1
3.
LUMINESCENCJA.
WYZNACZENIE
WYDAJNOŚCI
KWANTOWYCH
FLUORESCENCJI
1.
Wprowadzenie
Luminescencją nazywana jest każda emisja promieniowania, która nie jest świeceniem
ciał ogrzanych do wysokich temperatur. Luminescencja zachodzi z elektronowo -
oscylacyjnie wzbudzonej cząsteczki będącej w równowadze termicznej z otoczeniem. Różne
rodzaje luminescencji związane są z różnymi sposobami dostarczania energii wzbudzenia.
Fotoluminescencją nazywamy emisję atomów lub cząsteczek, które po zaabsorbowaniu
promieniowania ultrafioletowego, widzialnego lub podczerwonego o odpowiedniej energii
powracają do stanu podstawowego.
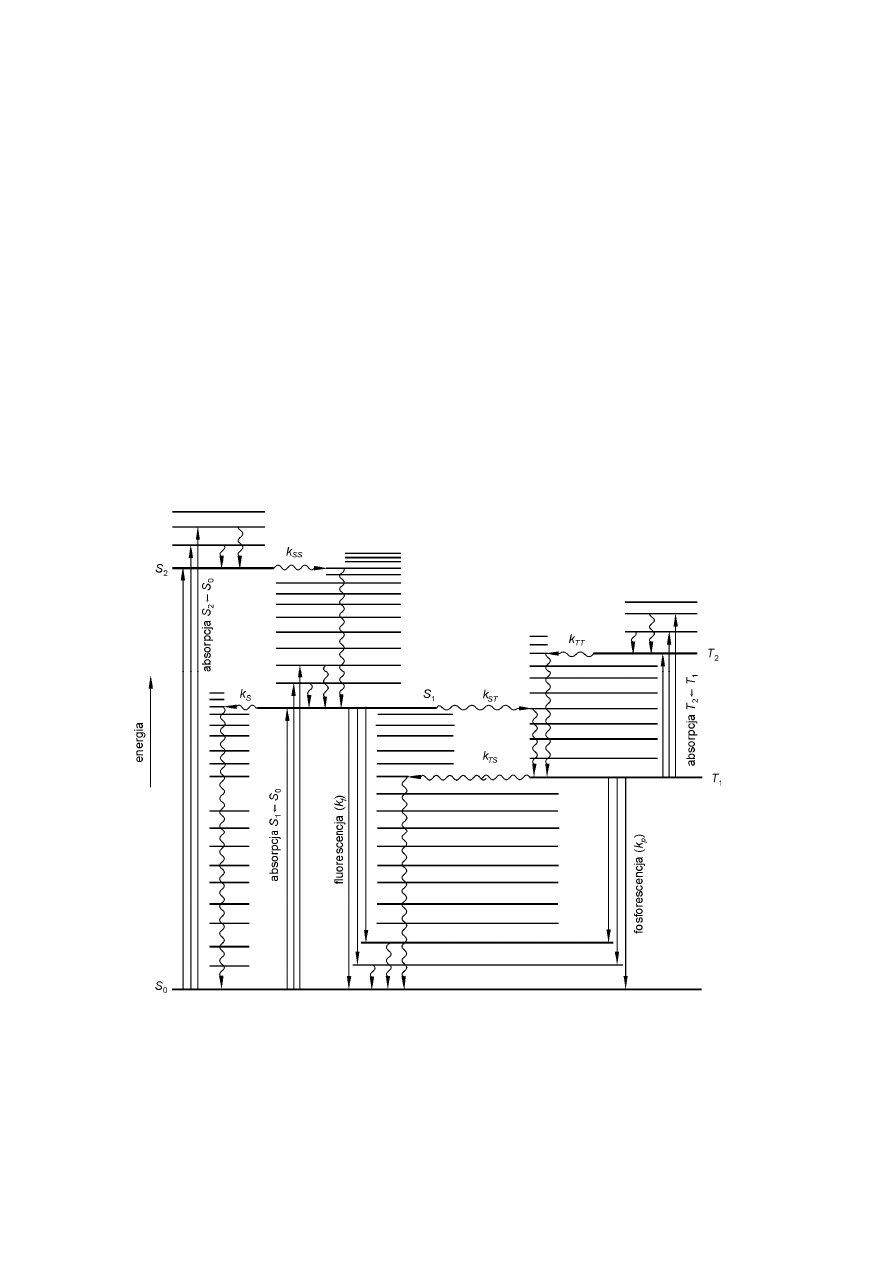
Schemat procesów fotofizycznych, związanych z absorpcją i emisją promieniowania w
cząsteczkach wieloatomowych o podstawowym stanie singletowym (S
0
), przedstawiono na
diagramie, nazywanym od nazwiska jego twórcy, diagramie Jabłońskiego (por. rys. 1).
Rys. 1. Diagram Jabłońskiego dla cząsteczki o singletowym podstawowym
stanie elektronowym. Strzałki proste oznaczają przejścia promieniste,
strzałki faliste – przejścia bezpromieniste
2
Na rys. 1 elektronowe przejścia promieniste zaznaczono strzałkami prostymi, elektronowe
przejścia bezpromieniste falistymi strzałkami poziomymi, procesy zaś relaksacji oscylacyjnej,
tj. utraty nadmiaru energii oscylacyjnej w danym stanie elektronowym, falistymi strzałkami
pionowymi. Literą k z odpowiednimi indeksami oznaczono stałe szybkości promienistych i
bezpromienistych przejść elektronowych i mają one wymiar [s
–1
].
Fotoluminescencja, która powstaje w wyniku przejścia z najniższego poziomu
oscylacyjnego wzbudzonego stanu elektronowego S
1
na dowolny oscylacyjno-rotacyjny
poziom stanu podstawowego S
0
. (por. przejście S
1
→
S
0
rys. 1) nazywana jest fluorescencją.
Bezpromieniste przejście elektronu z poziomu S
1
do metastabilnego poziomu trypletowego
T
1
powoduje pojawienie się emisji związanej z przejściem elektronowym T
1
→
S
0
, przesuniętej
ku dłuższym falom w stosunku do fluorescencji S
1
→
S
0
,
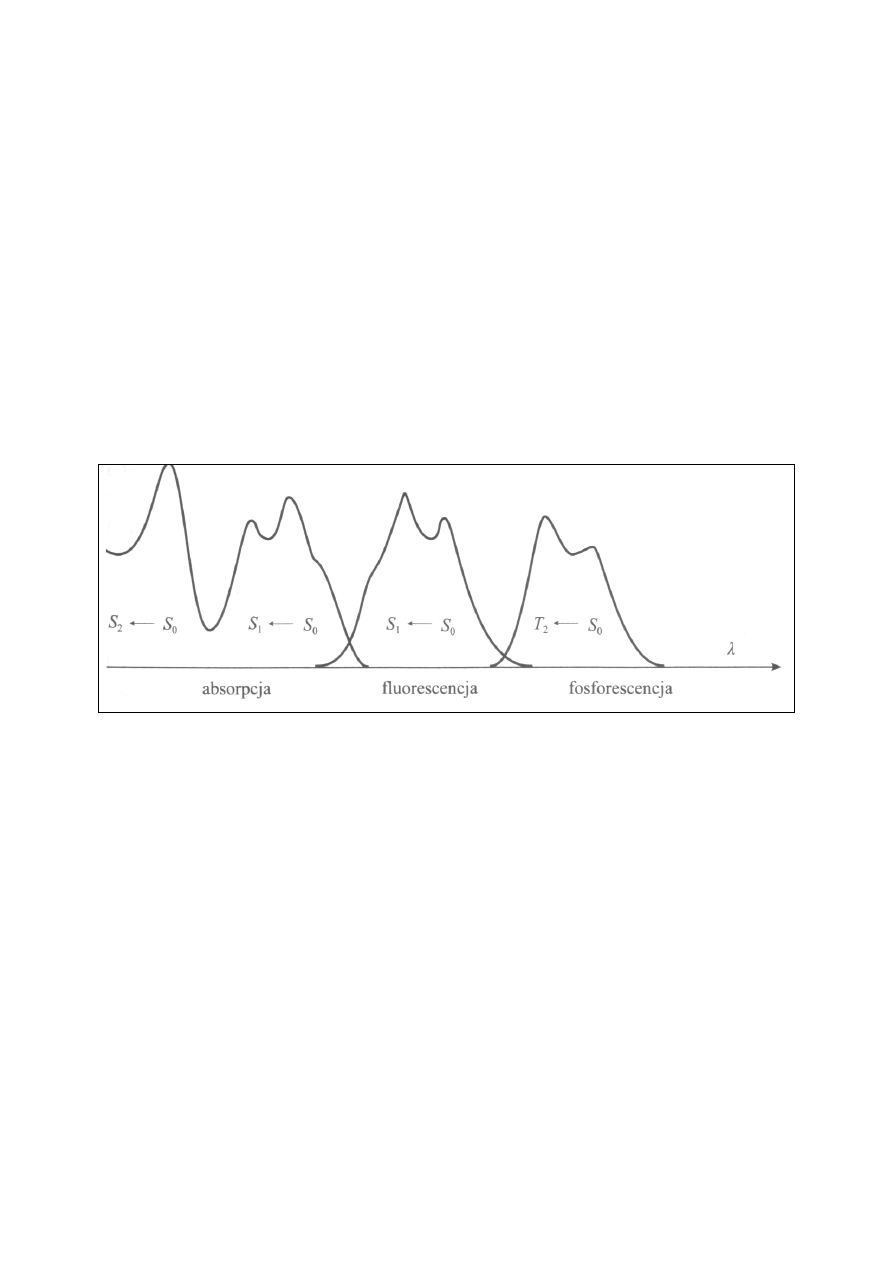
zwanej fosforescencją, (por. rys. 2).
Rys 2. Względne położenia widm: A – absorpcji, B – fluorescencji, C – fosforescencji.
Przejścia bezpromieniste pomiędzy stanami elektronowymi o jednakowej krotności
(S
2
~~
>
S
1
, T
2
~~
>
T
1
) nazywamy konwersją wewnętrznej, natomiast przejścia bezpromieniste,
które zachodzą pomiędzy stanami o różnej krotności (S
1
~~
>
T
1
i T
1
~~
>
S
0
na rys. 1)
nazywamy przejściami międzysystemowymi. Wymienione przejścia bezpromieniste następują
wówczas, gdy wielowymiarowe hiperpowierzchnie energii potencjalnej odpowiednich stanów
elektronowych dostatecznie zbliżają do siebie w otoczeniu pewnego punktu albo nawet
przecinają się ze sobą, a konwersja zarówno S
1
~~
>
S
0
(T
2
~~
>
T
1
),
jak i S
1
~~
>
T
1
(T
1
~~
>
S
0
)
zachodzą bez zmiany konfiguracji i pędów jąder. Przejście cząsteczki ze stanu S
1
na wysoki
poziom oscylacyjny stanu S
0
jest równoznaczne z zamianą jej energii wzbudzenia
elektronowego na energię oscylacji w stanie niewzbudzonym. Energia ta zostaje następnie
odprowadzona przez zderzenia z innymi cząsteczkami w szybkim, trwającym 10
–11
–10
–13
s,
procesie relaksacji oscylacyjnej, a więc ostatecznie zamieniona na energię kinetyczną

3
cząsteczek. Proces ten jest bardzo szybki i podobnie jak relaksacja oscylacyjna następuje po
wzbudzeniu cząsteczki do wyższych stanów wibronowych.
1.1. Prawa absorpcji. Prawo Bouguera-Lambert i Lamberta-Beera
W myśl prawa sformułowanego przez P. Bouguera J. H. Lamberta, zmniejszenie natężenia
monochromatycznej wiązki przechodzącej przez bardzo cienką warstwę jednorodnego
ośrodeka, złożonego z pochłaniających promieniowanie cząsteczek, jest proporcjonalne do
natężenia wiązki wchodzącej do tej warstwy i do grubości warstwy.
Natężeniem wiązki promieniowania (I) nazywamy energię przepływającą w jednostce
czasu przez jednostkowy przekrój wiązki, prostopadły do kierunku jej biegu. Wymiarem
natężenia promieniowania jest [W· m
–2
].
Gdy natężenie wiązki padającej prostopadle na warstwę elementarną o grubości dx
wynosi I, to:
dI
kIdx
− =
(1)
Oznaczając przez I
0
natężenie wiązki wchodzącej do absorbującej warstwy o skończonej
grubości d, natomiast przez I
p
natężenie, jakie ma ta wiązka po przejściu przez warstwę,
obliczamy rozdzielając zmienne w równaniu i całkując jego lewą stronę w granicach od I
= I
0
do I = I
p
, zaś prawą w granicach od x = 0 do x = d:
0
0
ln
ln
p
p
I
I
kd
I
I
−
=
=
,
0
e
kd
p
I
I
−
=
(2)
Współczynnik k w równaniach (2) nazywamy
naturalnym współczynnikiem absorpcji; ma
on wymiar [m
–1
]. Wartość k dla danej substancji jest zależna od długości fali absorbowanego
promieniowania i od parametrów określających gęstość absorbującej substancji (T, p).
Przechodząc z logarytmów naturalnych na dziesiętne, ze wzoru (2) otrzymujemy:
0
log
p
I
ad
I
=
(3)
Współczynnik a = k/2,303 nazywamy
współczynnikiem absorpcji. Zarówno równanie
różniczkowe (1), jak i jego scałkowane postacie (2,3) przedstawiają różne sposoby zapisu
prawa
Bouguera-Lamberta.
Badając absorpcję substancji rozpuszczonych w ciekłych rozpuszczalnikach, które same
pochłaniają promieniowanie w innym obszarze widma, prawdopodobieństwo, że foton
penetrujący warstwę roztworu napotka cząsteczkę, przez którą zostanie zaabsorbowany,
zależy od iloczynu grubości warstwy i stężenia roztworu. Oznaczając przez c stężenie
roztworu, wyrażone w mol
⋅
dm
–3
, zastępujemy k we wzorze (1) iloczynem 2,303
ε
c, w którym
4
współczynnik
ε
ma wymiar [cm
–1
·(mol·dm
–3
)
–1
] i nazywany jest
molowym współczynnikiem
absorpcji.
Otrzymujemy wówczas równanie wyrażające w postaci różniczkowej
prawo
Lamberta–Beera, a po scałkowaniu równanie to przybiera postać:
0
log
p
I
cd
A
I
ε
=
≡
,
0
0
10
10
cd
A
p
I
I
I
ε
−
−
=
=
(4)
Wyrażenie log (I
0
/I
p
) nazywamy
absorbancją i oznaczamy symbolem A.
Maksymalna wartość molowego współczynnika absorpcji,
ε
maks
, jest miarą
intensywności przejścia. Ponieważ pasmo absorpcji rozciąga się na pewien zakres liczb
falowych,
1
ν
λ
=
%
[cm
−
1
], podanie wartości
molowego współczynnika absorpcji tylko dla jednej
liczby falowej może nie być prawdziwym wskaźnikiem intensywności pasma.
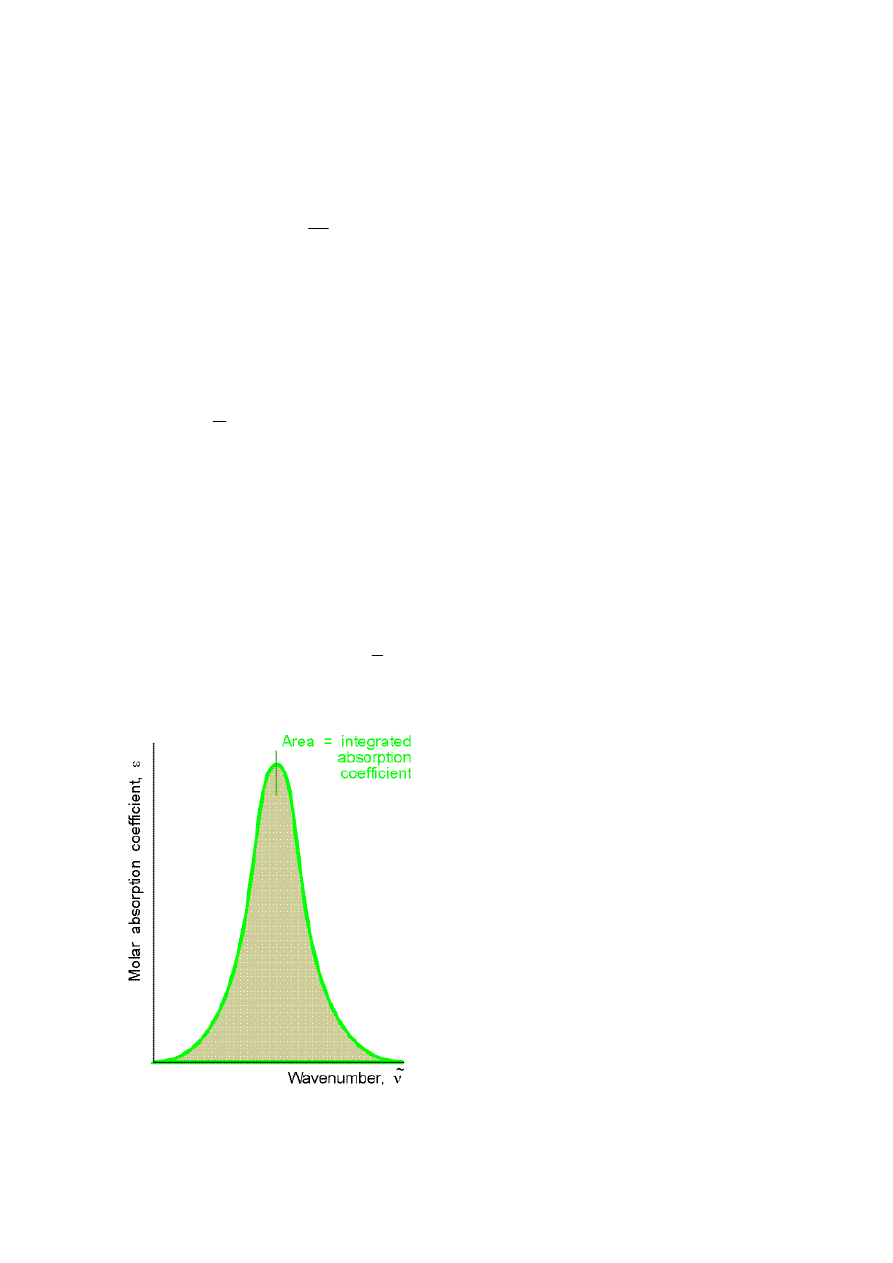
Integralny współczynnik absorpcji jest sumą (całką) współczynników absorpcji w
całym paśmie i odpowiada polu pod krzywą, obrazującą zależność molowego współczynnika
absorpcji od liczby falowej (por. rys. 3).
2
1
int
( ) d
ν
ν
ε
ε ν ν
=
∫
%
%
%
%
(5)
Rys. 3. Zależność molowego współczynnika absorpcji od liczby falowej promieniowania
zaabsorbowanego przez substancję.

5
Inną
wielkością
charakteryzującą
absorpcję
promieniowania
w
ośrodku
pochłaniającym jest stosunek
I
p
/
I
0
zwany
przepuszczalnością (transmitancją), podawany
także w procentach:
0
I
I
T
p
=
lub
100
%
0
I
I
T
p
=
(6)
Pomiędzy absorbancją a przepuszczalnością zachodzi prosty związek:
T
A
log
−
=
lub
%)
log(
2
T
A
−
=
(7)
1.2.
Moment przejścia. Moc oscylatora
Dane przejście jest obserwowane w widmie absorpcji, jeżeli spełnione są następujące
warunki na absorpcję promieniowania elektromagnetycznego o natężeniu E=E
0
cos(
ω
t):
1. Warunek konieczny –
warunek rezonansu:
Energia promieniowania padającego h
ν
musi być równa różnicy energii pomiędzy badanymi
stanami:
n
m
h
E
E
ν
= −
(8)
2. Warunek wystarczający -
warunek niezerowego momentu przejścia
O szybkości i intensywności absorpcji decyduje
moment przejścia (moment dipolowy
przejścia pomiędzy stanami m i n).
Tylko jego niezerowa wartość umożliwia cząsteczce
„opuszczenie” stanu.
*
0
mn
m
n
d
ψ ψ τ
=
≠
∫
M
µ
(9)
E
0
-amplituda pola elektrycznego promieniowania,
ω
ω
ω
ω
- częstość kołowa promieniowania (w s
-1
)
µµµµ
- operator momentu dipolowego cząsteczki:
∑
∑
−
=
atomy
A
elektrony
i
i
A
A
e
e
r
R
Z
µµµµ
Z
A
- ładunek jądra, wektory
R
A
i
r
i
- położenie jąder i elektronów.
ψ
m
i
ψ
n
–pełne funkcje falowe obu stanów cząsteczki, między którymi zachodzi przejście.
Szybkość przejścia w
mn
(szybkość zmiany prawdopodobieństwa przejścia)
2
2
0
2
6
mn
mn
mn
dt
dP
w
M
E
h
π
=
=
(10)

6
Kwadrat momentu przejścia jest miarą intensywności absorpcji, opisywaną przez
wielkość nazywaną
mocą lub siłą oscylatora przejścia:
(11)
B
mn
-
współczynnik absorpcji wymuszonej Einsteina
B
mn
=
2
2
0
6
1
mn
M
h
ε
(12)
(m
e
i e - masa i ładunek elektronu).
Siła (moc) oscylatora –jest bezwymiarową wielkością i podaje efektywną liczbę elektronów
biorących udział w określonym przejściu. W warunkach doświadczalnych, przy zadanej
częstości promieniowania padającego, w miejsce „ostrego” przejścia między stanami m i n
otrzymujemy rozmyte pasmo odpowiadające
integralnemu współczynnikowi absorpcji.
Wówczas doświadczalnie wyznaczoną moc oscylatora określa wyrażenie:
(13)
1.3. Czas życia cząsteczek w stanie wzbudzonym
Przejścia elektronów w cząsteczce z niższych poziomów na wyższe zachodzi dzięki
absorpcji
promieniowania,
odwrotnym
przejściom
towarzyszy
emisja.
Jednakże
prawdopodobieństwa tych przejść są różne i różne w związku z tym natężenia linii widmowych.
Emisja samorzutna (spontaniczna) polega na wyemitowaniu fotonu
h
νννν
przez cząsteczkę
niezależnie od działania nań czynników zewnętrznych.
Pomiędzy wymuszoną absorpcją i spontaniczną emisją istnieje ścisły związek:
(14)
A
nm
-współczynnik emisji spontanicznej Einsteina,
B
mn
-
współczynnik absorpcji wymuszone Einsteina.
Jeśli wzbudzone cząsteczki B* ulegają dezaktywacji tylko przez emisję fluorescencji, to
natężenie fluorescencji, wynoszące z chwilą przerwania wzbudzania I
0
, maleje z upływem
czasu wykładniczo, zgodnie z równaniem:
0
/
0
0
e
e
f
k t
t
t
I
I
I
τ
−
−
=
=
(15)
Symbolem
k
f
oznaczona została stałą szybkości promienistego zaniku fluorescencji (por.
rys. 1) i stała ta jest tożsama ze
współczynnikiem emisji spontanicznej Einsteina A
nm
. Chwi-
lowe natężenie emisji jest proporcjonalne do chwilowej szybkości zaniku wzbudzonych
cząsteczek B*, wyrażonej pochodną (-d[B*]/dt). W równaniu (15)
τ
0
= 1/k
f
oznacza
2
0
2
2
2
2
4
3
e
e
mn
mn
mn
mn
mn
m
m
f
M
B
e
e
ε
ω
ω
=
=
h
h
h
( )
9
12
4, 33 10
f
d
ε ν ν
−
=
⋅
∫
%
%
3
8
nm
mn
h
A
B
c
π ν
=

7
naturalny średni czas życia cząsteczek B
*
w stanie wzbudzonym i jest zależny jedynie od
prawdopodobieństwa samorzutnego przejścia promienistego B*
→
B. Korzystając z
zależności pomiędzy współczynnikiem emisji spontanicznej
Einsteina
A
nm
i współczynnikiem
absorpcji wymuszone
j Einsteina B
mn
(14) i związku pomiędzy
B
mn
z integralnym
współczynnikiem absorpcji można wnosić, że wartość
τ
0
musi być odwrotnie proporcjonalna
do
int
ε
.Uproszczony wzór okreslający tę zależność, po podstawieniu liczbowych wartości
stałych fizycznych, przybiera postać:
2
1
8
0
2
3, 44·10
( )
m
d
ν
ν
τ
ν ε ν ν
=
∫
%
%
%
%
%
(16)
m
ν
~ - liczba falowa odpowiadająca maksimum pasma absorpcji przejścia S
1
←
S
0
, a
∫
2
1
~
~
~
)
~
(
ν
ν
ν
ν
ε
d
jest wyznaczonym eksperymentalnie molowym współczynnikiem absorpcji scałkowany po
całym paśmie absorpcji.
Ze schematu przedstawionego na rys. 1 wynika, że wzbudzony stan elektronowy
odpowiedzialny za fluorescencję ulega dezaktywacji również na inne sposoby, co prowadzi
do zależności:
τ
t
o
e
I
I
−
=
(17)
gdzie:
τ
−
średni mierzony doświadczalnie najkrótszy czas zaniku, po którym wartość
o
I
I
maleje do
wartości
e
1
i wynosi:
(
)
ISC
IC
f
k
k
k
+
+
=
1
τ
(18)
k
IC
−
stała szybkości konwersji wewnętrznej (S
1
S
o
)
i może zmieniać się w szerokich
granicach, często oceniana jest na 10
5
-10
7
s
-1
;
k
ISC
−
stała szybkości konwersji międzysystemowej i jej wartość jest zwykle zawarta w
granicach 10
5
-10
10
s
-1
;
k
f
−
stała szybkości emisji fluorescencji i wynosi 10
8
-10
9
s
-1
,
jeśli przejście S
1
→
S
o
(por. rys.
1) jest dozwolone wszystkimi regułami wyboru.

8
1.4. Wydajność kwantowa fluorescencji
Wydajność kwantową fluorescencji
ϕ
f
definiuje się jako stosunek liczby
wyemitowanych fotonów do liczby fotonów promieniowania wzbudzającego, pochłoniętych
przez substancję w tym samym czasie i tej samej objętości. Wartość wydajności kwantowej
(
ϕ
f
) jest niemal zawsze mniejsza od jedności i na ogół zmienia się proporcjonalnie do
τ
(wyjątek stanowią rozrzedzone gazy),
f
f
o
f
IC
ISC
k
k
k
k
τ
φ
τ
=
=
+
+
(19)
o
τ
−
naturalny średni czas życia cząsteczek w stanie wzbudzonym i można go w przybliżeniu
wyznaczyć korzystając z relacji (16).
2. Pomiary spektrofotometryczne i spektrofluorymetryczne
2.1. Pomiar widma absorpcji
Pomiar widm absorpcji roztworów w obszarze widzialnym i nadfiolecie, czyli
zależności
A(
λ
) [lub
T(
λ
)] względnie
A(
ν
% ) [lub T(
ν
% )], przeprowadzamy przy użyciu
klasycznych spektrofotometrów UV-VIS.
Każdy spektrofotometr składa się z czterech zasadniczych elementów:
1.
źródła promieniowania polichromatycznego,
2.
monochromatora, rozszczepiającego wiązkę promieniowania polichromatycznego na
szereg wiązek monochromatycznych,
3.
detektora czułego na dany zakres promieniowania i zamieniającego sygnał świetlny
na elektryczny,
4.
elementu pomiarowego odczytującego i przetwarzającego sygnał wysyłany z detektora
na właściwą wielkość pomiarową (
A lub T) oraz zapisującego go w funkcji długości
fali lub liczby falowej. Zwykle zadaniem tego przyrządu jest też sterowanie
eksperymentem, np. powodowanie zmiany długości fali, przy której mierzona jest
absorpcja po każdym kolejnym odczycie.
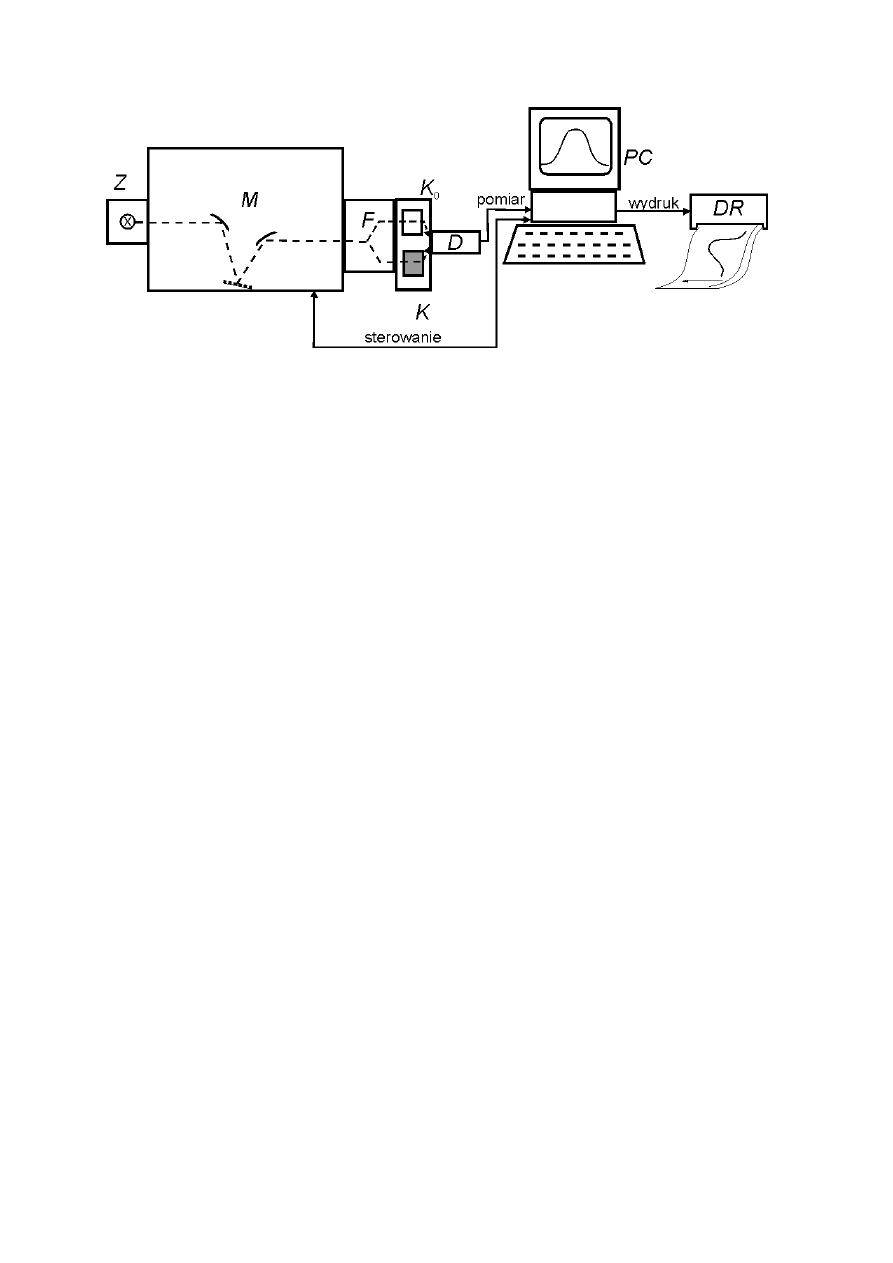
Schemat blokowy typowego spektrofotometru przedstawiono na rys. 3.
9
Rys. 3. Schemat blokowy spektrofotometru absorpcyjnego.
Z – źródło promieniowania
polichromatycznego,
M – monochromator, F – fotometr, K
0
,
K, – kuwety z odnośnikiem i
badaną próbką,
D – detektor promieniowania, PC – komputer lub inne urządzenie sterujące i
zapisujące wyniki.
DR – urządzenie peryferyjne, np. drukarka
Ponieważ celem pomiaru jest wyznaczenie absorbancji
A lub przepuszczalności T,
należy zmierzyć zarówno
I
0
jak i
I [patrz wzory (4) i (6)]. Realizuje się to przepuszczając
ś
wiatło opuszczające monochromator na przemian przez kuwetę
K z badaną substancją i
przez identyczną kuwetę porównawczą (odnośnik)
K
0
. W przypadku pomiarów absorpcji
roztworów w obszarze widzialnym lub nadfiolecie, kuweta
K
0
jest wypełniona
rozpuszczalnikim, w którym rozpuszczono substancję badaną w kuwecie
K. Kuwety są to
naczynia o płasko–równoległych wypolerowanych ścianach, wykonanych z kwarcu (lub ze
szkła, jeśli pomiar wykonuje się tylko w zakresie widzialnym).
2.2. Pomiar widma fluorescencji
Pomiar widma fluorescencji i widma wzbudzenia fluorescencji roztworów w obszarze
widzialnym i nadfiolecie, czyli zależności
intensywność fluorescencji = f(
λ
) przeprowadzamy
przy użyciu klasycznych spektrofluorymetrów.
Spektrofluorymetr wyposażony jest w dwa monochromatory zazwyczaj siatkowe, w
lampę ksenonową oraz fotopowielacz pracujący w reżimie zliczania pojedynczych fotonów.
Schemat blokowy takiego spektrofluorymetru przedstawiono na rysunku 4.
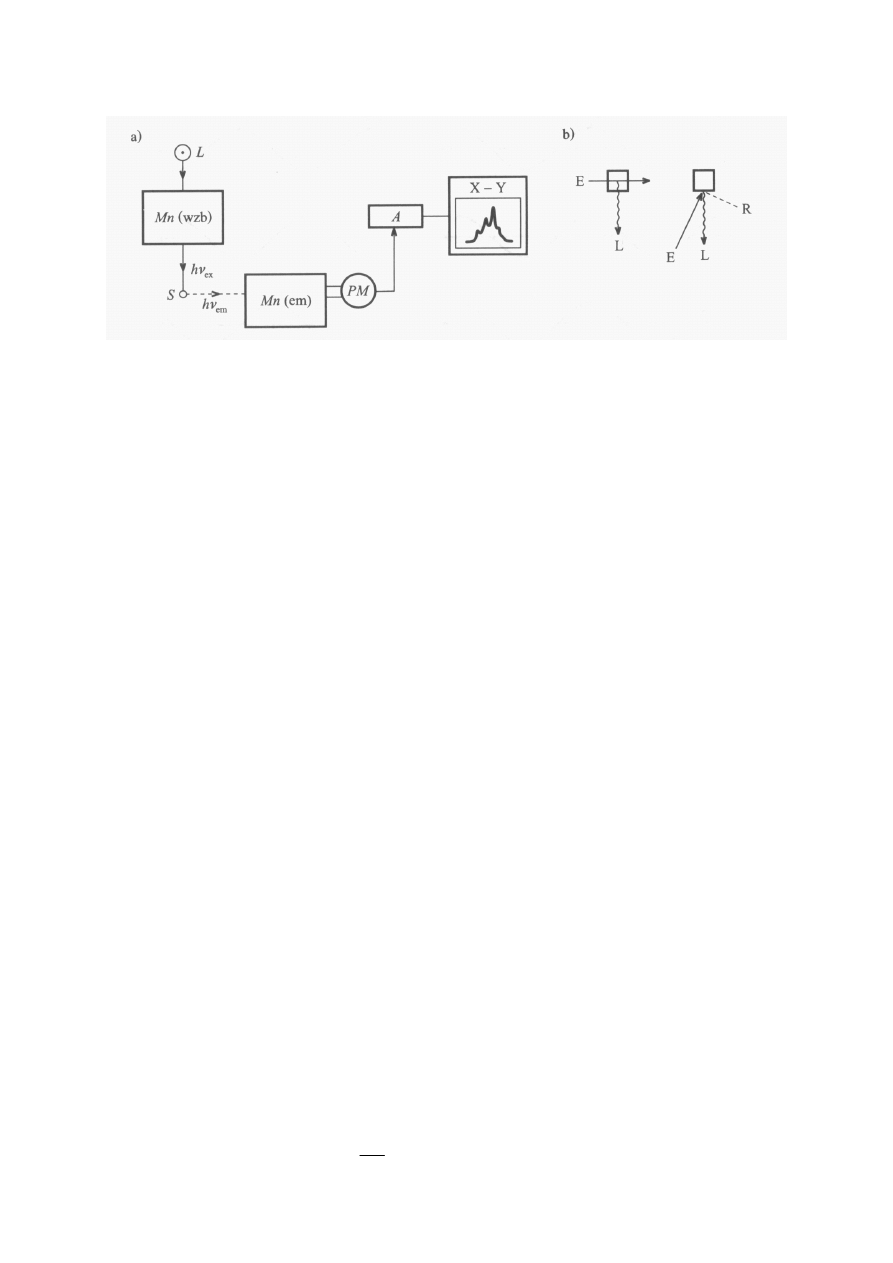
10
Rys. 4. (a) Schemat blokowy spektrofluorymetru.
L – źródło światła, Mn(wzb) –
monochromator wybierający światło wzbudzające,
S – próbka, Mn(em) –
monochromator do analizy widma emisji,
PM – fotopowielacz, A – wzmacniacz, X-
Y –rejestrator.
(b)
Obserwacja pod kątem prostym (po lewej) i obserwacja czołowa (po prawej).
E – wiązka wzbudzająca, L – luminescencja, R – rozproszenie.
2. Pomiary wydajności kwantowych
Pomiary bezwzględnej wydajności kwantowej luminescencji są pomiarami trudnymi,
dlatego też wykonywane są najczęściej względem wzorców luminescencyjnych. W
pomiarach aktynometrycznych dla określenia bezwzględnej wydajności luminescencji
mierzymy natężenie promieniowania wzbudzającego i natężenie emisji próbki aktynometrem
chemicznym, a stosunek natężeń emisji do wzbudzenia stanowi bezwzględną wydajność
kwantową. Całkowite natężenie wiązki wyemitowanej przez próbkę jest mierzone roztworem
aktynometru chemicznego. Metoda ta została szczegółowo opisana w [1,2].
2.1. Wyznaczenie wydajności kwantowej z pomiaru czasu zaniku fluorescencji
Bezwzględną wydajność kwantową można wyznaczyć korzystając z relacji (19), jako
stosunek zmierzonego czasu życia fluorescencji
τ
do wyznaczonego z integralnego
współczynnika absorpcji, naturalnego średniego czasu życia cząsteczek w stanie
wzbudzonym
τ
o
.
f
o
τ
φ
τ
=
(20)

11
Wartości wydajności kwantowych uzyskane tą metodą obarczone są znacznym
błędem. Błąd wynika z niedokładności oznaczania wartości
τ
o
,
tj. pola powierzchni pod
krzywą pasma absorpcji odpowiadającej pierwszemu przejściu elektronowemu S
0
→
S
1
.
Zazwyczaj każde pasmo w widmie absorpcji jest obwiednią kilku nakładających się na siebie
pasm, dlatego też dokładniejsze informacje o składowych poszczególnych pasm można
uzyskać przez ich rozkład.
Również wyznaczony doświadczalnie czas życia fluorescencji próbki obarczony jest
ok. dziesięcioprocentowym błędem.
2.1.1. Pomiary czasów zaniku fluorescencji
Wyznaczenie czasu życia fluorescencji polega na zarejestrowaniu czasu zaniku emisji
badanego luminoforu po jego krótkotrwałym wzbudzeniu.
Do
pomiarów
czasów
ż
ycia
fluorescencji stosujemy między innymi lampy błyskowe, dające dostatecznie krótkie
(nanosekundowe) i stromo opadające impulsy świetlne.
Emisję próbkujemy po każdym błysku wzbudzającym za pomocą szybkiego
fotopowielacza. Po uwzględnieniu czasowego zaniku impulsu wzbudzającego, można
otrzymać czasowy zanik emisji substancji badanej. Rozwinięciem tej metody jest metoda
zliczania pojedynczych fotonów. W metodzie tej część światła wzbudzającego (lasera
impulsowego lub impulsowej lampy np. deuterowej) kierowana jest do fotodiody (lub
fotopowielacza)
PD, która ustala zero w skali czasu. Impuls ten wyzwala liniowo narastające
napięcie uzyskane z przetwornika czasu na napięcie (lub na amplitudę). W chwili zauważenia
przez fotopowielacz
PM pierwszego wyemitowanego fotonu, wzrost napięcia jest zatrzymany,
a impuls z przetwornika zostaje zarejestrowany w odpowiednim kanale analizatora
wielokanałowego. W analizatorze wielokanałowym każdy kanał jest określony czasem, jaki
upłynął od chwili rozbłysku impulsu wzbudzającego, a dla każdego impulsu wzbudzającego
zliczany jest tylko pierwszy foton. Eksperyment jest powtarzany wielokrotnie i w ten sposób
zostaje zarejestrowana cała krzywa zaniku emisji. Uproszczony schemat układu do zliczania
pojedynczych fotonów przedstawiony jest na rys. 5.
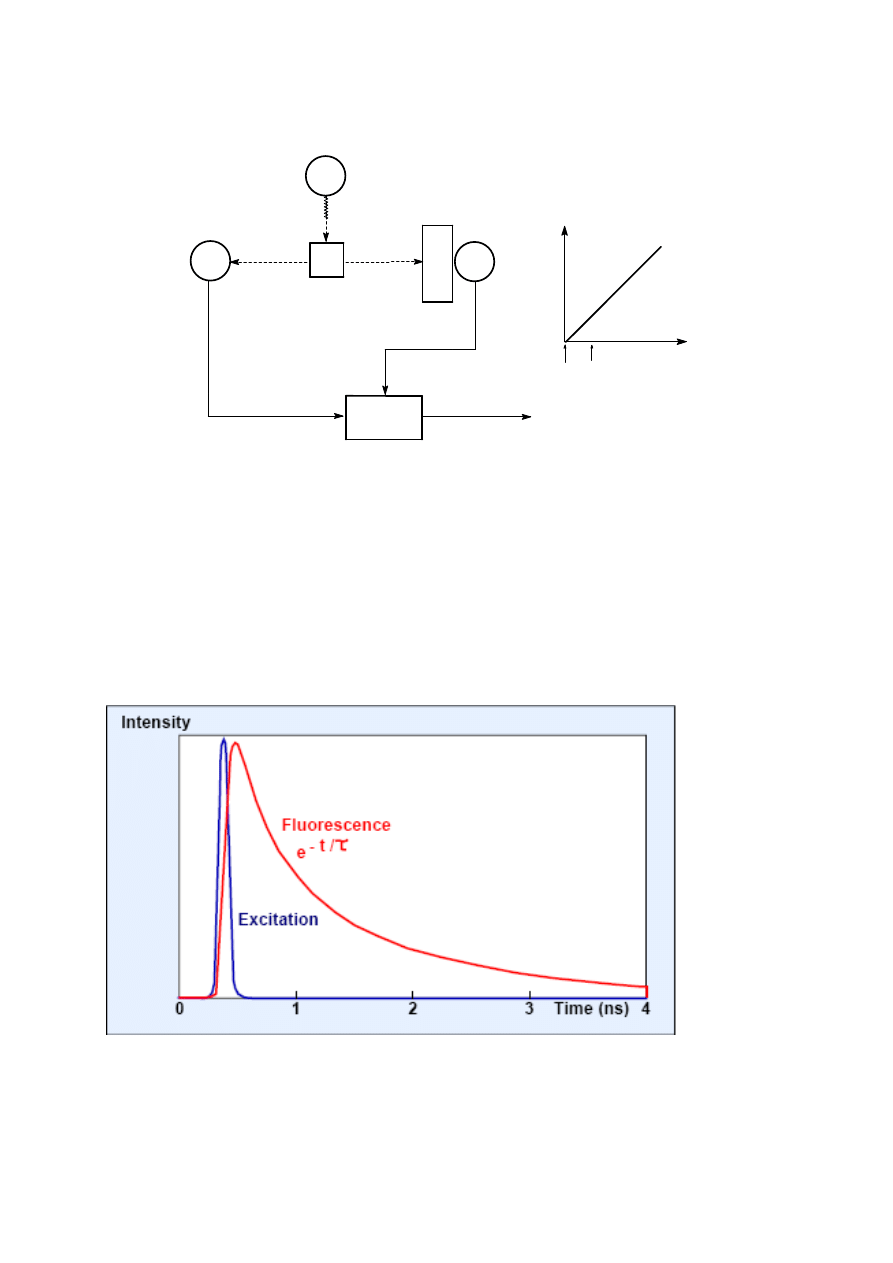
12
Rys.5. (a) Schemat układu do zliczania pojedynczych fotonów: L - impulsowe źródło światła,
PD - fotodioda, S- próbka, F- filtr interferencyjny lub monochromator, PM- fotopowielacz, R-
generator liniowo narastającego napięcia (1-start, 0-stop), AW- analizator wielokanałowy.
(b) liniowa zmiana napięcia w czasie.
Analizator AW zlicza impuls wyjściowy w kanałach odpowiadających poszczególnym
wartościom napięcia impulsów wyjściowych. Wynikiem pomiaru jest krzywa rozkładu liczby
fotonów, proporcjonalna do czasu zaniku natężenia fluorescencji (por. rys. 6).
Rys.6. Czas trwania emisji fluorescencji (
czerwony
) po wzbudzeniu krótkim impulsem lasera
(
niebieski
).
L
S
F
PM
R
AW
PD
V
czas
1
0
V
1
0
(a)
(b)

13
2.2 Wyznaczenie względnej wydajności kwantowej metodą porównawczą
W metodzie porównawczej względną wydajność kwantową oblicza się z zależności:
2
2
2
2
2 2 2
2
2
2
2
2
2
1
1
1 1 1
1
1
1
1
o
o
F
c l I
n
A
n
F
c l I
n
A
n
ϕ ε
ϕ
ϕ ε
ϕ
=
⋅
⋅
=
⋅
⋅
(21)
gdzie:
F
2
i
F
1
oznaczają powierzchnię pod krzywymi rozkładu natężeń fluorescencji w
widmach emisji próbki i wzorca,
ϕ
1
i
ϕ
2
są odpowiednio bezwzględne wydajności kwantowe
wzorca o znanej wydajności kwantowej i próbki,
ε
2
i
ε
1
- molowe współczynniki absorpcji
próbki i wzorca przy długości fali światła wzbudzającego (takiej samej dla wzorca i próbki),
c
2
i
c
1
- stężenie badanej próbki i wzorca,
l
2
i
l
1
- długości drogi optycznej w warstwach
roztworów próbki i wzorca,
I
0
- intensywność początkowa,
n
2
i
n
1
- współczynniki załamania
ś
wiatła rozpuszczalnika próbki i wzorca,
A
2
i
A
1
-
absorbancja próbki i wzorca przy długości
fali światła wzbudzającego fluorescencję (istotne jest, aby absorbancja próbki i wzorca nie
przekraczała wartości 0,1 tj. w zakresie, w którym występuje liniowa zależność natężenia
fluorescencji od stężenia).
Zastosowany wzorzec powinien posiadać pasmo absorpcji oraz fluorescencji w tym
samym zakresie spektralnym jak i badana próbka. Ponadto, aby pomiar wydajności
kwantowej fluorescencji był najdokładniejszy, wzbudzenie próbki i wzorca powinno odbywać
się w identycznych warunkach. Współczynniki załamania światła w równaniu (21) pozwalają
uwzględnić różnicę w stosowanych rozpuszczalnikach.
Aby wydajność kwantowa była poprawnie wyznaczona, wymagany jest prawidłowy
dobór wzorca. Najczęściej stosowane wzorce fluorescencyjne oraz ich wydajność kwantowa
fluorescencji zostały przedstawione w tabeli 1.
Tabela1. Wzorce fluorescencyjne oraz ich wydajność kwantowa fluorescencji
Związek
Rozpuszczalnik
λ
ex
[nm]
φ
f
Sulfonowana chinina
0.1 M H
2
S0
4
350
366
0.577
0.53
±
0.023
Fenol
Woda
275
0.14
±
0.01
Rodamina 6G
Etanol
488
0.94
Rodamina 101
Etanol
450-465
1.0
Chlorofil a
Metanol
644
0.21
±
0.02
Chlorofil a
Eter etylowy
_
0.20
±
0.02
Chlorofil b
Metanol
_
0.048
±
0.007
Chlorofil b
Eter etylowy
_
0.074
±
0.007
Ftalocyjanina Zn
Toluen
610
0.30
±
0,02
14
3. Wykonanie ćwiczenia i opracowanie wyników
Celem ćwiczenia jest wyznaczenie wydajności kwantowej fluorescencji wodnego
roztworu ftalocyjaniny, wykorzystując zmierzony czasu życia fluorescencji (
τ
) oraz obliczony
naturalny czasu życia fluorescencji (
τ
0
).
3.1 Opracowanie widm absorpcji roztworu badanego związku
Wykonanie ćwiczenia polega na pomiarze widm absorpcji badanych związków przy
użyciu spektrofotometru UV – VIS, którego schemat blokowy przedstawiono na rysunku 7.
Spektrofotometr wyposażony jest w dwa źródła światła: lampę deuterową (w zakresie
promieniowania UV) i lampę halogenową (w zakresie widzialnym). W układzie
monochromatora znajdują się szczeliny do regulacji szerokości wiązki światła, siatka
dyfrakcyjna i modulator.
Rys.7.
Schemat
blokowy
spektrofotometru
(UV-2101PC)
UV-VIS
(Scanning
Spectrophotometr Shimadzu).
Widmo absorpcji zapisane jest w postaci pliku danych z rozszerzeniem
.ASC, w
którym w pierwszej kolumnie jest długość fali [nm], w drugiej - absorbancja przy danej
długości fal. Przykładowe widmo przedstawiono na rys. 8.
Rys. 8. Widmo absorpcji wodnego roztworu ftalocyjaniny (c= 1,25
⋅
10
−
6
mol
⋅
dm
−
3
)
Ź
RÓDŁO
Ś
WATŁA
MONOCHROMATOR
PRÓBKA
ODNO
Ś
NIK
FOTOPOWIELACZ
KOMPUTER
MIERNIK
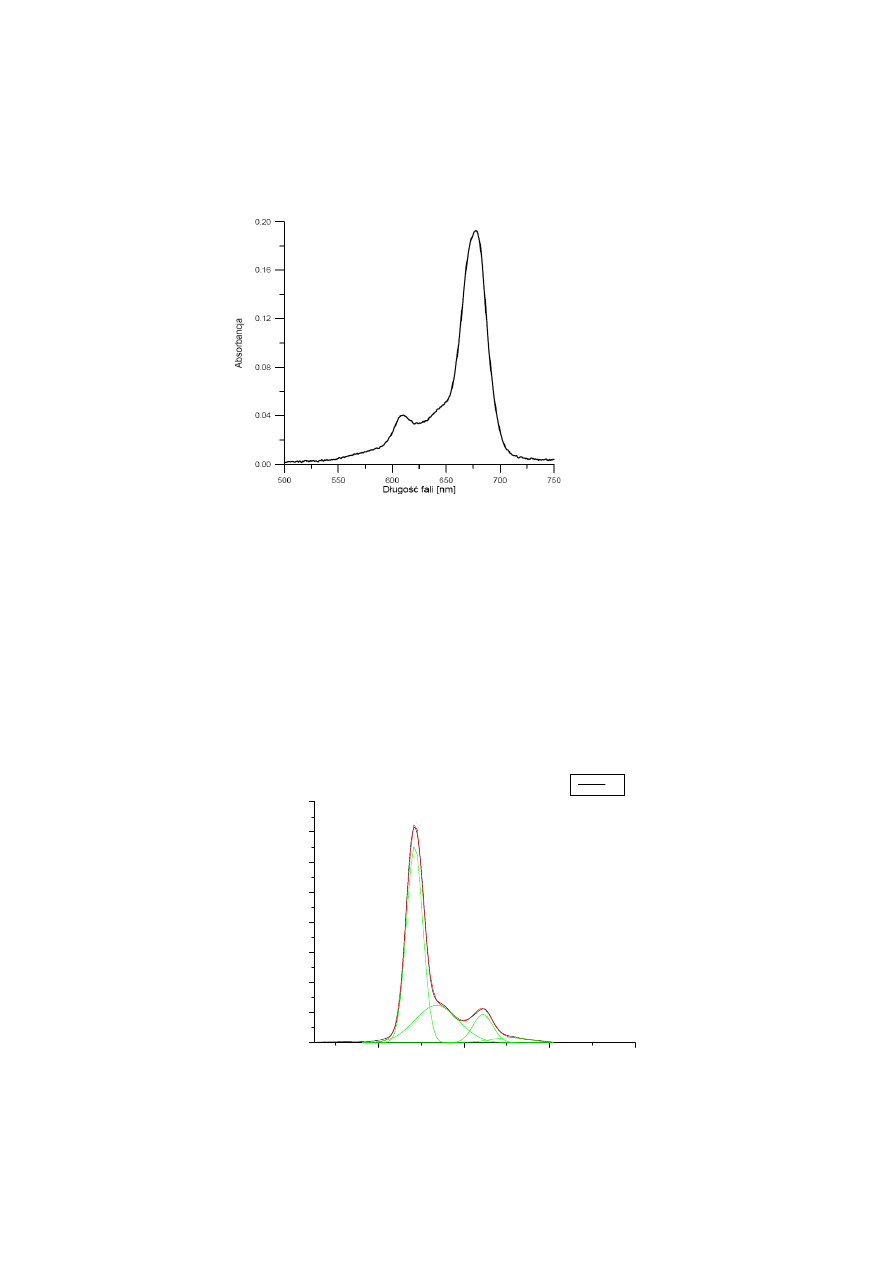
15
Z widma zarejestrowanego w całym mierzonym zakresie spektralnym należy wybrać pasmo
odpowiadające pierwszemu przejściu absorpcyjnemu S
1
←
S
0
. W przypadku roztworów
ftalocyjaniny należy wybrać fragment 500-750 nm (por. rys. 9).
Rys. 9. Fragment widma absorpcji wodnego roztworu ftalocyjaniny (c= 1,25
⋅
10
−
6
mol
⋅
dm
−
3
)
Znając stężenie roztworu i stosując
prawa Lamberta – Beera zmieniamy skalę absorbancji na
skalę molowego współczynnika absorpcji. W tym celu należy podzielić absorbancję przez
stężenie substancji w roztworze i grubość warstwy (1 cm).
Również skalę długość fali należy zamienić na skalę liczb falowych wyrażoną w cm
−
1
. Na
rys.10 przedstawiono fragment widma absorpcji po dokowaniu tych zamian.
14000
16000
18000
20000
0,0
2,0x10
5
4,0x10
5
6,0x10
5
8,0x10
5
1,0x10
6
1,2x10
6
1,4x10
6
1,6x10
6
M
o
lo
w
y
w
s
p
ó
łc
z
y
n
n
ik
a
b
s
o
rp
c
ji
Liczba falowa
D
Rys. 10. Fragment widma absorpcji wodnego roztworu ftalocyjaniny (c= 1,25
⋅
10
−
6
mol
⋅
dm
−
3
)
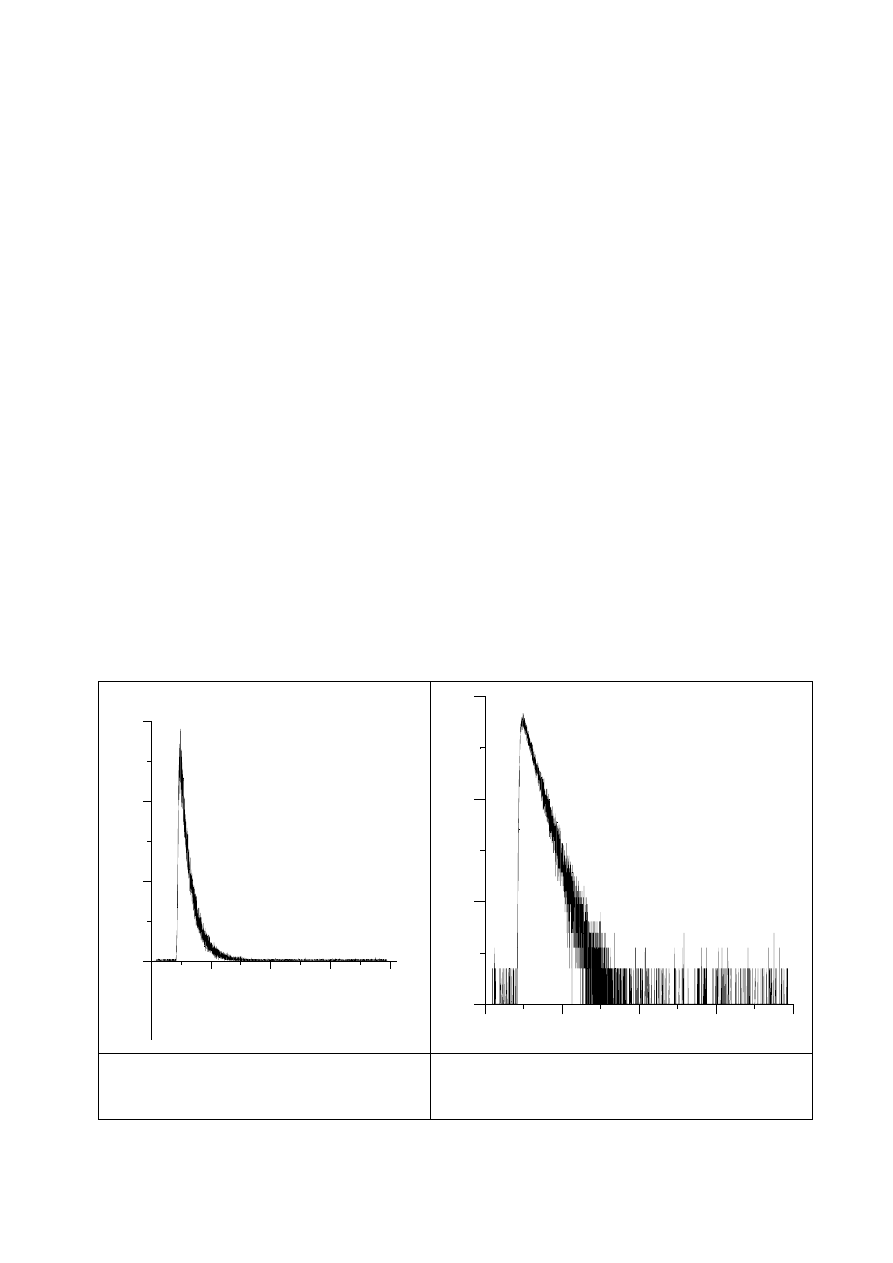
16
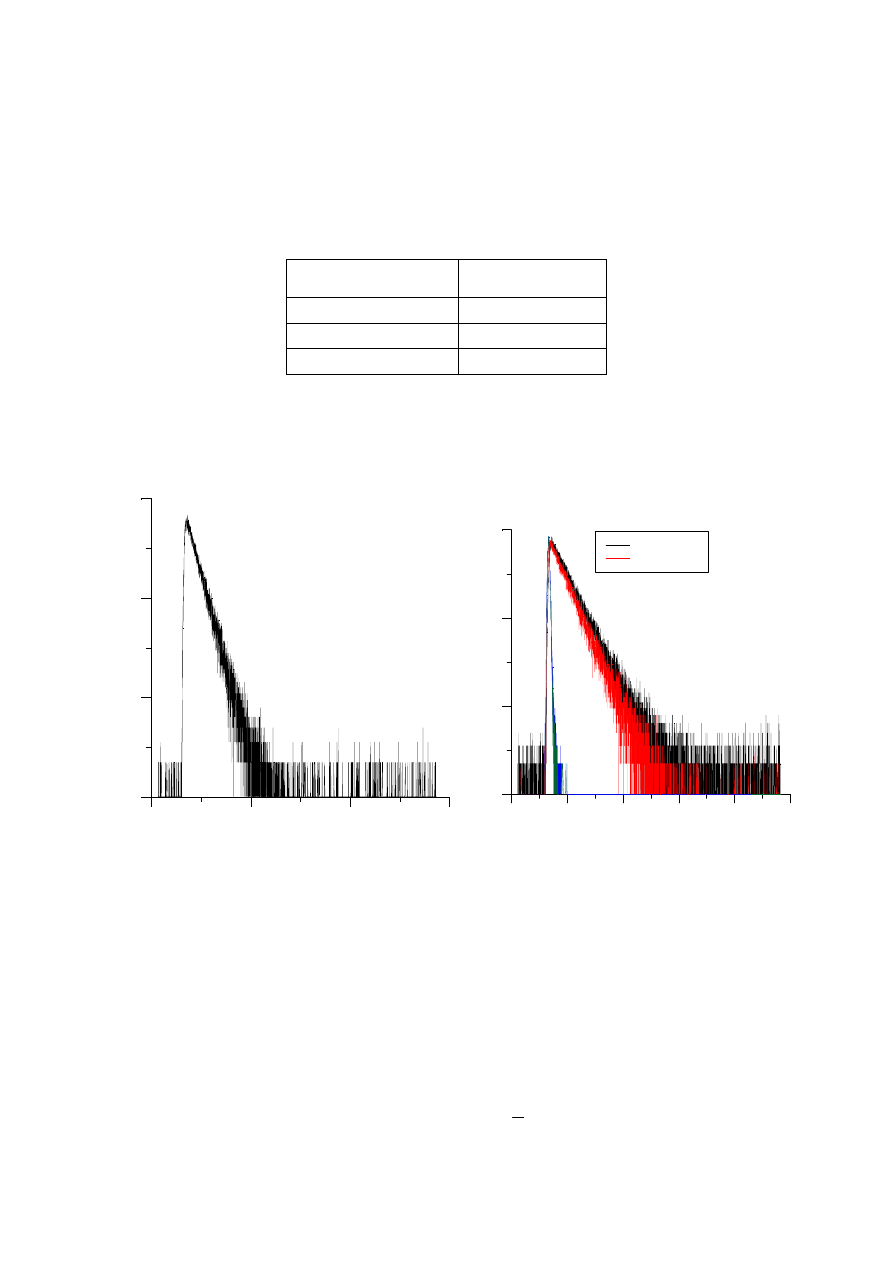
Kolejnym krokiem jest rozkładanie widma w tym zakresie na kilka pasma (np. programem
ORIGIN), których obwiednią jest funkcja Gaussa (Gaussiany). Liczba pasm powinna być
dobrana tak, aby powierzchnia pod każdym pasmem była dodatnia, dopasowanie sumaryczne
do kształtu pasma zmierzonego było najbliższe jedności i aby najintensywniejsze, pierwsze
pasmo, było odtworzone najwierniej jedną funkcją Gaussa (por. rys.10).
Z tak rozłożonego widma należy odczytać integralny współczynnik absorpcji pierwszego,
najintensywniejszego pasma oraz liczbę falową określającą maksimum tego pasma, a
następnie korzystając z zależności (13) i (16) obliczyć siłę oscylatora przejścia (
f) i naturalny
czas życia fluorescencji (
τ
0
).
3.2.Opracowanie wyników pomiaru czasu zaniku fluorescencji
Dane z pomiaru czasu zaniku fluorescencji otrzymujemy w postaci pliku z
rozszerzeniem
.TXT, gdzie w pierwszej kolumnie jest numer kanału, w drugiej – liczba
pojedynczych zliczonych fotonów w odpowiednim kanale. Krzywa rozkładu liczby fotonów
w kolejnych kanałach w funkcji numeru kanału odpowiada czasowemu zanikowi
fluorescencji (por. rys 11a). Jest to zależność ekspotencjalna, natomiast w skali
logarytmicznej – otrzymujemy liniową zależność ln (liczby impulsów) =
f (nr kanału).
Zależność ta przedstawiona jest na rys.11b.
0
2000
4000
6000
8000
Numer kanału
0
100
200
300
L
ic
z
b
a
i
m
p
u
ls
ó
w
0
2000
4000
6000
8000
numer kanału
0
2
4
6
ln
(
lic
z
b
y
i
m
p
u
ls
ó
w
)
Rys. 11a. Liczba impulsów (fotonów) emisji
zliczona w każdym kanale analizatora
Rys. 11b. Logarytm naturalny z liczby impulsów
emisji w funkcji numeru kanału .
17
Przed każdym pomiarem dobierana jest skala podstawy czasu, tj. czas, w którym rejestrujemy
liczbę impulsów pojawiających się od chwili t = 0 do t = podstawie czasu. Skala podstawy
czasu wybierana jest w zależności od spodziewanego czasu zaniku fluorescencji.
W Tabeli 1 zestawiono wartości podstawy czas i czas przypadający na każdy kanał.
Tabela 1. Wartości podstawy czasu i ilość nanosekund przypadająca na jeden kanał.
podstawa czasu [ns]
ns/ kanał [ns]
100
0,01455
200
0,02919
500
0,06322
Iloczyn liczby kanałów użytych w pomiarze i czasu, jaki jest potrzebny na zarejestrowanie
impulsów w każdym kanale, zamienia skalę liczby kanałów na czas [w ns] (por. rys 12a).
(a) (b)
0
40
80
120
czas [ns]
0
2
4
6
ln
(
lic
z
b
y
i
m
p
u
ls
ó
w
)
ln (liczba zliczeń)
Rys. 12. (a) Wykres zależności ln (liczba zliczeń)=
f (czas). (b) Wykres zależności ln (ilości
zliczeń)=
f (czasu) dla próbki sulfonowanej ftalocyjaniny glinu w wodzie o stężeniu
c=6
⋅
10
-5
M. Pomiar zarejestrowany w kuwecie kwadratowej (
τ
=7,7ns) i w kuwecie trójkątnej
(
τ
=6,66ns). Linie zaznaczone kolorem zielonym i niebieskim odnoszą się do wzorca
fluorescencyjnego.
Zależność ln
I od czasu jest zależnością liniową, jeśli zanik emisji jest monoeksponencjalny,
a współczynnik kierunkowy prostej: ln(
I)=f (t);
ln
ln
o
t
I
I
τ
= −
+
jest równy
−
1/
τ
.
0
20
40
60
80
100
0.00
2.00
4.00
6.00
kuweta kwadratowa
kuweta trojkatna

18
Z zależności tej obliczmy czas zaniku fluorescencji.
3.3 Sprawozdanie z ćwiczenia.
Student otrzymuje dwa komplety danych. Jeden plik z rozszerzeniem .ASC zawiera
dane pomiaru widma absorpcji, który należy opracować tak, jak to zostało przedstawione w
punkcie 3.1. W sprawozdaniu należy umieścić wszystkie widma oraz obliczoną siłę
oscylatora pierwszego przejścia absorpcyjnego i obliczony naturalny czas życia fluorescencji.
Drugi plik, z rozszerzeniem .TXT zawiera dane mierzonego czasu zaniku fluorescencji, który
należy opracować tak, jak to zostało przedstawione w punkcie 3.2. W sprawozdaniu należy
przedstawić wszystkie krzywe zaniku oraz obliczony czas zaniku fluorescencji.
Iloraz
τ
/
τ
0
jest wielkością wydajności kwantowej fluorescencji, którą również należy
obliczyć.
Literatura:
[1] K. Pigoń, Z. Ruziewicz, „Chemia fizyczna”, PWN, Warszawa, (1986), (rozdz. 11.2.3, 11.3.4,
11.3.5).
[2] A. Kawski, „Fotoluminescencja roztworów”, PWN, Warszawa, (1992), (rozdz.1, 6).
Opracowała dr hab. Krystyna Palewska
Wyszukiwarka
Podobne podstrony:
Abolicja podatkowa id 50334 Nieznany (2)
4 LIDER MENEDZER id 37733 Nieznany (2)
katechezy MB id 233498 Nieznany
metro sciaga id 296943 Nieznany
perf id 354744 Nieznany
interbase id 92028 Nieznany
Mbaku id 289860 Nieznany
Probiotyki antybiotyki id 66316 Nieznany
miedziowanie cz 2 id 113259 Nieznany
LTC1729 id 273494 Nieznany
D11B7AOver0400 id 130434 Nieznany
analiza ryzyka bio id 61320 Nieznany
pedagogika ogolna id 353595 Nieznany
Misc3 id 302777 Nieznany
cw med 5 id 122239 Nieznany
D20031152Lj id 130579 Nieznany
mechanika 3 id 290735 Nieznany
więcej podobnych podstron