Postępy Biochemii 59 (4) 2013
357
Mateusz Tomczyk
Witold Nowak
Agnieszka Jaźwa
*
Zakład Biotechnologii Medycznej, Wydział
Biochemii, Biofizyki i Biotechnologii, Uniwer-
sytet Jagielloński, ul. Gronostajowa 7, 30-387
Kraków
*
Zakład Biotechnologii Medycznej, Wydział
Biochemii,
Biofizyki
i
Biotechnologii,
Uniwersytet Jagielloński; ul. Gronostajowa
7, 30-387 Kraków, e-mail: agnieszka.jazwa@
uj.edu.pl
Artykuł otrzymano 2 października 2013 r.
Artykuł zaakceptowano 25 listopada 2013 r.
Słowa kluczowe: angiogeneza, cukrzyca, dys-
funkcja śródbłonka, miażdżyca, stres oksyda-
cyjny
Wykaz skrótów: AGE (ang. advanced glycation
end-products) — końcowe produkty zaawanso-
wanej glikacji; BH
4
— tertrahydrobiopteryna;
EC (ang. endothelial cells) — komórki śródbłon-
ka; ECM (ang. extracellular matrix) — macierz
zewnątrzkomórkowa; ET — endotelina; NO
— tlenek azotu; NOS (ang. nitric oxide syntha-
se) — syntaza tlenku azotu; PGI
2
— prostacy-
klina; RFT — reaktywne formy tlenu; TXA
2
—
tromboksan A
2
; VEGF (ang. vascular endothelial
growth factor) — czynnik wzrostu śródbłonka
naczyń
Śródbłonek w fizjologii i patogenezie chorób
STRESZCZENIE
K
omórki śródbłonka wyścielają pojedynczą warstwą naczynia krwionośne, stanowiąc w
ten sposób pierwszą barierę pomiędzy krwią a tkankami. Komórki te pełnią liczne funk-
cje takie, jak regulacja napięcia ścian naczyń krwionośnych, kontrolowanie przepływu i ci-
śnienia krwi, utrzymywanie równowagi pomiędzy procesami krzepnięcia krwi i fibrynolizy,
udział w reakcjach układu odpornościowego oraz tworzenie nowych naczyń krwionośnych.
Zaburzenie którejkolwiek z tych funkcji śródbłonka może prowadzić do rozwoju różnych
chorób m.in. nadciśnienia tętniczego, miażdżycy i zakrzepicy naczyń, a także nowotworów.
WPROWADZENIE
Odkrycie i opisanie śródbłonka łączy się ściśle z odkryciem i poszerzaniem
wiedzy na temat układu krążenia. Historia zaczęła się w 1628 roku, kiedy opu-
blikowana została praca Williama Harveya „De Motu Cordis”, w której autor
przedstawił zjawisko krążenia krwi w naczyniach i wskazał serce jako narząd
wprawiający ją w ruch. Wkrótce tę wizję uzupełnił Marcello Malpighi, który opi-
sał krążenie w naczyniach włosowatych płuc żaby i fizyczne rozdzielenie krwi
od tkanek [1]. Kolejne istotne odkrycie miało miejsce w XIX wieku, kiedy Frie-
drich von Recklinghausen wykazał, że naczynia krwionośnie są wyścielone ko-
mórkami, a nie, jak uprzednio uważano, jedynie zanurzonymi w tkankach „ru-
rami”, którymi przepływa krew. U schyłku XIX wieku śródbłonek – pojedyncza
warstwa komórek wyścielających światło naczyń krwionośnych – opisywany
był zarówno jako aktywny układ komórek wydzielniczych, jak i selektywna, fi-
zyczna bariera oddzielająca mięśniówkę naczyniową od krwi [2]. Obecnie śród-
błonek jest coraz częściej nazywany „narządem rozproszonym”, stanowiącym
heterogenny, dynamiczny zespół komórek, który można też traktować jako gru-
czoł endokrynny. Funkcję fizycznej bariery w wyjątkowy sposób pełnią komórki
śródbłonka (EC, ang. endothelial cells) zlokalizowane w naczyniach mózgu, które
tworzą barierę krew-mózg. Jest to bariera fizyczna stworzona przez połączenia
ścisłe pomiędzy EC, która kieruje cały transport substancji z krwi na drogę trans-
komórkową, w odróżnieniu od pozostałych śródbłonków, gdzie taki transport
odbywa się głównie drogą parakomórkową [3].
U dorosłego człowieka śródbłonek liczy około dziesięć bilionów (10
13
) komó-
rek, tworząc tkankę o masie około 1 kg i polu powierzchni nawet do 7 m
2
[2].
Bierze on udział w wielu procesach fizjologicznych, takich jak regulacja przepły-
wu i ciśnienia krwi, synteza i wydzielanie czynników aktywnych biologicznie,
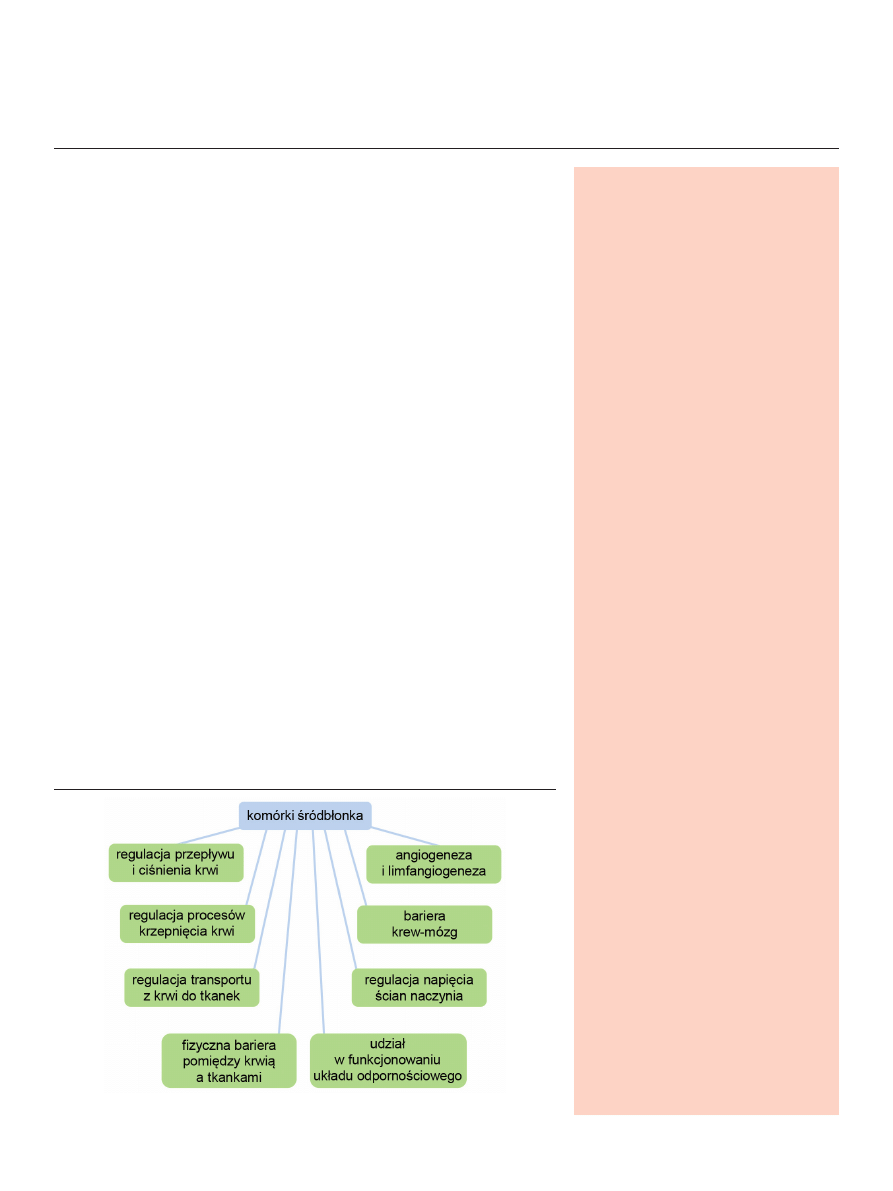
Rycina 1. Rola śródbłonka w utrzymaniu homeostazy naczyniowej.
358
www.postepybiochemii.pl
kontrola przepływu substancji odżywczych, angiogeneza
czy reakcje zapalne i odpornościowe (Ryc. 1) [2,4]. Komórki
śródbłonka zlokalizowane są w naczyniach krwionośnych,
które wyścielają od wewnątrz pojedynczą warstwą. Można
je również znaleźć w ścianach dużych naczyń, gdzie pełnią
funkcję vasa vasorum, tworząc układy mikrokrążenia zaopa-
trujące w krew komórki warstwy środkowej i zewnętrznej
naczyń [5]. Umiejscowienie komórek śródbłonka sprawia,
że to one tworzą pierwszą granicę z krwią i to im przypa-
da pierwszy kontakt z wędrującymi z krwi do tkanek ko-
mórkami i substancjami. Taka lokalizacja pozwala szybko
reagować na chemiczne i fizyczne bodźce i odpowiadać w
postaci zmiany syntezy molekuł adhezyjnych czy wydzie-
lania czynników regulujących między innymi zachowanie
komórek mięśniówki gładkiej naczyń (VSMC, ang. vascular
smooth muscle cells), ich proliferację i migrację, napięcie ścian
naczyń czy rozwój ogniska zapalnego [6]. Zaburzenie pra-
widłowego funkcjonowania komórek śródbłonka odgrywa
znaczącą rolę w powstawaniu i przebiegu wielu jednostek
chorobowych, takich jak miażdżyca, nadciśnienie, cukrzyca
czy choroba niedokrwienna serca i tętnic obwodowych [6].
Dysfunkcje śródbłonka mają również znaczenie w rozwoju
zmian nowotworowych [7].
ROLA ŚRÓDBŁONKA W UTRZYMANIU
HOMEOSTAZY NACZYNIOWEJ
ŚRÓDBŁONEK W REGULACJI NAPIĘCIA
ŚCIANY NACZYŃ KRWIONOŚNYCH
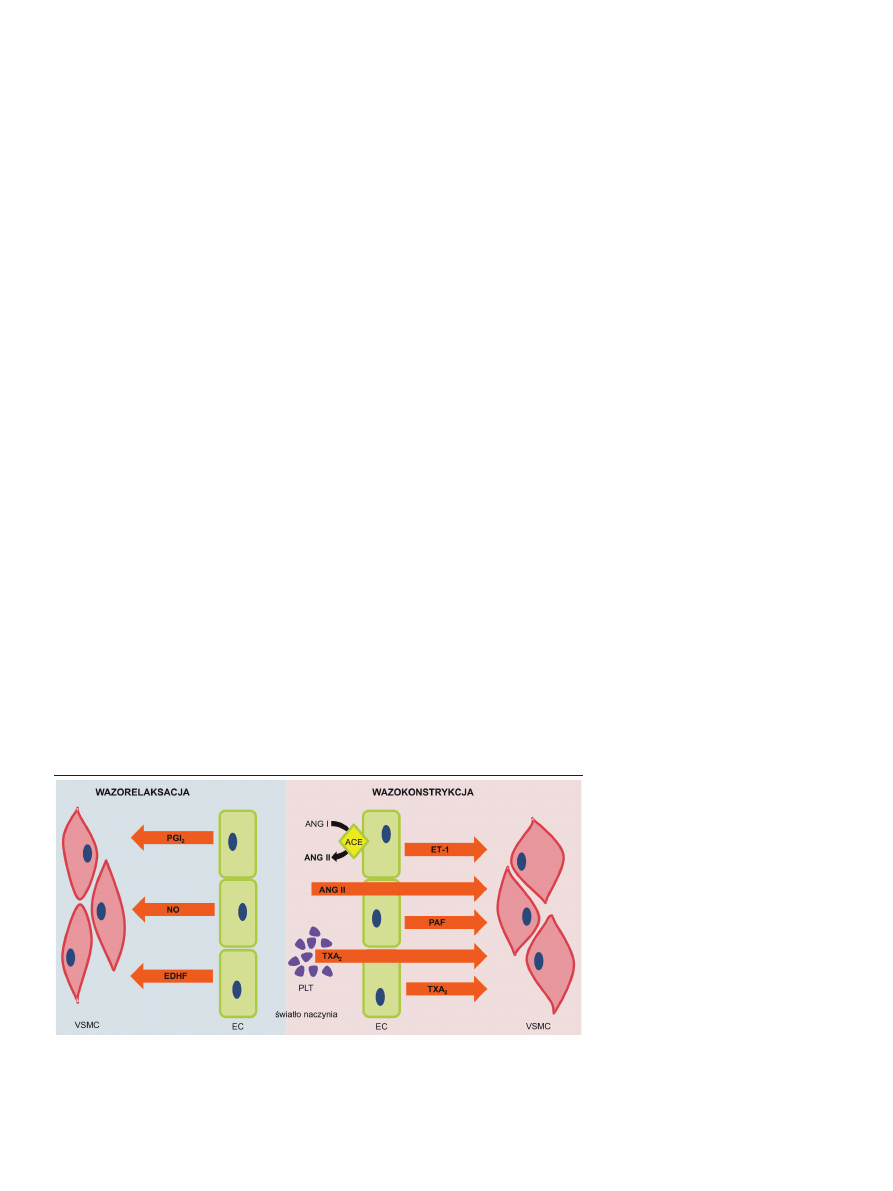
Bardzo ważną funkcją śródbłonka jest możliwość regula-
cji napięcia ściany naczyń krwionośnych poprzez wydziela-
nie różnych czynników powodujących wazorelaksację lub
wazokonstrykcję (Ryc. 2). Zdolność śródbłonka do wpływa-
nia na relaksację mięśniówki naczyniowej, została opisana
po raz pierwszy w 1980 roku, w pracy autorstwa Roberta
Furchgotta i Johna Zawadzkiego, w której autorzy postu-
lowali istnienie czynnika, któremu nadali nazwę śródbłon-
kowy czynnik rozluźniający (EDRF, ang. endothelial derived
relaxing factor) [8]. Niedługo później wykazano, że czynnik
ten to tlenek azotu (NO) [6], który jest wolnym rodnikiem o
aktywności biologicznej [2]. Może on kontrolować napięcie
ścian naczyń, stan naprężenia organów układu pokarmo-
wego, czy działać jako neuroprzekaźnik [9].
NO jest produktem reakcji katalizowanej przez syntazę
tlenku azotu (NOS, ang. nitric oxide synthase). Znane są trzy
izoenzymy NOS: neuronalna (nNOS), ulegająca ekspresji
w centralnym i obwodowym układzie nerwowym; indu-
kowalna (iNOS), która ulega ekspresji w wielu rodzajach
komórek pod wpływem różnych czynników, jak na przy-
kład czynnik martwicy nowotworu α (TNFα, ang. tumor
necrosis factor α) czy interleukina 1β (IL-1β) oraz śródbłon-
kowa (eNOS), której produkcję wykazano po raz pierw-
szy w komórkach śródbłonka, a później również między
innymi w komórkach wsierdzia i kardiomiocytach komór
serca [10,11]. Indukowalna syntaza tlenku azotu występuje
w formie dimeru, natomiast pozostałe dwa izoenzymy w
formach tetramerów, w których dodatkowymi podjednost-
kami są cząsteczki kalmoduliny, z których jedna przypada
na każdą cząsteczkę NOS. Ten fakt sprawia, że aktywność
nNOS i eNOS jest zależna, a iNOS jest niezależna od jonów
wapnia [10]. Każda cząsteczka NOS posiada stosunkowo
mocno związane kofaktory: (6R)-5,6,7,8-tetrahydrobiopte-
rynę (BH
4
), dinukleotyd flawinoadeninowy (FAD), mono-
nukleotyd flawinowy (FMN) oraz hem. W reakcji przez nie
katalizowanej dochodzi do utleniania azotu ugrupowania
guanidynowego L-argininy tlenem cząsteczkowym (O
2
), z
wykorzystaniem NADPH jako donora elektronów. Produk-
tami reakcji są L-cytrulina, NADP i tlenek azotu [10].
Śródbłonek uczestniczy w regulacji ciśnienia i przepływu
krwi przez uwalnianie związków o działaniu relaksacyj-
nym na mięśniówkę naczyń, takich jak NO, prostacyklina
(PGI
2
), śródbłonkowy czynnik hiperpolaryzujący (EDHF,
ang. endothelium derived hiperpolarizing factor) oraz czynni-
ków powodujących obkurczenie naczyń: endoteliny (ET),
tromboksanu A
2
(TXA
2
), angiotensyny II (ANG II), czynnika
aktywującego płytki (PAF, ang. platelet activating factor) czy
leukotrienów [2,4].
Interesującym faktem jest obecność w promotorze genu
Nos3 (kodującego białko eNOS) sekwencji rozpoznawanej
przez czynniki aktywowane przez naprężenia ścinające
(ang. shear stress). Dzięki temu prze-
pływ krwi wpływa na aktywację
transkrypcji genu Nos3 i w efekcie
prowadzi do wazodylatacji. Proces
ten wiąże się między innymi z me-
chanizmem regulacyjnym, odgrywa-
jącym istotną rolę w pozytywnych
skutkach treningu i ćwiczeń fizycz-
nych. Dochodzi wtedy do rozluźnie-
nia naczyń i zwiększenia przepływu
krwi [12].
Sam tlenek azotu działa na kilka
sposobów na układ naczyniowy.
NO jest nietrwały, a jego czas ży-
cia wynosi zaledwie kilka sekund.
Mimo to, bardzo szybko dociera do
komórek mięśniówki naczyniowej
ze względu na to, że jest silnie lipo-
filny, i po dotarciu na miejsce akty-
Rycina 2. Regulacja napięcia ściany naczyń krwionośnych. Śródbłonek za pośrednictwem wielu czynników wpły-
wa zarówno na relaksację, jak i skurcz mięśniówki naczyniowej. VSMC — komórki mięśniówki gładkiej naczyń;
PLT — płytki krwi; ACE — enzym konwertujący angiotensynę; ANG I i ANG II — angiotensyna I i II; EC — ko-
mórki śródbłonka.

Postępy Biochemii 59 (4) 2013
359
wuje rozpuszczalną cyklazę guanylanową, wiążąc się do
jej grupy prostetycznej, hemu [13]. W ten sposób, poprzez
stałe pobudzanie relaksacji mięśniówki naczyniowej, utrzy-
muje naprężenie ścian naczyń na podstawowym poziomie.
Ponadto, NO hamująco wpływa na przyleganie leukocytów
do śródbłonka. Upośledza również migrację i proliferację
komórek mięśni gładkich naczyń, jednocześnie stymulując
ruch i podziały komórek śródbłonka, co jest istotne podczas
odbudowy ściany uszkodzonego naczynia [2,14]. Kolejny
efekt, za który odpowiedzialny jest NO, to zapobieganie ak-
tywacji, agregacji i adhezji płytek krwi. Co więcej, wydaje
się, że NO może również pośrednio promować deagrega-
cję trombocytów poprzez upośledzanie aktywności kina-
zy 3-fosfatydyloinozytolu (PI3K). Zapobiega w ten sposób
zmianom konformacyjnym integryny α
IIb
β
IIIa
, umożliwia-
jącym oddziaływanie płytek krwi z fibrynogenem [2,14].
Dodatkowo, NO hamując syntezę P-selektyny w płytkach
krwi, blokuje napływ jonów wapnia do wnętrza trombocy-
tów i uniemożliwia zależną od jonów wapnia zmianę kon-
formacji integryny α
IIb
β
IIIa
[2].
Kolejnym czynnikiem powodującym relaksację naczyń
jest śródbłonkowy czynnik hiperpolaryzujący (EDHF). Po-
woduje on otwarcie kanałów potasowych komórek mięśni
gładkich, co prowadzi do zwiększonego wypływu jonów
potasu i hiperpolaryzacji ich błony komórkowej. Efekt wy-
wierany przez EDHF jest niezależny od tlenku azotu ani
prostacykliny, więc nie można go zablokować inhibitorami
NOS czy cyklooksygenazy, a jedynie za pomocą kombinacji
inhibitorów kanałów potasowych: apaminy z maurotoksy-
ną lub z TRAM-39 [15,16].
Kontrola napięcia mięśniówki gładkiej naczyń przez
śródbłonkową prostacyklinę (PGI
2
) jest również ściśle re-
gulowana. Prostacyklina nie jest stale produkowana, ani
przechowywana w komórkach, natomiast jej błyskawicz-
na synteza przez komórki śródbłonka jest odpowiedzią
na czynniki humoralne lub uszkodzenie mechaniczne [2].
PGI
2
jest eikozanoidem (lipidem, pochodną kwasu arachi-
donowego) o krótkim czasie życia, działającym parakryn-
nie na komórki. Receptor IP dla prostacykliny (sprzężony z
białkami G) jest obecny zarówno na płytkach krwi, jak i na
komórkach mięśni gładkich naczyń. PGI
2
może indukować
relaksację naczyń i hamować agregację płytek, jednak pełni
raczej funkcję koordynującą obkurczenie naczyń i agregację
płytek w uszkodzonych naczyniach, a nie regulującą pod-
stawowe naprężenie ścian naczyń [2,17]. Związkiem dzia-
łającym przeciwnie niż prostacyklina jest inny eikozanoid
- tromboxan A
2
(TXA
2
), produkowany głównie przez płytki
krwi, ale również przez komórki śródbłonka. TXA
2
powo-
duje silne obkurczenie naczyń, wzmożoną agregację płytek
krwi oraz rozrost mięśniówki naczyń [2,18].
Przeciwstawnie do prostacykliny działa również czynnik
aktywujący płytki (PAF, ang. platelet-activating factor). Jest
on fosfolipidem, nie podlega konstytutywnej produkcji, ani
nie jest przechowywany wewnątrz komórek, lecz podobnie
jak TXA
2
powstaje w odpowiedzi na czynniki humoralne i
stres mechaniczny. Działa on w sposób jukstakrynny, po-
zostaje przyłączony do powierzchni komórek śródbłonka i
tak oddziałuje z leukocytami. Receptor PAF jest receptorem
metabotropowym, sprzężonym z białkami G. PAF powodu-
je obkurczenie mięśniówki naczyniowej oraz promuje adhe-
zję leukocytów do śródbłonka [2,19].
Peptydy należące do rodziny endotelin pełnią wiele fizjo-
logicznych funkcji, takich jak modulowanie napięcia ścian
naczyń czy wpływ na proliferację. Znane są trzy endoteli-
ny: ET-1, ET-2 i ET-3, każda z nich jest peptydem liczącym
21 aminokwasów. Produkowane są przez wiele rodzajów
komórek [20]. Zdecydowanie najlepiej poznana jest ET-1,
powstająca w komórkach śródbłonka. W wyniku trans-
krypcji powstaje proendotelina-1, a późniejsza stymulacja
poprzez niedotlenienie czy naprężenia ścinające, prowadzi
do powstania aktywnego peptydu, przy udziale enzymu
konwertującego endotelinę [21]. Efekty ET-1 są wynikiem
oddziaływania z dwoma homologicznymi receptorami en-
dotelin A (ETA) i B (ETB). Są to powierzchniowe receptory
sprzężone z białkami G. Konsekwencją związania liganda
przez receptor jest wzrost wewnątrzkomórkowego stężenia
jonów wapnia w komórkach posiadających receptor [21].
Produkowana przez EC endotelina-1 w wyniku bodźca jest
wydzielana i wpływa na komórki mięśniówki naczynio-
wej przez receptor ETA, co prowadzi do obkurczenia na-
czynia. Co ciekawe, skurcz trwa przez jakiś czas nawet po
zakończeniu oddziaływania ET-1 z jej receptorem. Wynika
to ze znacznie dłużej utrzymującego się wewnątrz komórek
podniesionego stężenia Ca
2+
[2]. Z drugiej strony aktywacja
receptora ETB zlokalizowanego na komórkach śródbłonka
sprzyja wydzielaniu NO i PGI
2
przez śródbłonek. NO po-
woduje zmniejszenie uwalniania ET-1 w sprzężeniu zwrot-
nym, zaś ET-1 hamuje iNOS [4]. Efekt przedłużonego skur-
czu wynikający z podniesionego poziomu jonów wapnia
w komórkach mięśni gładkich jest zaburzany przez tlenek
azotu, który skutecznie obniża poziom wapnia wewnątrz
komórek. Oddziaływanie endoteliny-1 z jej receptorami
przyczynia się do utrzymywania podstawowego napięcia
mięśniówki naczyniowej, jednocześnie doprowadzając do
wazokonstrykcji jedynie w obrębie naczyń, gdzie doszło
do znacznego pobudzenia EC do wydzielania endoteliny-1.
ET-1 jest również mitogenem dla komórek mięśni gładkich
[2,4].
Komórki śródbłonka uczestniczą w przekształcaniu
angiotensyny I do angiotensyny II. Proces ten jest częścią
układu renina-angiotensyna-aldosteron. Przekształcenie
zachodzi na powierzchni śródbłonka wewnątrz naczyń,
dzięki aktywności konwertazy angiotensyny (ACE, ang.
angiotensin converting enzyme). ACE jest zlokalizowana w
błonie komórkowej śródbłonka, szczególnie w mocno una-
czynionych organach, takich jak płuca [22]. Powstała an-
giotensyna II między innymi powoduje skurcz mięśniówki
naczyniowej i proliferację komórek mięśni gładkich, syn-
tezę i wydzielanie aldosteronu przez korę nadnerczy, a w
konsekwencji wzrost ciśnienia krwi spowodowany wazo-
konstrykcją i aktywnością aldosteronu, który zwiększa re-
sorpcję Na
+
w kanalikach dystalnych w nerkach i prowadzi
do zatrzymania wody w organizmie [23,24].
ŚRÓDBŁONEK A HEMOSTAZA
Istotną rolą śródbłonka jest zaangażowanie w regulację
krzepnięcia krwi, poprzez udział w utrzymywaniu odpo-
wiedniej równowagi pomiędzy czynnikami pro- i antyko-

360
www.postepybiochemii.pl
agulacyjnymi [25], tak by w prawidłowych naczyniach nie
dochodziło do krzepnięcia, a w uszkodzonych nie docho-
dziło do powstawania nadmiernego zakrzepu [4]. Na po-
wierzchni śródbłonka i w warstwie podśródbłonkowej znaj-
duje się proteoglikan, siarczan heparanu, o właściwościach
antykoagulacyjnych, który może pełnić funkcję kofaktora
inhibitora proteaz serynowych, w tym obecnej w osoczu an-
tytrombiny III (ATIII). Ta produkowana w wątrobie gliko-
proteina hamuje aktywność enzymów układu krzepnięcia
między innymi trombiny i czynników IXa, Xa, XIa i XIIa.
Heparyna przyspiesza oddziaływanie ATIII z proteazami
serynowymi, co stało się podstawą przeciwzakrzepowej
terapii heparynowej. Lokalnie powstająca w naczyniach
trombina jest inaktywowana przez ATIII, której niewielkie
ilości znajdują się na proteoglikanie komórek śródbłonka.
Siarczan heparanu zlokalizowany jest w dużych ilościach
warstwie podśródbłonkowej, a naruszenie integralności
ściany naczynia może powodować jego odsłonięcie [26].
Na powierzchni ECs, szczególnie obficie w mikronaczy-
niach płuc, znajduje się integralne białko błonowe trombo-
modulina, która oddziałując z trombiną obecną w osoczu
przyczynia się do zmiany jej konformacji. Tak aktywowana
trombina może działać jako jedyny czynnik mogący akty-
wować białko C, które z kolei w obecności kofaktora, białka
S, inaktywuje czynniki Va i VIIIa i działa antykoagulacjnie
[25,26]. Ponadto, wydzielana przez komórki śródbłonka
prostacyklina powoduje rozluźnienie komórek mięśni gład-
kich naczynia i zaburza oddziaływanie monocytów ze śród-
błonkiem [26].
PGI
2
i NO hamują agregację i adhezję płytek krwi [27].
Na aktywację płytek krwi może również wpływać ATP i
ADP. Ektonukleazy wytwarzane przez komórki śródbłon-
ka (ektodifosfataza i ektotrifosfataza adenozyny) rozkładają
ADP do inozyny, a ATP do adenozyny [28], w ten sposób
usuwając czynniki aktywujące płytki krwi. Co więcej, prze-
ciwzakrzepowa aktywność komórek śródbłonka polega
również na produkcji, wraz z komórkami wątroby, inhibito-
ra zewnątrzpochodnej drogi układu krzepnięcia zależnej od
czynnika tkankowego (TFPI, ang. tissue factor pathway inhi-
bitor), który w obecności czynnika Xa jest zdolny do hamo-
wania aktywności kompleksu czynnika tkankowego (TF,
ang. tissue factor) i czynnika VIIa [25]. Co ciekawe, poziom
TFPI w osoczu w przypadkach ostrej niewydolności wątro-
by jest niezmieniony, w przeciwieństwie do przypadków
zespołu rozsianego wykrzepiania wewnątrznaczyniowego
(DIC), gdzie odnotowano obniżony poziom TFPI, co może
wskazywać właśnie na śródbłonek jako główne źródło tego
inhibitora [26].
Z drugiej strony, komórki śródbłonka są, wraz z mega-
kariocytami, miejscem syntezy czynnika von Willebranda
(vWF), który odgrywa szczególną rolę w procesie adhezji
i agregacji płytek krwi. Czynnik ten jest stale uwalniany do
krwioobiegu, ale pewna jego pula jest również przechowy-
wana w cytoplazmie komórek śródbłonka w ciałkach We-
ibel-Palade’a. Znaleźć go można również w ziarnistościach
α w płytkach krwi oraz w podśródbłonkowej warstwie
tkanki łącznej [29]. VWF jest ligandem umożliwiającym
adhezję płytek krwi w miejscu uszkodzenia warstwy śród-
błonka naczynia i odsłonięcia tkanki łącznej, ponieważ wią-
że on powierzchniowe glikoproteiny płytek krwi i białka
warstwy podśródbłonkowej np. kolageny. Może również
wiązać VIII czynnik krzepnięcia i w ten sposób stabilizować
jego aktywność. Zmniejszona ilość i aktywność czynnika
vWF wywołuje objawy skazy krwotocznej, w tym krwawe
wylewy podskórne, wylewy do stawów i mięśni, liczne i
obfite krwawienia z dziąseł, nosa, lub po ekstrakcji zębów i
zabiegach chirurgicznych, które opisywane są jako choroba
von Willebranda [26,29].
Śródbłonek jest zaangażowany także w procesy fibryno-
lizy. W komórkach śródbłonka zachodzi synteza proteazy
serynowej, tkankowego aktywatora plazminogenu (t-PA),
który jest uwalniany do krwiobiegu. Uwolniony t-PA po-
przez ograniczoną proteolizę aktywuje plazminogen w
niewielkim stopniu, aż do momentu, gdy ma możliwość
oddziaływania z włóknikiem. Obecność włóknika zwiększa
zdolność t-PA do aktywacji plazminogenu nawet tysiąc-
krotnie. Tak powstająca plazmina może degradować białka
tworzące skrzep [30].
ŚRÓDBŁONEK W POWSTAWANIU NOWYCH
NACZYŃ KRWIONOŚNYCH
Naczynia krwionośne powstają na różne sposoby, albo w
drodze waskulogenezy, czyli de novo głównie w okresie roz-
woju zarodkowego, albo angiogenezy, a więc z naczyń już
istniejących, poprzez tworzenie odgałęzień przez migrujące
dojrzałe komórki śródbłonka (ang. sprouting angiogenesis)
lub przez podział wzdłuż długiej osi naczyń (ang. intus-
susceptive angiogenesis). Początkowo panował pogląd, że w
dorosłych organizmach zachodzą jedynie procesy angio-
genezy, a waskulogeneza jest charakterystyczna wyłącznie
dla rozwoju zarodkowego. W roku 1997 opisano jednak w
szpiku kostnym komórki funkcjonujące jak progenitorowe
komórki śródbłonka (EPCs, ang. endothelial progenitor cell)
[31,32]. Asahara i współodkrywcy EPCs scharakteryzowa-
li je jako komórki jednojądrzaste, pobierające acetylowane
cząsteczki LDL, wiążące lektyny oraz zdolne do adhezji do
białek macierzy zewnątrzkomórkowej (ECM, ang. extra-
cellular matrix). W komórkach tych dochodzi do syntezy
CD31, CD34, eNOS, E-selektyny oraz receptora VEGF typu
2 (VEGFR-2) [33]. Wykazano też, że EPCs uwalniane są ze
szpiku kostnego, a krążąc we krwi mogą nie tylko wbudo-
wywać się w nowo powstające naczynia, ale również do
miejsc uszkodzenia [34]. Badania ostatnich lat pokazują jed-
nak, że EPC są raczej proangiogennymi monocytami, mogą-
cymi przyczyniać się do tworzenia nowych oraz regeneracji
uszkodzonych naczyń krwionośnych głównie w sposób pa-
rakrynny, wydzielając w miejscu uszkodzenia odpowiednie
czynniki wspomagające te procesy [35,36]. Stąd też ostatnio
częściej opisuje się je jako proangiogenne komórki pocho-
dzenia szpikowego (BMDC, ang. bone marrow-derived endo-
thelial cells).
Co ciekawe, Fang i współpracownicy opisali w 2012 roku
na łamach czasopisma PLoS Biology populację komórek
macierzystych śródbłonka naczyniowego rezydujących w
obrębie ściany naczyń [37]. Komórki te charakteryzują się
między innymi obecnością markerów śródbłonkowych
(CD31 i CD105), markerów komórek macierzystych (Sca-
1 i CD117 — c-kit), a jednocześnie nie wykazują obecności
markerów linii hematopoetycznych. Komórki śródbłonka

Postępy Biochemii 59 (4) 2013
361
wywodzące się z lokalnych komórek macierzystych śród-
błonka c-kit+ potrafią utworzyć funkcjonalne naczynia w
teście in vivo, oraz mają zdolność do samoodnowy. U my-
szy KitW-sh, u których synteza c-kit jest zaburzona, liczba
lokalnych komórek macierzystych śródbłonka jest niższa,
a wzrost i unaczynienie nowotworów niższe niż u myszy
typu dzikiego [37].
Proces angiogenezy przebiega w kilku etapach. Naj-
pierw, w odpowiedzi na NO i w wyniku działania czynni-
ka wzrostu śródbłonka naczyniowego (VEGF, ang. vascular
endothelial growth factor), dochodzi do wazodylatacji oraz
do zwiększenia przepuszczalności naczyń krwionośnych.
Powoduje to wydostanie się białek osocza poza naczynia.
Przeciwstawne do VEGF działanie wykazuje angiopoety-
na 1, która wiążąc swój receptor Tie-2 uszczelnia naczynia
krwionośne [38]. Następnym etapem w procesie tworzenia
nowych naczyń jest rozluźnienie połączeń pomiędzy ko-
mórkami śródbłonka i rozpoczęcie trawienia macierzy ze-
wnątrzkomórkowej, któremu towarzyszy uwalnianie czyn-
ników proangiogennych zgromadzonych w ECM, takich jak
VEGF czy zasadowy czynnik wzrostu fibroblastów (bFGF,
ang. basic fibroblast growth factor) [38]. Następnie rozpoczyna
się migracja i proliferacja komórek śródbłonka, stymulowa-
na przez oddziaływanie VEGF z VEGFR-2. Migrujące ECs
tworzą odgałęzienie, a po czasie wykształcają światło no-
wego naczynia. Gdy dojdzie do powstania funkcjonalnego
naczynia, a przede wszystkim, gdy rozpocznie się przepływ
krwi, następują kolejne procesy dojrzewania naczynia, w
których dochodzi do rekrutacji perycytów i komórek mięśni
gładkich oraz stabilizacji naczynia [38].
Powstawanie nowych naczyń krwionośnych jest bar-
dzo złożonym procesem, ważnym dla organizmu. Odgry-
wa istotną rolę między innymi w gojeniu ran czy podczas
odbudowy endometrium po menstruacji, jednak może być
również procesem patologicznym istotnym w przerzutowa-
niu nowotworów czy rozwoju retinopatii [38]. Badanie roli
śródbłonka w angiogenezie i dokładne zrozumienie me-
chanizmów tego procesu może pozwolić na opracowanie
nowych terapii schorzeń, w których powstawanie naczyń
odgrywa istotną rolę.
DYSFUNKCJA ŚRÓDBŁONKA
W CHOROBACH CYWILIZACYJNYCH
MIAŻDŻYCA
Zaburzenia metaboliczne takie jak hiperlipidemia i hiper-
glikemia, a także palenie tytoniu czy sam proces starzenia
się, mogą prowadzić do zmiany funkcji komórek śródbłon-
ka i powstawania schorzeń, w tym miażdżycy. Miażdżyca
to złożona jednostka chorobowa rozwijająca się w ścianach
tętnic w odpowiedzi na różne czynniki uszkadzające na-
czynie, indukujące nadmierny stan zapalny i powodujące
pogrubienie i włóknienie ściany naczynia [39]. Zaburzenie
funkcji komórek śródbłonka jest istotne w powstawaniu
zmian chorobowych.
Do uszkodzenia naczyń często przyczynia się podwyż-
szony poziom cholesterolu we krwi. Stan ten może wyni-
kać zarówno z nadprodukcji lipoprotein w wątrobie, nie-
prawidłowej diety [40], czy zaburzenia pobierania lipopro-
tein z krwi spowodowanego mutacjami receptorów lub
ligandów uczestniczących w tym procesie [41]. Tworzenie
zmian miażdżycowych przebiega na ogół w podobny spo-
sób, szczególnie, gdy rozpatruje się obszary podatne na
rozwój schorzenia, takie jak zastawki serca, tętnice szyjne i
wieńcowe oraz łuk aorty. Do zmian przyczynia się również
sprzyjający miażdżycy turbulentny przepływ krwi [42].
Komórki śródbłonka bezpośrednio narażone na kontakt z
krwią bogatą w cholesterol i triglicerydy podlegają zmia-
nie fenotypu na tzw. fenotyp wydzielniczy (powiększenie
siateczki śródplazmatycznej i aparatu Golgiego, zmiany w
strukturze cytoszkieletu) [43]. Zmianie tej towarzyszy roz-
rost błony podstawnej. Dochodzi do akumulacji lipoprotein
β pod warstwą komórek śródbłonka, w przerośniętej błonie
podstawnej i poza nią; tam mogą one oddziaływać z prote-
oglikanami i białkami ECM, czy ulegać utlenieniu i innym
modyfikacjom. Do przeniesienia lipoprotein dochodzi na
drodze transcytozy. W czasie transportu modyfikowanych
lipoprotein do warstwy podśródbłonkowej może dochodzić
do ich degradacji w komórkach śródbłonka [44]. W efekcie
w ECs odkładają się duże ilości lipidów, co prowadzi do
powstawania tzw. komórek piankowatych pochodzenia
śródbłonkowego [45]. W zaawansowanej miażdżycy taki
wygląd przyjmują również inne komórki tworzące płytkę
miażdżycową, takie jak makrofagi czy komórki mięśniówki
gładkiej [43].
Długotrwałe zaburzenia gospodarki lipidowej mogą
prowadzić do powstawania reaktywnych form tlenu (RFT),
takich jak anionorodnik ponadtlenkowy czy nadtlenek wo-
doru, które są w stanie aktywować komórki śródbłonka, co
jest istotne w początkowych etapach rozwoju miażdżycy.
Wytwarzanie RFT w komórkach naczyń przez różne en-
zymy utleniające (oksydazy NADPH, lipoksygenazę, cyto-
chrom P450, oksygenazę hemową-1 (HO-1)) oraz w reak-
cjach rozprzęgania łańcucha oddechowego i przez rozprzę-
żony eNOS, jest równoważone przez aktywność enzymów
antyoksydacyjnych (katalazy, dysmutazy ponadtlenkowej,
peroksydazy glutationowej) [40,46].
Bardzo niebezpieczne jest zmniejszenie ilości tlenku
azotu w wyniku obniżenia aktywności i ekspresji lub roz-
przęgania eNOS [47]. To ostatnie zjawisko jest szczególnie
istotne, może bowiem prowadzić do przekazywania elek-
tronów z NADPH na tlen zamiast na L-argininę, co skut-
kuje powstaniem anionorodnika ponadtlenkowego zamiast
tlenku azotu. Przyczyną takiego przebiegu reakcji może być
niedostępność L-argininy, a także oksydacyjna degradacja
tetrahydrobiopteryny, kofaktora eNOS. Pula tlenku azo-
tu może ulec dalszej redukcji w wyniku reakcji NO z O
2
·-
,
w której powstaje nadtlenoazotyn (ONOO
-
), bardzo moc-
ny utleniacz. Wykazano, że może on hamować aktywność
syntazy prostacykliny, a więc upośledzać wazorelaksację
nie tylko wywoływaną przez tlenek azotu, ale także przez
PGI
2
. Co więcej, ONOO
-
może przyczyniać się do utleniania
BH
4
, jeszcze bardziej rozprzęgając eNOS. Częściowo utle-
niona tetrahydrobiopteryna nie pełni roli kofaktora NOS,
ale współzawodnicząc z BH
4
dodatkowo zaburza produk-
cję NO [47].
Na rozwój miażdżycy znaczny wpływ ma również prze-
wlekły stan zapalny i nieprawidłowe działanie układu

362
www.postepybiochemii.pl
odpornościowego. W aktywowanych w toku choroby ko-
mórkach śródbłonka dochodzi do zwiększenia wydzielania
cytokin prozapalnych i produkcji molekuł adhezyjnych, co
prowokuje napływ komórek układu odpornościowego w
obszary zmienione miażdżycowo. Wnikanie makrofagów
czy limfocytów do uszkodzonego naczynia przyczynia
się do powiększania i destabilizacji blaszki miażdżycowej
[44,48]. Znaczny rozrost może powodować zaburzenia w
przepływie krwi przez naczynia. Co więcej, destabilizacja
blaszki może prowadzić do jej pęknięcia i powstawania za-
krzepów, których szkodliwość zależy od umiejscowienia. W
efekcie może dojść do niedrożności naczyń i niedotlenienia
obszarów zaopatrywanych w krew przez zatkane naczynia.
W najgorszym wypadku może to prowadzić do ostrego ze-
społu wieńcowego, uszkodzenia mięśnia sercowego i nawet
śmierci [5].
NADCIŚNIENIE TĘTNICZE
Rozwój nadciśnienia tętniczego jest bezpośrednio zwią-
zany z regulacją napięcia ścian naczyń krwionośnych przez
śródbłonek. Szczególnie ważne są czynniki powodujące
obkurczenie mięśniówki naczyniowej, takie jak endoteli-
na-1 czy TXA
2
[4]. Istotną rolę w patogenezie nadciśnienia
odgrywają reaktywne formy tlenu, zarówno poprzez pod-
noszenie ciśnienia krwi jak i wpływ na przebudowę ścian
naczyń. Zubożenie puli NO w warunkach stresu oksyda-
cyjnego prowadzi do zaburzenia funkcji śródbłonka, a RFT
mogą stymulować proliferację, migrację i apoptozę komó-
rek, wywołując jednocześnie stan zapalny [49]. Usunięcie
czynników wazorelaksacyjnych, czyli zaburzenie równo-
wagi pomiędzy mechanizmami relaksacji i skurczu naczyń
może prowadzić do przerostu ścian naczyń, utraty ich ela-
styczności i w konsekwencji do rozwoju zmian miażdżyco-
wych i nadciśnienia tętniczego [49].
CUKRZYCA
Przykładem schorzenia, w którego patologii istotną rolę
odgrywają komórki śródbłonka jest cukrzyca. Wyróżnić
można dwa główne rodzaje tej choroby: cukrzycę typu 1
(T1DM, ang. type 1 diabetes mellitus) i o wiele częstszą cu-
krzycę typu 2 (T2DM, ang. type 2 diabetes mellitus). U pod-
łoża T1DM leży autoreaktywność układu odpornościo-
wego, która prowadzi do zniszczenia komórek β trzustki,
produkujących insulinę i kontrolujących stężenie glukozy
we krwi [50]. Rozwój T2DM jest natomiast spowodowany
insulinoopornością, której wynikiem są zaburzenia meta-
bolizmu i wchłaniania glukozy. Zwiększona produkcja in-
suliny, mająca kompensować oporność, prowadzi do trwa-
łego uszkodzenia, a następnie apoptozy komórek trzustki
i nieodwracalnego obniżenia produkcji hormonu. Efektem
jest hiperglikemia prowadząca do powikłań cukrzycowych
takich jak stopa cukrzycowa, nefropatia, neuropatia, czy
retinopatia cukrzycowa [51]. Przy hiperglikemii glukoza
jest wykorzystywana do syntezy ATP, jednocześnie jednak
dochodzi do znacznie zwiększonej produkcji reaktywnych
form tlenu, co prowadzi do dysfunkcji komórek śródbłonka.
Istotną rolę w powstawaniu RFT odgrywa szlak sorbitolu,
w którym reduktaza aldozowa katalizuje przekształcenie
glukozy w sorbitol. W procesie tym zużywany jest NAD(P)
H pochodzący ze szlaku pentozofosforanowego. W warun-
kach hiperglikemii przemiany glukozy w sorbitol ulegają
intensyfikacji, a jednocześnie reduktaza aldozowa zużywa
więcej NAD(P)H. Wpływa to na pojemność przeciwutlenia-
jącą komórek poprzez wyczerpanie zapasów zredukowane-
go glutationu i w konsekwencji zahamowanie aktywności
peroksydazy glutationowej [52]. W cukrzycy, zwłaszcza
źle wyrównanej, nadmiar glukozy przyczynia się także do
niespecyficznej glikozylacji białek i tworzenia końcowych
produktów zaawansowanej glikacji (AGE, ang. advanced
glycation end-products). To właśnie RFT i AGE zaburzają
funkcjonowanie komórek śródbłonka poprzez ograniczenie
biodostępności tlenku azotu, wzmożoną produkcję wol-
nych rodników, czynników wzrostowych i cytokin proza-
palnych. Zwiększają także syntezę molekuł adhezyjnych, co
prowadzi do nadmiernej migracji komórek układu odpor-
nościowego i adhezji płytek krwi, przyczyniając się do po-
wstawania zmian miażdżycowych i zmniejszenia elastycz-
ności naczyń [31]. Ponadto, w wielu badaniach wykazano
upośledzenie mobilizacji i funkcji komórek EPC/BMDC
spowodowane hiperglikemią [53-57]. Komórki wykazywa-
ły mniejszą produkcję eNOS i VEGF, charakteryzowały się
upośledzoną migracją, osłabioną odpowiedzią na stromal-
ny czynnik wzrostu-1 (SDF-1, ang. stromal cell-derived factor
1), mniejszym potencjałem angiogennym oraz prozapalnym
fenotypem (zwiększoną produkcją IL-12 i wydajniejszą niż
w wypadku komórek osób zdrowych aktywacją limfocytów
T) [58]. To wszystko może przyczyniać się do powstawania
powikłań cukrzycowych, w tym powstawania trudno goją-
cych się ran i owrzodzeń [31].
Innym istotnym powikłaniem cukrzycy są mikroangio-
patie, takie jak retinopatia cukrzycowa. Kapilary w siatków-
ce oka zostają uszkodzone w dużej mierze w wyniku zabu-
rzenia oddziaływania pericytów z komórkami śródbłonka.
Niewłaściwe funkcjonowanie kapilar prowadzi do niedo-
tlenienia, zwiększającego produkcję VEGF w siatkówce.
W konsekwencji dochodzi do rozrostu niefunkcjonalnych
kapilar, które zaburzają strukturę siatkówki. Powstałe na-
czynia charakteryzują się zwiększoną przepuszczalnością,
co nasila procesy zapalne. Zmiany te mogą prowadzić do
utraty wzroku [59].
Nefropatie i neuropatie to także przykłady naczyniowych
powikłań cukrzycowych. W nefropatii dochodzi do niepra-
widłowej angiogenezy w kłębuszkach nerkowych spowo-
dowanej zwiększoną produkcją VEGF, co w konsekwencji
może prowadzić do przewlekłej niewydolności nerek [59].
W wypadku neuropatii AGE wpływają na metaloproteina-
zy macierzy zewnątrzkomórkowej, które mogą uszkadzać
włókna nerwowe oraz na monocyty i komórki śródbłon-
ka, powodując zwiększenie produkcji cytokin i cząsteczek
adhezyjnych. Prawdopodobnie pogrubienie i hialinizacja
ścian małych naczyń może powodować niedotlenienie ner-
wów. Wszystko to przyczynia się do uszkadzania neuro-
nów i rozwoju neuropatii objawiających się między innymi
bólem i zaburzeniami czucia [60].
NOWOTWORY
Procesy angiogenezy i limfangiogenezy prowadzą do
powstawania naczyń krwionośnych również w obrębie
guzów nowotworowych. Może to być warunek niezbędny

Postępy Biochemii 59 (4) 2013
363
do przyspieszenia wzrostu guza oraz ułatwienia przerzu-
towania drogą naczyniową bądź chłonną. Budowa naczyń
zaopatrujących w krew guzy jest na ogół inna niż w wy-
padku zwykłych naczyń. Mają one chaotyczny przebieg i
nieprawidłową morfologię, bardzo często charakteryzują
się zwiększoną przepuszczalnością. Komórki śródbłonka
mają nieregularne kształty i często tworzą wypustki sięga-
jące poza naczynie lub przechodzące przez jego światło. W
niektórych nowotworach istotną rolę może też odgrywać
mimikra naczyniowa, gdy komórki nowotworowe współ-
tworzą naczynia guza lub tworzą kanały przypominające
funkcjonalnie naczynia krwionośne [7].
PODSUMOWANIE
Śródbłonek w pełni zasługuje na miano organu. Spełnia
wiele funkcji służących utrzymaniu homeostazy, regulując
przepływ i ciśnienie krwi, syntezę i wydzielanie czynników
aktywnych biologicznie, kontrolując przepływ substancji
odżywczych, angiogenezę czy reakcje zapalne i odporno-
ściowe. To właśnie wielofunkcyjność śródbłonka sprawia,
że nieprawidłowości w jego działaniu przyczyniają się do
zaburzeń prowadzących do rozwoju różnych stanów choro-
bowych, takich jak choroba von Willebranda, nadciśnienie
tętnicze, choroba niedokrwienna serca, czy miażdżyca tęt-
nic obwodowych. Śródbłonek odgrywa też decydującą rolę
w rozwoju powikłań cukrzycy oraz w unaczynianiu guzów
nowotworowych. Pomimo, że wiedza na temat śródbłonka
jest już obecnie bardzo rozległa wciąż konieczne są badania
pozwalające lepiej poznać mechanizmy molekularne regu-
lujące poszczególne aktywności komórek śródbłonkowych.
Ich zrozumienie jest również niezbędne przy opracowywa-
niu nowych terapii i leków.
PIŚMIENNICTWO
1. Fishman AP (1982) Endothelium: a distributed organ of diverse capa-
bilities. Ann N Y Acad Sci 401: 1-8
2. Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McE-
ver RP, Pober JS, Wick TM, Konkle BA, Schwartz BS, Barnathan ES,
McCrae KR, Hug BA, Schmidt AM, Stern DM (1998) Endothelial cells
in physiology and in the pathophysiology of vascular disorders. Blood
91: 3527-3561
3. Abbott NJ, Rönnbäck L, Hansson E (2006) Astrocyte-endothelial inte-
ractions at the blood-brain barrier. Nat Rev Neurosci 7: 41-53
4. Wnuczko K, Szczepański M (2007) Śródbłonek - charakterystyka i
funkcje. Pol Merkur Lekarski 23: 60-65
5. Lerman A, Zeiher AM (2005) Endothelial function: cardiac events. Cir-
culation 111: 363-368
6. Deanfield JE, Halcox JP, Rabelink TJ (2007) Endothelial function and
dysfunction: testing and clinical relevance. Circulation 115: 1285-1295
7. Dudley AC (2012) Tumor endothelial cells. Cold Spring Harbor Per-
spectives in Medicine 2: a006536
8. Furchgott RF, Zawadzki JV (1980) The obligatory role of endothelial
cells in the relaxation of arterial smooth muscle by acetylcholine. Na-
ture 288: 373-376
9. Forstermann U, Kleinert H (1995) Nitric oxide synthase: expression
and expressional control of the three isoforms. Naunyn Schmiede-
bergs Arch Pharmacol 352: 351-364
10. Alderton WK, Cooper CE, Knowles RG (2001) Nitric oxide synthases:
structure, function and inhibition. Biochem J 357: 593-615
11. Felaco M, Grilli A, De Lutiis MA, Patruno A, Libertini N, Taccardi AA,
Di Napoli P, Di Giulio C, Barbacane R, Conti P (2001) Endothelial ni-
tric oxide synthase (eNOS) expression and localization in healthy and
diabetic rat hearts. Ann Clin Lab Sci 31: 179-186
12. Venema RC, Nishida K, Alexander RW, Harrison DG, Murphy TJ
(1994) Organization of the bovine gene encoding the endothelial nitric
oxide synthase. Biochim Biophys Acta 1218: 413-420
13. Bellamy TC, Wood J, Garthwaite J (2002) On the activation of soluble
guanylyl cyclase by nitric oxide. Proc Natl Acad Sci USA 99: 507-510
14. Pigazzi A, Heydrick S, Folli F, Benoit S, Michelson A, Loscalzo J (1999)
Nitric oxide inhibits thrombin receptor-activating peptide-induced
phosphoinositide 3-kinase activity in human platelets. J Biol Chem
274: 14368-14375
15. Hinton JM, Langton PD (2003) Inhibition of EDHF by two new com-
binations of K
+
-channel inhibitors in rat isolated mesenteric arteries.
British J Pharmacol 138: 1031-1035
16. Ozkor MA, Quyyumi AA (2011) Endothelium-derived hyperpolari-
zing factor and vascular function. Cardiol Res Pract 2011: 156146
17. Katusic ZS, Santhanam AV, He T (2012) Vascular effects of prostacyc-
lin: does activation of PPARδ play a role? Trends Pharmacol Sci 33:
559-564
18. Ting HJ, Murad JP, Espinosa EVP, Khasawneh FT (2012) Thromboxa-
ne A2 receptor: biology and function of a peculiar receptor that rema-
ins resistant for therapeutic targeting. J Cardiovasc Pharmacol Ther 17:
248-259
19. Pober JS, Sessa WC (2007) Evolving functions of endothelial cells in
inflammation. Nat Rev Immunol 7: 803-815
20. Drawnel FM, Archer CR, Roderick HL (2013) The role of the paracri-
ne/autocrine mediator endothelin-1 in regulation of cardiac contracti-
lity and growth. Br J Pharmacol 168: 296-317
21. Jain A (2013) Endothelin-1-induced endoplasmic reticulum stress in
disease. J Pharmacol Exp Ther 346: 163-172
22. Bernstein KE, Ong FS, Blackwell W-LB, Shah KH, Giani JF, Gonzalez-
-Villalobos RA, Shen XZ, Fuchs S, Touyz RM (2013) A modern under-
standing of the traditional and nontraditional biological functions of
angiotensin-converting enzyme. Pharmacol Rev 65: 1-46
23. Unger T (2002) The role of the renin-angiotensin system in the deve-
lopment of cardiovascular disease. Amer J Cardiol 89: 3A-9A
24. Briet M, Schiffrin EL (2010) Aldosterone: effects on the kidney and car-
diovascular system. Nature Rev Nephrol 6: 261-273
25. Aird WC (2004) Natural anticoagulant inhibitors: activated Protein C.
Best Pract Res Clin Haematol 17: 161-182
26. Wu KK, Thiagarajan P (1996) Role of endothelium in thrombosis and
hemostasis. Annu Rev Med 47: 315-331
27. Rubanyi GM (1993) The role of endothelium in cardiovascular home-
ostasis and diseases. J Cardiovasc Pharmacol 22 Suppl 4: S1-14
28. Yegutkin GG, Henttinen T, Samburski SS, Spychala J, Jalkanen S (2002)
The evidence for two opposite, ATP-generating and ATP-consuming,
extracellular pathways on endothelial and lymphoid cells. Biochem J
367: 121-128
29. Sadler JE (1998) Biochemistry and genetics of von Willebrand factor.
Annu Rev Biochem 67: 395-424
30. Dobrovolsky AB, Titaeva EV (2002) The fibrinolysis system: regulation
of activity and physiologic functions of its main components. Bioche-
mistry Mosc 67: 99-108
31. Kotlinowski J, Kozakowska M (2010) Cukrzyca a komórki progenito-
rowe śródbłonka. Post Biol Kom 37: 153-165
32. Carmeliet P (2003) Angiogenesis in health and disease. Nat Med 9: 653-
660
33. Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T,
Witzenbichler B, Schatteman G, Isner JM (1997) Isolation of putative
progenitor endothelial cells for angiogenesis. Science 275: 964-967
34. Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Ke-
arne M, Magner M, Isner JM (1999) Bone marrow origin of endothelial
progenitor cells responsible for postnatal vasculogenesis in physiolo-
gical and pathological neovascularization. Circ Res 85: 221-228
35. Rohde E, Malischnik C, Thaler D, Maierhofer T, Linkesch W, Lanzer G,
Guelly C, Strunk D (2006) Blood monocytes mimic endothelial proge-
nitor cells. Stem Cells 24: 357-367
364
www.postepybiochemii.pl
Endothelium in physiology and pathogenesis of diseases
Mateusz Tomczyk, Witold Nowak, Agnieszka Jaźwa
*
Department of Medical Biotechnology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, 7 Gronostajowa St., 30-387
Krakow, Poland
*
e-mail: agnieszka.jazwa@uj.edu.pl
Key words: angiogenesis, diabetes, dysfunction of endothelium, atherosclerosis, oxidative stress
ABSTRACT
Vascular endothelium is the layer of cells that line blood vessels and serve as the primary barrier between the blood and the tissues. These
cells play many important functions such as regulation of the vascular tone, blood flow and pressure control, maintaining the balance be-
tween thrombosis and fibrinolysis, participation in immune system reactions and new blood vessels formation. Disturbance in any of these
functions may contribute to the development of different diseases such as for e.g. hypertension, atherosclerosis and deep vein thrombosis, as
well as cancer.
36. Pearson JD (2010) Endothelial progenitor cells - an evolving story. Mi-
crovasc Res 79: 162-168
37. Fang S, Wei J, Pentinmikko N, Leinonen H, Salven P (2012) Generation
of functional blood vessels from a single c-kit+ adult vascular endothe-
lial stem cell. PLoS Biol 10: e1001407
38. Carmeliet P (2000) Mechanisms of angiogenesis and arteriogenesis.
Nat Med 6: 389-395
39. Libby P, Ridker PM, Maseri A (2002) Inflammation and atherosclero-
sis. Circulation 105: 1135-1143
40. Stapleton PA, Goodwill AG, James ME, Brock RW, Frisbee JC (2010)
Hypercholesterolemia and microvascular dysfunction: interventional
strategies. J Inflamm (Lond) 7: 54
41. Gill PJ, Harnden A, Karpe F (2012) Familial hypercholesterolaemia.
BMJ 344: e3228
42. Malek AM, Alper SL, Izumo S (1999) Hemodynamic shear stress and
its role in atherosclerosis. JAMA 282: 2035-2042
43. Simionescu M (2007) Implications of early structural-functional chan-
ges in the endothelium for vascular disease. Arterioscler Thromb Vasc
Biol 27: 266-274
44. Sitia S, Tomasoni L, Atzeni F, Ambrosio G, Cordiano C, Catapano A,
Tramontana S, Perticone F, Naccarato P, Camici P, Picano E, Cortigiani
L, Bevilacqua M, Milazzo L, Cusi D, Barlassina C, Sarzi-Puttini P, Tu-
riel M (2010) From endothelial dysfunction to atherosclerosis. Autoim-
mun Rev 9: 830-834
45. Yu X-H, Fu Y-C, Zhang D-W, Yin K, Tang C-K (2013) Foam cells in
atherosclerosis. Clin Chim Acta 424: 245-252
46. Bonomini F, Tengattini S, Fabiano A, Bianchi R, Rezzani R (2008) Athe-
rosclerosis and oxidative stress. Histol Histopathol 23: 381-390
47. Karbach S, Wenzel P, Waisman A, Münzel T, Daiber A (2013) eNOS
uncoupling in cardiovascular diseases - the role of oxidative stress and
inflammation. Curr Pharm Des, w druku
48. Sima AV, Stancu CS, Simionescu M (2009) Vascular endothelium in
atherosclerosis. Cell Tissue Res 335: 191-203
49. Schulz E, Gori T, Munzel T (2011) Oxidative stress and endothelial
dysfunction in hypertension. Hypertension Res 34: 665-673
50. Bluestone JA, Herold K, Eisenbarth G (2010) Genetics, pathogenesis
and clinical interventions in type 1 diabetes. Nature 464: 1293-1300
51. Fowler MJ (2008) Microvascular and macrovascular complications of
diabetes. Clin Diabetes 26: 77-82
52. Forbes JM, Cooper ME (2013) Mechanisms of diabetic complications.
Physiol Rev 93: 137-188
53. Tamarat R, Silvestre J-S, Le Ricousse-Roussanne S, Barateau V, Le-
comte-Raclet L, Clergue M, Duriez M, Tobelem G, Lévy BI (2004) Im-
pairment in ischemia-induced neovascularization in diabetes: bone
marrow mononuclear cell dysfunction and therapeutic potential of
placenta growth factor treatment. Am J Pathol 164: 457-466
54. Kränkel N, Adams V, Linke A, Gielen S, Erbs S, Lenk K, Schuler G,
Hambrecht R (2005) Hyperglycemia reduces survival and impairs
function of circulating blood-derived progenitor cells. Arterioscler
Thromb Vasc Biol 25: 698-703
55. Fadini GP, Miorin M, Facco M, Bonamico S, Baesso I, Grego F, Mene-
golo M, de Kreutzenberg SV, Tiengo A, Agostini C, Avogaro A (2005)
Circulating endothelial progenitor cells are reduced in peripheral va-
scular complications of type 2 diabetes mellitus. J Am Coll Cardiol 45:
1449-1457
56. Segal MS, Shah R, Afzal A, Perrault CM, Chang K, Schuler A, Beem
E, Shaw LC, Li Calzi S, Harrison JK, Tran-Son-Tay R, Grant MB (2006)
Nitric oxide cytoskeletal-induced alterations reverse the endothelial
progenitor cell migratory defect associated with diabetes. Diabetes 55:
102-109
57. Zhang W, Wang X, Hu R (2008) Effects of high glucose plus high in-
sulin on proliferation and apoptosis of mouse endothelial progenitor
cells. Inflamm Res 57: 571-576
58. Loomans CJ, Van Haperen R, Van Zonneveld AJ (2009) Differentiation
of bone marrow-derived endothelial progenitor cells is shifted into a
proinflammatory phenotype by hyperglycemia. Mol Med 15: 152-159
59. Xu L, Kanasaki K, Kitada M, Koya D (2012) Diabetic angiopathy and
angiogenic defects. Fibrogenesis Tissue Repair 5: 13
60. Deretic V (2012) Autophagy as an innate immunity paradigm: expan-
ding the scope and repertoire of pattern recognition receptors. Curr
Opin Immunol 24: 21-31
Wyszukiwarka
Podobne podstrony:
Abolicja podatkowa id 50334 Nieznany (2)
4 LIDER MENEDZER id 37733 Nieznany (2)
katechezy MB id 233498 Nieznany
metro sciaga id 296943 Nieznany
perf id 354744 Nieznany
interbase id 92028 Nieznany
Mbaku id 289860 Nieznany
Probiotyki antybiotyki id 66316 Nieznany
miedziowanie cz 2 id 113259 Nieznany
LTC1729 id 273494 Nieznany
D11B7AOver0400 id 130434 Nieznany
analiza ryzyka bio id 61320 Nieznany
pedagogika ogolna id 353595 Nieznany
Misc3 id 302777 Nieznany
cw med 5 id 122239 Nieznany
D20031152Lj id 130579 Nieznany
mechanika 3 id 290735 Nieznany
więcej podobnych podstron