
Dr EWA CZERWIENIEC
Katedra Inżynierii i Chemii Środowiska
Wydział Budownictwa i Inżynierii Środowiska
Politechnika Rzeszowska
Chemia
Materiały pomocnicze dla studentów
Kierunku Budownictwo
Rzeszów 2013

2
KOROZJA METALI
Korozja (łac. corrosio – zżeranie) - niszczenie materiałów pod wpływem działania
otaczającego je środowiska (atmosfery, opadów, wód), jak i czynników technologicznych
uwalnianych do atmosfery w wyniku działalności człowieka. Są to tlenki siarki, azotu,
dwutlenek węgla, kurz, itp. oraz wszelkiego rodzaju chemikalia. Najczęściej zjawisko korozji
odnosimy do metali i ich stopów, jednakże dotyczy ono również tworzyw niemetalowych,
takich jak beton, materiały ceramiczne, tworzywa sztuczne, itp.
Mnogość czynników wywołujących korozję, warunki i przebieg procesu korozyjnego
powoduje wiele różnych rodzajów korozji (m.in. korozja elektrochemiczna, chemiczna,
atmosferyczna, cierna, szczelinowa i in.). Najczęściej korozja przebiega według mechanizmu
elektrochemicznego i chemicznego.
Korozja chemiczna
Jest wynikiem reakcji zachodzących między metalem a środowiskiem agresywnym nie
mającym cech elektrolitu. Następuje w wyniku działania suchych gazów (tlen, wodór, chlor,
ditlenek węgla, siarkowodór, amoniak) lub bezwodnych cieczy nie przewodzących prądu
elektrycznego np. ropa naftowa i jej pochodne. Wyróżnia się korozję przez rozpuszczanie i
korozję przez utlenianie. Korozją chemiczną może być działanie tlenu na metale w
podwyższonej temperaturze, w rezultacie którego na powierzchni metalu powstaje warstwa
tlenku. Utlenianie metalu do jego tlenków nie zawsze jest procesem szkodliwym. Jeżeli
warstwa tlenku jest dostatecznie zwarta i mocno związana z powierzchnią metalu, chroni
(pasywuje) metal przed dalszym utlenianiem.
Pasywacja jest więc procesem, w którym substancja aktywna chemicznie w danym
środowisku wytwarza na swojej powierzchni powłokę, utworzoną z produktów reakcji tej
substancji z otoczeniem. Pasywacja jest wtedy, gdy powłoka ta jest całkowicie odporna na
dalsze reakcje chemiczne z tym środowiskiem i jednocześnie na tyle szczelna, że stanowi
barierę ochronną dla reszty substancji, którą otacza. Proces pasywacji odnosi się zasadniczo
do metali. Może być procesem naturalnym, wynikającym z właściwości danego metalu w
danym środowisku, lub też procesem sztucznie wywołanym przez człowieka. Nie wszystkie
metale ulegają naturalnej pasywacji, a ponadto pasywacja ta przebiega odmiennie dla

3
różnych środowisk. Glin jak wynika z jego położenia w szeregu napięciowym metali jest
metalem nieszlachetnym. Jednak ten metal i jego stopy odznaczają się w środowisku
obojętnym dużą odpornością na korozję, wynikającą z utworzenia się pasywnej, trudno
rozpuszczalnej warstwy tlenku AlOOH. Podobne tlenkowe warstewki ochronne tworzy cynk,
chrom i nikiel. W przypadku jednak gdy warstwa produktów korozji jest porowata i nie
przylega ściśle do powierzchni metalu, kontakt reagentów z powierzchnią metalu jest
ułatwiony i w efekcie korozja może doprowadzić do zniszczenia metalu. Przebieg tego
procesu zależy w dużej mierze od powinowactwa metali do tlenu. Szybkość korozji zależy
także od charakteru atmosfery oddziaływującej na metal. Do korozji chemicznej zalicza się
także czernienie przedmiotów srebrnych na powietrzu. Głównym składnikiem ciemnego
nalotu jest siarczek srebra powstający w reakcji srebra ze związkami siarki zawartymi w
powietrzu.
Korozja elektrochemiczna
Korozja elektrochemiczna jest najbardziej powszechnym rodzajem korozji. Należy do
niej zjawisko korozji atmosferycznej, która jest działaniem wilgotnego powietrza i zawartych
w nim zanieczyszczeń na metale. Korozja elektrochemiczna powstaje wskutek działania tzw.
krótkozwartych ogniw na styku metalu z elektrolitem. Ogniwa te powstają w rezultacie
niejednorodności chemicznej (lub fizycznej) metalu np. na styku różnych metali, bądź
wskutek niejednorodności krystalicznej w strukturze metalu. Korozja ta najczęściej objawia
się poprzez powierzchniowe ubytki metalu (plamy i wżery), bądź przez obniżenie
wytrzymałości metali. Najczęściej szybkość korozji określa się przez ubytek masy próbki
metalu pod wpływem działania czynnika korodującego na jednostkę powierzchni i czasu.
Podstawy teoretyczne korozji elektrochemicznej
Półogniwo stanowi układ składający się z metalu zanurzonego do roztworu jego soli.
Potencjału półogniwa nie można bezpośrednio zmierzyć, mierzy się natomiast różnicę
potencjału między tym półogniwem a innym układem o znanym, lub umownie przyjętym
potencjale. Takim układem jest elektroda wodorowa, której potencjał umownie równy jest
zero. Elektroda wodorowa składa się z platyny pokrytej czernią platynową (bardzo subtelnie
rozdrobniona platyna), która zanurzona jest w roztworze zawierającym jony H
+
o stężeniu 1
mol/dm
3
, oraz obmywana jest strumieniem gazowego wodoru pod ciśnieniem 1013 hPa w

4
temperaturze 25
o
C. Elektroda wodorowa to elektroda odniesienia dla określenia potencjału
każdego innego półogniwa.
Potencjał normalny stanowi różnicę potencjałów między elektrodą wodorową a
półogniwem składającym się z metalu zanurzonego w roztworze jego soli o stężeniu 1
mol/dcm
3
w temperaturze 25
o
C. Wartości normalnych potencjałów półogniw dla różnych
metali są różne. Jeżeli normalne potencjały różnych metali uszeregujemy w kolejności
wzrastających (lub malejących) wartości to otrzymamy szereg napięciowy metali. Szereg
napięciowy wybranych metali (i wodoru) wygląda następująco:
Na < Mg < Al < Zn < Fe < Cd < Co < Ni < Pb < H < Cu < Hg < Ag < Au
Każdy metal tego szeregu wypiera następne metale z roztworu ich soli. Wszystkie metale
występujące w szeregu przed wodorem posiadają ujemne potencjały normalne. Są to metale
“nieszlachetne”, które wypierają wodór z kwasów czyli rozpuszczają się w kwasach z
wydzieleniem wodoru. Wszystkie metale występujące w szeregu napięciowym za wodorem
posiadają dodatnie potencjały normalne. Są to metale “szlachetne”, które nie wypierają
wodoru z kwasów. Położenie metalu w szeregu napięciowym, a więc wartość jego potencjału
normalnego posiada bardzo istotne znaczenie dla podatności metalu na korozję
elektrochemiczną. Im bardziej ujemna jest wartość potencjału normalnego metalu tym
większą posiada on tendencję do przechodzenia do roztworu.
Stal oprócz żelaza zawiera także ok. 0,2 % węgla w postaci grafitu lub węgliku żelaza
Fe
3
C. Na powierzchni stali znajdują się więc obszary o różnym składzie chemicznym, które w
zetknięciu z roztworem elektrolitu przyjmują różne potencjały. Powstają w ten sposób
mikroogniwa, w których żelazo jest zawsze anodą, natomiast zarówno grafit jak i węglik
żelaza pozostają katodą. W rezultacie żelazo ulega utlenieniu i przechodzi do roztworu:
Fe → Fe
2+
+ 2 e
-
Reakcja ta może zachodzić tylko wówczas, gdy składniki roztworu będą ulegać redukcji. W
środowisku kwaśnym będzie zachodził równocześnie proces:
2H
+
+ 2e
-
→ H
2

5
Zaś w środowisku obojętnym w którym stężenie jonów wodorowych jest małe, redukcji ulega
rozpuszczony w wodzie tlen: 1/2 O
2
+ H
2
O + 2 e
-
→ 2 OH
-
.
Proces redukcji będzie zachodził na powierzchni grafitu względnie węgliku żelaza. Opisane
wyżej procesy redukcji i utleniania zachodzą równocześnie lecz nie w tym samym miejscu:
utlenianie: Fe → Fe
2+
+ 2 e
-
redukcja: 1/2 O
2
+ H
2
O + 2 e
-
→ 2 OH
-
anoda katoda
Reakcja sumaryczna:
Fe + 1/2 O
2
+ H
2
O → Fe(OH)
2
Według podanego zapisu w przestrzeni anodowej żelazo utlenia się do jonów Fe
2+
(utlenianie
jest reakcją anodową) zaś w przestrzeni katodowej zachodzi redukcja rozpuszczonego w
wodzie tlenu do jonów OH
-
(redukcja jest reakcją katodową). Końcowym produktem korozji
żelaza jest wodorotlenek żelaza(II), który jest dalej utleniany tlenem atmosferycznym do
uwodnionego tlenku żelaza(III) czyli rdzy:
4 Fe(OH)
2
+ O
2
→ 2 Fe
2
O
3
· H
2
O + 2 H
2
O
rdza
Szczególnym przypadkiem korozji elektrochemicznej są zjawiska korozji obserwowane na
styku dwóch różnych metali. W obecności wilgoci na styku metali powstaje lokalne ogniwo
składające się z dwóch półogniw. W półogniwie o mniejszym potencjale elektrochemicznym
będzie dominowała reakcja powodująca przejście metalu w formie jonowej do roztworu:
Me
1
→ Me
n+
+ n e
-
W półogniwie metalu o większym potencjale elektrochemicznym będzie dominowała reakcja
odwrotna, w rezultacie której jony metalu z roztworu będą przechodziły do powierzchni
metalu:
Me
2
n+
+ n e
-
→ Me
Zgodnie z tą zasadą metal mniej szlachetny zanurzony w roztworze soli metalu bardziej
szlachetnego będzie wypierał jony metalu bardziej szlachetnego, zgodnie z reakcją:

6
Me
1
+ Me
2
n+
→ Me
1
n+
+ Me
2
Typowym przykładem takiego oddziaływania jest zanurzenie blaszki cynkowej do roztworu
zawierającego jony miedzi Cu
2+
. W tym przypadku obserwuje się wydzielanie miedzi z
roztworu, która osadza się na blaszce cynkowej zgodnie z reakcją:
Cu
2+
+ Zn → Cu + Zn
2+
Ochrona metali przed korozją
W zależności od rodzaju korozji i charakteru chemicznego czynników korozyjnych
istnieje wiele sposobów zapobiegania lub zmniejszania skutków korozji:
1. Dobór odpowiedniego metalu lub stopu.
2. Osłabienie agresywności środowiska. Sposób ten można stosować, gdy ilość ośrodka
atakującego jest ograniczona:
• przez usuwanie tlenu z elektrolitów o odczynie obojętnym np. odpowietrzanie wody
kotłowej
• stosowanie inhibitorów (opóźniaczy).
3. Stosowanie ochrony katodowej i protektorowej
4. Stosowanie powłok ochronnych
Powszechnie stosowane powłoki ochronne:
- powłoki nieorganiczne: metalowe i niemetalowe
- powłoki organiczne: farby, lakiery, tworzywa sztuczne, smoła i smary.
Powłoki metalowe wytwarzane na skalę przemysłową dzielimy na dwie grupy: anodowe i
katodowe.
Powłoki anodowe są wykonane z metali o bardziej ujemnym potencjale elektrochemicznym
(mniej szlachetnych) niż metal chroniony. Pokrywanie metali powłokami anodowymi
zapewnia chronionemu metalowi ochronę katodową, gdyż powłoka z metalu mniej
szlachetnego działa w charakterze anody jako protektor. Jako przykład powłok anodowych
można wymienić cynk i kadm. Najważniejszym, praktycznym zastosowaniem powłok
anodowych jest pokrywanie stali powłoką cynkową (blachy ocynkowane). W przypadku

7
pokrywania powierzchni stalowych cynkiem w razie pojawienia się rysy lub szczeliny tworzy
się ogniwo w którym katodą jest żelazo zaś anodą cynk. W tej sytuacji do roztworu
przechodzą jony cynku a nie jony żelaza. Tak więc w przypadku pokrywania metali
powłokami anodowymi, powłoka pokrywająca nie musi być idealnie szczelna.
Powłoki katodowe są wykonane z metali bardziej szlachetnych niż metal chroniony.
Przykładem powłok katodowych są np. powłoki z miedzi, niklu, chromu, cyny lub srebra.
Powłoka katodowa jest skuteczna tylko wówczas, kiedy cała powierzchnia stalowa jest nią
szczelnie pokryta. Po utworzeniu szczeliny powstaje mikroogniwo, w którym żelazo jest
anodą i ono ulega rozpuszczeniu, co przyspiesza korozję, a metal szlachetny staje się katodą
ogniwa. W rezultacie uszkodzenia powłoki katodowej szybkość korozji w miejscu
uszkodzenia jest większa niż w przypadku braku powłoki katodowej.
Metaliczne powłoki ochronne mogą być nakładane przez: zanurzenie w ciekłym metalu,
platerowanie (zwalcowanie na gorąco), natryskiwanie roztopionego metalu na powierzchnię
chronioną i elektrolizę.
Niemetaliczne powłoki ochronne wywoływane są na powierzchni metali przez wytworzenie
na niej związku chemicznego w wyniku zabiegów chemicznych jak:
- utlenianie (oksydowanie) mające na celu wytworzenie na chronionym metalu pasywnych
warstewek tlenkowych
- fosforanowanie za pomocą kwasu fosforowego (tworzą się trudno rozpuszczalne fosforany
metali)
- chromianowanie za pomocą mieszaniny kwasu chromowego i siarkowego w wyniku którego
tworzą się powłoki chromianowe.
Do niemetalicznych powłok ochronnych zalicza się również emalie szkliste, które wyróżniają
się dobrą odpornością na działanie alkaliów, kwasów a także na działanie rozpuszczalników
organicznych i na działanie podwyższonych temperatur.
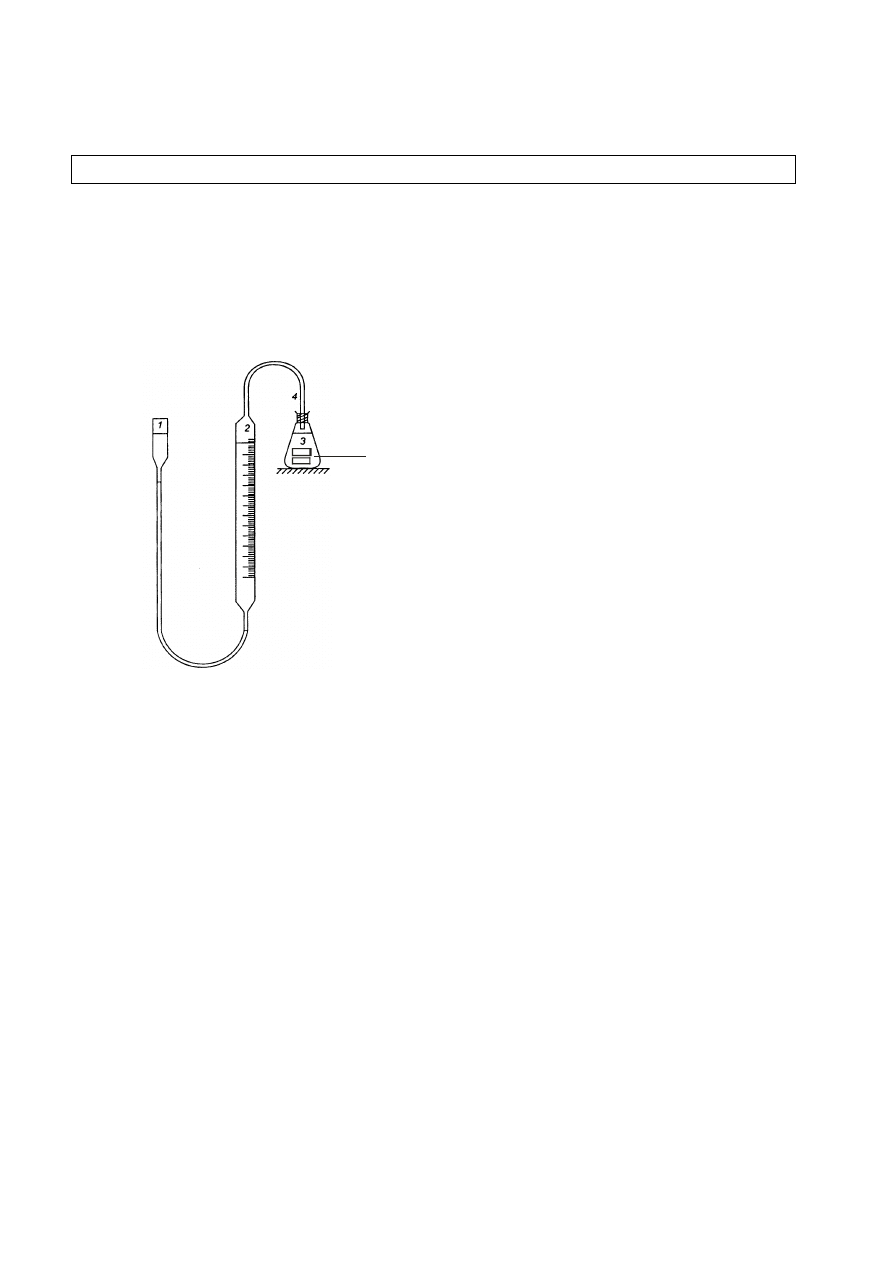
8
Ć W I C Z E N I A L A B O R A T O R Y J N E Z C H E M I I
BADANIE ODPORNOŚCI KOROZYJNEJ METALI
SPRZĘT I SZKŁO:
ODCZYNNIKI I MATERIAŁY:
zestaw pomiarowy jak na rysunku
blaszki
stalowe
1 – zbiornik, 2 – biureta, 3 – kolba stożkowa, 4 –
wąż gumowy.
H
2
SO
4
, roztwór 2M
blaszki stalowe.
ZASADA POMIARU:
W wyniku zachodzenia korozji próbek metalu następuje rozpuszczanie się metalu.
Towarzyszy temu wydzielanie gazowego wodoru. W odniesieniu do żelaza sumaryczną
reakcję można zapisać:
Fe + H
2
SO
4
FeSO
4
+ H
2
Z reakcji tej wynika, że rozpuszczeniu jednego mola żelaza towarzyszy wydzielanie się jednego
mola wodoru (22,4 dm
3
w temperaturze 25
C i pod ciśnieniem 1 atm.). Mierząc objętość
wydzielonego wodoru można określić ubytek masy dla danej próbki.

9
WYKONANIE ĆWICZENIA:
1)
Blaszki stalowe zmierzyć przy użyciu linijki i wyliczyć ich powierzchnie oraz objętość.
2)
Następnie zważyć je na wadze laboratoryjnej (zapisać masę - m).
3)
Zważone blaszki umieścić w kolbie stożkowej (3) i zalać 2m kwasem siarkowym tak,
aby zostały one całkowicie przykryte.
4)
Kolbę stożkową umieścić w statywie i zamknąć korkiem połączonym z pipetą.
5)
Odnotować czas i poziom cieczy w biurecie.
6)
Układ pozostawić na 40 min, co 10 min notować przyrost wydzielającego się wodoru.
7)
Po upływie zaplanowanego czasu wyjąć korek i wymontować kolbkę.
8)
Pozostały w kolbce roztwór wylać rozcieńczając go uprzednio wodą.
9)
Blaszki przepłukać wodą i osuszyć bibułą.
OBLICZANIE WYNIKÓW:
Uzyskane wyniki zestawić w tabeli:
Czas [min]
Obj. H
2
[cm
3
]
10
20
30
40
m [g]
V [cm
3
]
S [cm
2
]
d [g cm
-3
] = m
1
V
-1
7,8
V
C
[g cm
-2
doba
-1
]
V
p
[mm rok
-1
]
Stopień odporności korozyjnej
o
k
(wg. PN-78/H-04608)
1000
4
,
22
56
V
m
gdzie:
m – ubytek masy [g],
56 – masa jednego mola żelaza [g],

10
V – objętość wydzielonego wodoru [cm
3
],
22,4 – objętość jednego mola gazu [dm
3
]
t
S
m
V
C
gdzie:
V
C
– szybkość korozji [g cm
-2
doba
-1
],
m – ubytek masy [g]
t – czas trwania procesu [doba],
S – powierzchnia czynna próbki [cm
2
].
C
p
V
d
V
1000
365
gdzie:
V
C
– szybkość korozji [g cm
-2
doba
-1
],
V
P
– liniowa szybkość korozji [mm rok
-1
],
d – gęstość właściwa materiału [g cm
-3
].
Liniowa szybkość
korozji V
p
, mm/rok
Stopień odporności
korozyjnej,
k
Liniowa szybkość
korozji V
p
, mm/rok
Stopień odporności
korozyjnej,
k
V
p
0,001
1
0,1 < V
p
0,5
6
0,001 < V
p
0,005
2
0, 5 < V
p
1,0
7
0,005 < V
p
0,01
3
1,0 < V
p
5,0
8
0,01 < V
p
0,05
4
5,0 < V
p
10,0
9
0,05 < V
p
0,1
5
V
p
10
10
Otrzymane w ćwiczeniu wartości przedstawić w formie graficznej w postaci zmiany
ilości wydzielonego wodoru w zależności od czasu badania.

11
Analiza chemiczna
Metody analizy chemicznej
Badania chemiczne w budownictwie związane są z kontrolą jakości materiałów
budowlanych i ich składników, oceną prawidłowości przebiegu różnych operacji
technologicznych podczas wytwarzania i stosowania materiałów budowlanych, a także oceną
agresywności chemicznej środowiska użytkowania budowli i doborem odpowiedniego
materiału do ochrony budowli przed korozją. Badania chemiczne mają istotne znaczenie
podczas diagnostyki stanu konstrukcji żelbetowej (stopień skażenia betonu, stopień
karbonatyzacji kamienia cementowego, stopień skorodowania stali zbrojeniowej) oraz przy
prognozowaniu jej trwałości. Metody chemiczne są pomocne również przy ocenie
"kompatybilności" współpracujących ze sobą materiałów budowlanych (negatywny przykład
stanowi tu alkaliczna reakcja cementu z kruszywem) oraz przy ocenie przydatności
materiałów zbyt długo lub niewłaściwie składowanych. Metody te często służą do ustalania
przyczyn wystąpienia usterek budowlanych czy też uszkodzeń budowli. Wszystkie te sposoby
postępowania związane są z identyfikacją materiału i wchodzą w zakres analizy chemicznej.
Celem analizy chemicznej jest oznaczenie składu jakościowego i ilościowego oraz
określenie struktury danej substancji.
Podział analizy chemicznej:
•
analiza jakościowa - mająca na celu stwierdzenie obecności poszukiwanej substancji lub jej
braku, bądź identyfikacja substancji nie znanej w badanym materiale,
•
analiza ilościowa - polegająca na oznaczeniu stężenia określonej substancji w badanej
próbce;
Obok metod wagowych i objętościowych, stosuje się także metody fizykochemiczne,
polegające na pomiarze różnych właściwości fizyko-chemicznych (np. optyczne, elektryczne,
adsorpcja) zależnych od stężeń badanych składników; metody te wykorzystują często
skomplikowane przyrządy i nazywane są metodami instrumentalnymi.
Metody analityczne dzieli się również ze względu na najmniejszą wielkość próbki
koniecznej do badań, a także ze względu najmniejsze wykrywalne stężenie oznaczanej
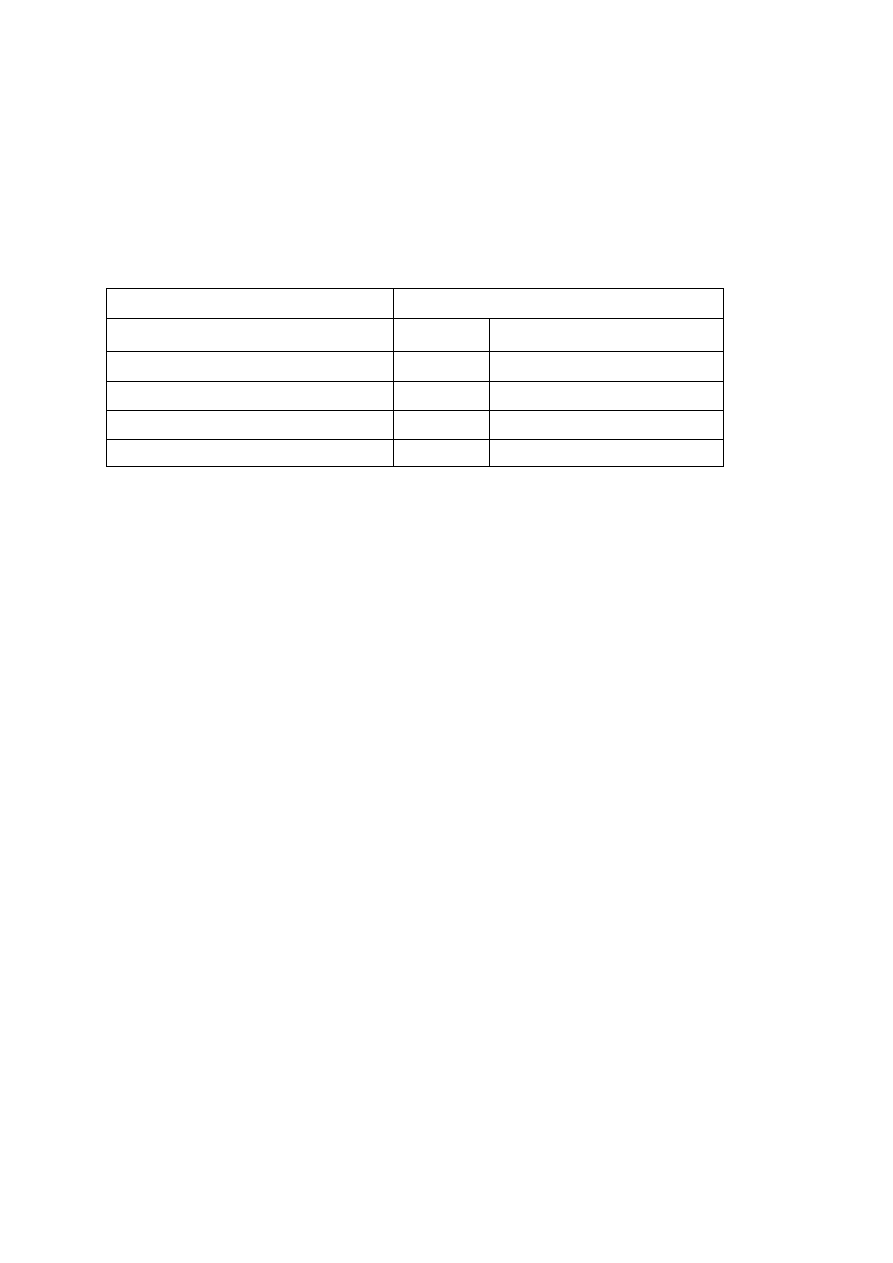
12
substancji – tabela 1. W budownictwie stosuje się zazwyczaj metody makro- analityczne.
Laboratoria budowlane z reguły nie zajmują się analizą chemiczną związków organicznych, a
to ze względu na ogromną liczbę tych związków oraz stopień skomplikowania samej ich
struktury.
Tabela 1. Skala metod analitycznych
Najmniejsza ilość analizowanej próbki
objętość [cm
3
]
masa [g]
Skala makro (decygramowa)
0,1
1
Skala półmikro (centygramowa)
0,01
0,1
Skala mikro (miligramowa)
0,001
0,01
Skala Ultramikro (mikrogramowa)
0,000001
0,00001
Klasyczne metody analizy jakościowej
Analiza półilościowa
W budownictwie stosunkowo często oprócz analizy jakościowej wystarczająco przydatne
są oznaczenia półilościowe, wykonywane zamiast pełnej analizy ilościowej. Są to oznaczenia
w zasadzie podobne do jakościowych, ale dające równocześnie przybliżoną ocenę zawartości
danej substancji w badanym materiale. Przeważnie dla oznaczenia tej zawartości posługuje
się porównaniem z odpowiednim wzorcem. Porównanie przeprowadza się wizualnie bez
specjalnych przyrządów.
Przykładem analizy półilościowej może być oznaczenie zawartości Cl
-
i SO
4
2-
w roztworach wodnych w aspekcie ich agresywności chemicznej wobec betonu:
Cl
-
+ Ag
+
→ AgCI
biały osad
SO
4
2-
+ Ba
2+
→ BaSO
4
biały osad
W analizie półilościowej ważny problem stanowi interpretacja uzyskanych wyników. Zwykle
służy ona jako kryterium podjęcia decyzji o dalszym postępowaniu analitycznym. Na
przykład, gdy półilościowe oznaczenie Cl
-
w ekstrakcie wodnym z betonu wykaże tylko ślady
jonów chlorkowych, to analizy ilościowej można zaniechać. Jeżeli powstanie obfity osad, to
należy podjąć odpowiednie kroki w zakresie ochrony przed korozją i również w tym

13
przypadku dalsza analiza ilościowa jest niekonieczna. Jeśli w próbie tej wystąpi zmętnienie,
to - w celu ustalenia, czy potrzebne są zabiegi ochronne - konieczne jest przeprowadzenie
analizy ilościowej. W analogiczny sposób można oszacować wstępnie agresywność
siarczanową wody w stosunku do betonu: zmętnienie - niepotrzebna analiza ilościowa ani
ochrona betonu, obfity osad - agresywność silna, potrzebna jest ochrona odpowiednia dla
takiego stopnia agresywności, silne zmętnienie i wytrącenie osadu po dłuższym czasie -
konieczne jest przeprowadzenie analizy ilościowej w celu dobrania właściwego sposobu
ochrony.
Analiza ilościowa
W toku klasycznej analizy ilościowej wyróżnia się 3 istotne etapy:
wyodrębnienie poszukiwanej substancji (pierwiastek, związek) z badanego materiału;
zabieg ten nie zawsze jest konieczny - można często wykonać oznaczenie bezpośrednio na
tak zwanej próbce pierwotnej,
oznaczenie ilościowe zawartości (stężenie) substancji,
obliczenie i sformułowanie wyników analizy.
Najczęściej stosowane metody analizy ilościowej to analiza wagowa - metody
grawimetryczne, i analiza objętościowa, do której zalicza się metody miareczkowe oraz
gazometryczne. Gazometria polega na pomiarze objętości gazu wydzielającego się w reakcji.
Przykładem jej zastosowania w budownictwie może być oznaczenie zawartości niedopału w
wapnie palonym poprzez pomiar objętości CO
2
wydzielającego się w wyniku reakcji węglanu
wapnia z kwasem solnym.
Wyniki analiz wyraża się w różny, często umowny sposób.
W analizie gazów (np. powietrza) stężenie składników podaje się zwykle w mg danej
substancji (CO
2
, SO
2
, C1
2
, HCI, HF, N
2
O
3
) w 1 m
3
powietrza, a także w ppm (parts per
million), czyli w mg/kg lub cm
3
/m
3
. W analizie wody (np. woda do celów budowlanych, woda
środowiskowa itp.) stężenie oznaczanych substancji podaje się w milimolach lub
miligramorównoważnikach (milivalach) jonu (Cl
-
, NO
3
-
, Ca
2+
, Mg
2+
, NH
4+
itp.) lub związku
(np. CO
2
) w 1 dm
3
wody, a także w miligramach (mg) danego jonu lub substancji w 1 dm
3
wody.
14
W materiałach stałych zawartość oznaczanej substancji podaje się często umownie w
przeliczeniu na zawartość wybranego składnika, np. zawartość alkaliów jako Na
2
(bez względu
na to, czy występują one jako związki sodu czy potasu). Zawartość fosforanów (sole kwasu
H
3
PO
4
) podaje się przeważnie jako %P
2
O
5
, a rzadziej jako %PO
4
3-
itp. Zawartość krzemianów
wyraża się w przeliczeniu jako udział SiO
2
, zawartość glinianów jako Al
2
O
3
soli Fe
2+
, Fe
3+
i
A1
3+
jako tak zwaną "sumę tlenków" Al
2
O
3
+ Fe
2
O
3
bez rozróżnienia obu tych składników itp.
Analiza objętościowa (miareczkowa)
Bardzo często stosowana metoda analizy ilościowej, ze względu na szybkość wykonania
oraz proste wyposażenie laboratoryjne.
Miareczkowanie to metoda analityczna polegająca na ilościowym oznaczaniu zawartości
substancji, przez:
stopniowe dodawanie do jej roztworu odczynnika o dokładnie znanym stężeniu;
stechiometryczny przebieg reakcji
wyznaczenie objętości odczynnika potrzebnej do osiągnięcia momentu w której ilość
dodawanego odczynnika do roztworu będzie chemicznie równoważna ilości substancji
badanej
PODSTAWOWE POJĘCIA
-
Substancja miareczkowana – substancja oznaczana metodą miareczkowania;
-
Substancja miareczkująca (= titrant, roztwór mianowany) dodawany odczynnik o
znanym stężeniu;
-
Punkt równoważnikowy – punkt odpowiadający istniejącej w roztworze równowadze;
-
Punkt końcowy – punkt wynikający z obserwacji zmian w roztworze, uznawany za
moment zakończenia miareczkowania.
Zawartość oznaczanej substancji po analizie oblicza się na podstawie dokładnie
zmierzonej (odczytanej z biurety) objętości zużytego roztworu miareczkującego V
2
, przy
znanym jego stężeniu Cn
2
oraz znanej objętości wody wziętej do badania V
1
:
Cn
1
V
1
= Cn
2
V
2
→ Cn
1
= Cn
2
V
2
/V
1
1-roztwór badany, 2 - titrant

15
Ć W I C Z E N I A L A B O R A T O R Y J N E Z C H E M I I
OZNACZANIE STĘŻENIA ROZTWORU HCl
Oznaczanie stężeń roztworów kwasów i zasad wykonuje się w oparciu o reakcje zobojętniania
(alkacymetria). W przypadku oznaczania stężenia kwasu solnego wykorzystuje się reakcję
zobojętnienia z wodorotlenkiem sodowym:
HCl + NaOH
NaCl + H
2
O
Dla reakcji jednowartościowych substratów (HCl i NaOH) można wykorzystać równanie
analityczne używając stężeń molowych:
C
1
V
1
= C
2
V
2
Gdzie C
1
i C
2
oznaczają stężenia molowe roztworów kwasu i zasady,
V
1
i V
2
– odpowiednio objętości roztworów w/w związków.
WYKONANIE ĆWICZENIA:
Przygotować stanowisko do miareczkowania (zgodnie z instrukcją na następnej stronie) stosując
roztwór miareczkujący NaOH o stężeniu 0,1 mol/l. Do kolby stożkowej o pojemności 250
cm
3
odmierzyć pipetą 10 cm
3
badanego roztworu HCl oraz około 20 cm
3
wody destylowanej,
a następnie wlać 1-2 krople oranżu metylowego. Miareczkować (zgodnie z instrukcją) do
zmiany zabarwienia z czerwonego na żółte. W momencie zmiany zabarwienia przerwać
dodawanie zasady, jeśli powróciła dawna barwa wskaźnika, dodać jeszcze tyle kropli
roztworu miareczkującego, aby zmiana zabarwienia roztworu kwasu była całkowita i trwała.
Odczytać poziom cieczy w biurecie i obliczyć objętość roztworu zużytego do
miareczkowania.
16
Powtórzyć miareczkowanie takiej samej objętości kwasu, używając jako wskaźnika 1-2 krople
roztworu fenoloftaleiny. Miareczkowanie zakończyć z pojawieniem się różowego
zabarwienia roztworu. Dane z doświadczenia zestawić w tabeli wyników.
C
NaOH
V
HCl
wskaźnik
V
NaOH
oranż metylowy
fenoloftaleina
średnia
C
HCl
=
OBLICZANIE WYNIKÓW:
Średnią objętość zasady użytą do zmiareczkowania kwasu należy wykorzystać do obliczenia
stężenia molowego badanego roztworu kwasu wg równania:
HCl
NaOH
NaOH
HCl
V
V
C
C
gdzie: C
NaOH
– stężenie r-ru NaOH, V
NaOH
– objętość r-ru NaOH użyta do zmiareczkowania
kwasu, V
HCl
– objętość HCl użyta do miareczkowania (10 cm
3
).
Instrukcja miareczkowania
Podstawowym przyrządem w analizie miareczkowej jest biureta. Jest to wąska, kalibrowana
rurka szklana, zakończona kranikiem (szklanym lub teflonowym), która umożliwia
precyzyjne odmierzanie roztworu niewielkimi porcjami (kroplami) oraz pomiar objętości
zużytego roztworu miareczkującego (titranta).
Przygotowanie i stosowanie biurety:
Biureta powinna być umocowana pionowo.

17
Kran biurety powinien być szczelny.
Biuretę napełnia się nieco powyżej kreski zerowej (jeśli wlewa się przez lejek – należy go po
napełnieniu biurety wyjąć).
Całkowicie usunąć powietrze z końcówki biurety.
Doprowadzić poziom titranta w biurecie dokładnie do kreski zerowej spuszczając nadmiar.
Ewentualną kroplę titranta na końcówce biurety usunąć przez dotknięcie do ścianki
podstawionego naczynia szklanego.
Wykonanie miareczkowania:
Analizowaną próbkę w kolbie stożkowej umieszcza się pod wylotem biurety. Wtedy, gdy to
konieczne dodaje się wskaźnik.
Każde miareczkowanie zaczyna się od zera.
Palcami lewej ręki delikatnie otworzyć kurek biurety, a prawą ręką cały czas mieszać ciecz w
kolbie stożkowej ruchem wirowym.
Aby zmiana barwy była lepiej zauważalna można podłożyć pod kolbę biały papier (ekran).
Roztwór mianowany spuszcza się z biurety początkowo dosyć szybko, a zbliżając się do
końca miareczkowania coraz wolniej, po kropli.
Cały czas należy obserwować roztwór w kolbie, a nie biuretę - poziom titranta w biurecie
odczytuje się zawsze po zakończeniu miareczkowania.
Całe miareczkowanie należy prowadzić przy jednorazowym napełnianiu biurety.
Miareczkowanie powinno się wykonywać w miejscu dobrze oświetlonym.
Oznaczenia wykonuje się zwykle trzykrotnie, przy czym różnica między miareczkowaniami
nie powinna być większa niż 0,15 ml.
Po zakończeniu oznaczenia należy opróżnić biuretę.
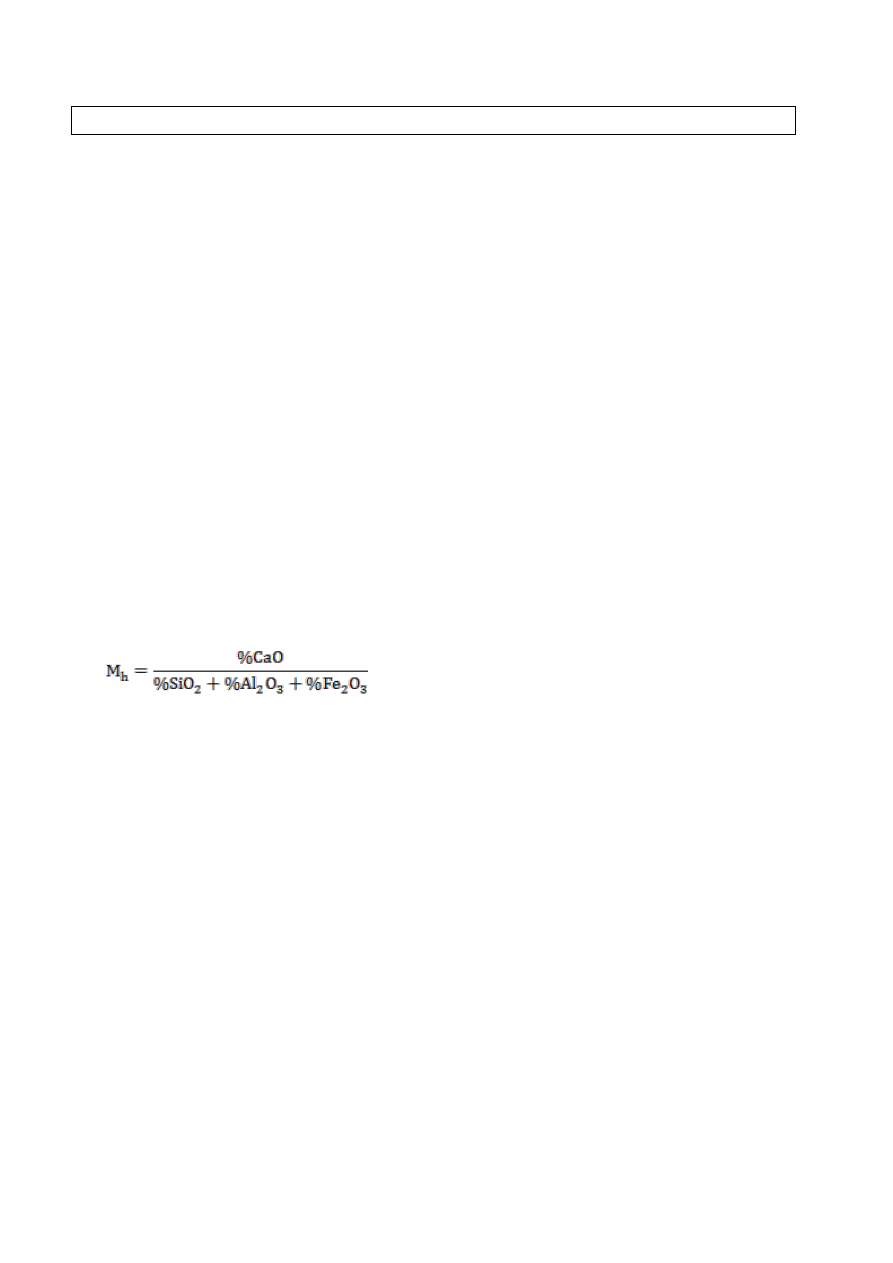
18
Ć W I C Z E N I A L A B O R A T O R Y J N E Z C H E M I I
ANALIZA CHEMICZNA CEMENTU – Oznaczanie zawartości tlenku wapnia i
obliczenia modułów.
Spoiwa mineralne to wypalone i sproszkowane surowce skalne, które po zarobieniu z wodą
tworzą plastyczny zaczyn dający się łatwo formować i dzięki reakcjom chemicznym wiążą i
twardnieją. Spoiwa mineralne są to materiały powstające przez wypalenie i rozdrobnienie na
proszek surowców skalnych. Po zarobieniu wodą (lepiszcza wiążą na drodze procesów
fizycznych – ostudzenia, odparowanie rozpuszczalnika itp.). Spoiwa składają się z mieszaniny
tlenków o charakterze kwaśnym (SiO
2
) lub amfoterycznym (Al
2
O
3
, Fe
2
O
3
) oraz zasadowym
(CaO, MgO), które reagując ze sobą po zarobieniu wodą dają nierozpuszczalne w wodzie
sole. Spoiwa można podzielić na powietrzne (które wiążą tylko przy dostępie powietrza i nie
są odporne na działanie wody) oraz hydrauliczne (które wiążą także pod wodą i są odporne na
wodę). Rodzaj spoiwa określa tzw. moduł hydrauliczny (zasadowości), który jest stosunkiem
ilości [% wag.] tlenku wapniowego do sumy tlenków krzemu, glinu i żelaza.
Moduł hydrauliczny
oblicza się na podstawie wzoru:
dla M
h
> 4,5 spoiwo jest powietrzne;
dla M
h
= 2,5 ÷ 4,5 jest to wapno hydrauliczne;
dla M
h
= 1,7 ÷ 2,4 jest to cement portlandzki.
Cement o M
h
poniżej 1,7 wykazują niedostateczną wytrzymałość mechaniczną, zaś cement o
M
h
powyżej 2,3 ma niedostateczną stałość objętości. Ze wzrostem M
h
wzrasta wytrzymałość,
zwłaszcza początkowa, ale rośnie też ilość ciepła potrzebna do wypału, a z kolei zmniejsza się
odporność na agresję chemiczną.
Wartość modułu hydraulicznego powinna być zawarta w granicach od 2,1 do 3,5.
Moduł glinowy
oblicza się na podstawie wzoru:
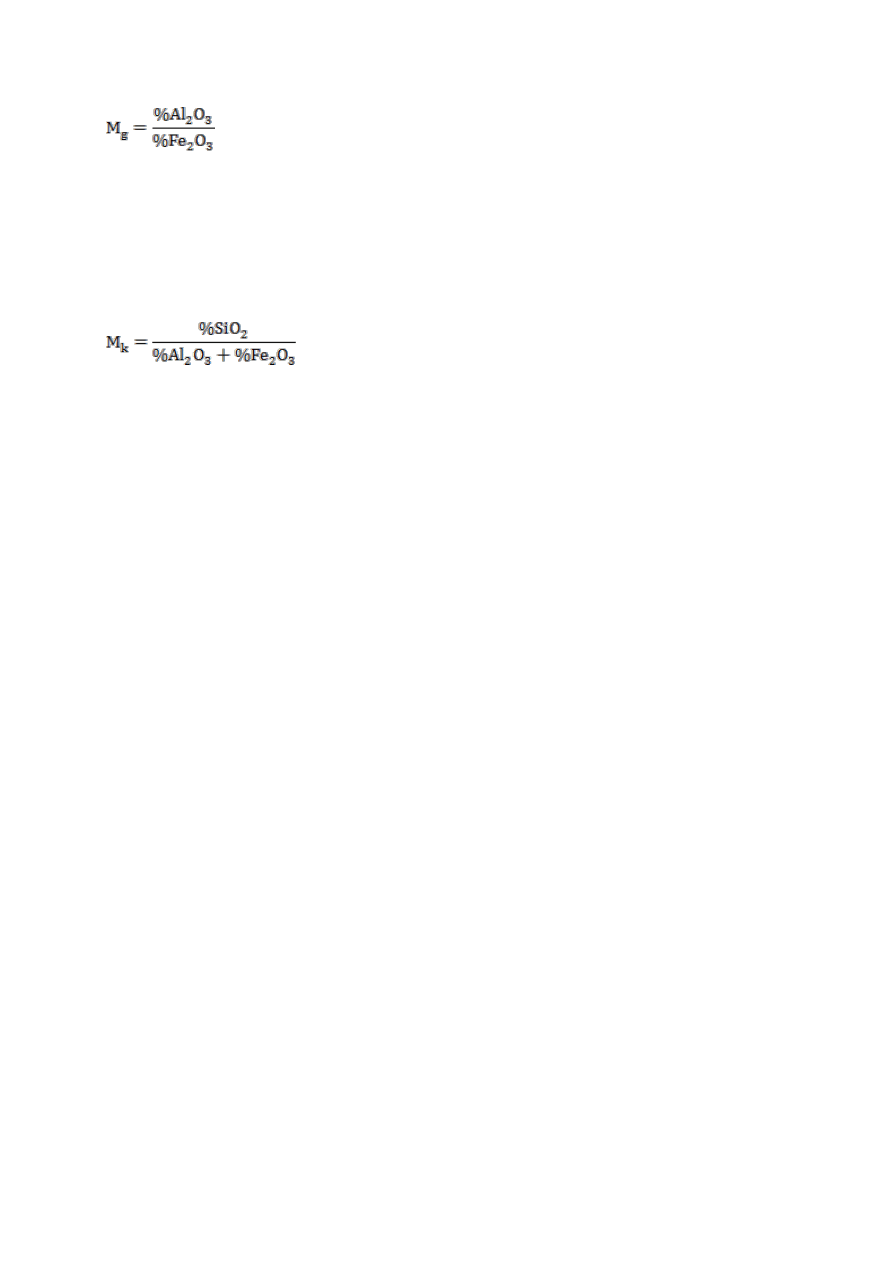
19
Wartość modułu glinowego powinna być zawarta w granicach 1,0-3,0.
Moduł krzemianowy
oblicza się na podstawie wzoru:
Wartość modułu krzemianowego powinna być zawarta w granicach 1,7-3,5.
Wykonanie ćwiczenia:
Sprzęt laboratoryjny: biureta, kolby Erlenmayera, zlewka, cylinder miarowy.
Odczynniki: 0,5 M KOH, 0,5 M HCl, fenoloftaleina.
Przeprowadzenie doświadczenia
1,0 g wysuszonego i dokładnie sproszkowanego cementu wsypać do kolby Erlenmeyera
(kolba stożkowa). Odmierzyć za pomocą cylindra miarowego 80 cm
3
wody destylowanej,
zagotować w zlewce i wlać do kolby stożkowej. Następnie dodać do niej 40 cm
3
0,5 M HCl i
gotować jej zawartość przez 3 minuty, celem odpędzenia dwutlenku węgla. Po przegotowaniu
dodać 3 – 5 kropel 1% roztworu fenoloftaleiny i na gorąco miareczkować 0,5 M roztworem
KOH, aż do wystąpienia różowego zabarwienia roztworu miareczkowanego. Zapisać ilość
cm
3
0,5M KOH zużytego do miareczkowania. Doświadczenie powtórzyć. Zapisać reakcje
zachodzące podczas dodawania do cementu roztworu kwasu solnego oraz podczas
miareczkowania.
Obliczenia
Zawartość wolnego CaO w próbce obliczamy wg. wzoru:
%CaO = (40 – A) · 0,014 ·100%

20
A – ilość cm
3
0,5 M KOH zużyta na zobojętnienie nadmiaru kwasu solnego, który nie
przereagował z wolnym CaO znajdującym się w 1 g cementu;
(40 – A) – ilość cm
3
0,5 M HCl, który przereagował z wolnym CaO znajdującym się w 1 g
cementu;
0,014 – ilość CaO wyrażona w gramach, z którą reaguje 1 cm
3
0,5 M HCl.
Obliczyć wartości modułów: M
h
, M
g
, M
k
Zawartość procentowa pozostałych tlenków:
SiO
2
– 22,12%
Al
2
O
3
– 4,73%
Fe
2
O
3
– 4,63%
ANALIZA INSTRUMENTALNA
Spośród metod instrumentalnych szczególnie często stosuje się metody optyczne,
zwłaszcza kolorymetryczne, z uwagi na szybkość i łatwość ich wykonania. Polegają one na
przeprowadzeniu oznaczanej substancji w związek barwny, a następnie na porównaniu
intensywności powstałego zabarwienia z intensywnością barwy wcześniej sporządzonych
wzorców o znanych stężeniach. Podstawą analizy kolorymetrycznej jest prawo Lamberta-
Beera:
I
t
= I
o
10
-abc
I
o
– natężenie promieniowania padającego na próbkę,
I
t
- natężenie promieniowania przechodzącego przez roztwór
b – grubość warstwy absorbującej
c – stężenie substancji absorbującej
a – współczynnik absorpcji
Wielkością mierzoną jest logarytm stosunku natężenia promieniowania padającego na
próbkę (I
o
) do natężenia promieniowania przechodzącego przez roztwór(I
t
). Wielkość ta nosi

21
nazwę absorbancji (A) lub ekstynkcji (E) i jest proporcjonalna do stężenia (c):
Znając wielkość absorbancji (ekstynkcji) z krzywej wzorcowej odczytuje się stężenie
analizowanej substancji lub oblicza wykorzystując znany kąt nachylenia (α) krzywej do osi
X:
Ć W I C Z E N I A L A B O R A T O R Y J N E Z C H E M I I
OZNACZANIE TLENKU ŻELAZA (III) W CEMENCIE
Zasada oznaczenia polega na rozpuszczeniu próbki cementu w kwasie siarkowym,
zredukowaniu jonów żelaza (III) do żelaza (II) za pomocą chlorowodorku hydroksyloaminy i
po doprowadzeniu pH do wartości 3-4, utworzeniu związku kompleksowego jonów żelaza
(II) z o-fenentroliną.
WYKONANIE ĆWICZENIA:
Odważyć 1g cementu. Odważkę wprowadzić do zlewki o pojemności 250 cm
3
, dodać 40 cm
3
wody destylowanej, 2.5 cm
3
20% roztworu kwasu siarkowego i gotować przez 5 minut.
Następnie sączyć przez twardy sączek do zlewki o pojemności 250 cm
3
. Osad na sączku
przemyć małymi porcjami wody destylowanej (łącznie 5-10 cm
3
). Otrzymany przesącz po
ostudzeniu
przenieść
ilościowo
do
kolby
miarowej
o pojemności 100 cm
3
, dodać 5 cm
3
roztworu chlorowodorku hydroksyloaminy
22
i 15 cm
3
cytrynianu sodu do otrzymania pH 3-4. Następnie dodać 5 cm
3
roztworu
o-fenantroliny, dopełnić wodą do kreski mierniczej i dokładnie wymieszać. Po upływie 10
minut
zmierzyć
absorbancję
barwnego
roztworu
przy
długości
fali
512 nm, wobec ślepej próby. Na podstawie zmierzonej absorbancji z krzywej wzorcowej
odczytać zawartość tlenku żelaza (III).
OBLICZANIE WYNIKÓW:
Zawartość tlenku żelaza (III) Fe
2
O
3
należy obliczyć w procentach wg wzoru:
100
1
m
m
x
gdzie:
m – odważka badanej próbki [g],
m
1
– zawartość tlenku żelaza, odczytana z krzywej wzorcowej [g].
Ć W I C Z E N I A L A B O R A T O R Y J N E Z C H E M I I
ELEKTROLITY – pomiar pH i wyznaczanie stałej dysocjacji
Instrukcja wykonania ćwiczenia:
Oznaczanie pH metodą potencjometryczną:
pH-metr CP-315
1.
Przed każdym pomiarem przepłukać ogniwo wodą destylowaną i osuszyć bibułą
(podczas osuszania bibułą nie wycierać elektrody tylko delikatnie usunąć krople płynu z
membrany).
2.
Włączyć przyrząd przyciskiem ON/OFF.
23
3.
Umieścić elektrodę w zlewce z badaną próbą.
4.
Nacisnąć przycisk pH i po ustabilizowaniu się wartości odczytać na wyświetlaczu
wartość mierzoną.
5.
Po zakończonym pomiarze przepłukać ogniwo i pozostawić elektrodę w zlewce z
wodą destylowaną.
6.
Wyłączyć pH-metr przyciskiem ON/OFF.
Zmierzyć trzykrotnie pH wody wodociągowej oraz destylowanej.
Następnie z otrzymanych roztworów kwasu i soli sporządzić roztwory buforowe odmierzając
starannie do małych kolbek stożkowych za pomocą pipet następujące objętości:
Kolbka
roztwór kwasu [cm
3
]
roztwór soli [cm
3
]
I
5
20
II
10
10
III
20
5
Roztwory starannie wymieszać.
Zmierzyć pH sporządzonych roztworów.
Obliczyć stałą dysocjacji oddzielnie dla każdego roztworu:
Gdzie:
[A
-
] – stężenie soli
[HA] – stężenie kwasu
Otrzymane wyniki wpisać do tabel:
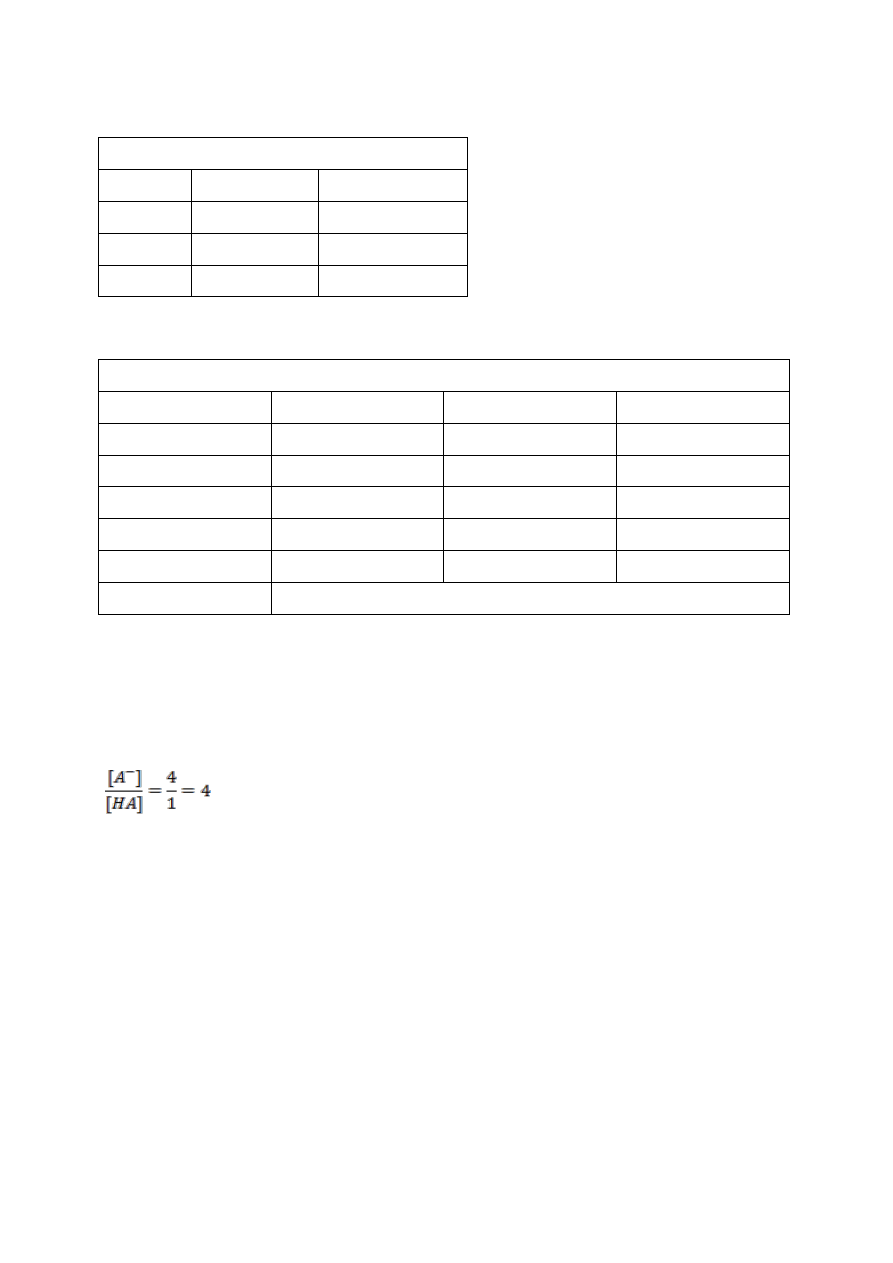
24
Tabela 1
Pomiar pH wody wodociągowej i destylowanej
Lp.
wodociągowa
destylowana
pomiar 1
pomiar 2
pomiar 3
Tabela 2
Wyznaczanie stałej dysocjacji
Roztwór I
Roztwór II
Roztwór III
C
soli
/C
kwasu
pH
log[H
+
]
[H
+
]
K
K
śr
Przykład obliczania:
W kolbce I zmieszano jedną porcję kwasu i cztery porcje jego soli.
Stosunek stężenia soli do kwasu wynosi:
Z pomiaru pH tej mieszaniny uzyskano wynik pH=5,6
Z wzoru na pH wylicza się stężenie kwasu [H
+
]:
pH = -log[H
+
] = 5,6
[H
+
] = 10
-5,6
[H
+
] = 2,5 · 10
-6
Stąd
K = 2,5 · 10
-6
· 4 = 1 · 10
-5

25
WODA W BUDOWNICTWIE
Woda stosowana w budownictwie do zarabiania cementu, zapraw cementowych,
cementowo-wapiennych, betonów oraz mas ceramicznych jest nazywana wodą zarobową.
Pełni ona zasadniczą rolę w kształtowaniu cech technologicznych mieszanek, a także w pro-
cesach wiązania i twardnienia spoiw budowlanych. Występujące w wodzie zarobowej związki
mineralne i substancje organiczne wpływają na procesy wiązania. W przypadku hydratacji
cementu substancje te mogą powodować obniżenie wytrzymałości betonu, występowanie
plam na powierzchni betonu, a także doprowadzić do korozji zbrojenia w żelbecie. Ponadto,
jako element środowiska naturalnego, woda oddziałuje na budowle, a tym samym ma wpływ
na ich trwałość. Woda taka powinna odpowiadać specjalnym wymaganiom. Stąd wodę przed
użyciem do zarabiania mieszanek należy poddać analizie chemicznej. Wyjątek stanowi woda
ze źródła objętego siecią wodociągową.
Woda powinna być przezroczysta, nie zawierać mułu, oleju, tłuszczu i glonów. Niedozwolone
jest używanie do celów budowlanych ścieków przemysłowych i miejskich, jak również wód
morskich. Agresywność wód w stosunku do cementu zależy zarówno od właściwości
cementu jak i wód.
Składnikami, które w sposób zasadniczy wpływają na jakość wody zarobowej są: jony
siarczanowe (SO
4
2-
), jony siarczkowe (S
2-
), kwasy (jony wodorowe; H
+
), cukier, substancje
humusowe, sole wapnia i magnezu.
Siarczany - jony siarczanowe reagują ze składnikami zaczynu cementowego, tworząc
związki znacznie zwiększające swoją objętość i przez to mogące być przyczyną rozsadzania
betonu. Jeżeli agresja siarczanowa jest słaba, to tworzący się w reakcji z wodorotlenkiem
wapnia gips (CaSO
4
·2H
2
O) - do pewnego stopnia - uszczelnia beton. W przypadku dużego
stężenia jonów siarczanowych w wodzie, gips reagując dalej tworzy związki krystalizujące, z
przyłączeniem dużej liczby cząsteczek wody, np. sól Candlota: 3CaO·A1
2
O
3
·3CaS0
4
·32H
2
O,
które powodują zniszczenie betonu.
Siarkowodór i jego sole - ulegają stosunkowo łatwo reakcji utleniania. Jony siarczkowe (S
2-
)
przechodzą w siarczanowe IV (siarczynowe) (SO
3
2-
), a następnie w siarczanowe VI (SO
4
2-
).
Szkodliwe działanie tych ostatnich zostało wyjaśnione wcześniej. Siarkowodór może

26
reagować również z wodorotlenkiem wapniowym tworząc wodorosiarczek wapniowy
Ca(HS)
2
, łatwo rozpuszczalny w wodzie i nie wykazujący właściwości wiążących.
Kwasy (jony H
+
) - są obecne w wodzie jako składniki naturalne, np. kwas węglowy, kwas
huminowy, produkty hydrolizy soli mocnych kwasów i słabych zasad. W wyniku
zanieczyszczenia w wodzie (ściekach) mogą występować również mocne kwasy mineralne,
jak solny czy siarkowy. Kwasy reagują ze składnikami cementu oraz z produktami jego
uwodnienia tworząc łatwo rozpuszczalne związki. Utrudnia to, a czasem uniemożliwia,
wiązanie spoiwa.
Cukier - jest związkiem organicznym zaliczanym do grupy węglowodanów. Głównym
składnikiem cukru jest sacharoza (C
I2
H
22
O
11
), która tworząc z wodorotlenkiem wapniowym
cukrzany wapniowe utrudnia proces wiązania betonu i obniża jego wytrzymałość. W
niektórych przypadkach obecność cukru może całkowicie uniemożliwić wiązanie.
Substancje humusowe - występują głównie w gruntach, glebach bagiennych i torfowiskach.
Powstają w wyniku rozkładu szczątków pochodzenia roślinnego i zwierzęcego. Zawierają
kwas huminowy, ulminowy i kreonowy. Kwas huminowy ma cztery grupy kwasowe zdolne
do reakcji i charakteryzuje się dużą masą cząsteczkową. Kwas ten reaguje z wodorotlenkiem
wapniowym obecnym w zaczynie cementowym tworząc nierozpuszczalny huminian wap-
niowy. Niebezpieczne działanie mogą wykazywać składniki towarzyszące kwasom
humusowym, np. jony SO
4
2-
powstałe przez utlenianie pirytów. Z tego względu stosowanie
do betonu wody zawierającej zawiesiny, grudki, kłaczki jest niewskazane.
Twardość – to cecha wody, która wynika z zawartości w niej związków wapnia i magnezu.
Jednostkami stosowanymi do wyrażania tej cechy wody są stopnie twardości, milimole lub
miligramorównoważniki (milivale) tlenków wapnia lub magnezu. Stopnie twardości wody
(niemiecki
o
n, francuski
o
fr lub angielski
o
ang) odpowiadają określonej zawartości CaO lub
CaCO
3
w 1 litrze wody. Twardość 1 mmol/dm
3
odpowiada obecności w wodzie 56 mg CaO
lub równoważnej ilości 40 mg MgO.
W tabeli poniżej zestawiono współczynniki do przeliczania różnych jednostek twardości
wody:

27
mval/l
o
n
o
fr
o
ang
mmol/l
28 mg CaO lub
50 mg CaCO
3
na 1 litr
10 mg
CaO na 1
litr
10 mg
CaCO
3
na 1 litr
14,3 mg
CaCO
3
na
1 litr
100 mg
CaCO
3
na
1 litr
mval/l
1
2,8
5
3,5
0,5
o
n
0,357
1
1,78
1,25
0,18
o
fr
0,2
0,56
1
0,7
0,1
o
ang
0,29
0,8
1,43
1
0,14
mmol/l
2
5,6
10
7,02
1
Ze względu na rodzaj związków wapnia i magnezu występujących w wodzie rozróżnia się:
- twardość przemijającą (węglanową), wywołaną obecnością wodorowęglanów wapnia i
magnezu. Twardość ta zanika w czasie ogrzewania wody, gdyż zawarte w niej
wodorowęglany ulegają rozkładowi wg reakcji:
Ca(HCO
3
)
2
→ CaCO
3
+ H
2
O + CO
2
Mg(HCO
3
)
2
→ Mg(OH)
2
+ 2CO
2
Po usunięciu twardości przemijającej pozostaje twardość stała.
- twardość stałą (niewęglanową), wywołaną obecnością innych soli wapniowych i
magnezowych, np. chlorków, siarczanów, azotanów itp. Całkowita twardość wody (twardość
ogólna) jest sumą twardości węglanowej i niewęglanowej i określa całkowitą zawartość
związków wapnia i magnezu w wodzie.
Woda o zbyt dużej twardości utrudnia proces wiązania zaczynu cementowego, ponieważ
zmniejsza rozpuszczalność składników cementu w wodzie.
Poniżej przedstawiono skalę twardości wody
SKALA TWARDOŚCI WODY
Twardość ogólna,
o
n
bardzo miękka
0-5
miękka
5-10
średnio twarda
10-20
twarda
20-30
bardzo twarda
powyżej 30

28
Ditlenek węgla (dwutlenek węgla)
Ditlenek węgla występuje prawie we wszystkich wodach naturalnych. Pochodzi on
głównie z atmosfery bądź z procesów mineralizacji substancji organicznych, a w wodach
wgłębnych z procesów geochemicznych. Ilość ditlenku węgla, który może pochodzić z
różnych procesów chemicznych i biologicznych w wodzie, zależy od zawartości w niej ciał
organicznych oraz intensywności i kierunku samego procesu. Z tych względów zawartość
CO
2
, szczególnie w wodach powierzchniowych stojących, może dać pewne wskazówki co do
stopnia zanieczyszczenia wody substancjami organicznymi oraz procesów w niej
zachodzących. Pochłanianie ditlenku węgla z atmosfery obserwuje się przede wszystkim w
morzach, bardzo rzadko w wodach powierzchniowych rzecznych, które zawierają ditlenek
węgla zwykle w większych ilościach, niż wymaga tego stan równowagi w atmosferze. Stąd w
wodach rzecznych obserwuje się na ogół proces odwrotny - wydzielanie się ditlenku węgla do
atmosfery. Na zmniejszanie sią zawartości ditlenku węgla w wodach naturalnych (oprócz
wydzielania się go do atmosfery) mają wpływ również procesy, w których ditlenek węgla
wchodzi w reakcje chemiczne (np. rozpuszczanie skal węglanowych) lub jest zużywany w
procesie fotosyntezy przez roślinność wodną.
Zawartość ditlenku węgla w wodach naturalnych waha się w bardzo szerokich granicach - od
kilku (w rzekach i jeziorach) do kilkuset, a nawet kilku tysięcy mg/dm
3
(wody mineralne). W
zależności od odczynu wody ditlenek węgla może występować całkowicie w postaci gazowej
(pH < 4,5), wyłącznie w postaci jonu wodorowęglanowego HCO
3
-
(pH = 8,4) lub
węglanowego CO
3
2-
(pH = 10,5). Ponieważ większość naturalnych wód podziemnych ma pH
w granicach 6,5-8,5, zatem w wodach tych ditlenek węgla występuje na ogół w całym zespole
związków kwasu węglowego. Poniżej podano schemat układu ditlenku węgla, który może
istnieć w wodach naturalnych.
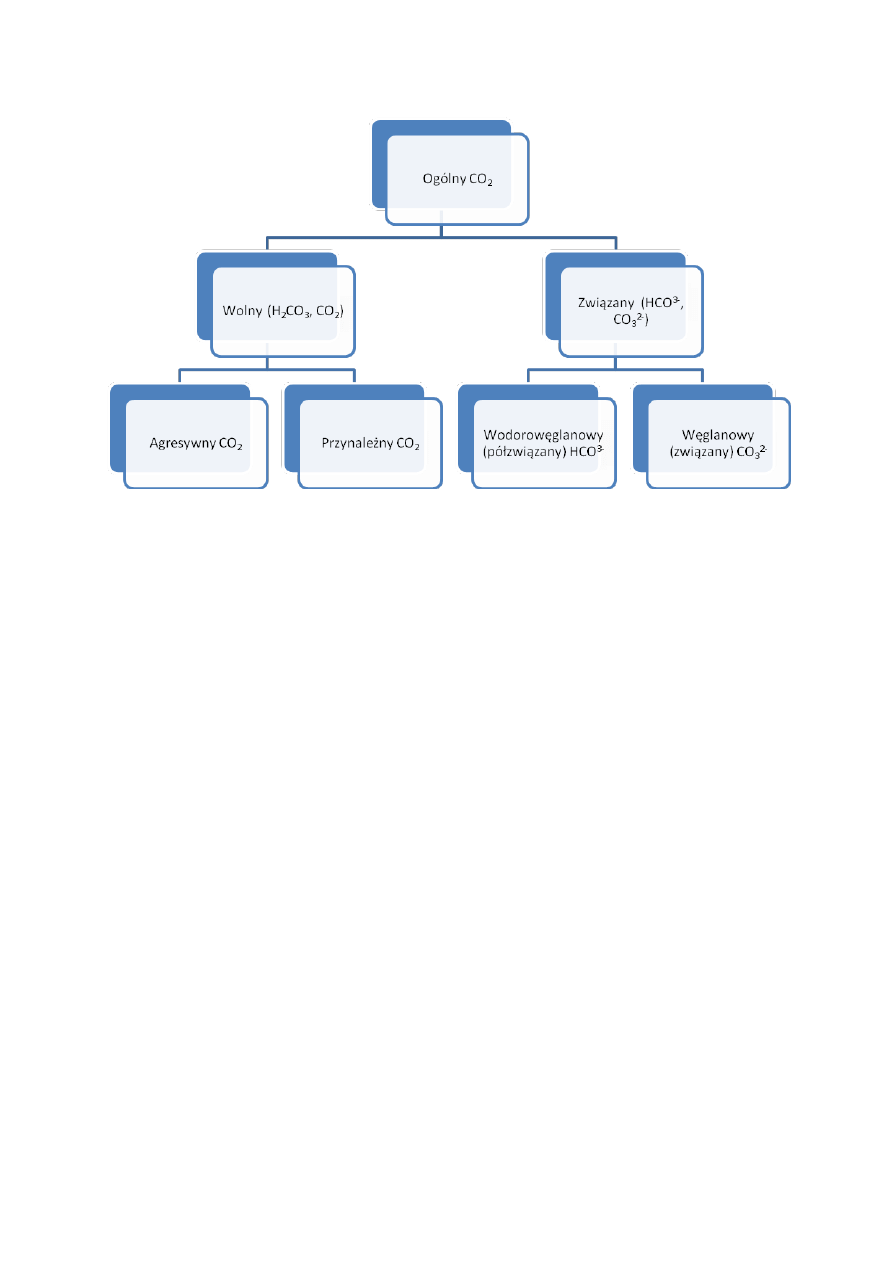
29
Ogólny ditlenek węgla dzielimy na wolny i związany. Ditlenek węgla związany znajduje się
w wodzie pod postacią wodorowęglanów HCO
3
-
i węglanów CO
3
2-
. Ditlenek węgla związany
odpowiada zawartości ditlenku węgla węglanowego i 1/2 wodorowęglanowego. W wodach
naturalnych niezawierających węglanów, lecz wodorowęglany równa się on połowie
zasadowości wodorowęglanowej, wyrażonej w CO
2
wodorowęglanów; jeśli zasadowość jest
obliczona w stopniach, to związany CO
2
równa się stopnie pomnożone przez 7,86 mg/dm
3
CO
2
, jeżeli w milivalach - równa się ilość mvali pomnożona przez 22 mg/dm
3
.
Wolny ditlenek węgla występuje w postaci rozpuszczonego CO
2
i H
2
CO
3
. W wodach
naturalnych prawie cały wolny ditlenek węgla znajduje się w postaci rozpuszczonej, a tylko
niespełna 1% występuje w wodzie w postaci kwasu węglowego.
Część wolnego ditlenku węgla niezbędna dla utrzymania w roztworze rozpuszczonego
wodorowęglanu wapniowego w myśl równania:
Ca(HCO
3
)
2
↔ CaCO
3
+ H
2
O + CO
2
nazywa się ditlenkiem węgla równowagi węglanowo-wapniowej lub ditlenkiem węgla
przynależnym (równowagi).
W miarę zwiększania się w wodzie stężenia wodorowęglanów (twardości węglanowej)
ilość niezbędnego ditlenku węgla przynależnego znacznie wzrasta. Pozostała część wolnego
ditlenku węgla w wodzie, czyli jego nadmiar w stosunku do stechiometrycznej ilości ditlenku
węgla przynależnego (równowagi) jest ditlenkiem węgla agresywnym. Może on powodować

30
działanie agresywne w stosunku do betonu i metali, jednakże w procesie tym nie bierze
udziału cały ponieważ jego część znowu musi utrzymywać w roztworze wodorowęglany
powstające jako skutek działania agresywnego ditlenku węgla. Nie można więc obliczyć
ilości ditlenku węgla agresywnego bezpośrednio z różnicy między całym CO
2
rozpuszczonym w wodzie a CO
2
równowagi.
Obecność ditlenku węgla w wodach naturalnych stwarza problemy gospodarcze i
techniczne ze względu na powodowanie korozji urządzeń metalowych i betonowych. Z
higienicznego punktu widzenia przy ocenie wód podziemnych ditlenek węgla nie ma
większego znaczenia. Przy ocenie wód powierzchniowych ditlenek węgla daje niekiedy
pewne wskazówki co do stopnia zanieczyszczenia wody substancjami organicznymi oraz
procesów w niej zachodzących. Oznaczanie wolnego i agresywnego ditlenku węgla wykonuje
się przeważnie w celu określenia agresywności wody w stosunku do betonu i rurociągów.
Karbonatyzacja betonu
Pod wpływem dwutlenku węgla z powietrza zachodzi w betonie zjawisko karbonatyzacji
Ca(OH)
2
+ CO
2
→ CaCO
3
+ H
2
O
w wyniku czego zasadowość betonu od wartości pH = 11÷12 maleje do pH ok. 9, przy tej
wartości w betonie może zachodzić już depasywacja stali.
Ć W I C Z E N I A L A B O R A T O R Y J N E Z C H E M I I
OZNACZANIE AGRESYWNEGO DWUTLENKU WĘGLA W WODZIE METODĄ
BEZPOŚREDNIĄ
Obecność dwutlenku węgla w wodach naturalnych stwarza poważne problemy gospodarcze i
techniczne ze względu na powodowanie korozji urządzeń metalowych i betonowych. Zasada
oznaczenia polega na reakcji agresywnego dwutlenku węgla z dodanym do wody marmurem
(CaCO
3
). Tworzy się rozpuszczalny wodorowęglan wapniowy, który powoduje wzrost

31
zasadowości wody. Zawartość agresywnego dwutlenku węgla oblicza się z różnicy
zasadowości wody po wytrząsaniu ze sproszkowanym marmurem a zasadowości wody bez
dodatku marmuru.
WYKONANIE ĆWICZENIA:
Oznaczyć zasadowość wody przeznaczonej do badania:
Przygotować stanowisko do miareczkowania stosując roztwór miareczkujący HCl o stężeniu
0,1 mol/l.
Do kolby stożkowej o pojemności 250 ml odmierzyć 100 ml badanej wody, dodać 2-3 krople
oranżu metylowego i miareczkować (zgodnie z instrukcją) 0,1-molowym roztworem kwasu
solnego do zmiany zabarwienia z żółtej na żółtoróżową. Obliczyć zasadowość m zgodnie z
wzorem.
Oznaczyć zasadowość badanej wody po reakcji z marmurem:
Do butelki o pojemności 500 ml wsypać 2-3 g sproszkowanego marmuru
a następnie wlać ok. 250-300 ml badanej wody. Zamknąć szczelnie naczynie
i wytrząsać energicznie przez ok. 15 min. Odstawić na 10 min po czym pobrać pipetą z górnej
warstwy 100 ml wody i ponownie oznaczyć w niej zasadowość. Obliczyć zasadowość m
1
.
OBLICZANIE WYNIKÓW:
Zasadowość ogólną [m i odpowiednio m
1
] próbek wody oblicza się zgodnie z wzorem:
m = (b
100) / V
gdzie:
b - objętość 0,1 molowego roztworu HCl zużyta na zmiareczkowanie próbki [ml],
V - objętość próbki wody użytej do oznaczenia [ml].

32
Zawartość agresywnego dwutlenku węgla CO
2agr
oblicza się w mg/l według wzoru:
CO
2agr
= (m
1
- m) · 22
gdzie:
m - zasadowość ogólna wody przed wytrząsaniem z marmurem
m
1
- zasadowość ogólna wody po wytrząsaniu jej z marmurem
Ć W I C Z E N I A L A B O R A T O R Y J N E Z C H E M I I
BADANIE TECHNICZNE WODY
OZNACZANIE TWARDOŚCI OGÓLNEJ WODY METODĄ WERSENIANOWĄ
Chemiczne oddziaływanie poszczególnych substancji zawartych w wodach naturalnych czy
przemysłowych wywołuje bardzo różnorodne i ważne skutki, dlatego też konieczna jest
dokładna znajomość składu chemicznego wody stosowanej w praktyce przemysłowej.
Analiza obejmuje różne oznaczenia, zależnie od przeznaczenia wody, tok postępowania
podają polskie normy.
Jednym z najbardziej podstawowych jest oznaczenie twardości ogólnej wody.
WYKONANIE ĆWICZENIA:
Przygotować stanowisko do miareczkowania (zgodnie z instrukcją na następnej stronie)
stosując roztwór miareczkujący EDTA o stężeniu 0,02 mol/l. 100 cm
3
wody wodociągowej
odmierzyć do kolby stożkowej, dodać 1 cm
3
roztworu buforu amoniakalnego oraz 7 kropli
roztworu czerni eriochromowej T (lub ¼ łyżeczki czerni sproszkowanej). Wodę
miareczkować (zgodnie z instrukcją) roztworem wersenianu sodowego do zmiany barwy z
fioletowej na niebieską (do uzyskania trwałego zabarwienia). Powtórzyć miareczkowanie
33
biorąc kolejno wodę wodociągową przegotowaną i wodę destylowaną. Uzyskane dane
zestawić w tabeli wyników:
Woda
Objętość wody
[ml]
Objętość
EDTA [ml]
Twardość
ogólna [
o
n]
Wodociągowa nie przegotowana
Wodociągowa przegotowana
Destylowana
OBLICZANIE WYNIKÓW:
Znając objętość wody badanej (V ml), objętość (V' ml) i stężenie molowe (C
m
) roztworu
wersenianu sodowego użytego do miareczkowania należy obliczyć twardość ogólną wody,
wyrażając ją w stopniach twardości wody [
o
n]:
T
og
= (C
m
V'
5,6 ·1000) / V
Klasyfikacja wody pod względem twardości
twardość wody [
o
n]
rodzaj wody
0 - 4
bardzo miękka
4 - 8
miękka
8 -20
twarda
ponad 20
bardzo twarda
Ć W I C Z E N I A L A B O R A T O R Y J N E Z C H E M I I
BADANIE TECHNICZNE WODY
OZNACZANIE ZAWARTOŚCI SIARCZANÓW METODĄ WINKLERA oraz
OZNACZANIE pH

34
W budownictwie stosuje się wodę do zarabiania betonu i zapraw z kruszyw i spoiw. Woda ta
musi odpowiadać określonym wymaganiom technicznym. Zawartość siarczanów (SO
4
2-
) musi
być mniejsza niż 600 mg/dm
3
, a dopuszczalny odczyn musi być większy niż 4 jednostki pH.
WYKONANIE ĆWICZENIA:
Oznaczenie zawartości jonów siarczanowych SO
4
2-
.
Do dwóch probówek wlać po 5 cm
3
wody wodociągowej. Do jednej probówki dodać 2,5 cm
3
10% roztworu HCl oraz 2,5 cm
3
10% roztworu BaCl
2
(z chwilą rozpoczęcia dodawania
BaCl
2
uruchomić sekundomierz). Druga probówka służy do porównania. Zmierzyć czas
upływający do wystąpienia zmętnienia wody po dodaniu odczynników (porównując z wodą
bez dodatku odczynników). Na podstawie tabeli 1 oszacować zawartość jonów siarczanowych
w wodzie (mg/dm
3
). Wynik wpisać do tabeli 2.
Wykonać tą samą metodą oznaczenie jonów siarczanowych w wodzie zanieczyszczonej. W
przypadku natychmiastowego zmętnienia cieczy, rozcieńczyć dwudziestokrotnie badaną
wodę wodą destylowaną, w której należy uprzednio sprawdzić zawartość jonów
siarczanowych i powtórzyć oznaczenie. Na podstawie danych tabeli 1 określić zawartość
siarczanów w próbce wody zanieczyszczonej, uwzględniając jej dwudziestokrotne
rozcieńczenie. Wyniki wpisać do tabeli 2.
Oznaczenie wykładnika stężenia jonów wodorowych, pH.
pH wody najszybciej określa się za pomocą papierków uniwersalnych. Papierek uniwersalny
należy zanurzyć w badanej wodzie na 15 s. Barwę, która przyjmuje papierek porównać ze
skalą wzorcową i odczytać pH.
Należy oznaczyć pH wody wodociągowej i wody zanieczyszczonej. Wyniki wpisać do tabeli
2.
35
WYNIKI:
Tabela 1.
Przybliżona zawartość jonu SO
4
2-
w wodzie (metoda Winklera).
Zmętnienie cieczy
po upływie czasu
[s]
0
obfity
osad
0
zmętnieni
e
5
7
10 15 20 30 45 60 120 300
Zawartość jonu
SO
4
2-
[mg dm
-3
]
powyżej
600
600
100 80 70 60 50 40 30 25
15
10
Tabela 2.
Woda wodociągowa
Woda zanieczyszczona
pH
zbadana w stosunku
do normy
zawartość
siarczanów
zbadana w stosunku
do normy
Ogólna ocena
przydatności wody
KINETYKA CHEMICZNA
Kinetyka zajmuje się badaniami szybkości przebiegu reakcji chemicznych. Znajomość
szybkości reakcji w określonych warunkach fizycznych ma podstawowe znaczenie zarówno
teoretyczne jak i praktyczne. Teoretyczne – gdyż pozwala na wyciągnięcie wniosków
dotyczących mechanizmu reakcji. Praktyczne – gdyż określa przebieg danej reakcji w
warunkach laboratoryjnych lub przemysłowych.
Intuicyjną miarą szybkości reakcji może być czas upływający od momentu rozpoczęcia
reakcji do chwili zaniku objawów jej przebiegu. Im większa jest szybkość tym krótszy jest
ten czas.
Różne reakcje przebiegają z różną szybkością. Niektóre z nich, np. rozkład substancji
wybuchowych, rozkład bromku srebra na kliszy fotograficznej, reakcje jonowe (np.
zobojętnienia, wytrącania osadów) przebiegają z tak dużą szybkością, że ich czas przebiegu
jest rzędu ułamków sekundy. Natomiast inne jak np. korozja metali trwa wiele lat zanim
przebiegnie całkowicie.

36
Każdą reakcję można przyśpieszyć albo zwolnić zmieniając warunki w których zachodzi.
Tak np. ta sama mieszanina wodoru i tlenu w normalnych warunkach przez wiele lat
pozostaje praktycznie bez zmiany. Pomimo, że reakcji syntezy wody towarzyszy duży ubytek
energii. Natomiast w temperaturze powyżej 1100 K lub po zainicjowaniu iskrą elektryczną
reakcja przebiega wybuchowo (czas przebiegu reakcji w ułamkach sekundy).
Z kinetycznego punktu widzenia celowy jest podział reakcji na jednorodne
(homogeniczne), które zachodzą całkowicie w jednej fazie (ciekłej, gazowej) oraz na
niejednorodne heterogeniczne, które przebiegają zawsze na granicy rozdziału faz.
Przykłady reakcji homogenicznej, gdzie wszystkie reagenty są w stanie gazowym:
3H
2
+ N
2
2NH
3
SO
2
+ O
2
SO
3
Przykład reakcji homogenicznej, gdzie wszystkie reagenty są w stanie ciekłym:
H
2
SO
4
+ 2NaOH → 2Na
+
+ SO
4
2-
+ 2H
2
O
Przykłady reakcji heterogenicznych (s – ciało stałe, g - gaz, c – ciecz):
CuO (s) + H
2
(g) → Cu (s) + H
2
O (g)
CaCO
3
(s) + 2HCl (c) = CaCl
2
(c) + CO
2
(g) + H
2
O (c)
C (s) + O
2
(g) = CO
2
(g)
Szybkość reakcji chemicznej
Reakcje chemiczne są procesami zachodzącymi z określoną szybkością v.
Szybkość reakcji:
A + B → AB
można zdefiniować jako zmianę stężenia produktów lub substratów w czasie:

37
gdzie c
A
, c
B
oznaczają stężenia molowe substratów A i B a c
AB
stężenie molowe produktu
AB. Ponieważ w wyniku reakcji stężenie substratów maleje, zmiana stężenia w czasie
oznaczona jest znakiem ujemnym (-dc
A
, -dc
B
). Stężenie produktu rośnie w czasie więc zmiana
stężenia AB ma znak dodatni (+dc
AB
).
Oczywiście szybkość reakcji chemicznej niekoniecznie musi być mierzona zmianami stężenia
molowego. Można użyć każdego innego sposobu wyrażania ilości substancji reagujących
(masę, stężenie molarne, ułamek molowy, ułamek atomowy itp.).
Definicja szybkości wyrażona równaniem przedstawia sposób pomiaru szybkości reakcji.
Wystarczy mierzyć odpowiednie stężenia w czasie i wykreślić odpowiednie zmiany za
pomocąs funkcji c = f(t).
Szybkość reakcji jest cechą charakterystyczną dla danego zespołu reagentów i zależy od
wielu czynników. Najważniejsze z nich to:
rodzaj i stężenie reagentów;
temperatura ;
ciśnienie (gdy reakcja przebiega w fazie gazowej);
promieniowanie elektromagnetyczne (gdy reakcja jest fotochemiczna) ;
rozwinięcie powierzchni (w przypadku reakcji powierzchniowych);
obecność katalizatora lub inhibitora.

38
Ć W I C Z E N I A L A B O R A T O R Y J N E Z C H E M I I
KINETYKA CHEMICZNA
BADANIE WPŁYWU STĘŻENIA SUBSTRATÓW NA SZYBKOŚĆ REAKCJI
CHEMICZNEJ
WYKONANIE ĆWICZENIA:
Do pięciu probówek odmierzyć pipetą podane w tabeli objętości roztworu 0,5 m tiosiarczanu
sodowego Na
2
S
2
O
3
, a następnie z drugiej biurety odpowiednie objętości wody - wymieszać.
Do przygotowanych roztworów kolejno dodawać z pipety po 1 cm
3
1m kwasu siarkowego –
zawartość probówek mieszać(!) i stoperem mierzyć czas t od chwili wprowadzenia kwasu do
wystąpienia wyraźnego, jednakowego we wszystkich probówkach zmętnienia roztworu.
Uzyskane wyniki zanotować w tabeli. Obserwacje prowadzić na ciemnym tle.
OBLICZANIE WYNIKÓW:
Wystąpienie opalescencji (zmętnienia) roztworu jest wynikiem wydzielania się siarki w
reakcji:
Na
2
S
2
O
3
+ H
2
SO
4
Na
2
SO
4
+ S + SO
2
+ H
2
O
Czas t, który upływa od chwili rozpoczęcia reakcji do wystąpienia opalescencji, jest
odwrotnie proporcjonalny do szybkości, z jaką przebiega reakcja i zależy od stężenia
substratów. Odwrotność wartości t można uważać za miarę szybkości reakcji wyrażoną w
umownych jednostkach. Stężenie substratów należy wyliczyć z odpowiednich danych w
umownych jednostkach a / a+b Na podstawie tych obliczeń oraz odpowiednich wartości 1/t
należy wykonać wykres zależności:
1/t = f(a / a+b)

39
Nr probówki Objętość
[cm
3
]
Stężenie
Czas
Szybkość
Na
2
S
2
O
3
H
2
O dest.
Na
2
S
2
O
3
reakcji
reakcji
a
b
a / a+b
t [s]
1/t [s
-1
]
1
6
0
2
5
1
3
4
2
4
3
3
5
2
4
STATYKA CHEMICZNA
Stan równowagi chemicznej
Wszystkie reakcje chemiczne są procesami odwracalnymi (odwracalność chemiczna
reakcji polega na możliwości przeprowadzenia jej w obu kierunkach). Dopiero w specjalnych
warunkach reakcje mogą przebiegać nieodwracalnie np. gdy jeden z produktów opuszcza
środowisko reakcji, reakcja wtedy biegnie aż do wyczerpania substratów.
Np. reakcja cynku z kwasem siarkowym:
Zn + H
2
SO
4
→ ZnSO
4
+ H
2
prowadzona w otwartym naczyniu, z którego wodór może się wydzielać swobodnie,
przebiega aż do wyczerpania Zn lub
H
2
SO
4
. Można jednak spowodować jej przebieg w
odwrotnym kierunku:
ZnSO
4
+ H
2
→ Zn + H
2
SO
4
wprowadzając wodór pod wysokim ciśnieniem do wodnego roztworu ZnSO
4
.
Ze zjawiskiem odwracalności reakcji wiąże się względny charakter terminów “substraty”,
“produkty”. W celu uniknięcia dwuznaczności przyjmuje się, że substraty to substancje
zapisane po lewej (przed reakcją), a produkty po prawej stronie równania chemicznego (po
reakcji).

40
Jednak kierunek reakcji zależy od warunków. Teoretycznie każda reakcja w odpowiednich
warunkach przebiega w obu kierunkach.
Jeżeli szybkość reakcji w prawo zdecydowanie dominuje nad szybkością reakcji w lewo, to
substraty niemal ilościowo przereagowują w produkty a proces uważa się za praktycznie
nieodwracalny, czyli biegnący do końca.
Zmiany stężeń substratów i produktów w czasie biegu reakcji chemicznej
Gdy szybkości reakcji w obu kierunkach są takie same wówczas wytwarza się stan
równowagi chemicznej (równoznaczny ze stanem równowagi termodynamicznej). W stanie
równowagi chemicznej ilość substratów i produktów nie ulega zmianie choć obie reakcje
zachodzą w dalszym ciągu.
Stan równowagi chemicznej wytwarza się w układach zamkniętych (czyli nie
wymieniających masy z otoczeniem).
Dla określenia, że dana reakcja osiąga w danych warunkach stan równowagi stosuje się w
równaniach chemicznych znak strzałki w obu kierunkach (
lub
) zwany znakiem
równowagi, np.:
3H
2
+ N
2
2NH
3
Jedną strzałkę stosuje się gdy:
- układ nie osiąga stanu równowagi np. podczas rozpuszczania metalu (cynku) w kwasie
siarkowym w naczyniu otwartym
- przy teoretycznym założeniu 100 % wydajności (np. prze obliczaniu efektów cieplnych)
ciepła reakcji.
- jeżeli proces rozpatruje się w jednym kierunku w celu podania równania kinetycznego na
szybkość reakcji w jedną stronę.

41
Prawo działania mas
Układ znajduje się w stanie równowagi jeśli nie zachodzi w nim zmiana stężeń jego
składników.
aA + bB ↔ cC + dD (T=const., p=const. dla gazów)
Po pewnym czasie trwania reakcji ustali się stan równowagi chemicznej, w którym szybkości
reakcji w obu kierunkach będą równe.
v
1
= k
1
• c
A
a
• c
B
b
= k
2
• c
C
c
• c
D
d
Iloraz dwóch stałych szybkości reakcji k
1
i k
2
jest w danej temperaturze stały.
k
1
/k
2
= c
C
c
• c
D
d
/c
A
a
• c
B
b
= K
K - stała równowagi chemicznej, odniesiona do stężeń molowych reagentów.
Wzór wyraża prawo działania mas - ilościowe ujęcie równowagi chemicznej (prawo
Guldberga-Waagego):
w stanie równowagi chemicznej stosunek iloczynu stężeń molowych produktów reakcji do
stosunku iloczynu stężeń molowych substratów w danej temperaturze i przy danym ciśnieniu
jest wielkością stałą:
b
B
a
A
d
D
c
C
c
c
c
c
K
Reguła Le Chateliera i Browna (Reguła przekory)
Zmiana parametrów wyznaczających stan równowagi chemicznej – stężenia reagentów,
temperatury i ciśnienia prowadzą do zaburzenia (przesunięcia) stanu równowagi. Układ
chemiczny dąży wtedy do nowego stanu równowagi wyznaczonego nowymi, zmienionymi
parametrami. Wpływ zmiany stężenia reagentów, ich ciśnienia oraz temperatury na stan
równowagi określa reguła Le Chateliera i Browna (reguła przekory).

42
Reguła ta mówi, że jeżeli zostanie zakłócony stan równowagi przez zmiany temperatury
(T), ciśnienia (p), stężenia (c), w układzie rozpoczyna się taka przemiana, która będzie
przeciwdziałała zakłóceniom prowadząc do osiągnięcia ponownego stanu równowagi.
Przemiany zainicjowane zakłóceniem równowagi trwają aż do ponownego zrównoważenia się
szybkości reakcji przebiegających w przeciwnych kierunkach. Po pewnym czasie równowaga
znów się ustala, ale już przy innych niż poprzednio stężeniach.
Reasumując reguła przekory brzmi:
Jeżeli układ będący w stanie równowagi poddamy działaniu zewnętrznemu, tj. zmianie
stężenia reagentów, zmianie ciśnienia lub zmianie temperatury, to w układzie tym przesuwa
się równowaga chemiczna w kierunku kompensacji tych zmian.
Wpływ zmiany stężenia na stan równowagi
Zmiana stężenia reagujące substancji wywołuje zmianę stężenia pozostałych
z zachowaniem stałej równowagi. Jeżeli do układu w stanie równowagi dodatkowo
wprowadzimy pewną ilość reagenta (substratu lub produktu) to stan równowagi przesunie się
w kierunku zmniejszenia jego wartości w układzie. Podobnie, jeżeli z układu usuniemy
pewną ilość jednego składnika, to zajdą przemiany, które
zmniejszą jego ubytek.
Przykład:
N2 + O2 ↔ 2 NO
Ze wzrostem stężeń substratów wydajność reakcji wzrośnie czyli równowaga przesunie się w
kierunku syntezy NO.
Stała K
c
pozostaje bez zmiany, ma tę samą wartość gdyż reakcja przebiega w stałej
temperaturze.
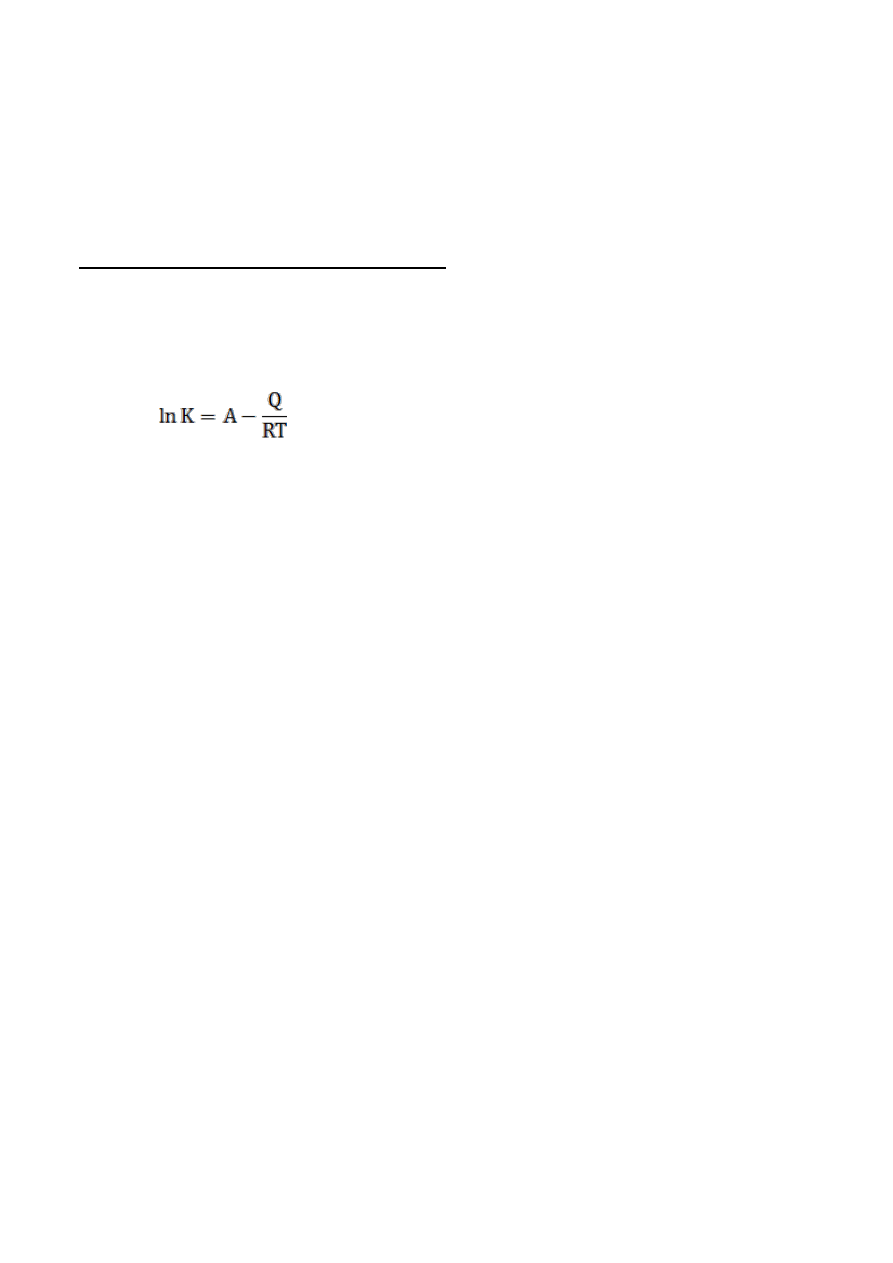
43
Wzrost stężenia substratów, w układzie powoduje przesunięcie równowagi z lewa na prawo,
w kierunku syntezy produktu, tym samym zwiększa wydajność procesu. I odwrotnie –
usunięcie jednego z substratów spowoduje zmniejszenie wydajności tej reakcji.
Wpływ zmiany temperatury na stan równowagi
Zależność stałej równowagi (K) od temperatury określa równanie:
gdzie:
A – stała charakterystyczna dla danej reakcji
Q – ciepło reakcji
R – stała gazowa
T – temperatura.
Wzrost temperatury przesuwa położenie równowagi chemicznej i równocześnie zmienia
wartość stałej równowagi, w odróżnieniu od zmian stężenia, które przesuwają równowagę
przy zachowaniu tej samej wartości K
.
Jeżeli reakcja jest endotermiczna tzn. towarzyszy jej pochłanianie ciepła to wzrost
temperatury, w myśl reguły przekory przesuwa równowagę w kierunku kompensacji tej
zmiany, czyli w kierunku pochłaniania ciepła ( tj. z lewej strony na prawą).
Wzrost temperatury w przypadku reakcji egzotermicznej, egzoenergetycznej (takiej której
towarzyszy wydzielanie ciepła) przesuwa ją w kierunku odwrotnym (z prawej strony na
lewą).
44
Przykład:
Wpływ temperatury na stan równowagi reakcji syntezy amoniaku:
3H
2
+ N
2
2NH
3
+ Q
p
Q
p
= -92,38 kJ
indeks (p) oznacza że reakcja biegnie przy stałym ciśnieniu.
Jest to reakcja egzotermiczna (ciepło wydziela się w czasie reakcji).
Wzrost temperatury (w myśl reguły przekory) przesuwa równowagę w kierunku kompensacji
wzrostu temperatury – w kierunku pochłaniania ciepła, czyli z prawej strony na lewą.
Oznacza to, że ze wzrostem temperatury wydajność procesu (ilość amoniaku) maleje.
Wpływ zmiany ciśnienia na stan równowagi
Zmiany ciśnienia wpływają na położenie stanu równowagi dla reakcji chemicznych
przebiegających w fazie gazowej, gdzie następuje zmiana objętości.
Rozpatrzyć należy trzy typy reakcji w stanie gazowym:
Typ 1. Reakcja syntezy, w wyniku której z kilku substratów powstaje jeden produkt, np.
reakcja otrzymywania amoniaku.
N
2
+ 3H
2
2NH
3
Dla p,T = const. objętość produktów (1+3) jest większa od objętość substratów (2) -
przemiana substratów w produkty powoduje więc zmniejszenie sumarycznej ilości reagentów.
Jeżeli zakłócimy stan równowagi układu zwiększając ciśnienie, to nastąpi przemiana
dodatkowej ilości substratów w produkty, a więc zmniejszy się sumaryczna ilość reagentów w
układzie, a tym samym ciśnienie.
Typ 2. Reakcja, gdzie z jednego substratu powstaje kilka produktów.

45
CH
3
-CH
2
-CH
3(g)
↔ CH
2
CH-CH
3(g)
+ H
2(g)
Efekt wzrostu ciśnienia jest odwrotny niż w reakcji I typu - stan równowagi zostaje
przesunięty na korzyść substratów.
Typ 3. Reakcja gdzie nie ma zmiany objętości.
CO
(g)
+ H
2
O
(g)
CO
2(g)
+ H
2(g)
Zmiany ciśnienia nie wywierają żadnego wpływu na położenie stanu równowagi.
Dla roztworów ciekłych - umiarkowane zmiany ciśnienia nie wpływają na położenie stanu
równowagi reakcji.
Wpływ katalizatorów na stan równowagi
Katalizatory – substancje zmieniające szybkość reakcji, a same pozostają niezmienione po
zakończeniu reakcji. Zjawisko wywołane katalizatorami nosi nazwę katalizy. Na wartość
stałej równowagi nie ma wpływu obecność katalizatora. Wpływ katalizatora ogranicza się
wyłącznie do zmiany szybkości reakcji. W reakcjach odwracalnych katalizator w
jednakowym stopniu zmienia szybkość reakcji właściwej, jak i reakcji odwrotnej.
Ć W I C Z E N I A L A B O R A T O R Y J N E Z C H E M I I
STATYKA CHEMICZNA
BADANIE WPŁYWU STĘŻENIA REAGENTÓW NA STAN RÓWNOWAGI
CHEMICZNEJ

46
WYKONANIE ĆWICZENIA:
Do probówki nalać 2 cm
3
roztworu chlorku żelazowego FeCl
3
(0,02n) i 2 cm
3
roztworu
rodanku potasowego KSCN (0,02n), wymieszać i rozdzielić do czterech probówek. Jedną z
nich (0) pozostawić jako wzorcową. Do drugiej (1) probówki dodać dwie krople nasyconego
roztworu rodanku potasowego, do trzeciej (2) nasyconego roztworu chlorku żelazowego, do
czwartej (3) - wrzucić kilka kryształków chlorku potasowego. Obserwować różnicę
intensywności zabarwienia w trzech probówkach w porównaniu z probówką wzorcową.
Wyniki obserwacji zanotować w tabeli wyników.
Nr probówki
Dodatkowy
odczynnik
Zmiana
intensywności
zabarwienia
Stała
równowagi
reakcji
Zmiana stęż.
reagentów
wzrost
obniżenie
0
-
-
-
-
1
2
3
OPRACOWANIE WYNIKÓW:
Reakcja między roztworem chlorku żelazowego i rodanku potasowego doprowadza
do
wytworzenia barwnego niezdysocjowanego rodanku żelazowego Fe(SCN)
3
i ustalenia się stanu równowagi zgodnie z równaniem:
- w postaci cząsteczkowej
FeCl
3
+ 3 KSCN
Fe(SCN)
3
+ 3 KCl
- w postaci jonowej
Fe
3+
+ 3Cl
-
+ 3K
+
+3SCN
-
Fe(SCN)
3
+ 3K
+
3Cl
-
- w postaci skondensowanej
Fe
3+
+ 3SCN
-
Fe(SCN)
3

47
Stan równowagi można przesunąć przez dodanie poszczególnych reagentów, co uwidacznia
się zmianą zabarwienia roztworu. W tabeli wyników zapisać, jaka jest zmiana stężenia
każdego z reagentów wywołana dodaniem jednego z nich.
LITERATURA:
Czerwieniec E. Chemia. Materiały pomocnicze. Oficyna Wyd. PRz. 2010
Czarnecki L., Broniewski T., Henning O.: Chemia w budownictwie, Arkady Warszawa 1994;
Bentkowska H., Grzędzicki K.: Chemia budowlana; skrypt PG, 1983;
Kiedryńska L., Papciak D., Granops M. Chemia sanitarna, SGGW, Warszawa 2006
Pajdowski L.: Chemia ogólna, PWN Warszawa 1981;
Ujma J, Mazanek A.: Chemia budowlana. Skrypt PCz, 1982.
Wojtaś R.: Chemia ogólna i budowlana. Skrypt Politechniki Świętokrzyskiej, 1988
Banaś J. A (red.) Chemia ogólna, Materiały dydaktyczne AGH – Uczelniana Platforma
E-learningowa
Bielański A.: Podstawy chemii nieorganicznej, PWN Warszawa 1987;
Liwski J.: Chemia budowlana. PWN Warszawa 1978
Minczewski A., Marczenko K.: Chemia analityczna, t.I,II PWN Warszawa 1987.
Pauling L, Pauling P., Chemia, PWN Warszawa 1983;
Penkala T.: Podstawy chemii ogólnej; PWN Warszawa 1975;
Chemia.Wirtualny podręcznik.Podstawy i zastosowania:
Kropidłowska A.: Materiały dydaktyczne do ćwiczeń rachunkowych z chemii
nieorganicznej; Wydział Chemiczny, Politechnika Gdańska
Wyszukiwarka
Podobne podstrony:
sprawozdanie chemia 3, Budownictwo UZ semestr I , II, Chemia budowlana, Sprawozdania od Seweryna
Opisowe - chemia, Budownictwo 2, Budownictwo, Chemia budowlana
ściaga+chemia, Budownictwo PK, Chemia Budowlana
e skrypt chemia inżynieria środowiska
Definicje - chemia, Budownictwo 2, Budownictwo, Chemia budowlana
odpow chemia, budownictwo PK, I rok, chemia
Chemia(1), budownictwo pk, sem 1, chemia
Chemia, Budownictwo-studia, chemia
ćwiczenie 1 chemia, Budownictwo-studia, chemia
2 sprawko chemia, Budownictwo III Semestr, Chemia budowlana
zaliczenie chemia, budownictwo pk, sem 1, chemia
Chemia1, Budownictwo UTP, semestr 1 i 2, budownictwo, SEMESTR ZIMOWY, fizyka, sprawozdania, Fizyka
Pytania chemia, Budownictwo PŚK, II semestr, chemia, testy+materiały na zal
sprawozdanie chemia 3, Budownictwo UZ semestr I , II, Chemia budowlana, Sprawozdania od Seweryna
Kopia Opis techniczny B, Skrypty, UR - materiały ze studiów, studia, studia, 4 STASZEK, Semestr II,
więcej podobnych podstron