CATECHOL
1
Catechol
1
OH
OH
[120-80-9]
C
6
H
6
O
2
(MW 110.11)
InChI = 1/C6H6O2/c7-5-3-1-2-4-6(5)8/h1-4,7-8H
InChIKey = YCIMNLLNPGFGHC-UHFFFAOYAA
(reagent for heterocycle synthesis; allylation catalyst; carbonyl
protecting group; transition metal ligand)
Alternate Name:
pyrocatechol.
Physical Data:
mp 104–105
◦
C; bp 119–121
◦
C/10 mmHg;
d
1.344 g cm
−3
; sublimable (vp 1.0 mmHg, 75
◦
C); steam
volatile.
Solubility:
sol benzene, acetone, ether, ethanol, 2.3 parts water,
pyridine, aqueous alkali; slightly sol chlorinated solvents.
Form Supplied in:
colorless needles (or plates).
Purification:
recrystallization from toluene; distillation.
Handling, Storage, and Precautions:
toxic (readily absorbed
through skin); irritant; causes burns. Light and air sensitive.
Explodes on contact with conc. HNO
3
.
Original Commentary
Bruce A. Barner
Union Carbide Corp., South Charleston, WV, USA
Alkylation and Heterocycle Synthesis. Catechol undergoes
many of the reactions characteristic of phenols, and as such is
used to prepare o-alkoxyphenols, o-dialkoxybenzenes, and
heterocycles.
1
The high yield preparation of five-membered
o
-phenylene heterocyclic systems from catechol is particularly
common, and includes 1,3,2-benzodioxaborole from diborane
(eq 1) (see Catecholborane),
2
methylenedioxybenzene from
diiodomethane,
3
o
-phenylene carbonate from phosgene,
4
1,3-
benzodioxoles from allenic derivatives,
5
o
-phenylene chloro- and
bromoboronates from the respective boron trihalides,
6,7
and
o
-phenylene phosphorochloridite from phosphorus trichloride.
8
Larger heterocyclic ring systems, such as 1,4-benzodioxanes
(eq 2)
9
and dibenzo- and dicyclohexyl-18-crown-6 polyethers
10
are prepared in good yields using alkylations of catechol.
OH
OH
O
B
O
H
BH
3
, THF
(1)
0–23 °C
80%
O
OTs
K
2
CO
3
, DMF
60 °C
75%
OH
OH
O
O
OH
(2)
Allylation Catalyst.
Bis(catecholato)allylsiliconates, pre-
pared in situ from catechol, triethylamine, and allyl(trialkoxy)-
silanes, are effective allylating agents for aldehydes (eq 3).
11
Good
yields of homoallylic alcohols are obtained in a regiospecific man-
ner.
PhCHO
Ph
OH
erythro
:threo = 1:9
catechol, Et
3
N
CHCl
3
, reflux
88%
Si(OMe)
3
(3)
Carbonyl Protecting Group.
Ketones and aldehydes
react with catechol under acidic conditions to form cyclic acetals
(eq 4).
12
The acetals formed are more stable to acid than ethylene
acetals.
13
Cleavage of the o-phenylene acetals of α-unbranched
and aromatic ketones with BBr
3
gives geminal dibromides and
vinyl bromides, respectively (eq 5).
14
t
-BuCHO
TMSCl, CH
2
Cl
2
91%
OH
OH
O
O
H
t
-Bu
(4)
BBr
3
CH
2
Cl
2
, 0 °C
83%
O
O
Et
Et
Et
Et
Br
Br
(5)
Transition Metal Ligand. Catechol binds readily to virtually
all transition metals, giving catecholato metal complexes.
15
The
σ
- and π-donating properties of catecholato ligands,
16
as well as
other hydroxo ligands,
17
are postulated to stabilize unsaturated
organometallic derivatives.
First Update
Aileen F. G. Bongat & Alexei V. Demchenko
University of Missouri - St. Louis, St. Louis, MO, USA
Acylation.
Condensation reactions between catechol and
carboxylic acids or their derivatives are very common. The
reactions of catechol with esters can either be catalyzed by a
base
18
or an acid
19
(eq 6). Carboxylic acids couple with catechol
in the presence of dicyclohexylcarbodiimide (DCC) and catalytic
4-dimethylaminopyridine (DMAP, eq 7).
20
O
O
O
O
O
rt, 3 h, 83%
(6)
OH
OH
p
-TsOH, DCM
OH
+
Avoid Skin Contact with All Reagents
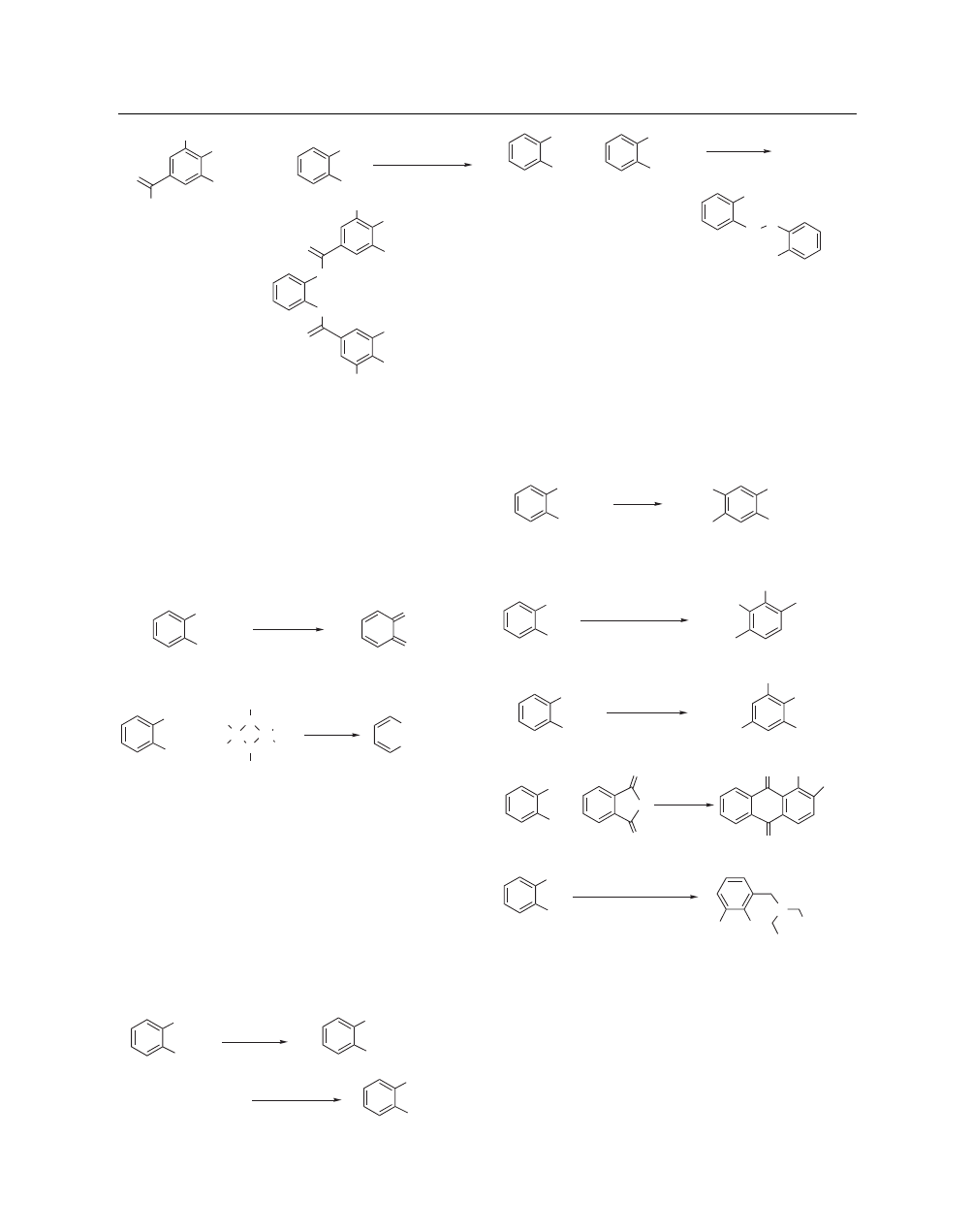
2
CATECHOL
OH
OH
OC
5
H
11
OC
5
H
11
OC
5
H
11
O
OH
+
OC
5
H
11
OC
5
H
11
OC
5
H
11
O
O
OC
5
H
11
OC
5
H
11
OC
5
H
11
O
O
DCC, DMAP
CH
2
Cl
2
, rt, 20 h, 90%
(7)
Oxidation.
Oxygen (O
2
) readily oxidizes catechol to o-
quinone in the presence of a suitable catalyst. In enzyme-catalyzed
transformations of this type, oxidases, such as tyrosinase
21
and laccase,
22
are often the catalysts of choice (eq 8). Salts
such as PbO
2
,
23
Ag
2
O,
24
or a combination of Pd(OAc)
2
-
Cu(OAc)
2
25
have also been employed as catalysts for this reaction.
Furthermore, catechol can be oxidatively opened to form useful
synthetic intermediates, for instance the (Z,Z)-monomethyl
muconate shown below (eq 9).
26
OH
OH
O
O
cat laccase, O
2
(8)
buffer, rt
O
2
, pyridine
80–85%
OH
OH
Cu
O
Cu
O
py
Cl
Cl
Py
Me
Me
COOH
COOCH
3
+
(9)
Functional Group Interconversion. Catechol can be trans-
formed into more reactive intermediates through selective or
global conversion of its hydroxyl moieties. In the example below
(eq 10), catechol was converted into 1,2-bis(nonaflyl)benzene
since the aromatic sulfonate functionality is more easily replaced
by a cyano group in the next step.
27
It was noted that catechol
reacted with polyfunctional 2-(chloroseleno)benzoyl chloride
in a different way than that of aliphatic alcohols. While in the
latter case faster O-acylation with the COCl moiety was
detected, reaction with catechol favored the O-selenylation
product (eq 11).
28
OH
OH
OSO
2
C
4
F
9
OSO
2
C
4
F
9
CN
CN
1. Et
3
N
2. C
4
F
9
SO
2
F
Pd(PPh
3
)
4
/Zn(CN)
2
(10)
DMF, 79%
DMF, 83%
OH
OH
+
C(O)Cl
SeCl
C(O)Cl
Se
HO
O
(11)
CH
3
CN, Et
3
N
rt, 71%
Electrophilic Aromatic Substitution Reactions. By virtue
of its aromatic nature, catechol typically undergoes electro-
philic aromatic substitution reactions such as halogenation,
29
carboxylation,
30
sulfonation,
31
and Friedel–Crafts acylation
32
(eqs 12–15). In addition to these common classes of aromatic
substitution reactions, a solvent- and catalyst-free regioselective
Mannich-type reaction of catechol resulting in the exclusive
formation of o-substituted aminomethylated derivatives has been
recently reported (eq 16).
33
OH
OH
OH
OH
Br
Br
Br
2
, CCl
4
(12)
80%
OH
OH
OH
HO
COOH
HOOC
(13)
NaOH then CO
2
, 80 atm
200
°C, 48 h, 54%
HSO
3
Cl
110
°C, 1.5 h, 37%
OH
OH
OH
OH
SO
2
Cl
ClO
2
S
(14)
OH
OH
O
O
O
O
O
OH
OH
(15)
+
AlCl
3
/NaCl
OH
OH
OH
HO
N
CO
2
Et
CO
2
Et
(16)
1 equiv ethyliminodiacetate
excess (CH
2
O)
n
, 50
°C, 70%
Formation of Zwitterionic Pentacoordinate Silicates.
Recently, catechol was found to mediate Si–C and Si–O bond
cleavage reactions in dialkoxy- (aminoorganyl)organylsilanes
to afford spirocyclic λ
5
Si–silicates with an SiO
4
C framework
(eqs 17 and 18).
34
These novel Si–C cleavage reactions were
applied to novel “traceless” silicon-based linkage formation and
cleavage strategies for the synthesis of aromatic compounds on
solid phase.
35
A list of General Abbreviations appears on the front Endpapers
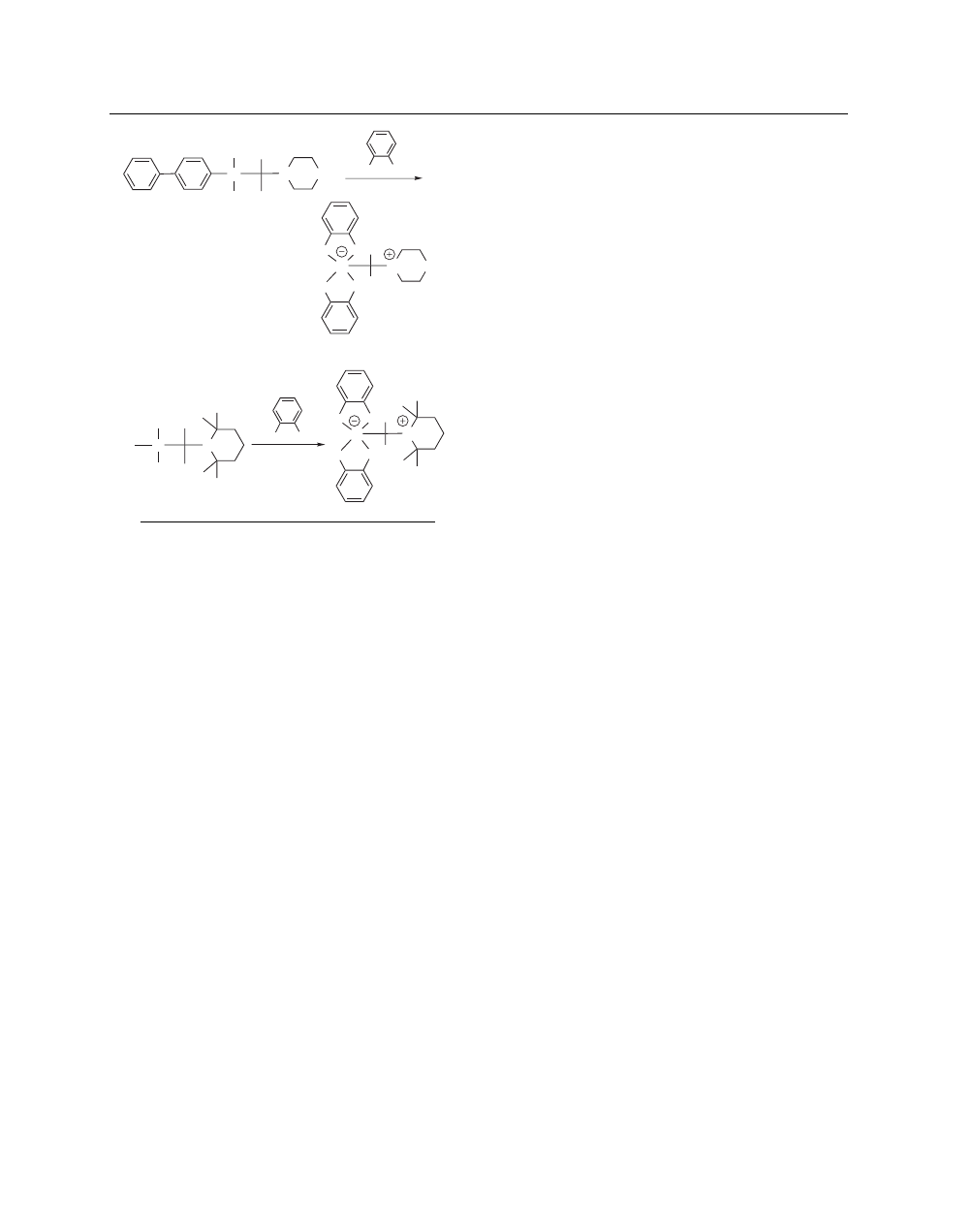
CATECHOL
3
Si
OEt
Ph
N
O
H
H
OH
HO
O
O
Si
O
O
NH
O
H
H
(17)
H
3
C
Si
CH
3
O
CH
3
O
N
H
H
OH
HO
O
O
Si
O
O
H
H
NH
(18)
1.
(a) Varagnat, J. Kirk-Othmer Encyclopedia of Chemical Technology, 3rd
ed.; Wiley: New York, 1981; Vol. 13, p 39. (b) Raff, R. A. V.; Ettling,
B. V. Kirk-Othmer Encyclopedia of Chemical Technology, 2nd ed.;
Wiley: New York, 1966; Vol. 11, p 462.
2.
Brown, H. C.; Gupta, S. K., J. Am. Chem. Soc. 1971, 93, 1816.
3.
Castillo, P.; Rodriguez-Ubis, J. C.; Rodriguez, F., Synthesis 1986, 839.
4.
Hanslick, R. S.; Bruce, W. F.; Mascitti, A., Org. Synth., Coll. Vol. 1963,
4
, 788.
5.
Cabiddu, S.; Cadoni, E.; Ciuffarin, E.; Fattuoni, C.; Floris, C., J.
Heterocycl. Chem. 1991
, 28, 1573.
6.
Gerrard, W.; Lappert, M. F.; Mountfield, B. A., J. Chem. Soc. 1959,
1529.
7.
Singleton, D. A.; Redman, A. M., Tetrahedron Lett. 1994, 35, 509.
8.
Crofts, P. C.; Markes, J. H. H.; Rydon, H. N., J. Chem. Soc. 1958, 4250.
9.
(a) Delgado, A.; Leclerc, G.; Lobato, M.-C.; Mauleon, D., Tetrahedron
Lett. 1988
, 29, 3671. (b) Martin, A. R.; Mallick, S. K.; Caputo, J. F.,
J. Org. Chem. 1974
, 39, 1808. (c) Koo, J.; Avakian, S.; Martin, G. J.,
J. Am. Chem. Soc. 1955
, 77, 5373.
10.
Pedersen, C. J., Org. Synth., Coll. Vol. 1988, 6, 395.
11.
(a) Hosomi, A.; Kohra, S.; Tominaga, Y., J. Chem. Soc., Chem. Commun.
1987, 1517. (b) Hayashi, T.; Matsumoto, Y.; Kiyoi, T.; Ito, Y.; Kohra, S.;
Tominaga, Y.; Hosomi, A., Tetrahedron Lett. 1988, 29, 5667.
12.
Nishida, Y.; Abe, M.; Ohrui, H.; Meguro, H., Tetrahedron: Asymmetry
1993, 4, 1431.
13.
Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis,
2nd ed.; Wiley: New York, 1991; p 197.
14.
Napolitano, E.; Fiaschi, R.; Mastrorilli, E., Synthesis 1986, 122.
15.
Pierpont, C. G.; Buchanan, R. M., Coord. Chem. Rev. 1981, 38, 45.
16.
Darensbourg, D. J.; Klausmeyer, K. K.; Mueller, B. L.; Reibenspies,
J. H., Angew. Chem., Int. Ed. Engl. 1992, 31, 1503.
17.
Lunder, D. M.; Lobkovsky, E. B.; Streib, W. E.; Caulton, K. G., J. Am.
Chem. Soc. 1991
, 113, 1837.
18.
Gondi, S. R.; Son, D. Y., J. Sulfur Chem. 2005, 26, 13.
19.
Wulferding, A.; Jankowski, J. H.; Hoffmann, M. R., Chem. Ber. 1994,
127
, 1275.
20.
Judele, R.; Laschat, S.; Baro, A.; Nimtz, M., Tetrahedron 2006, 62, 9681.
21.
Wagreich, H.; Nelson, J. M., J. Biol. Chem. 1936, 115, 459.
22.
Leutbecher, H.; Conrad, J.; Klaiber, I.; Beifuss, U., Synlett 2005, 3126.
23.
Stahl, P.; Kissau, L.; Mazitschek, R.; Huwe, A.; Furet, P.; Giannis, A.;
Waldmann, H., J. Am. Chem. Soc. 2001, 123, 11586.
24.
Aebisher, D.; Brzostowska, E. M.; Mahendran, A.; Greer, A., J. Org.
Chem. 2007
, 72, 2951.
25.
Kawamura, Y.; Imai, T.; Hosokawa, T., Synlett 2006, 3110.
26.
Rogic, M. M.; Demmin, T. R., J. Am. Chem. Soc. 1978, 100, 5472.
27.
Krempl, H.; Mattmer, R.; Hanack, M., Synlett 2000, 1705.
28.
Osajda, M.; Mlochowski, J., Tetrahedron 2002, 58, 7531.
29.
Liu, H.; Liu, Y.; Liu, M.; Chen, C.; Xi, F., Tetrahedron Lett. 2001, 42,
7083.
30.
Waters, S. P.; Kozlowski, M. C., Tetrahedron Lett. 2001, 42, 3567.
31.
Schwert, D. D.; Richardson, N.; Ji, G.; Raduechel, B.; Ebert, W.; Heffner,
P. E.; Keck, R.; Davies, J. A., J. Med. Chem. 2005, 48, 7482.
32.
Dhananjeyan, M. R.; Milev, Y. P.; Kron, M. A.; Nair, M. G., J. Med.
Chem. 2005
, 48, 2822.
33.
du Moulinet d’Hardemare, A.; Jarjayes, O.; Mortini, F., Synth. Commun.
2001, 34, 3975.
34.
Richter, I.; Penka, M.; Tacke, R., Organometallics 2002, 21, 3050.
35.
Tacke, R.; Ulmer, B.; Wagner, B.; Arlt, M., Organometallics 2000, 19,
5297.
Avoid Skin Contact with All Reagents
Wyszukiwarka
Podobne podstrony:
catechol sulfate eros rc034
benzyl chloride eros rb050
hydrobromic acid eros rh031
chloroform eros rc105
magnesium eros rm001
oxalyl chloride eros ro015
potassium permanganate eros rp244
peracetic acid eros rp034
p toluenesulfonic acid eros rt134
hexamethylenetetramine eros rh019
copper II chloride eros rc214
glyoxylic acid eros rg009
p methoxybenzaldehyde eros rm081
Rozdział V Eros
Eros i Tanatos
0844 fuego en el fuego eros ramazzotti XSLM5TN7TUAX4Y72WS55YKTPIHYGXX5ZORKV6YA
więcej podobnych podstron