
POLITECHNIKA GDAŃSKA
WYDZIAŁ CHEMICZNY
KATEDRA TECHNOLOGII CHEMICZNEJ
TECHNOLOGIA CHEMICZNA, sem. VII
TECHNOLOGIA ORGANICZNA
SYNTEZA I ZASTOSOWANIE KATALIZATORÓW
Gda
ń
sk, 2012

WSTĘP I PODSTAWOWE POJĘCIA
Reakcje chemiczne w większości przypadków prowadzi się w obecności katalizatorów
mających na celu przyspieszenie reakcji, umożliwienie otrzymania pożądanego produktu.
Katalizatory można podzielić na dwie zasadnicze grupy:
1. katalizatory chemiczne
2. oraz biokatalizatory
Katalizator – substancja, która zwiększa szybkość zbliżania się układu do stanu
równowagi i kieruje reakcję do określonego, możliwego z punktu widzenia termodynamiki
produktu, nie będąc w istotny sposób zużywana w tej reakcji. Katalizator nie zmienia
położenia równowagi chemicznej, wpływa jedynie na szybkość dochodzenia układu do tego
stanu. Substancja ta tworzy nietrwałe połączenia przejściowe z substratami reakcji, po czym
- pod koniec reakcji – zostaje odzyskana i pozostaje chemicznie niezmieniona.
Do podstawowych właściwości katalizatorów można zaliczyć:
1.
Aktywność (miara szybkości reakcji),
2.
Selektywność,
3.
Stabilność,
4.
Zakres temperaturowy efektywnego działania katalizatora.
Aktywność katalizatora (A
k
) określa się jako różnicę między szybkościami reakcji
chemicznej zachodzącej w obecności katalizatora (V
k
) i bez katalizatora (V):
A
k
= V
k
- V
Szybkość reakcji prowadzonej bez katalizatora jest zazwyczaj bardzo mała w porównaniu do
szybkości reakcji prowadzonej w obecności katalizatora, wobec tego miarą aktywności
katalizatora jest w praktyce szybkość.
Często również za miarę aktywności katalizatora przyjmuje się stopień przereagowania
substratów. Wtedy aktywność katalizatora charakteryzuje układ katalizator-reagenty, a nie
sam katalizator.
Opisując aktywność katalizatora, trzeba zaznaczyć w jakiego typu reakcji jest on aktywny.
Kolejną cechą opisującą katalizatory jest selektywność. Określa ona zdolność katalizatora do
tworzenia poszczególnych produktów. Im bardziej katalizator przyspiesza określoną reakcję
danego substratu tym bardziej jest selektywny.
Jeśli rozważymy reakcję, w której substrat A przereagowuje do kilku różnych produktów (Pi,
Pk itd), to selektywność w kierunku tworzenia produktu Pi można wyrazić jako stosunek
liczby moli substratu A przekształconego w związek Pi do całkowitej liczby przereagowanych
moli substancji A wyrażony w procentach.
Z danego substratu w obecności różnych katalizatorów można otrzymać różne produkty.
Dlatego mówi się, że dany katalizator jest selektywny w danej reakcji w kierunku tworzenia
jednego z produktów. W tabeli przedstawione zostały typy katalizatorów stosowane w
katalitycznej konwersji etanolu w temp. 300ºC.
KATALIZATOR
PRODUKTY
Cu
Aldehyd octowy, wodór

Al
2
O
3
Eten, woda
MgO-SiO
2
(Na
2
O) [1:1(0.1%mas)]
1,3-butadien, woda
Zeolit ZSM-5
Syntetyczna benzyna, woda
Bardzo często z opisem katalizy związane są takie pojęcia jak adsorpcja i desorpcja.
Adsorpcja polega na wiązaniu cząsteczek na powierzchni zewnętrznej i wewnętrznej ciała
stałego, który pełni rolę adsorbenta (katalizator). Oddziaływania te mogą mieć charakter
fizyczny lub/i chemiczny. W przypadku adsorpcji fizycznej następuje związanie za pomocą
oddziaływań Van der Waalsa, a w przypadku adsorpcji chemicznej - chemisorpcji następuje
chemiczne wiązanie cząsteczki z powierzchnią katalizatora.
Ze względu na fazy reakcji katalitycznej możemy ją podzielić na:
a.
Homogeniczną – jedna faza w układzie, katalizator nie wyróżnialny,
b.
Heterogeniczną – katalizator występuje w innej fazie niż substraty.
Częściej stosowane są katalizatory heterogeniczne ponieważ charakteryzuje je łatwość
opracowania i oddzielenia z mieszaniny reakcyjnej. Ze względu na te cechy są częściej
stosowane (ok. 80%), mimo iż są bardziej podatne na zatrucia i są mniej selektywne niż
katalizatory homogeniczne.
Dezaktywacją katalizatorów nazywamy częściowe lub całkowite obniżenie aktywności
katalizatora w wyniku zmian zachodzących na jego powierzchni. Dezaktywacja może zajść
wskutek:
•
działania trucizn katalizatora, które zwykle (w niewielkich ilościach) są wprowadzane
wraz z surowcami. Zatrucie następuje wskutek adsorpcji lub reakcji trucizny z
katalizatorem i powstania związku katalitycznie nieaktywnego. Zatrucie może być
odwracalne i nieodwracalne. Szczególnie wrażliwe są na zatrucia katalizatory metaliczne.
Truciznami katalizatorów są: siarkowodór, siarczki organiczne i nieorganiczne, związki
arsenu, fosforowodór, amoniak,
•
zmniejszenia powierzchni aktywnej w warunkach podwyższonej temperatury, w wyniku
rekrystalizacji lub spiekania,
•
pokrywania powierzchni katalizatora zanieczyszczeniami np: pyłem lub substancjami
stałymi powstającymi podczas procesu. Takim przykładem blokowania powierzchni jest
gromadzenie się związków węgla na powierzchni glinokrzemianów podczas krakingu
katalitycznego. Zregenerowanie katalizatora następuje przez wypalenie powstałego koksu.
Katalizator w fazie stałej składa się kilku elementów:
1.
Nośnik – jego zadaniem jest zwiększenie powierzchni fazy aktywnej i zwiększenie
wytrzymałości mechanicznej i termicznej katalizatora. Odpowiada za stabilność
katalizatora. Może być źródłem centrów aktywnych innego typu niż faza aktywna,
albo modyfikować jej strukturę elektronową. Przykłady nośników to tlenki glinu i
krzemu SiO
2
, γ-Al
2
O
3
, α-Al2O3, tlenki chromu Cr
2
O
3
, MgO, CaO, węgiel aktywny.
2.
Faza aktywna – jest źródłem centrów aktywnych jednego bądź różnych typów, na
których powstają kompleksy przejściowe. Przykłady fazy aktywnej to metale np. Fe,
Ni, Pt, Cu Pd, Ag, tlenki metali, np. NiO, ZnO, siarczki metali, halogenki metali.
3.
Promotory – modyfikują strukturę fizyczną i elektronową substancji aktywnej,
hamują jej niekorzystne przemiany fazowe i ułatwiają regenerację. Przykłady
promotorów to ZrO
2
, HCl, MgO, K
2
O, pierwiastki ziem rzadkich.
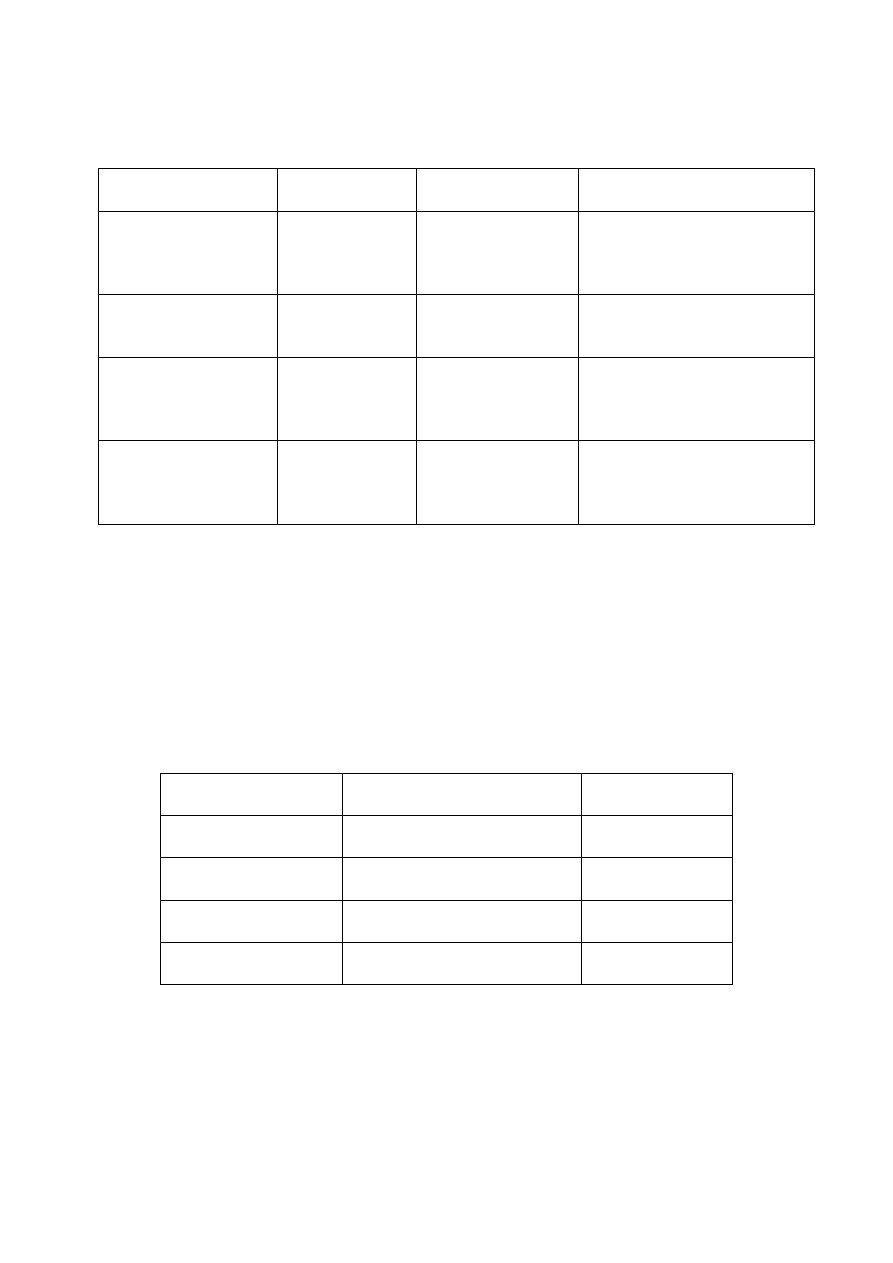
Tabela przedstawia klasyfikację składników aktywnych katalizatora.
SKŁADNIK
AKTYWNY
TYP REAKCJI
REAKCJE
PRZYKŁADY
Metale
(przewodniki)
Utlenienia-
redukcji
Uwodornienie
Hydrogenoliza
Utlenienie
-metale grup I,VI, VII, VIII
- Fe, Ni, Pt, Cu, Ag
Tlenki i siarczki
metali
(półprzewodniki)
Utlenienia-
redukcji
Uwodornienie
Hydrogenoliza
Utlenienie
- NiO, ZnO, CuO
- MoS
2
, WS
2
, CoS
2
Tlenki (izolatory)
Kwasowo-
zasadowa
Izomeryzacja
Alkilowanie
Kraking
dehydratacja
-glinokrzemiany
-zeolity
-Al.
2
O
3
, SiO
2
, MgO
Kompleksy metali
Mechanizm
koordynacyjny
Polimeryzacja
Karbonylowanie
hydroformylowanie
- katalizatory Zieglera-Natty
-karbonylki
Większość katalizatorów to układy metal(tlenek metalu)-nośnik. Najczęściej stosuje się
substancje o dobrze rozwiniętej powierzchni właściwej, które warunkują wysoką dyspersję
składników aktywnych, a co za tym idzie odpowiednią liczbę centrów aktywnych
odpowiedzialnych za przebieg reakcji.
Funkcją nośnika jest stabilizacja powierzchni, na której zdyspegrowany jest składnik
aktywny, w taki sposób, aby nie następowało łączenie krystalitów w aglomeraty. Nośnik
charakteryzuje się wysoką temperaturą topnienia, gdyż katalizatory stosuje się najczęściej w
wysokich temperaturach, w których krystality są bardziej ruchliwe, zderzają się ze sobą i
łączą w aglomeraty.
TYP
TLENEK
TEMP.
TOPNIENIA şC
Zasadowe
MgO,
CaO
3073
2853
Amfoteryczne
á-Al
2
O
3
,
TiO
2
2318
2113
Obojętne
MgAl
2
O
4
,
CaSiO
3
2408
1813
Kwasowe
ă
-Al
2
O
3
,
SiO
2
-Al
2
O
3
2318
1818
Promotory dodawane są często w małych ilościach i powodują wzrost aktywności i/lub
selektywności. Jednym z ważnych zadań promotora jest też kontrola stabilności układu
katalitycznego. W przypadku γ-Al
2
O
3
, ogrzewanie do temperatur powyżej 900 ºC prowadzi
do przejścia fazowego tlenku w formę o małej powierzchni - α-Al
2
O
3
. W tak wysokich
temperaturach zazwyczaj nie prowadzi się reakcji ale często regenerację katalizatora. Dodatek
promotora w postaci 1-2% wag. SiO
2
lub ZrO
2
podnosi temperaturę przejścia fazowego.

Etapy preparatyki katalizatora heterogenicznego.
Katalizatory i nośniki produkowane są w postaci proszków, a następnie w procesie
technologicznym przekształcane w różne formy, których kształt i wielkość zależą od
zastosowania. Najczęściej otrzymuje się tabletki, kulki, granulki, proszki oraz wytłoczki w
formie cylindrów, pierścieni, rurek i bardziej wyrafinowanych kształtów. Im większe cząstki
katalizatora, tym tańszy jest katalizator.
Ważnymcelem podczas produkcji katalizatorów jest uzyskanie jednolitego przepływu
reagentów, niewielkiego spadku ciśnienia i ograniczenie efektów dyfuzyjnych oraz odporność
cząstek katalizatora na mechaniczne kruszenie.
Znanych jest wiele sposobów formowania nośników i katalizatorów.Mają one na celu nadanie
katalizatorom odpowiednich kształtów i wytrzymałości mechanicznej. Jedną z najprostszych
metod jest formowanie przy użyciu tabletkarek. Szeroko rozpowszechnioną metodą jest
metoda wytłaczania przy pomocy specjalnych urządzeń mechanicznych (wytłaczarki
ś
limakowe). Po wysuszeniu i kalcynacji wytłoczki kruszone są do odpowiedniej długości;
stosownie do średnicy ziaren i wymagań technologicznych. W metodzie tej jako substancję
wiążącą stosuje się najczęściej rozcieńczone kwasy (azotowy, solny); w zależności od
stężenia roztworów można modyfikować strukturę nośnika. W procesie formowania stosuje
się także m.in. różnego rodzaju związki organiczne takie jak substancje powierzchniowo
czynne, żelatyna, polialkohole. Katalizatory i nośniki po procesie formowania poddaje się
obróbce termicznej, następuje suszenie i kalcynacja.
METODY NANOSZENIA SKŁADNIKÓW AKTYWNYCH NA NOŚNIK
1.
IMPREGNACJA
Metoda ta polega na wypełnieniu porów nośnika roztworem soli metalu aktywnego o
właściwym stężeniu dla określonego pokrycia powierzchni. W pierwszym etapie nośnik, w
formie wytłoczek lub proszku, jest suszony celu usunięcia wilgoci z porów, a następnie
wprowadzany jest roztwór soli w ilości wystarczającej do wypełnienia porów i zwilżenia
zewnętrznej powierzchni cząstek. Ilość roztworu można obliczyć z pomiarów chłonności
nośnika.
Rozkład składników aktywnych zależy od oddziaływania metali aktywnych z powierzchnia
nośnika; im to oddziaływanie jest silniejsze tym bardziej nierównomierny jest rozkład
składnika aktywnego na powierzchni nośnika. Szybkość osadzania się soli metali w porach
nośnika zależy od współczynnika dyfuzji. W preparatyce katalizatora tą metodą stosuje się
dwukrotną obróbkę termiczną (po impregnacji prowadzi się suszenie i kalcynację).
2.
STRĄCANIE
Strącanie polega na reakcji pomiędzy roztworem soli metalu (azotan, siarczan, chlorek) a
nośnikiem wobec dodatku alkalicznego roztworu np. KOH, NaOH, Na
2
CO
3
. W wyniku
reakcji otrzymuje się wodorotlenek lub węglan metalu na nośniku. Otrzymany proszek
(katalizator) odsącza się i przemywa w celu usunięcia jonów alkalicznych, anionów i
nadmiaru soli na zewnętrznej powierzchni cząstek nośnika. Tutaj również stosuje się
dwukrotną obróbkę termiczną - suszenie i kalcynacja. Strącanie jest metodą osadzania
preferowaną dla pokrycia wyższego niż 10-20 % wag.
3.
WYMIANA JONOWA

Jony o niższej wartościowości, takie jak Na
+
, wymieniają się z jonami o wyższym ładunku,
np. Ni
2+
, zgodnie z równaniem:
2PNa
+
+ Ni
2+
= Pni
2+
+ 2Na
+
Metodę tę powszechnie stosuje się przy modyfikacji zeolitów. Zeolity najczęściej produkuje
się w formie sodowej. Kationy sodu są łatwo wymienialne na wszystkie inne kationy, także
NH
4+
, z którego następnie przez aktywację otrzymuje się H
+
.
Po etapie otrzymywania katalizatorów prowadzi się procesy obróbki termicznej (suszenie i
kalcynacja). Dokonuje się tego po etapie nanoszenia składnika aktywnego oraz po procesie
formowania.
Suszenie prowadzi się w celu wykrystalizowania soli na powierzchni porów. Jeśli suszenie
nie jest przeprowadzone w sposób właściwy, może doprowadzić do nierównomiernego
rozłożenia składników aktywnych. Gdy jest zbyt wolne, może doprowadzić do osadzenia
substancji w dolnej lub środkowej części porów, jeśli zbyt szybkie - odparowanie może
doprowadzić do migracji soli na zewnątrz porów. Warunki suszenia dobiera się
eksperymentalnie w zależności od typu katalizatora. W czasie suszenia zachodzi także
usunięcie fizycznie związanej wody.
Kalcynacja. Wykrystalizowana sól może ponownie ulec rozpuszczeniu, jeśli jest wystawiona
na działanie wilgoci. Kalcynacja lub wygrzewanie w atmosferze redukcyjnej przekształca sól
w tlenek lub metal i ostatecznie decyduje o rozkładzie składnika aktywnego na nośniku.
Procesy zachodzące podczas kalcynacji polegają głównie na rozkładzie prekursorów.
Ostatnim etapem w produkcji naniesionych składników aktywnych jest aktywacja. Jeśli sam
tlenek jest składnikiem aktywnym, aktywacja nie jest potrzebna. Jeśli fazą aktywną w danej
reakcji są metale lub siarczki metali prowadzi się odpowiednio redukcję lud nasiarczanie.
BIOKATALIZATORY
Bardzo interesującą grupą katalizatorów są biokatalizatory, czyli enzymy. Enzymy są to
białka zdolne do katalizowania reakcji chemicznych. Miejscem gdzie zachodzi reakcja jest
centrum aktywne powstałe przez odpowiednie pofałdowanie łańcucha polipeptydowego.
Wiele enzymów należy do białek złożonych, w skład których oprócz części białkowej
wchodzi grupa prostetyczna. Grupę prostetyczną może stanowić:
a. kation metalu,
b. polisacharyd,
c. lipid
d. porfiryna.
W niektórych enzymach grupa prostetyczna związana jest w sposób odwracalny i wówczas
część białkowa nosi nazwę apoenzymu, a grupa prostetyczna – koenzymu.
Ź
ródłem enzymów mogą być mikroorganizmy (bakterie, grzyby mikroskopowe), komórki
roślinne oraz zwierzęce. Reakcje z wykorzystaniem enzymów jako katalizatorów można
prowadzić: z wydzielonym i oczyszczonym enzymem, jak również z naniesionym na stały
nośnik – mówimy wtedy o enzymie immobilizowanym. Trzecim sposobem prowadzenia

reakcji enzymatycznej jest wykorzystanie całych komórek mikroorganizmów. Naturalnym
ś
rodowiskiem, w którym znajdują się enzymy w organizmach żywych jest woda. Z tego też
powodu reakcje enzymatyczne należałoby prowadzić właśnie w roztworze wodnym, w
temperaturze około 20-40°C, i pH około 5,5-8. Wydzielenie enzymu a także jego
immobilizacja na stałym nośniku umożliwia przeprowadzanie reakcji w rozpuszczalnikach
organicznych, w temperaturze poniżej 20°C oraz powyżej 40°C, a także w innych zakresach
pH. Możliwość prowadzenia reakcji w rozpuszczalnikach organicznych zwiększyła zakres
zastosowania ich w reakcjach chemicznych oraz możliwość wielokrotnego wykorzystania.
Zastosowanie biotransformacji (reakcji enzymatycznych z wykorzystaniem czystych
enzymów lub całych komórek) umożliwiło prowadzenie wielu reakcji w łagodnych
warunkach jak również dało szanse na otrzymanie wielu związków niemożliwych do
otrzymania na drodze chemicznej. Każdy katalizator, więc również enzym działa w ten
sposób, że obniża energię aktywacji reakcji, nie wpływając na różnicę jej potencjału
termodynamicznego.
Enzym
zwykle
prowadzi
reakcję
przez
szereg
etapów,
charakteryzujących się stanami przejściowymi o niższych energiach. Efekty katalityczne
osiągane w reakcjach enzymatycznych są bardzo duże, reakcja jest przyspieszana od 109 do
1015 razy w stosunku do odpowiedniej niekatalizowanej reakcji chemicznej. Średnio około
1000 moli substratu reaguje z molem enzymu w ciągu jednej minuty, przy czym niektóre
enzymy osiągają wartość 107 moli substratu na mol enzymu.
W procesie katalizy enzymatycznej możemy wyróżnić:
•
procesy fizyczne takie jak dyfuzję substratu do centrum aktywnego enzymu oraz
uwolnienie produktu z centrum aktywnego.
•
proces chemiczny, czyli reakcję zachodzącą w centrum aktywnym enzymu prowadzącą do
otrzymania produktu.
Przyjmuje się, że reakcja enzymatyczna zachodzi według jednego z czterech poniższych
mechanizmów lub ich kombinacji. Tak więc, przyspieszenie reakcji enzymatycznej dokonuje
się poprzez:
1.
Katalizę przez zbliżenie reagujących cząsteczek
2.
Katalizę przez utworzenie kowalencyjnych produktów pośrednich z enzymem
3.
Katalizę kwasowo-zasadową
4.
Katalizę przez odpowiednią deformację cząsteczki substratu
Enzymy ze względu na katalizowane przez nie reakcje zostały podzielone na sześć klas:
1. Oksyreduktazy – katalizujące reakcje utleniania i redukcji.
2. Transferazy – katalizujące reakcje przenoszenia grup funkcyjnych z cząsteczki donora do
cząsteczki akceptora.
3. Hydrolazy – specjalna grupa transferaz, która katalizuje przenoszenie grupy funkcyjnej z
cząsteczki donora do cząsteczki wody; enzymy te katalizują hydrolizę wiązań estrowych,
eterowych, glikozydowych, amidowych itp.
4. Liazy – katalizują reakcję addycji wody, amoniaku lub dwutlenku węgla do wiązań
podwójnych lub reakcje odwrotne.

5. Izomerazy – katalizują wzajemne przekształcanie jednych izomerów w drugie (izomerów
optycznych, konstytucyjnych, geometrycznych, enolu w keton itp.).
6. Ligazy – katalizują reakcję łączenia dwóch substratów, w wyniku czego powstają wiązania
C – O, C – S, C – N, C – C.
KATALIZA MIĘDZYFAZOWA
Kataliza przeniesienia międzyfazowego (PTC, ang. Phase Transfer Catalysis) - technika ta
polega na prowadzeniu reakcji w układzie dwufazowym: ciecz-ciecz, lub ciało stałe-ciecz, w
której odpowiednie katalizatory poprzez tworzenie lipofilowej pary jonowej zapewniają
transport jednego z reagentów z fazy wodnej lub stałej do fazy organicznej.
Reakcje chemiczne zachodzące pomiędzy reagentami znajdującymi się w dwóch nie
mieszających się fazach są często hamowane z powodu braku kontaktu. Konwencjonalne
sposoby rozwiązania tego problemu polegają na użyciu intensywnego mieszania lub
zastosowaniu odpowiednich rozpuszczalników, w których oba reagenty się rozpuszczają.
Jeżeli reakcja przebiega na granicy faz, można się spodziewać, że szybkie mieszanie
zwiększy powierzchnię kontaktu faz, a tym samym przyśpieszy reakcję.
Rozpuszczalniki, w których w pewnym zakresie rozpuszczają się równocześnie związki
organiczne o małej polarności, takie jak halogenki alkilowe i sole nieorganiczne, np. cyjanek
potasu, są rozpuszczalnikami protonowymi wśród których wyróżnić można: metanol, etanol
i ich mieszaniny z wodą, lub też dipolarnymi, aprotonowymi takimi jak acetonitryl,
sulfotlenek dimetylowy lub dimetyloformamid. Jakkolwiek użycie takich rozpuszczalników w
pewnym zakresie może rozwiązywać problemy związane z rozpuszczalnością substratów, to
jednak ma wiele wad. W rozpuszczalnikach protonowych obserwuje się często konkurencyjną
hydrolizę i obniżenie aktywności nukleofili na skutek solwatacji, natomiast rozpuszczalniki
aprotonowe są często toksyczne, drogie i trudno z nich wydzielić produkt.
Pierwsze doniesienia na temat katalizy przeniesienia międzyfazowego pochodzą z roku 1965,
aczkolwiek rozwój tej techniki syntezy datuje się od roku 1971, kiedy to Starks, Mąkosza i
Brändstöm przedstawili jej fundamenty.
Technika ta oferuje wiele znaczących korzyści takich jak:
- przyspieszenie tempa reakcji w łagodnych warunkach,
- użycie niedrogich i łatwo dostępnych katalizatorów,
- wykorzystanie tanich, nietoksycznych i odzyskiwalnych rozpuszczalników,
- możliwość użycia tanich zasad nieorganicznych, służących do generowania anionów,
- możliwość prowadzenia reakcji bez używania rozpuszczalnika,
- polepszenie wydajności i enancjoselektywności produktu,
- możliwość prowadzenia reakcji na dużą skalę.
Zasada działania PTC polega na ciągłym generowaniu par jonowych składających się z
reagującego anionu organicznego lub nieorganicznego oraz lipofilowego kationu,
takiego jak amoniowy, fosfoniowy lub kationu metalu alkalicznego skompleksowanego
eterem koronowym. Aniony mogą być dostępne w postaci soli sodowych lub potasowych,
ewentualnie należy je wytworzyć poprzez działanie odpowiedniej zasady – najczęściej KOH.

PTC jest szczególnie skuteczną metodą syntezy różnych związków na drodze substytucji
nukleofilowej, w wyniku czego otrzymuje się halogenoalkany, nitryle, azydki, aminokwasy i
wiele innych.
Klasyczną katalizę przeniesienia fazowego można podzielić na: katalizę w układzie ciecz-
ciecz (LL-PTC) oraz na katalizę w układzie ciało stałe–ciecz (SL-PTC). Fazą ciekłą może być
rozpuszczalnik bądź ciekłe substraty. Faza stała lub wodna jest z kolei źródłem reagujących
anionów organicznych bądź nieorganicznych, albo też zasady.
CZĘŚĆ DOŚWIADCZALNA
Ćwiczenie 1. Otrzymywanie Al(OH)
3
i Al
2
O
3
Sprzęt laboratoryjny:
Wkraplacze o pojemności 250 ml, statyw laboratoryjny, zlewka o poj. 500 ml,
Wykonanie ćwiczenia:
Na statywie zamocować dwa wkraplacze o pojemności około 250 ml tak aby wypływające z
nich ciecze łączyły się w powietrzu w jedną strugę. Pod wkraplaczami umieszcza się na
kuchence elektrycznej zlewkę o pojemności 500 ml zawierającą 100 ml wody destylowanej.
W jednym wkraplaczu umieszcza się 150 ml 15 % roztworu AlCl
3
, w drugim 25 % roztwór
wody amoniakalnej. Następnie z obu wkraplaczy dozuje się równomiernie w/w roztwory.
Natężenie strumieni należy tak wyregulować aby pH roztworu było równe 6; pH roztworu
kontroluje się stosując papierki wskaźnikowe. Otrzymany żel wodorotlenku glinu sączy się na
gorąco na lejku Büchnera, a następnie przemywa się gorącą wodą destylowaną aż do
wymycia jonów chlorkowych co kontroluje się stosując azotan srebra (AgNO
3
). Przemyty
osad wodorotlenku glinu przenosi się z lejka na tackę porcelanową i suszy w temperaturze
110 ºC przez 12 godzin. Wysuszony osad rozciera się w moździerzu porcelanowym i
przesiewa przez sito 0,3 mm.
Formowanie nośnika
Wysuszony wodorotlenek glinu zarabia się na pastę za pomocą 3 % kwasu azotowego. W tym
celu należy odważyć na wadze laboratoryjnej przesiany i wysuszony wodorotlenek glinu i
umieścić w moździerzu porcelanowym. Następnie wkraplać powoli 3 % HNO3(V) z biurety i
zarabiać aż do momentu uzyskania plastycznej masy. Odczytać ilość zużytego HNO
3
(V).
Otrzymaną plastyczną masę rozprowadza się na płytce ebonitowej. Otrzymane wytłoczki
suszy się początkowo w temperaturze pokojowej przez 24 godziny. Następnie wytłoczki
suszy się w suszarce: 30, 50 i 70 ºC (po 0,5 h) i 110 ºC (24 h). Kalcynację prowadzi się w
następującym reżimie temperaturowym: 200, 300 i 400ºC (po 1 h) i 450 ºC (3 h).
Sporządzanie katalizatora niklowego (3 %mas. NiO/Al2O3)
Katalizator niklowy sporządza się stosując metodę suchej impregnacji nośnika wodnym
roztworem azotanu niklu ). Ilość roztworu azotanu potrzebnego do impregnacji nośnika
określa się na podstawie oznaczenia chłonności nośnika.
W celu oznaczenia chłonności odważa się dwie próbki po około 1 g uformowanego nośnika i
zalewa wodą destylowaną w naczynkach wagowych. Po 20 minutach zlewa się wodę z
naczynek i przesypuje nośnik na bibułę. Nadmiar wody odprowadza się przez mokrą bibułę
nakładają na nią kolejne suche bibuły. Suchą bibułę zmienia się do czasu aż pozostaną na niej
pojedyncze mokre ślady. Następnie nośnik przenosi się na szkiełko zegarkowe i waży.
Chłonność uformowanego nośnika oblicza się wg wzoru:
Chłonność= przyrost masy/naważka
Ilość wody potrzebnej do sporządzenia roztworu azotanu niklawego oblicza się mnożąc przez
masę nośnika jaką poddajemy impregnacji i dodając 10 % więcej wody niż to wynika z
obliczonej chłonności.
Ćwiczenie 2. Otrzymywanie katalizatora przeniesienia międzyfazowego – chlorek
trietylobenzyloamoniowy oraz wykorzystanie zsyntezowanego produktu w reakcji.
CZĘŚĆ I
Aparatura i szkło:
1. kolba okrągłodenna, jednoszyjna poj. 250 cm3,
2. chłodnica zwrotna,
3. cylinder pomiarowy poj. 50 cm3,
4. kosz grzejny.
5. lejek szklany ze spiekiem
6. kolba ssawkowa
7. szalka Petry’ego
Surowce i odczynniki:
1. chlorek benzylu, M = 126,5,
2. trietyloamina, M = 79, d = 0,978,
3. alkohol butylowy
4. 2-propanol
5. eter dietylowy
Cl
N
N
Cl
+
+
-
Sposób wykonania ćwiczenia:
W kolbie kulistej zaopatrzonej w chłodnicę zwrotną i rurkę w chlorkiem wapniowym
umieszcza się 5g (6,9 ml, 0,05mola) trietyloaminy, 8,2 g (7,5 ml, 0,065 mola) chlorku
benzylu oraz 4 ml mieszaniny 2-propanolu i 1-butanolu w stosunku objętościowym 1:1.
Zawartość kolby ogrzewa się przez 1 godz. utrzymując mieszaninę w temperaturze wrzenia.
Po ochłodzeniu lotną frakcję odparowuje się na wyparce obrotowej, a pozostałość rozdrabnia
się, zadaje 20 ml eteru dietylowego, zamyka szczelnie korkiem i wstawia na pół godz. do
zamrażalnika. Wytrąconą sól diazoniową odsącza się na lejku Buchnera i przemywa
trzykrotnie zimnym eterem dietylowym. Osad suszy się na wyparce rotacyjnej w temp. 30ºC.
Otrzymuje się produkt z wydajnością około 70% w postaci białych, drobnych kryształków.
Produkt należy przechowywać w szczelnie zamkniętym naczyniu ze względu na znaczną
higroskopijność.
Zagadnienia:
Mechanizm reakcji trietyloaminy z chlorkiem benzylu
Jaki wpływ na przebieg reakcji wywierają dodawane alkohole?
Jaką rolę spełnia eter dietylowy?
CZĘŚĆ II
Synteza
N-benzyloftalimidu
w
warunkach
bezrozpuszczalnikowej
katalizy
międzyfazowej.
Odczynniki i sprzęt laboratoryjny:
Chlorek benzylu, DMF, K
2
CO
3
, ftalimid, glikol polietylenowy-400, reaktor mikrofalowy
NH
O
O
Cl
N
O
O
+
+ HCl
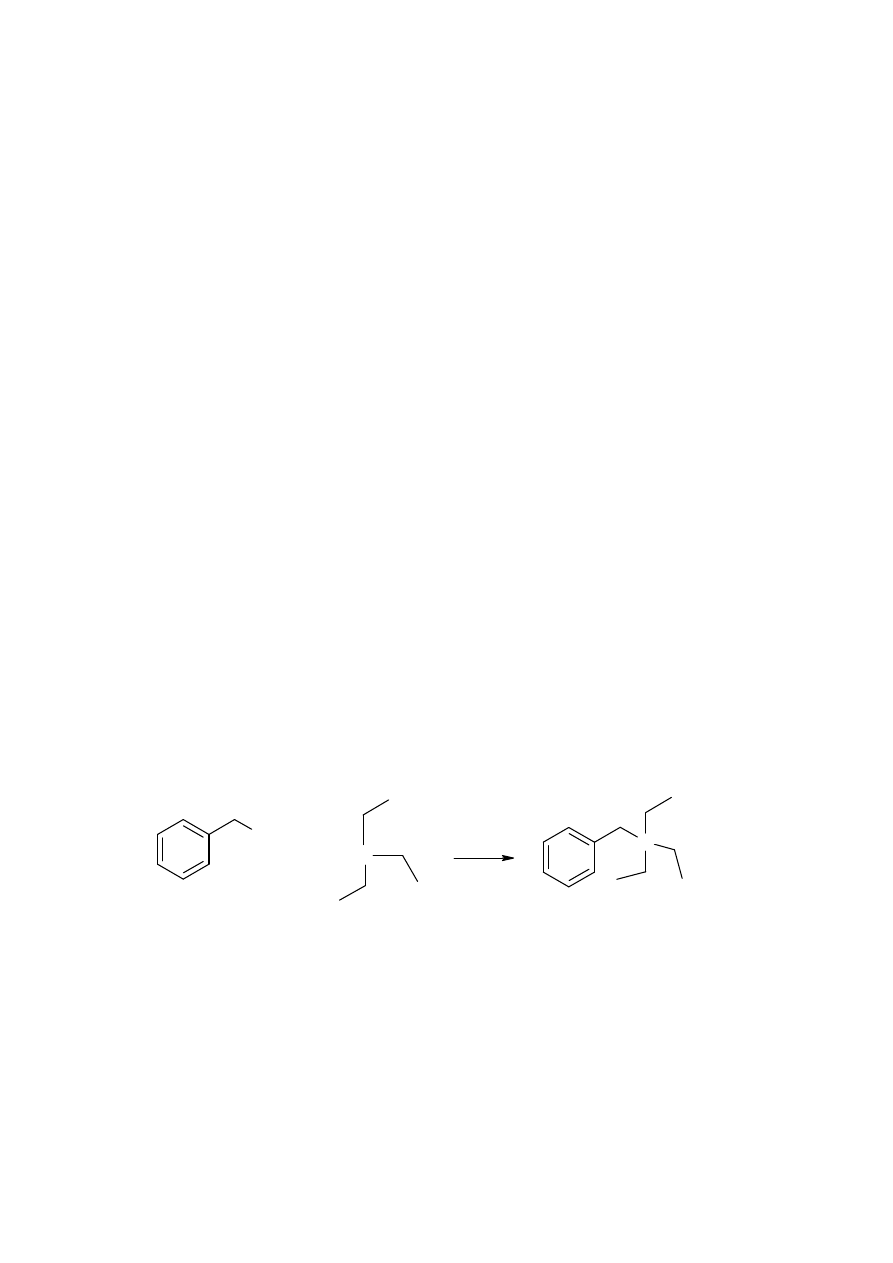
W moździerzu utrzeć 1.47g ftalimidu (10mmol), 4,14 g bezwodnego K
2
CO
3
(30mmol), 0,32g
TEBA do uzyskania pudrowej konsystencji. Dodać 1,2 ekwiwalentu chlorku benzylu i
pozostawić reakcję na 15 min. w temperaturze pokojowej. W tym czasie obserwuje się
wzrost temp. do 60ºC (reakcja egzotermiczna). Po oziębieniu do temp. otoczenia dodać 100
ml wody destylowanej i odsączyć surowy produkt. Biały produkt suszyć w piekarniku (105 º
C) przez 2 godziny, po oziębieniu zbadać temperaturę topnienia otrzymanego związku.
Wykonać widma w podczerwieni otrzymanego produktu i porównać z widmem wzorca.
W drugim naczyniu wykonać podobną reakcję ale bez dodatku TEBA. Porównać wydajność
otrzymanych produktów.
Ćwiczenie 3. Otrzymywanie acetyloactanu niklu (II)
Aparatura:
3 zlewki 250 ml, 1 kolba 100 ml, cylinder 100 ml, pipeta 10ml, krystalizator, łyżeczka,
mieszadło, płyta grzewcza, zestaw do sączenia pod próżnią, wyparka, waga analityczna.
Odczynniki:
Chlorek niklu (NiCl
2
· 6H
2
O), chlorek kobaltu (CoCl
2
· 6H
2
O), acetyloaceton, metanol, octan
sodu, woda destylowana, lod.
Wykonanie ćwiczenia
Do zlewki o pojemności 250 ml dodać 50 ml wody destylowanej a następnie 0.05 mola NiCl
2
·
6H
2
O,a następnie mieszać na płytce grzewczej z mieszadłem aż do rozpuszczenia (roztwór
A). w drugiej zlewce o pojemności 250 ml rozpuścić 0.1 mola acetyloacetonu w 20 ml
metanolu i dodać do roztworu A, ciągle mieszając (roztwor B). W kolbie o pojemności 250
ml rozpuścić 0.1 mola octanu sodu w 30 ml wody destylowanej i dodać do roztworu B, ciągle
mieszając (roztwor C). Mając przygotowany roztwor C należy go szybko podgrzać, a
następnie schłodzić w łaźni lodowej przez ok. 1 godzinę. Schłodzony roztwór C umieścić w
zestawie do sączenia pod próżnią. W trakcie sączenia (po uprzednim wyłączeniu pompy)
należy produkt przemyć kilka razy wodą destylowaną. Następnie umieścić proszek w kolbce i
suszyć w wyparce przez ok. 1 godzinę. Wysuszony katalizator zważyć.
Analogiczne operacje stosuje się w przypadku preparatyki katalizatora kobaltowego,
zastępując sól niklu solą kobaltu CoCl
2
· 6H
2
O.
Opracowanie wyników
Powstały katalizator należy zważyć i obliczyć jego wydajność.
Ćwiczenie 4a. Otrzymywanie niklu Raneya
Aparatura:
2 zlewki 250 ml, 1 zlewka 100 ml, cylinder 100 ml, mieszadło magnetyczne, płyta grzewcza,
łaźnia wodna.
Odczynniki:
Wodorotlenek sodu NaOH, stop Raneya, woda destylowana, lod.
Do intensywnie mieszanego roztworu 19g NaOH w 75 ml wody, umieszczonego w zlewce o
poj 200 ml i oziębionego do temp. 10 ºC w lodzie, dodaje się małymi porcjami 15 g stopu
Raneya z taką szybkością, aby temperatura mieszaniny nie wzrosła powyżej 25 ºC. Po
dodaniu całości pozostawia się mieszaninę do osiągnięcia temp. pokojowej. Gdy zmniejszy
się szybkość wydzielania wodoru, ogrzewa się zlewkę na łaźni wodnej aż do całkowitego
ustania wydzielania pęcherzyków gazu. Objętość cieczy utrzymuje się na stałym poziomie
dodając wody destylowanej. Następnie pozostawia się katalizator do odstania , po czym ciecz
dekantuje się, a katalizator przemywa się przez dekantację i przenosi do zlewki opoj. 100 ml.
Wodę zlewa się i dodaje roztwór 2,5 g wodorotlenku sodowego w 25 ml wody. Po dokładnym
wymieszaniu pozostawia się do odstania, po czym zlewa zasadę. Katalizator przemywa się
przez dekantację wodą do odczynu obojętnego. Przemywanie kontynuuje się stosując 3 razy
po 10 ml 95% etanolu oraz trzy razy za pomocą absolutnego etanolu. Silnie piroforyczny
katalizator przechowuje się w naczyniu napełnionym po brzegi absolutnym etanolem w
lodówce.
Ćwiczenie 4b.
Redukcja nitroakrydonu do imidazoakrydonu
z wykorzystaniem niklu
Raneya
N
H
O
Cl
NO
2
N
O
Cl
N
Al/Ni
HCOOH

Mieszaninę 1-chloro-4-nitroacrydonu ( 2 mmol), 3g katalizatora Raneya oraz 30 ml 96%
kwasu mrówkowego ogrzewać do wrzenia z intensywnym mieszaniem roztworu przez 2
godziny. Po zakończeniu reakcji dodać 100 ml metanolu, odsączyć katalizator oraz sole
nieorganiczne powstałe w czasie reakcji. Rozpuszczalnik (metanol) odparować na wyparce
obrotowej. Pozostałość po odparowaniu rozpuścić w roztworze metanolu z 1%dodatkiem
kwasu metasulfonowego, dodać węgiel aktywny i ogrzewać przez kilka minut. Odsączyć
węgiel aktywny, a przesącz odparować. Następnie dodać aceton lub eter dietylowy w celu
wytrącenia produktu. Czystość otrzymanego związku sprawdzić metodą
TLC w układzie
:
Ćwiczenie 5. Otrzymywanie dichlorobis(trifenylofosfina)nikiel – katalizator wielu
reakcji chemicznych, substrat do otrzymywania różnych kompleksów niklu.
Odczynniki:
Uwodniony chlorek niklu NiCl
2
· 6H
2
O, trifenylofosfina, eter dietylowy, etanol, 2-propanol
Wykonanie ćwiczenia:
W kolbie kulistej o poj 250 ml zaopatrzonej w chłodnicę zwrotną umieszcza się 2,8 g
trifenylofosfiny i 30 ml 2-propanolu. W kolbce stożkowej przygotowuje się roztwór NiCl
2
·
6H
2
O w 15 ml etanolu. Tak przygotowany roztwór wlewa się przez chłodnicę do wrzącego
roztworu trifenylofosfiny. Obserwuje się natychmiastowe wypadanie osadu produktu. Po
ochłodzeniu osad odsącza się na lejku Buchnera i przemywa trzykrotnie etanolem (porcje po
10 ml) a następnie zimnym eterem dietylowym. Osad suszy się na powietrzu. Wydajność
około 70%.
Zagadnienia
Jak powstaje wiązanie metal-fosfina?
Jakie może być zastosowanie tego kompleksu?
Ćwiczenie 6. Kataliza enzymatyczna: oksydaza, katalaza, peroksydaza
Cel ćwiczenia
Zapoznanie się z działaniem enzymów różnych klas (oksydazy, katalazy, peroksydazy) za
pomocą enzymów zawartych w wyciągu z ziemniaka.
Przebieg ćwiczenia
Przygotowanie wyciągu ziemniaczanego
Kilka ziemniaków umyć, obrać i utrzeć na tarce. Następnie włożyć je do płóciennego
woreczka i mocno ściskając ręką wycisnąć z nich sok. Kubek z sokiem ziemniaczanym
pozostawić na kilka minut w celu osadzenia się skrobi i resztek z ziemniaka.
Oksydazy
Korzystając z wyciągu z ziemniaka zbadać aktywność enzymów z klasy oksydaz. Sprawdzić,
które substancje mogą zostać rozłożone przez enzymy zawarte w wyciągu z ziemniaka i jak
zmienia się przebieg reakcji w czasie.

Wykonanie doświadczenia
Przygotować 3 probówki i dodać do każdej po 5ml wyciągu z ziemniaka. Do pierwszej
probówki dodać 10 kropli 1% fenolu, do drugiej 10 kropli 1% pirokatechiny, a do trzeciej 10
kropli 1% roztworu pirogalolu. Zawartość probówek zamieszać i zaobserwować zmiany
zabarwienia związków w czasie.
Katalaza
Katalaza katalizuje reakcję rozkłady nadtlenku wodoru do tlenu i wody. W tej części
ć
wiczenia korzystamy z 3% wody utlenionej i sprawdzamy jak enzymy zawarte w wyciągu z
ziemniaka działają w jej obecności.
Wykonanie doświadczenia
Do dwóch probówek dodać po 5ml wyciągu z ziemniaka. Do jednej z nich dodać 1ml 3%
wody utlenionej (H
2
O
2
) i obserwować zmiany. Drugą z probówek zagotować a następnie
również dodać 1ml 3% wody utlenionej.
Peroksydaza
Peroksydazy działają podobnie jak katalazy, jednakże nadtlenek wodoru nie jest rozkładany
lecz wykorzystywany w reakcjach utlenienia wielu substratów np. fenoli, amin
aromatycznych. W doświadczeniu sprawdza się wpływ nadtlenku wodoru na reakcje
katalizowane przez peroksydazy.
Wykonanie doświadczenia
Przygotować 3 probówki i dodać do każdej po 5ml wyciągu z ziemniaka. Do pierwszej
probówki dodać 10 kropli 1% fenolu, do drugiej 10 kropli 1% pirokatechiny, a do trzeciej 10
kropli 1% roztworu pirogalolu. Następnie do każdej z nich dodać po 10 kropli 3% nadtlenku
wodoru i zabserwować zmiany.
Literatura:
1. M. Ziółek, I. Nowak, „Kataliza heterogeniczna – wybrane zagadnienia”, Wyd. Naukowe,
1999.
2. B. Grzybowska-Świerkosz, „Elementy katalizy heterogenicznej”, PWN, Warszawa 1993.
3. H. Maciejewski, J. Guliński Ćwiczenia laboratoryjne z chemii nieorganicznej UAM
Poznań, 2003
4. J.T. Wróbel, Preparatyka i elementy syntezy organicznej PWN Warszawa 1983
5. T. Milk, „Kataliza i katalizatory”, Wyd. Szkolne i Pedagogiczne., 1975.
6. J.E. Germain, „Kataliza w układach niejednorodnych”, PWN, 1962.
7. J. Barcicki, „Podstawy katalizy heterogenicznej”, Wyd. UMCS Lublin ,1998.
8. P. Kafarski, B. Lejczak, „Chemia bioorganiczna” PWN Warszawa 1994
9. F.Pruchnik, "Kataliza homogeniczna", PWN,1993
10. B.Grzybowska-Świerkosz, "Elementy katalizy heterogenicznej" PWN 1993.
11. G.C.Bond, "Kataliza heterogeniczna – podstawy i zastosowania", PWN 1979

Załącznki:
Wzór pierwszej strony sprawozdania
POLITECHNIKA GDAŃSKA
WYDZIAŁ CHEMICZNY
KATEDRA TECHNOLOGII CHEMICZNEJ
SPRAWOZDANIE Z ĆWICZEŃ LABORATORYJNYCH
NAZWA ĆWICZENIA
PROWADZĄCY:
NAZWISKA OSÓB WYKONUJĄCYCH
Ć
WICZENIE:
1.
2.

3.
4.
5.
KIERUNEK STUDIÓW:
GRUPA:
DATA WYKONANIA ĆWICZENIA:
DATA ODDANIA SPRAWOZDANIA:
GDAŃSK ROK
Wyszukiwarka
Podobne podstrony:
Kraking katalityczny id 250043 Nieznany
Abolicja podatkowa id 50334 Nieznany (2)
4 LIDER MENEDZER id 37733 Nieznany (2)
katechezy MB id 233498 Nieznany
metro sciaga id 296943 Nieznany
perf id 354744 Nieznany
interbase id 92028 Nieznany
Mbaku id 289860 Nieznany
Probiotyki antybiotyki id 66316 Nieznany
miedziowanie cz 2 id 113259 Nieznany
LTC1729 id 273494 Nieznany
D11B7AOver0400 id 130434 Nieznany
analiza ryzyka bio id 61320 Nieznany
pedagogika ogolna id 353595 Nieznany
Misc3 id 302777 Nieznany
cw med 5 id 122239 Nieznany
D20031152Lj id 130579 Nieznany
więcej podobnych podstron