
1.
BEZPIECZEŃSTWO
REAKTORÓW
CHEMICZNYCH
Reaktor chemiczny to aparat do prowadzenia reakcji chemicznych,
w których mogą brać udział gazy, ciecze i ciała stałe. Reakcja chemiczna z kolei, to
przemiana, w czasie której zachodzi zmiana rodzaju cząsteczek substancji
reagujących.
1.1. PODZIAŁ REAKTORÓW
Reaktory chemiczne klasyfikuje się najczęściej według następujących
kryteriów:
Pierwszym kryterium jest metoda pracy reaktora.:
reaktor chemiczny okresowy- jest to zbiornik lub kocioł zwykle wyposażony
w urządzenia mieszające oraz elementy do wymiany ciepła. W reaktorze tym
wprowadzenie całej ilości substancji oraz usuwanie produktów odbywa się
cyklicznie,
reaktor chemiczny przepływowy- jest to reaktor, w którym doprowadzanie
substratów i odbiór produktów są jednoczesne i ciągłe. Ze wzglądu na kształt
reaktory przepływowe dzieli się na: rurowe, wieżowe, zbiornikowe,
reaktory chemiczne półprzepływowe- jest to reaktor typu zbiornikowego
z mieszadłem, pracujący metodą kombinowaną, tzn. albo w sposób ciągły
doprowadza się do niego część reagentów (pozostałe były załadowane
wcześniej), albo odprowadza się jeden z produktów lub doprowadza część
reagentów w sposób ciągły i jednocześnie odprowadza jeden z produktów.
Ze względu na charakter fazowy reagującego układu, wyróżnia się:
reaktory do procesów homogenicznych,
reaktory do procesów heterogenicznych,
reaktory kontaktowe jako szczególny przykład reaktorów heterogenicznych.
Uwzględniając warunki temperaturowe:
reaktory izotermiczne,
reaktory adiabatyczne
reaktory politermiczne (pracujące w warunkach nieizotermicznych
i nieadiabatycznych).
Kolejnym kryterium stanowią własności konstrukcyjne:
reaktory zbiornikowe,
reaktory rurowe,
reaktory wieżowe (kolumnowe),
reaktory półkowe,
reaktory fluidyzacyjne,
reaktory specjalne.
2. ANALIZA KINETYCZNA PROCESU CHEMICZNEGO
Przedmiotem kinetyki chemicznej są zagadnienia szybkości przebiegu
reakcji chemicznej lub ogólniej- szybkości procesów chemicznych. Szybkość
łączenia się różnych cząsteczek jest bardzo ważną własnością, ponieważ od niej
zależy możliwość realizacji danego procesu w praktyce.

2
Analiza danych kinetycznych umożliwia wyznaczenie optymalnych warunków
przebiegu procesu w skali laboratoryjnej i przemysłowej, dobranie najlepszych
wartości parametrów, wybór odpowiednich katalizatorów i nadanie właściwego
i pożądanego kierunku reakcjom chemicznym. W zakres kinetyki chemicznej
wchodzą zagadnienia związane z samą szybkością reakcji chemicznych oraz z ich
mechanizmem.
2.1. SZYBKOŚĆ REAKCJI CHEMICZNEJ
2.1.1. POJĘCIE SZYBKOŚCI REAKCJI
Szybkość reakcji chemicznej określa się jako zmianę liczby moli reagenta
odniesioną do jednostki czasu oraz jednostki objętości masy reakcyjnej bądź
objętości reaktora (reakcje homogeniczne), powierzchni na której przebiega reakcja,
bądź wreszcie masy katalizatora w fazie stałej (reakcje heterogeniczne).
Definicja szybkości reakcji chemicznej w postaci ogólnej wyraża się wzorem:
kg)s
,
m
,
(m
mol
dτ
dn
W
1
ν
1
r
2
3
i
i
r
(2.1.1)
gdzie: W– wielkość odniesienia (objętość, powierzchnia, masa),
Tak określona szybkość jest wielkością zawsze dodatnią i nie zależy od wyboru
reagenta. Równanie to opisuje szybkość reakcji właściwą reagującemu układowi,
bez względu na warunki w jakich zachodzi proces chemiczny. W zagadnieniach
bilansowych, z jakimi mamy do czynienia w inżynierii reaktorowej, bardziej
użyteczne jest pojęcie szybkości reakcji zdefiniowanej tylko dla wybranego
reagenta A:
dt
dn
W
1
r
A
A
(2.1.2)
Znak (-), gdy A jest substratem, a znak (+)-gdy A jest produktem.
2.1.2. SZYBKOŚĆ REAKCJI W PROCESIE OKRESOWYM
Proces okresowy (prowadzony w reaktorze okresowym) charakteryzuje się
warunkami nieustalonymi. Szybkość reakcji obliczamy bezpośrednio z równania
definicyjnego (2.1.1) , przyjmując jako wielkość odniesienia objętość masy
reagującej. Równanie to ma postać:
dt
dn
V
1
r
A
m
A
(2.1.3)
gdy V
m
= const, co można przyjąć dla reakcji biegnących w środowisku ciekłym,
wtedy:
dt
dc
r
A
A
(2.1.4)

3
W warunkach nieustalonych można również prowadzić reakcje w fazie gazowej,
stosując reaktory zamknięte o stałej objętości . Wtedy do wzoru zamiast V
m
należy
wprowadzić objętość reaktora V
R
=const.
2.1.3. SZYBKOŚĆ REAKCJI W PROCESIE PRZEPŁYWOWYM
W procesach przepływowych, stacjonarnych, prowadzonych w warunkach
ustalonych, wszystkie wielkości związane z masą reagującą są funkcjami położenia,
a nie czasu. Zmienną niezależną jest objętość reaktora lub współrzędna położenia
mierzona wzdłuż osi reaktora w kierunku przepływu, bądź czas zastępczy,
wyrażający wielkość obliczeniową.
Szybkość reakcji chemicznej zachodzącej w przepływającym strumieniu
w warunkach ustalonych wyraża wzór:
s
m
mol
dV
n
d
r
3
R
A
A
(2.1.5)
Gdzie:
A
n
– strumień substancji A
2.2. RÓWNANIE KINETYCZNE
Szybkość reakcji chemicznej lub procesu chemicznego zależy od zmiennych
związanych z masą reagującą. Największe znaczenie ma temperatura, stężenie
reagentów, a także ciśnienie. Specyficzny wpływ na szybkość reakcji wywiera
obecność katalizatorów, które w tych samych warunkach mogą wielokrotnie
zmienić jej wartość.
Równanie kinetyczne wyraża zależność szybkości procesu od jego parametrów,
określoną dla ustalonych warunków katalitycznych w najogólniejszym przypadku
funkcją:
)
p
,...,
c
,
c
,
T
(
f
r
B
A
A
(2.2.1)
Zaniedbując ciśnienie, zależność tę można wyrazić w postaci:
,...)
c
,
c
,
T
(
)
T
(
k
r
B
A
A
(2.2.2)
która rozróżnia funkcję temperatury k(T) wyrażającą odwrotność oporu i nazywaną
stałą szybkości oraz funkcję (T,c
A
,c
B
,...)mającą własności siły napędowej,w której
parametr T jest związany ze stałą równowagi K
c
reakcji odwracalnych. Jeżeli więc
reakcja jest nieodwracalna, bądź T=const, to siła napędowa (T,c
A
,c
B
,...) nie zależy
od temperatury. Do równania kinetycznego można wprowadzić ciśnienie
cząstkowe, gdy reagentami są gazy. Gdy natomiast reakcja biegnie w środowisku
ciekłym, to ciśnienie, jako niezależny parametr kinetyczny, praktycznie nie ma
większego znaczeni i z zasady jest pomijane.
2.2.1. WPŁYW STĘŻENIA
Postać funkcji (T,c
A
,c
B
,...), występującej w równaniu (2.2.2), zależy od
mechanizmu procesu, ale dla licznych reakcji, szczególnie homogenicznych, może
być z góry postulowana na podstawie prawa działania mas (Guldberg, Waage),
zgodnie z którym szybkość reakcji chemicznej zachodzącej w stałej temperaturze

4
jest proporcjonalna do aktualnych stężeń reagentów w odpowiednich potęgach. Jest
to podstawowe twierdzenie kinetyki chemicznej, stosujące się do wszystkich
prostych przemian chemicznych, a także do wielu reakcji złożonych
przebiegających w środowisku jednorodnym.
Zgodnie z prawem działania mas zależność szybkości reakcji od stężenia reagentów
wyraża funkcja potęgowa, bądź ogólniej kombinacja liniowa funkcji potęgowych:
i
n
i
j
j
A
c
k
r
(2.2.3)
gdzie: c
i
– stężenie molowe reagenta,
n
i
– wykładnik liczbowy,
k
j
– współczynnik zależny od temperatury.
Jeżeli reakcja przebiega w fazie gazowej, to stężenie molowe może być zastąpione
ciśnieniami cząstkowymi reagentów. Wtedy zależność (2.2.3) ma postać:
n
i
p
n
i
A
p
k
c
k
r
(2.2.4)
Stałe szybkości k i k
p
mają różne wymiary oraz wartości. Przyjmując, że zachowują
się jak gazy doskonałe, istnieje zależność:
i
n
p
n
n
,
RT
k
k
(2.2.5)
2.2.2. WPŁYW TEMPERATURY
Temperatura jest drugim obok stężenia parametrem, który ma zasadnicze
znaczenie dla szybkości reakcji chemicznych. Zwykle szybkość reakcji rośnie
w miarę wzrostu temperatury, ale zależność ta może mieć różny charakter.
W równaniu kinetycznym temperatura występuje zawsze jako parametr niezależny
stałej szybkości. Dla większości reakcji homogenicznych i wielu reakcji
heterogenicznych stała szybkość jako funkcja temperatury jest dana równaniem
Arrheniusa, które w postaci logarytmicznej przedstawia wzór:
T
A
B
RT
E
k
ln
k
ln
0
(2.2.6)
Zgodnie z ujęciem Arrheniusa zależność stałej szybkości od temperatury jest
funkcją
wykładniczą
określoną
przez
dwie
stałe,
tj.
współczynnik
przedwykładniczy k
0
i wielkość E, nazywaną energią aktywacji. Z równania (2.2.6)
wynika, że im wyższa jest energia aktywacji, tym niższa jest stała szybkości przy
założeniu, że k
0
=const. Energia aktywacji wyraża nadwyżkę energii cząsteczek
reagujących z sobą ponad przeciętny poziom energii substratów. Nadwyżka ta jest
niezbędna do zajścia reakcji chemicznej.
2.3. STABILNOŚĆ TERMODYNAMICZNA
Pojęcie stabilności termodynamicznej wiąże się z trwałością równowagi
układu, w którym mogą zachodzić przemiany chemiczne bądź fazowe. Jeżeli układ
jest stabilny w stosunku do danego zakłócenia, to zakłócenie to nie zmieni w sposób
trwały jego równowagi pierwotnej i po ustąpieniu bodźca układ wróci do stanu

5
równowagi pierwotnej. Układ jest niestabilny, jeżeli zakłócenie spowoduje trwałe
przesunięcie jego równowagi.
Każde zakłócenie równowagi układu można scharakteryzować chwilową zmianą
liczby postępu reakcji
d
oraz warunkami (ustalonymi), w których ta zmiana
nastąpiła. Zakłócenie może polegać na zaburzeniu równowagi chemicznej lub na
powstaniu nowej fazy w układzie pierwotnie jednorodnym. W tym ostatnim
przypadku mówi się o stabilności fazowej układu. Powstanie nowej fazy może być
spowodowane działaniem różnych bodźców i może mieć różny charakter.
Rozróżnia się więc stabilność fazy względem dyfuzji oraz stabilność cieplną
i mechaniczną fazy.
2.3.1. STABILNOŚĆ CHEMICZNA
Ogólne kryterium stabilności układu wynika bezpośrednio z nierówności de
Dondera i można je wyrazić następująco:
0
Ad
s
Td
i
(2.3.1)
Zgodnie z zależnością (2.3.1) układ jest stabilny wówczas, gdy przesunięcie
d
określone liczbą postępu reakcji jest związane z ujemną produkcją entropii.
2.3.2. STABILNOŚĆ WZGLĘDEM DYFUZJI
W układach wieloskładnikowych pierwotnie jednorodnych mogą tworzyć się
fazy różniące się tylko składem. Są to roztwory o ograniczonej rozpuszczalności
ciekłe lub stałe. Na przykład mieszaniny dwóch cieczy tworzących układy
o ograniczonej rozpuszczalności rozpadają się w określonym zakresie stężeń
(zależnym od temperatury) na dwie fazy ciekłe, których składy są stałe i niezależne
od stężenia sumarycznego. Zatem tylko dla pewnych składów faza ciekła jest
stabilna względem faz sąsiednich. Stabilność tego rodzaju nazywa się stabilnością
względem dyfuzji. Zakłócenie stabilności fazy sprowadza się więc do wystąpienia
niejednorodności w zakresie stężenia fazy pierwotnie jednorodnej. Proces ten
można rozważać jako przemianę dyfuzyjną polegającą na wymianie składników
między dwoma dowolnymi elementami objętości układu pierwotnego.
2.3.3. STABILNOŚĆ CIEPLNA (TERMICZNA)
Zgodnie z regułą faz każda wieloskładnikowa faza ma dwa wolne
parametry, np. T i p, T i V, p i V. Zakres zmienności tych parametrów jest jednak
ograniczony i po przekroczeniu określonych wartości powstaje w układzie druga
faza. Pojawienie się nowej fazy należy uważać za zaburzenie równowagi fazy
pierwotnej i w tym kontekście rozważa się warunki termodynamiczne stabilności
fazowej układu. Stabilność cieplna dotyczy warunków trwałości fazy w przypadku
zmian energii wewnętrznej spowodowanych ogrzewaniem układu, a więc zmianą
jego temperatury w warunkach V=const lub p=const. Ogrzewanie fazy stałej lub
ciekłej z reguły prowadzi do powstania nowej fazy.
2.3.4. STABILNOŚĆ MECHANICZNA
Stabilność mechaniczna odnosi się do trwałości fazy, której równowaga jest
zakłócona zmianami objętości lub ciśnienia układu (T=const), co w ogólnym
przypadku może prowadzić do powstania nowej fazy w układzie. W tym przypadku
faza pierwotna jest trwała, jeżeli przejście od układu jednofazowego do

6
dwufazowego byłoby procesem nienaturalnym, a zatem wiązało się z dodatnim
przyrostem energii swobodnej układu (
0
df
).
3. ZAGROŻENIA DLA REAKTORÓW CHEMICZNYCH
Podstawowymi
parametrami
wpływającymi
na
szybkość
procesu
chemicznego są: temperatura i stężenie (w przypadku fazy ciekłej), bądź ciśnienie
gdy mamy do czynienia z fazą gazową. Stanowią one jednocześnie poważne
zagrożenie dla bezpieczeństwa reaktorów chemicznych. Gwałtowny wzrost
temperatury jak i podwyższenie ciśnienia (w przypadku procesów zachodzących
w fazie gazowej) mogą stać się przyczyną poważnej awarii chemicznej (pożaru,
wybuchu czy niekontrolowanej reakcji prowadzącej na przykład do wydzielenia
niebezpiecznych substancji). Poniżej omówione zostaną kryteria, jakie powinny
spełniać powyższe wielkości dla optymalnego przebiegu procesu chemicznego.
3.1. TEMPERATURA
Dla pojedynczej reakcji nieodwracalnej należy stosować możliwie najwyższą
temperaturę. Wtedy bowiem szybkość reakcji jest maksymalna, co w konsekwencji
daje maksymalną jednostkową zdolność produkcyjną reaktora i minimalną objętość
przestrzeni reakcyjnej. O maksymalnej wartości temperatury decydują najczęściej
techniczne możliwości realizacji procesu, koszty dostarczania ciepła, a także rodzaj
katalizatora dla reakcji katalitycznych. W przypadku endotermicznej reakcji
odwracalnej sytuacja jest analogiczna jak dla reakcji nieodwracalnej, tzn. że
optymalną jest temperatura najwyższa z możliwych. Jeżeli natomiast reakcja
odwracalna jest egzotermiczna, to ze wzrostem temperatury zwiększa się jej
szybkość, ale równocześnie maleje jej stała równowagi i związany z tym
równowagowy stopień przemiany. W tej sytuacji wyznacza się, zależnie od typu
reaktora bądź optymalną temperaturę, bądź też optymalny profil temperatury.
3.2. CIŚNIENIE (FAZA GAZOWA) LUB STĘŻENIE (FAZA
CIEKŁA)
Wzrost ciśnienia całkowitego wpływa na szybkość pojedynczej reakcji
chemicznej zachodzącej w fazie gazowej, gdyż wzrasta wtedy stężenie reagentów.
Wpływ ciśnienia jest tym silniejszy, im wyższy jest rząd reakcji. W reakcjach
odwracalnych w fazie gazowej od ciśnienia zależy ponadto równowagowy stopień
przemiany. Gdy reakcja przebiega ze zmniejszeniem całkowitej liczby moli, to
należy stosować w całym okresie reakcji możliwie wysokie ciśnienie. Wtedy
jednostkowa zdolność produkcyjna będzie maksymalna. Jeżeli zaś reakcja
przebiega ze zwiększeniem liczby moli to w początkowym okresie reakcji należy
stosować wysokie ciśnienie, przez co uzyskuje się wysoki stopień przemiany.
W miarę jednak postępu reakcji ciśnienie powinno zmniejszać się ze względu na
niekorzystny wpływ na równowagowy stopień przemiany.
W reakcjach zachodzących w fazie ciekłej analogiczny wpływ jak ciśnienie
dla reakcji gazowych spełnia stężenie. Gdy reakcja odwracalna przebiega w fazie
ciekłej ze wzrostem liczby moli, to w okresie początkowym należy stosować
maksymalne stężenie substratów, po czym w miarę postępu reakcji rozcieńczać
mieszaninę reakcyjną za pomocą substancji inertnych. Jeżeli natomiast reakcja

7
odwracalna przebiega ze zmniejszeniem liczby moli, to korzystne jest
utrzymywanie w całym okresie dużego stężenia substratów w mieszaninie
reakcyjnej.
4. RODZAJE ZAGROŻEŃ CHEMICZNYCH
4.1. NAJCZĘSTSZE PRZYCZYNY I SKUTKI WYSTĘPOWANIA
AWARII CHEMICZNYCH
Ogromna większość katastrof stałych obiektów przemysłowych to poważne
awarie chemiczne związane z przetwarzaniem, produkcją, magazynowaniem oraz
operacjami przeładunku niebezpiecznych substancji toksycznych, palnych
i wybuchowych, a także z wystąpieniem niekontrolowanych reakcji chemicznych,
które mogą towarzyszyć takim procesom z użyciem niekoniecznie substancji
niebezpiecznych.
Analiza poważnych awarii przemysłowych, wykonana na zlecenie Komisji UE
w ramach działań koordynowanych przez Biuro zagrożeń Poważnymi Awariami
(MAHB) wykazała, że w wielu przypadkach w trakcie ich przebiegu powstały
niebezpieczne substancje chemiczne, nie występujące w normalnych warunkach
procesu lub magazynowania. Szczególną uwagę zwrócono na zdarzenia awaryjne,
w których wystąpiły:
–
pożary,
–
reakcje niekontrolowane,
–
reakcje niepożądane,
prowadzące do powstania substancji niebezpiecznych, nie występujących
w normalnych warunkach danego procesu. Analiza danych wykazała, że spośród
wymienionych trzech głównych scenariuszy awarii, najbardziej znaczącą rolę
odgrywają pożary i stanowią ok. 49% wszystkich przypadków, następnie reakcje
niekontrolowane - ok. 32% i wreszcie reakcje niepożądane - ok. 19% (rys. 4.1).
0
50
100
150
200
250
liczba awarii (sztuk)
reakcje niepożądane
reakcje niekontrolowane
pożary
Rys. 4.1. Scenariusze awarii prowadzące do powstania substancji niebezpiecznych
[5]
W przypadku reakcji chemicznych głównym scenariuszem, w którym
powstawały substancje niebezpieczne, były reakcje niekontrolowane (ok. 60%
zdarzeń). Z kolei reakcje niepożądane (nieplanowane) były najczęstszą przyczyną
powstawania nowych substancji niebezpiecznych przede wszystkim podczas
operacji z cieczami i ich transportu (ok. 38%) (rys. 4.2)

8
Rys. 4.2. Liczba awarii (%) podczas różnych operacji technologicznych w zależno-
ści od scenariuszy awarii [5]
Na podstawie przeprowadzonych analiz sformułowane zostały następujące
wnioski:
niekontrolowane reakcje chemiczne wiążą się przede wszystkim z reakcjami
chemicznymi oraz destylacją. Zachodzą one w urządzeniach reakcyjnych i są
zazwyczaj wynikiem ogrzania się systemów chemicznych. Charakterystycznym
zjawiskiem jest to, że zachodzą one najczęściej w trakcie normalnych operacji
i procesów w reaktorach o działaniu okresowym- około 85% takich
przypadków
niepożądane reakcje są związane głównie z postępowaniem z substancjami
ciekłymi i stałymi. Są one najczęściej powodowane przez błędy, które prowadzą
do przypadkowego kontaktu niebezpiecznych substancji.
0
10
20
30
40
50
60
70
80
liczba awarii (%)
pożary
reakcje niekontrolowane
reakcje niepożądane
Reakcja chemiczna
Destylacja
Transport/ operacje
z płynami
Mieszanie
Transport/operacje
z
ciałami stałymi
Magazynowanie
Inne
Nieznane

9
0
10
20
30
40
50
60
70
80
90
100
liczba awarii (sztuk)
konserwacja
operacje nietypowe
procesy okresowe w normalnych warunkach
procesy ciągłe w normalnych warunkach
uruchamianie, wyłączanie
nieznane
Rys. 4.3. Liczba awarii w zależności od rodzaju operacji (awarie z udziałem
procesów niekontrolowanych) [5]
4.2. PRZYKŁADY POWAŻNYCH AWARII CHEMICZNYCH
W STAŁYCH OBIEKTACH PRZEMYSŁOWYCH
–
wybuch warnika w cukrowni „Głogów”, 1991
2 listopada 1991r. w cukrowni „Głogów” warnik o pojemności 60t i ciężarze
własnym 36t został wyrzucony siłą wybuchu do góry. Przebił dach hali
produkcyjnej i runął w odległości ok. 60m od posadowienia na podwórze cukrowni.
Cukrzyca wyrzucona z warnika pod dużym ciśnieniem spowodowała śmierć
7 pracowników, a 10 doznało niebezpiecznych dla życia poparzeń i urazów. Straty
ekonomiczne oszacowano na ok. 7 mln PLN. Bezpośrednią przyczyną wybuchu
było zbyt długie przetrzymywanie gorącej cukrzycy w warniku, spowodowane
brakiem opróżniania mieszadeł (błąd w technologii organizacji). Do zaistnienia
awarii przyczyniły się wady konstrukcyjne i wykonawcze warnika, powierzenie
obsługi pracownikom bez odpowiednich kwalifikacji oraz błędy organizacyjno-
decyzyjne.
–
awaryjne uwolnienie tetrachlorodwubenzo-p-dioksyny, Seveso, Włochy, 1976
10 lipca 1976r. otworzył się zawór bezpieczeństwa w reaktorze chemicznym
w zakładach ICMESA w Seveso, w wyniku nie kontrolowanego rozwoju reakcji
egzotermicznej w trakcie wytwarzania trójchlorofenolu. Wyrzucone pary substancji
rozprzestrzeniały się w otoczeniu zakładu. Około 1500 ha gęsto zaludnionego
obszaru zostało skażone. Znacznie ucierpiały zwierzęta, rośliny, a ludzie byli
narażeni na działanie toksycznej chmury. Skażenie gleby dioksyną było odczuwalne
przez następne 10 lat.
4.3. OGRANICZANIE ZAGROŻEŃ POWAŻNYMI AWARIAMI
W PAŃSTWACH UNII EUROPEJSKIEJ
Ograniczanie zagrożeń poważnymi awariami przemysłowymi, związanymi
z niebezpiecznymi substancjami chemicznymi w instalacjach produkcyjnych oraz
10
obiektach magazynowych, ma szczególne znaczenie w państwach europejskich.
Wiąże się to z dużą ilością zakładów użytkujących lub wytwarzających takie
substancje oraz z dużą gęstością zaludnienia, szczególnie w aglomeracjach
i zespołach miejsko – przemysłowych.
Do najważniejszych międzynarodowych rozwiązań z tego zakresu należy
Dyrektywa Unii Europejskiej z 1982 r., zwana Dyrektywą Seveso.
Stosownie do wymagań Dyrektywy EWG Komisja opublikowała raport
zawierający informacje dotyczące zarówno wdrożenia do prawodawstwa państw
członkowskich przepisów Dyrektywy Seveso jak i jej wykonywania w okresie
1994-1996. Raport ten zawiera m. in. dane o poważnych awariach przemysłowych,
jakie miały miejsce w państwach członkowskich UE. Dyrektywa Seveso II (weszła
w życie 3.02.1997), wprowadziła liczne zmiany. Obecnie dyrektywa ta jest
najbardziej precyzyjnym i najbardziej rygorystycznym międzynarodowym
dokumentem
prawnym
spośród
wszystkich
dotyczących
przedmiotowej
problematyki. Jej wymagania są bardziej precyzyjne, ostrzejsze, nastąpiły zmiany
w sferze kryteriów kwalifikacyjnych. Między innymi obiektem niebezpiecznym wg
Dyrektywy Seveso II jest instalacja a nie-jak w przypadku poprzedniej –teren
zakładu.
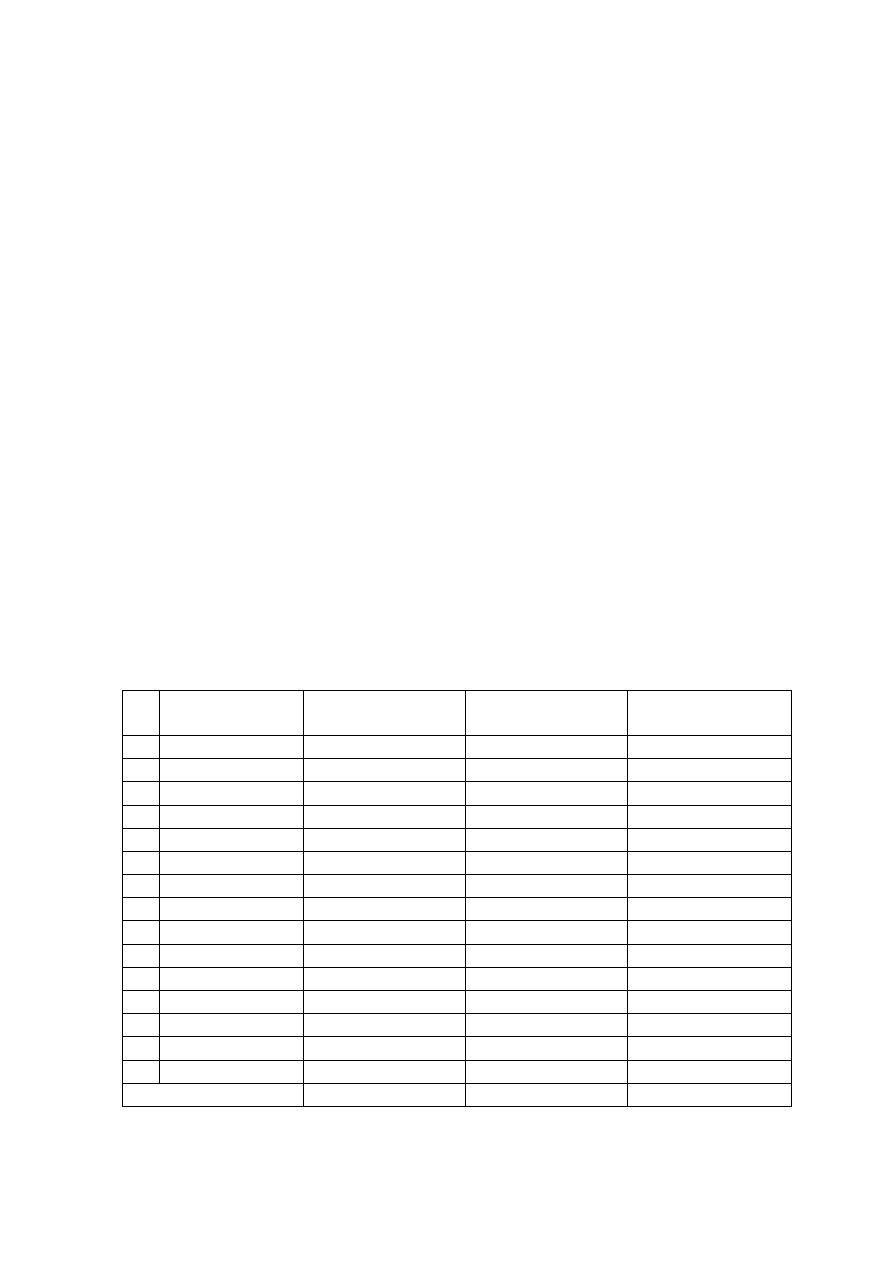
Potencjał zagrożeń poważnymi awariami przemysłowymi, mierzonymi
liczbą raportów bezpieczeństwa (tabela 1) jest w państwach UE, szczególnie
w tych największych, bardzo duży. Mimo zastosowania odpowiednich zasad
bezpieczeństwa oraz procedur, liczba poważnych awarii, które zdarzyły się
w krajach członkowskich UE w latach 1994-1996, wynosiła 92.
Tabela 1. Dane o wykonywaniu procedur Dyrektywy Seveso oraz o wielkości
zagrożeń poważnymi awariami w państwach UE [4]
Lp. Państwo
członkowskie
Liczba zakładów
Raporty
bezpieczeństwa
Liczba poważnych
awarii
1 Austria
140
100
0
2 Belgia
85
162
4
3 Dania
21
41
1
4 Finlandia
69
74
2
5 Francja
392
720
38
6 Grecja
52
52
0
7 Hiszpania
147
150
4
8 Holandia
124
119
4
9 Irlandia
20
19
0
10 Luksemburg
4
4
0
11 Niemcy
1828
1909
20
12 Portugalia
42
42
0
13 Szwecja
69
69
1
14 Wielka Brytania
308
493
13
15 Włochy
430
430
5
Razem UE
3731
4384
92

11
4.4. STAN ZAGADNIENIA W POLSCE
Z porównania oszacowanego potencjału zagrożeń w Polsce z wielkością
zagrożeń w państwach członkowskich UE wynika, że Polskę należy zaliczyć do
„czołówki” państw europejskich, jeśli chodzi o zagrożenia poważnymi awariami.
Wprowadzenie i funkcjonowanie w Polsce przepisów dotyczących systemu
przeciwdziałania poważnym awariom przemysłowym i wykonywania zadań
i procedur tego systemu przez zakłady stwarzające zagrożenia awariami oraz przez
odpowiednie władze spełniające funkcje nadzoru oraz wykonujących także szereg
innych zadań jest kwestią o nader ważnym znaczeniu. Wynika to z potrzeb
harmonizacji polskich przepisów z prawem unijnym. Przede wszystkim jednak
potrzeba wprowadzenia w Polsce tych przepisów i ich wykonywania wynika
z wielkości występujących w Polsce zagrożeń poważnymi awariami. Dokładne
określenie liczby zakładów oraz instalacji stwarzających zagrożenie wielkimi
awariami
chemicznymi
(wybuchy,
pożary,
uwolnienia
do
otoczenia
niebezpiecznych substancji) nie było dotychczas możliwe. W Polsce nie było
dotychczas prawnie usankcjonowanych kryteriów kwalifikacyjnych. W związku
z tym w prowadzonych od kilku lat przez Państwową Inspekcję Pracy (PIP),
Państwową Straż Pożarną (PSP) oraz przez Inspekcję Ochrony Środowiska (IOŚ)
rozpoznaniach i ocenach mających na celu zidentyfikowanie obiektów
stwarzających zagrożenie wielkimi awariami, instytucje te zastosowały kryteria
różniące się miedzy sobą i różne od ustalonych w Dyrektywie SevesoII. Tym
niemniej na podstawie danych PIP, PSP i IOŚ było możliwym dokonanie
oszacowania liczby obiektów stwarzających wielkie zagrożenia w Polsce.
W Centralnym Instytucie Ochrony Pracy zostały wykonane analizy danych PIP,
PSP oraz IOŚ z uwzględnieniem kryteriów kwalifikacyjnych Dyrektywy SevesoII.
Na tej podstawie oszacowano liczbę obiektów, dla których niezbędne jest
opracowanie raportów o bezpieczeństwie. Może ona wynosić w Polsce 200–250
(lub nawet więcej). Jest to poważny potencjał zagrożeń, który odniesiony do danych
dotyczących państw członkowskich UE, sytuuje Polskę pod względem wielkości
zagrożeń po Niemczech, Francji, Wielkiej Brytanii i Włoszech.
Począwszy od 1 października 2001 r. całokształt zagadnień przeciwdziałania
poważnym awariom przemysłowym będą regulować w Polsce przepisy ustawy
Prawo ochrony środowiska, przepisy ustawy o wprowadzeniu ustawy Prawo
ochrony środowiska, ustawy o odpadach oraz o zmianie niektórych ustaw, oraz
rozporządzenia Ministra Gospodarki i Ministra Środowiska.
LITERATURA
1. Szarawara J., Skrzypek J.: Podstawy inżynierii reaktorów chemicznych. Warszawa.
WNT, 1980.
2. Szarawara J.: Termodynamika chemiczna stosowana. Warszawa. WNT, 1997.
3. Koradecka D.: Bezpieczeństwo pracy i ergonomia, tom 2. Warszawa. Centralny
Instytut Ochrony Pracy, 1999.
4. Michalik J.S.: Wykonywanie przepisów Dyrektywy Seveso oraz zagrożenia
poważnymi awariami w państwach UE. Bezpieczeństwo pracy 10/2000.

12
5. Michalik J.S., Gajek A.: Substancje niebezpieczne powstające podczas poważnych
awarii przemysłowych. Bezpieczeństwo pracy 10/2002.
6. Berezowski M.: Stabilność reaktora chemicznego z recyklem. Inżynieria chemiczna
i procesowa 2/1993.
7. Michalik J.S.: Ograniczanie zagrożeń poważnymi awariami przemysłowymi. Nowe
przepisy polskie, Łódź. Centralny Instytut Ochrony Pracy,2001.
WYKAZ OZNACZEŃ
A, B
–stałe charakterystyczne dla danej reakcji
r
r
–szybkość reakcji, mol (m
-3
, m
-2
, kg
-1
) s
-1
r
A
–szybkość reakcji odniesiona do reagenta A, mol m
-3
s
-1
n
A
, n
i
–liczba moli reagenta (składnika), mol
t,
–czas, s
V
m
–objętość masy reagującej, m
3
V
R
–objętość reaktora, m
3
A
n
–strumień substancji A, mol s
-1
T
–temperatura bezwzględna, K
p, p
i
–ciśnienie cząstkowe, Pa
k
i
–współczynnik zależny od temperatury
n
i
–wykładnik liczbowy
c
A
, c
i
–stężenie molowe reagenta (składnika), mol m
-3
k
p
–stała szybkości reakcji, s
-1
R
–stała gazowa, J (mol K)
-1
E
–energia aktywacji
Wyszukiwarka
Podobne podstrony:
Wszystkiego Najlepszego chłopaki z okazji świąt reaktor chemiczny
2010-05-17, bezpieczeństwo publiczne
projekt reaktory chemiczne
Zadania Analiza finansowa, mikroekonomia makroekonomia ,bezpieczeństwo wewnetrzne, chemiczne,terrory
Ekonomia 1 PRZEDMIOT EKONOMII, mikroekonomia makroekonomia ,bezpieczeństwo wewnetrzne, chemiczne,te
audyt i kontrola ODPOWIEDZI 1, mikroekonomia makroekonomia ,bezpieczeństwo wewnetrzne, chemiczne,ter
INŻYNIERIA REAKTORÓW CHEMICZNYCH (2 termin - zadania) - 9.03.2012, PK - technologia chem, Rok V, Rea
INŻYNIERIA REAKTORÓW CHEMICZNYCH (2 termin zadania) 9 03 2012
17-borowceTECH, Technologia chemiczna PG, Chemia, I ROK, WYKŁADY, WYKŁADY
BEZPIECZEŃSTWO instalacji chemicznych
reaktor chemiczny2
Reaktor chemiczny(1)
reaktory chemiczne
Bezpieczeństwo chemiczne
inzynieria chemicza 1 17
Zarządzanie w sytuacjach kryzysowych - wykłady z 02.10.- 17.12. - 15.01, Sudia - Bezpieczeństwo Wewn
więcej podobnych podstron