Wykład nr 7 (22.04.2010)
ZINTEGROWANY SYSTEM ZARZĄDZANIA
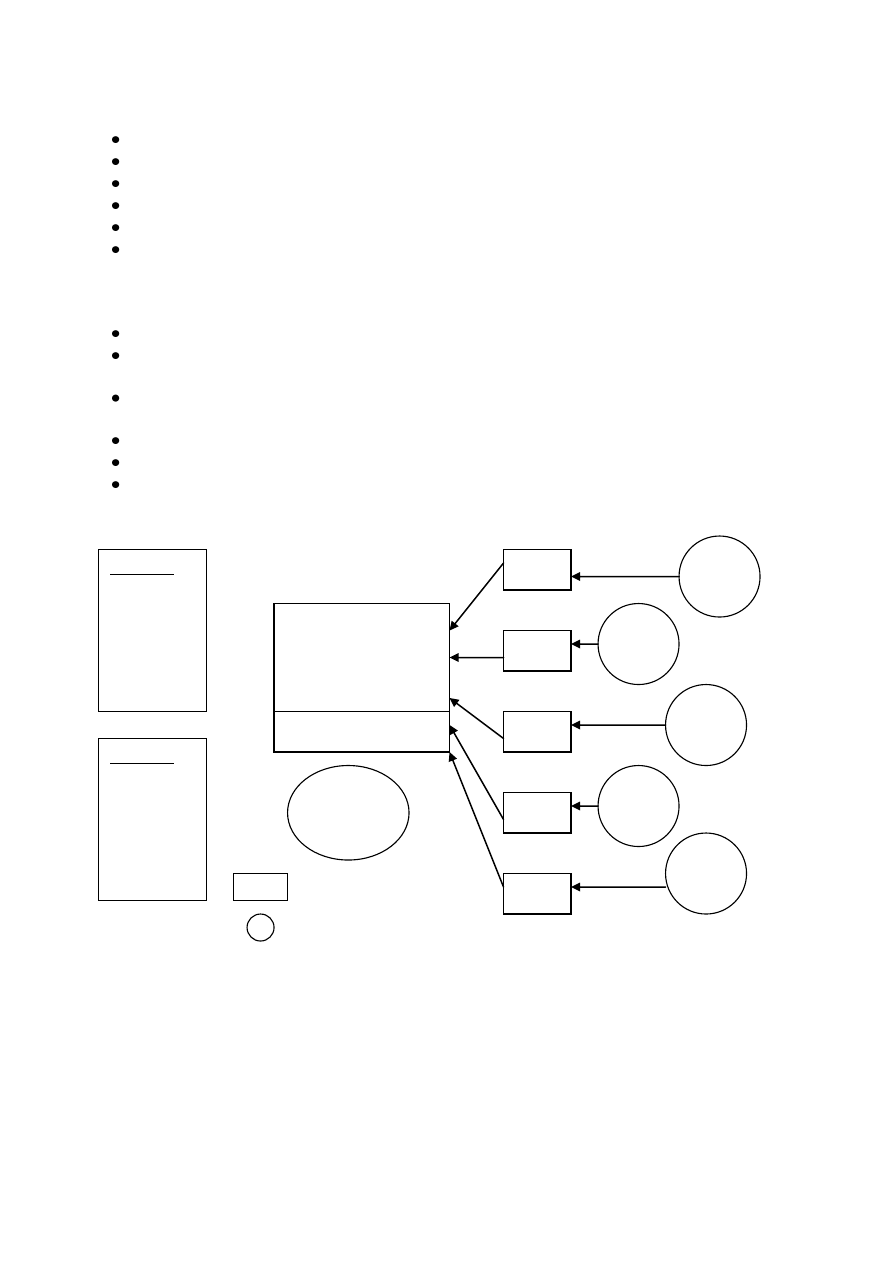
ELEMENTY (PODSYSTEMY) tworzące Zintegrowany System Zarządzania to:
o
System Zarządzania Jakością – wg Polskiej Normy PN – EN ISO 9000:2001
o
System Zarządzania Środowiskiem – wg Polskiej Normy PN – EN ISO – 14001:1998
o
System Zarządzania Bezpieczeństwem i Higieną Pracy – wg Polskiej Normy PN – N
18001:2004
<=Podsystemy mogą funkcjonować samodzielnie; Kryterium wspólnych cech
FILOZOFIA TQM
Ciągłe ulepszanie wszystkich elementów działalności organizacji:
o
Identyfikacja kluczowych zagadnień działalności organizacji
o
Wyznaczanie celów i standardów jakości działań
o
Dobór metod do realizacji celów według określonych standardów oraz procedur
o
Podejmowanie działań dla wykonywania założonych celów
o
Mierzenie uzyskanych osiągnięć
o Ocena i podejmowanie odpowiednich działań naprawczych lub korygujących
POLITYKA OCHRONY
ŚRODOWISKA
POLITYKA
JAKOŚCI
POLITYKA
BEZPIECZEŃSTWA
OPERACYJNE
PROCEDURY
SYSTEMOWE
INSTRUKCJE
ROBOCZE I
STANOWISKOWE
KSIĘGA
OCHRONY
ŚRODOWI
SKA
KSIĘGA
JAKOŚCI
KSIĘGA
BHP
OPERACYJNE PROCEDURY SYSTEMOWE
INSTRUKCJE ROBOCZE STANOWISKOWE
TPM – TOTAL PRODUCTIVE MAINTENANCE
Eliminacja 6 Dużych Strat poprzez prace w wielofunkcyjnych zespołach – Focused Improvement
1. Formalne włączenie pracowników produkcji w pomoc przy utrzymaniu ruchu Autonomous
Maintenance
2. Zbudowanie systemu planowanych przeglądów, konserwacji i prewencji – Planned
Maintenance
3. Podnoszenie wiedzy i umiejętności operatorów i pracowników Działu Utrzymania Ruchu
poprzez specjalistyczne szkolenia
4. Zbudowanie systemu zapewniającego projektowanie/ zakup/ produkcję łatwego w obsłudze
i utrzymaniu sprzętu - Early Equipment Management
REGULACJE PN – N – 18001:2004
Rozporządzenie Ministra Pracy I Polityki Socjalnej z dnia 26 września 1997 r., w sprawie ogólnych
przepisów bezpieczeństwa i higieny pracy (Jednolity tekst Dz. U. z 2003 r. Nr 169, poz. 1650 z
późniejszymi zmianami) - §39, §39a, §39b I §39c.
MODEL
TQM
Koncentracja na
klientach
Rozwój procesów
Planowanie procesów
Uczestnictwo
wszystkich
Zarządzanie procesem
-6 -5 -4 -3 -2 -1 0 1 2 3 4 5
WPROWADZENIE
PO WPROWADZENIU
6 LAT
1 ROK
PRZED
1 ROK
PO
4 LATA
PO
TQM
S - TPM
ISO 9001
Postęp w procesach na
drodze standaryzacji
Zwiększenie wydajności
maszyn
Maksymalizacja zadowolenia
klienta

Nowelizacja ww. rozporządzenia (w sprawie ogólnych przepisów bezpieczeństwa i higieny pracy)
obowiązująca od 22 czerwca 2007 r. precyzowała co powinna zawierać dokumentacja oceny ryzyka
zawodowego, sporządzona przez pracodawcę.
REGULACJE PN – N – 18001:2004
1. Ocenianie i dokumentowanie ryzyka zawodowego związanego z wykonywaną pracą (art.
226 pkt 1)
2. Stosowanie niezbędnych środków profilaktycznych zmniejszających ryzyko zawodowe, a w
szczególności:
- zapewnienia organizacji pracy (w tym organizacji stanowisk pracy) zabezpieczającej
pracowników przed zagrożeniami wypadkowymi
- oddziaływaniem czynników środowiska pracy (art. 227 §1);
3. Likwidacja zagrożeń dla zdrowia i życia pracowników poprzez, np. zmianę technologii,
wymianę maszyn i urządzeń technicznych, zmianę materiałów i substancji
4. Informowanie pracowników o ryzyku zawodowym, związanym z wykonywana przez nich
pracą, zasadach ochrony przed zagrożeniami środowiska pracy wynikającymi z
przeprowadzonej oceny ryzyka zawodowego (art, 226, pkt 2)
5. konsultowania z pracownikami lub ich przedstawicielami wszystkich działań związanych z
bezpieczeństwem i higieną pracy, w szczególności dotyczących oceny ryzyka zawodowego
przy wykonywaniu określonych prac (art. 23711a § 1 pkt 2).
RYZYKO ZAWODOWE PN – N – 18001:2004
PRAWDOPODOBIEŃSTWO Z JAKIM KTOŚ MOŻE ZOSTAĆ POSZKODOWANY W
ZWIĄZKU Z ISTNIEJĄCYM ZAGROŻENIEM W ŚRODOWISKU PRACY.
o Czynniki chemiczne
o
Hałas i drgania mechaniczne
o
Ręczne prace transportowe
o Czynniki biologiczne
CZYNNIKI CHEMICZNE
Wymagania zawarto w rozporządzeniu Ministra Zdrowia z dnia 30 grudnia 2004 r.
1. Listy wszystkich chemicznych substancji i preparatów obecnych stanowiskach pracy oraz
stanowiskach sąsiadujących.
2. Informacje- konieczna jest identyfikacja:
o niebezpiecznych właściwości czynników chemicznych,
o
zagrożeń związanych z występowaniem czynnika chemicznego (np. z karty charakterystyki);
informacje od dostawcy,dotyczących
o
scenariuszy narażenia
o
rodzaju, poziomu i czasu trwania narażenia
o
wartości najwyższych dopuszczalnych stężeń w środowisku pracy (co jest możliwe dla
czynników, dla których je ustalono),
o
wartości dopuszczalnych stężeń w materiale biologicznym – grupa czynników dla których
zostały ustalone jest bardzo mała, obejmuje np. ołów, styren
o
efektów działań zapobiegawczych,
o
wyników oceny stanu zdrowia pracowników - jeśli została przeprowadzona,
o
warunków pracy przy użytkowaniu czynników chemicznych, z uwzględnieniem ilości tych
czynników
SZACOWANIE POZIOMU RYZYKA ZAWODOWEGO – CZYNNIKI CHEMICZNE
NDS/NDN (najwyższe dopuszczalne stężenie / natężenie) określają stężenie toksycznego związku
chemicznego lub innego czynnika szkodliwego w danym okresie pracy.
NDSP – wartość stężenie toksycznego związku chemicznego, która ze względu na zagrożenie
zdrowia lub życia pracownika nie może być w środowisku pracy przekroczona w żadnym
momencie.
NDSCh – najwyższe dopuszczalne stężenie chwilowe – wartość średnia stężenia określonego,
toksycznego związku chemicznego, które nie powinno spowodować ujemnych zmian w stanie
zdrowia pracownika, jeżeli występuje w środowisku pracy nie dłużej niż 15 minut i nie częściej niż
2 razy w czasie zmiany roboczej w odstępie czasu nie krótszym niż 1 godzina.
Dla czynników nieposiadających ustalonych wartości dopuszczalnych - stosuje się metodę
opracowaną przez Komisję Bezpieczeństwa, Higieny i Ochrony Zdrowia w Pracy Europejskiej
Komisji Zatrudnienia i Spraw Socjalnych; uwzględniane trzy zmienne:
- podstawowe zagrożenie daną substancją chemiczną (na podstawie zwrotów R)
- skłonność do przedostawania się substancji do środowiska (lotność/tworzenie pyłów)
- ilość substancji użyta w ocenianej operacji
Prawdopodobieństwo
Ciężkość następstw
Małe
Średnie
Duże
Mało prawdopodobne
Małe
1
Małe
1
Średnie
2
Prawdopodobne
Małe
1
Średnie
2
Duże
3
Wysoce prawdopodobne
Średnie
2
Duże
3
Duże
3
ZAPEWNIENIE BEZPIECZNYCH WARUNKÓW PRACY
1. Ograniczanie ryzyka zawodowego
2. Przeprowadzenie oceny ryzyka zawodowego
3. Likwidowanie zagrożeń u źródeł ich powstawania
Ryzyko zawodowe
małe
Ryzyko zawodowe
duże
Ryzyko zawodowe
średnie
P
s
P
ch
P
p
NIE
TAK
TAK
NIE
Czy
P
s
›NDS lub
P
ch
›NDSCh
lub P
p
›NDSP
P
s
‹0,5NDS i
P
ch
‹0,5NDSCh lub
P
s
‹0,5NDS i
P
p
‹0,5NDSP?
lub P
s
‹0,5NDS
4. Dostosowanie warunków i procesów pracy do możliwości pracownika - odpowiednie
projektowanie i organizowanie stanowisk pracy, dobór maszyn, narzędzi pracy, a także
metod produkcji i pracy z uwzględnieniem zmniejszania uciążliwości pracy
5. Stosowanie nowych rozwiązań technicznych
6. Zastępowanie niebezpiecznych procesów technologicznych urządzeń, substancji –
bezpiecznymi lub mniej niebezpiecznymi
7. Nadawanie priorytetu środkom ochrony zbiorowej przed środkami ochrony indywidualnej
8. Instruowanie pracowników w zakresie bezpieczeństwa i higieny pracy
DOKUMENTACJA OCENY RYZYKA ZAWODOWEGO
Opis ocenianego stanowiska pracy, w tym wyszczególnienie:
- stosowanych maszyn
- narzędzi i materiałów
- wykonywanych zadań,
- występujących na stanowisku niebezpiecznych, szkodliwych i uciążliwych czynników środowiska
pracy
- stosowanych środków ochrony zbiorowej i indywidualnej,
- osób pracujących na tym stanowisku;
Wyniki przeprowadzonej oceny ryzyka zawodowego dla każdego z czynników środowiska pracy
oraz niezbędne środki profilaktyczne zmniejszające ryzyko; datę przeprowadzonej oceny oraz
osoby dokonujące oceny
CZYNNIKI CHEMICZNE
niebezpieczne właściwości czynnika chemicznego,
- otrzymane od dostawcy informacje dotyczące zagrożenia czynnikiem chemicznym (np. zawarte w
karcie charakterystyki),
- rodzaj, poziom i czas trwania narażenia,
- wartości najwyższych dopuszczalnych stężeń w środowisku pracy,
- wartości dopuszczalnych stężeń w materiale biologicznym,
- efekty działań zapobiegawczych,
- wyniki oceny stanu zdrowia pracowników,
- warunki pracy przy użytkowaniu czynników chemicznych, z uwzględnieniem ilości tych
czynników.
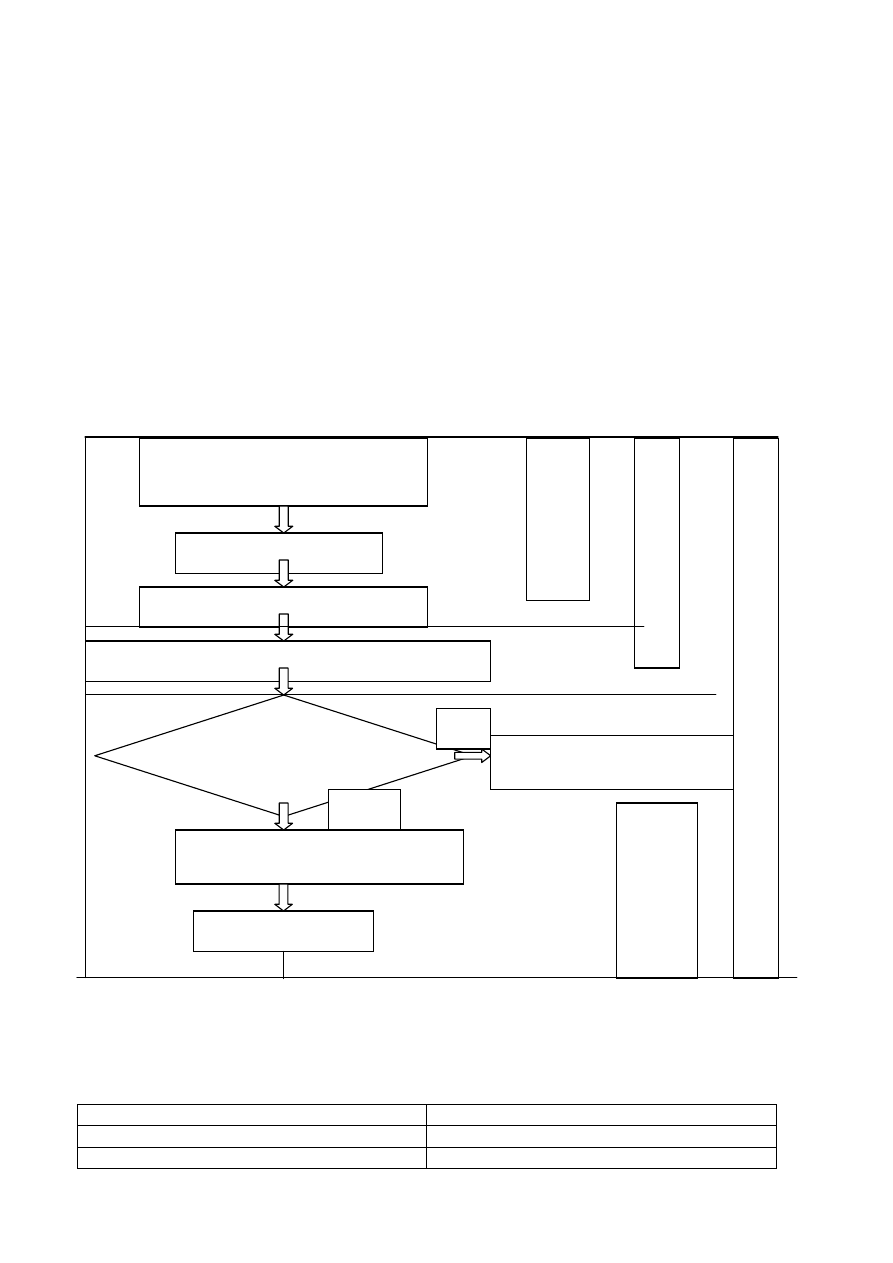
Identyfikacja zagrożeń w wyniku zebrania
niezbędnych informacji
Szacowanie poziomu ryzyka zawodowego
Działania korygujące i/lub zapobiegające
Okresowa ocena ryzyka zawodowego
Informowanie pracowników o ryzyku zawodowym
Dokume
ntowa
nie oc
eny
ryz
yka
za
wodow
ego

WYKŁAD 8 (29.04.2010)
SZACOWANIE POZIOMU RYZYKA ZAWODOWEGO – CZYNNIKI CHEMICZNE
Najwyższe dopuszczalne stężenie (NDS) - wartość średnia ważona stężenia, którego oddziaływanie
na pracownika w ciągu 8-godzinnego dobowego i przeciętnego tygodniowego wymiaru czasu
pracy, określonego w Kodeksie pracy, przez okres jego aktywności zawodowej nie powinno
spowodować ujemnych zmian w jego stanie zdrowia oraz w stanie zdrowia jego przyszłych pokoleń
Wartości NDS stanowią wytyczne dla projektantów nowych i modernizowanych technologii i
wyrobów, kryteria oceny warunków pracy oraz podstawę do prowadzenia działalności
profilaktycznej w zakładach pracy.
Do wykazu wartości najwyższych dopuszczalnych stężeń wprowadzono oznakowania, które
dostarczają istotnej informacji o kierunku działania substancji chemicznej.
Substancje o działaniu żrącym - C,
Substancje o działaniu drażniącym - I,
Substancje o działaniu uczulającym- A,
Substancje o działaniu rakotwórczym- kat 1 lub 2,
Substancje o działaniu toksycznym na płód - Ft
Substancje o działaniu wchłaniającym się przez skórę-Sk
Ps - wskaźnik narażenia umożliwiający ocenę stężenia średniego ważonego dla całej zmiany
roboczej
Zależność Ps<0,5 NDS ma zastosowanie w przypadku substancji chemicznych, dla których nie
określono wartości NDSCh i NDSP
Pch - wskaźnik narażenia umożliwiający ocenę stężeń chwilowych
Pp - wskaźnik narażania umożliwiający ocenę stężeń pułapowych
NDS – Najwyższe Dopuszczalne Stężenie
NDSCh – Najwyższe Dopuszczalne Stężenie Chwilowe
NDSP – Najwyższe Dopuszczalne Stężenie Pułapowe
ā=W1A1+w2a2+…wnan/w1+w2+…+wn
Poziom ryzyka zawodowego w granicach 10^3 – 10^4 oznacza możliwość przyrostu dodatkowych
zachorowań (związanych z kontaktem z danym odczynnikiem) w liczbie 1 przypadek na 1000 osób
narażonych – 1 przypadek na 10000 osób.
NORMNA PN – N – 18001:2004 – ZAGROŻENIA
3.1. Zagrożenie-stan środowiska pracy mogący spowodować wypadek lub zagrożenie zdrowia
3.2. Zagrożenie znaczące - mogące spowodować poważne uszkodzenie zdrowia lub śmierć,
występujące w szczególności podczas wykonywania prac szczególnie niebezpiecznych lub w
sytuacjach poważnych awarii
3.3. Zdarzenie potencjalnie wypadkowe- niebezpieczne zdarzenie związane z wykonywana pracą
podczas którego nie dochodzi do urazów lub pogorszenia stanu zdrowia

NORMA PN – N – 18001:2004
Pkt. 3.20 SYSTEM ZARZĄDZANIAHIGIENĄ I BEZPIECZEŃSTWEM PRACY to część
ogólnego systemu zarządzania organizacją, która obejmuje strukturę organizacyjną, planowanie,
odpowiedzialność, zasady postępowania, procedury, procesy i zasoby potrzebne do opracowania,
wdrażania, realizowania, przeglądu i utrzymywania polityki bezpieczeństwa i higieny pracy
Systemowe zarządzanie bezpieczeństwem jest metodą, która pozwala uporządkować i
usystematyzować wszystkie działania związane z bezpieczeństwem i higieną pracy w firmie, a jej
ideą jest pełne i udokumentowane zaangażowanie zarówno kierownictwa, jak i każdego pracownika
w rzeczywiste działania na rzecz bezpieczeństwa pracy
DZIAŁANIA W OBSZARZE BHP
1. opracowanie i ogłoszenie załodze polityki oraz celów w zakresie BHP, będących wyrazem
zaangażowania najwyższego kierownictwa
2. określenie odpowiedzialności i uprawnień poszczególnych pracowników zakresie działań na
rzecz BHP
3. zapewnienie właściwych szkoleń oraz motywowanie pracowników do bezpiecznej i higienicznej
pracy
4. organizowanie sprawnego systemu komunikowania się w zakresie BHP
5. ocenianie ryzyka zawodowego
6. opracowanie zasad reagowania na wypadki przy pracy
7. wdrażanie działań korygujących i zapobiegawczych związanych z bezpieczeństwem i higieną
pracy
8. monitorowanie i auditowanie systemu zarządzania BHP
POLITYKA BHP
Pkt. 3.16 POLITYKA BHP - co to jest ?
Deklaracja organizacji dotycząca jej intencji i zasad odnoszących się do ogólnych efektów
działalności w zakresie bezpieczeństwa i higieny pracy, określająca ramy do działania i ustalania
celów organizacji dotyczących zarządzania bezpieczeństwem i higieną pracy
POLITYKA
Kierownictwo firmy mając na względzie zdrowie i bezpieczeństwo pracowników wdrożyło,
utrzymuje i doskonali System Zarządzania Bezpieczeństwem i Higiena Pracy zgodny z:
□ Wymaganiami normy PN-N-18001:2004
□ Obowiązującymi przepisami prawnymi i wymaganiami BHP dotyczącymi (FIRMA)
□ Własnymi planami i programami doskonalenia BHP
Ciągłe
doskonalenie
Zaangażowanie
kierownictwa oraz
polityka BHP
Przegląd
zarządzania
Planowanie
Sprawdzanie oraz
działania
korygujące i
zapobiegawcze
Wdrażanie i
funkcjonowanie
Na rys: Model zarządzania
bezpieczeństwem i higieną pracy
PN – N – 18001 styczeń 2004
(zastępuje PN – N – 18001:1999)
Systemy zarządzania
bezpieczeństwem i higieną pracy.
Wymagania
Norma zbieżna z normą
międzynarodową Guidelines on
occupational safety and Heath
management system (ILO – OSH
2001)
Kierownictwo firmy deklaruje, że:
□ Będzie dokonywać przeglądu polityki BHP, celów i zadań w celu weryfikacji w stosunku do
zmieniających się warunków prowadzonej działalności
□ Analizuje wyniki auditów i prowadzonych działań zapobiegawczych oraz korygujących a także
nadzoruje ustanowione zapisy i procedury
POLITYKA
* Dba o podnoszenie poziomu świadomości i kwalifikacji pracowników dotyczących BHP poprzez
szkolenia oraz informowanie o możliwych zagrożeniach i związanym z nimi ryzykiem zawodowym
* Prowadzi dialog z pracownikami mający na celu ich angażowanie na rzecz BHP
* Prowadzi ciągłe działania doskonalące w zakresie BHP
Niniejsza polityka jest znana, zrozumiała i realizowana przez pracowników firmy oraz udostępniana
zainteresowanym stronom zewnętrznym.
* Zapewni odpowiednie środki finansowe, technologiczne, techniczne i organizacyjne dla realizacji
polityki BHP, celów i ustalonych zadań
* Identyfikuje zagrożenia mające wpływ na bezpieczeństwo i zdrowie pracowników, monitoruje ich
stopień i zapobiega wypadkom przy pracy oraz chorobom zawodowym
* Ma opracowane procedury awaryjne w przypadku wystąpienia zdarzenia dotyczącego BHP
WARTOŚCIOWANIE RYZYKA
Wskaźnik ryzyka W=SxP
S – stopień szkód
P – prawdopodobieństwo szkód zdarzenia
Stopień szkód – S
Poziom
Charakterystyka
Znikome urazy
Lekkie szkody
Lekkie obrażenia
Wymierne szkody
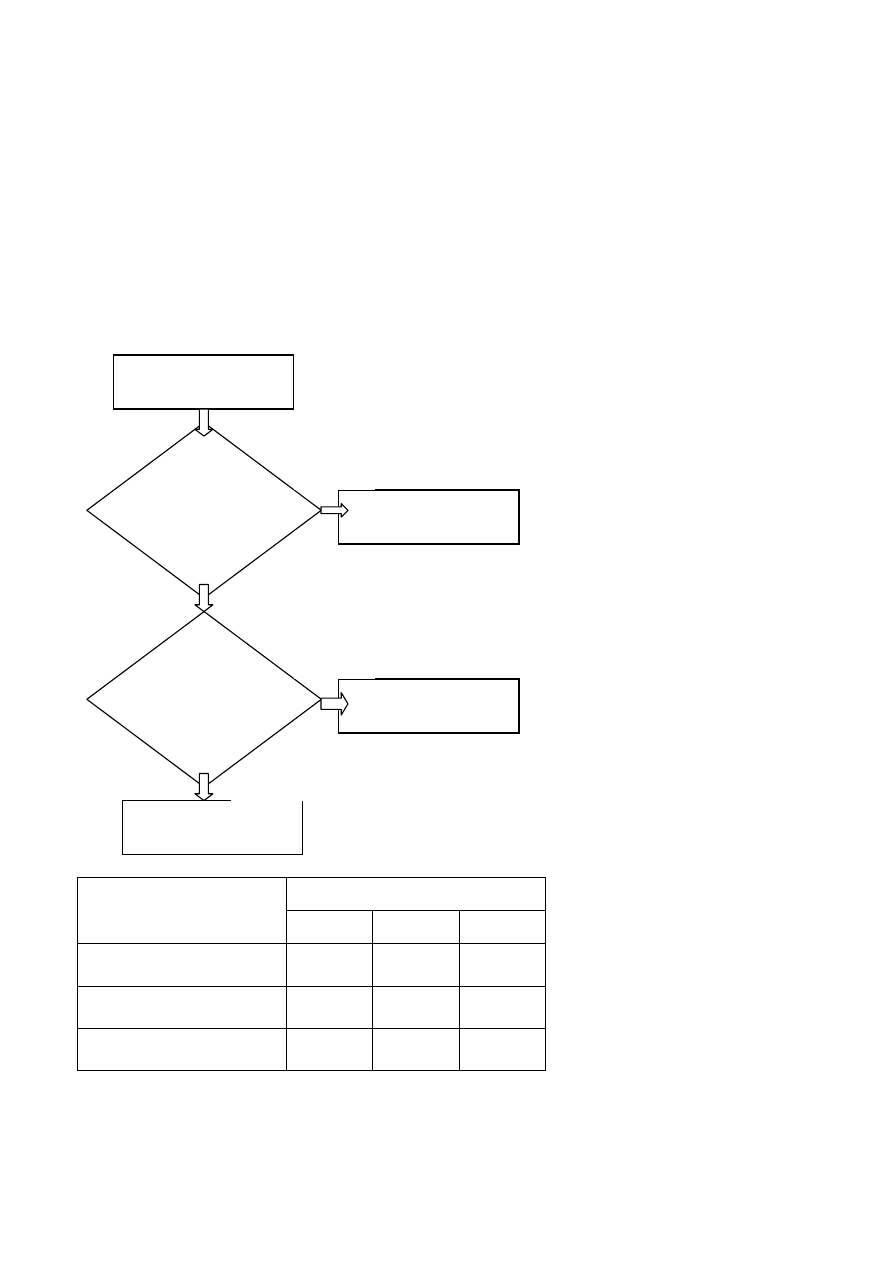
Zebranie informacji potrzebnych do
oceny ryzyka zawodowego
Identyfikacja zagrożeń
Oszacowanie ryzyka zawodowego
Wyznaczenie dopuszczalności ryzyka zawodowego
Opracowanie planu działań
korygujących i/lub zapobiegających
Realizacja planu
Czy są potrzebne
działania korygujące
i/lub zapobiegawcze
Okresowe przeprowadzanie
oceny ryzyka zawodowego
NIE
TAK
Ana
li
za
r
y
zyk
a
za
wodow
ego
Oc
ena
ryz
yka
z
awodo
we
go
ZARZĄ
DZA
NI
E RYZ
Y
KI
EM ZA
W
O
DO
W
YM
Ogr
anicz
enie lub
eli
mi
nowa
nie r
yz
yka
za
wodow
ego

Ciężkie obrażenia
Znaczne szkody
Pojedyncze wypadki śmiertelne
Ciężkie szkody
Zbiorowe wypadki śmiertelne
Szkody na bardzo dużą skalę na terenie
zakładu
Zbiorowe wypadki śmiertelne
Szkody na dużą skalę poza terenem zakładu
Prawdopodobieństwo szkód – P
Poziom
Charakterystyka
Bardzo nieprawdopodobne
Mało prawdopodobne
Zdarzające się raz na 10 lat
Doraźne wydarzenie
Zdarzające się raz w roku
Dosyć częste wydarzenia
Zdarzające się raz w miesiącu
Częste, regularne wydarzenia
Zdarzające się raz w tygodniu
Duże prawdopodobieństwo wydarzenia
WARTOŚCIOWANIE RYZYKA
Matryca wartościowania ryzyka
1. ryzyko akceptowalne
2. dopuszczalna akceptacja ryzyka po ocenie ryzyka
3. ryzyko niedopuszczalne – wymagane zmniejszenie ryzyka
Risk score
Jest metodą jakościową, wskaźnikową w której wartościowanie ryzyka opisuje wyrażenie
R=SxExP
S – potencjalne skutki zagrożenia
E – ekspozycja na zagrożenie
P – prawdopodobieństwo wystąpienia zagrożenia
Ryzyko zaniedbywane
Żadne działanie nie jest potrzebne
Ryzyko akceptowalne
Działania profilaktyczne nie są potrzebne
Ryzyko średnie
Działania profilaktyczne są wskazane, ale należy wziąć pod uwagę koszty i uzyskane efekty
(powinno zostać ograniczone w przeciągu 3-6 miesięcy)
Ryzyko poważne
W tej sytuacji praca nie może zostać rozpoczęta. W przypadku prac już wykonywanych ryzyko
powinno zostać zredukowane w okresie 1-3 miesięcy w zależności od liczby osób narażonych
Ryzyko nieakceptowalne
Praca nie może zostać rozpoczęta ani kontynuowana dopóki ryzyko nie zostanie zredukowane do
poziomu akceptowalnego
KORZYŚCI Z OCENY RYZYKA
o
Gwarancja spełnienia wymagań przepisów prawa pracy
o
Pełna świadomość tego:
- jakie zagrożenia występują na stanowiskach pracy
- jakie jest ryzyko zawodowe
- czy zastosowane środki profilaktyczne są wystarczające przy uwzględnieniu podejmowanego
ryzyka wypadku, a co się z tym wiąże, strat finansowych dla organizacji (przedsiębiorstwa)
o
Ulgi po wprowadzeniu zróżnicowanej składki ubezpieczeniowej
o
Ulgi przy ubezpieczeniach majątkowych i ewentualnie dodatkowych dla pracowników. W
Polsce już działają komercyjnie firmy ubezpieczeniowe, które przy ustalaniu wysokości składek
biorą pod uwagę kategorię ryzyka opierając się na ocenie systemu bezpieczeństwa w organizacji

o
Celowe i w pełni kontrolowane zarządzanie poziomem ryzyka w miejsce działan opartych
wyłącznie na intuicji
o
Lepszą pozycję przy ewentualnych pertraktacjach lub procesach sądowych o odszkodowanie z
tytułu, np. wypadku przy pracy
Wykład nr 9
Branżowe normy z Zarządzania
Skąd się wzięły branżowe normy ISO?
Fenomen ISO serii 9000 jest określeniem spotkanym w opracowaniach dotyczących zarządzania i
związany jest z powszechnością i liczbą certyfikowanych systemów. Do końca 2005r. wydano
około 800000 certyfikatów ISO 9001. Zgodnie z założeniami ISO 9001 może być stosowana w
dowolnych organizacjach i stanowić podstawę systemowych rozwiązań. Jednak w przypadku
niektórych branż wymagania ogólne ISO 9001 pozostawiają dużą swobodę doboru rozwiązań, co
zostało uznane za niewystarczające. Niektóre z branż stosują swoiste dla siebie, ale ich główny
szkielet opiera się na normie ISO 9001. Spowodowało to również trochę inne spojrzenie na
definicję systemu zintegrowanego zarządzania.
Systemy zarządzania jakością w Informatyce – ISO/ICE 27000:
Informacja ma dla wielu organizacji znaczenie strategiczne. Od jej dostępności i jakości
zależy trafność i szybkość podejmowania decyzji
Bezpieczeństwo informacji oznacza zachowanie poufności, integralności oraz dostępności
Poufność – to zapewnienie dostępu do informacji tylko osobom upoważnionym
Integralność – to zapewnienie dokładności i kompletności informacji oraz metod jej przetwarzania
takich, by zapewnić, że informacja pochodzi z wiarygodnego źródła i nie wprowadzano
niepożądanych zmian
Dostępność – to zapewnienie, że osoby upoważnione mają do niej swobodny dostęp.
Bezpieczeństwo informacji może być osłabione w wyniku tzw. podatności zasobów na działanie
zagrożeń. Zagrożenia te mogą pochodzić od:
Człowieka: np. skasowanie pliku, hakerzy w systemie, niewiedza i niefrasobliwość
pracowników
Ze środowiska, np. zalanie, pożar, itp.
Od sprzętu, np. awaria, niewłaściwe skonfigurowanie, brak odpowiednich programów
antywirusowych
Stały wzrost poziomu zagrożenia związanego z bezpiecznym przetwarzaniem informacji jest
motorem podjęcia wdrożenia odpowiednich norm dotyczących systemu zarządzania
bezpieczeństwem informacji
System zarządzania bezpieczeństwem informacji (SZBI) ISO/IEC 27001:2007 – Technika
Informatyczna – Technika Bezpieczeństwa – Systemy Zarządzania Bezpieczeństwem Informacji –
Wymagania
System ten opiera się na podejściu procesowym i jest zgodny z modelem PDCA i polega na:
Zrozumieniu biznesowych wymagań bezpieczeństwa informacji oraz potrzeby ustanowienia
polityki i celów BI
Wdrażaniu i eksploatowaniu zabezpieczeń związanych z zarządzaniem ryzykiem
Monitorowaniu i przeglądzie wydajności i skuteczności systemu
Ciągłym doskonaleniu na podstawie obiektywnego pomiaru
Wymagania ISO/IEC 27001 zawarte są w punktach normy:
4. System zarządzania bezpieczeństwem informacji (SZBI)
5. Odpowiedzialność kierownictwa
6. Audyt wewnętrzny SZBI
7. Przegląd SZBI realizowany przez kierownictwo

8. Doskonalenia SZBI
Polityka SZBI powinna określać wymagania biznesowe, prawne, nadzoru, wynikające z umów z
klientami oraz kryteria według których ma być szacowane ryzyko dotyczące bezpieczeństwa.
Norma nie narzuca sposobu szacowania ryzyka, ale określa czynności jakie powinna w tym celu
wykonać organizacja. Proces szacowania ryzyka poprzedza:
Określenie zasobów oraz właścicieli tych zasobów
Określenie podatności, które mogą być wykorzystane przez zagrożenia
Określenie skutków, jakie mogą wystąpić w stosunku do aktywów w przypadku utraty
poufności, integralności i dostępności
Na szacowanie skutków wystąpienia ryzyka składa się:
Wyznaczenie poziomu ryzyka (w przyjętej skali)
Stwierdzenie, czy poziom ryzyka jest akceptowalny, czy też wymaga podjęcia działań
korygujących
Dokumentem podsumowującym proces szacowania ryzyka jest tzw. Deklaracja stosowania (SoA),
która dokumentuje ustalone przez organizacje cele stosowania zabezpieczeń oraz zakładane
zabezpieczenia. Ich zakres określa załącznik A normy. Deklaracja SoA jest obowiązkowym
dokumentem systemu, a szczegółowe informacje dotyczące wyrobu i zastosowania zabezpieczeń
podano w ISO/IEC 17799 – Praktyczne zasady zarządzania bezpieczeństwem informacji.
Po oszacowaniu ryzyka należy opracować „Plan postępowania z zagrożeniami”. Plan ten powinien
określać metodę postępowania z zagrożeniami, wdrożone zabezpieczenia, proponowane
zabezpieczenia, czas na działanie oraz zakresy obowiązków pracowników
Norma wymaga wdrożenia odpowiednich procedur umożliwiających wykrywanie naruszenia
bezpieczeństwa informacji. Wiążę się to z opracowaniem planów awaryjnych, planowaniem
ciągłości działania. Plany awaryjne powinny zawierać definicje potencjalnych zdarzeń
kryzysowych, listę osób odpowiedzialnych za ogłaszanie stanu kryzysu z zakresem ich
obowiązków.
System zarządzania bezpieczeństwem informacji jest stosunkowo nowym dynamicznie
rozwijającym się systemem w związku z ciągłym rozwojem IT, jak i nowymi zagrożeniami. Seria
norm ISO 27000 jest aktualnie rozbudowywana i obejmuje coraz to nowe normy. Normy te
obejmują: zasady zarządzania bezpieczeństwem informacji, przewodnik do wdrożenia SZBI,
zarządzanie ryzykiem, przewodnik dla akredytowanych jednostek do przeprowadzenia SZBI
Systemy zarządzania jakością w branży medycznej
Pierwsza europejska norma dotycząca przemysłu medycznego ukazała się w 1993r. W 200r.
pojawiło się najważniejsze wydanie normy EN ISO 13485 – Systemy jakości – Wyroby medyczne
– Szczególne wymaganie, które są bardziej specyficzne niż w normie ISO 9001. Norma ta ma
zastosowanie gdy konieczna jest ocena jakości dostawcy wyrobów medycznych. Łącznie z normą
9001 definiuje się wymagania dotyczące systemów jakości, odnoszące się do projektowania, prac
rozwojowych, produkcji, instalowania i serwisu wyrobów medycznych.
W branży medycznej obowiązują również następujące normy:
ISO 13485 – Systemy jakości – wyroby medyczne – wymagania dla celów przepisów
prawnych
ISO/TR 14949 Wyroby medyczne - Systemy zarządzania jakością – Wytyczne do
stosowanie ISO 13845
ISO 15189 Laboratoria medyczne – Szczególne wymagania dotyczące jakości i kompetencji
ISO 15195 Medycyna laboratoryjna, wymagania dla referencyjnych laboratoriów
pomiarowych (norma do akredytacji)
IWA 1 (International Workshop Agrement) – Wytyczne dla doskonalenia w organizacjach
służby zdrowia – Wytyczne opierają się na ISO 9004 i są poszerzone o zagadnienia
specyficzne dla służby zdrowia
Systemy zarządzania jakością w branży motoryzacyjnej:
Unormowania dotyczące branży motoryzacyjnej są bardzo restrykcyjne i producenci samochodów
stwierdzili, że wymagania norm ISO serii 9000 są obowiązujące ale niewystarczające. Od wielu lat
najwięksi przedstawiciele przemysłu motoryzacyjnego dążyli do wprowadzenia bardziej
restrykcyjnych warunków jakie powinni spełnić współpracujący z nimi dostawcy. Przemysł ten
okazał się wiodącym w zakresie rozwoju programów certyfikacji dostawców. Do znanych w tym
zakresie standardów należą:
System jakości QS 9000
System jakości VDA 6.1
System jakości ISO/TS 16949
System jakości QS 9000
Za pewną tendencję rynkową uznaje się unifikację wymagań w zakresie kształtowania jakości
przybierającą postać coraz to liczniejszych aktów normatywnych, które stanowią wymagania
stawiane dostawcom. Wiodące organizacje na rynku motoryzacyjnym ustanowiły własne programy
doskonalenia jakości, narzucając określone wymagania dostawom. Najważniejszym z nich jest
system „Wielkiej Trójki” – Daimler-Chrysler Corp., Ford Motor Corp. i General Motors Corp.
standard QS 9000. Standard ten ma zastosowanie do wszystkich dostawców wewnętrznych i
zewnętrznych tych koncernów.
Standard QS 9000 dotyczy w szczególności dostawców:
Materiałów produkcyjnych
Części i podzespołów międzyoperacyjnych oraz części zamiennych
Odróbki cieplnej, malowania, chromowania itp., a także wszelkich innych prac
wykończeniowych
Ponadto opracowane zostały wymogi dotyczące dostawców narzędzi, maszyn oraz innych
urządzeń operacyjnych
Standard QS powinien pozwolić zminimalizować uciążliwość wielokrotnej oceny rzetelności
potencjalnych i obecnych dostawców oraz zapewnić, że produkowane samochody będą
odpowiadały oczekiwanej przez użytkowników jakości.
Dostawca chcący sprostać QS 9000 powinien ustanowić krótko i długofalowe biznes plany w
zakresie określających przedsięwzięć, tj. plany jakości, wskazujące jednoznacznie etapy oceny i
weryfikacji jakości przy realizacji nowych wyrobów, począwszy od weryfikacji jakości dostaw do
prototypu. Standard ten wymaga również ustanowienia systemu ciągłej poprawy w kluczowych
obszarach działalności firmy, wskazującego mechanizmy identyfikacji szans oraz monitoring
stopnia spełnienia bieżących i identyfikacji przyszłych potrzeb klientów. QS 9000 stawia także
wymagania wobec organizacyjnych i technicznych zdolności do realizacji „dostaw na czas” (Just in
time)
System QS składa się z trzech sekcji podstawowych
Sekcja 1 – obejmuje wszystkie 20 podstawowych wymagań znajdujących się także w
normie ISO 9001 oraz formułuje podstawy systemu QS 9000
Sekcja 2 – obejmuje tzw. specyficzne wymagania sektorowe, przewyższające znacznie
wymagania opisane w normie ISO 9001, a dotyczące takich zagadnień jak: proces
zatwierdzania części produkcyjnych, ciągłe doskonalenie oraz zdolność produkcyjna.
Sekcja 3 – obejmuje unikalne, tzw. specyficzne wymagania klienta, jakie stawiają
niezależnie od siebie przedstawiciele „Wielkiej Trójki” wobec poddostawców. Za każdym
razem dostawca musi przedyskutować te wymagania, zarówno w odniesieniu do aktualnych
jak i przyszłych kontraktów. Dodatkowe wymagania odnośnie systemu jakości poddostawcy
pochodzą z własnej dokumentacji opracowanej przez poszczególnych producentów
„Wielkiej Trójki” oraz producentów ciężarówek, m.in. Mac Tracks, Peterbilt Trucks
Norma ISO 9001 stanowi pewnego rodzaju nadkładkę dla wymagań poszczególnych koncernów.
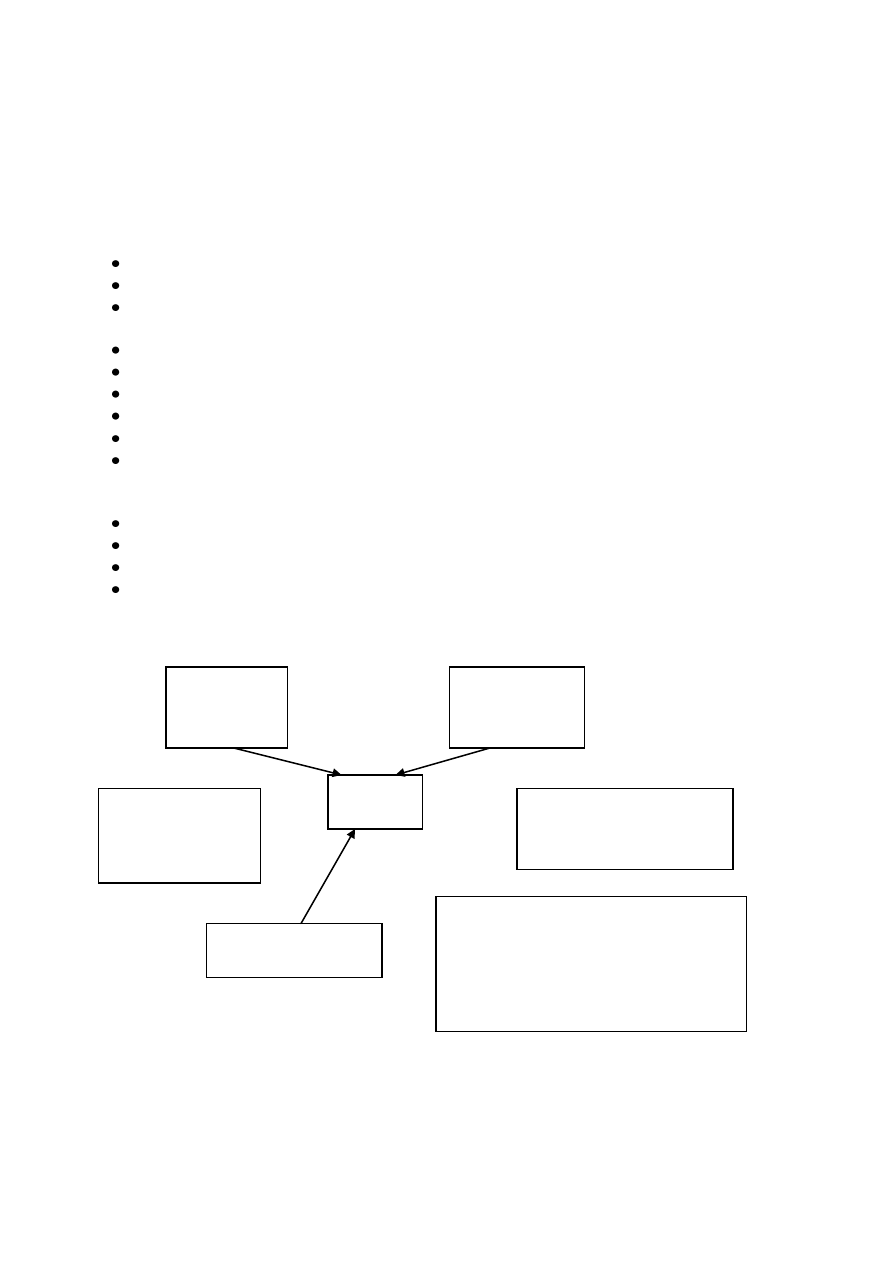
Z zasady niedopuszczalne są jakiekolwiek niezgodności w stosunku do wymagań QS 9000, co
może oznaczać że niezgodność ujawniona podczas auditu certyfikującego jest jednoznaczna z nie
uzyskaniem certyfikatu.
Wymagania QS 9000 obligują do dokonywania wizyty i przeprowadzenia bezpośredniej
wystarczalności systemu jakości dla spełnienia uzgodnionych wymagań kontraktowych u
dostawców
Założenia systemu QS 9000, wynikające z ogólnych wymagań przemysłu samochodowego w USA,
przedstawione są w części drugiej systemu i składają się z następujących elementów:
Proces aprobowania części produkcyjnych i zmian w dokumentacji
Proces ciągłych ulepszeń, celem podnoszenia jakości i efektywności
Zdolność procesów wytwórczych
W ramach normy QS 9000 zapisane są w zbiorze podręczników wymagania:
APQP – zaawansowane planowanie jakości
FMEA – analiza potencjalnych wad i skutków
MSA – analiza systemów pomiarowych
PPAP – proces zatwierdzania części do produkcji
SPC – statystyczne sterowanie procesem
QSA – ocena systemu jakości
Wymagania systemu QS wynikające ze specyficznych wymagań koncernów samochodowych w
USA przedstawione w części trzeciej stanowią:
Wymagania koncernu Chrysler Co.
Wymagania koncernu Ford Motor Co.
Wymagania koncernu General Motors Co.
Wymagania producentów ciężarówek
Organizacja systemów jakości QS 9000 – III edycja:
System VDA 6.1
Wymagania niemieckiego sektora motoryzacyjnego zostały sprecyzowane przez Związek
Niemieckiego Przemysłu Motoryzacyjnego (Verband der Automobilindustrie e.V.) w porozumieniu
z większymi przedsiębiorstwami i dostawcami. W efekcie stworzono serię norm VDA 6. Norma
ISO 9000
Wymagania
podstawowe
Indywidualne
wymagania
klientów
QS 9000
III edycja
20 podstawowych
wymagań oraz
dodatkowe stanowiące
podstawy systemu QS
9000
Koncernu Chrysler Co.
Koncernu Ford Motor Co.
Koncernu General Motors Co.
Producentów ciężarówek
Podręczniki związane z
normą QS 9000
PPAP – proces zatwierdzania części do
produkcji
APQP – zaawansowane planowanie jakości
MSA – analiza systemów pomiarowych
SPC – statystyczne sterowanie procesem
QSA – ocena systemu jakości
FMEA – analiza potencjalnych wad i skutków
VDA 6.1 – I wydanie – została opublikowana w 1991r. Norma ta opiera się na ISO 9001. Jest
jednak organizowana na dwóch specyficznych obszarach: kierownictwo oraz wyrób i proces.
Norma ta obowiązuje wszystkich dostawców koncernów VW Group (VW, Audi, Seat, Skoda),
Porsche, Adam Opel, Ford-Were.
Aktualnie funkcjonuje III wydanie normy VDA 6.1. Składa się ona z 23 elementów, w tym
załącznik A1 (strategia przedsiębiorstwa), w którym przyporządkowano zestaw pytań auditowych
wraz z wyczerpującym i jednoznacznym komentarzem. Stanowi to bazę do przeprowadzania
auditów Systemu Zarządzania Jakością w relacji: producent-dostawca. Uważa się, że VDA 6.1 jest
bardziej wymagająca niż QS 9000. Wiele koncernów stosuje jeszcze bardziej surowe wymagania.
Należy do nich Mercedes-Benz, który oprócz VDA 6.1 stosuje siedem dodatkowych wymagań od
swoich dostawców.
Dodatkowe wymagania technologiczne Mercedesa to:
Prace rozwojowe nad materiałami
Prace rozwojowe nad systemami produkcyjnymi
Współpraca z zakładami Mercedesa
Powtórne wykorzystanie produktów
Aspekty ochrony środowiska u dostawcy
Standardy Jakości ISO/TS 16949
Podstawowym celem powołania nowego standardu było opracowanie jednolitych wymagań dla
rynku motoryzacyjnego, stanowiących podstawę do certyfikacji, który uzyska akceptację na rynku
europejskim i amerykańskim. Norma ISO/TS 16949 została opracowana przez grupę IATF
(Internationa Automotive Task Force) oraz reprezentantów ISO serii 9000. Drugie wydanie ISO/TS
16949:2002 zawierało uzgodnienia pomiędzy IATF oraz stowarzyszeniem Japońskich Producentów
(JAMA). Wielkoseryjny charakter w przemyśle samochodowym, wymuszająca kooperację
powodują, że jakoś wyrobu końcowego zależy od wielu specyficznych uwarunkowań.
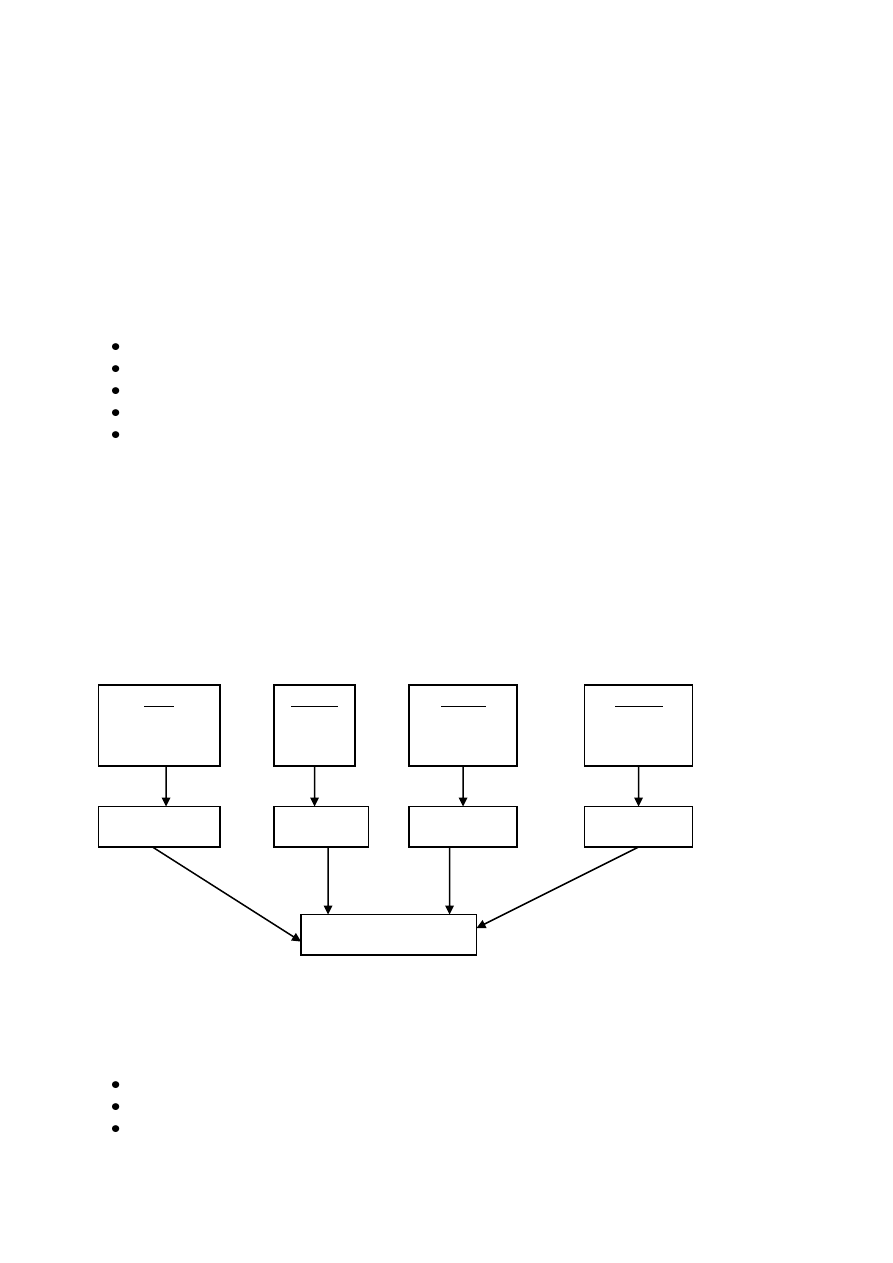
Schemat harmonizacji norm światowego przemysłu motoryzacyjnego:
System jakości oparty o standard ISO/TS 16949 obejmuje swym zakresem cały cykl tworzenia
wyrobu, a więc od organizacji firmy i systemu jakości poprzez działania poznawania rynku, etapy
projektowania, wytwarzanie, kontrole i badania, podejmowanie działań korygujących i
zapobiegawczych, aż do wysyłki wyrobu do klienta.
Standard zbudowany jest tak aby:
W każdym punkcie najpierw przywołana była norma ISO 9001
Następnie podane są dodatkowe wymagania
Zharmonizowanie dla wcześniejszych standardów przemysłu motoryzacyjnego
USA
Ford
General Motors
Chrysler
Niemcy
Audi
BMW
VW
Francja
PSA
Renault
Włochy
Fiat
Iveco
QS 9000
VDA 6.1
EAQF
AVSQ
ISO/TS 16949
Dodatkowe wymagania dotyczą zarówno kwestii inżynierskich jak i zagadnień związanych ze
skutecznością I efektywnością systemu. Te szczególne wymagania dotyczą:
Dokumentacji technicznej
Planowanej jakości
Zakupów
Analizy systemów pomiarowych
Badania wyrobów niespełniających wymagań
Przeprowadzenia audytów
Oraz innych obszarów i działań, uznawanych za ważna w uzyskaniu skutecznych i efektywnych
procesów wytwarzania.
Standard ISO/TS 16494 odróżnia od ISO 9001:
Podkreślenie znaczenia klienta i jego satysfakcji
Normatyw ISO/TS 16494 ukierunkowany jest na zapobieganie niezgodnościom, a nie
likwidowanie ich skutków
Działania w sposób wyraźny ukierunkowane są na ciągłe doskonalenie i minimalizowanie
strat i kosztów
Zwraca uwagę na znaczenie odpowiedzialności producenta za wyrób
Podkreśla znaczenie tworzenia planów jakości i kontroli
Zobowiązuje do ścisłej współpracy z klientem, szczególnie na etapie tworzenia wyrobów
Norma ISO/TS 16494, a inne wymagania:
Standard jakościowe w przemyśle lotniczym AS 9000 i D1-9000
W opracowaniu nowych wymagań brału dział takie firmy jak: Allied Signal, Allson Enigne
Company, Boeing, General Electric Engines, McDonnel Douglas oraz inne światowe koncerny.
Standard AS 9000 opiera się na 20 punktach ISO 9001, a dodatkowo uwzględnia 28 specyficznych
wymagań lotniczych, opracowanych przez Federal Aviation Administration (FAA) oraz przez
Departament of Demence (DOD)
W 1997r. AS 9000 uznany został w USA za normę krajową i stanowi ważny krok w kierunku
normalizacji wymagań w przemyśle lotniczym.
Do najistotniejszych korzyści można zaliczyć:
QS 9000:
FMEA
APQP
SPC
PPAP
MSA
QSA
QS 9000:
FMEA
APQP
SPC
PPAP
MSA
QSA
ISO/TS 16494:2002
Na podstawie
ISO 9001:2000
Zasady certyfikacji
CSR
CSR
CSR
CSR
General
Motors
Ford
Motor Co
CSR
Daimler
Chrysler
VW
Volvo
IATF
(AIAG, VDA)
- dokument
- organizacja

Wykazanie zgodności z wymaganiami norm ISO serii 9000 oraz specyficznymi
wymaganiami branżowymi
Wprowadzenie atmosfery porozumienia wśród producentów przemysłu lotniczego przez
zunifikowanie funkcjonujących do tej pory wymagań
Zarządzanie, którego celem jest osiągnięcie bezpieczeństwa i niezawodności wytwarzanych
wyrobów
Standard D1-9000 jest zespołem wymagań opracowanym przez firmę Boeing Co., która pomimo
istnienia standardu AS 9000, od swoich poddostawców wymaga wypełnienia własnych kryteriów
jakościowych, ponieważ własne wymagania uznał za wyższe i skuteczniejsze, szczególnie w
odniesieniu do problemu bezpieczeństwa pasażerów.
System zarządzania JAR-145
Wspólne Władze Lotnictwa Cywilnego – JAA (Joint Aviation Authorities), czyli 23 kraje
europejskie w tym Polska, uzgodniły wspólne, ogólne i szczegółowe przepisy lotnicze w postaci
norm, zwane Joint Aviation Requirements (JAR). Celem tych przepisów jest:
Ułatwienie przedsiębiorstwom certyfikacji w przypadku joint venture, ułatwienie eksportu i
importu wyrobów lotniczych
Zapewnienie, by obsługa dokonana w jednym z krajów europejskich była uznawana przez
władze lotnictwa cywilnego innego kraju
Zgodnie z przepisami JAR-145 żaden samolot, śmigłowiec lub sterowiec nie może być
dopuszczony do lotów bez poświadczenia o zdatności wydanego przez organizację obsługową
mającą stosowne orzeczenie upoważniające do obsługi.
Wymagania w przemyśle zbrojeniowym AQAP:
W przemyśle zbrojeniowym państw NATO przyjęto politykę jakości, która zakłada:
Pełną odpowiedzialność wszystkich zaangażowanych stron (użytkownika, zamawiającego,
dostawcy, personelu) za jakość wyrobu w ramach GQA (Government Quality Assurance,
Rządowe Zapewnienie Jakości)
Szacowanie ryzyka związanego z realizacją kontraktu, na podstawie którego podejmuje się
decyzję o przeprowadzeniu GQA
W 1987 państwa NATO podjęły decyzję o przyjęciu normy ISO 9001 jako podstawy formułowania
wymagań dotyczących zapewnienia jakości przez dostawców wyposażenia i sprzętu na cele
wojskowe.
Obowiązujące dokument AQAP oparte na normie ISO 9001 to:
AQAP 2110, wyd.2 – Wymagania NATO dotyczące zapewnienia jakości w
zaprojektowaniu, pracach rozwojowych i produkcji
AQAP 2120, wyd. 2 – Wymagania NATO dotyczące zapewnienia jakości w produkcji
AQAP 2130 wyd. 2 – Wymagania NATO dotyczące zapewnienia jakości w kontroli i
badaniach
AQAP 2210, wyd. 1. – Wymagania uzupełniające NATO do AQAP 2110 dotyczące
zapewnienia jakości oprogramowania
AQAP 2105 wyd. 1 – Wymagania NATO dotyczące planów jakości dla wyrobu
zamawianego
W celu nadzorowania i monitorowania systemu jakości dostaw obowiązują:
STANG 4107 – który ustala zasady nadzorowania systemu zarządzania jakością dostaw,
zwanego procesem rządowego zapewniania jakości (GOA)
AQAP – 2000 – polityka NATO dotyczące zintegrowanego systemowego podejścia do
jakości w cyklu życia wyrobu
Poszerzenia wymagań AQAP w stosunku do wymagań ISO 9001 objemują:
Umożliwienie zamawiającemu dostępu do wszystkich dokumentów związanych z umową
Konieczności opracowania do każdej umowy planu jakości

Uzyskania w zakresie nadzorowania wyposażenia do monitorowania i pomiarów zgodności
z normą ISO 10012
Uzupełnienia wymagań związanych z realizacją wyrobu o zarządzanie konfiguracją i
zagadnienia niezawodności i możliwości obsługiwania wyrobu (ARMP – Publikacje
Standaryzacyjnych Niezawodności i Obsługiwalności)
Objęcia programem auditów wewnętrznych wszystkich wymagań zwartych w umowie i
przekazywanie informacji o wszystkich stwierdzonych niezgodnościach zamawiającemu
Obecności zamawiającego lub przedstawiciela wojskowego w trakcie odbioru wyrobu
Akceptacji przez zamawiającego wszelkich napraw, przeróbek i dopuszczeń w zakresie
nadzoru nad wyrobem niezgodnym.
Wykład 10.
1.
PCA – Polskie Centrum akredytacyjne
2.
PCA jest członkiem organizacji skupiającej:
- jednostki akredytujące laboratoria,
-jednostki certyfikujące,
- jednostki kontrolujące w Europie EA(European co-operation for Accreditation)
PCA jest członkiem międzynarodowej organizacji skupiającej instytucje akredytujące jednostki
certyfikujące i kontrolujące IAF (international accreditation forum – inc.)
PCA jest członkiem międzynarodowej organizacji skupiającej laboratoria badawcze i wzorcujące
ILAC (international laboratory accreditation cooperation)
Akredytacje PCA są uznawane w Europie i na świecie
3.
PCA zachęca laboratoria do stosowania metod badawczych wynikających z najnowszych zdobyczy
wiedzy oraz zgodnych z opisanymi w normach europejskich i międzynarodowych.
4.
PCA udziela akredytacji laboratoriom tylko na taką działalność, do prowadzenia której ma ono
kompetencje.
PCA nie udziela akredytacji na badania zlecane podwykonawcom.
Ponadto PCA nie udziela akredytacji na badania, co do których laboratorium ma zawartą umowę na
dorywcze wykorzystanie kluczowego wyposażenia pomiarowo – badawczego i/lub pomieszczeń
laboratoryjnych
W wypadku ubiegania się o akredytację organizacji mającej laboratoria w wielu lokalizacjach,
wnioskująca organizacja może złożyć jeden wniosek obejmujący wszystkie lokalizacje lub odrębne
wnioski dla każdej lokalizacji.
5.
Dokumentami zawierającymi opis realizacji metod badawczych wymienionych w zakresie
akredytacji mogą być:

-normy polskie, regionalne i międzynarodowe
-normy zagraniczne publikowane przez krajowe jednostki normalizacyjne państw członkowskich
ISO
-przepisy prawne polskie i UE
-kodeksy postępowania
-ogólnie dostępne publikacji metodyczne
-własne procedury badań.
6.
Zasadą jest, że akredytowane laboratoria badawcze samodzielnie wykonują badania w ramach
posiadanego zakresu akredytacji.
Wyjątkowo, laboratorium badawcze może podzielić własne akredytowane badania oraz badania
spoza własnego zakresu akredytacji innemu laboratorium akredytowanemu, którego zakres
akredytacji obejmuje podzlecane badania, zgodnie z własną procedurą dotyczącą podwykonawstwa.
7.
Laboratorium zlecające badania podwykonawcy:
-powinno poinformować klienta o zamiarze podzlecenia badań przed tym działaniem i uzyskać jego
pisemną zgodę na to zlecenie,
-powinno uzyskać od laboratorium będącego podwykonawcą sprawozdanie z badań obejmujące
wyniki zleconego badania
-może zamieścić w swoim sprawozdaniu z badań wyniki otrzymane od podwykonawcy (pod
warunkiem wyraźnego ich oznaczenia) lub załączyć sprawozdanie otrzymane od podwykonawcy
-w przypadku zamieszczenia w swoim sprawozdaniu z badań wyników utrzymanych od
podwykonawcy powinno uzyskać jego pisemną zgodę na publikację
-laboratorium zlecając badania podwykonawcy każdorazowo powinno monitorować jego zakres
akredytacji.
8.
PCA (DA – 05)
Badanie biegłości (PT)1 – określenie za pomocą porównań międzylaboratoryjnych zdolności
laboratorium do przeprowadzania wzorcowań lub badań, lub zdolności jednostki inspekcyjnej do
przeprowadzania badań.
Porównanie międzylaboratoryjne (ILC)2 – zorganizowanie, wykonanie i ocena wzorcowań/badań
tego samego lub podobnych obiektów wzorcowań/badań przez co najmniej dwa laboratoria,
zgodnie z uprzednio ustalonymi warunkami. W tekście używa się oznaczeń skrótowych PT i ILC.
9.
PCA (DA – 05)
PCA traktuje PT/ILC jako jeden z podstawowych elementów wykazania kompetencji technicznych
akredytowanych laboratoriów.
PCA nie wymaga by laboratoria objęły uczestnictwem w programach PT/ILC wszystkie
badania/pomiary objęte zakresem akredytacji.
W takich wypadkach laboratoria powinny przedstawić inne wiarygodne i skuteczne środki
wykazania swojej biegłości. Polityka laboratoriów dotycząca takich wypadków powinna być
określona i udokumentowana w Księdze Jakości,
10.
PCA (DA – 05)

Kryteria akceptacji uzyskanych rezultatów stosowane przy ocenie osiągnięć laboratoriów
badawczych:
- żaden wynik dotyczący badanych cech/pomiarów nie może znaleźć się w grupie wyników
niezadowalających,
- w wypadku wykorzystywania wskaźnika z przy ocenie uzyskanych rezultatów dopuszcza się20%
wyników w grupie wątpliwych (2<|z|<3).
11.
PCA (DA – 05)
Organizatorami programów PT/ILC akceptowanymi przez PCA mogą być (lecz nie ograniczając się
tylko do nich)>
- krajowe jednostki akredytujące,
- komercyjnie organizatorzy programów PT/ILC
- regionalne lub międzynarodowe organizacje zrzeszające jednostki akredytujące
- organy stanowiące (wskazane przez nie laboratoria referencyjne)
- przemysł lub organizacje/zrzeszenia producentów.
ORGANIZATOR PROGRAMU PT/ILC POWINIEN MIEĆ WDROŻONY SYSTEM
ZARZĄDZANIA JAKOŚCIĄ.
12.
PCA (DA – 07)
Wyposażenie pomiarowe stosowane do wzorcowań, badań i inspekcji, mające istotny wpływ na
niepewność pomiaru związaną z wynikami tych działań, powinno być wzorcowanie przez krajową
instytucję metrologiczną ( w Polsce – Główny Urząd Miar)1 albo przez akredytowane laboratoria
wzorcujące 2.
Wzorce odniesienia akredytowanych laboratoriów wzorcujących powinny być wzorcowane w
GUM-ie lub akredytowanych laboratoriach wzorcujących o odpowiedniej najlepszej możliwości
pomiarowej.
Jednym z elementów spójności pomiarowej jest niepewność pomiaru związana z wzorcowaniem
przyrządów pomiatowych.
13.
PCA (DA – 07)
Zmiany zakresu akredytacji
Zakres akredytacji laboratorium badawczego można rozszerzyć o:
- nowe obiekty/grupy obiektów
- nowe cechy
- nowe zakresy już akredytowanych metod badawczych
- nowe metody badawcze
-nowe działy techniczne / nowe lokalizacje
14.
EN 45000
W 1989r. Europejski Komitet Normalizacyjny CEN ustanawia grupę norm mających ścisły związek
z normami serii 9000, normy serii EN 45000. Normy te określają zasady działania jednostek
certyfikujących i laboratoriów badawczych.
15.
PN-EN ISO/IEC 17025:2001

Ogólne wymagania dotyczące kompetencji laboratoriów badawczych i wzorcujących zastępuje
wytyczne ISO 25 i EN 45001 (grudzień 1999r.)
Standard ten można traktować jako techniczne uzupełnienie do ISO 9001:2000.
Instytut posiada 19 laboratoriów akredytowanych przez (PCA) na zgodność z normą PN-EN
ISO/IEC 17025:2005 wchodzących w struktury zakładów badawczych.
Certyfikat akredytacji Laboratorium Badawczego nr AB 170 metody badań fizykochemiczncyth i
użytkowych benzyn silnikowych, olejów napędowych, benzyn lotniczych.
16.
PN – EN ISO/IEC 17025:2001
Wymagania w sferze zarządzania:
- uregulowania funkcjonowania całego laboratorium
- zarządzania (polityka, komunikacja wewnątrz laboratorium)
- doskonalenia (zaangażowanie najwyższego kierownictwa)
- dokumentacja zapisów
- podwykonawstwa badań
- obsługi klientów (łącznie ze skargami)
- nadzoru nad niezgodnymi badaniami i/lub wzorcowaniami
- działań korygujących i zapobiegawczych
- auditów i przeglądów zarządzania
17.
PN – EN ISO/IEC 17025:2001
Wymagania techniczne:
Personel
Warunki lokalowe i środowiskowe
Metody badań i/lub wzorcowań wraz z ich walidacją szacowaniem niepewności
Nadzorowanie wyposażenia
Zapewnienie spójności pomiarowej
Pobieranie próbek i postępowanie z obiektami do badań
Zapewnienie jakości badań i/lub wzorcowań
Przedstawienie wyników badań i/lub wzorcowań.
18.
Audit pierwszej strony
Przeprowadzany jest z inicjatywy kierownictwa danej firmy dla własnych potrzeb i zwykle przez
własnych pracowników.
W trakcie audytu pierwszej strony następuje weryfikacja poszczególnych elementów systemu
jakości w tym zwłaszcza ocena stopnia przestrzegania wcześniej założonych ustaleń. Ustalenia te
muszą być spisane i udokumentowane między innymi w postaci procedur i instrukcji.
Najważniejszym dokumentem który jest podstawowym odnośnikiem w trakcie przeprowadzania
wszystkich rodzajów audytów systemu jakości jest tzw. Księga Jakości. Opisane są w niej ogólne
dyspozycje przyjęte przez przedsiębiorstwo w celu osiągnięcia wymaganej jakości wyrobów i
usług. Inaczej mówiąc jest to dokument opisujący system zapewnienia jakości w przedsiębiorstwie.
19.

Audit drugiej strony
Dane przedsiębiorstwo występując w roli zamawiającego przeprowadza audyt jakości u swoich
aktualnych lub potencjalnych podwykonawców wykorzystuje do tego najczęściej własny zespół
audytorów lub zleca wykonanie audytów wyspecjalizowanej firmie.
Zgodnie z postanowieniami norm PN ISO serii 9000 firma zobowiązana jest do pełnienia
skutecznego nadzoru nad systemami jakości swoich podwykonawców.
Ponadto dzięki prowadzeniu audytów drugiej strony przedsiębiorstwa mogą nie tylko dokonać
wyboru najlepszych podwykonawców ale także pomóc im w poprawie ich własnych systemów
jakości.
20.
Audit trzeciej strony
Wniosek o jego przeprowadzenie zgłasza do upoważnionej instytucji przedsiębiorstwo.
Celem takiego audytu jest pragnienie podbudowania renomy lub ugruntowanie pozycji na rynku.
Audyt może być także skutkiem żądań potencjalnego kontrahenta.
Pozytywny rezultat audytu trzeciej strony pozwala na nadanie certyfikatu systemowi zapewnienia
jakości w tym przedsiębiorstwie. Jest to oficjalny dokument stwierdzający, że system obowiązujący
w danej firmie odpowiada wymaganiom jednej z norm podlegających certyfikacji.
Docelowym rezultatem pomyślnie zakończonego audytu trzeciej strony ma być wzrost zaufania
zleceniodawców do danego przedsiębiorstwa.
Wykład 11
Zarządzanie jakością w laboratorium według ISO 17025
PN-EN ISO/IEC 17025:2000 - Ogólne wymagania dotyczące kompetencji laboratoriów
badawczych i wzorcujących
• Norma ta została opracowana na podstawie wcześniejszej EN 45001 oraz ISO/IEC Guide 25.
Zawiera ona wszystkie wymagania jakie powinny spełniać laboratoria badawcze i wzorcujące i
stanowi podstawę do ich akredytacji. Zawiera wszystkie wymagania normy ISO 9001. Posiadanie
certyfikatu ISO 9001 nie jest równoznaczne z uzyskaniem akredytacji przez laboratorium.
Zakres normy
W rozdziale tym w punktach od 1 do 6 omówiono zakres normy oraz układ i powiązania z
Systemem Zarządzania Jakością.
• Normy powołane
Do norm powołanych aktualnie obowiązujących należy norma ISO 9001 oraz aktualne normy
związane i przewodniki zawarte w bibliografii
• Terminy i definicje
Do celów tej normy stosuje się terminy i definicje zawarte w normie ISO 9000 i ISO/IEC guide 2
oraz VIM
• Wymagania dotyczące zarządzania
Organizacja

Laboratorium lub organizacja w ramach której działa powinno mieć odpowiedzialność prawną.
Badania wykonywane przez laboratorium powinny spełniać wymagania zawarte w normie i
satysfakcję klienta.
System Zarządzania Laboratorium obowiązuje wszystkie prace wykonywane zarówno w
laboratorium jak i poza stałą siedzibą. W celu wykluczenia konfliktu interesów, w przypadku
laboratoriów, które są częścią organizacji, należy ściśle sprecyzować zakres odpowiedzialności
personelu.
Laboratorium powinno:
a. Zatrudniać personel kierowniczy i techniczny posiadający odpowiednie uprawnienia i środki do
realizacji zadań
b. Mieć ustalenia zapewniające niezależność
c. Posiadać procedury i politykę zapewniające poufność
d. Mieć politykę i procedury umożliwiające angażowania się w działania zmniejszające zaufanie
klientów
e. Określić strukturę organizacyjną i zarządzania laboratorium
f. Zapewnić przez kompetentne osoby nadzór nad personelem wykonującym badania
g. Zatrudniać kierownictwo techniczne, które ponosi odpowiedzialność za wyposażenie i
działalność techniczną laboratorium
h. Wyznaczyć kierownika ds. jakości i ustalić dla niego adekwatny zakres obowiązków i praw.
i. Wyznaczyć zastępców dla ważnego personelu
System jakości
-„Laboratorium powinno ustanowić, wdrożyć i utrzymywać system jakości właściwy dla zakresu
jego obowiązków".
- Laboratorium powinno udokumentować swoją politykę, systemy, programy, procedury i
instrukcje w stopniu niezbędnym do zapewnienia jakości wyników badań
- Dokumentacja systemu powinna być podana do wiadomości całego personelu, zrozumiana,
dostępna i wdrażana
- Polityka i cele jakości powinny być określone w księdze jakości
- Księga jakości powinna przedstawiać strukturę, dokumentację, przywoływać lub zawierać
procedury
- Księga jakości powinna zawierać dane odnośnie roli i odpowiedzialności kierownictwa
technicznego i kierownika jakości łącznie z odpowiedzialnością dotyczącą zapewnienia zgodności z
normą
Nadzór nad dokumentami
Laboratorium powinno opracować i utrzymywać procedury nadzorowania wszystkich dokumentów,
takich jak: przepisy, normy, metody badań, wzorcowań, rysunki, oprogramowanie, specyfikacje itp.
Dokumenty Systemu Jakości powinny być jednoznacznie zidentyfikowane, przeglądane i
zatwierdzane przez upoważnione osoby.
Procedura nadzoru nad dokumentami powinna być tak opracowana, by uniemożliwić stosowanie
dokumentów nieważnych.
Dokumenty przechowywane w archiwum powinny być odpowiednio oznakowane.
Należy opracować procedurę w jaki sposób wprowadza się zmiany w dokumentach. Zmiany do
dokumentów powinny być przeglądane i zatwierdzane w tym samym miejscu gdzie przegląd
pierwotny.
Przegląd zamówień, ofert i umów
Laboratorium powinno ustanowić i utrzymywać procedury dotyczące przeglądu zamówień, ofert i
umów.

Wszelkie różnice pomiędzy zamówieniem a umową powinny być wyjaśnione przed rozpoczęciem
badań.
Z przeglądów natęży utrzymywać zapiski, również rozmów z klientami.
Przeglądy powinny obejmować prace podzlecane przez laboratorium.
Klient powinien być informowany o wszystkich odstępstwach od umowy.
Podwykonawstwo badań i wzorcowań
Laboratorium powinno posiadać rejestr podwykonawców i podzlecać pracę tylko podwykonawcy
stosującemu się do niniejszej normy.
O fakcie zatrudnienia podwykonawcy należy niezwłocznie poinformować klienta.
Laboratorium odpowiada za jakość pracy podwykonawcy, za wyjątkiem kiedy został on wskazany
przez klienta.
Zakupy usług i dostaw
W punktach od 1 do 4 zawarto wymagania dotyczące zakupów, a pozwalające na zapewnienie
jakości badań.
Do najważniejszych należy posiadanie polityki oraz procedury dotyczącej wyboru i zakupu
wykorzystywanych przez siebie usług i dostaw.
Posiadanie listy kwalifikowanych dostawców.
Należy posiadać procedury dotyczące nabywania, przyjmowania i magazynowania odczynników i
materiałów pomocniczych.
Obsługa klienta
Laboratorium powinno umożliwić klientom taką współpracę, aby byli nią usatysfakcjonowani, ale
jednocześnie nie naruszać interesów i poufności badan innych.
Skargi
Laboratorium powinno posiadać procedurę i politykę rozpatrywania skarg.
Należy utrzymywać zapisy z ich rozpatrywania i podjętych działań korygujących.
Nadzorowanie niezgodnych z wymaganiami badań, wzorcowań
Laboratorium powinno mieć politykę i procedury postępowania, gdy wynik pracy nie odpowiada
własnym procedurom lub uzgodnieniom z klientem. Należy wówczas ustalić odpowiedzialność,
ocenę znaczenia niezgodności oraz podjąć działania korygujące, powiadomić o fakcie klienta.
Działania korygujące
Laboratorium powinno ustanowić politykę i procedury działań korygujących w przypadku
stwierdzenia niezgodności.
Postępowanie korygujące powinno opierać się na: analizie przyczyn, wyborze, wdrożeniu i
monitorowaniu działań korygujących oraz auditach dodatkowych
Działania zapobiegawcze
Laboratorium powinno posiadać procedury działań zapobiegawczych. Należy określić możliwe
ulepszenia i potencjalne źródła niezgodności technicznych i systemu. Jeżeli to konieczne to
opracować i wdrożyć plany działań zapobiegawczych i monitorować je.
Nadzór nad zapisami
Laboratorium powinno posiadać procedurę nadzoru nad zapisami- identyfikacji, gromadzenia,
dostępu, oznaczania, katalogowania, przechowywania, utrzymywania i niszczenia zapisów. Zapisy
dotyczące jakości powinny obejmować raporty z auditów wewnętrznych, przeglądów zarządzania,
działań korygujących i zapobiegawczych. Laboratorium powinno posiadać procedurę dotyczącą
ochrony i tworzenia kopii bezpieczeństwa zapisów
elektronicznych. Przez ustalony czas laboratorium powinno przechowywać zapisy techniczne
dotyczące

wykonywanych badań, tak oznakowane, aby umożliwić ich identyfikację i identyfikowalność.
Audity wewnętrzne
Laboratorium powinno okresowo i zgodnie z wcześniej ustalonym programem i procedurą
przeprowadzać
audity wewnętrzne. Planowanie i organizowanie auditów wewnętrznych należy do kierownika ds.
jakości.
Audity powinny być organizowane przez odpowiednio przeszkolony personel. Nie należy
auditować własnej pracy.
Wszystkie działania związane z auditem, a szczególnie zlecone działania korygujące powinny być
zapisywane. W trakcie działań poauditowych należy sprawdzić skuteczność podjętych działań
korygujących.
Wymagania techniczne
Postanowienia ogólne
Do czynników wpływających na prawidłowość i wiarygodność badań należą:
Czynnik ludzki
Warunki lokalowe i środowiskowe
Metody badań i wzorcowań oraz ich walidacja
Wyposażenie
Spójność pomiarowa
Pobieranie próbek
Postępowanie z obiektami badań i wzorcowań
Personel
Kierownictwo laboratorium musi zapewnić, że osoby wykonujące badania mają odpowiednią
wiedzę i kompetencje. Do obsługi specjalnego wyposażenia konieczne są osobiste certyfikaty.
Kierownictwo powinno upoważnić personel do poboru próbek. Laboratorium powinno mieć
procedury dotyczące szkoleń personelu, sformułować cele dotyczące wykształcenia.i umiejętności
pracowników.
Do prac laboratorium może wykorzystywać swój personel lub z którym zawarto odpowiednią
umowę. Laboratorium powinno posiadać zakresy obowiązków całego personelu
Zapisy dotyczące wykształcenia, kwalifikacji, szkoleń doświadczenia itp. powinny być
potwierdzone i zawierać datę potwierdzenia.
Warunki lokalowe i środowiskowe
Laboratorium powinno monitorować, nadzorować i rejestrować warunki środowiskowe zgodnie z
wymaganiami określonych specyfikacji, metod i procedur oraz tam, gdzie warunki środowiskowe
mogą wpłynąć na wyniki badań.
Należy podjąć środki zabezpieczające przed wzajemnym zanieczyszczeniem próbek np. przez
skuteczne rozgraniczenie poszczególnych obszarów.
Dostęp do i wykorzystanie obszarów powinno być nadzorowane, należy określić zakres nadzoru dla
konkretnych warunków lokalowych.
Należy zapewnić środki do utrzymania czystości, jeśli to konieczne opracować specjalną procedurę.
Wyposażenie
Laboratorium powinno być wyposażone w urządzenia i aparaty do pobierania próbek, pomiarów i
badań, niezbędne do prawidłowego przeprowadzenia badań czy wzorcowań.
Wyposażenie i jego oprogramowanie powinno zapewniać wymaganą dokładność oraz spełniać
odpowiednie specyfikacje dotyczące badań.
Wyposażenie powinien obsługiwać upoważniony personel.

Instrukcje obsługi, konserwacji, przechowywania powinny być aktualne i łatwo dostępne.
Wyposażenie powinno być jednoznacznie zidentyfikowane.
Zapisy dotyczące wyposażenia powinny zawierać:
identyfikację wyposażenia i oprogramowania, nazwę producenta, typ, numer seryjny, sprawdzenie
czy urządzenie odpowiada specyfikacji,
aktualną lokalizację,
instrukcje producenta lub informacje o miejscu ich przechowywania,
daty wzorcowań oraz ich wyniki i świadectwa, plan konserwacji,
informacje o każdym uszkodzeniu, wadliwym działaniu naprawie czy modernizacji.
Urządzenie niesprawne powinno być oddzielone od pozostałych
Wszędzie gdzie to możliwe należy urządzenie nadzorowane opatrzyć etykietami, kodem, datą
następnego sprawdzenia Jeżeli urządzenie znajdzie się poza nadzorem laboratorium to przed
użyciem należy je sprawdzić. Jeżeli to konieczne to przeprowadza się zgodnie z procedurą
sprawdzania pośrednie. Wyposażenie powinno być zabezpieczone przed adiustacjami, które mogą
unieważnić wyniki.
Spójność pomiarowa
Laboratorium powinno mieć opracowany program i procedurę dotyczącą wzorcowania swoich
wzorców odniesienia.
Wzorce odniesienia powinny być wykorzystywane tylko do wzorcowania. Wzorce należy
wzorcować przed i po adiustacji.
Materiały odniesienia powinny być powiązane z jednostkami SI lub certyfikowanym materiałem
odniesienia.
Sprawdzanie bieżące powinno być przeprowadzane zgodnie z procedurą i programem.
Pobieranie próbek
Laboratorium powinno mieć plan i procedury pobierania próbek oraz procedurę zapisywania
odpowiednich danych i czynności związanych z pobieraniem próbek.
Jeżeli to możliwe to plan ten i procedury powinny się opierać na metodach statystycznych.
Jeżeli klient zażąda odstępstw od planu i procedury pobierania próbek to powinno to być
odpowiednio zapisane.
Postępowanie z obiektami badań i wzorcowań
Laboratorium powinno mieć procedury postępowania dotyczące transportowania, przejmowania,
magazynowania, pogorszenia własności obiektów. Należy stosować system identyfikacji badanych
obiektów.
Zapewnienie jakości wyników badania i wzorcowania
Laboratorium powinno mieć procedury sterowania jakością w celu monitorowania podejmowanych
badań. Uzyskane dane powinny być zapisywane w taki sposób, aby umożliwić śledzenie kierunków
zmian. Należy przy tym stosować możliwe techniki statystyczne. Monitorowanie powinno być
planowane i poddawane przeglądom.
Przedstawianie wyników
Wyniki badań, wzorcowań, serii badań powinny być podane dokładnie, jasno, jednoznacznie,
obiektywnie i zgodnie ze wszystkimi specyficznymi instrukcjami zawartymi w metodach badań i
wzorcowań.
Sprawozdanie z badań i świadectwo wzorcowań powinno zawierać:
1. Tytuł
2. Nazwę i adres laboratorium oraz miejsce wykonania badań, jeżeli jest inne niż laboratorium
3. Niepowtarzalną identyfikację sprawozdania i oznaczenie każdej strony sprawozdania
4. Nazwę i adres klienta
5. Identyfikację zastosowanej metody
6. Opis, stan i jednoznaczną identyfikację obiektów badania lub wzorcowania
7. Datę przyjęcia obiektów badania i datę wykonania badania, wówczas gdy jest to istotne dla
miarodajności wyników i ich zastosowania.
8. Odwołanie się do planu pobierania próbek i procedur wykorzystywanych w laboratorium.
9. Wyniki badań i wzorcowań wraz, z tym gdzie to właściwe, jednostkami miar.
10. Nazwiska, funkcje i podpisy osób autoryzujących sprawozdanie z badania.
11. Oświadczenie, gdy to istotne, że wyniki odnoszą się wyłącznie do badanych obiektów.
Opinie i interpretacje - jeżeli zamieszcza się opinie i interpretacje, to laboratorium powinno
udokumentować dane, które są podstawą opinii czy interpretacji.
Wyniki badań i wzorcowań uzyskane od podwykonawców należy wyraźnie zidentyfikować.
Laboratorium powinno opracować formularze do każdego rodzaju badań, tak aby były czytelne i
pozwalały właściwie wykorzystywać zawarte w nich dane. Szczególne warunki opisane punktem
5.4.7 normy odnoszą się do przekazywania wyników badań drogą elektroniczną.
Wykład 12. (01.06.2010)
Zarządzanie jakością żywności
Zarządzanie jakością w agrobiznesie i przemyśle żywnościowym jest szczególnie ważne ze
względu na emocjonalny stosunek klienta do produktów żywnościowych oraz możliwość
zagrożenia bezpieczeństwa zdrowotnego.
Aktualnie w Polsce obowiązują
obligatoryjnie przepisy HACCP (Hazard Analysis and Critical Control Point) - Analiza
Zagrożeń i Krytyczny Punkt Kontroli - system opracowany w celu rozpoznania i kontroli
zagrożeń, które mogą się pojawić w jakimkolwiek momencie procesu produkowania i
składowania żywności
Norma PN-EN ISO 2200 - Systemy zarządzania bezpieczeństwem żywności. Wymagania
dla każdej organizacji należącej do łańcucha żywnościowego.
Do najczęściej stosowanych systemów przy zapewnieniu jakości żywności należą:
GMP - Dobra Praktyka Produkcyjna
GHP - Dobra Praktyka Higieniczna
GLP - Dobra Praktyka Laboratoryjna
System HACCP
System Zarządzania Jakością ISO 9001
Systemy Zarządzania Bezpieczeństwem Żywności ISO 22000
Wdrażanie systemu HACCP w małych lub słabo przygotowanych firmach może napotkać na szereg
trudności. Warto wówczas, wprowadzając zasady HACCP, zachować dużą elastyczność,
odpowiednią do możliwości i sytuacji, jaka występuje w zakładzie.
Postawa kierownictwa decyduje o efektywności i skuteczności działania systemu. To właśnie
kierownictwo powinno być inicjatorem i siłą napędową przy podejmowaniu działań dotyczących
systemu, będąc jednocześnie całkowicie przekonanym co do słuszności i celowości wdrażania.

Zaangażowanie i postawa kierownictwa wobec HACCP powinna być oficjalnie przedstawiona
załodze.
Określony powinien być podział obowiązków, odpowiedzialności i uprawnień w tym zakresie.
Zaangażowanie kierownictwa związane jest ze stworzeniem załodze warunków do podnoszenia
wiedzy z zakresu praktycznej realizacji zasad systemu oraz organizowaniem szkoleń, instruktażu.
Działania o charakterze edukacyjno-szkoleniowym mają na celu doprowadzenie do zmian w
mentalności załogi oraz zwiększenia odpowiedzialności.
Do zadań kierownictwa należy określenie zakresu HACCP, przy uwzględnieniu:
1. Poszczególnych produktów przetwarzanych w zakładzie
2. Etapów postępowania z tymi produktami
Kierownictwo powinno w przybliżeniu określić:
1. ramy czasowe wdrażania systemu
2. szacunkowe koszty,
3. Inwestycje
4. zmiany organizacyjne
Przed przystąpieniem do wdrażania systemu HACCP należy przejrzeć aktualnie obowiązujące
zasady dotyczące stanu technicznego i higienicznego oraz możliwości zapewnienia w nim pełnego
bezpieczeństwa żywności.
Działania z tego zakresu to tzw. Programy
Warunków Wstępnych obejmujące zasady GHP i GMP.
Zasady GHP i GMP obejmują:
prawidłowość linii technologicznych i prowadzenia procesów
wyposażenie w maszyny i urządzenia oraz ich funkcjonowanie
wyposażenie w sprzęt kontrolno-pomiarowy
wielkość, funkcjonalność i usytuowanie pomieszczeń
procesy mycia i dezynfekcji
stan sanitarno-higieniczny zakładu i otoczenia
organizację pracy
higienę i sposób postępowania załogi
kwalifikacje personelu
zabezpieczenie przed szkodnikami
jakość wody
jakość sprowadzanych surowców i materiałów pomocniczych
magazynowanie żywności
warunki jej transportu
Zasady GHP i GMP odnoszą się do:
Pomieszczeń zakładu i procesu produkcji, czyli tzw. obszaru bezpośrednio produkcyjnego
Otoczenia zakładu oraz dostaw surowcowi ich dystrybucji, czyli tzw. obszaru
przyprodukcyjnego
Stan sanitarno-higieniczny zakładu i otoczenia:
1. Kto jest odpowiedzialny za utrzymanie porządku, w jaki sposób je wykonuje oraz kto
nadzoruje te czynności
2. Czy zakład nie oddziałuje negatywnie na środowisko

3. Czy wokół zakładu nie ma czynników, które mogłyby zagrażać bezpieczeństwu żywności
Wielkość, funkcjonalność i usytuowanie pomieszczeń:
1. Czy wielkość zakładu i poszczególnych pomieszczeń jest odpowiednia do wykonywanych
czynności
2. Czy poszczególne czynności czy procesy odbywaja się w odpowiednio przystosowanych
pomieszczeniach
3. Czy pomieszczenia te utrzymywane sa w odpowiednim porządku i czystości
4. Czy w pomieszczeniach jest odpowiednia wentylacja
5. Czy w poszczególnych pomieszczeniach jest zapewniona temperatura odpowiednia do
prowadzonych w nich procesów
prawidłowość linii technologicznych i prowadzenia procesów:
1. Czy proces technologiczny jest możliwie prostoliniowy ( nie ma krzyżowania się dróg
czynności „brudnych" i „czystych")
2. Czy wydajność linii technologicznych koreluje z możliwościami składowania
3. I wyrobów gotowych
4. Czy wydzielone są w zakładzie strefy „brudne" i „czyste"
5. Czy przestrzegane są i nadzorowane istotne parametry procesów technologicznych
Wyposażenie w maszyny i urządzenia oraz ich funkcjonowanie:
1. Czy w zakładzie jest dostateczna liczba odpowiednich maszyn i urządzeń
2. Czy są one systematycznie kontrolowane z punktu widzenia ich funkcjonalności
3. Czy prowadzone są okresowe przeglądy maszyn i urządzeń
4. Czy maszyny i urządzenia są na bieżąco konserwowane i remontowane
5. Czy istnieją harmonogramy konserwacji i remontów maszyn i urządzeń
6. Czy prowadzone są stosowne zapisy dotyczące nadzoru nad maszynami i urządzeniami
Wyposażenie w sprzęt kontrolno-pomiarowy:
1. Czy sprzęt i urządzenia kontrolno-pomiarowe są odpowiednio kalibrowane i wzorcowane
2. Czy istnieją harmonogramy wzorcowania urządzeń kontrolno-pomiarowych
3. Czy istnieją zapisy z kalibracji i wzorcowania
Organizacja pracy
1. Czy w zakładzie wyznaczone są osoby odpowiedzialne za bezpieczeństwo i jakość
zdrowotną żywności
2. Czy na poszczególnych stanowiskach określone są działania w aspekcie zapewnienia
bezpieczeństwa żywności
Procesy mycia i dezynfekcji:
1. Czy są opracowane procedury mycia i dezynfekcji poszczególnych pomieszczeń i urządzeń
w zakładzie (z określeniem sposobu mycia i stężenia stosowanych środków myjących i
dezynfekujących)
2. Czy są osoby wyznaczone do prowadzenia procesów mycia i dezynfekcji
3. Czy osoby te mają odpowiednie uprawnienia i przeszkolenie
4. Kto prowadzi nadzór na prawidłowością procesów mycia i dezynfekcji
5. Czy środki i narzędzia stosowane do mycia dezynfekcji są odpowiednio oznakowane,
przechowywane i czy są dopuszczone do kontaktu z żywnością
Higiena i sposób postępowania załogi:
1. Czy pracownicy są nadzorowani z uwagi na spełnianie zasad higieny (czystość osobista,
higieniczne zachowanie, niepalenie tytoniu itp..)

2. Czy pracownicy dysponują odpowiednią liczbą odzieży ochronnej, zgodnie z potrzebami
3. Czy sprawdzany jest stan zdrowia pracowników pracujących w kontakcie z żywnością
4. Czy pracownicy mają warunki do zachowania higieny - np. mycia rąk na poszczególnych
stanowiskach
Kwalifikacje personelu:
1. Czy pracownicy posiadają odpowiednie do ich stanowiska kwalifikacje formalne
(świadectwa szkolne, odbyte kursy itp..)
2. Czy prowadzone są systematyczne szkolenia lub instruktaż w zakresie higieny
3. Czy prowadzone szkolenia i kursy są odpowiednio udokumentowane
4. Czy kierownictwo egzekwuje od załogi znajomość zasad pracy na stanowiskach
Zabezpieczenie przed szkodnikami:
1. Czy w zakładzie jest program zabezpieczenia przed szkodnikami
2. Czy kontrolowana jest skuteczność zabezpieczenia
3. Czy prowadzony jest nadzór nad urządzeniami i pułapkami służącymi do oceny stopnia
inwazyjności szkodników i czy są one odpowiednio wymieniane
Jakość wody:
1. Czy przechowywane są aktualne wyniki badania wody
2. Czy zapewniony jest skuteczny rozdział wody technologicznej od wody do celów
technicznych
3. Czy istnieją dwa oddzielne rurociągi w tym celu i czy są oznakowane
Jakość sprowadzanych surowców i mat. pomocniczych
1. Czy sprawdzane są atesty i specyfikacje
2. Czy prowadzona jest ocena przyjmowanych surowców i materiałów pomocniczych
3. Czy ocenia się stan opakowań przyjmowanych surowców i materiałów pomocniczych
4. Czy, o ile to potrzebne, dokonuje się pomiaru
Magazynowanie żywności:
1. Czy kontrolowane są warunki przechowywania surowców, półproduktów i produktów
gotowych (np. w zakresie temperatury i wilgotności, jeżeli to konieczne)
2. Czy w magazynach zachowana jest segregacja surowców od wyrobów gotowych itp.. Czy
prowadzona jest kontrola terminów przydatności do spożycia
3. Czy prowadzona jest kontrolowana rotacja zapasów w magazynach
4. Czy wszystkie produkty spożywcze przechowywane są w odpowiednich magazynach (a nie
np. w pomieszczeniach produkcyjnych)
Warunki transportu żywności:
1. Czy dokonywana jest ocena warunków sanitarnych i higienicznych środków transportu
żywności
2. Czy prowadzona jest dokumentacja w tym zakresie
3. Czy są wyznaczone osoby odpowiedzialne za ocenę warunków transportu żywności
4. Czy kierowcy/konwojenci posiadają odpowiednie przeszkolenia
5. Czy są nadzorowani z punktu widzenia zachowania warunków higienicznych (odzież
ochronna, czystość osobista)
Definicja -
HACCP jest systemowym postępowaniem mającym na celu zapewnienie
bezpieczeństwa Zdrowotnego żywności poprzez identyfikację i oszacowanie skali zagrożeń
bezpieczeństwa żywności z punktu widzenia jej jakości zdrowotnej oraz ryzyka wystąpienia tych
zagrożeń podczas przebiegu wszystkich etapów produkcji i dystrybucji żywności. Jest to również
system mający na celu określenie metod ograniczania tych zagrożeń oraz ustalenie działań
naprawczych.
System HACCP działa w oparciu o 7 podstawowych zasad
1. Analiza zagrożeń - zidentyfikowanie i ocena zagrożeń oraz ryzyka ich wystąpienia, a także
ustalenie środków kontroli i metod przeciwdziałania tym zagrożeniom
2. Ustalenie Krytycznych Punktów Kontroli - KPK (CCP) - w celu wyeliminowania lub
zminimalizowania występowania zagrożeń
3. Ustalenie dla każdego KPK wymagań (parametrów), jakie powinien spełniać oraz określenie
granic tolerancji (limitów krytycznych)
4. Ustalenie i wprowadzenie systemu monitorowania KPK
5. Ustalenie działań korygujących, jeśli KPK nie spełnia ustalonych wymagań
6. Ustalenie procedur weryfikacji w celu potwierdzenia, że system jest skuteczny i zgodny z
planem
7. Opracowanie i prowadzenie dokumentacji systemu HACCP dotyczącej etapów jego
wprowadzania, a także ustalenie sposobu rejestrowania i przechowywania danych oraz
archiwizowania systemu.
Zasady wdrażania HACCP
Przy wdrażaniu systemu HACCP zaleca się postępowanie zgodne z 12 sekwencyjnymi zaleceniami
(krokami) Kodeksu Żywnościowego:
1. Powołanie zespołu HACCP - system powinien być wdrażany przez kilkuosobowy zespół (w
małych przedsiębiorstwach może wystarczyć jeden wyszkolony pracownik) dobrze wyszkolonych i
przygotowanych wysoko wykwalifikowanych specjalistów posiadających wiedzę z dziedziny i
mikrobiologii, inżynierii, technologii, higieny : produkcji, zarządzania jakością itp. Dobrze gdy
mają - kontakt z bieżącą praktyką produkcyjną.
2. Opisanie produktu W zakładzie powinien być opracowany dokładny opis poszczególnych
produkowanych wyrobów, z uwzględnieniem składu surowcowego, stosowanej technologii, cech
fizykochemicznych, cech mikrobiologicznych, sposobu pakowania i znakowania, metod
dystrybucji, transportu, magazynowania. W zakładach o różnorodnym profilu działania lub
wieloprodukcyjnych np. w zakładach żywienia zbiorowego, zaleca sie grupowanie produktów o
podobnych cechach
3. Określenie przewidywanego sposobu wykorzystania produktu przez konsumenta Należy określić
zakładane wykorzystanie lub zastosowanie produktu przez konsumenta. Określić czy będzie
spożywany bezpośrednio, czy po ewentualnej obróbce termicznej, oraz jak konsument może
postępować z produktem podczas jego przechowywania i przygotowania do konsumpcji. Rozważyć
także należy do jakiej grupy konsumentów skierowany jest produkt, ze zwróceniem szczególnej
uwagi na najbardziej wrażliwe grupy konsumentów (niemowlęta, małe dzieci, chorzy w szpitalach).
4. Opracowanie schematu blokowego procesu technologicznego
Schemat procesu technologicznego powinien być przygotowany w postaci diagramu i powinien
obejmować wszystkie fazy procesu produkcji ( przyjmowanie surowców, proces produkcji,
dystrybucja, obsługa klienta). Na każdym z etapów należy ustalić podstawowe parametry (np. temp.
obróbki termicznej, czas trwania procesów, warunki składowania, pH i aktywność wodną produktu,
wilgotność produktu i otoczenia, sposoby monitorowania). Prawidłowe opracowanie schematu

technologicznego ułatwia zrozumienie przepływu surowców i półproduktów, pozwala na
zidentyfikowanie krzyżowania się dróg i jest podstawą podejścia systemowego HACCP.
5. Weryfikacja schematu procesu technologicznego
Zasadniczą sprawą jest, aby szczegółowe dane przedstawione na schemacie technologicznym
odzwierciedlały rzeczywisty stan linii technologicznych i całego procesu produkcji. Weryfikacja
powinna dotyczyć wszystkich normalnych warunków prowadzenia procesu. Schemat
technologiczny powinien być systematycznie uaktualniany.
6. Sporządzenie listy wszystkich ewentualnych zagrożeń związanych z każdym etapem produkcji
oraz listy wszelkich prewencyjnych środków kontroli danego zagrożenia (Zasada 1) Na każdym
etapie produkcji należy sporządzić listę wszystkich możliwych zagrożeń biologicznych,
chemicznych, fizycznych. Powinny być one podane w sposób precyzyjny. Każde zagrożenie
powinno być opisane osobno. Każdemu zagrożeniu należy przypisać określone środki kontroli.
Jako środki kontroli danego zagrożenia określa się działania, czynności lub warunki, jakie są
wymagane do eliminacji zagrożeń lub zmniejszenia do poziomu akceptowalnego.
7. Określenie Krytycznych Punktów Kontroli CCP (Zasada 2)
Jako środek pomocniczy proponuje się tutaj tzw. drzewko decyzyjne, tj. logiczną sekwencję pytań i
odpowiedzi w odniesieniu do każdego surowca i etapu produkcji, pozwalającą na określenie
najbardziej istotnych miejsc i etapów procesu produkcyjnego. Ważnym aspektem drzewka
decyzyjnego jest to, że naprowadza ono użytkownika do stosowania jedynie minimalnej liczby
krytycznych punktów kontroli decydujących o bezpieczeństwie produktu.
8. Określenie celów i granic tolerancji dla każdego punktu krytycznego (Zasada 3)
Dla każdego KPK (CCP) należy ustalić tzw. wartości docelowe wraz z dopuszczalnymi
tolerancjami oraz graniczne wartości określonych parametrów tzw. wartości krytyczne. Wartości te
powinny gwarantować skuteczną eliminację zagrożenia. Kryteria te obejmują najczęściej czas,
temperaturę, wilgotność, aktywność wodną itp., ale także cechy o charakterze sensorycznym. Przy
ustalaniu wartości zaleca się posługiwanie danymi literaturowymi lub przyjęcie wartości
określonych w przewodnikach.
9.Opracowanie systemu monitorowania każdego KPK (Zasada 4)
Metody monitorowania muszą być ściśle zdefiniowane, procedura monitoringu powinna być
wiarygodna. Opracowując je należy określić jakie parametry są mierzone, jakie są granice
krytyczne dla dokonywanych pomiarów, w jaki sposób, kiedy i z jaka częstotliwością prowadzi się
monitoring. Instrukcje monitorowania powinny pozwalać na szybkie wykrywanie w każdym z
ustalonych KPK ewentualnych odchyleń poza przyjęte granice tolerancji. Częstotliwość
monitoringu musi być tak dobrana, aby dać pewność bezpieczeństwa produktu. Należy ustalić
personel odpowiedzialny za dokonywanie pomiarów.
10. Ustalenie działań korygujących (Zasada 5)
Dla każdego punktu krytycznego należy ustalić działania korygujące. Powinny one umożliwić
natychmiastowe usunięcie odchyleń w wartościach przyjętych parametrowi zapewnić, że KPK
znajduje się pod kontrolą.
Działania korygujące obejmują:
sposoby przywrócenia kontroli nad KPK
sposoby postępowania z wyrobem niepewnym
Działania korygujące powinny być możliwie proste i łatwe do przeprowadzenia przez przeszkolony.
Działania korygujące muszą być dokumentowane. Informacja o ich podjęciu powinna być
przekazana kierownictwu.
11. Ustalenie procedury weryfikacji (Zasada 6)

Weryfikacja ma za zadanie ustalić, czy procedury dają pożądany rezultat oraz wykryć ewentualne
iratedociągnięcia. Czynności weryfikacyjne mogą obejmować badanie produktu gotowego, analizę
v wyników, przeglądy rejestrów wraz ze stwierdzonymi! odchyleniami, ustalenie poziomów
tolerancji itp. Weryfikacja nie powinna być prowadzona przez osoby związane z przedmiotem
weryfikacji. Weryfikację należy prowadzić okresowo, w sposób planowany tak, aby zapewnić
skuteczną realizacje planów HACĆP.
12. Prowadzenie dokumentacji i zapisów (Zasada 7)
Właściwie sporządzona dokumentacja jest podstawowym dowodem efektywności działania
systemu i podstawą do oddalenia ewentualnych reklamacji.
Zapisy powinny dotyczyć planu HACCP, etapów procesu technologicznego, potencjalnych
zagrożeń, wskazanych KPK oraz przyjętych dla nich kryteriów i tolerancji, procedury monitoringu i
działań korygujących, odpowiedzialności personelu itp.
Dokumentacja powinna być możliwie najprostsi
Prawidłowa dokumentacja systemu HACCP składa się z dwóch członów:
1. Planu HACPP
2. Dokumentów potwierdzających funkcjonowanie systemu
PLAN HACCP
Dokument powołujący zespół HACCP i lista jego członków
Zakres stosowania systemu
Opis produktu i jego przeznaczenie
Schemat produkcyjny
Dokument stwierdzający zgodność schematu z praktyką
Systemy Zarządzania Jakością Zawartość tematyczna
1. Jakość, geneza, podstawowe pojęcia, definicje
2. Koncepcje i modele zarządzania przez jakość(Deming, Juran, Crosby, Feigenbaum)
3. Kompleksowe Zarządzanie Jakością - TQM
4. Systemy zarządzania jakością według ISO serii 9000
5. Wymagania norm ISO 9000, 9001, 9004
6. Zarządzanie środowiskiem - ISO serii 14000
7. Zarządzanie środowiskiem - ISO 14001, Analiza Cyklu Życia
8. Systemy zarządzania bezpieczeństwem i higieną pracy - ISO serii 180(M
9. Systemy zarządzania bezpieczeństwem i higieną pracy - ISO 18001, ocena ryzyka
zawodowego
10. Zarządzanie chemikaliami
11. Systemy zarządzania bezpieczeństwem żywności - HCCP, ISO 22000 I
12. Dokumentacja w systemach zarządzania jakością - ISO/TR 10013
Według Pani Hoffman na zaliczeniu obowiązują tylko punkty, które zostały opracowane na
wykładzie – czyli wszystko bez Analizy Cyklu Życia oraz punktu 12.
Wyszukiwarka
Podobne podstrony:
10,11,12
24 05 2010 B&K, Bazy Danych 10 11 12
klima pytania, 7 8 9 10 11 12
24.05.2010 B&K Bazy Danych 10 11 12
pkmiu zadania 1 2 3 4 6 7 8 10 11 12, ściągi III OP
2013 2014 ZARZADZANIE ZASOBAMI LUDZKIMI wyklad 10 11 12
prezentacja ze szkolenia 10 11 12 2011 coaching
10,11,12
10,11,12
10, 11, 12
Język jako narzedzie komunikacji wykł 10 11.12.07
10, 11, 12, 10
10, 11, 12, 10
2010 10 11(2),12,22 zbiory, ciągi
10,11,12
10,11,12
MGiF2b cz1 bez3,10,11,12
więcej podobnych podstron