Wydział Geologii, Geofizyki i Ochrony Środowiska
AkAdemii Górniczo–Hutniczej
Kraków | 2011
Ma
teriał
y do ćwiczeń
dl
A
student
ó
w
kieru
nku Ochr
O
na Śr
O
d
O
W
isk
a
tomasz Bajda
Maciej Manecki
Jakub Matusik
Grzegorz rzepa
cHemiA
Geo
[50]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
rozdział Piąty:
GeOchemia sTrefy hiperGenicznej
Strefa hipergeniczna obejmuje przypowierzchniowe środowiska ziemskie, w których dominującą rolę od‑
grywają procesy wietrzeniowe. Minerały odporne chemicznie ulegają przede wszystkim wietrzeniu fizycz‑
nemu, sprowadzającemu się głównie do rozdrabniania i ścierania ziarn. Jednak większość pierwotnych faz
obecnych w skałach magmowych i metamorficznych jest podatna na przemiany chemiczne, a zatem ulega
przeobrażeniom prowadzącym do powstania nowych substancji. Na charakter wietrzenia bardzo wyraźnie
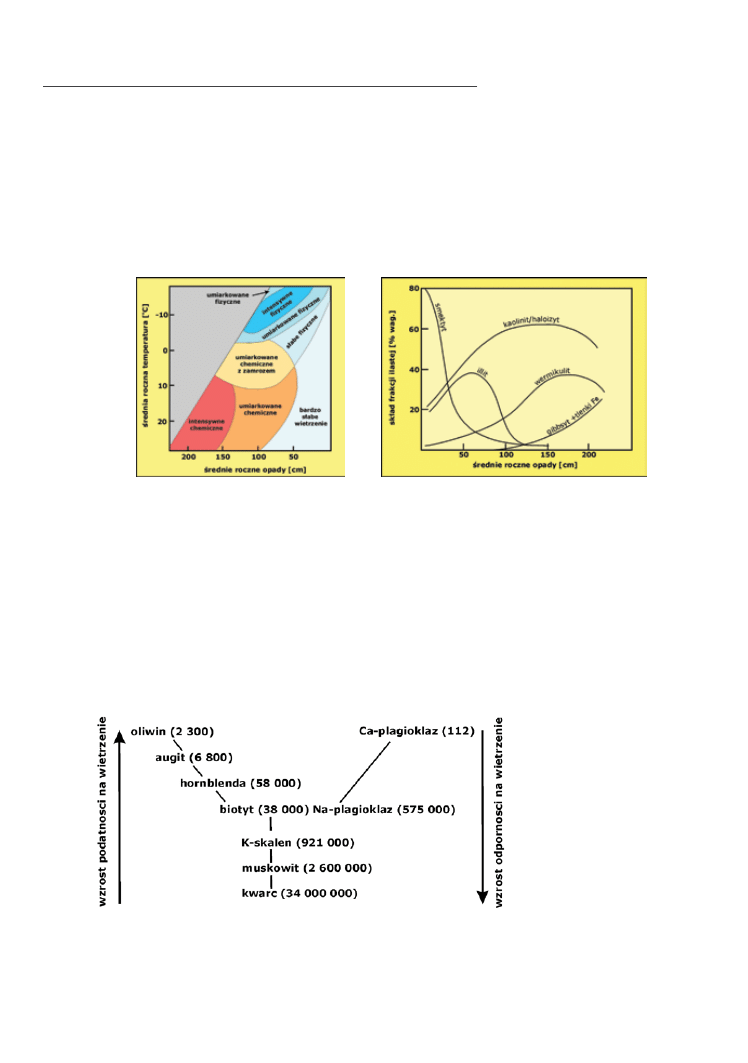
wpływają czynniki klimatyczne, przede wszystkim średnia temperatura i wielkość opadów (Fig. 5.1).
fig. 5.1a,b. Wpływ klimatu na charakter wietrzenia. Wietrzenie chemiczne zachodzi najintensywniej w strefach gorących i wilgot‑
nych (lewy rysunek). Niskie temperatury powstrzymują wietrzenie chemiczne, powodując wzrost znaczenia procesów
fizycznych (np. cykle zamarzania – rozmarzania). Typ klimatu wpływa również na skład mineralny zwietrzelin – np. na
rodzaj minerałów ilastych (prawy rysunek)
Wśród pospolitych pierwotnych minerałów skałotwórczych sekwencja wzrastającej podatności na wie‑
trzenie przypomina szereg krystalizacyjny Bowena (Fig. 5.2) – najłatwiej wietrzeją ciemne krzemiany (oliwi‑
ny, pirokseny, amfibole, biotyt) i plagioklazy bogate w wapń. Znacznie odporniejsze są skalenie alkaliczne,
muskowit i kwarc, dzięki czemu mają one szansę na nagromadzenie się w okruchowych skałach osado‑
wych. Oczywiście, substancje łatwo rozpuszczalne w wodzie, jak pospolite chlorki czy niektóre siarczany,
będą podlegały wietrzeniu chętniej niż najmniej nawet odporne krzemiany. Z drugiej strony, istnieje wiele
minerałów, które są stabilniejsze w strefie hipergenicznej niż kwarc.
Wietrzenie chemiczne to
szereg
skomplikowanych,
współdziałających ze sobą
procesów,
zachodzących
głównie na skutek działania
wody, powietrza i mikroorga‑
nizmów. Do najważniejszych
z nich należą: rozpuszczanie,
hydratacja, hydroliza, karbo‑
natyzacja, reakcje redoksowe
oraz procesy adsorpcyjne.
fig. 5.2. Podatność krzemianów na wietrzenie chemiczne naśladuje szereg Bowena. Ciemne
minerały i plagioklazy bogate w wapń rozkładają się najszybciej. Bardziej odporne są
skalenie alkaliczne, muskowit i kwarc. W nawiasach podana jest szacunkowa trwa‑
łość (w latach) kostki minerału o wielkości 1 mm (wg Goldicha 1938, zmienione)
[51]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
rozpuszczanie to przejście pierwiastków do roztworu bez zmiany składu chemicznego substancji rozpusz‑
czanej. Do roztworu przechodzą jony proste (np. Na
+
, K
+
, Ca
2+
, Cl
–
) lub złożone (np. SO
4
2–
, CO
3
2–
, HCO
3
–
).
Procesy rozpuszczania zależą przede wszystkim od natury substancji rozpuszczanej (głównie od charakteru
wiązań chemicznych), temperatury i ciśnienia oraz właściwości rozpuszczalnika (jest nim praktycznie zawsze
woda, ale może mieć bardzo różny skład). Woda jest rozpuszczalnikiem polarnym, więc oddziałuje elek‑
trycznie na cząsteczki minerałów. Jeżeli energia tego oddziaływania jest większa niż energia sieci krystalicz‑
nej, następuje odrywanie jonów z tej sieci i ich przejście do roztworu (z otoczkami hydratacyjnymi). Nie ma
wielu pospolitych minerałów, które byłyby łatwo rozpuszczalne – należą do nich niektóre chlorki (np. halit,
sylwin), azotany (np. nitronatryt) czy siarczany (np. heksahydryt, melanteryt, thenardyt, glauberyt). Miarą
ich podatności na ten proces jest rozpuszczalność, czyli maksymalna ilość substancji, jaka może znaleźć
się w danej ilości roztworu (jest to więc stężenie roztworu nasyconego). Wyraża się ją w g/100 g roztworu,
czasem w g/dm
3
lub w mol/dm
3
. Dla substancji słabo rozpuszczalnych, a takimi jest większość minerałów
skałotwórczych, podaje się zwykle iloczyn rozpuszczalności KSP, czyli iloczyn stężeń (aktywności) jonów
w roztworze nasyconym (pozostającym w równowadze z osadem). Iloczyn rozpuszczalności to nic innego,
jak stała równowagi dla reakcji rozpuszczania danego minerału. Im mniejszy jest iloczyn rozpuszczalności
minerału, tym jest on słabiej rozpuszczalny (Tabela 5.1). Warto zapamiętać, że często nierozpuszczalne lub
słabo rozpuszczalne związki tworzą kationy o dużym ładunku (+2, +3, +4) z anionami o wyższych ładun‑
kach (SO
4
2–
, CO
3
2–
, PO
4
3–
) oraz grupami hydroksylowymi. Praktycznie nierozpuszczalne są ponadto siarczki
pierwiastków chalkofilnych (Pb, Cd, Hg, Fe etc.).
Ponieważ rozpuszczaniu sprzyjają ruchy termiczne atomów (jonów) w sieci krystalicznej minerałów i w wo‑
dzie, dlatego rozpuszczalność wielu minerałów wzrasta wraz z temperaturą (ale nie wszystkich! – patrz np.
węglany). Przeciwna tendencja występuje w przypadku gazów – ich rozpuszczalność wydatnie spada przy
wzroście temperatury.
tabela 5.1. Wartości iloczynu rozpuszczalności (KSP) oraz wykładników iloczynu rozpuszczalności
(pKSP = –log KSP) dla wybranych minerałów
minerał
KSP
pKSP
halit NaCl
10
1,58
–1,58
anhydryt CaSO
4
10
–4,5
4,5
kalcyt CaCO
3
10
–8,35
8,3
baryt BaSO
4
10
–10,0
10,0
fluoryt CaF
2
10
–10,4
10,4
galena PbS
10
–27,5
27,5
hematyt Fe
2
O
3
10
–43,3
43,3
goethyt FeOOH
10
–44,0
44,0
fluoroapatyt Ca
5
(PO
4
)
3
F
10
–60,4
60,4
bizmutynit Bi
2
S
3
10
–96,8
96,8
hydratacja polega na przyłączaniu cząsteczek wody przez powierzchnię minerału. Jeżeli w jej wyniku utwo‑
rzone zostaną wiązania chemiczne pomiędzy H
2
O a minerałem, wówczas dochodzi do tworzenia hydratów:
CaSO
4
(anhydryt) + 2H
2
O ⇒ CaSO
4
· 2H
2
O (gips)
albo do dalszej hydratacji już istniejących:
MgSO
4
· 6H
2
O (heksahydryt) + H
2
O ⇒ MgSO
4
· 7H
2
O (epsomit)
Oczywiście, w warunkach niskiej wilgotności i przy – najczęściej – podwyższonych temperaturach, może
dochodzić do usuwania wody, czyli dehydratacji. Hydratacji ulegają również cząsteczki i jony występujące
w roztworach wodnych. To zjawisko zostanie przybliżone nieco dalej.
[52]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
hydroliza to reakcja rozkładu minerału pod wpływem zdysocjowanych cząsteczek wody, które w postaci
jonów H
3
O
+
i OH
–
są włączane do produktów reakcji. Hydroliza następuje przy działaniu wody na sole sła‑
bych kwasów i słabych zasad połączonych z dowolnymi zasadami i kwasami. Zwykle jeden z produktów hy‑
drolizy jest łatwo rozpuszczalny i zostaje wyługowany, drugi zaś, nierozpuszczalny, pozostaje jako minerał
wtórny. W przypadku wietrzenia glinokrzemianów częstym produktem reakcji są minerały ilaste (również
będące glinokrzemianami lub krzemianami glinu) lub wodorotlenki Al. Przebieg tych reakcji zależy m.in.
od klimatu (patrz Fig. 5.1b).
Mg
2
SiO
4
+ 4H
2
O ⇒ 2Mg
2+
+ 4OH
–
+ H
4
SiO
4
4KAlSi
3
O
8
+ 6H
2
O ⇒ Al
4
(OH)
8
Si
4
O
10
+ 8H
2
O + 4K
+
+ 4OH
–
(kaolinityzacja)
KAlSi
3
O
8
+ 2H
2
O ⇒ Al(OH)
3
+ 3SiO
2
+ K
+
+ OH
–
(laterytyzacja)
3KAlSi3O8 + 14H2O ⇒ KAl3Si3O 10(OH)2 + 6H4SiO4 + 2K
+
+ 2OH
–
(illityzacja)
Jak widać, w wyniku hydrolizy krzemianów powstaje kwas krzemowy lub koloidalny SiO2. Powyższe reakcje
nie uwzględniają jednakże obecności w wodzie rozpuszczonego CO2, który tworząc zdysocjowany kwas
węglowy modyfikuje przebieg procesu hydrolizy:
Mg
2
SiO
4
+ 4H
2
CO
3
⇒ 2Mg
2+
+ 4HCO
3
–
+ H
4
SiO
4
2NaAlSi
3
O
8
+ 2H
2
CO
3
+ 9H
2
O ⇒ 2Na
+
+ 4H
4
SiO
4
+ Al
2
Si
2
O
5
(OH)
4
+ 2HCO
3
–
Konsekwencją takiego przebiegu reakcji jest powstawanie jonów wodorowęglanowych, co jest jedną
z przyczyn ich dominacji w wodach śródlądowych. Taki proces, w którym na minerał działa dwutlenek
węgla lub kwas węglowy, nazywa się karbonatyzacją (uwęglanowieniem). Jest on zarazem jednym z głów‑
nych czynników regulujących globalny obieg węgla.
PRzyKłAD 1: Oblicz stężenie ołowiu w nasyconym roztworze PbSO
4
.
PbSO
4
⇔ Pb
2+
+ SO
4
2–
K = 10
–7,8
PbSO
4
jako czysta faza stała ma aktywność = 1
K = [Pb
2+
][SO
4
2–
] = 10
–7,8
= 1,58 · 10
–8
[Pb
2+
] = [SO
4
2–
] = x
x
2
= K
[Pb
2+
] = x = K
1/2
= 10
–3,9
[mol/dm
3
]
1 mol Pb = 207,2 g
[Pb
2+
] = 26,08 mg/dm
3
PRzyKłAD 2: Oblicz stężenie ołowiu w nasyconym roztworze CaSO4 i PbSO4.
CaSO
4
⇔ Ca
2+
+ SO
4
2–
K1 = 10
–4,5
PbSO
4
⇔ Pb
2+
+ SO
4
2–
K2 = 10
–7,8
[Ca
2+
] + [Pb
2+
] = [SO
4
2–
]
z porównania stałych równowagi K
1
i K
2
wynika, że CaSO
4
jest znacznie bardziej rozpuszczalny niż PbSO
4
,
zatem stężenie Ca
2+
w roztworze będzie znacznie większe niż stężenie Pb
2+
[Ca
2+
] >> [Pb
2+
], czyli [Ca
2+
] + [Pb
2+
] = [Ca
2+
]
[Ca
2+
] + [Pb
2+
] = [SO
4
2–
]
[Ca
2+
] = [SO
4
2–
] = x
[53]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
K
1
= [Ca
2+
][SO
4
2–
] = 10
–4,5
= 3,16 · 10
–5
x = [SO
4
2–
] = K1
1/2
= 10
–2,25
= 5,62 · 10
–3
mol/dm
3
otrzymaną wartość wstawiamy do równania na K
2
K
2
= [Pb
2+
][SO
4
2–
] = [Pb
2+
] · 5,62 · 10
–3
[Pb
2+
] · 5,62 · 10
–3
= 1,58 · 10
–8
[Pb
2+
] = 2,81 · 10
–6
mol/dm
3
[Pb
2+
] = 0,58 mg/dm
3
Widać, że w drugim przypadku stężenie (a zatem i rozpuszczalność) ołowiu jest niższe. Zatem obecność
CaSO
4
zmniejsza rozpuszczalność ołowiu. Zjawisko to nazywa się efektem wspólnego jonu.
Jeżeli minerał zawiera pierwiastki, które mogą występować na różnych stopniach utlenienia, wówczas dużą
rolę w jego wietrzeniu odgrywają procesy redoksowe (utleniania–redukcji). utlenianie to proces oddawa‑
nia przez atom (jon) elektronów, w wyniku czego podwyższa od swój stopień utlenienia. Przeciwnym pro‑
cesem jest redukcja, wiążąca się z przyłączaniem elektronów, a tym samym obniżaniem stopnia utlenienia.
Ponieważ procesy te są związane z transferem elektronów, zawsze zachodzą razem. Jon lub atom, który utle‑
nia się nazywany jest reduktorem, gdyż dostarcza do układu elektronów, które zostają „wyłapane” przez inny
atom (jon), umożliwiając tym samym jego redukcję. Przeciwnie – jon lub atom, który się redukuje nazywamy
utleniaczem, gdyż przyłączając elektrony umożliwia utlenianie innej substancji. Dla geochemii najważniejsze
są procesy redoksowe, którym podlegają żelazo, siarka i mangan (Tabela 5.2), a także węgiel i azot.
tabela 5.2. Najważniejsze stopnie utlenienia żelaza, manganu i siarki oraz ich przykładowe nośniki
pierwiastek
stopień utlenienia
przykłady substancji
Fe
0
żelazo rodzime
+ 2
fayalit Fe
2
SiO
4
, syderyt FeCO
3
, biotyt K(Mg,Fe)
3
[(OH)
2
AlSi
3
O
10
], piryt FeS
2
,
+ 3
hematyt Fe
2
O
3
, goethyt FeOOH, jarosyt KFe
3
[(OH)
6
|(SO
4
)
2
]
Mn
+ 2
rodochrozyt MnCO
3
, ciemne (zawierające Fe
2+
) krzemiany
+ 3
manganit MnOOH
+ 4
piroluzyt MnO
2
, birnessyt, todorokit, wernadyt
S
– 2
(– 1)
siarczki – np. pirotyn FeS, chalkopiryt CuFeS
2
, H
2
S
piryt FeS
2
0
siarka rodzima
+ 4
SO
2
+ 6
SO
3
, siarczany np. anhydryt CaSO
4
, ałunit KAl
3
[(OH)
6
|(SO
4
)
2
]
Jednym z powodów, dla których ciemne krzemiany ze skał magmowych tak łatwo ulegają wietrzeniu che‑
micznemu, jest fakt, że zawierają żelazo dwuwartościowe. Utlenianie Fe
2+
do Fe
3+
destabilizuje strukturę
minerału przyspieszając jego rozpad.
Fe2SiO4 + ½O2 + 3H2O ⇒ 2FeOOH + H4SiO4
1. produKty wIetrzenIa
W wyniku działania powyższych procesów powstają trzy główne rodzaje produktów wietrzenia:
1. odporne minerały pierwotne, ulegające praktycznie tylko ścieraniu i kruszeniu. Ze strefy wietrze‑
nia są wynoszone (głównie przez wodę, w mniejszym stopniu wskutek działania wiatru i grawitacji)
[54]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
w formie okruchów. Taki transport wymaga dużej siły nośnej, jest więc zwykle stosunkowo mało
efektywny i działa na względnie małe odległości. Do minerałów takich należą najodporniejsze pier‑
wotne krzemiany skałotwórcze (kwarc i muskowit) oraz wiele tzw. minerałów ciężkich – np. złoto
rodzime, platynowce, kasyteryt SnO
2
, cyrkon ZrSiO
4
, chromit FeCr
2
O
4
, magnetyt Fe
3
O
4
, rutyl TiO
2
,
diament, wolframit (Fe, Mn)WO
4
, granaty, ilmenit FeTiO
3
, turmaliny etc.
2. Nierozpuszczalne w wodzie minerały wtórne, będące wynikiem procesów rozpuszczania niekon-
gruentnego (takiego, w którym powstają fazy nierozpuszczalne), przede wszystkim produkty hy‑
drolizy krzemianów – minerały ilaste, amorficzna krzemionka, tlenki i wodorotlenki Fe, Mn i Al.
Wszystkie te substancje są bardzo drobnoziarniste, co powoduje, że w wodzie tworzą trudno sedy‑
mentującą zawiesinę. Pełnią one ważną rolę w transporcie wielu produktów wietrzenia, gdyż cha‑
rakteryzują się dużymi powierzchniami właściwymi (do kilkuset m
2
/g) z licznymi centrami aktywny-
mi, na których dochodzi do wiązania wielu kationów i anionów obecnych w wodzie.
tabela 5.3. Uproszczone oraz uwzględniające hydratację formy zapisujonów w roztworach wodnych
uproszczony zapis
rzeczywisty jon
Al
3+
Al(H
2
O)
6
3+
Al(OH)
2
+
Al(H
2
O)
5
(OH)
2
+
Cu
2+
Cu(H
2
O)
6
+
Zn
2+
Zn(H
2
O)
4
2+
H
+
H(H
2
O)
n
+
3. rozpuszczalne i lotne produkty wietrzenia. Należą tu jony proste (głównie litowce, berylow‑
ce i chlorowce: Na
+
, K
+
, Ca
2+
, Mg
2+
, Cl
–
, F
–
etc.) i złożone (np. SO
4
2–
, HCO
3
–
, PO
4
3–
, HPO
4
2–
, NO
3
–
),
kompleksy organiczne i nieorganiczne oraz gazy (np. CO
2
, SO
2
, H
2
S). Praktycznie wszystkie jony
występujące w wodzie ulegają solwatacji (hydratacji), czyli przyłączają dipole wody (Tabela 5.3).
O przypuszczalnej formie otoczki hydratacyjnej wiele może powiedzieć wielkość potencjału jo-
nowego (stosunek ładunku jonu do jego promienia). Kationy o niskich potencjałach (duże kationy
o małych ładunkach – np. K
+
, Na
+
, Li
+
, Rb
+
, Ag
+
, Ba
2+
) występują w formie prostych jonów z niewiel‑
ką zwykle otoczką hydratacyjną. Wokół kationów o większych potencjałach pole elektryczne jest
silniejsze – silne przyciąganie może spowodować, że jeden z wodorów dipolu wody oddzieli się.
Kationy zostają więc otoczone grupami hydroksylowymi, tworząc wodorotlenki, które mogą poli‑
meryzować i ulegać wytrącaniu. Jeszcze wyższe wartości potencjału jonowego powodują całkowite
rozbicie dipoli wody, czego efektem jest otoczenie kationu tlenami – powstają złożone aniony, jak
SO
4
2–
, PO
4
3–
, WO
4
2–
, CrO
4
2–
etc.
2. ph–eh
Najważniejszymi i najczęściej wykorzystywanymi parametrami opisującymi warunki panujące w strefie
wietrzenia są pH (wykładnik stężenia jonów wodorowych) i Eh (potencjał redoks).
Woda jest słabym elektrolitem ulegającym procesowi autojonizacji. Dysocjuje wg następującego równania:
H
2
O ⇔ H
+
+ OH
–
Z zapisu tego równania wynika, że stężenia jonów wodorotlenowych i wodorowych są sobie równe. Do‑
świadczalnie wykazano, że wynoszą one w temperaturze pokojowej 10
–7
mol/dm
3
. Zatem, w rezultacie
niskiej wartości iloczynu jonowego wody przynajmniej jedno ze stężeń [H
+
] lub [OH
–
] wyraża się bardzo

[55]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
małą liczbą. W celu ułatwienia posługiwania się tak małymi wartościami wprowadzono tzw. wykładnik
stężenia jonów wodorowych ph.
pH = –log[H
+
]
W roztworach bardziej stężonych, gdy nie można pominąć oddziaływań międzyjonowych, stosuje się bar‑
dziej ogólną zależność:
pH = –log aH+
w czystej chemicznie wodzie [H
+
] = [OH
–
] = 10
–7
M, czyli pH = 7
w środowisku kwaśnym [H
+
] >[OH
–
] czyli pH < 7
w środowisku zasadowym [H
+
] < [OH
–
] czyli pH > 7
Skala wartości pH jest skalą logarytmiczną, co oznacza, że np. spadek pH o jednostkę odzwierciedla wzrost
stężenia jonów wodorowych o rząd wielkości. Czyli w roztworze o pH = 5,0 jest 100 razy więcej jonów wo‑
dorowych niż w roztworze obojętnym, o pH = 7,0.
Zapisane powyżej równanie dysocjacji wody sugeruje, że tworzy się w jej wyniku jon wodorowy, czyli pro‑
ton. Jednak wobec faktu, że wytwarza on wokół siebie pole elektryczne o wysokim natężeniu, musi on
ulegać hydratacji. Natężenie pola elektrycznego wokół protonu, którego promień jest o około pięć rzędów
wielkości mniejszy niż promień przeciętnego jonu, jest około 10 rzędów większe. Jest całkowicie niepraw‑
dopodobne występowanie wolnego protonu w roztworze wodnym. Dlatego uważa się, że proces dysocja‑
cji wody można traktować jako przeniesienie protonu między dwiema cząsteczkami wody:
H
2
O + H
2
O → H
3
O
+
+ OH
–
Jon H
3
O
+
nosi nazwę jonu hydroniowego, który traktujemy jako proton związany koordynacyjnie z czą‑
steczką wody. Tak więc bardziej prawidłowe jest wyrażanie pH przy pomocy wzoru:
pH = –log[H
3
O
+
] lub pH = –log a
H
3
O
+
To wyrażenie jest powszechnie używane, chociaż wiadomo, że jon hydroniowy ulega dalszej bardzo silnej
hydratacji przyłączając 3 cząsteczki wody i tworząc kompleks:
[H
3
O]
+
(H
2
O)
3
lub [H(H
2
O)
4
]
+
W rzeczywistości ten trwały kompleks posiada jeszcze labilną otoczkę utworzoną z cząsteczek wody.
PRzyKłAD 3: Oblicz pH 0,01 molowego roztworu HNO
3
.
Kwas azotowy(V) jest mocnym elektrolitem, więc całkowicie dysocjuje na jony:
HNO
3
⇔ H
+
+ NO
3
–
z tego powodu [H
+
] = [HNO
3
] = 0,01 mol/dm
3
z definicji pH = –log[H
+
] = –log (0,01) = 2

[56]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
PRzyKłAD 4: Oblicz rozpuszczalność Pb(OH)
2
przy pH = 2,0, pH = 7,0 i pH = 9,0.
Pb(OH)
2
⇔ Pb
2+
+ 2OH
–
K = [Pb
2+
][OH
–
]
2
= 5,01 · 10
–16
przy pH = 2,0 stężenie jonów wodorowych wynosi 10
–2
mol/dm
3
, zatem [OH
–
] = 10
–12
mol/dm
3
[Pb
2+
] = K/[OH
–
] = (5,01 · 10
–16
)/(10
–12
)
2
= 5,01 · 10
8
mol/dm
3
co oznacza, że w tych warunkach Pb(OH)2 jest w całości rozpuszczony
przy pH = 7,0 stężenie jonów wodorowych wynosi 10
–7
mol/dm
3
, zatem [OH
–
] = 10
–7
mol/dm
3
[Pb
2+
] = K/[OH
–
] = (5,01 · 10
–16
)/(10
–7
)
2
= 5,01 · 10
–2
mol/dm
3
= 10381 mg/dm
3
czyli Pb(OH)
2
w tych warunkach jest znacznie słabiej rozpuszczalny niż przy pH = 2,0
przy pH = 9,0 stężenie jonów wodorowych wynosi 10
–9
mol/dm
3
, zatem [OH
–
] = 10
–5
mol/dm
3
[Pb
2+
] = K/[OH
–
] = (5,01 · 10
–16
)/(10
–5
)
2
= 5,01 · 10
–6
mol/dm
3
= 1,04 mg/dm
3
czyli Pb(OH)
2
w tych warunkach jest jeszcze słabiej rozpuszczalny
Z punktu widzenia geochemii istotny jest fakt, że różne metale wytrącają się z wody (w postaci wodoro‑
tlenków) przy różnych wartościach pH. Generalnie wysokie wartości pH (a więc przewaga OH
–
) sprzyjają
procesom wytrącania, niskie wartości pH (a więc przewaga H
+
) sprzyjają utrzymywaniu się większości pier‑
wiastków w roztworze. Kwaśne wody są zwykle bardziej agresywne i rozpuszczają więcej minerałów. Jednym
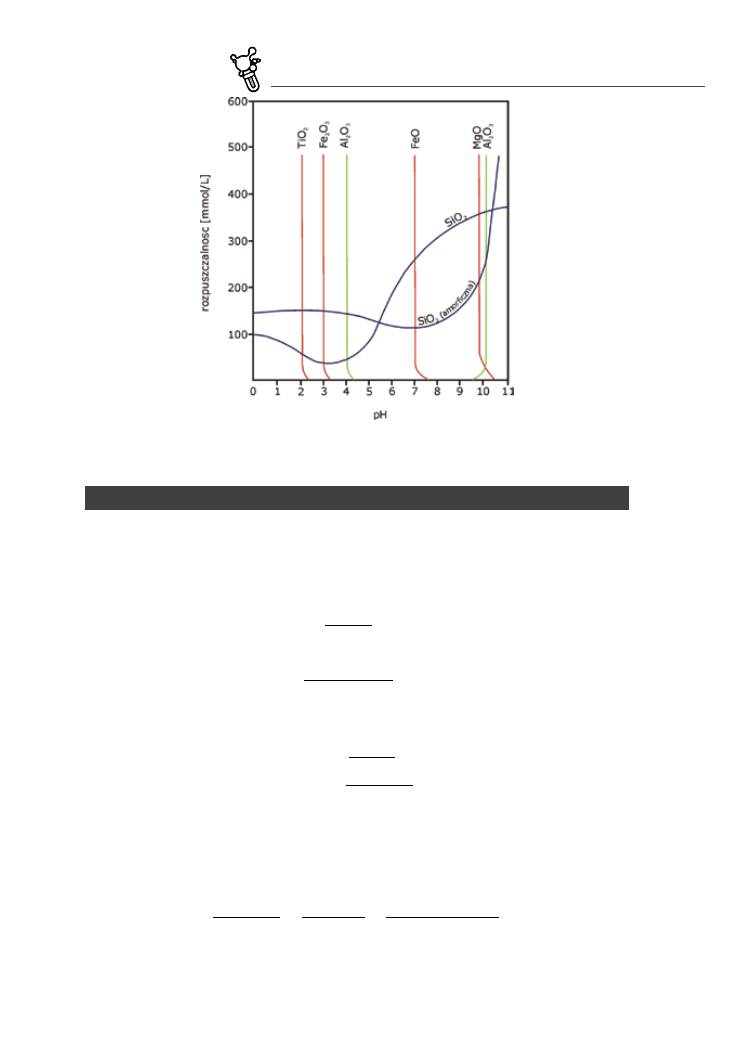
z wyjątków jest rozpuszczalność SiO2, która rośnie ze wzrostem pH (dlatego nie można przechowywać roz‑
tworów mocnych zasad w naczyniach szklanych) (Fig. 5.3). Tłumaczy to powstawanie laterytów i boksytów.
KAlSi
3
O
8
+ 2H
2
O ⇒ Al(OH)
3
+ 3SiO
2
+ KOH
NaAlSi
3
O
8
+ 2H
2
O ⇒ Al(OH)
3
+ 3SiO
2
+ NaOH
Powstające ługi (KOH i NaOH) bardzo silnie alkalizują środowisko. W takich warunkach krzemionka jest
rozpuszczana i odprowadzana ze strefy wietrzenia, natomiast wodorotlenki glinu (i żelaza) pozostają na
miejscu, gdyż zasadowy odczyn sprzyja ich wytrącaniu.
Ponieważ większość minerałów to sole słabych kwasów i mocnych zasad, ich reakcja z wodą powoduje
wzrost pH (powstają jony OH
–
, patrz reakcje powyżej). Jednak reakcja niektórych minerałów (będących
solami mocnych kwasów i słabych zasad) z wodą powoduje spadek pH:
KFe
3
(SO
4
)
2
(OH)
6
+ 3H
2
O ⇒ K
+
+ 3Fe(OH)
3
+ 2SO
4
2–
+ 3H
+
Spadek pH może być również związany z utleniającym wietrzeniem pierwotnych minerałów zawierają‑
cych Fe
2+
:
Fe
2+
+ ½O
2
+ 1½H
2
O ⇒ FeOOH + H
+
[57]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
fig. 5.3. Rozpuszczalność różnych tlenków w zależności od pH. W większości przypadków wzrost pH zmniejsza rozpuszczalność.
Jednym z wyjątków jest SiO2 – stąd jego odprowadzanie ze strefy wietrzenia laterytowego (wg Rosler, Lange 1979)
PRzyKłAD 5: Oblicz pH czystej wody, znajdującej się w równowadze z powietrzem.
Na odczyn wody, będącej w równowadze z powietrzem wpływ ma rozpuszczanie atmosferycznego CO
2
,
tworzącego z wodą słabo zdysocjowany kwas węglowy.
CO
2(g)
+ H
2
O ⇔ H
2
CO
3
(1)
H
2
CO
3
⇔ H
+
+ HCO
3
–
(2)
K
1
=
[H
2
CO
3
]
= 10
–1,47
P
CO
2
K
2
=
[H
+
][HCO
3
–
]
= 10
–6,35
[H
2
CO
3
]
z równania (2) wynika, że [H
+
] = [HCO
3
–
], zatem:
K
2
=
[H
+
]
2
[H
2
CO
3
]
[H
+
] = √ K
2
· [H
2
CO
3
]
[H
2
CO
3
] wyliczamy, przekształcając równanie (1) i wstawiając PCO
2
= 10
–3,5
bar (stężenie CO
2
w powietrzu
atmosferycznym, wyrażone jako ciśnienie parcjalne):
[H
2
CO
3
] = K1 · PCO
2
= 10
–1,47
· 10
–3,5
[H
+
] = √ K
2
· [H
2
CO
3
] = √ K
2
· K
1
· P
CO
2
= √ 10
–6,35
· 10
–1,47
· 10
–3,5
= 10
–5,66
ph = – log[h
+
] = 5,66
[58]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
Z przykładu tego wynika, że naturalne czyste wody (np. deszczówka) powinny być lekko kwaśne.
Również pozostawiona w laboratorium woda destylowana po krótkim czasie staje się kwaśna (bar‑
dziej niż woda wodociągowa, gdyż pozbawiona jest działających buforująco kationów Ca
2+
, Mg
2+
i al‑
kaliów). W śródlądowych wodach powierzchniowych i podziemnych CO
2
pochodzi przede wszyst‑
kim z procesów rozkładu substancji organicznej, a także obecności innych substancji, głównie kwa‑
sów organicznych.
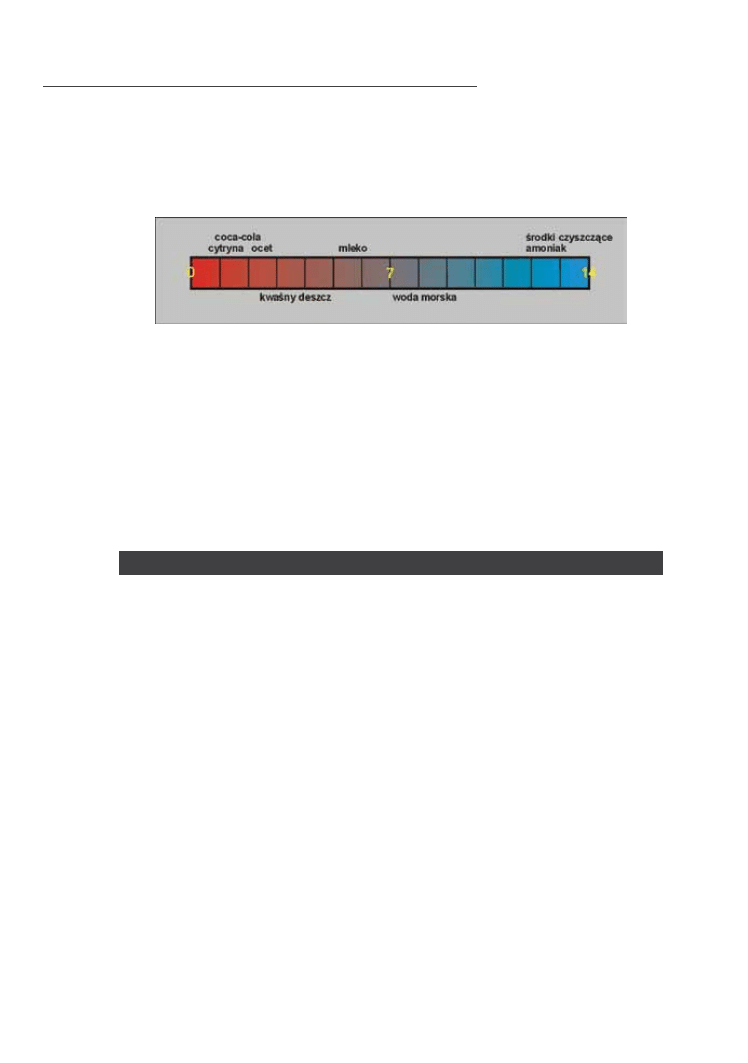
fig. 5.4. Typowe wartości pH substancji spotykanych w życiu codziennym
W codziennym życiu spotykamy się z substancjami o bardzo różnych wartościach pH. Do najbardziej kwa‑
śnych należą kwas w akumulatorach samochodowych, ocet czy sok z cytryny. Z kolei silnie zasadowe są
najczęściej środki czystości stosowane w gospodarstwie domowym (Fig. 5.4).
Dopuszczalne zakres pH dla wody pitnej wynosi 6,5–8,5, większość „normalnych” wód charakteryzuje się
odczynem 4–9. Zdarzają się jednak sytuacje, gdy pH przyjmuje ekstremalnie niskie lub wysokie (rzadziej,
np. silnie zasadowe wody słonych jezior na suchych obszarach) wartości, a czasem wręcz „wychodzi” poza
zwykły zakres 0–14. Typowym przykładem są środowiska kwaśnych wód kopalnianych – amd (acid
mine drainage).
PRzyKłAD 6: Oblicz pH wynikające z reakcji 0,1 g pirytu z 1 dm
3
wody.
FeS
2
+ 7/2O
2
+ H
2
O ⇒ Fe
2+
+ 2SO
4
2–
+ 2H
+
utlenianie 1 mola FeS
2
generuje 2 mole H
+
, zatem:
119,85 g FeS
2
– 2 mole H
+
0,1 g FeS
2
– x moli H
+
x = 1,67 · 10
–3
mol ⇒ pH = –log [H
+
] = 2,78
Minerały siarczkowe, chociaż są praktycznie nierozpuszczalne w wodzie, bardzo łatwo wietrzeją, gdyż pod
wpływem tlenu (lub innych utleniaczy) rozkładają się. Siarka z formy siarczkowej przechodzi w siarczano‑
wą, a minerały siarczanowe są znacznie łatwiej rozpuszczalne. Proces utleniania siarczków jest problemem
np. przy składowaniu na powietrzu węgla zawierającego piryt – utlenianie FeS
2
jest reakcją egzotermiczną,
która może spowodować samozapłon węgla na hałdzie. Do podobnej sytuacji dochodzi, gdy złoża siarcz‑
kowych kruszców zostaną odsłonięte wskutek eksploatacji. Dostęp tlenu atmosferycznego powoduje za‑
chodzenie reakcji, które w uproszczeniu można zapisać następująco:
2FeS
2
+ 2H
2
O + 7O
2
⇒ 2FeSO
4
+ 2H
2
SO
4
4FeSO
4
+ 6H
2
O + O
2
⇒ 4FeOOH + 4H
2
SO
4
Wskutek tego przepływające wody drenujące rejon wietrzenia siarczków są często mniej lub bardziej stę‑
żonymi roztworami kwasu siarkowego. To właśnie w takim środowisku zanotowano najniższe pH natu‑
ralnych wód wynoszące (sic!) –3,6 (kopalnia Richmond, północna Kalifornia, USA). W większości miejsc

[59]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
występowania zjawisk typu AMD pH wód jest
znacznie wyższe (zwykle 2–4), ale i tak zbyt niskie
dla normalnego funkcjonowania flory i fauny.
Środowiska tego rodzaju noszą nazwę acydotro‑
ficznych, z kwaśnolubnymi (acydofilnymi) orga‑
nizmami. Szczególnie charakterystyczne są tutaj
bakterie żelaziste, np. Acidithiobacillus ferrooxi-
dans, które czerpią energię z procesów utleniania
żelaza i siarki. Napędzają one bieg procesów re‑
doksowych, gdyż np. utlenianie Fe
2+
przy niskich
wartościach pH zachodzi bardzo wolno, a udział
bakterii przyspiesza je o czynnik rzędu 10
6
razy.
fig. 5.5. Zalane wyrobisko w starej kopalni pirytu w Wieściszowi‑
cach. Wypełniająca je woda jest bardzo kwaśna (pH < 3),
bogata w żelazo (do około 700 mg/L), siarczany (nawet
ponad 3500 mg/L), mangan (do 10 mg/L), glin (do 90
mg/L) i niektóre metale ciężkie (np. Cu, Ni, Co) (fot.
Bożena Gołębiowska)
fig. 5.6a–d. W rejonie Łęknicy środowisko AMD związane jest z zaniechaną eksploatacją węgla brunatnego zawierającego piryt. Był
on tutaj wydobywany w kopalniach odkrywkowych i podziemnych. Osiadanie terenu nad starymi wyrobiskami spowodo‑
wało zalanie kwaśną wodą (pH 2–4) sporych obszarów i degradację lasów (widoczne kikuty drzew). Żółty kożuch widocz‑
ny na powierzchni wody (prawa górna fotografia) to kolonie bakterii Acidithiobacillus ferrooxidans. Duże stężenia żelaza
skutkują powstawaniem charakterystycznych filmów na powierzchni wody (dolna lewa fotografia). Grunty występujące
na tym terenie są tak kwaśne, że miejscami niemal zupełnie pozbawione roślinności. Skutkuje to nasiloną erozją (dolna
prawa fotografia) (fot. Grzegorz Rzepa)

[60]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
W Polsce tego typu kwaśne środowiska występują np. na Dolnym Śląsku – w Radzimowicach i Wieściszowi‑
cach, w Górach Świętokrzyskich (Wiśniówka Mała) i w rejonie Łuku Mużakowa na granicy polsko–niemieckiej.
Kwaśne środowisko ułatwia wietrzenie otaczających skał, w związku z czym wody środowisk AMD są bar‑
dzo silnie zmineralizowane. Mineralizacja tych wód (czyli suma rozpuszczonych składników stałych) może
sięgać kilkudziesięciu czy nawet kilkuset gramów w litrze (dla porównania woda wodociągowa charaktery‑
zuje się zwykle mineralizacją rzędu stu kilkudziesięciu do kilkuset mg/dm
3
, a butelkowane wody mineralne
rzędu co najwyżej kilku gramów w litrze). Co gorsza, poza typowymi kationami występującymi w „nor‑
malnych” wodach (Ca
2+
, Mg
2+
, Na
+
, K
+
) pojawiają się w bardzo dużych stężeniach jony tzw. metali ciężkich
(np. Fe
2+
, Pb
2+
, Mn
2+
, Cu
2+
, Cd
2+
, Hg
2+
) i innych toksycznych pierwiastków (np. Al
3+
, Zn
2+
, As
3+
, As
5+
etc.). To
kolejny powód, dla którego zjawiska AMD są ważnym problemem środowiskowym.
Ilościową miarą warunków reakcji redoksowych jest potencjał oksydacyjno–redukcyjny (eh). Pozwala
on na stwierdzenie, czy dana reakcja będzie przebiegała w kierunku utleniania, czy redukcji. Podaje się go
w woltach lub miliwoltach w stosunku do wzorcowego potencjału reakcji:
2H
+
+ 2e
–
⇔ H
2
który w normalnych warunkach (t = 25°C, p = 1 atm., a = 1) jest przyjmowany jako 0,0 V. Potencjały innych
reakcji ustala się eksperymentalnie (Tabela 5.4). Gdy dotyczą warunków normalnych, oznacza się je jako E
0
,
dla dowolnych innych, potencjał redoks określany jest zależnością:
E
h
= E
0
–
R·T
ln
[red]
n·F
[utl]
gdzie:
R – stała gazowa
T – temperatura [K]
n – ilość elektronów biorących udział w reakcji
F – liczba Faradaya
[red] i [utl] – aktywności formy utlenionej i zredukowanej
tabela 5.4. Przykładowe potencjały redoks różnych reakcji
reakcja
E
0
[V]
Au
+
+ e
–
⇒ Au
+ 1,68
Pt
2+
+ 2e
–
⇒ Pt
+ 1,20
Ag
+
+ e
–
⇒ Ag
+ 0,80
Cu
+
+ e
–
⇒ Cu
+ 0,52
2H
+
+ 2e
–
⇒ H2
0,00
Fe
2+
+ 2e
–
⇒ Fe
– 0,44
Mg
2+
+ 2e
–
⇒ Mg
– 2,37
Mn
3+
+ e
–
⇔ Mn
2+
+ 1,51
MnO2 + 4H
+
+ 2e
–
⇔ Mn
2+
+ 2H2O
+ 1,28
O2 + 4H
+
+ 4e
–
⇔ 2H2O
+ 1,23
Fe
3+
+ e
–
⇔ Fe
2+
+ 0,77
Cu
2+
+ e
–
⇔ Cu
+
+ 0,15
Mn(OH)3 + e
–
⇔ Mn(OH)2 + OH
–
+ 0,10
Fe(OH)2 + 2H
+
+ 2e
–
⇔ Fe + 2H2O
– 0,05
[61]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
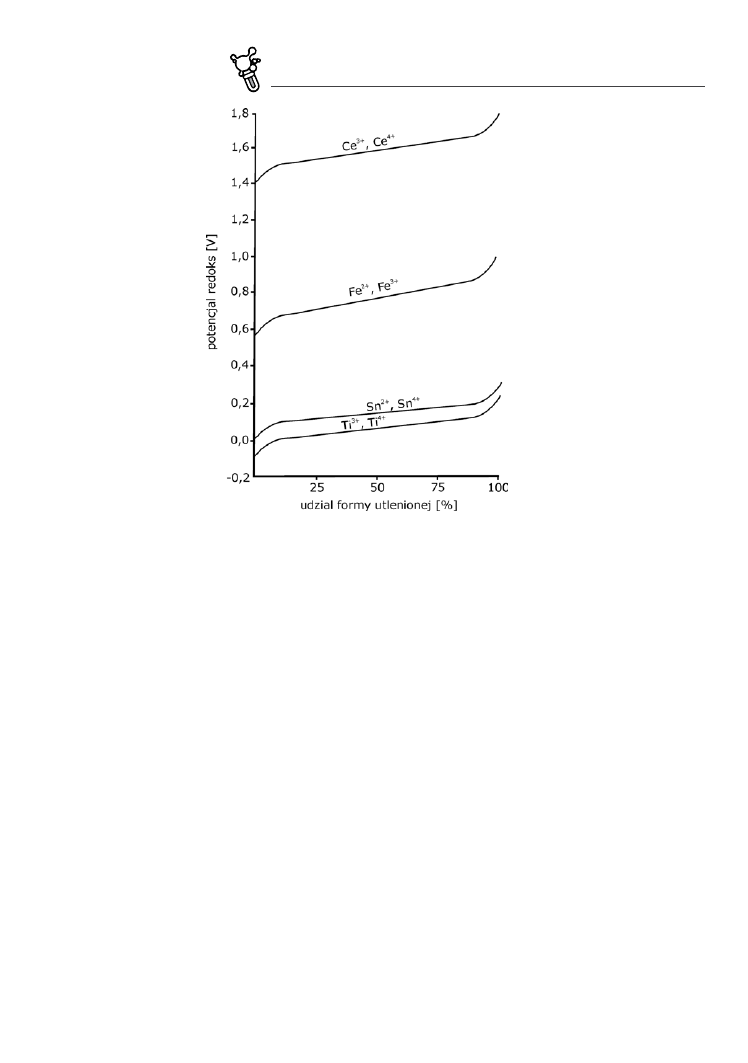
fig. 5.7. Wpływ wielkości potencjału redoks na proporcje pomiędzy zredukowaną a utlenioną formą kilku przykładowych pier‑
wiastków (Polański, Smulikowski 1969)
Patrząc na to równanie i powyższą tabelkę warto zapamiętać kilka ogólnych zależności:
– gdy [red] = [utl], wówczas Eh = E
0
(ln 1 = 0);
– wysokie wartości Eh sprzyjają utlenianiu, niskie sprzyjają redukcji;
– wyższe wartości E
0
danej reakcji oznaczają wyższe wartości Eh potrzebne do utlenienia (czyli środo‑
wisko musi być bardziej utleniające), tak więc w przyrodzie łatwiej przebiegają reakcje charaktery‑
zujące się niższymi E
0
. Dobrym przykładem ilustrującym tą prawidłowość jest oddzielenie żelaza od
manganu w strefie wietrzenia. Gdy migrujące w wodzie Fe
2+
i Mn
2+
dostają się w strefę oddziaływa‑
nia warunków utleniających (np. przy wypływie zmineralizowanej wody źródlanej na powierzch‑
nię), jako pierwsze będą się wytrącać związki Fe
3+
, gdyż potencjał redoks utleniania Fe
2+
do Fe
3+
jest
mniej więcej dwukrotnie niższy niż dla reakcji utleniania Mn
2+
do Mn
3+
. Konsekwencją tego może
być dalsza „samotna” migracja manganu do strefy gdzie będą panować silniej utleniające warunki;
– w przypadku metali, im wyższy jest E
0
jego jonizacji, tym metal jest bardziej „szlachetny” (Au > Pt
> Ag > Cu > Fe), czyli tym trudniej go utlenić. W skałach znajdujących się na powierzchni Ziemi
znajdujemy rodzime złoto, platynę i srebro, znacznie rzadziej miedź, natomiast występowanie np.
magnezu w formie metalicznej jest niemożliwe;
– pierwiastki o niższych E
0
wypierają pierwiastki o wyższych E
0
z roztworów ich soli, np.:
CuSO
4
+ Fe ⇒ FeSO
4
+ Cu
Bardzo istotny geochemicznie jest fakt silnej zależności potencjału redoks od pH. Np. w przypadku reakcji
utleniania żelaza potencjał redoks w środowisku silnie kwaśnym (pH < 3) wynosi około +0,77 V, a w warun‑
kach silnie alkalicznych (pH = 14) spada do około –0,56 V. Zatem, żeby utlenić obecne w roztworze Fe
2+
do
Fe
3+
można
zwiększyć eh
lub
podwyższyć ph
(Fig. 5.8):
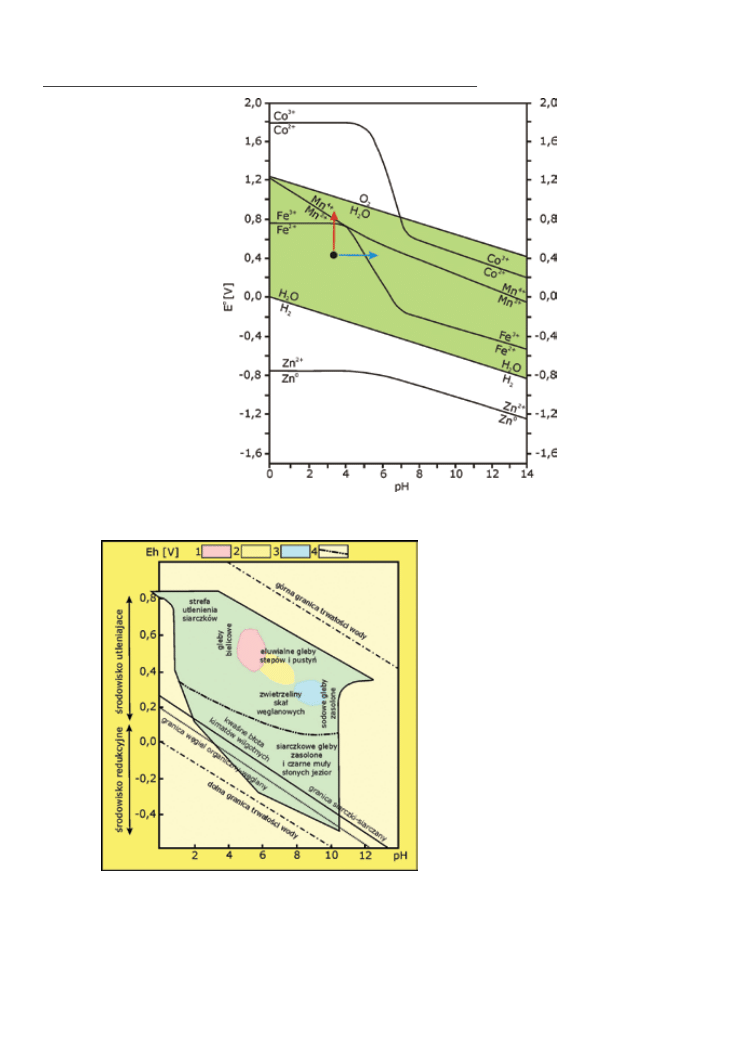
[62]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
fig. 5.8. Diagram pH–Eh dla wybranych par jonów (Polański, Smulikowski 1969). Każda linia oddziela pole trwałości utlenionej
(powyżej linii) i zredukowanej (poniżej) formy pierwiastka
Diagram zmienności pH–Eh w wybra‑
nych środowiskach na powierzchni Ziemi
przedstawiono na Fig. 5.9. Zwróć uwagę,
że w przyrodzie nie ma możliwości wystę‑
powania wszystkich kombinacji pH i Eh,
np. wykluczone są środowiska bardzo sil‑
nie redukcyjne i jednocześnie bardzo kwa‑
śne oraz bardzo silnie utleniające i jedno‑
cześnie zasadowe. Wynika to z trwałości
wody, która w tak skrajnych warunkach
ulega rozkładowi.
fig. 5.9. Zakres zmienności pH i Eh w środowiskach hipergenicznych (na
podstawie Perelmana 1966 i Brookinsa 1988). Pokolorowane typo‑
we wartości tych pH i Eh dla wód 1 – deszczowych, 2 – rzecznych,
3 – oceanicznych. Przerywana linia (4) oddziela warunki utlenia‑
jące od redukcyjnych. Powyżej linii „granica siarczki–siarczany”
siarka jest trwała w formie jonów SO4
2–
, poniżej jako H2S lub HS
–
.
Analogiczna zasada panuje dla granicy węgiel organiczny–węglany
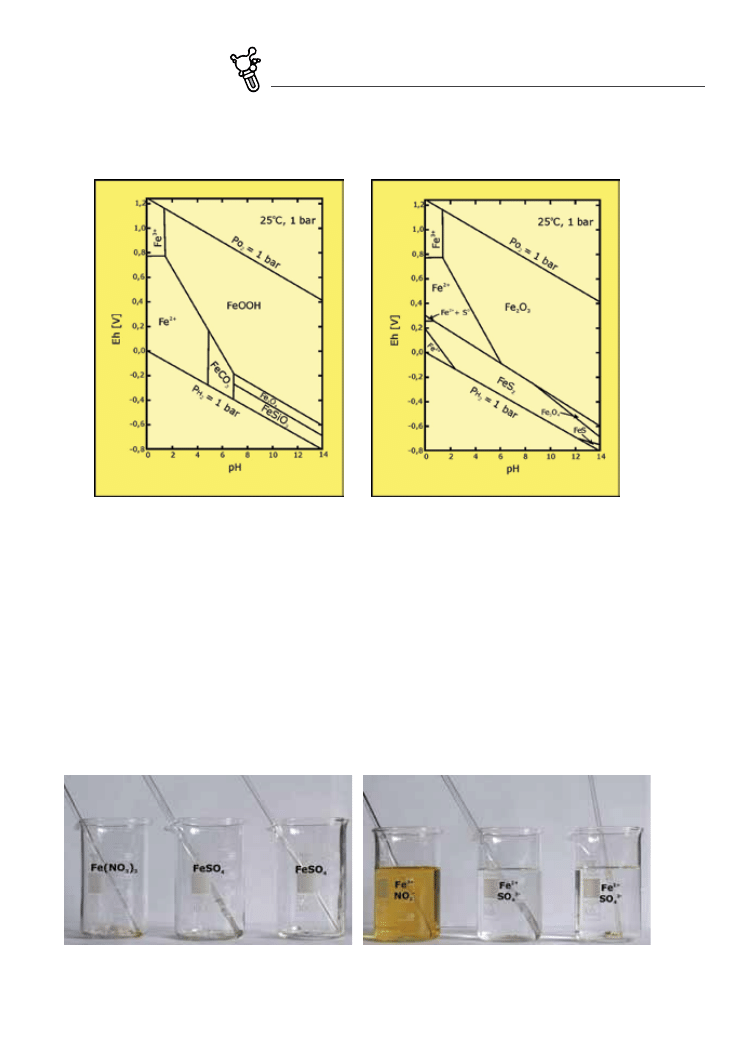
[63]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
Tak więc do podanych wyżej prawidłowości dochodzą jeszcze dwie dodatkowe:
– wyższe pH sprzyja reakcjom utleniania;
– w przyrodzie nie są możliwe wszystkie kombinacje Eh–pH. Zakres dopuszczalnych warunków jest
ograniczony trwałością wody.
fig. 5.10a,b. Diagramy pH–Eh dla żelaza w układach Fe–C–Si–O–H (po lewej) oraz Fe–S–O–H (po prawej. Przyjęto następujące
aktywności rozpuszczonych specjacji: Fe = 10
–6
, Si = 10
–3
, C = 10
–3
, S = 10
–3
(wg Brookinsa 1988)
Diagramy pH–Eh , ilustrujące zachowanie w środowisku wodnym różnych pierwiastków i ich związków, są
bardzo użytecznym narzędziem, powszechnie stosowanym w geochemii. Pozwalają przewidzieć formy wy‑
stępowania (specjacje) danego pierwiastka w roztworze w różnych warunkach środowiska. Z drugiej stro‑
ny, umiejętność odczytywania tych diagramów ułatwia wnioskowanie o warunkach paleośrodowiska na
podstawie składu mineralnego skał, o których wiadomo, że powstały w środowisku wodnym. Warto przy
tym zdawać sobie sprawę, że w trakcie konstruowania takiego diagramu bierze się pod uwagę nie tylko
samą zmienność pH i Eh, ale również ciśnienie, temperaturę, spodziewane stężenia (aktywności) związków
danego pierwiastka oraz obecność innych substancji w układzie (Fig. 5.10). Z tego powodu wykreślone dla
tego samego pierwiastka diagramy mogą się różnić zależnie od przyjętych warunków.
Na koniec, zachowanie żelaza w strefie hipergenicznej można zilustrować wykonując na ćwiczeniach
prosty eksperyment:
fig. 5.11 a–b. Objaśnienia w tekście (fot. Grzegorz Rzepa)

[64]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
fig. 5.11 c–d.
Objaśnienia w tekście (fot. Grzegorz Rzepa)
1. Do trzech zlewek (Fig. 5.11a) wsypujemy po kilkaset miligramów soli żelaza – azotanu(V) żelaza(III)
oraz siarczanu(VI) żelaza(II).
2. Wlewamy wodę destylowaną. Związki umieszczone w zlewkach są łatwo rozpuszczalne, więc po
chwili w całości przechodzą do roztworu (Fig. 5.11b). Zauważmy, że sole żelaza(III) znacznie inten‑
sywniej barwią roztwór niż ma to miejsce w przypadku żelaza(II). Odmienne jest też samo zabar‑
wienie – żółte lub pomarańczowe dla Fe(III) i bladozielonkawe lub słomkowe dla Fe(II). Ponieważ
biorące udział w eksperymencie związki są solami słabych zasad i mocnych kwasów, w wodzie hy‑
drolizują kwaśno, a zatem spodziewać się powinniśmy niskich wartości pH.
3. Do dwóch pierwszych zlewek wsypujemy kilka granulek stałego NaOH. Dodatek tego wodorotlen‑
ku wyraźnie zwiększa pH. Ponieważ rozpuszczalność związków Fe w środowisku alkalicznym jest
niewielka (pamiętamy: „wysokie wartości pH sprzyjają procesom wytrącania, niskie wartości pH
sprzyjają utrzymywaniu się większości pierwiastków w roztworze”), konsekwencją jest natychmia‑
stowe wytrącenie osadów (Fig. 5.11c).
fig. 5.12 a–b.
Objaśnienia w tekście (fot. Grzegorz Rzepa)
4. Po kilku sekundach osady wypełniają niemal całą zlewkę (Fig. 5.11d). Teraz jest chwila czasu, żeby
się im przyjrzeć. Różnią się one wyraźnie barwą – w zlewce po lewej stronie powstał wodorotlenek
żelaza(III), w środkowej hydroksosiarczan żelaza(II,III). Zapamiętajmy, że minerały (szczególnie tlen‑
ki) zawierające żelazo trójwartościowe są zazwyczaj żółte, brązowe (np. goethyt), pomarańczowe
(np. lepidokrokit) lub czerwone (np. hematyt), natomiast jeśli w jednym związku współwystępują
Fe(II) i Fe(III), wówczas zabarwienie jest często intensywnie zielone (np. krzemian glaukonit) lub
niebieskie (np. częściowo utleniony fosforan wiwianit). Wytworzone przez nas w zlewkach związki
mają swoje odpowiedniki w przyrodzie – szybkie wytrącanie związków Fe(III) sprzyja powstawaniu
czerwonobrązowego ferrihydrytu, który jest pospolitym składnikiem gleb i wielu młodych osadów.
Ten bardzo słabo krystaliczny minerał ulega transformacji, w zależności od warunków, w goethyt
lub hematyt i dlatego nie spotyka się go w starszych skałach. Z kolei w trakcie utleniania soli Fe(II)

[65]
o5
r
ozdzi
A
ł
p
iĄ
T
y
GeocheMia
strefY
hiperGenicznej
zazwyczaj tworzą się nietrwałe połączenia typu hydroksosoli, które znane są – z uwagi na charak‑
terystyczne zabarwienie – pod ogólną nazwą „zielonych rdzy”. Często występują one w glebach
i osadach. Z czasem przekrystalizowują w goethyt lub lepidokrokit.
5. Do utlenienia żelaza nie jest oczywiście konieczna zmiana pH. Żeby to zilustrować, do trzeciej zlew‑
ki kroplami dodajemy perhydrol, czyli 30% H
2
O
2
. Ponieważ nadtlenek wodoru jest silnym utlenia‑
czem, powoduje błyskawiczne utlenienie Fe(II) do Fe(III).
6. Po chwili cały roztwór w zlewce przybiera pomarańczowe zabarwienie, świadczące o utlenieniu
żelaza (Fig. 5.12a). Osad się nie wytrąca, ponieważ nie nastąpiła wyraźna zmiana pH. Roztwór jest
ciągle silnie kwaśny, a w takich warunkach związki żelaza są rozpuszczalne (Fig. 5.9). Aby doprowa‑
dzić do powstania osadu musimy zwiększyć pH – znów dodając NaOH.
7. Podobnie jak w zlewce pierwszej, i tym razem wytrąca się wodorotlenek żelaza(III) (Fig. 5.12b).
literatura pomocnicza:
1. Macioszczyk A.,
1987:
Hydrogeochemia.
Wydawnictwa Geologiczne, Warszawa: 20–66, 314–359.
2. Macioszczyk A., Dobrzański D.,
2007:
Hydrogeochemia. Strefy aktywnej wymiany wód
podziemnych.
Wydawnictwo Naukowe PWN, Warszawa: 42–110.
3. Migaszewski Z., Gałuszka A.,
2007:
Podstawy geochemii środowiska.
Wydawnictwa Naukowo–
Techniczne, Warszawa, 103–134, 362–372.
4. Polański A., Smulikowski K.,
1968:
Geochemia.
Wydawnictwa Geologiczne, Warszawa: 62–73, 128–146,
218–227.
5. Skowroński A.,
2007:
Zarys geochemii poszukiwawczej.
Skrypt AGH nr 1693, Kraków: 25–45.
[66]
o6
r
ozdzi
A
ł
sz
ó
sT
y
elementY
hydrOGeOchemii
rozdział SzóSty:
elemenTy hydrOGeOchemii
Woda jest jedną z najważniejszych substancji wykorzystywanych przez człowieka. Poza oczywistą
podstawową funkcją – wody pitnej, jest stosowana na wiele innych sposobów, np. w hodowli ryb,
w basenach, w przemyśle etc. Każde z tych zastosowań wymaga wody o odpowiedniej jakości, re‑
gulowanej stosownymi rozporządzeniami prawnymi. Jakość wody jest oczywiście przede wszystkim
związana z jej składem chemicznym, ale nie tylko – ważne są też inne czynniki, np. woda pitna lub
wykorzystywana w kąpieliskach nie może być zanieczyszczona biologicznie czy zbyt mętna, a woda
stosowana w przemyśle musi mieć odpowiednią temperaturę i niską mineralizację itd. Niespełnienie
tych wymogów nie musi być związane z niekorzystnym wpływem działalności człowieka, często jest
wynikiem procesów naturalnych.
tabela. 6.1. Ogólna charakterystyka wód podziemnych związanych z obszarami występowania różnych skał
(wg Brownlowa 1996)
Rodzaj skał
Mineralizacja
Dominujące jony
pH
Zawartość SiO
2
granity, ryolity
niska
Na
+
, HCO
3
‑
6,3–7,9
średnia–wysoka
gabra, bazalty
średnia
Ca
2+
, Mg
2+
, HCO
3
–
6,7–8,5
wysoka
piaskowce
wysoka
Ca
2+
, Mg
2+
, Na
+
, HCO
3
‑
5,6–9,2
niska–średnia
mułowce, iły
wysoka
Na
+
, Ca
2+
, Mg
2+
, HCO
3
–
, SO
4
2–
, Cl
–
4,0–8,6
niska–średnia
wapienie, dolomity, margle
wysoka
Ca
2+
, Mg
2+
, HCO
3
–
7,0–8,2
niska
łupki metamorficzne, gnejsy niska–średnia
HCO
3
–
, Ca
2+
, Na
+
5,2–8,1
niska
Na skład chemiczny wód powierzchniowych i podziemnych wpływ ma szereg różnorodnych czynni‑
ków. Należą do nich: rzeźba terenu, klimat, geologia rejonu, oraz działalność organizmów wodnych
i człowieka. Ponieważ woda przepływając wśród skał, może reagować z ich składnikami, wśród czyn‑
ników czysto geologicznych najważniejszy jest właśnie rodzaj skał występujących w danym rejonie
(Tabela 6.1). Np. wody na obszarach zdominowanych przez skały węglanowe zawierają duże ilości
wapnia i magnezu, są więc „twarde” (często smaczniejsze, ale nie najlepiej nadające się do np. prania
i powodujące wytrącanie się „kamienia”, czyli CaCO
3
np. na grzałkach czajników elektrycznych), a ich
pH jest zazwyczaj wysokie. Wody drenujące masywy „kwaśnych” skał magmowych są słabiej zminera‑
lizowane (ponieważ takie skały stosunkowo wolno wietrzeją), a wśród kationów jest względnie dużo
sodu i potasu (z wietrzejących skaleni). Wody na obszarach torfowiskowych są kwaśne i zawierają
dużo substancji organicznych itd.
Aby ocenić przydatność wody do określonego celu trzeba zbadać jej skład chemiczny oraz parametry
fizykochemiczne i pewne inne właściwości. Do najważniejszych parametrów fizykochemicznych wody
należą temperatura, pH, Eh, przewodność elektrolityczna, obecność zawiesin, barwa i zapach. Znacze‑
nie pH i Eh zostało omówione w poprzednim rozdziale. Bardzo istotnym parametrem, pozwalającym
szybko otrzymać użyteczne informacje o wodzie jest przewodność elektrolityczna właściwa (pew,
ec – electrolytic conductivity). Za przewodzenie prądu elektrycznego w roztworach wodnych odpo‑
wiada przepływ jonów. Jak pamiętamy, czysta chemicznie woda prawie nie dysocjuje na jony, a więc
słabo przewodzi prąd elektryczny. Zatem zdolność do jego przewodzenia będzie proporcjonalna do
obecności jonów powstałych z rozpuszczonych w wodzie substancji. Im większe są stężenia kationów
i anionów, tym więcej nośników ładunku i większa przewodność (mniejsza oporność elektryczna). Tak
więc pomiar przewodności od razu informuje nas o tym czy woda jest słabo czy silnie zmineralizowa‑
na. Jednostką przewodnictwa jest odwrotność ohma – siemens (1 S = 1/Ω), a przewodności właściwej
S/m. Przewodność właściwą wody podaje się zwykle w mS/cm lub w µS/cm. Nie ma niestety jedno‑

[67]
o6
r
ozdzi
A
ł
sz
ó
sT
y
elementY
hydrOGeOchemii
znacznego przełożenia przewodności na mineralizację, gdyż wpływa na to zbyt wiele różnych czynni‑
ków. Często przyjmuje się jednak, że przewodność mierzona w µS/cm jest mniej więcej równa sumie
rozpuszczonych w wodzie kationów i anionów (w mg/dm
3
), co z kolei można w wielu przypadkach
sprowadzić do mineralizacji wody. Inna podawana zależność, wiążąca sumę anionów (lub kationów)
z przewodnością ma postać:
SA = SK (w meq/dm
3
) ≈ przewodność/100 (w µS/cm)
Obecność w wodzie zawiesin związana jest z unoszonymi przez nią cząstkami mułu, iłu oraz różnych
organicznych i nieorganicznych substancji koloidalnych. Najdrobniejsze zawieszone w wodzie cząstki
wpływają na jej mętność. Parametr ten wyznacza się porównując badaną wodę z odpowiednio przy‑
gotowanymi roztworami zawierającymi zawiesinę koloidalnej krzemionki i stąd jego wartość podaje się
w mg SiO
2
/dm
3
(chociaż obecnie przyjmowaną jednostką jest NTU – nephelometric turbidity unit, licz‑
bowo równoważna mg SiO
2
/dm
3
). barwa naturalnej wody może być lekko żółtawa, co wiąże się z obec‑
nością rozpuszczonych kwasów organicznych. Intensywność zabarwienia mierzy się kolorymetrycznie,
poprzez porównanie próbki ze skalą wzorców otrzymanych przez rozpuszczenie związków platyny i ko‑
baltu. Dlatego wynik podawany jest w mg Pt/dm
3
. Z kolei zapach określa się organoleptycznie, oceniając
jego charakter (np. roślinny, gnilny, specyficzny) i moc (np. słabo wyczuwalny, bardzo wyraźny etc.) lub
krotność. Krotność oznacza stopień rozcieńczenia próbki, przy którym zapach przestaje być wyczuwal‑
ny. Niektóre rodzaje zapachów – np. gnilne, specyficzne – wykluczają możliwość wykorzystania wody
do celów pitnych.
Składniki mineralne rozpuszczone w wodzie tworzą jej mineralizację. W sposób przybliżony ogólną mi‑
neralizację wód wyznacza się na podstawie masy suchej pozostałości, czyli masy substancji pozostałych
po odparowaniu wody (TDS – total dissolved solids). Wody podziemne na podstawie mineralizacji ogólnej
dzielimy na następujące grupy:
– wody zwykłe (słodkie) – mineralizacja < 500 mg/dm
3
;
– akratopegi (wody o podwyższonej mineralizacji) – mineralizacja 500–1000 mg/dm
3
;
– wody mineralne (słone) – mineralizacja > 1000 mg/dm
3
.
Przy badaniu składu chemicznego wody ważne są dwie grupy składników. Główne kationy i aniony wy‑
stępują w największych ilościach, decydując o mineralizacji wody oraz o jej typie hydrogeochemicznym.
Z kolei składniki występujące w podrzędnych lub nawet śladowych ilościach często warunkują możliwość
wykorzystania wody do określonego celu – np. jako wody pitnej. Głównymi kationami występującymi w
wodach śródlądowych są ca
2+
, mg
2+
, na
+
i K
+
, natomiast podstawowe aniony to hco
3
–
, so
4
2–
i cl
–
(czę‑
sto, choć nie zawsze w takiej kolejności).
W podrzędnych ilościach pojawiają się w wodach związki azotu i fosforu. Mają one jednak bardzo duży
(korzystny lub niekorzystny) wpływ na jakość wody, gdyż są niezbędnymi biopierwiastkami wykorzysty‑
wanymi przez organizmy wodne, a zatem wpływają na ich rozwój. Podstawowymi formami, w jakich te
pierwiastki występują w wodach są jony: no
3
–
, no
2
–
, nh
4
+
i po
4
3–
oraz związki organiczne. Ich nadmiar
związany np. ze spłukiwaniem do wód nadmiernych ilości nawozów, może prowadzić do eutrofizacji
zbiorników. Chociaż najbardziej rzucającym się w oczy przejawem tego zjawiska są tzw. zakwity wód, czyli
powstawanie utrzymującego się na powierzchni wody kożucha fitoplanktonu, to eutrofizacja prowadzi do
daleko idących zmian właściwości wody w całym zbiorniku, gdyż mikroorganizmy tworzące ten kożuch
odcinają dopływ światła do niżej położonych stref i zużywają cały rozpuszczony tlen, przyczyniając się do
wytworzenia środowiska anaerobowego, a często wydzielają również trujące produkty metabolizmu.

[68]
o6
r
ozdzi
A
ł
sz
ó
sT
y
elementY
hydrOGeOchemii
W przypadku azotu, proporcje różnych jego form pozwalają niekiedy wnioskować o źródłach zanieczysz‑
czenia wód. Ogólnie przyjmuje się, że w wodach gruntowych zanieczyszczonych odpadami bytowymi bądź
hodowlanymi obecność jonów amonowych, przy braku azotanów i azotynów, wskazuje na zanieczyszczenie
świeże, pochodzące z bliskiego ogniska. Współwystępowanie wszystkich form mineralnych azotu wskazuje
na trwałe zanieczyszczenie. Występowanie jedynie azotanów, przy nieznacznej domieszce azotynów wska‑
zuje na odległe w czasie lub przestrzeni zanieczyszczenie wód podziemnych.
Pozostałe nieorganiczne jony naturalnych wód to mikroskładniki – pojawiają się w stężeniach rzędu ułamków
mg/dm
3
do kilku mg/dm
3
. Należą do nich m.in. Sr
2+
, Ba
2+
, Fe
2+
, Mn
2+
, B
3+
, Al
3+
, Zn
2+
, Cu
2+
, Li
+
, F
–
, Br
–
, I
–
. W jesz‑
cze mniejszych ilościach występują szczególnie toksyczne pierwiastki, jak As
3+
, As
5+
, Cr
6+
, Pb
2+
, Cd
2+
, Hg
+
.
Pamiętamy, że w wyniku procesów hydrolizy krzemianów powstaje koloidalna krzemionka lub kwas krze‑
mowy. Można zatem spodziewać się obecności znacznych ilości si w wodach – często rzędu kilkudziesięciu
a nawet kilkuset mg SiO
2
/dm
3
. Koncentracje i formy, w jakich krzem występuje zależą od wielu czynników,
takich jak pH, temperatura czy stężenia innych jonów (głównie fluorków).
Badając jakość wód powierzchniowych czy podziemnych, nie można zapomnieć o substancji organicznej,
gdyż bardzo często to właśnie ona jest głównym czynnikiem zanieczyszczającym. Pod tym pojęciem rozu‑
miemy zarówno mikroorganizmy obecne w wodach, jak również produkty ich metabolizmu oraz rozkła‑
du, a także różnorakie substancje pochodzenia antropogenicznego. Do tych ostatnich należą np. fenole,
węglowodory aromatyczne (btX – benzen, toluen, ksylen), wielopierścieniowe węglowodory aromatyczne
(wwa), pestycydy i inne. Ze względu na trudności analityczne oraz tak szeroki wachlarz możliwych sub‑
stancji, bardzo często na etapie badań wstępnych szacuje się stopień zanieczyszczenia wód substancjami or‑
ganicznymi w sposób uproszczony, wprowadzając pewne umowne wskaźniki. Należą do nich ogólny węgiel
organiczny, oraz chemiczne i biochemiczne zapotrzebowanie na tlen.
Ogólny węgiel organiczny (owo, albo toc – total organic carbon) to stężenie węgla organicznego (czyli
zawartego we wszystkich związkach organicznych w wodzie, a nie związanego w jonach węglanowych CO
3
2–
czy wodorowęglanowych HCO
3
–
). Podczas jego oznaczania, w celu wyeliminowania węgla nieorganicznego
(tIc – total inorganic carbon) próbkę wody zakwasza się, ponieważ powoduje się tym samym usunięcie wę‑
glanów, w głównej mierze będących nośnikami węgla nieorganicznego. Wartości OWO wahają się w bardzo
szerokich granicach, dochodząc w szczególnych przypadkach do kilkuset mg/dm
3
.
Chemiczne zapotrzebowanie na tlen (chzt, albo cod – chemical oxygen demand) oznacza ilość moc‑
nego utleniacza (dwuchromianu potasu K
2
Cr
2
O
7
lub manganianu(VII) potasu KMnO
4
) niezbędną do roz‑
kładu substancji organicznych występujących w wodzie. Parametr ten wyznacza się miareczkując próbkę
wody dwuchromianem lub nadmanganianem w obecności kwasu siarkowego i katalizatora, a wynik podaje
w przeliczeniu na tlen cząsteczkowy, czyli w mg O
2
/dm
3
. Aby oszacować ilość mikroorganizmów w wodzie
określa się jeszcze jeden wskaźnik – biochemiczne zapotrzebowanie na tlen (bzt albo bod – biochemi-
cal oxygen demand). Idea jego oznaczania opiera się na spostrzeżeniu, że organizmy wodne zużywają roz‑
puszczony w wodzie tlen. Zatem jego ilość w zamkniętej porcji wody powinna z czasem maleć. Analiza
jest dwuetapowa. Mierzy się stężenie tlenu rozpuszczonego w wodzie przed (tuż po pobraniu próbki) i po
pięciodniowej (stąd zwykle BZT
5
) inkubacji w laboratorium, w ciemności i stałej temperaturze. W takich
warunkach ubytek tlenu jest wynikiem zużywania go przez mikroorganizmy wodne. Np. jeśli początkowe
stężenie O
2
w wodzie wynosiło 8,3 mg/dm
3
, a po pięciu dniach spadło do 5,0 mg/dm
3
, to BZT
5
jest równe
3,3 mg/dm
3
.

[69]
o6
r
ozdzi
A
ł
sz
ó
sT
y
elementY
hydrOGeOchemii
Poza tlenem, woda zawiera rozpuszczone inne gazy, przy czym najważniejszym z nich jest CO
2
. Wody o znacz‑
nej jego zawartości (500–1000 mg/dm
3
) są nazywane wodami kwasowęglowymi, natomiast jeszcze silniej
nasycone CO
2
(powyżej 1000 mg/dm
3
) noszą nazwę szczaw.
Bardzo istotne z punktu widzenia użytkowania wody, szczególnie do celów pitnych, są ponadto wskaźniki
biologiczne, a szczególnie stan bakteriologiczny. Biologiczna ocena jakości wody opiera się na fakcie, że skład
i właściwości wody wpływają na rodzaj i liczebność zamieszkujących ją organizmów. Zwykle wykorzystuje się
do tego celu tzw. gatunki wskaźnikowe, które dzieli się na trzy główne grupy:
– saprokseniczne – występują tylko w wodach czystych, a unikają zanieczyszczonych;
– saprofilne – występują w wodach czystych, ale w zanieczyszczonych masowo;
– saprobiontyczne – występują tylko w wodach zanieczyszczonych.
Zatem badając populacje biologiczne w wodach poprzez szacowanie liczebności i proporcji gatunkowych,
można wnioskować o stanie czystości wód. Spośród wskaźników mikrobiologicznych, wskazujących na stan
bakteriologiczny wody, najczęściej stosuje się ilość pałeczek okrężnicy (Escherichia coli) w określonej objętości
próbki. Można ją też podawać jako miano coli, czyli najmniejszą objętość wody (w cm
3
), na jaką przypada
jedna bakteria okrężnicy.
Często wspominaną cechą wody jest jej twardość. Jest to pojęcie umowne określające zużycie mydła w wo‑
dzie bez wytwarzania piany przy skłócaniu. Wiąże się to z obecnością w wodzie rozpuszczonych jonów dwu‑
wartościowych, głównie Ca
2+
, Mg
2+
, Sr
2+
i Ba
2+
(a także Fe
2+
, Zn
2+
) i trójwartościowych (Al
3+
, Fe
3+
). Jeśli metale
te występują w postaci wodorowęglanów, węglanów lub wodorotlenków, wówczas mówimy o tzw. twardo-
ści węglanowej (przemijającej). Zwykle zanika ona w trakcie gotowania wody przez wytrącenie minerałów
węglanowych (głównego składnika „kamienia kotłowego”):
Ca(HCO
3
)
2
⇒ H
2
O + CO
2
+ CaCO
3
⇓
Gdy w wodzie obecne są siarczany i chlorki tych metali, wówczas mamy do czynienia z twardością niewęgla-
nową (trwałą). Taka twardość nie zanika w trakcie gotowania, lecz wymaga stosowania specjalnych metod
chemicznych. Suma twardości węglanowej i niewęglanowej daje twardość ogólną.
Jednostkami twardości wody są mval/dm
3
(milirównoważnik), lub stopnie twardości wg skali niemieckiej
(°N, 1 stopień odpowiada 10 mg CaO w dm
3
), francuskiej (°Fr, 1 stopień odpowiada 1 g CaCO
3
w 100 dm
3
),
angielskiej (°Ang, 1 stopień odpowiada 1 g CaCO
3
w 70 dm
3
) oraz amerykańskiej (°USA) i WHO (1° = 500
mg CaCO
3
/dm
3
). W Polsce stosowane są najczęściej mval/dm
3
, stopnie skali niemieckiej lub jednostki WHO.
W zależności od twardości ogólnej wyróżniamy (1 mval/dm
3
= 2,8°N):
– wodę bardzo miękką;
< 0,5°N
– wodę miękką;
5–10°N
– wodę średnio twardą;
10–20°N
– wodę twardą;
20–30°N
– wodę bardzo twardą
> 30°N
Twardość wody ma wpływ na możliwości jest wykorzystania. Z wód twardych łatwo wytrąca się kamień, więc
nie powinno się ich wykorzystywać np. w sieciach centralnego ogrzewania. Nie powinno się jej również stoso‑
wać w gospodarstwie domowym, gdyż zwiększa użycie mydła i środków piorących, a niekiedy nawet zmienia
smak mięsa, kawy i herbaty. Może też powodować podrażnienia skóry. Z kolei wody zbyt miękkie sprzyjają
np. zachodzeniu procesów korozyjnych w rurach wodociągowych oraz zwiększają ryzyko chorób serca.

[70]
o6
r
ozdzi
A
ł
sz
ó
sT
y
elementY
hydrOGeOchemii
Twardość wody można wyznaczyć eksperymentalnie lub obliczyć z wyników analizy chemicznej
wody. W takim przypadku mnoży się stężenia kationów powodujących twardość (podane w mg/
dm
3
) przez odpowiednie, wynikające ze stechiometrii, współczynniki przeliczeniowe (Tabela 6.2)
otrzymując wyniki w mval/dm
3
, które następnie sumuje się.
tabela 6.2. Współczynniki przeliczeniowe do obliczania twardości wody
Kation
Mnożnik
Kation
Mnożnik
Ca
2+
0,04990
Fe
3+
0,05372
Mg
2+
0,08224
Al
3+
0,1112
Sr
2+
0,02282
Zn
2+
0,03059
Fe
2+
0,03581
Mn
2+
0,07281
1. sposoby poboru I utrwalanIa próbeK wód
Przy pobieraniu próbek wód do analiz chemicznych i fizykochemicznych musimy zdawać sobie spra‑
wę z faktu, że właściwości wody ulegają bardzo szybkim zmianom. Zmiany te mogą być związane
m.in. z takimi zjawiskami jak:
– zużywanie niektórych składników pokarmowych przez bakterie, glony itp., wydzielanie pro‑
duktów metabolizmu, reakcje fotosyntezy;
– utlenianie substancji organicznej, Fe(II), siarczków;
– wytrącanie niektórych składników (CaCO
3
, Fe(OH)
3
, Mg
3
(PO
4
)
2
etc.);
– ulatnianie się niektórych składników (O
2
, H
2
S, CN
–
, Hg, związków organicznych);
– adsorpcja CO
2
z powietrza – zmiana pH, przewodności, zawartości CO
2
etc.;
– adsorpcja na ściankach naczyń lub na obecnych w wodzie zawiesinach – np. adsorpcja fosfo‑
ranów na wytrącającym się koloidalnym Fe(OH)
3
;
Z tego względu niektóre oznaczenia powinno wykonywać się od razu, w terenie. Dotyczy to m.in.
temperatury, pH, Eh, przewodności, zawartości rozpuszczonego tlenu. W innych przypadkach moż‑
na wodę analizować po przetransportowaniu do laboratorium, ale wymaga to odpowiedniego spo‑
sobu poboru, przechowywania, a w wielu przypadkach również tzw. utrwalania próbek. Zapobiega‑
nie zmianom składu i właściwości próbek obejmuje:
– odpowiedni dobór i przygotowanie przyrządów i naczyń (np. nie można przechowywać w
naczyniach szklanych próbek do oznaczania śladowych zawartości Si, Al, Na, B, a w plastiko‑
wych próbek, w których oznaczane będą niektóre składniki organiczne – np. WWA);
– sączenie w przypadku próbek przeznaczonych do badania składników rozpuszczonych;
– schładzanie/zamrażanie próbek (wstrzymuje to proces rozmnażania bakterii oraz sprzyja
zatrzymaniu substancji lotnych w próbce – pamiętamy, że rozpuszczalność gazów rośnie ze
spadkiem temperatury);
– całkowite napełnianie naczyń (ogranicza wstrząsy w trakcie transportu, zapobiega wymianie
składników z powietrzem, spowalnia reakcje redoksowe);
– dodawanie odpowiednich środków utrwalających
Rodzaj dodawanych środków utrwalających zależy od tego, jakie oznaczenia chcemy wykonać. Bar‑
dzo często może się więc okazać, że do laboratorium przywozimy kilka różnych naczyń zawierają‑
cych próbkę tej samej wody, ale utrwaloną w różny sposób. Do najczęściej stosowanych „utrwala‑
czy” zaliczamy:

[71]
o6
r
ozdzi
A
ł
sz
ó
sT
y
elementY
hydrOGeOchemii
– stężone kwasy (gł. HNO
3
, H
2
SO
4
– oznaczanie metali, N–NH
4
, Norg, ChZT, fenoli); HNO
3
utrzy‑
muje metale w stanie rozpuszczonym, H2SO4 działa jako bakteriocyd i tworzy siarczany z lotny‑
mi zasadami (aminy, amoniak);
– chloroform (oznaczanie N–NO
3
, N–NO
2
, detergentów anionowych, cukrów, WWA, zawiesin);
– NaOH (oznaczanie CN
–
, fenoli lotnych); tworzy sole sodowe lotnych kwasów;
– HgCl
2
(bakteriocyd – do próbek zawierających biodegradowalne związki organiczne oraz różne
formy azotu i fosforu).
Należy pamiętać, że utrwalenia próbki dokonuje się po pomiarach wstępnych (pH, przewodnictwo
itd.)! Warto również podkreślić, że utrwalenie próbki wody pozwala jedynie na niewielką zwłokę w wy‑
konaniu analizy. Większość składników powinno się oznaczyć przed upływem 24 lub 48 h. Stosunkowo
najodporniejsze na zmiany są (oczywiście przy odpowiednim utrwaleniu i przechowywaniu próbki)
tzw. metale ciężkie – w niektórych przypadkach dopuszcza się nawet kilkumiesięczny okres przecho‑
wywania próbek. Z podanych powyżej powodów często warto pokusić się o analizę przynajmniej części
składników w terenie. Umożliwiają to terenowe wersje urządzeń pomiarowych – jak wielofunkcyjne
mierniki pozwalające na podłączenie rozmaitych elektrod pomiarowych – pH, konduktometrycznych,
Eh, jonoselektywnych itp. Często korzysta się również z przenośnych spektrofotometrów i automatycz‑
nych titratorów, przy pomocy których można w warunkach polowych wykonać praktycznie pełną ana‑
lizę chemiczną wody (Fig. 6.1–6.2).
fig. 6.1a,b. Pomiary parametrów fizykochemicznych wody w warunkach polarnych przy użyciu miernika wielofunkcyjnego
(po lewej). Przenośny spektrofotometr o bateryjnym zasilaniu, przeznaczony do polowych oznaczeń składu
chemicznego wód (po prawej) (fot. Maciej Manecki, Monika Kwaśniak–Kominek)
2. sposoby przedstawIanIa wynIKów
analIz chemIcznych wód
Analizy chemiczne wód przedstawia się w różnych jednostkach. Najczęściej stosuje się mg/dm
3
(mg/L)
i µg/dm
3
(µg/L), rzadziej mg/kg czy µg/kg. Dla wód mineralnych, zawierających duże ilości rozpuszczo‑
nych substancji, często ich ilość wyraża się w g/dm
3
(g/L). W przypadku substancji o szczególnie niskich
stężeniach (np. WWA) wykorzystuje się bardzo małe jednostki, jak ng/dm
3
(ng/L).

[72]
o6
r
ozdzi
A
ł
sz
ó
sT
y
elementY
hydrOGeOchemii
fig. 6.2a,b. Oznaczanie zasadowości wody przy wykorzystaniu automatycznego titratora (po lewej). Jednorazowe testy służące
do oznaczania składu chemicznego wód (po prawej). (fot. Monika Kwaśniak–Kominek)
Większość składników rozpuszczonych w wodach występuje w formie jonowej. Sumaryczny dodatni ła‑
dunek kationów musi być równoważony przez sumaryczny ładunek ujemny anionów. Z tego względu, do
celów prezentowania składu wód oraz ich klasyfikacji, zawartość (stężenie) każdego kationu i anionu przed‑
stawia się w formie równoważnikowej (np. mval/dm
3
, meq/L), uwzględniającej jego masę molową i warto‑
ściowość. Masę równoważnikową dla danego jonu otrzymujemy dzieląc jego masę molową przez ładunek.
PRzyKłAD 1: Poniższe wyniki analizy chemicznej wody zestawione w mg/dm
3
przedstaw
w mmol/dm
3
i mval/dm
3
.
Na
+
– 13,70
HCO
3
–
– 79,9
K
+
– 1,18
Cl
–
– 31,2
Ca
2+
– 42,50
SO
4
2–
– 39,0
Mg
2+
– 3,21
NO
3
–
– 1,3
1 mol Na ma masę 22,99 g, czyli 1 mmol Na to 22,99 mg. Zatem jeśli stężenie sodu w wodzie wynosi 13,70
mg/dm
3
, to z proporcji otrzymujemy:
1 mmol Na – 22,99 mg
x mmol Na – 13,70 mg
x = 0,60 mmol
Stężenie sodu wynosi 0,60 mmol/dm
3
, a ponieważ sód występuje w postaci kationu jednowarto‑
ściowego, więc 1 mmol = 1 mval, czyli 0,60 mmol/dm
3
= 0,60 mval/dm
3
.
W przypadku wapnia masa molowa wynosi 40,08 g, więc stężenie molowe Ca w tej wodzie to
42,50/40,08=1,06 mg/dm
3
. Ale wapń występuje w postaci kationu dwuwartościowego, więc równoważnik
jest dwukrotnie większy, czyli 1,06 mg/dm
3
´ 2 = 2,12 mval/dm
3
.
W ten sposób przeliczamy wszystkie kationy i aniony na „stężenia ładunków” wyrażone w mval/dm
3
otrzy‑
mując (Tabela 6.3):
[73]
o6
r
ozdzi
A
ł
sz
ó
sT
y
elementY
hydrOGeOchemii
tabela 6.3. Dane i wyniki do przykładu 1
Jon
mg/dm
3
Masa molowa
mmol/dm
3
Ładunek
mval/dm
3
Na
+
13,70
22,99
0,60
1
0,60
K
+
1,18
39,1
0,03
1
0,03
Ca
2+
42,50
40,08
1,06
2
2,12
Mg
2+
3,21
24,31
0,13
2
0,26
HCO
3
–
79,9
61,02
1,31
1
1,31
SO
4
2–
39,0
96,06
0,41
2
0,82
Cl
–
31,2
35,45
0,88
1
0,88
NO
3
–
1,3
62,00
0,02
1
0,02
PRzyKłAD 2: Oblicz twardość wody z przykładu 1.
W przypadku tej wody mamy tylko dwa kationy powodujące twardość (Ca
2+
i Mg
2+
). Ich stężenia w mval/
dm
3
wynoszą, odpowiednio, 2,12 i 0,26 mval/dm
3
. Zatem twardość ogólna wynosi 2,12 + 0,26 = 2,38 mval/
dm
3
. Ponieważ 1 mval/dm
3
= 2,8°N, więc w naszym przypadku twardość w stopniach skali niemieckiej
wynosi 6,66°n. Jest to zatem woda miękka.
Przedstawiając wyniki analizy chemicznej można od razu w prosty sposób sprawdzić jej poprawność. Po‑
nieważ sumaryczny dodatni ładunek kationów musi być równoważony przez sumaryczny ładunek
ujemny anionów, więc suma (mili)równoważników kationów powinna być (mniej więcej) równa sumie
(mili)równoważników anionów. Wyraźna różnica pomiędzy tymi wartościami może wynikać albo z błędu
analitycznego (błędne oznaczenie któregoś ze składników), albo z faktu, że badana woda ma nietypowy
skład chemiczny – zawiera jakieś składniki, których rutynowo się nie oznacza. Ten drugi przypadek ma
często miejsce przy badaniach wód silnie zanieczyszczonych i ścieków.
PRzyKłAD 3: Na podstawie bilansu jonowego sprawdź poprawność analizy chemicznej wody
z Przykładu 1.
Sumujemy stężenia równoważnikowe kationów i anionów:
∑K = 0,60 + 0,03 + 0,26 + 2,12 = 3,01
∑A = 0,88 + 1,31 + 0,82 + 0,02 = 3,03
Widać, że ∑K ≈ ∑A, ale lepiej to wyrazić obliczając błąd analizy (jego dopuszczalna wielkość zależy od ogól‑
nej mineralizacji, patrz Tabela 6.4):
x =
∑K – ∑A
·100%
∑K + ∑A
w naszym przypadku błąd analizy jest bardzo mały i wynosi:
x = (3,01–3,03)/(3,01+3,03) · 100% = – 0,3%
tabela 6.4. Dopuszczalne względne błędy analizy wody w zależności od mineralizacji (wg PN–89/C–04638)
Sumaryczne stężenie jonów w wodzie [mval/dm
3
]
Dopuszczalny błąd względny [%]
> 15
2
5–15
2–5
3–5
5–10
< 3
nie ustalono
[74]
o6
r
ozdzi
A
ł
sz
ó
sT
y
elementY
hydrOGeOchemii
3. grafIczne sposoby prezentacjI sKładu chemIcznego wód
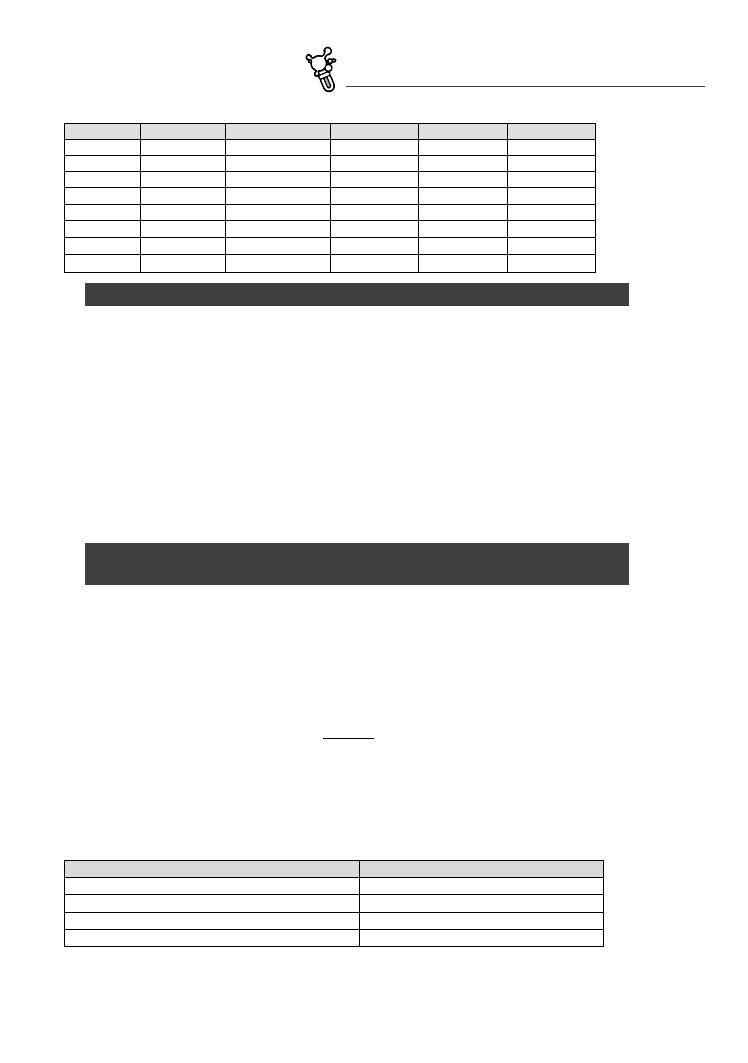
W celu szybkiego zorientowania się w typie hydrogeochemicznym danej grupy wód, ich składy chemiczne
często przedstawia się graficznie – na diagramach kołowych, słupkowych, trójkątnych i innych. Do najpopu‑
larniejszych i najczęściej stosowanych sposobów takiej prezentacji należą diagramy Pipera, Stiffa i Udlufta.
diagram pipera składa się z trzech części – dwóch trójkątów
i rombu (Fig. 6.3). Lewy trójkąt podaje
względne proporcje głów‑
nych kationów (Ca
2+
, Mg
2+
oraz sumy Na
+
i K
+
) przeliczone z wy‑
ników analiz wyrażonych w mval/dm
3
, a prawy przedstawia w ten
sam sposób główne aniony (HCO
3
–
+ CO
3
2–
, SO
4
2–
i Cl
–
). Punkty od‑
powiadające proporcjom kationów i anionów rzutuje się następnie
prostymi na diagram rombowy, uzyskując w ich przecięciu punkt
projekcyjny reprezentujący badaną wodę. Dla pełniejszego zobra‑
zowania jej składu zwykle różnicuje się średnicę kółka w zależności
od wielkości mineralizacji wody.
fig. 6.3. Diagram Pipera z naniesioną pozycją
wody z Przykładu 1
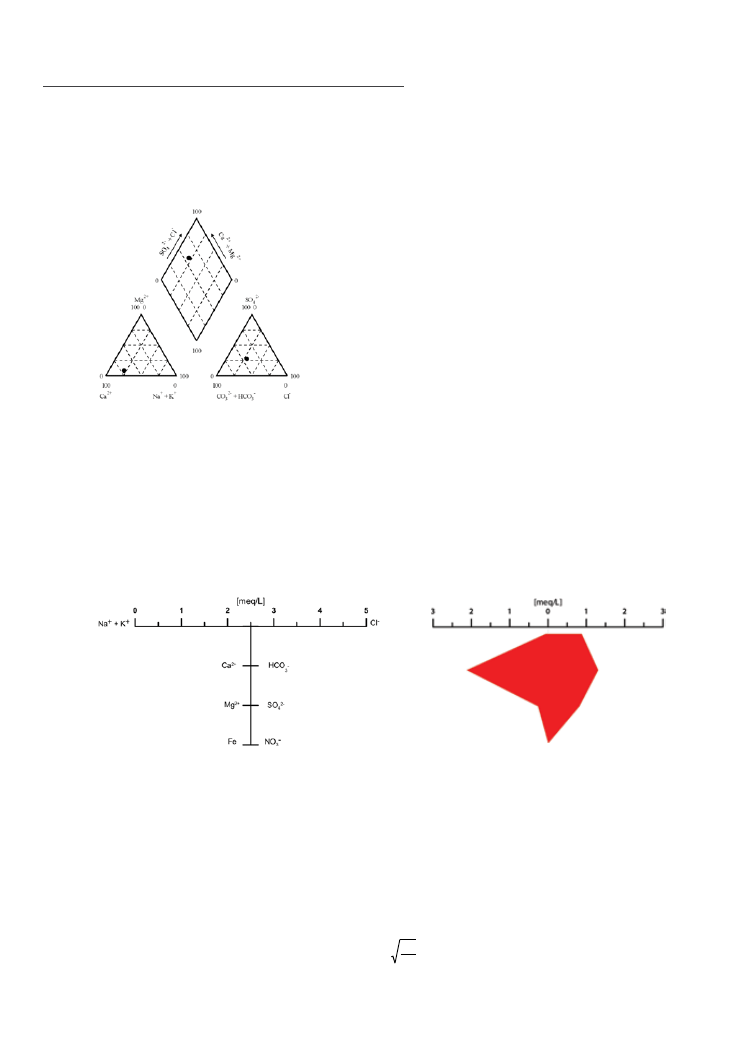
diagram stiffa często stosuje się do przedstawiania składu chemicznego wód podziemnych w profilach
pionowych. Na tym wykresie pionową oś pomocniczą (przy profilu wyskalowaną jako głębokość otworu)
przecinają cztery (najczęściej) równoległe osie, dające w efekcie osiem półosi, na których nanosi się stężenia
kationów (po lewej stronie osi pionowej) i anionów (po jej prawej stronie). Zawsze tym samym osiom przy‑
porządkowuje się określone jony, a stężenia, podobnie jak w diagramie Pipera nanosi się w mval/dm
3
(Fig.
6.4), przypisując pionowej osi pomocniczej stężenie zerowe. W oryginalnej wersji diagramu lewym półosiom
przypisuje się (idąc od góry): Na
+
+ K
+
, Ca
2+
, Mg
2+
i Fe
2+
, zaś prawym półosiom, odpowiednio, Cl
–
, HCO
3
–
,
SO
4
2–
i CO
3
–
. Naniesione na półosiach punkty stężeń łączy się ze sobą otrzymując nieregularny wielobok.
fig. 6.4a,b. Przyporządkowanie jonów do półosi oraz przykładowy diagram Stiffa
diagram udlufta jest wykresem kołowym, uwzględniającym nie tylko główne kationy i aniony, ale również
substancje gazowe rozpuszczone w wodzie (Fig. 6.5). Przy jego sporządzeniu przyjmujemy, że pole koła wy‑
raża sumaryczną ilość składników stałych i gazowych zawartych w wodzie (M). W zależności od przyjętej
skali wykresu, 1 mm
2
powierzchni koła może odpowiadać np. 1, 4, 9, 16 itd. mg/dm
3
sumy tych składników.
Przyjmując jednostkową zależność 1 mg/dm
3
= 1 mm
2
otrzymujemy:
M = πr
2
stąd:
r =
M
π
[75]
o6
r
ozdzi
A
ł
sz
ó
sT
y
elementY
hydrOGeOchemii
gdzie:
M – łączna masa składników stałych i gazowych rozpuszczonych w 1 litrze wody
r – promień koła [mm] obrazującego skład chemiczny wody
Składniki stałe i nie zdysocjowane zaznacza się w po‑
staci współśrodkowych kół, których promień oblicza
się zgodnie z powyższym wzorem, przyjmując zamiast
sumarycznego M masę składników gazowych i nie‑
zdysocjowanych. W górnej połowie koła lub pierście‑
nia przedstawia się wycinkami procenty miligramo‑
równoważników podstawowych kationów, w dolnej
połowie podstawowych anionów. Jony występujące
w podrzędnych ilościach zaznacza się w formie pro‑
mieni, biegnących od obwodu w kierunku środka koła.
Ich długość zależy od zawartości, przy czym w tym
celu przyjmuje się odrębną skalę (np. 1 r = 0,5% mval).
Podobnie, jak w przypadku głównych jonów, promie‑
nie odpowiadające kationom umieszcza się w górnej
części diagramu, a anionom w dolnej.
fig. 6.5. Przykładowy diagram Udlufta
4. wody mIneralne
Wody mineralne są specyficznym rodzajem wód, a ich nazewnictwo oraz wymagania jakościowe są re‑
gulowane odrębnymi rozporządzeniami prawnymi. Poza pewnymi warunkami ogólnymi, jakie powinny
spełniać takie wody (jak np. stałość składu chemicznego w czasie, brak zanieczyszczeń mikrobiologicznych,
szczególnie toksycznych i promieniotwórczych pierwiastków śladowych, odpowiednia wydajność źródła
etc.), muszą one charakteryzować się również specyficznym składem chemicznym, który jest również pod‑
stawą ich klasyfikacji i oznakowania (Tabela 6.5).
tabela 6.5. Kryteria klasyfikacji chemicznej stosowane w znakowaniu naturalnych wód mineralnych
(wg Rozporządzenia Ministra Zdrowia z 31.03.2011 r.)
składnik
stężenie [mg/dm
3
]
nazwa (określenie) wody
mineralizacja
< 50
bardzo niskozmineralizowana
mineralizacja
< 500
niskozmineralizowana
mineralizacja
> 1500
wysokozmineralizowana
mineralizacja
500–1500
średniozmineralizowana
CO
2
naturalny
> 250
kwasowęglowa
Mg
2+
> 50
zawiera magnez
Ca
2+
> 150
zawiera wapń
HCO
3
–
> 600
zawiera wodorowęglany
SO
4
2–
> 200
zawiera siarczany
Cl
–
> 200
zawiera chlorki
F
–
> 1
zawiera fluorki
Na
+
> 200
zawiera sód
Na
+
< 20
odpowiednia dla diety ubogiej w sód
Fe
2+
> 1
zawiera żelazo
Na
+
(Cl
–
)
F
–
NO
2
‑
NO
3
–
< 20
< 0,7
< 0,02
< 10
odpowiednia dla przygotowania żywności dla niemowląt

[76]
o6
r
ozdzi
A
ł
sz
ó
sT
y
elementY
hydrOGeOchemii
Osobno regulowana jest kwestia zawartości (stopnia nasycenia) i pochodzenia dwutlenku węgla. Biorąc
pod uwagę stężenie CO
2
, wody mineralne dzieli się na nienasycone dwutlenkiem węgla (niegazowane), ni‑
skonasycone (< 1500 mg CO
2
/dm
3
), średnionasycone (1500–4000 mg CO
2
/dm
3
) i wysokonasycone (> 4000
mg CO
2
/dm
3
). Na butelce powinna się również znaleźć informacja, czy zawartość tego gazu jest naturalne
(pochodzi ze źródła), czy też woda została nim nasycona później.
literatura pomocnicza:
1. Hermanowicz W., Dojlido J., Dożańska W., Koziorowski B., Zerbe J.,
1999:
Fizyczno–chemiczne bada-
nie wody i ścieków.
Wydanie drugie. Wydawnictwo Arkady, Warszawa: 37–294.
2. Macioszczyk A.,
1987:
Hydrogeochemia.
Wydawnictwa Geologiczne, Warszawa: 20–66, 314–359
.
3. Macioszczyk A., Dobrzyński D.,
2007:
Hydrogeochemia. Strefy aktywnej wymiany wód podziem-
nych.
Wydawnictwo Naukowe PWN, Warszawa.
4. Namieśnik J., Łukasiak J., Jamrógiewicz Z.,
1995:
Pobieranie próbek środowiskowych do analizy.
Wydawnictwo Naukowe PWN, Warszawa: 41–66; 109–147.
5. Namieśnik J., Jamrógiewicz Z., Pilarczyk M., Torres L.,
2000:
Przygotowanie próbek środowiskowych
do analizy.
Wydawnictwa Naukowo–Techniczne, Warszawa: 123–140
.
6. Rozporządzenie Ministra Środowiska z dnia 24 lipca 2006 r. w sprawie warunków, jakie należy
spełnić przy wprowadzaniu ścieków lub wód do ziemi, oraz w sprawie substancji szczególnie
toksycznych dla środowiska wodnego.
Dz. Ust. 137, poz. 984.
7. Rozporządzenie Ministra Środowiska z dnia 10 listopada 2005 w sprawie substancji szczególnie
szkodliwych dla środowiska wodnego, których wprowadzanie w ściekach przemysłowych do
urządzeń kanalizacyjnych wymaga pozwolenia wodnoprawnego.
Dz. Ust. 233, poz. 1988.
8. Rozporządzenie Ministra Środowiska z dnia 27 listopada 2002 w sprawie wymagań, jakim po-
winny odpowiadać wody powierzchniowe wykorzystywane do zaopatrzenia ludności w wodę
przeznaczoną do spożycia.
Dz. Ust. 204, poz. 1738.
9. Rozporządzenie Ministra Środowiska z dnia 23 lipca 2008 r. w sprawie kryteriów i sposobu
oceny stanu wód podziemnych.
Dz. Ust. 143, poz. 896.
10. Rozporządzenie Ministra Środowiska z dnia 30 sierpnia 2008 r. w sprawie sposobu klasyfikacji
stanu jednolitych części wód powierzchniowych.
Dz. Ust. 162, poz. 1008.
11. Rozporządzenie Ministra Środowiska z dnia 13 maja 2009 r. w sprawie form i sposobu prowa-
dzenia monitoringu jednolitych części wód powierzchniowych i podziemnych.
Dz. Ust. 81, poz. 685.
12. Rozporządzenie Ministra Środowiska z dnia 22 lipca 2009 r. w sprawie klasyfikacji stanu eko-
logicznego, potencjału ekologicznego i stanu chemicznego jednolitych części wód powierzchnio-
wych.
Dz. Ust. 122, poz. 1018.
13. Rozporządzenie Ministra Zdrowia z dnia 31 marca 2011 r. w sprawie naturalnych wód mine-
ralnych, wód źródlanych i wód stołowych.
Dz. Ust. 85, poz. 466.
14. Rozporządzenie Ministra Zdrowia z dnia 19 listopada 2002 r. w sprawie wymagań dotyczących
jakości wody przeznaczonej do spożycia przez ludzi.
Dz. Ust. 203, poz. 1718.
15. Rozporządzenie Ministra Zdrowia z dnia 29 marca 2007 r. w sprawie jakości wody przeznaczo-
nej do spożycia przez ludzi.
Dz. Ust. 61, poz. 417.
16. Rozporządzenie Ministra Zdrowia z dnia 20 kwietnia 2010 r. zmieniające rozporządzenie
w sprawie jakości wody przeznaczonej do spożycia przez ludzi.
Dz. Ust. 72, poz. 466.
17. Szczepaniec–Cięciak E., Kościelniak P.
(red.)
,
1999:
Chemia środowiska, ćwiczenia i seminaria, cz. 1.
Wydawnictwo UJ, Kraków: 379–435; 519–529.
Wyszukiwarka
Podobne podstrony:
Abolicja podatkowa id 50334 Nieznany (2)
4 LIDER MENEDZER id 37733 Nieznany (2)
katechezy MB id 233498 Nieznany
metro sciaga id 296943 Nieznany
perf id 354744 Nieznany
interbase id 92028 Nieznany
Mbaku id 289860 Nieznany
Probiotyki antybiotyki id 66316 Nieznany
miedziowanie cz 2 id 113259 Nieznany
LTC1729 id 273494 Nieznany
D11B7AOver0400 id 130434 Nieznany
analiza ryzyka bio id 61320 Nieznany
pedagogika ogolna id 353595 Nieznany
Misc3 id 302777 Nieznany
cw med 5 id 122239 Nieznany
D20031152Lj id 130579 Nieznany
mechanika 3 id 290735 Nieznany
więcej podobnych podstron