobrzusze, żylaki przełyku, splenomegalia, przekrwienie i zawał jelit.
Zespół Bantiego – zakrzepica żyły wrotnej (noworodkowe zapalenie pępka <omphalitis> lub
cewnikowanie żyły pępkowej), po latach włóknisty kanał naczyniowy, splenomegalia, żylaki
przełyku.
Zawał rzekomy Zahna – w ostrej zakrzepicy wewnątrzwątrobowych odgałęzień żyły wrotnej.
Stwardnienie wątrobowo-wrotne (sclerosis hepatoportalis) – postępujące stwardnienie w
układzie żyły wrotnej, np. schorzenia mieloproliferacyjne (z nadkrzepliwością i zapaleniem
otrzewnej), działanie związków arsenu.
57

Zaburzenia przepływu krwi przez wątrobę:
1) marskość wątroby,
2) zamknięcie światła zatok (niedokrwistość sierpowatokrwinkowa, DIC) – może pojawić się
martwica miąższu, np. w rzucawce porodowej,
3)
układowa niewydolność krążenia (prawokomorowa – przewlekłe przekrwienie bierne wątroby,
lewokomorowa lub wstrząs – martwica niedokrwienna hepatocytów strefy środkowej zrazików,
prawokomorowa + lewokomorowa – charakterystyczna martwica krwotoczna części środkowych
zrazików,
wątroba muszkatołowa <hepar moschatum>; może pojawić się tzw. stwardnienie
sercowe),
4)
peliosis hepatis – prosowate wylewy krwawe, pierwotne poszerzenie naczyń zatokowych
związane z używaniem steroidów anabolicznych, doustnych środków antykoncepcyjnych,
danazolu; może spowodować krwotok do jamy brzusznej lub niewydolność wątroby.
Blokada przepływu krwi przez żyły wątrobowe:
1)
zakrzepica żył wątrobowych (
zespół Budda i Chiariego) – hepatomegalia, utrata masy
ciała, wodobrzusze, bóle brzucha; przyczyny – czerwienica prawdziwa, choroby
mieloproliferacyjne, ciąża, połóg, doustne środki antykoncepcyjne, napadowa nocna
hemoglobinuria, rak wątrobowokomórkowy; mechaniczna blokada przepływu spowodowana
przez ropnie lub torbiele pasożytnicze,
2)
choroba wenookluzyjna – po przeszczepieniu szpiku (chemioterapia, radioterapia) lub
alkaloidach pirolizydynowych, obrzęk podśródbłonkowy i zarastanie światła małych
odgałęzień żył wątrobowych.
XIX. Nowotwory i zmiany nowotworopodobne wątroby
Nowotwory łagodne:
1)
naczyniak jamisty (haemangioma cavernosum),
2)
ogniskowy rozrost guzkowy (hyperplasia nodularis focalis) – zmiana dobrze odgraniczona,
ale słabo otorebkowana, z centralnie położoną włóknistą blizną; najczęściej u ludzi dorosłych w
młodym i średnim wieku; nie rozwija się w nowotwór złośliwy,
3)
gruczolak wątrobowokomórkowy (adenoma hepatocellulare) – u kobiet w wieku rozrodczym
stosujących doustne środki antykoncepcyjne, cofa się po ich odstawieniu; dobrze odgraniczony,
często tuż pod torebką; zbudowany z wysp i sznurów komórek przypominających normalne
hepatocyty lub ujawniających pewną różnorodność, brak przestrzeni wrotnych, widoczne
naczynia tętnicze i żylne; ryzyko pęknięcia, zwłaszcza w czasie ciąży; może rozwinąć się rak
wątrobowokomórkowy.
Nowotwory złośliwe (pierwotne):
1)
rak wątrobowokomórkowy – związany z zakażeniem HBV, nadużywaniem alkoholu,
przewlekłymi chorobami wątroby, HCV, aflatoksynami (Aspergillus flavus), częściej u mężczyzn
oraz u osób rasy czarnej; w rejonach o wysokiej zachorowalności zwykle w 20-40 r.ż., na
obszarach o niskiej zachorowalności w 50-60 r.ż.; rozwija się u 40% osób z wrodzoną
tyrozynemią; nowotwór jednoogniskowy, wieloogniskowy lub rozlegle inwazyjny; słabo
odgraniczony, z tendencją do naciekania kanałów naczyniowych (przerzuty, wrastanie do żyły
wrotnej lub żyły głównej dolnej i osiąganie prawego serca); wysoko dojrzały (sznury lub wyspy
hepatocytów, kuleczki żółci w cytoplazmie i kanalikach rzekomych) lub nisko (olbrzymie,
58

anaplastyczne komórki wielojądrowe); mogą być obecne wtręty przypominające
ciałka
Mallory’ego; z reguły niewielka ilość zrębu,
2)
rak włóknisto-blaszkowy (fibrolamellar carcinoma) – wariant raka wątrobowokomórkowego,
występuje z równą częstością u młodych mężczyzn i kobiet (20-40 r.ż.), brak związku z
czynnikami ryzyka raka wątrobowokomórkowego, lepsze rokowanie; zwykle pojedynczy, duży,
twardy guz (carcinoma scirrhosum) zbudowany z wielobocznych, wysoko dojrzałych komórek
rosnących w gniazdach lub beleczkach, gęsto upakowane wiązki kolagenu,
3)
rak z nabłonka dróg żółciowych (cholangiocarcinoma) – może być związany z pierwotnym
zapaleniem dróg żółciowych ze stwardnieniem, przewlekłym zakażeniem dróg żółciowych przez
przywry z rodzaju Opisthorchis sinensis, narażeniem na Thorotrast; gruczolakorak; obfity,
włóknisty zrąb (desmoplazja), z reguły widoczne struktury gruczołowe i cewkowe wysłane
sześciennym lub walcowatym nabłonkiem, cechy nieznacznej anaplazji; brak barwników
żółciowych i szklistych wtrętów,
4)
wątrobiak płodowy (hepatoblastoma) – agresywny, występuje w dzieciństwie,
5)
mięsak naczyniowy (angiosarcoma).
Objawy kliniczne pierwotnych raków wątroby:
- hepatomegalia,
- u pacjentów z marskością – gwałtowne powiększanie się wątroby, nagłe zwiększenie się
wodobrzusza, pojawienie się krwistego płynu w jamie otrzewnowej, gorączka, bóle,
- ↑ stężenia α-fetoproteiny,
- rozsiew do okolicznych węzłów chłonnych, płuc, kości, nadnerczy,
- niepomyślne rokowanie (kacheksja, krwawienie z żołądka, jelit lub żylaków przełyku,
niewydolność wątroby ze śpiączką wątrobową, pęknięcie guza z krwotokiem prowadzącym do
zgonu).
Przerzuty:
- wątroba i płuca są najczęściej zajętymi narządami przez rozsiew nowotworowy,
- guz pierwotny zwykle w okrężnicy, płucu, sutku,
- wieloogniskowość.
XX. Choroby pęcherzyka żółciowego
Kamica pęcherzyka żółciowego
Patogeneza:
- przesycenie żółci cholesterolem,
- zastój w pęcherzyku żółciowym,
- 2 rodzaje kamieni:
cholesterolowe (80%) i barwnikowe (sole wapniowe bilirubiny),
-
kamienie cholesterolowe – Europa Północna i Ameryka, Indianie Pima, zaawansowany wiek,
hormony żeńskie (płeć kobieca, doustne środki antykoncepcyjne, ciąża), otyłość, hiperlipidemia, nagły
spadek masy ciała, zastój w pęcherzyku żółciowym, wrodzone zaburzenia metabolizmu kwasów
żółciowych,
-
kamienie barwnikowe – Azja, rejony wiejskie, przewlekła hemoliza, zakażenia dróg żółciowych,
schorzenia przewodu pokarmowego (choroby jelita krętego, np. choroba Leśniowskiego-Crohna),
wycięcie jelita krętego lub zespolenia omijające, niewydolność trzustki w przebiegu mukowiscydozy.
Morfologia:
-
kamienie cholesterolowe – najczęściej liczne, o gładkiej, płaskiej powierzchni, w większości
przepuszczalne dla Rtg,
59

-
kamienie barwnikowe – czarne (żółć jałowa, małe oraz liczne, zwykle nieprzepuszczalne dla Rtg) i
brązowe (przy zakażeniu, pojedyncze lub jest ich mało, przepuszczalne dla Rtg).
Objawy kliniczne:
- u wielu pacjentów brak objawów,
- rozdzierający ból (ciągły lub kolkowy),
- powikłania: ropniak (empyema), przedziurawienia, przetoki, zmiany zapalne w drzewie żółciowym,
zastój żółci, zapalenie trzustki, niedrożność jelit (z ucisku).
Zapalenie pęcherzyka żółciowego
Typy:
-
ostre kamicze – zamknięcie odpływu żółci, podrażnienie chemiczne,
-
ostre niekamicze – w stanach pooperacyjnych, po ciężkich urazach, w ciężkich stanach po
oparzeniach, w przebiegu posocznicy; odwodnienie, zastój w pęcherzyku, zwolniony przepływ krwi,
zakażenie bakteryjne,
-
przewlekłe – prawie zawsze związane z kamicą.
Morfologia:
-
ostre zapalenie – pęcherzyk powiększony i napięty, błona surowicza pokryta włóknikiem lub
wysiękiem ropnym, kamienie obecne w 90% przypadków, światło wypełnione mętną żółcią; może dojść
do ropniaka pęcherzyka żółciowego lub zgorzelinowego zapalenia (cholecystitis gangraenosa);
histologicznie obrzęk, przekrwienie, nacieki leukocytarne, tworzenie ropni lub zgorzel,
-
przewlekłe zapalenie – zwykle kamicze, pęcherzyk różnej wielkości, błona podśluzowa i warstwa
podsurowicówkowa często pogrubiałe wskutek włóknienia, nacieki z limfocytów.
Objawy kliniczne:
-
ostre kamicze – niezauważalne lub ostry ból w górnej części jamy brzusznej (często promieniujący
do prawej łopatki), gorączka, nudności, leukocytoza, krańcowe wyczerpanie, tkliwość prawej okolicy
podżebrowej na ucisk, skurcz mięśni brzucha,
-
ostre niekamicze – objawy często maskowane przez ogólnie ciężki stan pacjenta,
-
przewlekłe – nawracające napady ciągłego lub kolkowego bólu w nadbrzuszu lub w prawym górnym
kwadrancie, nudności, wymioty, nietolerancja tłustych pokarmów,
-
powikłania: zapalenie dróg żółciowych, posocznica, perforacja, ropień, rozlane zapalenie otrzewnej,
utworzenie przetoki żółciowo-jelitowej, niedrożność jelit, nasilenie już istniejących chorób,
dekompensacja.
XXI. Choroby pozawątrobowych dróg żółciowych
Kamica przewodu żółciowego wspólnego (choledocholithiasis) oraz wstępujące zapalenie
dróg żółciowych (cholangitis ascendens):
- przebiegają równolegle,
- zakażenie → E. coli, Klebsiella, Clostridium, Bacteroides, Enterobacter, streptokoki grupy D, w
niektórych populacjach Fasciola hepatica, Schistosoma, Clonorchis sinensis, Opisthorchis viverrini,
- gorączka, dreszcze, ból w jamie brzusznej, żółtaczka,
- ryzyko rozwoju posocznicy.
Zarośnięcie dróg żółciowych:
- jedna z przyczyn cholestazy noworodków,
- zapalenie i zwłóknienie dróg żółciowych,
- włóknienie okołowrotne i rozwój marskości w ciągu pierwszych 3-6 miesięcy życia,
- nieco częściej u dziewczynek.
60

XXII. Nowotwory
Rak pęcherzyka żółciowego:
- nieco częściej u kobiet,
- w siódmej dekadzie życia,
- często obecne kamienie,
- związek z zakażeniami,
- wzrost egzofityczny (włóknisty, twardy guz) lub naciekający,
- z reguły gruczolakorak (może tworzyć brodawkowate rozrosty, może być nisko lub zupełnie
niedojrzały), mniejszość to raki płaskonabłonkowe i rakowiaki,
- rozsiew do wątroby, przewodu pęcherzykowego, przewodów żółciowych, węzłów chłonnych wnęki
wątroby, otrzewnej, przewodu pokarmowego, płuc,
- początkowy przebieg podstępny, bóle brzucha, żółtaczka, jadłowstręt, nudności i wymioty.
Rak pozawątrobowych dróg żółciowych (cholangiocarcinoma), w tym brodawki Vatera:
- podstępny przebieg → żółtaczka, której nie towarzyszą objawy bólowe,
- w późniejszym wieku, nieco częściej u mężczyzn,
- czynniki ryzyka: zakażenie przywrami, pierwotne zapalenie dróg żółciowych ze stwardnieniem,
zapalenia idiopatyczne jelit,
- większość to gruczolakoraki, które mogą wydzielać śluz,
- obfity, włóknisty zrąb,
- żółtaczka, ciemny mocz, odbarwienie stolców, nudności, wymioty, utrata masy ciała,
- hepatomegalia,
- wyczuwalny, powiększony pęcherzyk,
- ↑ stężenia fosfatazy zasadowej i aminotransferaz, wydłużenie czasu protrombinowego,
-
guz Klatskina – w miejscu rozdwojenia prawego i lewego przewodu żółciowego,
-
rak brodawki Vatera – trudny do klinicznego odróżnienia od raka głowy trzustki.
CHOROBY TRZUSTKI:
Do najważniejszych chorób trzustki należą: Choroby części zewnątrzwydzielniczej: ostre i przewlekłe
zapalenia oraz rak trzustki i choroby części wewnątrzwydzielniczej cukrzyca i nowotwory.
1. Ostre zapalenie trzustki
a) czynniki etiologiczne zapalenia trzustki:
do najważniejszych należy alkohol oraz kamienie żółciowe, które łącznie odpowiedzialne są za 80%
przypadków. Ponadto wyróżniamy czynniki metaboliczne takie jak: hiperlipidemia, hiperkalcemia, leki,
predyspozycje genetyczne, czynniki mechaniczne: uraz, operacje, kamienie, czynniki naczyniowe:
wstrząs, zmiany miażdżycowo zatorowe oraz infekcje: świnka, Coxsackie, Mycoplasma pneumoniae.
b) Patogeneza.
Zwykle dochodzi do zatkania przewodu żółciowego wspólnego przez kamienie żółciowe, złogi z płynu
bogatobiałkowego, którego wydzielanie indukowane jest alkoholem. Zamknięcie przewodu powoduje
wzrost ciśnienia hydrostatycznego wewnątrz przewodów tym bardziej, że sok trzustkowy jest stale
produkowany. Obrzęk osłabia przepływ krwi przez narząd, dochodzi do niedokrwienia komórek trzustki.
W konsekwencji następuje aktywacja trypsynogenu wewnątrz trzustki (fizjologicznie powinna zachodzić
w obrębie przewodu pokarmowego. Aktywacji trypsynogenu sprzyja niskie ph, wysoki poziom wapnia.
Proces zachodzi albo na zasadzie autoaktywacji wewnątrz ziarna zymogenu albo ziarna stają się
niewłaściwym celem lizosomów i enzym jest aktywowany przez katapepsynę B (lizosomalną). Komórka
pęcherzykowa zostaje uszkodzona przez trypsynę. Uwalniane zostają również inne enzymy trawienne -
lipazy, elastazy itd. Enzymy wywołują martwice nienzymatyczne - miąższu trzustki innych komórek
pęcherzykowych oraz tkanki tłuszczowej trzustki i około trzustkowej. Uszkodzone komórki uwalniają
kininy i czynnik XII, w naczyniach trzustki rozwija się zakrzepica, zastój co sprzyja występowaniu
krwotoków. Wydzielane są TNF, PAF, IL1, NO wywołujące odpowiedź zapalną - zapalenie śródmiąższowe
trzustki.
61

c) Morfologia.
Cztery podstawowe objawy: proteolityczne zniszczenie miąższu trzustki, martwica naczyń
krwionośnych z krwotokami, martwica tłuszczu (lipazy) oraz ostra reakcja zapalna.
Martwica tłuszczu: zwakuolizowane komórki tłuszczowe wypełnione różowym nieprzejrzystym,
ziarnistym osadem pochodzącym z hydrolizy tłuszczu. Wolne kwasy tłuszczowe tworzą sole z wapniem
– wytrącają się w postaci złogów. Ogniska martwicy tłuszczowej znaleźć można także poza trzustką, a
surowiczy, mętny płyn w jamie otrzewnowej przyjmuje brązowy kolor z kuleczkami tłuszczu.
Ostre krwotoczne zapalenie trzustki charakteryzuje się występowaniem niebiesko czarnych obszarów
krwotoków obok obszarów rozmiękania i martwicy tłuszczowej.
d) Objawy
- bóle brzucha
- wstrząs wynikający z krwotoków oraz z wydzielania kinin i prostaglandyn
- wzrost stężeń amylazy i lipazy w surowicy
- glukozuria, żóltaczka, hiperglikemia, hipokalcemia
2. Przewlekłe zapalenie trzustki.
Choroba występuje najczęściej w u mężczyzn w średnim wieku, nadużywających alkoholu. Polega na
zaniku miąższu trzustki z zastąpieniem go przez tkankę włóknistą. Czynnikami sprzyjającymi są
hiperlipidemia i hiperkalcemia, niedożywienie, choroby dróg żółciowych nie odgrywają tak istotnej roli
jak w przypadku ostrego zapalenia trzustki.
a) patogeneza:
- rola alkoholu: alkohol obniża poziom litostatyn zapobiegających wytrącaniu się złogów białkowych, co
przy osłabionej zdolności do rozcieńczenia soku t. prowadzi do wytrącania się białkowych czopów, to z
kolei sprzyja procesom atrofii trzustki
- progresja ostrego zapalenia trzustki
- mutacja genu CFTR (nie zawsze występuje) powoduje spadek ph, dwuwęglanów, ilości soki
trzustkowego, wzrasta lepkość, gęstość płynu, wytrącają się złogi białkowe, dochodzi do zatykania
przewodów i atrofii trzustki zwykle nie występuje mukowiscydoza, lecz jeśli wystąpi choroba ma cięższy
przebieg (gen CFTR to gen, którego mutacja jest odpowiedzialna za mukowiscydozę, w tym wypadku
zazwyczaj nie ma zmian typowych dla tej choroby np. pot nie zmienia składu
b) Morfologia:
Miąższ trzustki staje się gęsty i włóknisty, gruczoły zewnątrzwydzielnicze zanikają, wyspy zwykle są
oszczędzone. Obecne są nacieki zapalne, trzustka może stać twarda przewody poszerzone, mogą być
obecne zwapniałe zrosty, a także pseudotorbiele.
c) Objawy:
- ból brzucha łagodny nawracający, stały albo średnio nasilony w postaci ataków
- niewielki wzrost stężenia amylaza i lipazy w osoczu
- w późnej fazie cukrzyca
- żółtaczka, niestrawność
3. Nowotwory komórek wysp.
Są rzadsze w porównaniu z nowotworami części zewnątrzwydzielniczej. Wyglądem przypominają
rakowiaki przewodu pokarmowego i podobnie jak one wywodzą się z komórek neuroendokrynnych.
Mają tendencję do wydzielania hormonów.
a) Insulinoma
Guz wydziela insuliną, może wywoływać hipoglikemię: nerwowość, dezorientację, otępienie. Zwykle
guz występuje w trzustce, (choć niekoniecznie), jest mały, otorebkowany, przypominają duże wyspy
trzustkowe z zachowanymi pasmami i normalnym ułożeniem komórek. Komórki B zawierają ziarnistości
z insuliną. Są to najczęściej guzy łagodne, choć zdarzają się też raki.
U noworodków matek chorych na cukrzycę może dojść do hiperplazji komórek wysp.
62

b) Zespół Zollingera-Ellisona = gastronoma
Zbudowane z komórek wydzielających gastrynę. Umiejscowione w trzustce, tkance okołotrzustkowej
lub w ścianie dwunastnicy. Histologicznie wyglądają na łagodne, ale szybko dają przerzuty ( dziwne to
jest, ale tak jest napisane w książce). Nadmierna produkcja gastryny wywołuje wydzielanie dużych
ilości kwasu żołądkowego, co z kolei przyczynia się do powstania wrzodów żołądka i dwunastnicy, które
mogą być mnogie. Czasem wrzody występują w nietypowej lokalizacji w jelicie cienki, wówczas należy
podejrzewać gastrinomę, a także wtedy gdy wrzody są oporne na tradycyjne sposoby leczenia.
4. Rak trzustki z części zewnątrzwydzielniczej.
Występuje najczęściej pomiędzy 60 a 80 rokiem życia, do jego wystąpienia bardzo mocno przyczynia
się palenie papierosów oraz wrodzone zapalenie trzustki. Nowotwór jest bardzo złośliwy, rokowanie
niekorzystne. Wiąże się z mutacjami w genach KRAS CDKN2A.
Morfologia:
Zwykle jest to gruczolakorak wywodzący się z nabłonka przewodów gruczołowych. Występuje w postaci
niezróżnicowanej lub też tworzy małe, nieregularne, anaplastyczne gruczoły. 60 – 70 % przypadków
wywodzi się z głowy trzustki i wówczas zatyka brodawkę Vatera blokując odpływ żółci, co wywołuje
żółtaczkę mechaniczną i rozdęcie dróg żółciowych. 10 % dotyczy trzonu, a 10-15% ogona trzustki. W
tych przypadkach nowotwór mimo wysokiego stopnia złośliwości nie zajmuje przewodów żółciowych i
pozostaje klinicznie niemy przez długi okres czasu. Rak nacieka sąsiadujące struktury: nerwy,
przestrzeń zaotrzewnową, nadnercza, kręgosłup, poprzecznicę i żołądek. Daje przerzuty do okolicznych
węzłów chłonnych i do wątroby kości oraz płuc.
Objawy:
Ból często jest pierwszym objawem, występuje jednak w późnym stadium rozwoju nowotworu,
świadczy o naciekaniu nerwów. Żółtaczka mechaniczna występuje, jeśli rak dotyczy głowy trzustki.
Podwyższony poziom CEA w surowicy i wielu innych enzymów oraz wędrujące zakrzepowe zapalenie
żył, czyli zespół Trousseau.
CUKRZYCA
Kryteria rozpoznania cukrzycy:
- glikemia na czczo powyżej 126 mg/dl
- losowo powyżej 200
- w doustnym teście obciążenia glukozą (75g) powyżej 200 przez dwie godziny
Pomiary dotyczą krwi żylnej (Robins)
Cukrzyca 1 – są dwa typy 1A autoimmunologiczna i 1B idiopatyczna.
Typ 1 występuje zwykle u dzieci, nie kojarzy się z otyłością, może być powikłany kwasicą ketonową,
wiąże się z obniżeniem stężenia insuliny we krwi.
W patogenezie cukrzycy 1A biorą udział czynniki Genetyczne jest związana z genami układu HLA,
niektóre z nich chronią przed cukrzycą inne sprzyjają jej wystąpieniu, z czynnikami środowiskowymi,
które mogą spełniać rolę wyzwalaczy np. wirus Coxsackie, wirusy świnki, odry, różyczki, mononukleoza
zakaźna (albo poprzez molekularną mimikrę odpowiedź immunologiczna przeciwko białku wirusa, które
jest podobne do białka GAD występującego w trzustce albo przez aktywację limfocytów T), oraz
autoimmunizacja, w której podstawową rolę spełniają limfocyty cytotoksyczne TCD8+, niszczące
komórki B wysp trzustkowych (indukcja apoptozy, uwalnianie ziarnistości cytotoksycznych), oraz
wtórnie powstające przeciwciała przeciw GAD, insulinie i innym białkom cytoplazmatycznym.
Innymi słowy u osób, które posiadają predyspozycje genetyczne, po zadziałaniu czynników
środowiskowych, rozpoczyna się proces autoimmunologicznego niszczenia komórek B w trzustce.
Obserwujemy spadek poziomu insuliny – jest to cukrzyca insulinozależna.
Cukrzyca 2= insulinoniezależna.
63

Pacjenci zwykle są dorośli, otyli, poziom insuliny zmniejszony lub prawidłowy, nie ma przeciwciał
przeciw insulinie i innym białkom trzustki.
U podstaw choroby leżą dwa zaburzenia: zaburzenia wydzielania insuliny oraz isnulinoporność.
Defekt wydzielania insuliny.
Początkowo poziom insuliny może utrzymywać się na prawidłowym poziomie, ale następuje utrata
pulsacyjnej struktury wydzielania insuliny – stępieniu ulega wydzielanie insuliny inicjowane
podwyższeniem GLC we krwi, może też nastąpić przejściowy wzrost stężenia insuliny we krwi jako
kompensacja insulinooporności. W dalszej fazie choroby poziom insuliny we krwi ulega zmniejszeniu z
trzech powodów:
- spadek liczby komórek B o 20 -50% nie jest to jednak najważniejsza przyczyna
_ defekt w rozpoznawaniu glukozy przez komórki B (wysoki poziom białka UCP
_ hiperinsulinomia powoduje wzrost odkładania amyliny, która otacza komórki B zamykając dostęp
glukozie (głównie glc powoduje wydzielanie insuliny), oraz niszczy komórki B
Insulinooporność
może występować niezależnie od cukrzycy w ciąży i otyłości (maskują subkliniczną postać cukrzycy).
Nadmiernie obfita tkanka tłuszczowa występująca u pacjentów z cukrzycą wydziela większej ilości
wolnych kwasów tłuszczowych, rezystywny i TNF, oraz mniejsze ilości leptyny, to wywołuje
insulinoporność między innymi w tkance tłuszczowej, mięśniach szkieletowych i wątrobie. Insulina jest
hormonem zwiększającym zużycie glukozy, zmniejszającym glukoneogenezę i zwiększającym
glikogenogenezę, skoro insulina nie działa podwyższa się poziom glukozy we krwi i mamy
hiperglikemię.
Patogeneza powikłań cukrzycy.
Nieenzymatyczna glikacja = łączenie glukozy z grupami aminowymi białek, produkty glikacji np.
kolagenu wchodzą w serię przemian i tworzą tzw. nieodwracalne produkty glikacji AGE, które:
- przyczyniają się do wychwytu z nieglikozylowanych białek osocza oraz białek zrębowych, LDL, co
powoduje wzrost cholesterolu w błonie wewnętrznej tętnic – przyspiesza miażdżycę
- wiążą się z receptorami komórek: śródbłonka, monocytów, makrofagów, limfocytów, komórek
mezangialnych, co powoduje migrację monocytów, uwalnianie cytokin i czynników wzrostu, wzrost
przepuszczalności śródbłonka, proliferację fibroblastów syntezę macierzy pozakomórkowej
Wewnątrzkomórkowa hiperglikemia zaburza szlak polyolu.
Dzieje się tak w komórkach nie wymagających insuliny do transportu glukozy, w tych (nerwy soczewka
naczynia krwionośne Ś). Glukoza jest w tych komórkach metabolizowana do sorbitolu i fruktozy, które
się akumulują, wzrasta osmolarność, napływa woda, komórka obrzęka. Sorbitol uszkadza również
pompę jonową, serycyty, komórki Schwanna.
Te dwa procesy wywołują najważniejsze powikłania cukrzycy: przyspieszenie miażdżycy â•
pierwsza
przyczyna zgonów u cukrzyków to zawał serca, mikroangiopatię, retinopatię, neuropatię, nefropatię.
MORFOLOGIA
Trustka:
Zmniejszenie liczby i wielkości wysp – głównie typ1
Naciek wysp przez leukocyty głównie 1A
Utrata ziarnistości komórek B – spadek insuliny w uszkodzonych komórkach gł. 1A
Zastępowanie wysp amyloidem w cukrzycy 2 – złogi różowej bezpostaciowej substancji wokół kapilar i
pomiędzy komórkami
U niecukrzycowych noworodków, których matki cierpią na cukrzycę obserwujemy wzrost liczby i
wielkości wysp.
Układ naczyniowy
Wcześniejszy początek i szybszy przebieg miażdżycy szczególnie tętnic wieńcowych i kończyn dolnych
(zgorzel)
Mikroangiopatia cukrzycowa
= zastąpienie błony podstawnej naczyń krwionośnych koncentrycznymi warstwami szklistego materiału
gł kolagenu IV = pogrubienia błon podstawnych, zmiana ta powoduje wzrost przepuszczalności naczyń
krwionośnych dla naczyń krwionośnych i jest podstawą rozwoju nefropatii i niektórych form neuropatii.
Nefropatia cukrzycowa. Obejmuje: zmiany kłębuszkowe, zmiany w naczyniach nerkowych – gł.
miażdżycę tętniczek i odmiedniczkowe zapalenie nerek z martwicą brodawek nerkowych.
64

Zmiany w nerkach:
- rozlane stwardnienie kłębuszków nerkowych â↑
rozlane zwiększenie macierzy mezangialnej z
proliferacją komórek mezangium + pogrubienie błony podstawnej. Prowadzi do białkomoczu ( nasilona
postać)
- Guzkowe stwardnienie k.n. - tworzenie kulistych warstw depozytów macierzy wewnątrz
mezangialnego rdzenia, guzki tworzą się na obwodzie kłębuszka,. Spychają pętle włośniczek na obwód
– tworzy się halo wokół guzka takie zmiany to zespół Kimmelstiel-Wilson. Złogi zawierają lipidy,
mukopolisacharydy, kolagen i są PAS +.
- niedokrwienie kanalików i z włóknienie śródmiąższowe (zajęcie naczyń zaopatrujących kanaliki)
- szkliste stwardnienie tętniczek do i odprowadzających
- Odmiedniczkowe Z.N. + martwica brodawek nerkowych
Retinopatia
Objawy cukrzycy.
-poliuria
-polidypsja
-utrata masy ciała
-polifagia
-hiperglikemia
-w przypadku 1 może dojść do śpiączki ketonowej, w przypadku dwójki do śpiączki hiperosmolalnej
wynikającej z odwodnienia
65

NARZĄDY PŁCIOWE MĘSKIE
1. PRĄCIE
A) Wady rozwojowe:
Spodziectwo (hypospadias)
o Częściej niż wierzchniactwo
o U 1/300 żywych noworodków płci męskiej
o Nieprawidłowe położenie ujścia cewki moczowej na brzusznej powierzchni prącia
o Ujście cewki często ulega zwężeniu lub zaciśnięciu, to daje niedrożność i wzrost ryzyka
zakażeń ukł. moczowego
o Czasem związane z innymi wadami rozwojowymi układu płciowego (przepuklina
pachwinowa, wnętrostwo)
Wierzchniactwo (epispadias)
o Nieprawidłowe położenie ujścia cewki na powierzchni grzbietowej prącia
o Często jest przyczyną zastoju moczu, czasem może też powodować nietrzymanie moczu
B) Zapalenia
Balanitis (lub balanoposthitis)
o Zapalenie charakterze miejscowym, dotyczy żołędzi prącia lub odpowiednio żołędzi i
napletka
o Gł. u mężczyzn nieobrzezanych, gdzie zła higiena prowadzi do gromadzenia się mastki pod
napletkiem
/mastka= drażniąca mieszanina złuszczonych kom. nabłonkowych, wydzieliny gruczołów
potowych oraz szczątków tkankowych/
o Zaczerwienienie, obrzęk i tkliwość dystalnej części prącia, czasem z obecnością treści
ropnej
Stulejka (phimosis)
66

o Jest to niemożność odprowadzenia napletka z żołędzi prącia
o Może być wrodzona, ale częściej jest następstwem bliznowacenia, wtórnego do
miejscowego zapalenia
o W większości przypadków towarzyszą tu cechy zapalenia dystalnej części prącia
o Siłowe odprowadzenie zwężonego napletka może dać upośledzenie krążenia krwi w obrębie
żołędzi prącia → przekrwienie, obrzęk i bolesność dystalnej części prącia (zadzierzgnięcie
napletka =paraphimosis)
o W zaawansowanych przypadkach zatrzymanie odpływu moczu
Infekcje grzybicze (np. kandydoza)
o Mogą objąć skórę prącia i moszny
o Sprzyja im wysoka temp, wilgotność i zaniedbania higieniczne
o Może być u osób zdrowych, ale najczęściej u chorych na cukrzycę
o Wrzodziejące zmiany + świąd + bolesność żołędzi, moszny
o Bioptaty zawierają pączkujące postaci drożdżaka i pseudostrzępki widoczne w
powierzchownych warstwach naskórka
C) Nowotwory
Rak płaskonabłonkowy (carcinoma planoepitheliale penis)
o Jest to większość nowotworów prącia
o 0,25% nowotworów złośliwych
o Większość >40r.ż. u nieobrzezanych
o Przyczyny: brak higieny (mastka), zakażenia HPV (16,18)
/smoła węglowa, sadza – rozwój raka skóry moszny, ale raczej nie prącia/
o Wygląd: szara, grudkowata zmiana, pokryta strupem, położona na żołędzi lub w obrębie
napletka
o Gdy nacieka leżącą głębiej tkankę łączną, powstaje twardy wrzodziejący naciek o nieostrych
granicach
o Istnieją formy brodawkowate = rak brodawkujący (c.verrucosum) – ma mniejszy stopień
cytologicznej atypii oraz rozprężający front naciekania w głębi tkanek
o Mikro: obraz inwazyjnego raka płaskonabł., o rozproszonym charakterze naciekania
o Większość wykazuje miejscową złośliwość
o U 25% przy rozpoznaniu są przerzuty do węzłów pachwinowych
o Dosyć rzadko powstają przerzuty odległe
o Średnie przeżycie 5-letnie = 70%
o
Poprzedzony rakiem in situ
67

Choroba Bowena
Pojedyncza, płaska zmiana na trzonie prącia
Komórki z morfologicznymi cechami złośliwości ograniczone do naskórka, nie
naciekają podścieliska
Może występować też na innych śluzówkach (jama ustna, pochwa)
W 10% progresji do raka naciekającego + wzrost ryzyka nowotworów złośliwych
innych narządów
Erytroplazja Queyrata = Rumieniowata plama w obrębie żołędzi i innych śluzówek
Bowenoid papulosis = zmiana przenoszona drogą płciową o etiologii wirusowej,
zlokalizowana na trzonie prącia
2.MOSZNA, JĄDRO, NAJĄDRZE
Skóra moszny: procesy zapalne, infekcje grzybicze, układowe choroby skóry
Nowotwory worka mosznowego są rzadkie, gł. rak płaskonabłonkowy (kiedyś często u
kominiarzy)
Wodniak (hydrocele):
•
najczęstsza przyczyna powiększenia moszny
•
jest to zbiornik surowiczego płynu w obrębie osłonki pochwowej
•
idiopatyczny lub odczynowy w reakcji na zakażenie lub nowotwory w sąsiednich
tkankach
inne przyczyny powiększenia jądra: krwiak śródosłonkowy (hematocele), zbiornik chłonki
(chylocele), słoniowacizna (elephantiasis) = karykaturalne powiększenie kończyny dolnej i
moszny wywołane przez poważne zaburzenia odpływu chłonki, np. w filariozach
choroby jąder: wrodzone, zapalne lub nowotworowe; mogą być przyczyną niepłodności, zaniku
lub powiększenia jąder, oraz źródłem bólu
A) Wnętrostwo i zanik jądra
o Wnętrostwo = niezstąpienie jądra do worka mosznowego (prawidłowo jądra zstępują do miednicy
z pierwotnej jamy ciała pod koniec 3.miesiąca życia płodowego, a w ostatnich 2 miesiącach
przedostają się do moszny przez kanał pachwinowy; jednak nie u wszystkich noworodków ten
proces jest całkiem dokonany już zaraz po urodzeniu)
o Przyczyny złego zstępowania: zaburzenia hormonalne, zaburzenia w budowie i/lub
funkcjonowaniu jądra, problemy mechaniczne (np.. niedrożność kanału), może też być częścią
zespołów wad wrodzonych (np. zespół Pradera-Williego), najczęściej jednak przyczyna nieznana
o Występuje u 0,7 – 0,8% populacji mężczyzn
o Trochę częściej w prawym jądrze, w 25% obustronnie
o We wczesnym okresie życia takie jądro może mieć prawidłową wielkość, ale potem dokonują się
zmiany zanikowe
W wieku 5-6 lat są mikroskopowe cechy zaniku kanalików nasieniotwórczych
68

U osób dojrzałych płciowo – szkliwienie
Gdy ↓ liczba kanalików nasieniotwórczych, często jest też rozrost komórek
śródmiąższowych (Leydiga)
W obrębie niezstąpionych jąder często są ogniska wewnątrzkanalikowego nowotworu
zarodkowego jądra (IGCN), które są stanem przednowotworowym
o Zmiany zanikowe w zstąpionych jądrach powstają pod wpływem: przewlekłego niedokrwienia,
urazów, promieniowania lub chemioterapii, także w przebiegu procesów związanych z ↑
aktywności estrogenów w surowicy (np. marskość wątroby)
o Obustronne wnętrostwo daje bezpłodność
o Wnętrostwu 1-stronnemu czasem towarzysza zmiany zanikowe w drugim jądrze, co też daje
bezpłodność
o Wnętrostwo daje 4-krotny ↑ ryzyka rozwoju nowotworu złośliwego jądra, a przy jednostronnym też
↑ ryzyka nowotworu w drugim (prawidłowym) jądrze
o Orchiopexia = zabieg chirurgicznego sprowadzenia jądra do worka mosznowego, jeśli wykonany
przed okresem dojrzewania, może zapobiec zanikowi jądra, ale nie gwarantuje uniknięcia
bezpłodności, nie wiadomo też, czy zabieg ten ma wpływ na ↓ ryzyka nowotworu
B) Zapalenia
o Procesy zapalne częściej dotyczą najądrza niż jądra
o
Nieswoiste zapalenie jądra i najądrza (orchitis et epididymitis non-specifica):
Zazwyczaj charakter wtórny do zakażenia ukł. moczowego i rozwija się jako zakażenie
wstępujące drogą nasieniowodu lub naczyń chłonnych powrózka nasiennego
Jądro jest obrzęknięte i tkliwe
Obecne nacieki zapalne z neutrofilów
o Zajęcie jądra przy ostrym zapaleniu przyusznic (świnka)
Jest powikłaniem infekcji wirusem nagminnego zapalenia ślinianek
Rozwija się u 20% dorosłych mężczyzn, rzadko występuje u dzieci
Jądro obrzęknięte i przekrwione
Nacieki zapalne z limfocytów i plazmocytów
Gdy ciężki przebieg → zniszczenie nabłonka nasieniotwórczego → zanik cewek i
włóknienie → niepłodność
o Ziarniniakowe zapalenie jądra (gł. gruźlica)
Gruźlica jądra rozwija się zazwyczaj w najądrzu, z wtórnym zajęciem jądra
Zapalenie ziarniniakowe z martwicą serowatą
C) Nowotwory jąder
o Są najczęstszą przyczyną bezbolesnego, twardego powiększenia jąder
o U 2/100000 mężczyzn
69

o Szczyt zachorowalności 15-34r.ż.
o 95% z nich rozwija się z komórek zarodkowych
o Prawie wszystkie są złośliwe (łagodne są guzy z kom. Leydiga i Sertoliego, ale one są rzadkie)
o Często są przyczyną zaburzeń hormonalnych, bo mają zdolność produkcji hormonów
steroidowych
o Ryzyko: wnętrostwo, dysgenezja jąder (zesp. feminizujących jader, zesp. Kinefeltera), u bliźniąt
chorych, obecność nowotworu w 1 jądrze, częściej rasa czarna
o Często powstanie izochromosomu krótkiego ramienia chromosomu 12
o Nowotwory nienasieniakowe dają wcześniej przerzuty (zarówno drogą krwionośną – głównie
wątroba i płuca, i chłonną)
o Ogniska przerzutowe mogą być o takim samym utkaniu jak pierwotne lub zawierają elementy
innego nowotworu zarodkowego
o Stopień zaawansowania klinicznego:
Stopień I – nowotwór ograniczony do jądra
Stopień II – przerzuty ograniczone do węzłów zaotrzewnowych poniżej przepony
Stopień III – przerzuty zlokalizowane w innych węzłach niż zaotrzewnowe
NASIENIAK (seminoma)
a) Klasyczny
50% wszystkich nowotworów zarodkowych jądra
Histologicznie: jak dysgerminoma jajnika i germinoma w OUN
Dobrze odgraniczone, duże guzy z miękkiej homogennej, szaro-kremowej tkanki,
uwypuklają się ponad powierzchnię przekroju zajętego jądra
Zwykle nie przekracza osłonki białawej otaczającej jądro
Mogą być ogniska martwicy skrzepowej, ale brak wylewów krwi
Duże, dobrze odgraniczone komórki o jasnej cytoplazmie, z dużą ilością glikogenu, jądra
z wyraźnymi jąderkami
Często komórki układają się w gniazda oddzielone od siebie przegrodami
łącznotkankowymi
Często też obecny naciek zapalny z limfocytów lub odczyn zapalny o charakterze
ziarniniakowym
W 7-24% obecne kom. olbrzymie (podobne do kom. syncytiotrofoblastu) – dają wynik
dodatni na HCG
U 10% podwyższony poziom AFP
Przerzuty najczęściej w węzłach biodrowych i przyaortalnych w górnych okolicach lędźwi
Przerzuty drogą krwionośną są bardzo rzadkie
Są promienioczułe oraz wrażliwe na chemioterapię (opartej na platynie)
70

b) Spermatocytowy
Rzadszy niż klasyczny i u starszych pacjentów
Mieszana populacja dużych komórek 1- lub wielojądrowych, komórek średnich i małych
kom. podobnych do spermatocytów II rzędu
Bardzo rzadko daje przerzuty
RAK ZARODKOWY (carcinoma embryonale)
o Wiek: 20-30r.ż
o naciekający nowotwór, słabo odgraniczony od otaczających tkanek
o często ma ogniska martwicy i wylewów krwi
o guz pierwotny może być niewielki, nawet gdy są już przerzuty
o większe guzy naciekają najądrze i powrózek nasienny
o komórki nowotworowe wyglądają prymitywnie: są duże, z zasadochłonną cytoplazmą, mają
niewyraźne granice i duże jądra z wyraźnymi jąderkami
o kom. nowotworowe leżą w litych polach, niekiedy zawierających struktury gruczołowe lub
brodawkowate, często są elementy woreczka żółtkowego i komórki choriocarcinoma
o w większości występują z domieszką (np. yolk sac tumor, teratoma, choriocarcinoma)
o postaci czyste stanowią 1-3%
o w świetle kanalików nasieniotwórczych na obrzeżu guza obecność wewnątrzkanalikowego
nowotworu zarodkowego (intratubular germ cell neoplasia)
o w 90% podwyższony poziom HCG i/lub AFP
GUZ ZATOKI ENDODERMALNEJ
o najczęstszy pierwotny nowotwór jądra u chłopców <3r.ż.
o u dorosłych towarzyszą najczęściej rakowi zarodkowemu
o komórki pluripotencjalne różnicujące się w kierunku zatoki endodermalnej
o zwykle duży, dobrze odgraniczony guz
o komórki sześcienne lub walcowate (przypominające komórki śródbłonka), tworzące struktury
brodawkowate i gruczołowe oraz układy ławicowe i drobnotorbielowate, często zawierające
kwasochłonne ciałka szkliste
o obecność ciałek Schillera-Duvala (struktury przypominające prymitywne kłębuszki)
o w 100% obecność AFP
NABŁONIAK KOSMÓWKOWY ZŁOŚLIWY (choriocarcinoma)
o wiek 20-30r.ż
o z pluripotencjalnych komórek różnicujących się w kierunku trofoblastu
o guz pierwotny mały (nawet jak są przerzuty) i niewyczuwalny palpacyjnie
71

o ławice prostokątnych komórek (kom. cytotrofoblastu) wymieszanych z olbrzymimi,
wielojądrowymi kwasochłonnymi kom.(kom.syncytiotrofoblasu), które często tworzą „czapeczki”
pokrywające kom. drobne
o nie tworzy w pełni wykształconych kosmków łożyskowych
o w 100% podwyższony poziom HCG (gł. przez kom. syncytiotrofoblastu)
POTWORNIAK (teratoma)
o jest to nowotworowy rozrost kom. różnicujących się w kierunku kom. somatycznych
o guzy o dużej spoistości, z widocznymi na przekroju torbielami i obszarami wyglądem
przypominającymi chrząstkę
a)potworniaki dojrzałe
mają dojrzałe tkanki pochodzące z 1 lub różnych listków zarodkowych (np. tkanka
nerwowa, chrząstka, tk. tłuszczowa, kość, nabłonki)
elementy tkankowe ułożone w sposób bezładny
b)potworniaki niedojrzałe
mają niedojrzałe elementy somatyczne przypominające tkanki płodowe
c)potworniaki z wtórną transformacją nowotworową
na bazie elementów potworniaka rozwija się nowotwór złośliwy (zwykle rak
płaskonabłonkowy lub gruczolakorak)
o potworniaki złośliwe występują zazwyczaj u dorosłych
o czyste potworniaki występujące u chłopców przed okresem dojrzałości są zwykle łagodne
o potworniaki u dorosłych mężczyzn występują zazwyczaj jako domieszka innego nowotworu, więc
niemal wszystkie nowotwory u dorosłych są złośliwe
o w 50% podwyższony poziom HCG i/lub AFP
MIESZANE GUZY ZARODKOWE
o stanowią 60% nowotworów jąder
o może być dowolna kombinacja utkania, ale najczęściej: teratoma, yolk sac tumor, carcinoma
embryonale
o w 90% podwyższony poziom HCG i/lub AFP
3.GRUCZOŁ KROKOWY
ZAPALENIE GRUCZOŁU KROKOWEGO (prostatitis)
o objawy: trudności w oddawaniu moczu, częstomocz, dolegliwości bólowe okolicy lędźwiowo-
krzyżowej oraz rozlany ból w miednicy lub okolicy nadłonowej
o powiększenie i tkliwość gruczołu (gł. w zapaleniu ostrym), często też objawom miejscowym
towarzyszy gorączka i leukocytoza
o przewlekłe zapalenie może być rezerwuarem dla mikroorganizmów dających zakażenia układu
moczowego (najważniejsza przyczyna nawrotowych zakażeń układu moczowego u mężczyzn)
•
Ostre
najczęściej wywołane przez E.coli i inne pałeczki Gram – ujemne
72

zwykle towarzyszy mu zakażenie cewki moczowej i pęcherza moczowego (urethrocystitis
acuta)
bakterie dostają się przez ciągłość (z cewki lub pęcherza) lub drogą naczyń z bardziej
oddalonych lokalizacji
nacieki z neutrofilów (we wczesnych fazach szczególnie liczne w świetle cew gruczołowych),
przekrwienie oraz obrzęk podścieliska gruczołu
dochodzi do niszczenia gruczołu oraz rozprzestrzeniania się zapalenia w obrębie stercza, z
tworzeniem mikroropni w jego obrębie
rzadko występują makroskopowo widoczne ropnie (współistniejąca cukrzyca) – rozlegle
niszczą tkankę stercza
•
przewlekłe
może być następstwem epizodu zapalenia ostrego lub rozwijać się samoistnie
czasem udaje się wyizolować bakterie występujące w zapaleniu ostrym (
przewlekłe bakteryjne
zapalenie gruczołu krokowego)
w innych przypadkach brak bakterii, ale jest podwyższona ilość leukocytów w wydzielinie
stercza (
zapalenie niebakteryjne) – większość przypadków
potencjalne niebakteryjne czynniki wywołujące nierzeżączkowe zapalenie cewki bierze się pod
uwagę: Chlamydia trachomatis, Ureaplasma urealuticum
obraz mikroskopowy przeważnie niecharakterystyczny
nacieki zapalne o różnym nasileniu (z komórek limfoidalnych), cechy uszkodzenia elementów
gruczołowych stercza, często jednoczesne zmiany o charakterze zapalenia ostrego
warunek rozpoznania: niszczenie tkanek stercza, rozplem fibroblastów, obecność innych
komórek zapalnych (najczęściej neutrofilów)
liczba izolowanych ognisk kom. limfoidalnych w sterczu wzrasta z wiekiem, więc nie jest to
podstawa do rozpoznania zapalenia
•
ziarniniakowi
jest szczególną postacią zapalenia przewlekłego
rodzaj reakcji odczynowej na działanie czynników uszkadzających
stwierdza się u pacjentów z układowymi chorobami zapalnymi przebiegającymi z odczynem
ziarniniakowym (rozsiana gruźlica, sarkoidoza, zakażenia grzybicze, ziarniniak Wegenera)
może też być jako nieswoista reakcja na zagęszczoną wydzielinę stercza oraz po przez
cewkowych zabiegach usunięcia części stercza
wielojądrowe komórki olbrzymie, kom. piankowate (histiocyty) oraz niekiedy granulocyty
kwasochłonne
martwica serowata występuje tylko w przypadku gruźlicy
ROZROST GUZKOWY GRUCZOŁU KROKOWEGO (hyperplasia nodularis prostatae)
73

o dotyczy głównie wewnętrznej części strefy pośredniej oraz strefy centralnej, natomiast
większość gruczolakoraków (70-80%) powstaje w strefie obwodowej
o polega na rozroście elementów gruczołowych i/lub zrębowych (proliferacja elementów
nabłonkowych i zrębu z następowym powiększeniem gruczołu oraz z czasowym utrudnieniem
odpływu moczu)
o częstość występowania rośnie z wiekiem (od 40 r.ż., pod koniec 80 r.ż. osiąga 90%)
o przyczyna nie jest do końca znana, ale możliwe, że znaczenie ma synergistyczne działanie
androgenów i estrogenów
o niezbędnym warunkiem rozwoju tego rozrostu jest obecność prawidłowo funkcjonującego jądra,
nie wystąpi nigdy u osoby wykastrowanej przed osiągnięciem dojrzałości płciowej
o największy wpływ mają: dihydrotestosteron (DHT – powstaje z testosteronu pod wpływem 5α-
reduktazy) i jego metabolit 3α-androstendiol
DHT wiąże się z receptorami w jądrach komórek stercza, pobudzając syntezę RNA, DNA,
czynników wzrostu i innych białek cytoplazmatycznych
W leczeniu objawów rozrostu stosuje się inhibitory 5α-reduktazy
Jednak objawy stwierdza się najczęściej u starszych osób, które mają już niski poziom
testosteronu, a podanie egzogennego testosteronu nie daje nasilenia objawów
Dlatego sądzi się, że rozwojowi rozrostu sprzyja wzrost stężenia estrogenów (związany z
wiekiem), które powodują zwiększenie ilości receptorów dla DHT na komórkach stercza,
wzmacniając przez to działanie DHT
o Najczęściej strefa około cewkowa, szczególnie powyżej poziomu wzgórka nasiennego
o Masa stercza może osiągnąć nawet 300g
o Makroskopowo: dosyć dobrze odgraniczone guzki uwypuklające się ponad powierzchnię
przekroju
o Zmiany guzowate mogą zajmować różne regiony, ale głównie strefa pośrednia i centralna
o Guzki są lite lub mają torbielowate przestrzenie (poszerzone struktury gruczołowe)
o Rozrost daje zwężenie lub niemal całkowite zamknięcie cewki moczowej, czasem guzki rosnące
bezpośrednio pod śluzówką proksymalnej cewki – uwypuklają się do światła pęcherza
moczowego, powodując utrudnienie przepływu cewkowego (zawór kulkowy)
o Mikroskopowo: wymieszane w różnych proporcjach proliferujące struktury gruczołowe oraz
włóknisto-mięśniowe podścielisko
Gruczoły wyścielone wysokim nabłonkiem walcowatokomórkowym oraz leżącą
obwodowo warstwą spłaszczonych komórek podstawnych
Stłoczone proliferujące kom. nabłonkowe powodują powstawanie struktur
brodawkowatych uwypuklających się do światła gruczołów
W świetle gruczołów często jest zagęszczona wydzielina białkowa tworząca ciała
piaszczakowate (corpora amylacea)
74

Zawsze musi być proliferujące podścielisko (choćby skąpe)
Niektóre guzki składają się tylko z wrzecionowatych komórek zrębu oraz tkanki łącznej
W zaawansowanych przypadkach: ogniska zawałów z ogniskową metaplazją
płaskonabłonkową w sąsiadujących strukturach gruczołowych
o Objawy występują u 10% osób z rozrostem
Trudności z inicjacją wypływu moczu
Przerywany strumień moczu podczas mikcji
Czasem bolesne rozdęcie pęcherza moczowego lub nawet wodonercze (gdy całkowite
zamknięcie dróg moczowych)
Objawy podrażnienia pęcherza moczowego: uczucie parcia na pęcherz, częstomocz,
nykturia
Częstszy rozwój zakażeń dróg moczowych
RAK GRUCZOŁU KROKOWEGO
o najczęstszy narządowy nowotwór złośliwy u mężczyzn
o druga (po raku płuca) nowotworowa przyczyna zgonu u mężczyzn >50 r.ż.
o szczyt zachorowań 65-75 r.ż.
o bardzo częste są raki nieme klinicznie (u ponad połowy mężczyzn >80 r.ż.)
o przyczyna nie jest znana, ale wpływ mają czynniki hormonalne, genetyczne i środowiskowe
nie występuje u mężczyzn poddanych kastracji przed osiągnięciem dojrzałości (rola
androgenów w rozwoju raka – dlatego też orchiektomia lub podanie egzogennych
estrogenów, np. dietylstilbestrolu)
zwiększone ryzyko występuje u krewnych pacjentów z tym nowotworem
u osób rasy czarnej objawowy rak pojawia się częściej i jest stwierdzany w młodszym
wieku niż u innych ras (ale rasa ma najprawdopodobniej tylko wpływ na postęp
rozwiniętego raka), różnice rasowe w ilości powtórzeń sekwencji CAG w genie receptora
androgenowego
znaczenie mogą mieć loci na ch. 1 oraz 10 (tam jest gen PTEN)
szczególnie częsty w Skandynawii, rzadki w Japonii i niektórych państwach azjatyckich
(czynniki środowiskowe – dieta bogata w tłuszcze zwierzęce)
o 70-80% rozwija się w strefie obwodowej, więc rak w postaci nieregularnych, twardych guzków
jest wyczuwalny w badaniu per rectum
75

o We wczesnych stadiach zaawansowania rzadko jest przyczyną zwężenia dolnego odcinka dróg
moczowych (bo lokalizacja obwodowa)
o Wczesne fazy: słabo odgraniczone zmiany guzkowe bezpośrednio pod torebką narządową
o Na przekroju: lite, spoiste szarobiaławe lub szarożółtawe nacieki o nieostrych granicach
o Przerzuty do okolicznych węzłów chłonnych miednicy mogą nastąpić bardzo wcześnie
o Przypadki zaawansowane miejscowo: naciekanie pęcherzyków nasiennych, około cewkowej
strefy stercza, tkanek miękkich otoczenia prostaty oraz ściany pęcherza moczowego (istnieje
naturalna bariera utrudniająca szerzenie się ku tyłowi = powięź Denonvilliersa – płat tkanki
łącznej oddzielający struktury dolnego odc. układu moczowego od ściany odbytnicy, więc
naciekanie odbytnicy bardzo rzadko)
o Mikroskopowo: większość to gruczolakoraki o różnym stopniu dojrzałości
raki bardziej dojrzałe: małe cewki gruczołowe nieregularnie naciekające podścielisko,
cewki nie są otoczone kolagenem i komórkami zrębu, lecz często przylegają do siebie i
wydają się ostro penetrować podścielisko
cewki wyścielone tylko 1 warstwą komórek sześciennych o jądrze z wyraźnym jąderkiem,
brak zewnętrznej warstwy komórek podstawnych (obecnych w gruczołach prawidłowych
i hiperplastycznych)
raki nisko dojrzałe: nieregularne struktury gruczołowe naciekające w sposób rozproszony
struktury sitowate i brodawkowate, skrajne przypadki mają postać ławic i litych nacieków
z komórek nowotworowych
w gruczołach leżących w sąsiedztwie raka często są cechy atypii komórek nabłonka
gruczołowego = węwnątrznabłonkowy rozrost nowotworowy gruczołu krokowego (PIN)
PIN współistnieje często z inwazyjnym rakiem stercza, więc uważa się go za zmianę
przedrakową
PIN podzielono na niskozróżnicowany (low-grade) i wysokozróżnicowany (high-grade)
Określenie stopnia złośliwości histologicznej: stopień zróżnicowania gruczołowego,
architektonika cew gruczołowych, stopień anaplazji komórek i aktywność mitotyczna
Powszechnie używa się skali Gleasona do oceny stopnia złośliwości – wykazuje korelację
ze stopniem zaawansowania oraz z rokowaniem
o często jest bezobjawowy, zwłaszcza gdy jest niezaawansowany
o ok. 20% raków ograniczonych do stercza stwierdzanych jest przypadkowo w materiale
tkankowym usuwanym z powodu objawowego łagodnego rozrostu stercza
o w badaniach sekcyjnych raka stwierdza się u 60% mężczyzn >80 r.ż.
o większość raków wykrywa się w badaniu per rectum (bo obwodowa lokalizacja)
o zaawansowane przypadki: uczucie dyskomfortu oraz zaburzenia odpływu moczu
o badanie przedmiotowe: lity, nieruchomy względem otoczenia naciek
76

o czasem pierwsze objawy związane są z ogniskami przerzutowymi (które są częste): szczególnie
często przerzuty do kości (głównie do szkieletu osiowego) – charakter osteolityczny lub częściej
osteosklerotyczny
o diagnostyka: PSA, badanie per rectum, USG przez odbytnicze, biopsja igłowa
o we wczesnej diagnostyce wykorzystuje się PSA:
fizjologicznie wydzielany w dużych stężeniach do zrazików stercza, skąd trafia do płynu
nasiennego, upłynniając ejakulat i przez to zwiększając ruchliwość plemników
produkowany przez komórki nabłonkowe prawidłowego stercza oraz komórki
nowotworowe
wartość graniczna = 4,0 ng/l
wzrost we krwi : rak gruczołowy, łagodny rozrost guzkowy, zapalenie stercza
w przypadku raka zazwyczaj wyższe poziomy niż w rozroście
szczególnie przydatny do monitorowania pacjentów po leczeniu raka (nawrót lub rozsiew
nowotworu)
o ocena stopnia zaawansowania: istotna w diagnostyce i wyborze metod leczenia
ocena za pomocą badania przedmiotowego, inwazyjnej diagnostyki chirurgicznej, badań
obrazowych oraz na podstawie stopnia histologicznej złośliwości i poziomu markerów
nowotworowych
zaawansowane miejscowo: leczenie operacyjne lub radioterapia
przypadki zaawansowane: głównie hormonoterapia (większość wrażliwa na androgeny,
dlatego stosuje się blokadę wpływu tych hormonów)
w praktyce klinicznej stosuje się: kastrację chirurgiczną lub farmakologiczną, estrogeny
lub leki blokujące receptory androgenowe
rokowanie dobre, gdy ograniczony do stercza, gdy rozsiany rak 10-letnie przeżycie u
10-40% pacjentów
19
20
CHOROBY KOŚCI
WRODZONE I DZIEDZICZNE CHOROBY KOŚCI
- zmiany izolowane lub zespoły; zaburzenia strukturalne, obecność dodatkowych kości, nieprawidłowe
zrastanie
- prowadzące do zmian w całym kośćcu- wpływ na wzrost, utrzymanie prawidłowej macierzy
- choroby metaboliczne wpływające na macierz kostną – zesp. Hurlera
77

ACHONDROPLAZJA
najczęstsza wrodzona osteodyschondrodysplazja, gł. przyczyna karłowatości
upośledzenie dojrzewania chrząstki w rozwijającej się płytce wzrostowej
mutacja autosom. dominująca genów rec.3 dla czynnika wzrostu fibroblastów (FGFR3), krótkie
ramię chrom.4
→
przetrwała aktywacja FGFR3
→
hamowanie prawidłowej proliferacji chrząstki w
pytce wzrostowej
20% występowania rodzinnego
homozygoty (rzadko)
→
niedorozwój klatki piersiowej
→
zgon w przebiegu niewydolności odd.
heterozygoty (najczęstsza choroba nieletalna)
→
znaczne nieproporcjonalne skrócenia części
prox. Kończyn, łukowate wygięcie nóg, uwydatnienie lordoz (łękowatość)
Morfologia – płytki chrzęstne wzrostowe zawierają hipoplastyczne lub bezładnie rozmieszczone
grupy chondrocytów.
SAMOISTNA KRUCHOŚĆ KOŚCI (osteogenesis imperfecta)
nieprawidłowy kolagen typu I (obecny w skórze, stawach, gałce ocznej, podstawowy składnik
osteoidu)
mutacje genów syntezy i wydzielania prokolagenu
α
1 i
α
2 oraz składania kolagenu
autosom. domin.
4 postacie IO
objawy: - mnogie złamania kości
- ciężkie postacie- zgony in utero/ tuż po urodzeniu
- może się ujawnić we wczesnym dzieciństwie (podejrzenie zesp. dziecka
maltretowanego)
- łagodne postacie nie skracają życia
- opóźnione wyrzynanie zębów
- utrata słuchu
- błękitne zabarwienie twardówek oka (
↓
ilości kolagenu)
MARMURKOWATOŚĆ KOŚCI (osteopetrosis)
niedostateczna aktywność osteoklastów
autosom. domin./rec., heterogenne
pogrubiałe, wysoce zmineralizowane i nadmiernie łamliwe kości
z powodu zmniejszenia przestrzeni dla szpiku i hematopoezy: niedokrwistość,
↓
PLT,
↑
podatność
na zakażenia
pogrubiałe kości mogą uciskać pnie nerwowe
→
porażenia nerwów czaszkowych
ZRZESZOTNIENIE KOŚCI (osteoporosis)
↓
masy kości i zaburzenia mikroarchitektury
→
↑
kruchości i podatności na złamania
↓
liczby i grubości blaszek kostnych, która nie łączą się w beleczki
1. miejscowe (po długotrwałym unieruchomieniu)
2. uogólnione:
a) pierwotne: - pomenopauzalne (najczęstsza postać)
- starcze (obie płcie, ciężkość wzrasta z wiekiem,
↓
gęstości kości)
- idiopatyczne
b) wtórne : - zaburzenia endokrynologiczne, nowotwory, związane z przewodem
pokarmowym, uogólnione choroby reumatyczne, związane z lekami i inne.
mechanizm:
Osteoklasty powstają z makrofagów pod wpływem szlaku sygnalizacyjnego RANK/ ligand
RANK. Ligand RANK związany z błoną komórkową osteoblastu lub komórki zrębowej łączy się z
receptorem RANK na powierzchni makrofaga (aktywacja ścieżki transkrypcyjnej w jądrze).
Dołącza się czynnik M-CSF ( stymulujący kolonię makrofagów). Makrofag przekształca się w
osteoklast.
Osteoprotegryna (OPG), wydzielana przez komórki zrębu i osteoblasty może działac jak
receptorowa pułapka dla ligandu RANK, uniemożliwiając łączenie się z rec.RANK na
makrofagach. Przez hamowanie różnicowania osteoklastów zapobiega resorpcji kości.
patogeneza:
nieprawidłowa regulacja współdziałania RANK, ligandu RANK i OPG (np. z
powodu
↓
estrogenów)
→
↑
aktywność osteoblastów i resorpcja kości
>
↓
aktywności osteoblastów i tworzenia kości
78

ryzyko zachorowania – zależy od szczytu masy kostnej u młodych dorosłych (wpływ mają:
aktywność fiz., genetyka, dieta, stężenie hormonów); gęstość kości jest większa o mężczyzn i
rasy czarnej
współczynnik utraty masy kostnej- 0,7% rocznie
największa utrata: kręgi, szyjka kości udowej
po 30 r.ż. każda powstała masa kości nie kompensuje już utraty
Czynniki wpływające na rozwój choroby:
ESTROGENY
a)
ich spadek powoduje
↑
produkcji IL1, IL6, TNF przez monocyty i inne kom. szpiku
→
↑
puli
prekursorów osteoklastów
→
↑
produkcji ligandu RANK i M-CSF
→
↑
resorpcji i
↓
formowania kości
b) stymulują wytwarzanie OPG
TESTOSTERON
w wieku starszym powoduje
↑
produkcji cytokin
→
↑
metabolizmu kości
GENETYCZNE
odmiany genu VDR (rec.wit.D) determinują gęstość kości
MECHANICZNE
Spadek aktywności fiz., unieruchomienie, siedzący tryb życia
DIETA
Przyswajanie Ca i wit.D (znaczenie mniejsze u dorastających dziewcząt)
Morfologia:
utrata masy kostnej największa tam, gdzie najwięcej kości beleczkowej (gąbczastej)
beleczki cienkie, oddalone od siebie, poszerzone kanały Haversa
nieznacznie nasilona akt. osteoklastów
w pozostałych częściach kości prawidłowa zawartość minerałów i macierzy białkowej
Klinicznie:
na początku- bezobjawowe
wczesna diagnostyka: dwufotonowa absorpcjometria rtg; później rtg
złamania: trzonów kręgów, miednicy, uda, (wymagają unieruchomienia u starszych, zgon z
powodu zatorowości płucnej, zapalenia płuc)
Leczenie:
suplementacja estrogenów
↓
tempo utraty kości i Ca, ale nie odwraca istniejących już zmian
przyjmowanie Ca w diecie przed 30 r.ż. zmniejsza ryzyko
suplementacja Ca u starszych nieznacznie
↓
tempo zmian
bifosfoniany
↓
resorpcję kości
SERMs – wybiórcze modulatory rec. estrogenu
Jeśli nie można przyjmować estrogenów – kalcytonina
↓
częstość złamań
KRZYWICA (rahitis) I ROZMIĘKANIE KOŚCI (osteomalacia)
z powodu
↓
wit.D
↓
mineralizacji kości, nadmiar osteoidu
CHOROBY KOŚCI TOWARZYSZĄCE NADCZYNNOŚCI PRZYTARCZYC
Skutki działania PTH:
- stymulacja osteoklastów,
↑
resorpcji kości i
↑
uwalniania Ca (pośrednio, przez
↑
ligandu RANK)
-
↑
resorpcji Ca w cewkach nerkowych
-
↑
syntezy akt. postaci wit.D
→
↑
resorpcji Ca w jelicie i uwalniania Ca z kosci
•
pierwotna nadczynność przytarczyc – PTH jest bezpośrednio odpowiedzialny za zmiany kostne
•
wtórna nadczynność przytarczyc (zwykle choroby nerek)
→
zniszczenie miąższu nerek w przewlekłej niewydolności i hiperfosfatemia hamująca akt.
nerkowej
α
hydroksylazy powodują
→
↓
poziomu wit.D ,
↓
absorpcji Ca w jelitach , kwasicę metaboliczną, odkładanie Al. w kościach
→
to wszystko powoduje nieprawidłową mineralizację osteoidu
Morfologia:
warstwa korowa i beleczkowa zanikają
resorpcja kości najsilniejsza podokostnowo- najlepiej widoczna na rtg II i III palca po stronie
promieniowej, środkowe paliczki
79

erozja powierzchni kości
w jamach szpikowych dużo luźnej tkanki łącznej i naczyń
złogi hemosyderyny
+
osteoklasty
+
reaktywne kom. olbrzymie
=
guz brunatny
zmiany torbielowate
ROPNE ZAPALENIE KOŚCI I SZPIKU (osteomyelitis purulenta)
rozsiew bakterii do kości – drogą krwionośną ( najczęściej)
- bezpośrednio z ostrego ogniska zakażenia w przyległych tkankach
- uraz, złamania otwarte, operacje ortopedyczne
najczęstsze bakterie: S. Aureus, dwoinki zapalenia płuc, paleczki G-, u noworodków paciorkowce
grB i E.Coli, Salmonella u chorych na anemię sierpowatą; w 50% przyczyna pozostaje nieznana
Klinicznie:
obj. ogólne (u dorosłych mogą nie wystąpić): gorączka, osłabienie, leukocytoza
obj. miejscowe ( u dzieci nieznaczne): ból, obrzmienie, zaczerwienienie
rtg- zmiany mogą być niewidoczne przez pierwsze 7 dni
badanie radioizotopowe!
powikłania: złamania patologiczne, bakteriemia, zapalenie wsierdzia
(rzadziej: skrobawica uogólniona, przetoki drenujące skórę lub okoliczne
tkanki, rak płaskonabłonkowy w kanale przewlekłej przetoki, mięsaki)
Morfologia – ostre:
obfity naciek z granulocytów w miejscu wniknięcia bakterii
dzieci- gł. przynasady kości długich (bo tam wolniejszy przepływ krwi)
dorośli- trzony kręgów (bo dobrze unaczynione)
martwica w ciągu kilku dni z powodu
↑
enzymów i ciśnienia w jamie szpikowej oraz z powodu
oddzielenia okostnej
podokostnowe ropnie (gł. u dzieci, bo u nich okostna przywiera słabiej do kory), szerzące się do
stawów i nasad
może szerzyć się na tkanki miękkie i wzdłuż kości długich
Morfologia – przewlekłe:
napływ komórek przewlekłego zapalenia może wywołać reakcje naprawcze: aktywacja
osteoklastów, proliferacja fibroblastów, powstawanie nowej kości
martwiak (sequestrum) – pozostałość martiwczej kości, może być resorbowany przez
osteoklasty
otoczka/ trumienka (involutrum) – większy martwaiak otoczony rąbkiem odcznowym
ropień Brodiego – ropień dobrze odgraniczony przez sklerotycznie zmienioną kość (bakterie
mogą wewnątrz przetrwać wiele lat)
GRUŹLICZE ZAPALENIE KOŚCI I SZPIKU (osteomyelitis tuberculosa)
jako powikłanie 1-3% gruźlicy płuc
szerzenie przez ciągłość lub drogą krwionośną (kości długie i kręgi)
•
choroba Potta – zapalenie kręgów- ich zniekształcenie i zapadanie może powodować
zaburzenia neurologiczne. Szerzy się na otaczające tkanki miękkie, powstaje ropień
zimny (opadowy) w mięśniu biodrowo- lędźwiowym.
zmiany mnogie u osób z niedoborami immunologicznymi
początek zakażenia najczęściej w maziówce (bo
↑
st. tlenu)
później w nasadach ( zapalenie ziarniniakowe z martwicą serowatą i niszczeniem kości)
CHOROBA PAGETA (osteitis deformans)
- Fazy:
1)osteolityczna-
↑
akt. osteoklastów, ilości komórek, niszczenie kości
2)mieszana- proliferacja osteoklastów i osteoblastów
3)osteosklerotyczna- gęsta, zmineralizowana kość,
↓
akt. komórek
efekt: kość bardziej gęsta, ale słabsza
→
zniekształcenia i złamania
Patogeneza (hipoteza!):
powolne zakażenie Paramyxowirusem :
80

→
ekspresja rec. RANK
→
powstają osteoklasty o
↑
akt. resorpcyjnej
→
stymulacja osteoblastów
→
↑
IL6 i akt. osteoblastów
Morfologia:
zmiany pojedyncze lub mnogie (90%)
najczęściej kręgosłup, czaszka, kości miednicy
faza1) zastępowanie szpiku przez luźną tkankę łączną, bogato unaczynioną,
beleczki wyścielone olbrzymimi osteoklastami wielojądrowymi
faza2) resorpcja + tworzenie
faza3) odkładaie nieprawidłowej kości, brak prawidłowych blaszek
znaczne pogrubienie kości zbitej i beleczkowej
elementy nowej kości tworzą przypadkową mozaikę!!
Klinicznie:
zwykle
>
40 r.ż., mężczyźni, zdarzają się przypadki rodzinne
- predysponuje do mięsaka kości (1%), rokowanie wtedy b. niepomyślne
zwykle bezobjawowa
↑
fosfatazy zasadowej (bo
↑
akt. osteoblastyczna)
czasem gdy wczesne zmiany obficie unaczynione- ucieplenie tkanek powyżej kości
gdy zaawansowane -
↑
rzutu serca (bardzo rzadko zastoinowa niewydolność krążenia)
w fazie2 bóle głowy, powiększenie obwodu głowy, zaburzenia widzenia i głuchota
( spowodowane zniekształceniem kości i uciskiem na nerwy czaszkowe)
bóle kręgosłupa, złamania kręgów, ucisk pni nerwowych – kalectwo
odkształcanie kości długich (poprzeczne złamania)
w fazie3
↑
wydalania Ca z moczem ( w osoczu Ca i P w normie)
NOWOTWORY KOŚCI
częściej przerzutowe (z: prostaty, piersi, płuc, nerek, przew.pokarmowego, tarczycy)
osteolityczne częściej niż blastyczne, ale w jednym ognisku mogą występować oba typy
czynniki ryzyka: choroba Pageta, przewlekłe zapalenie kości, promieniowanie
dziedziczne zesp. nowotworowe: zesp. Gardnera (mnogie kostniaki), rodzinny retinoblastoma
(mięsaki kościopochodne)
wszystkie grupy wiekowe i każda okalizacja, trudna diagnostyka
Kościotwórcze:
•
łagodne: - kostniak
- kostniak kostninowy
- kostniak zarodkowy
•
złośliwe: - kostniakomięsak pierwotny.
- kostniakomięsak wtorny
Chrzęstnotwórcze:
•
łagodne: - wyrośl chrzęstno- kostna
- chrzęstniak
- chrzęstniakomięsak
KOSTNIAK(osteoma)
- 40-50 r.ż.
glowa, szyja, zatoki,
pojedyncze, twarde, egzofityczne rozrosty na powierzchni kości
zbudowane z kości niedojrzałej i blaszkowatej, wyglądają jak prawidowa kość
nie naciekają, nie złośliwieją
KOSTNIAK KOSTNINOWY (osteoid osteoma)
przynasady k. udowej i piszczelowej, 20-30 r.ż., mężczyźni
średnica do 2 cm
miejscowy ból, mija po ASA
Morfologia:
przeplatające się dojrzewające beleczki kostne otoczone osteoblastami
rtg- dobrze odgraniczone, częściej w korze, w środku przejaśnienie – indus- ale może ulegać
mineralizacji i sklerotyzacji, obecny rąbek sklerotyczny
zrąb z tkanki łącznej
81

wyst. komórki olbrzymie
KOSTNIAK ZARODKOWY (osteoblastoma)
10-20 r.ż. , kręgosłup
większe niż kostninowe
ból trudniejszy do zlokalizawania, nie reaguje na ASA
może odrastać
Morfologia jak wyżej
KOSTNIAKOMIĘSAK
najczęstszy pierwotny złośliwy mezenchymalny,
kom. nowotworowa produkuje osteoid
Pierwotny:
najczęściej w okolicy stawu kolanowego , przynasada dalsza kości udowej, bliższe kości
piszczelowej i ramiennej
10-20r.ż., mężczyźni
mutacje genu supresorowego nowotworu TP53, nadekspresja MDM2, mutacje genów dla
retinoblastoma, utrata heterozygotyczności w 3p, 13q, 17p, 18q
Wtórny
jako powiklanie innych prcesów
kość udowa, ramieniowa, miednica
>
40 r.ż.
Morfologia:
rośnie na zewnątrz, unosi okostną (trójkąt Codmana), wrasta do jamy szpikowej
może być obecna chrząstka
duży guz o zatartych granicach
może być polimorfizm, kom. olbrzymie
Klinicznie:
szybko rośnie, boli
pierwszy objaw to często złamanie
agresywne, przerzuty drogą krwionośną często do płuc
WYROŚL KOSTNOCHRZĘSTNA(osteochondroma) / egzostoza
przynasady kości długich, narośla o szerokiej podstawie
10-30 r.ż.
zawiera dojrzałą kość i chrzęstną pokrywę
1/3 wszystkich łagodnych nowotworów kości
przestają się rozwijać wraz z ustaniem wzrostu osobniczego
zwykle pojedyncze, mnogie w przypadkach rodzinnych ( i takie mogą przekształcic się w
mięsaka)
CHRZĘSTNIAK(chondroma)
małe kości rąk i stóp, 30-50 r.ż.
dobrze odgraniczone, przypominają prawidłową chrząstkę, rosną w jamie szpikowej
zbudowane z dojrzałej chrząstki szklistej z nielicznymi chondrocytami
rzadko mnogie lub dziedziczne
choroba Olliera- mnogie, po jednej sronie ciała (atypia)
zesp. Maffucciego – mnogie (atypia) + naczyniaki tkanek miękkich
1/3 – przemiana w chrzęstniakomięsaka
CHRZĘSTNIAKOMIĘSAK
z komórek mezenchymalnych które prod. macierz chrzęstną, nie produkują osteoidu
najczęstsze po kostniakomięsakach
40-60 r.ż., mężczyźni
bark, miednica, żebra, cz.bliższa kości udowej
czasem bolesne
przerzuty drogą krwi, najczęściej do płuc
Morfologia:
rozwija się w jamie szpikowej najczęściej, ekspansywny rozrost, niszczy korę
82

obraz histologiczny bardzo zróżnicowany
GUZ OLBRZYMIOKOMÓRKOWY KOŚCI (osteoclastoma)
nasady kości długich (dalsza kości udowej i promieniowej, bliższa piszczelowej i ramiennej)
20-40 r.ż.
zmiany lityczne niszczące korę; podobne do osteoklastów komórki olbrzymie oraz owalne lub
wrzecionowate komórki jednojądrzaste(=nowotworowe)
20% wszystkich łagodnych zmian kości
Klinicznie:
ból, mylony z zapaleniem stawu
biologia nieprzewidywalna: może dawać wznowy, złośliwieć wskutek naświetlania, złośliwe de
novo, histologicznie łagodne mogą przerzutować do płuc
z grupy MIĘSAKa EWINGA (EWS)
1) mięsak kostny Ewinga (najczęściej)
2) postac pozakostna
3) prymitywny nowotwór neuroektodermalny (PNET) (najczęściej)
4)
neuroepithelioma
5) guz Askina
pochodzenie neuroektodermalne
translokacje- fuzja genów EWS na chrom.22q12 z ETS (gł. FLI 11q24 i ERG 21q22)
chimeryczne białka stymulują transkrypcję, rozregulowanie różnicowania i dojrzewania kom.
dzieci i młodzież, szczyt 20-30 r.ż.
trzony kości długich i przynasady (kość udowa, piszczel, miednica)
Morfologia:
w jamie szpikowej, miękki, ekspansywny guz, wnika pod okostną (obraz „łupin cebuli”)
małe niebieskie komórki (zawierają glikogen) – marker MIC2 służy do rozróżnienia z innymi neo
o takim obrazie: neuroblastoma, mięśniakomięsak prążkowanokomórkowy, chłoniak.
Klinicznie:
ból, zapalenie, gorączka
DYSPLAZJA WŁÓKNISTA
łagodna zmiana nowotworopodobna
zamiast kości beleczkowej- tkanka łączna i wyspy nieprawidłowej kości
Postacie:
1) zajęcie jednej kości (70%)
prawdopodobnie to zaburzenie rozwojowe
pojawia się w czasie dorastania, potem się uspokaja
żebra, kość udowa, piszczel, żuchwa, szczęka, sklepienie czaszki
powoduje miejscową deformację, złamania
2) zajęcie wielu kości (25%) (rzadko przechodzi w mięsaka)
pojawie się wcześniej, powikłania nawet w wieku dorosłym
wtarzoczaszka, miednica, udo, bark
deformacje, złamania, ogranicza ruch
3) wiele kości + zaburzenia endokrynologiczne (rzadko przechodzi w mięsaka)
u kobiet
zmiany po jednej stronie ciała, przebarwienia typu kawa z mlekiem, przedwczesne dojrzewanie
zesp. McCune’a – Albrighta
może współwystępować z nadczynnością tarczycy i Cushingiem
Morfologia:
przepuszcza prom rtg, rąbek sklerotyczny
fibroblasty, kolagen, wyspy młodej kości
CHOROBY STAWÓW
CHOROBA ZWYRODNIENIOWA STAWÓW(osteoarthritis)
głównie zmiany w chrząstce, wtórnie w kości
w starszym wieku, w stawach najbardziej obciążonych
83

mogą powstawać wtórnie w wyniku zaburzeń metabolicznych (hemochromatoza, cukrzyca) i
uprzednich deformacji, otyłości
rola czynników genetycznych,
↑
estrogenów, ryzyko proporcjonalne do gęstości kości
wczesna faza: chrząstka zawiera więcej wody, mniej proteoglikanów,
↓
produkcji i
↑
degradacji
kolagenu II,
↑
IL1, TNF i NO
↓
wytrzymałości na ściskanie i sprężystości
w odpowiedzi na to głębiej położone chondrocyty produkują kolagen i proteoglikany aż przestają
nadążać
Morfologia:
powiększenie i nieregularne rozmieszczenie chondrocytów, fibrylizacja (rozszczepienie) –
scieńczała chrząstka z pionowymi szczelinami
chrząstka może zostać całkowicie zniszczona, poniżej kość słoniowa (sklerotyzacja
podchrzęstna)
fragmenty mogą się odrywać, powstają torbiele wewnątrz kości (płyn stawowy)
wzrost proliferacji na obrzeżach- wyrośla kostne (osteophyton), kolce kostne,
zapalenie
Klinicznie:
stawy biodrowe, kolanowe, międzykręgowe lędźwiowe, szyjne, międzypaliczkowe, I
nadgarstkowo-śródręczny i skokowo-śródstopny
usztywnienie i głęboki rwący ból (RANO) nasilający się podczas ruchu
trzeszczenie, obrzęk, wysięk
guzki Heberdena u kobiet na dalszych międzypaliczkowych
zniekształcenie, bez zarastania stawu( w przeciwieństwie do RZS)
DNA MOCZANOWA (diathesis urica)
nadmierne gromadzenie w tkankach kwasu moczowego
powtarzające się epizody ostrego zapalenia stawów, odkładanie guzków dnawych, przewlekłe
zniekształcenia stawów
nie u wszystkich z hiperurykemią dochodzi do rozwoju dny
najczęściej występuje pierwotna nadprodukcja kw.moczowego z lub bez zwiększonego
wydalania (przyczyny niejasne), rzadziej przyczyną choroby jest zmniejszone wydalanie
moczanów
choroba Lescha- Nyhana – brak enzymu HGPRT (katalizuje łączenie wolnych puryn z PRPP na
szlaku rezerwowym syntezy nukleotydów) sprzężony z X
- tylko u mężczyzn, wzrost wydalania kw.moczowego, zaburzenia neuro, upośledzeine
umysłowe, samookaleczenia
- nagromadzenie PRPP i wzrost syntezy de novo puryn
- w rezultacie
↑
kw.moczowego
→
mniej nasilony niedobór- bóle w wieku dorastania, zab. neuroloogiczne
dna wtórna -
↑
wytwarzania moczanów wskutek rozpadu komórek (leczenie nowotworów)
-
↓
wydalanie w niewydolności nerek, diuretyki tiazydowe
Mechanizm:
wytrącają się kryształy moczanów jednosodowych( blade igły), które aktywują dopełniacz,
powstaje C3a i C5a, nagromadzenie neutrofilów i makrofagów w jamie szpikowej i maziówce
fagocytoza kryształow przez neutrofile
→
uwalnianie wolnych rodników i leukotrienów
→
rozpad
neutrofilów
→
uwalnianie enzymów lizosomalnych
→
uszkodzenie tkanki i zapalenie
fagocytoza przez makrofagi
→
uwolnienie IL1, IL6, IL8, TNF
→
reakcja zapalna, wydzielenie
proteaz
→
uszkodzenie
zapalenie mija w ciągu kilku dni/tyg.
Klinicznie:
ostre zapalenie stawów,
artropatia dnawa,
guzki dnawe (bezbolesne) w tkankach miękkich (nieregularne kredowobiałe złogi, przy granicy
torebki stawowej, powodują przewlekłe zapalenie ziarniniakowe)
nefropatia dnawa – kryształy zatykają cewki nerkowe, tworzą się kamienie nerkowe
84

u mężczyzn
>
30 r.ż.
Fazy:
1)bezobjawowa hiperurykemia
2)ostry napad dny z zapaleniem stawów
3)okres międzynapadowy
4)przewlekła dna z guzkami dnawymi
miejscowy nagły i silny ból jedno-, później wielostawowy
paluch, śródstopie, pięta, kolano, nadgarstek
powoduje zniekształcenia, owrzodzenia
nefropatia, nawracające zapalanie odmiedniczkowe, przewlekła niewydolność nerek
ZAKAŻENIA STAWÓW
OSTRE ROPNE ZAPALENIE STAWÓW
najczęściej gonokoki, gronkowce, paciorkowce, H. influenzae, G-, Salmonella
w wyniku urazu, bakteriemii, przez ciągłość, w niedoborach immunolog.
Ból, gorączka, naciek nautrofilów
CHOROBA Z LYME (borelioza)
Borrelia burgdorferi na ludzi od gryzoni za pośrednictwem kleszczy
1) namnożenie krętków w miejscu ukąszenia- zaczerwienienie z twardym, bladym
cetrum (erythema chronicum migrans, ustępuje po kilku tyg.) + gorączka i
powiększenie węzłów.
2) Wczesny okres rozsiewu drogą krwionośną- obrączkowe zmiany skórne,
powiększenie węzłów, wędrujące zapalenie stawów, bóle mięsni, zaburzenia
rytmu serca, zapalenie osierdzia i mięsnia sercowego, zapalenie opon, zajęcie
nerwów czaszkowych (powstają przeciwciała, ale czasem brak)
3) Późny okres rozsiewu, po 2-3 latach od ukąszenia- przewlekłe zapalenie stawów i
zapalenie mózgu, niedowład nerwów czaszkowych, „skóra pergaminowa”
najczęściej kolano, bark, łokieć
Morfologia:
początkowo przypomina RZS
zapalenie tętnic! z obrazem „łupin cebuli”
później ubytki w chrząstce
gdy zapalenie opon- w płynie m-rdz dużo komórek – limfocyty, plazmatyczne i p/ciała
p/krętkowe
CHOROBY MIĘŚNI SZKIELETOWYCH
NEUROGENNY ZANIK MIĘŚNI
Morfologia:
włókna mięśniowe są kanciaste, atroficzne, zaburzenia aktywności niektórych enzymów
zmiany we włóknach szybkich (typu II) i wolnych (typu I)
często jako zanik grupowy
w przypadku regeneracji nerwu, morfologia może wrócic do normy
grupowanie typów włókien – zanik naprzemiennego występowania włókien różnych typów
choroba Werdinga- Hoffmana – wrodzony rdzeniowy zanik mięsni typu I – rozległy zanik grup
włókien, między nimi pojedyncze włókna hipertroficzne
Klinicznie:
osłabienie siły mięśniowej- nieznaczne aż do niewydolności oddechowej
→
u noworodków- uogólniona hipotonia „zesp. wiotkości niemowląt”
ZANIK WŁÓKIEN MIĘŚNIOWYCH TYPU II
jedno z najczęstszych zaburzeń mięśni szkieletowych
wskutek nie używania mięśni, stosowania glikokortykosterydów i endogennym podwyższonym
poziomem kortyzolu
Morfologia:
włókna kanciaste i atroficzne, ale brak zaniku grup
MIASTENIA (nużliwość mięśni)
nabyta, autoimmunologiczna,
zaburzenia transmisji nerwowo- mięśniowej powodują osłabienie mięśni
85
w każdym wieku, młodsze częściej kobiety, starsi mężczyźni
przeciwciała p/receptorowi dla Ach, w złączu nerwowo-mięśniowym obecne Ig oraz składowe
dopełniacza
współwystępowanie z SLE, RZS, zesp. Sjogrena, choroby grasicy
Morfologia:
znikome zmiany
skupiska limfocytów- lymphorrhagies
„uproszczenie” budowy złącza
Klinicznie:
początek podstępny lub nagły
objawy nasilają się w ciągu dnia, bo osłabienie następuje po pobudzeniach
zajęte mięśnie gałek ocznych: ptosis (opadanie powieki) i diplopia (podwójne widzenie)
gdy wypowiedź pacjenta się przedłuża przyjmuje postać nosowej
powoli postępuje
może prowadzić do niewydolności oddechowej
poziom osłabienia nie zawsze jest proporcjonalny do ilości przeciwciał
ZESPÓŁ MIASTENICZNY LAMBERTA- EATONA
zwykle w towarzystwie raka drobnokomórkowego płuca
wyprzedza rozpoznanie nowotworu
p/ciała p/presynaptycznej części złącza
MIOPATIE ZAPALNE
zapalenie wielomięśniowe, skórno- mięśniowe, wtrętowe,
w przebiegu toksoplazmozy, wągrzycy, włośnicy
wirus grypy, Coxackie, HIV
→
nieswoiste pobolewania aż do piorunującej martwicy z
mioglobinurią i niewydolnością nerek
DYSTROFIA MIĘŚNIOWA DUCHENNE’A (DMD)
sprzężona z X (Xp21- jeden z największych genów człowieka),
brak dystrofiny (białko strukturalne, łączy części sarkomeru z błoną komórkową
obj. pojawiają się ok.5 r.ż., śmierć ok.20 r.ż. (niewydolność oddechowa, zapalenie płuc,
zastoinowa niewydolność krążenia, zaburzenia rytmu)
DYSTROFIA MIĘŚNIOWA BACKERA (BMD)
sprzężona z X
dystrofina o nieprawidłowej budowie
objawy mniej nasilone niż w DMD, występują później
Morfologia (DMD i BMD):
znaczne różnice w wielkości włókien (współistnienie przerostu i zaniku)
zmiany degeneracyjne, martwica oraz cechy regeneracji
dużo tkanki łącznej , włóknienie śródmiąższowe
Klinicznie (DMD i BMD):
zwykle mężczyźni
wczesne objawy: ogólna niezgrabność ruchowa, niewydolność mięsni obręczy barkowej i
biodrowej, niektóre mięsnie mogą przerastać (łydki)
INNE MIOPATIE
- wywołane wrodzonymi chorobami metabolicznymi, zab. mitochondrialne, subst. toksyczne
NOWOTWORY
TŁUSZCZAK
najczęstszy nowotwór tkanek miękkich
pojedyncze (przypadki dziedziczone – mnogie), sporadyczne, rosną wolno,
boli tylko angiolipoma
Morfologia:
miękkie, żółte, położone powierzchownie są otorebkowane, wewnątrzmięśniowe- nie
odgraniczone
zbudowane zwykle z dojrzałej tkanki
86

często ulega zmianom krwotocznym – ostry brzuch
TŁUSZCZAKOMIĘSAK
50-6O r.ż.
położone głęboko w jamie brzusznej i kończynach dolnych
typ śluzowaty i okrągłokomórkowy – translokacje chromosomalne
Morfologia:
dobrze odgraniczony
typy: wysoko dojrzały, śluzowaty (oba rosną powoli), okrągłokomórkowy, polimorficzny (oba
bardziej złośliwe- wznowy i przerzuty drogą krwi do płuc), odróżnicowany
GUZKOWE ZAPALENIE POWIĘZI (fasciitis nodularis)
odczynowy i samoustępujący rozrost fibroblastów
u młodych dorosłych, rośnie w ciągu kilku tygodni, czasem boli
zwykle kończyny górne i tułów
zmiana nieotorebkowana, powerzchowne dobrze odrganiczone
<
3 cm.
pękate fibroblasty na śluzowym podścielisku, obecne figury podziału, ale nie
nieprawidłowe
WŁÓKNIAKOWATOŚĆ (fibromatoza)
stały miejscowy rozrost fibroblastyczny
nacieka okoliczne tkanki, nawroty, możliwa miejscowa złośliwość, ale nie przerzuty
nie poddaje się leczeniu chirurgicznemu
Typy:
powierzchowna – rozwija się w powięziach powierzchownych, zniekształca, łagodna
fibromatoza dłoniowa (przykurcz Dupuytrena)
fibromatoza prącia (choroba Peyronie)
głęboka –skłonność do nawrotów i miejscowa złośliwość
włókniec (desmoid tumor) w jamie brzusznej, mięśniach tułowia i kończyn może występować w
przebiegu zesp. Gardnera (polipy gruczolakowate w j.grubym, kostniaki w kościach oraz
głębokie fibromatozy)
Morfologia:
zależy od lokalizacji
zmiana może być różnie odgraniczona
WŁÓKNIAKOMIĘSAK
złośliwy, pojedynczy, w tkankach głębokich: udo, kolano, przestrzeń zaotrzewnowa
przerzuty, wznowy, rośnie wiele lat
WŁÓKNIAK HISTIOCYTARNY
w skórze i tkance podskórnej guzki łagodne, dobrze odgraniczone, ruchome
nacieka, ale nie głęboko
fuzja genów – prowadzi do autokrynowej pętli wytwarzania PDGF
β
WŁÓKNIAKOMIĘSAK HISTIOCYTARNY
50-70 r.ż.
w mięśniach głębokich kończyn i zaotrzewnowo
wznowy miejscowe, przerzuty
MIĘŚNIAKOMIĘSAK PRĄŻKOWANOKOMÓRKOWY (rhabdomyosarcoma)
niemowlęta, dzieci i młodzież (najczęstszy mięsak u dzieci)
typ zarodkowy : głowa i szyja, drogi moczowo- płciowe, zaotrzewnowo
(translokacje i fuzje, chimeryczny brak PAX3-FKHR:)
typ pęcherzykowy: w wieku dorastania, kończyny, przewody nosowe i zatoki
typ polimorficzny u dorosłych
Morfologia:
obraz zmienny
mięsak groniasty ( sarcoma botryoides)- powierzchowne, miękie, galaretowate
warstwa kambialna- skupiane komórek nowotworowych tuż pod śluzówką
MAZIÓWCZAK ZŁOSLIWY (sarcoma synoviale)
87

z tkanki mezenchymalnej, zwykle w okolicy stawów
translokacja chromosomalna
Morfologia:
różne rozmiary i odgranczenie
komponent nabłonkowy tworzy gruczoły + sznury kom. przypominających fibroblasty
SKÓRA
Pojęcia
Plamka /macula/ - płaski obszar różniący się od skóry
Grudka /papula/ - wyniosłe ognisko <5mm
Guzek /nudulus/ - wyniosłe ognisko >5mm
Blaszka /lamella/ - wyniosły, płaski obszar >5mm
Pęcherzyk /vesicula/ - ognisko wypełnione płynem <5mm
Pęcherz /bulla/ - pęcherzyk >5mm
Krosta /postula/ - ognisko wypełnione ropą
Łuska /squama/ - jak płytka, nadmiernie zrogowaciała
Ostre zapalne choroby skóry
Pokrzywka
degranulacja mastocytów-> wzrost przepuszczalności naczyń -> zmiany na skórze (bąble)
różny obraz, szybki przebieg
etiologia - alergiczne (zależne od IgE) lub spowodowana przez substancje bezpośrednio działające na
mastocyty (leki)
Ostre wysypkowe zapalenie skóry (wyprysk)
etiologia - często reakcja na jad owadzi lub inne kontaktowe antygeny np. detergenty
czerwone, grudkowo-pęcherzykowe, sączące zmiany, które strupieją może wystąpić nadmierne
rogowacenie
typy: zapalenie kontaktowe skóry, zapalenie atopowe skóry, zapalenie wypryskowe atopowe , wykwity
fotouczuleniowe, pierwotne podrażnienie zapalne
mikro: gąbczastość (gromadzenie się płynu w obrębie naskórka - wygląd gąbki), nacieki limfocytarne,
eozynofile
Rumień wielopostaciowy
etiologia - ch. zakaźne, leki, nowotwory złośliwe, choroby autoimmunologiczne naczyń
rzadki, heterogenny obraz zmian: od plamek do pęcherzy, charakterystyczne jasne, pęcherzykowe
przejaśnienie w centrum
u dzieci z gorączką z nadżerkami błon śluzowych- z. Stevensa-Johnsona
mikro - naciek limfocytarny szczególnie w obrębie połączenia skórno-naskórkowego
Przewlekłe zapalenia skóry
Łuszczyca
1-2% ludności, często z zapaleniem stawów, miopatią, enteropatią
etiopatogeneza - wieloczynnikowa: genetyczno-immunologiczno-środowiskowa, limf T wnikając do
skóry i wydzielając cytokiny warunkują nieprawidłowy wzrost skóry
zajmuje skórę: łokci, kolan, głowy owłosionej, okolicy krzyżowo-lędźwiowej, żołędzi; w 30% zajmuje
paznokcie
płytki koloru różowego pokryte białosrebrzystymi łuskami
mikro -
↑
rogowacenia, długie sople naskórkowe, ścieńczenie lub brak warstwy ziarnistej, poszerzone,
kręte naczynia (krwawienia punktowe)
Liszaj płaski
płaskie, grudki, często łączące się w płytki, nadmierna pigmentacja (purpurowe) z
charakterystycznymi białymi kropkami lub liniami ( prążek Whickmana)
samoustępujący po ok. 2 latach
występowanie - kończyny, żołądź, jama ustna
mikro - naciek limfocytarny na złączu naskórkowo-skórnym (charakterystyczny piłowaty obraz),
pogrubienie warstwy ziarnistej i rogowej
88

Choroby pęcherzowe
Pęcherzyca /pemphigus/
etiopatogeneza - autoimmunologiczna , IgG przeciw desmogleinie -> zerwanie połączeń
międzykomórkowych w naskórku (acantholysis)
dotyczy osób w wieku średnim i starszym, zajmuje błonę śluzową i skórę
pęcherze -> pęknięcie -> płytkie nadżerki ze strupem -> blizna
typy: zwykła (80%), budująca, liściasta, rumieniowa
mikro - acantholysis, okrągłe keranocyty
Zmiany pęcherzycopodobne /pemphigoid bullosus/
etiopatogeneza - autoimmunologiczna , IgG przeciw składnikom hemidesmosomów
chorują ludzie starsi
pęcherze wypełnione jasnym płynem na niezmienionej lub różowej skórze, trudno pękające, nie
powstają blizny
mikro - podnaskórkowe, nieakantolityczne pęcherze
Opryszczkowe zapalenie skóry
etiopatogeneza - autoimmunologiczna, współistnieje z celiakią, IgA przeciw retikulinie
3,4 dekada życia, częściej mężczyźni
swędząca pokrzywka + zgrupowania pęcherzyków jak w Herpes
Nowotwory i zmiany nienowotworowe
Rogowacenie łojotokowe /keratosis saborrheica/
nowotwór łagodny u osób w starszym wieku , często mnogi
okrągłe płaskie płytki, jak monety koloru brunatnego
mikro - ostro odgraniczony, torbiele wypełnione keratyną, nadmierne rogowacenie
Rogowiak /keratoacanthoma/
u ludzi powyżej 50 rż, częściej u mężczyzn rasy białej, na odsłoniętej skórze
cielisty guzek zawierający rogowy czop w części centralnej (od 1 do kilku cm)
gwałtownie rosnący (przypomina raka kolczystokomórkowego), goi się samoistnie
Brodawki (kłykciny)
etiologia - HPV, u osób z prawidłowym ukł. immunologicznym nie ma progresji do raka
ustępują samoistnie po 0,5 do 2 lat
typy: pospolita (głównie na rękach, jasna grudka), płaska (twarz, grzbiet ręki, chropowata, łuszcząca
się zmiana), kończysta (krocze, kalafiorkowe masy)
mikro - brodawkowaty rozrost naskórka, koilocytoza
Rogowacenie popromienne /keratosis actinica/
stan przedrakowy, po przewlekłej ekspozycji na promieniowanie słoneczne, dyplazja
zwykle mniejsza niż 1cm; kolor brunatny, czerwony, cielisty
mikro - atypia komórkowa w dolnych warstwach, jądra w warstwie rogowej,
↑
wł. elastycznych w skórze
Rak kolczystokomórkowy /ca spinocellulare/
starsze osoby po długotrwałym działaniu promieni słonecznych, częściej u mężczyzn, osób z
immunosupresją, inne przyczyny: kancyrogeny przemysłowe, przewlekłe owrzodzenie, zapalenie kości
z przetokami, blizny po oparzeniach, arsen, promieniowanie jonizujące
na początku czerwona płytka -> naciekająca zmiana guzowata, rogowaciejąca
mikro - in situ: atypia na całej długości naskórka; naciekający: różna dojrzałość: wygląd komórek,
rogowacenie
przerzuty w 5% (w węzłach), rokowanie dobre
Rak podstawnokomórkowy /ca basocellulare/
występowanie jak rak kolcz., u osób o słabo pigmentowanej skórze, często w Xeroderma pigmentosum
perłowe grudki z teleangiektazjami, czasem z melaniną, owrzodzenia, nieleczony głębokie owrzodzenia
(wrzód drążący)
mikro - 2 typy: ogniskowa (powierzchniowe w naskórku), guzkowa (głębiej w skórze, sznury lub wyspy);
palisady z komórek
b. rzadko przerzuty
Znamię barwnikowe
prawidłowe melanocyty znajdują się w warstwie podstawnej nabłonka, mają małą aktywność
mitotyczną
89

w znamionach brzeżnych układają się w gniazda na granicy naskórkowo-skórnej, część z nich wrasta do
skóry właściwej - znamię złożone, w wypadku całkowitego oderwana od naskórka - znamię skórne
Rodzaj
Cechy budowy
Cechy
charakterystyczne
Klinika
wrodzone
głęboko w skórze, tk.
podskórnej
-
obecne przy urodzeniu,
ryzyko czerniaka
błękitne
naciekanie nietworzące
gniazd, często włóknienie
dendrytyczne,
mocno
pigmentowane
czarno-niebieskie guzki,
mylone z czerniakiem
wrzecionowate
(Spitza)
wzrost pasmowy
duże pulchne
komórki,
wrzecionowate
u dzieci, różowe guzki,
mylone z naczyniakiem
halo
nacieki limfocytarne wokół
zmiany
-
reakcja immunologiczna na
znamię
dysplastyczne
duże gniazda śródnabłonkowe,
rosnące wzdłuż łącza
naskórkowo-skórnego (plamy
soczewicowe), utrata
melaniny, naciek limfocytarny
atypia
prekursor czerniaka,
nieregularne okonturowane,
pstrokate, chropowate,
często >5mm, czasem
występowanie rodzinne
Czerniak
etiologia - skóra często wystawiana na działanie promieni słonecznych, znamiona dysplastyczny, cz.
genetyczne (rzadko p53)
występowanie - oprócz skóry : błona śluzowa jamy ustnej, krocze, przełyk, opony
objawy: zmiany kształty i/lub koloru znamienia, nieregularne granice; swędzenie, ból znamienia;
powstanie nowej zmiany, różnice w kolorze
najpierw wzrost poziomy (powierzchowny) - bez przerzutów i angiogenezy -> wzrost pionowy (w głąb
skóry) - bez cech dojrzewania, pojawienie się guzka, przerzuty do węzłów, wątroba, płuca, mózg,
często wiele lat później
mikro - duże komórki, atypia, czerwone jąderka, staging na podstawie długości wzrostu pionowego,
stopni Clarka i TNM
grubość Breslow’a
przeżycia pięcioletnie
<1mm
95-100%
1-2mm
80-96%
2.1-4mm
60-75%
>4mm
< 50%
czerniak oka - z melanocytów błony naczyniowej (tęczówka), komórki wrzecionowate(mało agresywne)
i nabłonkowate(wysoce złośliwe)
OUN
Obrzęk mózgu
zwiększona obecność wody w mózgowiu, 2 typy, najczęściej mieszany:
naczyniopochodny - zniszczona bariera krew-mózg, zapalenie, proces nowotworowy
cytotoksyczny - wewnątrzkomórkowy, spowodowany uszkodzeniem komórek: niedokrwienie, trucizny
duży miękki mózg, spłaszczone zakręty, zwężone bruzdy, uciśnięte komory
może doprowadzić do wklinowania
Wklinowanie /herniatio/
po wzroście ciśnienia wewnątrzczaszkowego, 3 typy:
podnamiotowe - hipokamp (hak) dociskany do namiotu móżdżku -> ucisk na nerw III -> upośledzenie
ruchu gałki ocznej i rozszerzenie źrenicy po stronie uszkodzenia; czasem ucisk t. tylnej mózgu ->
zaburzenia widzenia (pł. skroniowy)
podsierpowe - niesymetryczne powiększenie półkul -> zakręt obręczy pod sierp mózgu -> ucisk na t.
przednią mózgu -> zab. ruchowo-czuciowe w kończynie dolnej
podpotyliczne - migdałki móżdżku do otworu wielkiego -> ucisk na pień mózgu -> zagrożenie życia,
wtórne krwotoki (Dureta) do pnia mózgu
Wodogłowie /hydrocephalia/
najczęściej z powodu nieprawidłowego odpływu płynu m-rdz, 2 rodzaje:
niekomunikujące - przeszkoda w układzie komorowym (guzy, zapalenia)
niekomunikujące - przeszkoda poza układem komorowym (po zapaleniach opon i krwawieniach do
przestrzeni podpajęczynówkowej -> zniszczenie ziarnistości pajęczynówki)
rzadko z nadmiernej produkcji płynu - guzy splotu naczyniówkowego
90

jeśli przed zarostem szwów -> poszerzenie głowy
hydrocephalus ex vacuo - wtórne poszerzenie układu komorowego w wyniku zaniku mózgu
Choroby naczyniowe
IV przyczyna zgonów w Polsce, 3 rodzaje
zawały - 80%
krwotoki (wylewy)
uogólniony zmniejszony przepływ krwi
Uogólniona encefalopatia niedotleniowo-niedokrwienna
przy ciśnieniu skurczowym <50 mmHg zawodzą mechanizmy autoregulacji naczyń mózgowych ->
spadek perfuzji mózgu
największa podatność wykazują neurony hipokampa, gałki bladej, kom. Purkinjego móżdżku
ekscytotoksyczność - wtórne do niedokrwienia
↑
aktywność układu glutaminergicznego ->
↑
aktywność
neuronów ->
↑
utrata neuronów
przyczyny: spadek dopływu krwi do mózgu (wstrząs, zatrzymanie krążenia, zab. rytmu serca)
w zależności od wieku pacjenta, czasu trwania niedokrwienia, temperatury pacjenta może spowodować
od przejściowych zab. neurologicznych do śmierci
morfologia - na początku bez zmian, po 1-2 dniach obrzęk, ogniska małych krwotoków w istocie szarej,
warstwowa martwica kory (linijne odbarwienia w jej obrębie), zatarcie granicy istota biała-szara - mózg
respiratorowy (tylko nazwa)
mikro - po 12-24 h - obrzmienie neronów, eozynofilia cytoplazmy, martwica, poszerzenie przestrzeni
okołonaczyniowych i międzykomórkowych, mały odczyn zapalny
Zawały
70-85% udarów, częściej po 70 r.ż. u mężczyzn
etiologia - miażdżyca (czynniki ryzyka), zmiany głównie t. szyjna wew., t. środkowa mózgu i t.
podstawnej - zakrzepica lub zator przy rozerwaniu blaszki
anastomozy (koło tętnicze Willisa) chronią tyko przy zamknięciu naczyń proksymalnych
TIA - przemijające napady niedokrwienia mózgu /transient ischemic attacks/ - przejściowe zaburzenia
neurologiczne do 24h, u 1/3 osób zawał w ciągu 5 lat
miejsce zamknięcia:
t. środkowa mózgu - najczęściej, zaburzenia motoryczne, czucia i widzenia po przeciwległej stronie,
afazja
t. szyjna wew. - jak wyżej + utrata wzroku (t. oczna), ale często koło tętnicze zapewnia odpowiednią
perfuzję
tt. kręgowe i podstawna - niebezpieczne dla życia - pień mózgu, utrata wzroku, ataksja
morfologia - jak w uogólnionej encefalopatii niedokrwiennej tylko skupione w jednym ognisku + naciek
z granulocytów i makrofagów, po 6 miesiącach jama
Krwotoki, wylew
czasem po reperfuzji obszaru dotkniętego zawałem
Pierwotny krwotok śródmózgowy
osoby starsze
etiologia - nadciśnienie tętnicze -> osłabienie ściany naczyń (czasem tętniaki Charcota-Boucharda) ->
pękniecie naczynia; inne: zaburzenia krzepnięcia, zabiegi na otwartym sercu, zapalenia naczyń,
tętniaki, malformacje naczyniowe, ch. Alzhaimera
ostry początek - wzrost ciśnienia śródczaszkowego, wklinowania -> nagła utrata przytomności ->
śpiączka, duże zniszczenia
najczęściej: jądra podstawy, wzgórze, istota białą mózgu, most, móżdżek
Krwotok podpajęczynówkowy
u osób <50r.ż. częściej u kobiet
najczęstsza przyczyna tętniak workowaty/aneurysyma sacciforme/ - u 1% populacji, częściej u
osób z torbielowatością nerek, koarktacją aorty, malformacjami naczyniowymi; występują najczęściej w
rozwidleniach tętnic: t. środkowa mózgu, t. przednia mózgu ; często wrodzone, większe pękają rzadziej
nagły początek, wzrost ciśnienia wewnątrzczaszkowego , bóle głowy, wymioty, utrata przytomności,
objawy oponowe, krwisty płyn m-rdz; powikłania zawały, wklinowania, wodogłowie (ostre i przewlekłe)
ok. 50% umiera w ciągu kilku dni
morfologia tętniaka - brak bł. środkowej, dużo kolagenu, często skrzeplina wewnątrz
Wrodzone wady (malformacje) naczyń
zaburzenia angiogenezy, 4 typy:
malformacje tętniczo-żylne: najczęściej, konglomerat krętych naczyń, zwapnienia, krwotok po 10 r.ż.
91

inne (najczęściej nieobjawowe): naczyniaki jamiste, teleangiektazje włośniczkowe, naczyniaki żylne
Urazy OUN
przyczyna 25% zgonów po wypadkach
Krwiak nadtwardówkowy /hematoma epidurale/
etiologia - pęknięcie t. oponowej (głównie środkowej)w skutek pęknięcia kości czaszki
wzrost ciśnienia -> wklinowanie + obrzęk mózgu pod krwiakiem
często po urazie interwał jasny, po którym następuje stopniowa utrata świadomości
Krwiak podtwardówkowy / h. subdurale/
pęknięcie żył mostkowych (z powierzchni mózgu do zatok opony twardej) przy szybkiej zmianie
położenia głowy -> krwotok do przestrzeni podtwardówkowej
ostre - jednostronne, u niemowląt często obustronne, z powodu urazu, najczęściej w okolicy czołowo-
ciemieniowej, wolne narastanie objawów, spłaszczenie zakrętów, obrzęk, wzrost ciśnienia
wewnątrzczaszkowego, wklinowania, może przejść w przewlekły
przewlekły - rzadko towarzyszy urazom, często w zaniku mózgu, obustronne, wypełnione żółtawo
podbarwionym płynem, narastające objawy ogniskowe i zmiany psychiczne
Uszkodzenia śródmiąższowe
wstrząśnienie /commotio/ - utrata przytomności, drgawki i inne objawy neurologiczne mijające w
ciągu kilku dni, brak widocznych zmian, częściowa utrata pamięci
rozlane uszkodzenie aksonów - rozerwanie wypustek neuronów, otępienie pourazowe, przetrwały
stan wegetatywny; mikro - obrzmienie aksonów istoty białej (sferoidy aksonalne)
stłuczenie (contusio):
krwotoki do powierzchniowych warstw mózgowia spowodowane tępymi urazami, bieguny czołowe,
skroniowe, potyliczne, móżdżek; małe narzędzie
uraz w miejscu uderzenia (coup), szeroka powierzchnia; zmiany po przeciwległej stronie -
przeciwderzenie (contrecoup);
krwotoki podpajęczynówkowe, rozczłonkowanie, krwotoczno martwicze zmiany mózgowia, zniszczona
warstwa powierzchniowa (drobinowa) kory; stare zmiany - zapadnięte, twarde obszary (padaczki)
przy urazach mogą pojawić się też krwotoki śródmiąższowe i obrzęk mózgu
Wady cewy nerwowej (z. dysrafii)
zamknięcie cewy 22-28 dzień
etiologia: niedobór folianu, czynniki genetyczne
bezmózgowie /anencephalia/ - 1:500 urodzeń, brak sklepienia czaszki, „żabia” twarz, letalne, często z
wielowodziem i podwyższonym stężeniem
α
FP i acetylocholino esterazy
przepuklina mózgowa (mózgowo-oponowa) /encephalocele/ - uwypuklenie się przez ubytek kości
czaszki (gł. kości potylicznej) opon i mózgu [czaszkowa przepuklina oponowa /meningocele/ - to samo
ale bez mózgowia
rozszczep kręgosłupa /spina bifida/ - okolica krzyżowo-lędźwiowa, brak łuków kręgów:
przepuklina oponowo-rdzeniowa /meningomyelocele/ - worek przepuklinowy z oponami i rdzeniem,
często z wodogłowiem, z. Arnolda-Chiariego; przepuklina oponowa - to samo bez rdzenia
utajony rozszczep kręgosłupa /spina bifida occulta/ - ubytek łuków kręgów bez przepukliny
Wady związane z wodogłowiem
Z. Arnolda-Chiariego - spłycony dolny tył czaszki, przemieszczenie rdzenia przedłużonego i robaka do
kanału kręgowego, przepuklina oponowo-rdzeniowa
Z.Dandy’ego-Walkera - aplazja robaka, rozdęcie komory IV, powiększenie tylnego dołu czaszki,
przepuklina
Zaburzenia przodomózgowia
holoprosencephalia - brak podziału na półkule, cyklopia, trisomia 13, 15;
drobnozakrętowość /polimycrogyria/, gładko mózgowie /lissencephalia/
Uszkodzenia okołoporodowe
spowodowane przez hipoksje, hiperkapnie, kwasice
krwotok do macierzy zarodkowej - dokomorowe, zbliznowacenie -> wodogłowie
martwica istoty białej (leukomalacja)- opóźnienie rozwoju, porażenie czterokończynowe
zmiany w istocie szarej - mózgowe porażenie dziecięce /paralysis celebralis infantum/
Zakażenia OUN
Ostre ropne zapalnie opon /leptomeningitis acuta/
bakterie drogę krwi i nosogardzieli
patogeny:
92

noworodki - E. coli, paciorkowce grupy B
dzieci - H. influenzae, S. pneumonie
młodzież, młodzi dorośli - N. meningitidis
starsi - S.pneumoniae, pałeczki Gram -
u chorych z zastawkami odprowadzającymi płyn do jamy otrzewnej - S. aureus, pałeczki Gram -
morfologia - przekrwienie opony, dużo włóknika i naciek neutrofilów, wysiękowy płyn w przestrzeni
podpajęczynówkowej, mózg i rdzeń obrzęknięte, rzadko przejście na mózg
objawy oponowe, zmiana płynu m-rdz
Ostre limfocytarne (wirusowe) zapalenie opon
wirusy: ECHO, Coxsackie, świnki, HIV ; czasem z zapaleniem mózgu, dużo limfocytów
Przewlekłe zapalenie opon
bakteryjne lub grzybicze - M.tuberculosis, C. neoformans (AIDS), T.pallidum, Brucella
przewlekły ból głowy, wodogłowie, wiąd rdzenia (zaburzenia czucie i chodu), porażenie postępujące
morfologia - wysięk, zrostu pajęczynówki, pogrubienie opon, naciek limfocytarny, plazmocyty,
włóknienie naczyń
Zapalenia mózgu
Ropień mózgu /abscessus cerebri/
gronkowce, paciorkowce, beztlenowe - drogą krwi (zap. wsierdzia, ropnie płuc) lub przez ciągłość
najczęściej w półkulach, mogę być mnogie, jama otoczona łącznotkankową torebką
objawy: wzrost ciśnienia śródczaszkowego, ogniskowe
Gruźlica i toksoplazmoza
rozsiane zmiany
gruźlica - mózg i opony, gruźliczaki - twarde guzki z martwicą w środku
toksoplazmoza - w AIDS, istota szara, obszary martwicy
Wirusowe zapalenia mózgu
często z zapaleniem opon, okołonaczyniowe nacieki zapalne z limfocytów, plazmocytów i makrofagów,
skupiska kom. glejowych (guzki mikroglejowe), fagocytoza neuronów, czasem wtręty
arbowirusy - np. kleszczowe, kalifornijskie; przenoszone przez stawonogi (szczyt występowania wiosna,
lato)
HSV (herpes simplex) - typ 1 wirusa (typ 2 w AIDS), zmiany krwotoczne i martwicze w płatach
skroniowych i w dolnych zakrętach czołowych
CMV - noworodki i osoby z obniżoną odpornością, dużo wtrętów jądrowych
HIV - oprócz torowaniu drogi dla zakażeń oportunistycznych jest zdolny także do zakażenia OUN,
zapalenie mózgu, podostre zapalenie mózgu -> otępienie (encefalopatia), guzki mikroglejowe,
wielojądrowe komórki olbrzymie, miel opatia wakuolarna - rozpad mieliny w rdzeniu
Leukoencefalopatia wieloogniskowa postępująca PML - wirus JC, u osób z obniżoną odpornością,
zakażenia oligodendrocytów
Encefalopatie gąbczaste
powodowane przez priony - nieprawidłowe białka zmieniające konformację przestrzenną zdrowych
białek -> wakuole wewnątrz istoty szarej -> otępienie
ch. Creutzfelda-Jakoba i jej nowy wariant przenoszony po zjedzeniu wołowiny zwierząt chorych na
encefalopatię gąbczastą bydła (BSE), kuru, śmiertelna rodzinna bezsenność
NOWOTWORY OUN
2-3% nowotworów dorosłych
wszystkie mogą powodować, w zależności od umiejscowienia:
wzrost ciśnienia wewnątrzczaszkowego - bóle głowy, nudności
ogniskowe objawy neurologiczne
objawy uogólnione np. padaczki
zawsze jeśli możliwe leczenie operacyjne, radioterapia, chemioterapia
bardzo rzadko przerzuty
Glejaki
Gwiaździaki /astrocytomata/
WŁÓKIENKOWE (NACIEKAJĄCE, ROZLANE)
osoby dorosłe, mutacje p53, rokowanie zależne od typu, złe
93
częstość w %:
glejaki
50-6
0
meta
25-3
5
oponiaki
15-2
0
nerwiaki
5-10
przys.
4-8
rdzeniak
2-6

gwiaździaki wysoko dojrzałe - słabo odgraniczone; dojrzałe, naciekające astrocyty; nagromadzenia
płynu (mikrocysty)
gwiaździaki anaplastyczne - większa komórkowość, polimorfizm jąder i aktywność mitotyczna
glejak wielopostaciowy /glioblastoma multiforme/ - nieregularny, krwotoki, martwice, torbiele;
proliferacja drobnych naczyń, ,martwica palisadowa, leczenie blokerami rEGFR
WŁOSOWATOKOMÓRKOWE /PILOCYTICUM/
głównie u dzieci, częściej podnamiotowo, dość łagodny
dobrze odgraniczony, wydłużone wypustki astrocytów, włókna Rosenthala, szkliste ciałka ziarniste
Skąpodrzewiak /oligodendroglioma/
dorośli, dość odgraniczone, często zwapnienia, halo wokół jąder, skupianie wokół neuronów, różna
dojrzałość
Wyściółczak /ependymoma/
wewnątrzkomorowe (<20r.ż.), w rdzeniu (u dorosłych), często w komorze IV (wodogłowie), komórki o
wydłużonych jądrach tworzące pseudorozety naczyniowe i rozety ependymalne
PNET - Prymitywne nowotwory neuroepitelialne (rdzeniak /medulloblastoma/)
u dzieci (<20r.ż.), w robaku lub półkulach móżdżku, przerzuty przez płyn m-rdz, „małe, niebieskie”
komórki, rozetki Homera-Wrighta, nadciśnienie wewnątrzczaszkowe
inne : szyszyniak, wyściółczak zarodkowy, nabłoniak zarodkowy, neuroblastoma, mięsak Ewinga
Nowotwory neuronalne
z neuronów, druga dekada życia, często powodują napady padaczkowe u dzieci
nowotwory komórek zwojowych - zwojakoglejaki lub zwojaki, większość łagodna
dysembrioblastyczny guz neuroepitelialny DNT - dojrzałe neurony z oligodendrocytami, łagodny
Nerwiak ośrodkowy - w okolicy komory bocznej, okrągłe jądra z halo, łagodny
Inne nowotwory pierwotne śródmózgówe
pierwotny chłoniak OUN - z HIV +, jak inne chłoniaki , wokół naczyń, często wirus EBV
nowotwory germinalne - dzieci i młodzież, często w okolicy szyszynki i spoidła, zarodczak /germinoma/
(jak nasieniak) lub potworniak
naczyniak krwionośny zarodkowy/haemangioblastoma/ - móżdżek
Oponiak /meningioma/
z komórek pajęczynówki, poza miąższem, dorośli, częściej kobiety w kanale kręgowym, u osób z
nerwiakowłókniakowatością typu II
twarda masa przytwierdzona do opony, typ syncycialny i fibroblastyczny, zwapnienia - ciałka
piaszczakowate
Przerzuty
raki płuc, piersi, białaczki, czerniaki
często mnogie, rzadziej do opon, komórki obecne w płynie m-rdz
Choroby mieliny (demielinizacyjne)
Stwardnienie rozsiane SM
najczęstsza, głównie młodzi dorośli
etiologia: autoimmunologiczna - limfocyty CD4+ i CD8+ przeciw białku zasadowymi mieliny i innym
białkom mieliny, czynniki środowiskowe i genetyczne
morfologia: liczne, dobrze odgraniczone obszary demielinizacji zwane blaszkami (plaki), mikro:
okołonaczyniowe nacieki limfocytarne, komórki obładowane lipidami, aksony pozostają
nagły początek, podstępny, różne objawy: zaburzenia widzenia, mowy, chodu, parestezje,
spastyczność kończyn, zaburzenia psychiczne; naprzemienne rzuty i remisje
Inne
ostre rozsiane zapalenie mózgu i rdzenia - reakcja immunologiczna na zakażenia wirusowe (odra, ospa,
różyczka) lub szczepienia, jednofazowy, nagły przebieg,
mielinoliza środkowa mostu - u alkoholików i po szybkim skorygowaniu hiponatremii
Choroby zwyrodnieniowe
II przyczyna otępienia wśród ludzi (I choroby naczyniowe)
94

Choroba Alzheimera
najczęściej >50r.ż., ryzyko rośnie z wiekiem, postępująca demencja (5-15 lat)
etiopatogeneza:
cz. genetyczne - mutacje chromosomu:21 -białko prekursowe amyloidu (APP) (częściej z z.Downa), 14 -
preseniliny, apolipoproteina E
odkładanie się złogów amyloidu - produkt rozpadu APP -
β
-amyloid (A
β
), toksyczny dla neuronów
hiperfosfolyracja białka
τ
(tau) - uczestniczy w formowaniu cytoszkieletu
ekspresja alleli apolipoproteiny E - ApoE wiąże się A
β
morfologia: zanik mózgu, powiększenie układu komorowego, głębokie, szerokie zakręty; mikro: w
cytoplazmie neuronów - sploty neurofibrylarne (z białkiem
τ
), blaszki starcze - poskręcane, grube
neuryty z rdzeniem z A
β
, amyloid także wokół naczyń, ciałka Lewy’ego
Parkinsonizm
zaburzenie czynności motorycznych, charakteryzujące się wzmożonym napięciem mięśniowym,
maskowatą twarzą, zaburzeniami chodu, spowolnieniem, drżeniem spoczynkowym
etiologia: uszkodzenie dróg dopaminergicznych łączących istotę czarną z jądrami podstawy w wyniku:
ch. Parkinsona, urazów, zapaleń, toksyn, ch. naczyniowych
ch. Parkinsona (parkinsonizm idiopatyczny)
po 60 r.ż. , wieloczynnikowa, przebieg ok. 10 lat, czasem towarzyszy otępienie, śmierć głównie z
przyczyn urazowych
zwyrodnienie neuronów istoty czarnej i miejsca sinawego, odbarwienie, glioza, czasem ciałka Lewy’ego
ch. Huntingtona
genetyczna, dziedziczona autosomalnie dominująco, 4-5 dekada życia, objawy: mimowolne ruchy
skrętne, pląsawica, dalej zab. neuropsychiatryczne - depresja, otępienie; śmierć po 15-20 latach
etiopatogeneza: zwiększona liczba powtórzeń CAG -> nieprawidłowy gen huntingtyna (chromosom 4) -
> degeneracja neuronów
morfologia: zanik j. ogoniastego, skorupy, gałki bladej i innych struktur, poszerzenie komór
Stwrdnienie zanikowe boczne SLA
zwyrodnienie górnych i dolnych motoneuronów, postępujący zanik, spastyczność i osłabienie
mięśni(drżenie pęczkowe, żywe odruchy ścięgniste /babińskiego/), zgon po ok. 5 latach w skutek
niewydolności oddechowej lub zakażeń
morfologia: utrata motoneuronów w rogach przednich i korze czołowej, a także włókien korowo-
rdzeniowych w konarach mózgu, glioza, ciałka Lewy’ego
inne podobne choroby: z. Werdniga-Hoffmanna (z. wiotkiego dziecka)
Nabyte zaburznia metaboliczne i niedoborowe
niedobory
tiaminy B1 - z. Wernickiego-Korsakowa - encefalopatia Wernickiego (splątanie, porażenie mięśni oka,
ataksja) może przejść w z. amnestyczny Koesakowa (zaburzenia pamięci długo i krótkotrwałej) +
neuropatia obwodowa; u alkoholików zanik móżdżku
kobalaminy B12 - niedokrwistość + zwyrodnienie sznurów tylnych i bocznych rdzenia (zaburzenia
motoryczno-sensoryczne), splątanie
Zaburznia metaboliczne i zatrucia
encefalopatia wątrobowa - zaburzenia świadomości, grubofaliste drżenia kończyn, podobnie z
ch.Wilsona
zatrucia - metale (As, Hg, Pb), związki organiczne (fosforany, metanol), tlenek węgla, leki (metotreksat,
metronidazol, izoniazyd), promieniowanie jonizujące - radioterapia
Neuropatie obwodowe
zwyrodnienie aksonalne - uszkodzenie aksonu na skutek urazu, niedokrwienia, toksyny
demielinizacja odcinkowa - uszkodzenie osłonki z zachowaniem aksonu, leukodystrofie, z. Guillaina-
Barrego - po zakażeniach wirusowych i innych, głównie zaburzenia motoryczne, naciek nerwów przez
limfocyty i makrofagi
Nowotwory obwodowego układu nerwowego
nerwiak osłonkowy /schwannoma/ - na przebiegu nerwów (często VIII), dobrze odgraniczone, obszary
zagęszczone (Antoni A - palisady - ciałka Verocaya) i luźne (Antoni B), powiększone, hiperchromatyczne
jądra
nerwiakowłókniak /neurofibroma/ - skóra, tkanka podskórna, pnie nerwowe, kom. Schwanna i
fibroblasty
złośliwy nowotwór osłonek nerwów obwodowych MPNST - mięsak, często razem z
nerwiakowłókniakowatością typu I, guz trytoni (razem z rhabdomiosarcoma)
95

96
Document Outline
- CHOROBY UWARUNKOWANE GENETYCZNIE
- WRODZONE I DZIEDZICZNE CHOROBY KOŚCI
- CHOROBA PAGETA (osteitis deformans)
- NOWOTWORY KOŚCI
- Pokrzywka
- Ostre wysypkowe zapalenie skóry (wyprysk)
- Rumień wielopostaciowy
- Łuszczyca
- Liszaj płaski
- Pęcherzyca /pemphigus/
- Zmiany pęcherzycopodobne /pemphigoid bullosus/
- Opryszczkowe zapalenie skóry
- Rogowacenie łojotokowe /keratosis saborrheica/
- Rogowiak /keratoacanthoma/
- Brodawki (kłykciny)
- Rogowacenie popromienne /keratosis actinica/
- Rak kolczystokomórkowy /ca spinocellulare/
- Rak podstawnokomórkowy /ca basocellulare/
- Znamię barwnikowe
- Czerniak
- Obrzęk mózgu
- Wklinowanie /herniatio/
- Wodogłowie /hydrocephalia/
- Uogólniona encefalopatia niedotleniowo-niedokrwienna
- Zawały
- Pierwotny krwotok śródmózgowy
- Krwotok podpajęczynówkowy
- Wrodzone wady (malformacje) naczyń
- Krwiak nadtwardówkowy /hematoma epidurale/
- Krwiak podtwardówkowy / h. subdurale/
- Uszkodzenia śródmiąższowe
- Wady cewy nerwowej (z. dysrafii)
- Wady związane z wodogłowiem
- Zaburzenia przodomózgowia
- Uszkodzenia okołoporodowe
- Ostre ropne zapalnie opon /leptomeningitis acuta/
- Ostre limfocytarne (wirusowe) zapalenie opon
- Przewlekłe zapalenie opon
- Ropień mózgu /abscessus cerebri/
- Gruźlica i toksoplazmoza
- Wirusowe zapalenia mózgu
- Encefalopatie gąbczaste
- Gwiaździaki /astrocytomata/
- Skąpodrzewiak /oligodendroglioma/
- Wyściółczak /ependymoma/
- Stwardnienie rozsiane SM
- Inne
- Choroba Alzheimera
- Parkinsonizm
- ch. Huntingtona
- Stwrdnienie zanikowe boczne SLA
- niedobory
- Zaburznia metaboliczne i zatrucia
- Neuropatie obwodowe
- Nowotwory obwodowego układu nerwowego
Wyszukiwarka
Podobne podstrony:
patomorfologia opracowane pytania opisowe egzamin
ROZDZIAŁ 20 - Układ dokrewny, Medycyna, Patomorfologia, Opracowanie Robbins
Patomorfologia opracowany sylabus
Patomorfologia opracowanie uzupe nienie
patomorfologia opracowane pytania opisowe egzamin cz2
ROZDZIAŁ 23 - Oun, Medycyna, Patomorfologia, Opracowanie Robbins
Patomorfologia opracowanie 2
patomorfologia opracowane pytania opisowe egzamin
więcej podobnych podstron