ZINC BOROHYDRIDE
1
Zinc Borohydride
1
Zn(BH
4
)
2
[17611-70-0]
B
2
H
8
Zn
(MW 95.09)
InChI = 1/2BH4.Zn/h2*1H4;/q2*-1;+2
InChIKey = PTJGRTOJBSRNJP-UHFFFAOYAM
(mild reducing agent for carbonyl groups;
1
can be used in the
presence of base-sensitive functional groups; stereoselective
reducing agent
2
)
Solubility:
sol ether, DMF, CH
2
Cl
2
, toluene, THF.
Preparative Method:
commercially available anhydrous Zinc
Chloride (ca. 10 g) in a 200 mL flask was fused three or
four times under reduced pressure and then anhydrous ether
(ca. 100 mL) was added. The mixture was refluxed for 1–2 h
under argon and allowed to stand at 23
◦
C. The supernatant sat.
solution of ZnCl
2
(0.69 M) in ether (80 mL; 55 mmol) was
added to a stirred suspension of Sodium Borohydride (4 g;
106 mmol) in anhydrous ether (300 mL). The mixture was
stirred for 2 d and stored at rt under argon. The supernatant
solution was used for reduction.
3
Handling, Storage, and Precautions:
the solutions are sensitive
to moisture and must be flushed with N
2
or argon. However, it
is preferable to use freshly prepared reagent.
Mild Reducing Agent. Zn(BH
4
)
2
is a mild reducing agent
and only aldehydes, ketones, and azomethines
4
are reduced to
the corresponding alcohols and amines under normal condi-
tions. Moreover, the ether solutions are almost neutral and thus
can be used for the chemoselective reduction of aldehydes and
ketones in the presence of nitrile,
5
ester,
5,6
γ
-lactone,
7
aliphatic
nitro,
8
and base-sensitive functional groups (eqs 1 and 2).
5,9
Selective reduction of saturated ketones and conjugated aldehy-
des over conjugated enones can also be effected with Zn(BH
4
)
2
in DME (eq 3).
10
NHCHO
AcO
CN
C
5
H
11
O
Zn(BH
4
)
2
NHCHO
AcO
CN
C
5
H
11
OH
6
6
(1)
a mixture of epimeric alcohols
diglyme
25 °C
O
OCO
2
CH
2
CCl
3
O
O
O
Zn(BH
4
)
2
OH
OCO
2
CH
2
CCl
3
O
O
O
(2)
ether, rt
73%
(3)
O
O
O
OH
100% selectivity
Zn(BH
4
)
2
DME, –78 °C
Although Zn(BH
4
)
2
is usually unreactive towards carboxylic
acids and esters, activated esters (eq 4)
11
and thiol esters (eq 5)
12
undergo reduction, giving alcohols. Even carboxylic acids can
be reduced to alcohols with this reagent in the presence of
Trifluoroacetic Anhydride (TFAA) (eq 6)
13
and acid chlorides
undergo reduction by the addition of N,N,N
′
,N
′
-Tetramethyl-
ethylenediamine (eq 7).
14
Acetals are reductively cleaved to
ethers when Chlorotrimethylsilane is added (eq 8).
15
MeO
O
O
CO
2
H
O
Ar
O
HO
1. Im
2
CO
(4)
2. Zn(BH
4
)
2
DME, –20 °C
>45%
COSPh
Zn(BH
4
)
2
OH
(5)
ether, rt
99%
OH
O
( )
16
OH
1 equiv Zn(BH
4
)
2
( )
16
(6)
1 equiv TFAA
DME
92%
1 equiv Zn(BH
4
)
2
(7)
O
Cl
OH
1 equiv TMEDA
ether, 0 °C
93%
O
O
O
OH
( )
7
0.5 equiv Zn(BH
4
)
2
(8)
( )
8
( )
2
1.2 equiv TMSCl
ether, rt
97%
Reduction of aliphatic carboxylic esters takes place under
ultrasonic activation to give alcohols.
16
The reducing ability of
this system is enhanced by the addition of a catalytic amount
of N,N-dimethylaniline and thus aromatic esters which are un-
affected under the normal conditions undergo reduction (eqs 9
and 10).
16
O
O
CO
2
Me
Zn(BH
4
)
2
O
O
OH
(9)
sonication
DME
100%
CO
2
Me
Zn(BH
4
)
2
OH
(10)
sonication
DME
N,N
-dimethylaniline
100%
Unsymmetrical epoxides are reductively cleaved to the less
substituted alcohols by the use of silica gel-supported Zn(BH
4
)
2
(eq 11).
17,18
The same reagent is effective for regioselective
1,2-reduction of conjugated ketones and aldehydes to give
allylic alcohols (eq 12).
19
Zn(BH
4
)
2
supported on cross-linked
Poly(4-vinylpyridine) (XP4) reduces aldehydes in the presence
of ketones with high chemoselectivity (eqs 13 and 14).
20
This
Avoid Skin Contact with All Reagents
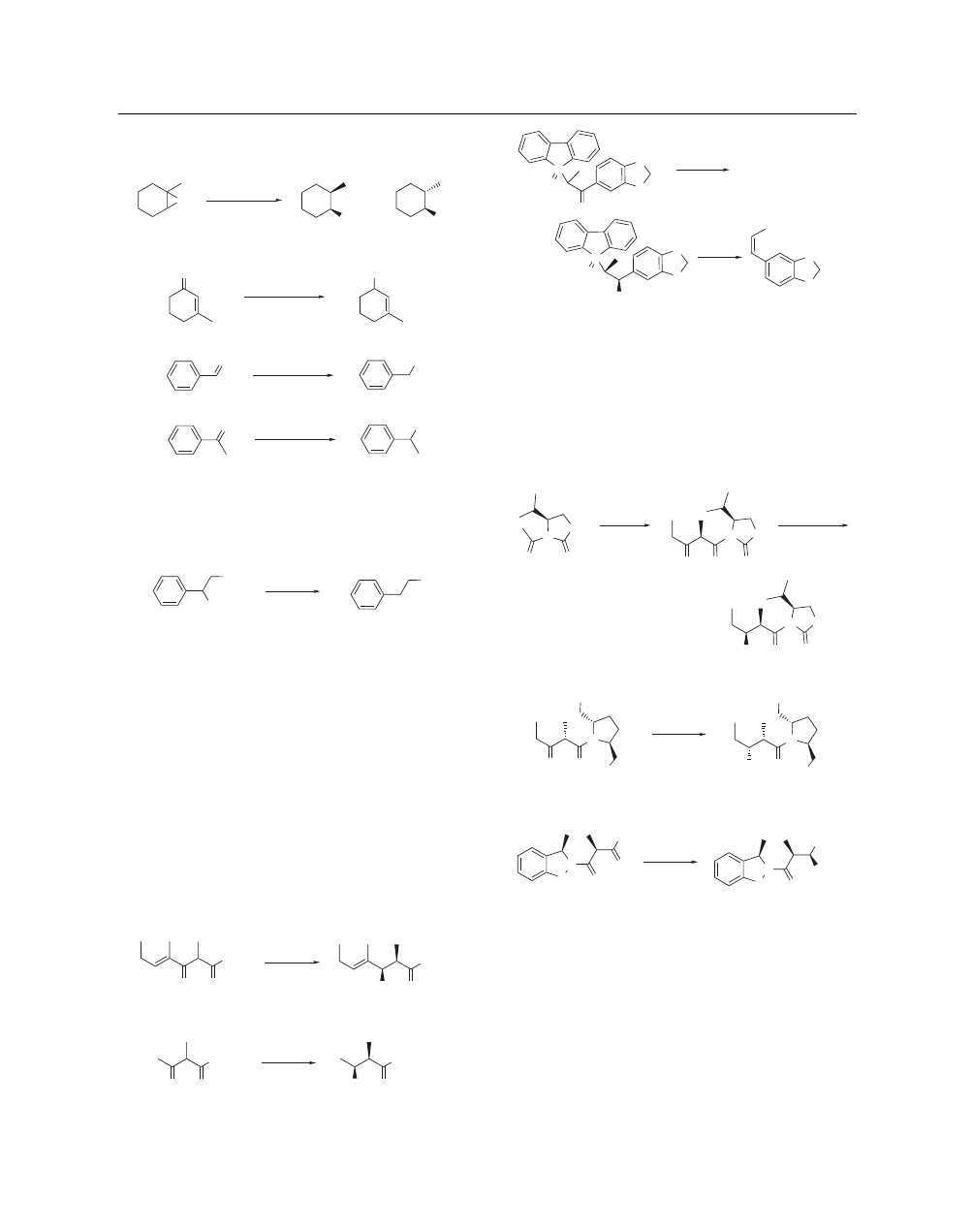
2
ZINC BOROHYDRIDE
polymer-supported reagent can be stored at rt without appre-
ciable change in its reactivity.
O
OH
OH
+
Zn(BH
4
)
2
/SiO
2
cis
:trans = 90:10
(11)
THF, rt
85%
(12)
O
OH
Zn(BH
4
)
2
/SiO
2
THF
–5 to –10 °C
80%
(13)
Zn(BH
4
)
2
/XP
4
O
OH
EtOH
80%
(14)
Zn(BH
4
)
2
/XP
4
O
OH
EtOH
0%
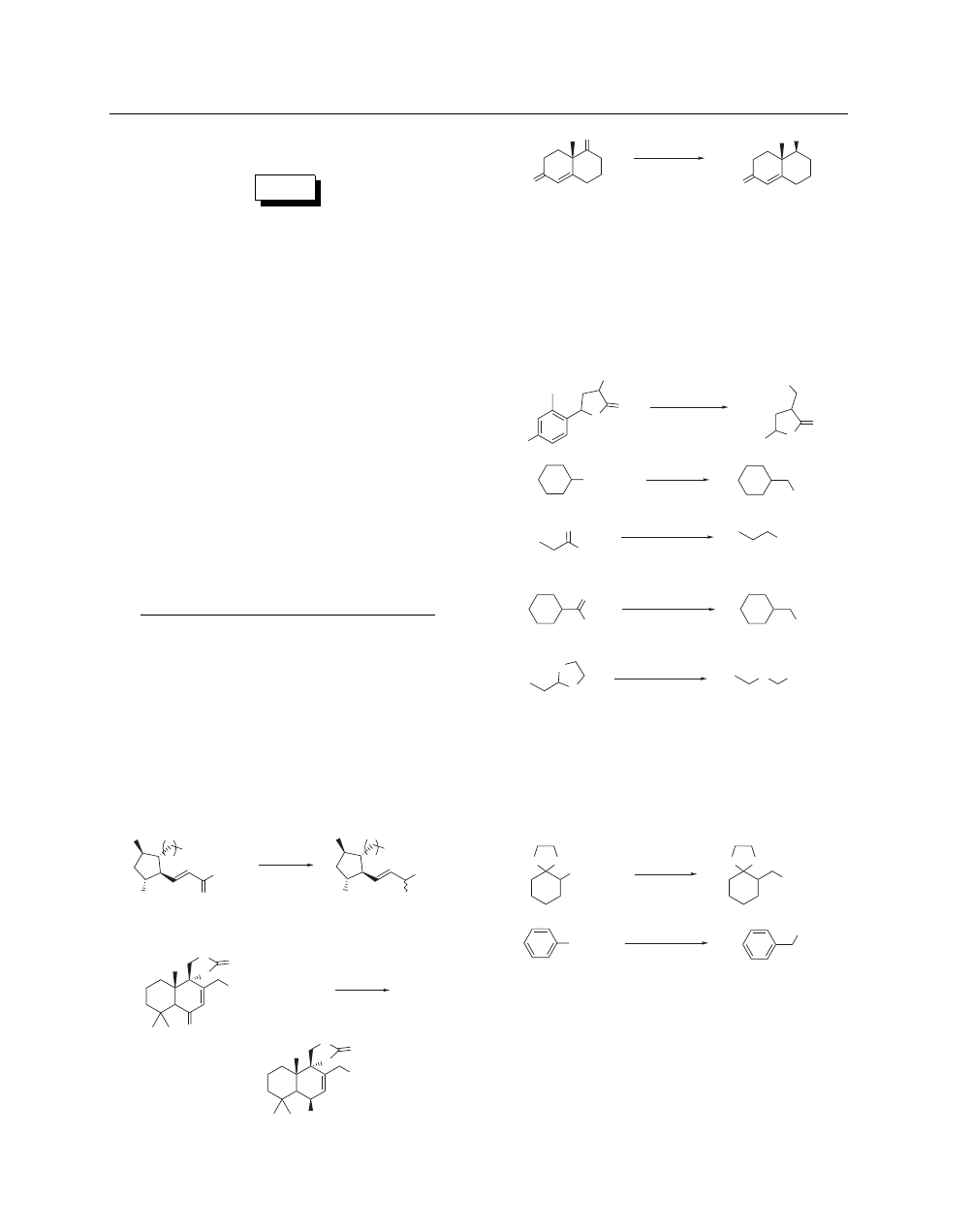
Tertiary and benzylic halides are reductively dehalogenated
with Zn(BH
4
)
2
(eq 15).
21
This process has been applied for the
selective reduction of the distant double bond(s) in geranyl farne-
syl and geranyl geranyl derivatives.
22
(15)
Br
Br
Br
Zn(BH
4
)
2
ether, rt
81%
Stereoselective Reductions. syn-α-Methyl-β-hydroxy esters
or their equivalents which repeatedly appear in the framework
of polyoxomacrolide antibiotics are synthesized stereoselectively
by the reduction of the corresponding α-methyl-β-keto esters
23,24
or α-methyl-β-hydroxy ketones
25
with Zn(BH
4
)
2
in ether. Excel-
lent selectivities are obtained when the carbonyl group is con-
jugated with phenyl or vinyl groups (eq 16)
23
–
25
or the esters
in α-methyl-β-keto esters are replaced by the amides (eq 17).
26
Ketones having a phosphine oxide group in place of esters or
amides produce syn products by the Zn(BH
4
)
2
reduction, while
reduction with Lithium Triethylborohydride gives the anti isomer
stereoselectively (eq 18).
27
The syn-directing reduction is pre-
sumed to proceed through a metal-mediated cyclic transition state
and thus the use of a complex hydride like Zn(BH
4
)
2
, whose metal
possesses a high coordinating ability, is advantageous for
producing excellent selectivity.
OBn
O
O
Zn(BH
4
)
2
OBn
OH
O
(16)
syn
:anti = >99:1
ether
0 °C
85%
NHPh
O
O
Zn(BH
4
)
2
NHPh
OH
O
(17)
syn
:anti = 98:2
ether
–78 °C
99%
P
O
O
O
O
P
O
OH
O
O
O
O
(18)
syn
:anti = 98:<2
Zn(BH
4
)
2
ether
–78 °C
95%
Acylation of chiral N-propionyloxazolidinones gives chiral α-
methyl-β-keto imides, whose Zn(BH
4
)
2
reduction affords opti-
cally active syn-α-methyl-β-hydroxy derivatives with virtually
complete stereoselectivity (eq 19).
28,29
In the same way, chiral
carboxamides (eq 20)
30
and (R)-N-acylsultams (eq 21)
31
also
afford chiral syn products with high selectivities.
N
O
O
O
N
O
O
O
O
N
O
O
O
OH
1. LDA
*
(19)
Zn(BH
4
)
2
2. EtCOCl
CH
2
Cl
2
–Et
2
O
0 °C
>95%
N
O
O
OMOM
OMOM
Zn(BH
4
)
2
N
OH
O
OMOM
OMOM
syn
:anti = 99:1
(20)
*
96%
S
O
2
N
Ph
O
O
Zn(BH
4
)
2
S
O
2
N
Ph
O
OH
syn
:anti = 99.1:0.9
(21)
*
ether
–10 °C
82%
Selectivity of Zn(BH
4
)
2
reductions of β-hydroxy.
32,33
or N-
aryl-β-amino
34
ketones lacking α-substituents is generally unsat-
isfactory. A case where an excellent result is obtained is shown
in eq 22.
32
For the stereoselective preparation of syn- and anti-
1,3-diols the use of other reagents is recommended.
35
However,
in the reduction of β-keto esters, with chiral ester units, the syn
selectivity is improved significantly (eq 23).
36
Reduction of the
same keto ester with DIBAL-BHT (Diisobutylaluminum 2,6-
Di-tert-butyl-4-methylphenoxide) affords the diastereomer with
high selectivity (eq 24).
36
A list of General Abbreviations appears on the front Endpapers
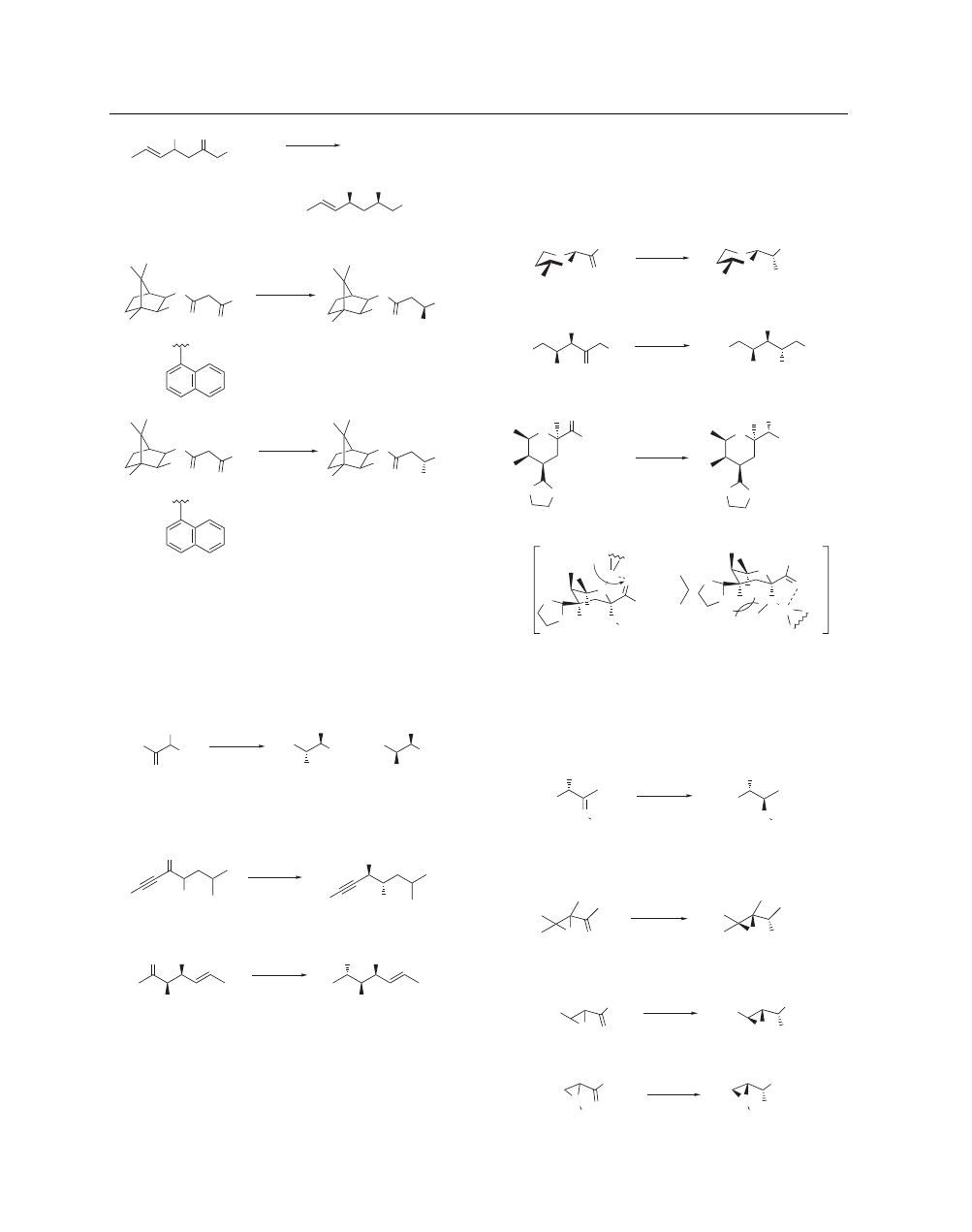
ZINC BOROHYDRIDE
3
CO
2
Me
OH
O
Zn(BH
4
)
2
CO
2
Me
OH
OH
syn
:anti = 91:9
(22)
ether
–20 °C
69%
O
Ar
R
O
O
ZnCl
2
Zn(BH
4
)
2
toluene
–78 °C
94%
O
Ar
R
OH
O
Ar =
*
syn
:anti = 92:8
(23)
, R = (CH
2
)
2
CH=CHMe
2
O
Ar
R
O
O
O
Ar
R
OH
O
DIBAL-BHT
Ar =
*
syn
:anti = 4:96
(24)
, R = (CH
2
)
2
CH=CHMe
2
toluene
–78 °C
82%
Zn(BH
4
)
2
reduction of α-hydroxy ketones gives anti products
predominantly over syn products. The selectivity is dependent on
the substitution pattern of the α-hydroxy ketones. When R
1
is
phenyl or R
2
is a sterically demanding group, anti selectivity is
excellent (eq 25).
37
This is reasonably explained by considering
a zinc-chelated five-membered transition state.
1,37
Other highly
selective examples of Zn(BH
4
)
2
reductions
38
–
42
of α-hydroxy
ketones are shown in eqs 26 and 27.
38,41
R
1
R
2
OH
O
R
1
R
2
OH
OH
R
1
R
2
OH
OH
+
(25)
R
1
= Ph, R
2
= Me
R
1
= Pr, R
2
= i-Pr
98:2
96:4
anti
syn
Zn(BH
4
)
2
ether
0 °C
OBn
O
R
Zn(BH
4
)
2
OBn
OH
R
(26)
R = (CH
2
)
2
OTHP
anti
:syn = 95:5
ether
–30 °C
OH
O
Bu
Zn(BH
4
)
2
OH
OH
Bu
(27)
anti
:syn = 98.5:1.5
ether
–50 °C
90%
In the cases where two functional groups are present on the α-
or β-position of the keto group, reduction proceeds through the
more stable transition state. When alkoxy and alkylthio functions
are present on the α-position of the keto group, Zn(BH
4
)
2
coor-
dinates preferentially with the former (eq 28).
43
Reduction of a
ketone having two alkoxy groups on the α- and β-positions pro-
duces the anti-2-alkoxy alcohol almost exclusively, showing that
a five-membered transition state involving the α-alkoxy group is
contributing far more than six-membered one (eq 29).
44
There is
also a case where the three-dimensional structure of the ketone
governs the selection of the transition state (eq 30).
45
O
S
Et
O
Zn(BH
4
)
2
O
S
Et
OH
(28)
anti
:syn = 99:1
THF
–78 °C
>98%
BnO
R
MOMO
OMOM
O
Zn(BH
4
)
2
BnO
R
MOMO
OMOM
OH
1
2
R = p-MeOC
6
H
4
(29)
1,2-anti:1,2-syn = >99:1
91%
O
CO
2
Me
O
MeO
S
S
Zn(BH
4
)
2
O
H
H
O
CO
2
Me
O
Zn
S
S
Me
O
H
H
O
O
CO
2
Me
Me
S
S
Zn
O
CO
2
Me
OH
MeO
S
S
α-OH:β-OH = 17:1
(30)
ether
–78 °C
100%
H
–
Optically active α-hydroxy imines are reduced with Zn(BH
4
)
2
to give anti-hydroxy amines (eq 31).
46
α
,β-Epoxy ketones pro-
duce anti-epoxy alcohols with high selectivity, irrespective of the
substitution pattern of the epoxide (eq 32).
47,48
The correspond-
ing aziridino ketones and imines are also reduced with Zn(BH
4
)
2
to the anti isomer with high selectivity (eqs 33 and 34).
49
Ph
OH
N
Me
Zn(BH
4
)
2
Ph
OH
HN
Me
(31)
Ephedrine
anti
:syn = 97:3
ether
–76 °C
>75%
(32)
anti
:syn = >99:1
O
O
O
OH
Zn(BH
4
)
2
ether
0 °C
86%
N
H
Ph
Ph
O
Zn(BH
4
)
2
N
H
Ph
Ph
OH
(33)
ether
100%
N
Ph
t
-Bu
NH
Zn(BH
4
)
2
N
Ph
t
-Bu
NH
2
(34)
ether
100%
Avoid Skin Contact with All Reagents

4
ZINC BOROHYDRIDE
1.
Pelter, A.; Smith, K.; Brown, H. C. Borane Reagents; Academic: London,
1988.
2.
Oishi, T.; Nakata, T., Acc. Chem. Res. 1984, 17, 338.
3.
(a) Gensler, W. J.; Johnson, F.; Sloan, A. D., J. Am. Chem. Soc. 1960, 82,
6074. (b) Nakata, T.; Tani, Y.; Hatozaki, M.; Oishi, T., Chem. Pharm.
Bull. 1984
, 32, 1141. (c) Crabbe, P.; Garcia, G. A.; Rfus, C., J. Chem.
Soc., Perkin Trans. 1 1973
, 810.
4.
Kotsuki, H.; Yoshimura, N.; Kadota, I.; Ushio, Y.; Ochi, M., Synthesis
1990, 401.
5.
Corey, E. J.; Andersen, N. H.; Carlson, R. M.; Paust, J.; Vedjs, E.; Vlattas,
I.; Winter, R. E. K., J. Am. Chem. Soc. 1968, 90, 3245.
6.
(a) Guzman, A.; Crabbe, P., Chem. Lett. 1973, 1073. (b) Rozing, G. P.;
Moinat, T. J. H.; de Koning, H.; Huisman, H. O., Heteroatom Chem.
1976, 4, 719.
7.
Crabbe, P.; Guzman, A.; Vera, M., Tetrahedron Lett. 1973, 3021.
8.
Ranu, B. C.; Das, A. R., Tetrahedron Lett. 1992, 33, 2361.
9.
(a) Naito, T.; Nakata, T.; Akita, H.; Oishi, T., Chem. Lett. 1980, 445.
(b) Sierra, M. G.; Olivieri, A. C.; Colombo, M. I.; Ruveda, E. A., J.
Chem. Soc. (C) 1985
, 1045.
10.
Sarkar, D. C.; Das, A. R.; Ranu, B. C., J. Org. Chem. 1990, 55,
5799.
11.
Yamada, K.; Kato, M.; Hirata, Y., Tetrahedron Lett. 1973, 29, 2745.
12.
Kotsuki, H.; Yoshimura, N.; Ushio, Y., Chem. Lett. 1986, 1003.
13.
Ranu, B. C.; Das, A. R., J. Chem. Soc., Perkin Trans. 1 1992, 1561.
14.
Kotsuki, H.; Ushio, Y.; Yoshimura, N.; Ochi, M., Bull. Chem. Soc. Jpn.
1988, 61, 2684.
15.
Kotsuki, H.; Ushio, Y.; Yoshimura, N.; Ochi, M., J. Org. Chem. 1987,
52
, 2594.
16.
Ranu, B. C.; Basu, M. K., Tetrahedron Lett. 1991, 32, 3243.
17.
Ranu, B. C.; Das, A. R., J. Chem. Soc., Chem. Commun. 1990, 1334.
18.
Ranu, B. C.; Das, A. R., J. Chem. Soc., Perkin Trans. 1 1992, 1881.
19.
Ranu, B. C.; Das, A. R., J. Org. Chem. 1991, 56, 4796.
20.
Firouzabadi, H.; Tamami, B.; Goudarzian, N., Synth. Commun. 1991,
21
, 2275.
21.
Kim S.; Hong C. Y.; Yang, S., Angew. Chem., Int. Ed. Engl. 1983, 22,
562.
22.
Julia, M.; Roy, P., Tetrahedron 1986, 42, 4991.
23.
Nakata, T.; Oishi, T., Tetrahedron Lett. 1980, 21, 1641.
24.
Nakata, T.; Kuwabara, T.; Tani, Y.; Oishi, T., Tetrahedron Lett. 1982, 23,
1015.
25.
Nakata, T.; Tani, Y.; Hatozaki, M.; Oishi, T., Chem. Pharm. Bull. 1984,
32
, 1411.
26.
Ito, Y.; Yamaguchi, M., Tetrahedron Lett. 1983, 24, 5385.
27.
Elliott, J.; Warren, S., Tetrahedron Lett. 1986, 27, 645.
28.
(a) Evans, A. D., Aldrichim. Acta 1982, 15, 23. (b) Evans, D. A.; Ennis,
M.; Le, T., J. Am. Chem. Soc. 1984, 106, 1154.
29.
Dipardo, R. M.; Bock, M., Tetrahedron Lett. 1983, 24, 4805.
30.
Ito, Y.; Katsuki, T.; Yamaguchi, M., Tetrahedron Lett. 1984, 25,
6015.
31.
Oppolzer, W.; Rodriguez, I.; Starkemann, C.; Walther, E., Tetrahedron
Lett. 1990
, 31, 5019.
32.
Kathawala, F. G.; Prager, B.; Prasad, K.; Repic, O.; Shapiro, M. J.;
Stabler, R. S.; Widler, L., Helv. Chim. Acta 1986, 69, 803.
33.
Kashihara, H.; Suemune, H.; Fujimoto, K.; Sakai, K., Chem. Pharm.
Bull. 1989
, 37, 2610.
34.
Pilli, R. A.; Russowsky, D.; Dias, L. C., J. Chem. Soc., Perkin Trans. 1
1990, 1213.
35.
Oishi, T.; Nakata, T., Synthesis 1990, 635.
36.
Taber, D. F.; Deker, P. B.; Gaul, M. D., J. Am. Chem. Soc. 1987, 109,
7488.
37.
Nakata, T.; Tanaka, T.; Oishi, T., Tetrahedron Lett. 1983, 24, 2653.
38.
Takahashi, T.; Miyazawa, M.; Tsuji, J., Tetrahedron Lett. 1985, 26,
5139.
39.
Jarosz, S., Chem. Rev. 1988, 183, 201.
40.
Pikul, S.; Raczko, J.; Ankner, K.; Jurczak, J., J. Am. Chem. Soc. 1987,
109
, 3981.
41.
Fujisawa, T.; Kohama, H.; Tajima, K.; Sato, T., Tetrahedron Lett. 1984,
25
, 5155.
42.
Sayo, N.; Nakai, E.; Nakai, T., Chem. Lett. 1985, 1723.
43.
Matsubara, S.; Takahashi, H.; Utimoto, K., Chem. Lett. 1992, 2173.
44.
Iida, H.; Yamazaki, N.; Kibayashi, C., J. Org. Chem. 1986, 51,
1069.
45.
Nakata, T.; Nagao, S.; Oishi, T., Tetrahedron Lett. 1985, 26, 6465.
46.
Jackson, W. R.; Jacobs, H. A.; Matthews, B. R.; Jayatilake, G. S.; Watson,
K. G., Tetrahedron Lett. 1990, 31, 1447.
47.
Nakata, T.; Tanaka, T.; Oishi, T., Tetrahedron Lett. 1981, 22, 4723.
48.
Banfi, S.; Colonna, S.; Molinari, H.; Julia, S., Synth. Commun. 1983, 13,
901.
49.
Bartnik, R.; Laurent, A.; Lesniak, S., J. Chem. Res. (S) 1982, 287.
Takeshi Oishi
Meiji College of Pharmacy, Tokyo, Japan
Tadashi Nakata
The Institute of Physical and Chemical Research (RIKEN),
Saitama, Japan
A list of General Abbreviations appears on the front Endpapers
Wyszukiwarka
Podobne podstrony:
zinc bromide eros rz005
zinc amalgam eros rz003
sodium borohydride eros rs052
tin IV chloride zinc chloride eros eros rt115
zinc borohydride
zinc eros rz001
zinc acetic acid eros rz002
benzyl chloride eros rb050
hydrobromic acid eros rh031
chloroform eros rc105
magnesium eros rm001
oxalyl chloride eros ro015
potassium permanganate eros rp244
peracetic acid eros rp034
więcej podobnych podstron