
1
Kłębuszkowe choroby nerek – udział
mechanizmów immunologicznych w procesie
zapalnym
Marian Klinger
Katedra i Klinika Nefrologii i Medycyny Transplantacyjnej
Akademii Medycznej we Wrocławiu
Immunopatogeneza kłębuszkowych zapaleń
nerek - mechanizmy
• Limfokina – produkt limfocytów T zwiększająca przepuszczalność
kłębuszkowej ściany naczyniowej (T cell derived vascular
permeability factor – VPF) – submikroskopowe KZN i ogniskowe
segmentowe stwardnienie kłębuszków (FSGS)
• Odłożone z krążenia lub powstające in situ w kłębuszkach złogi
immunologiczne (kompleksy antygen-przeciwciało): błoniaste KZN,
nefropatia IgA, błoniastorozplemowe KZN, popaciorkowcowe KZN,
toczniowe KZN
• Przeciwciała wobec antygenu błony podstawnej kłębuszków (anty-
GBM): gwałtownie postępujące KZN – choroba Goodpasture’a
• Przeciwciała wobec cytoplazmatycznych antygenów granulocytów
(ANCA) – ziarniniak Wegenera, mikroskopowe zapalenie naczyń
Immunopatogeneza
⇓
Czynniki genetyczne
⇒
Zapalenie
⇓
Zróżnicowana dynamika progresji
⇓
Ustępowanie zmian lub bliznowacenie
⇓
Rozwój niewydolności nerek
Efektorowe drogi zapalenia w KZN
• Układy dopełniacza i krzepnięcia, układ renina -
angiotensyna – aldosteron
• Granulocyty obojętnochłonne i monocyty/makrofagi
• Cytokiny i czynniki wzrostowe (m.in.PDGF, MCP-1,
TGF-
β)
• Metaloproteinazy odpowiedzialne za degradację
pozakomórkowej macierzy mezangialnej
Udział czynników genetycznych i
mechanizmów mediatorowych w patogenezie
i postępującym przebiegu kłębuszkowych
zapaleń nerek na przykładzie nefropatii IgA
Nefropatia
Nefropatia
IgA
IgA
-
-
patogeneza
patogeneza
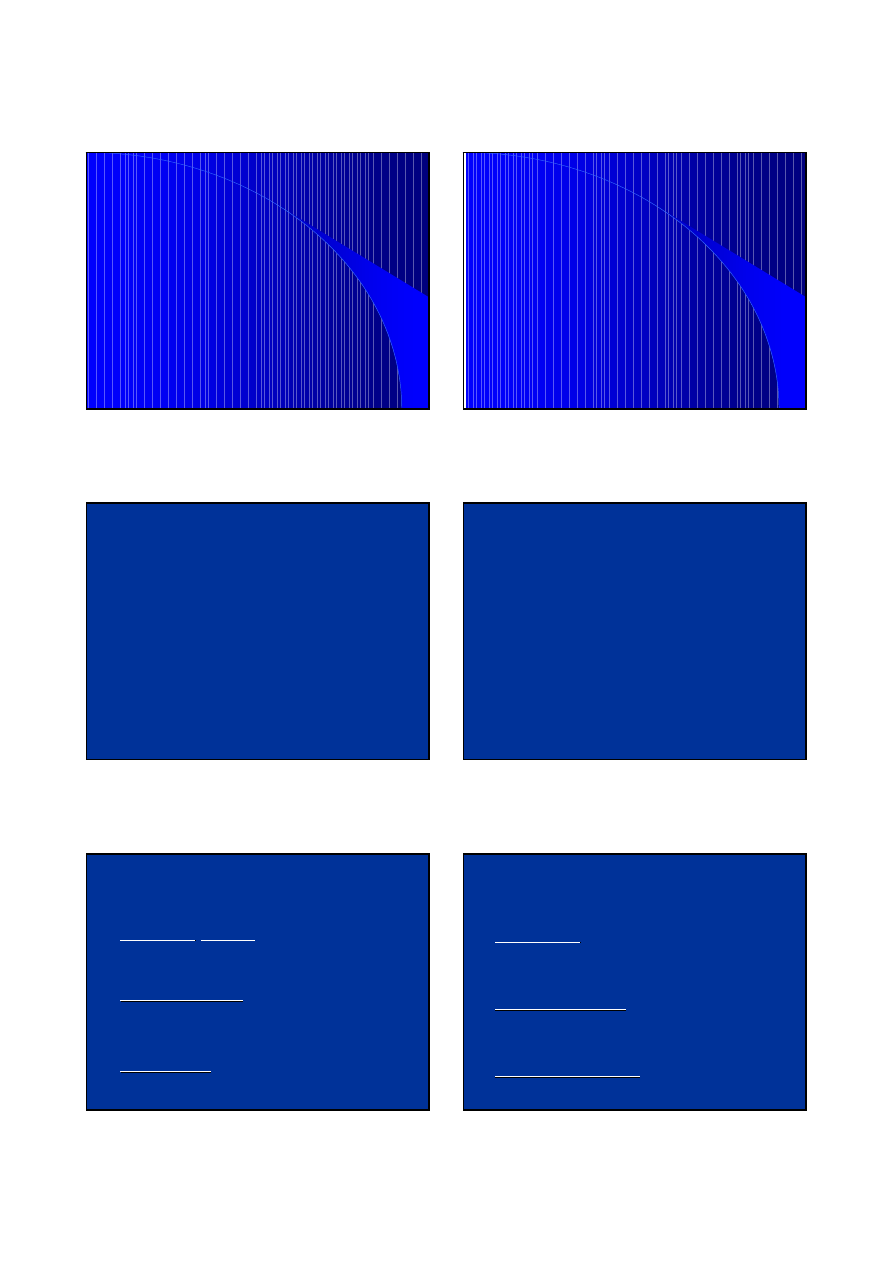
IgAN-choroba kompleksów immunologicznych –
polimeryczna IgA1 jako przeciwciało
z
Aktywacja ukł. odpornościowego błon śluzowych
→ wzrost produkcji
pIgA1
z
Polimery powstają z monomerów przez połączenie ich
łańcuchem J i w takiej postaci mogą przedostawać się przez śluzówkę
do światła układu oddechowego czy pokarmowego
IgA z upośledzoną galaktozylacją lepią się do
macierzy pozakomórkowej, która składa się m.in.
z fibronektyny, lamininy kolagenu typu IV
(białka obecne także w mezangium)
W IgAN-upośledzona galaktozylacja
CARB OHYD RA TE S
CARB OHYD RA TE S
TAIL
HINGE D RE GION
Fc
Fab
V
L
C
L
C a 2
Ca 3
Ca 1
VH
Monomer IgA1
Monomer IgA1
2
Nefropatia IgA
Nefropatia IgA
–
–
patogeneza (2)
patogeneza (2)
z
Fizjologicznie IgA-katabolizm w wątrobie po
związaniu z receptorem dla asjaloglikoprotein
(ASPGR),obecnym również w mezangium -po
glikozylacji przez łańcuchy glikozylowe
z
U chorych z IGAN- cząsteczki IgA < galaktozy na
łańcuchach glikanów i kw. sjalowego º
autoagregacja º trudność wychwytu przez
ASPGRº wiązanie z fibronektyną, lamininą i
kolagenem IV w mezangiumº inicjacja kzn
z
Niedobór 1,3 beta-galaktozotransferazy -
potencjalny czynnik patogenetyczny
Nefropatia IgA
Nefropatia IgA
–
–
patogeneza (3)
patogeneza (3)
z
Depozyty pIgA1 w mezangium
→
aktywacja układu dopełniacza
drogą alternatywną / złogi properdyny i C3, brak C1q i C4
→
komórki
mezangialne i napływowe /neutrofile, monocyty/
→
generowanie WRT,
uwalnianie enzymów lizozomalnych,cytokin- m.in. PDGF, TGFB
→
proliferacja komórek mezangium, wzrost produkcji macierzy
mezangium
→
szkliwienie i włóknienie
z
Antygeny wewnętrzne:
fragmenty bł.podstawnej i macierzy
mezangium,komórki mezangium, fibonektynna i sama IgA1- nie w pełni
galaktozylowana
z
Antygeny zewnętrzne:
czynniki infekcyjne (wirusowe,bakteryjne),
alergeny pokarmowe
Nefropatia IgA – patogeneza (4)
• Niedobór vascular endothelial growth factor (VEGF) jako
czynnik rozwoju zmian wstecznych w przebiegu nefropatii
IgA (Schrijvers i wsp., Kidney Int., 2004, 65, 203),
komórki jednojądrzaste krwi obwodowej chorych na
nefropatię IgA uwalniały do podłoża hodowlanego VEGF
pod wpływem stymulacji IL-2, IL-12, IL-15,IL-18,
natomiast hamująco oddziaływała stymulacja TGF-
β1,IL-
4, IL-10, IL-13.Dodanie nieprawidłowo glikozylowanej
IgA do hodowla komórek ludzkiego mezangium
→ spadek
ekspresji mRNA dla VEGF i zmniejszone uwalnianie do
podłoża.
Nefropatia IgA – patogeneza (5)
• Hamowanie aktywności VEGF w modelu doświadczalnym
mezangialnego KZN
→ rozwój stwardnienia kłębuszków.
• Doniesiono o występowaniu genotypu niskiej produkcji VEGF z
postępującym przebiegiem nefropatii IgA.
• Lokalna aktywacja układu krzepnięcia w nefropatii IgA (Hertig i
Rondau, J.Am. Soc. Nephrol., 2004, 15, 844), stymulowane przez
TNF-
α komórki mezangium wytwarzają TF (tkankowy czynnik
aktywacji krzepnięcia torem zewnątrzpochodnym), a w hybrydyzacji
in situ wysoka ekspresja mRNA dla czynnika V w komórkach
mezangium oraz wzmożona synteza inhibitora aktywatora
plazminogenu – PAI-1.
Postacie kliniczne kłębuszkowych zapaleń nerek - I
Sk
Sk
ą
ą
poobjawowa
poobjawowa: białkomocz (0.15-3.0 g/24h), krwinkomocz
> 2 erytrocytów w dużym polu widzenia w odwirowanym
osadzie moczu lub > 10 x 10 komórek/L, erytrocyty
→
zwykle dysmorficzne
Makroskopowy krwiomocz
Makroskopowy krwiomocz: brązowo-czerwonawy kolor
moczu (bez skrzepów i dolegliwości bólowych), typowo
współistniejący z zakażeniem, najczęściej górnych dróg
oddechowych, w okresach między atakami
→
krwinkomocz
± białkomocz
Zesp
Zesp
ó
ó
ł
ł
nerczycowy:
nerczycowy: białkomocz > 3.5 g/dobę dobę (dorośli),
> 40 mg/h/m² (dzieci), hipoalbuminemia < 35g/L,
obrzęki, hipercholesterolemia, lipiduria
Postacie kliniczne kłębuszkowych zapaleń nerek - II
Zesp
Zesp
ó
ó
ł
ł
nefrytyczny
nefrytyczny: skąpomocz, krwinkomocz z wałeczkami
erytrocytarnymi, białkomocz
→ zwykle < 3 g/dobę,
umiarkowane obrzęki, nadciśnienie, nagły początek, przebieg
samoograniczający: popaciorkowcowe kzn
Gwa
Gwa
ł
ł
townie post
townie post
ę
ę
puj
puj
ą
ą
ce
ce
kzn
kzn: rozwój niewydolności nerek w
ciągu dni/tygodni, białkomocz
→ zwykle < 3g/dobę,
krwinkomocz z wałeczkami erytrocytarnymi, ciśnienie
tętnicze
→ często prawidłowe, objawy zapalenia naczyń
Zaawansowane przewlek
Zaawansowane przewlek
ł
ł
e
e
kzn
kzn
:
: nadciśnienie, białkomocz
~3g/dobę, ubytek filtracji, zmniejszona wielkość nerek
3
Klasyfikacja patomorfologiczna
kłębuszkowych zapaleń nerek - I
Opiera się na badaniach świetlno-mikroskopowych,
immunohistochemicznych lub immunofluorescencyjnych oraz
elektronowo-mikroskopowych i wyróżnia:
1. Zapalenia rozplemowe:
a) wewnątrzwłośniczkowe
b) zewnątrzwłośniczkowe
c) mezangialne
Klasyfikacja patomorfologiczna
kłębuszkowych zapaleń nerek - II
2. Zapalenie mezangialno-włośniczkowe lub
błoniasto-rozplemowe:
typ I ze złogami podśródbłonkowymi
typ II z gęstymi złogami wewnątrz błony podstawnej
typ III ze złogami podśródbłonkowymi i
podnabłonkowymi
3. Zapalenie błoniaste
4. Zapalenie submikroskopowe
5. Ogniskowe segmentowe stwardnienie kłębuszków
6. Inne rzadkie postacie: włókienkowa, immunotaktoidalna,
lipoproteinowa
Pathology of glomerular disease. Comprehensive Clinical Nephrology
Edited by RJ Johnson, J Feehally; Mosby, Edinburgh 2003
Submikroskopowe KZN
Pathology of glomerular disease. Comprehensive Clinical Nephrology
Edited by RJ Johnson, J Feehally; Mosby, Edinburgh 2003
Ogniskowe segmentowe stwardnienie kłębuszków - FSGS
Pathology of glomerular disease. Comprehensive Clinical Nephrology
Edited by RJ Johnson, J Feehally; Mosby, Edinburgh 2003
Rozlany rozplem mezangium – nefropatia IgA
Pathology of glomerular disease. Comprehensive Clinical Nephrology
Edited by RJ Johnson, J Feehally; Mosby, Edinburgh 2003
Zajęcie nerek w zapaleniu naczyń – segmentowa martwica

4
Pathology of glomerular disease. Comprehensive Clinical Nephrology
Edited by RJ Johnson, J Feehally; Mosby, Edinburgh 2003
Gwałtownie postępujące KZN z przeciwciałami
anty-GBM – komórkowe „półksiężyce”
Dostępne środki terapeutyczne
• Glikortykosteroidy: prednizon, metyloprednizolon
• Cyklofosfamid
• Chlorambucil
• Cyclosporyna
• Tacrolimus
• Mykofenolan mofetilu
• Plazmaferezy
• Immunoglobuliny
• Przeszczepy komórek macierzystych
Submikroskopowe kłębuszkowe zapalenie nerek
Steroidy
Steroidy
→
→
remisja
remisja
↓
Wczesne nawroty lub
Wczesne nawroty lub
steroidooporno
steroidooporno
ść
ść
↓
Cytostatyk alkiluj
Cytostatyk alkiluj
ą
ą
cy
cy
lub
lub
CsA
CsA
,
,
Lewamizol
Lewamizol
, MMF
, MMF
Miejsce glikortykosteroidów w leczeniu KZN
• Pierwsze doniesienie: Luetschera i Deminga (J Clin Invest,
1950, 29, 1576): u 6 spośród 11 chorych leczonych
kortizonem zmniejszenie białkomoczu i poprawa GFR
• Od razu widoczna istota problemu: kortykoidy skuteczne u
części chorych
• U kogo stosować, jak długo, kiedy rozpoznawać
steroidooporność, zjawiska częstej nawrotowości i
steroidozależności remisji
• Początkowa monoterapia prednizonem: submikroskopowe,
KZN, FSGS, nefropatia IgA, błoniastorozplemowe KZN –
typ1
Ogniskowe segmentowe stwardnienie kłębuszków
Biopsja nerki
Biopsja nerki
→
→
typ FSGS,
typ FSGS,
czynniki rokownicze
czynniki rokownicze
Bia
Bia
ł
ł
komocz
komocz
subnerczycowy
subnerczycowy
Zesp
Zesp
ó
ó
ł
ł
nerczycowy
nerczycowy
ACEI lub/i ATB steroidy
Oporno
Oporno
ść
ść
CsA i prednizon,
tacrolimus i prednizon,
cytostatyk alkilujący i prednizon
Oporno
Oporno
ść
ść
MMF
Comprehensive Clinical Nephrology
Edited by RJ Johnson, J Feehally; Mosby, Edinburgh 2003
5
Renin
Renin
-
-
Angiotensin System
Angiotensin System
Renin inhibitors
Renin inhibitors
Angiotensinogen
Angiotensinogen
Renin
Renin
Angiotensin
Angiotensin
I
I
Non
Non
-
-
Renin
Renin
•
•
Tonin
Tonin
•
•
Cathepsin
Cathepsin
ACE inhibitors
ACE inhibitors
ACE/
ACE/
BPF
BPF
Angiotensin
Angiotensin
II
II
Non
Non
-
-
ACE
ACE
•
•
Chymase
Chymase
AT II
AT II
•
•
Vasodilation
Vasodilation
•
•
Anti proliferation
Anti proliferation
•
•
↑
↑
Kinins
Kinins
•
•
↑
↑
NO
NO
AT I
AT I
•
•
Vasoconstriction
Vasoconstriction
•
•
Cell growth
Cell growth
•
•
Na
Na
+
+
/H
/H
2
2
O retention
O retention
•
•
SNS activation
SNS activation
Cross talk
Cross talk
RAAS MODULATORS
RAAS MODULATORS
:
:
Spironolactone
Spironolactone
Eplerenone
Eplerenone
Beta blockers
Beta blockers
Bradykinin
Bradykinin
Inactive
Inactive
Peptides
Peptides
BKR
BKR
•
•
Vasodilation
Vasodilation
•
•
↓
↓
Ischemia
Ischemia
•
•
↓
↓
Platelet agg
Platelet agg
•
•
⊕
⊕
inotrope
inotrope
NO
NO
↑
↑
Enzymatic activity
Enzymatic activity
Enzymatic blockade
Enzymatic blockade
Product/receptor stimulation
Product/receptor stimulation
Układ RAAS podwójna blokada IEK + antagonista AT1
CB-8
IEK i antagoniści AT1 w celu obniżenia
nerczycowego białkomoczu
• Dawki wyższe niż wymagane dla optymalnej kontroli
ciśnienia tętniczego
• Lisinopril w dawce 20 mg/dobę przez 6 tygodni spadek
białkomoczu o 45%, w dawce 40 mg/dobę spadek
białkomoczu o 75%
• Korzyści z podwójnej blokady: lisinopril 20 – 40 mg/dobę
+ losartan 50 – 100 mg/dobę spadek białkomoczu o 85%
(Laverman G.D. i wsp., Kidney Int. , 2002,62, 1020)
CB-13
COOPERATE Trial
42
Doubling of serum creatinine
or ESRD
Primary outcome
Run-in to determine maximum
antiproteinuric ACEi dose
(trandolapril 3 mg) then
randomization to trandolapril
3 mg, losartan 100 mg, or both
Study design
263 pts with nondiabetic
kidney disease
Study population
Nakao N et al. Lancet 2003;361:117-124
CB-14
Proportion of Patients Reaching Endpoint
COOPERATE Trial
Nakao N et al. Lancet 2003;361:117-124
0
6
12
18
24
30
36
30
25
20
15
10
5
0
P
roport
io
n
re
ac
h
in
g
e
n
d
p
o
int
, %
(ren
al
f
ail
u
re o
r Cr
C×
2)
Combination
Months after randomization
Losartan
Trandolapril
42
At risk, n
Losartan
89
88
84
79
65
59
47
Trandolapril
86
85
83
75
72
63
58
Combination
88
87
86
83
76
73
67
p
= 0.018
Błoniaste kłębuszkowe zapalenie nerek
Różnicowanie między postacią idiopatyczną i wtórnymi
Biopsja nerki i ocena czynników rokowniczych
Białkomocz subnerczycowy Zespół nerczycowy
ACEI lub/i ATB Steroidy i cytostatyk alkilujący
Spadek białkomoczu po 2 miesiącach leczenia <1g/dobę
Kontynuacja leczenia
nadal białkomocz
→
rozważenie leczenia immunosupresyjnego CsA
Oporność
→
MMF
Postępowanie terapeutyczne w chorobie
Goodpasture’a
•
Pulsy metyloprednizolonu w dawce 7-15 mg/kg/dobę (maksymalnie
1g/dobę) przez 3 dni
→ 60 mg prednizonu codziennie przez 7 dni→
co tydzień
↓ dawki do 45, 30,20,15,10 i 5 mg
•
U chorych < 55 r.ż. jednocześnie cyklofosfamid – 3mg/kg/dobę, u
chorych > 55 r.ż. 2 mg/kg/dobę
•
Jednocześnie codzienne zabiegi plazmaferezy z wymianą 4 l. osocza
podczas seansu do czasu zniknięcia p/ciał anty-GBM – zwykle 14
dni
•
Po zniknięciu przeciwciał
↓ dawki cyklofosfamidu o 25 mg co 14
dni do 50 mg/dobę, zakończenie kuracji po 12 tygodniach
•
Profilaktyka przeciwinfekcyjna od początku terapii
immunosupresyjnej: płukanie j. ustnej nystatyną
sulfametoksazol/trimetoprim
→ zapobieganie Pneumocystis carinii
6
Gwałtownie postępujące kłębuszkowe zapalenie
nerek z obecnością przeciwciał ANCA
• Początek terapii
→ pulsowe dawki metyloprednizolonu po 500
mg przez 3 dni, następnie prednizon 1mg/kg/dobę
w
połączeniu z cyklofosfamidem 2 mg/kg/dobę. Alternatywa dla
doustnego cyklofosfamidu
→ comiesięczne wlewy dożylne
0.75g/m², dorównujące terapii doustnej w indukcji remisji, a
ustępujące pod względem ich trwałości
• Prednizon podawany przez okres ok. roku, po 3 miesiącach
redukcja do 12.5 – 15 mg/dobę, natomiast cytostatyk przez 12
miesięcy od uzyskania remisji
• Najcięższe przypadki zajęcia płuc i nerek (bezmocz lub stężenie
kreatyniny > 6 mg/dl) immunosupresja i zabiegi plazmaferezy
→
7 wymian osocza
→ 3-4 l w ciągu 14 dni
Plazmaferezy w leczeniu najcięższego zajęcia
nerek i płuc w przebiegu ziarniniaka
Wegenera
• Wskazania: gwałtownie postępujące KZN, w którym
szybko rozwija się wymagająca dializ niewydolność nerek
i ciężkie zajęcie płuc z krwotokami, oporne na leczenie
prednizonem i cyklofosfamidem
• Z klinicznej kazuistyki: 45-letnia T.T. przyjęta do Kliniki z
rozpoznaniem ziarniniaka Wegenera, stężenie kreatyniny
w sur. 7mg/dl, mocznika 35 mmol, Hb–7.2 g/dl, krwinki
białe 16 G/l, przeciwciała cANCA 122 IU/ml, przeciwciała
anty-GBM 21 IU/ml. Przed przekazaniem do Kliniki
otrzymała w oddziale pulmonologicznym 3g
metyloprednizolonu, a następnie przez tydzień
prednizolon 75 mg/die i.v. i cyklofosfamid 100 mg/die p.o.
Ziarniniak Wegenera z klinicznego archiwum cd.
• Wobec braku poprawy czynności nerek rozpoczęto
leczenie hemodializami, a po kolejnych 3 – dniach ze
względu na nasilenie zmian płucnych (krwotoki)
plazmaferzy: 5 codziennych 2-litrowych wymian osocza i
następnie po 3 dniowej przerwie 3 dalsze co 2 dzień. Po 5
zabiegach plazmaferezy można było zaprzestać
hemodializ. Na zakończenie każdego cyklu plazmaferez
podano 200 mg cyklofosfamidu i.v.
• W 2 miesiące od rozpoznania choroby - stężenie
kreatyniny w sur. 1.16 mg/dl, przeciwciała cANCA 12
IU/ml, leczenie prednizon 60 mg naprzemiennie z 40
mg/dobę, cyklofosfamid 100mg/dobę.
Przyjęcie do Kliniki
Przed plazmaferezami
Po zakończeniu programu plazmaferez

7
Leczenie kłębuszkowych zapaleń nerek: przyszłe
możliwości
• Tolerancja immunologiczna na antygen – LJP 394
(abetimus sodium); polimer DNA na rusztowaniu z
trietylenu glikolu, krzyżowe wiązanie z receptorem dla
dsDNA na nie uczulonych limfocytach B, bez udziału
komórek T, stan tolerancji u chorych na toczniowe
zapalenie nerek, stabilizacja remisji, międzynarodowe
badanie III fazy w toku
• Podobne podejście w KZN o mechanizmie przeciwciał
anty GBM
→ dobrze scharakteryzowany antygen próby na
zwierzętach
KZN – nowe metody terapeutyczne
• Usunięcie limfocytów B – rytuksymab, humanizowane
przeciwciało monoklonalne
→ oporne TZN → ziarniniak
Wegenera
→ nefropatia błoniasta
• Blokowanie kostymulacji – dwa preparaty CTLA4 (Ig i
LEA29Y) – skuteczność w RZS; planowane próby w
toczniu
• Ecluizumab
→ przeciwciało anty C5→ badanie II fazy w
USA u 122 chorych na nefropatię błoniastą, brak wpływu
krótkoterminowej terapii na białkomocz
KZN – co jeszcze w terapii przed klinicznym
progiem?
• Imatinib (Glivec) – inhibitor kinazy tyrozynowej;
hamowanie procesów komórkowych wzbudzanych przez
PDGF, przygotowywana próba kliniczna w nefropatii IgA
• Hamowanie szlaku TGF-
β1 za pomocą BMP-7
• Pierwotna skrobiawica: autologiczny przeszczep szpiku;
hamowanie wiązania glikozaminoglikanów do
prekursorowych białek NC –503 (sól dwusodowa kwasu
propanodwusiarkowego) przemiotem międzynarodowej
próby klinicznej II fazy
Zalecena literatura
1.
Klinger M., Zmonarski S. : Zespół nerczycowy. W:
„Interna”, Januszewicz W., Kokot F. red. PZWL,
2002,t.2
2.
Klinger M., Kazimierczak K: Pierwotne kłębuszkowe
zapalenia nerek, tamże
3.
Klinger M., Magott- Procelewska M.: Nerki w
chorobach układowych, tamże
4.
Rutkowski B., Klinger M. red.: Kłębuszkowe choroby
nerek. MAKmed, 2003
5.
Klinger M., Krajewska M.: Niezapalne choroby
kłębuszków nerkowych. W „Nefrologia”, Książek A.,
Rutkowski B. red., Czelej, 2004
Wyszukiwarka
Podobne podstrony:
immunologia kzn 2015
SEMINARIUM IMMUNOLOGIA Prezentacja
Testy immunologiczne
Seminarium 6 Immunologia transplantacyjna farmacja 2
Osteoporaza diag i lecz podsumow interna 2008
Cw 7 IMMUNOLOGIA TRANSPLANTACYJNA
Immunologia nowotworów
Immunoterapia1
KZN 2015
immunologiczne ostrzerzenie przed choroba
Mechanizmy swoistej immunoterapii alergii 3
W7 IMMUNOLOGIA INFEKCJI
immunodiagnostyka chorób z autoagresji 2008
więcej podobnych podstron