
Zespół Pradera-Williego
1
Zespół Pradera-
Williego
Zespół
Pradera-Williego
syndroma Prader-Willi
Q87.1
Obraz hiszpańskiego malarza barokowego Juana
Carreño de Mirandy (1616–1685)
przedstawiający Doñę Eugenię Martinez Vallejo
w wieku około 6 lat (circa 1680)
Zespół Pradera-Williego (zespół Pradera-Labharta-Williego, ang.
Prader-Willi syndrome, Prader-Labhart-Willi syndrome, PWS) –
zespół wad wrodzonych spowodowany aberracją chromosomalną,
najczęściej częściową utratą (delecją) długiego ramienia chromosomu
15, pochodzącego od ojca. Na obraz kliniczny choroby składają się
niski wzrost, upośledzenie umysłowe, niedorozwój narządów
płciowych (hipogonadyzm) oraz otyłość spowodowana mniejszym niż
u zdrowych ludzi zapotrzebowaniem energetycznym przy
jednoczesnym ciągłym niepohamowanym uczuciu głodu. Uważa się,
że zespół Pradera-Williego jest najczęstszą genetycznie uwarunkowaną
przyczyną otyłości
[1]
.
Historia
Przypuszczalnie jednym z pierwszych opisów choroby był ten
pozostawiony przez Johna Langdona Downa
[3]
[4]
, który w 1864
[5]
i
ponownie w 1887 roku w słynnej monografii "Mental Afflictions of
Childhood and Youth" opisał pacjentkę o inicjałach E.C.,
pensjonariuszkę Earlswood Asylum for Idiots, w wieku 14 lat ważącą
196 funtów (84 kg) przy wzroście 4 stóp i 4 cali (1,32 m). Jej
chrapanie w nocy przeszkadzało innym pensjonariuszom, a w dzień
była senna. Pacjentka miała także objawy hipogonadyzmu i
charakterystycznie małe stopy i dłonie. W autopsji zmarłej przedwcześnie kobiety stwierdzono hipoplastyczne
jajniki i macicę. Langdon Down dla opisu choroby użył terminu "polysarcia"
[6]
, podobnie jak I.D. Hopkins w opisie
z 1861 roku
[7]
. Zachowane dane nie pozwalają jednak na ocenę tego ostatniego przypadku według przyjętych dziś
kryteriów.
Nazwa choroby upamiętnia szwajcarskich lekarzy Andreę Pradera, Heinricha Williego i Alexisa Labharta, którzy w
1956 roku na łamach Schweizerische medizinische Wochenschrift opisali nowy zespół u dziewięciorga pacjentów z
niskorosłością, opóźnieniem umysłowym, otyłością, oraz małymi dłońmi i stopami
[8]
. W 1961 roku Prader i Willi
opublikowali kolejną pracę, w której szczegółowiej przedstawili fenotyp nowo opisanego zespołu
[9]
. Nazwa zespołu
Pradera-Williego jest spotykana obecnie częściej niż nazwa zespołu Pradera-Williego-Labharta. W starszym
piśmiennictwie spotyka się też zarzuconą nazwę zespołu HHHO (akronim głównych objawów zespołu:
hypotonia-hypomentia-hypogonadism-obesity)
[10]
[11]
.
Mikrodelecje chromosomu 15 odkrył w 1981 David Ledbetter i wsp.
[12]
. W 1983 Merlin Butler stwierdził, że
delecja dotyczyła fragmentu chromosomu pochodzącego od ojca
[13]
, a w 1989 Robert Nicholls wykazał obecność
jednorodzicielskiej disomii w tych przypadkach, w których nie dało się stwierdzić delecji chromosomu ojcowskiego
i zaproponował mechanizm imprintingu (piętnowania genomowego)
[14]
.

Zespół Pradera-Williego
2
Epidemiologia
Częstość występowania zespołu ocenia się na 1:10 000-1:25 000 żywych urodzeń. Nie donoszono o różnicach w
częstości PWS w różnych grupach etnicznych, jednak w jednej pracy wykazano, że fenotyp zespołu u
Afroamerykanów może być mniej wyraźnie zaznaczony: dysmorfia twarzy łagodniej wyrażona, stopy i dłonie
prawidłowej wielkości, a wzrost mniej wyraźnie obniżony. Autorzy pracy zasugerowali, że w populacji
afroamerykańskiej PWS może być niedodiagnozowany
[15]
. PWS jednakowo często dotyka obie płci
[16]
.
Oszacowania częstości występowania PWS
Autor
Kraj
Częstość/żywych urodzeń
Butler et al.[1]
1:25 000
Akefeldt et al.[17]
1:8 000
Ehara et al.[18]
1:15 000
Oiglane-Shlik et
al.[19]
1:30 000
Vogels[20]
1:27 000
Smith[21]
1:25 000
Whittington et al.[22]
Wielka Brytania 1:45 000

Zespół Pradera-Williego
3
Etiologia
Ideogram chromosomu 15 (po lewej) i region
krytyczny zespołu (PWCR, po prawej);
zaznaczono loci genów i charakter ich ekspresji
(legenda na dole). Wg Bittela i Butlera[23]
Zespół Pradera-Williego spowodowany jest nieprawidłowością w
regionie 15q11-q13, określanym jako region krytyczny zespołu
Pradera i Williego (PWCR, Prader-Willi critical region) powstałą na
skutek jednego z kilku mechanizmów:
• delecji fragmentu ojcowskiego chromosomu 15q11.2-q13 (około
70%)
• uniparentnej (jednorodzicielskiej – matczynej) heterodisomii (UPD,
uniparental disomy) chromosmomu 15
• mutacji imprintingowej (mikrodelecji w miejscu imprintingowym)
przekazanej na chromosomie ojcowskim
• zrównoważonej rearanżacji chromosomalnej w obrębie 15q11.2-q13
(mniej niż 1%).
Nieprawidłowości w obrębie PWCR dotyczą ponad 99% pacjentów z
fenotypem PWS.
Zespół Pradera-Williego był pierwszym opisanym u człowieka
przykładem zjawiska imprintingu. Zespół Angelmana jest schorzeniem
spowodowanym utratą matczynego fragmentu chromosomu
obejmującego region PWCR. Duplikacje PWCR powodują opóźnienie
PWS należy do grupy tzw. zespołów przyległych genów (contiguous
gene syndromes), ponieważ fenotyp wynika z delecji kilku genów o
sąsiadujących loci. W PWS delecji ulegają ojcowskie kopie genu
SNRPN kodującego dwa białka: czynnik splicingowy SmN i
polipeptyd SNURF (SNRPN upstream reading frame). SNRPN
przepisywany jest też na długi, podlegający alternatywnemu splicingowi RNA, zawierający liczne, małe jąderkowe
RNA (snoRNA, small nucleolar RNAs)
[24]
. W PWCR znajduje się też gen NDN kodujący nekdynę.
Model zwierzęcy
Dziesięciodniowe myszy z jednego miotu;
pierwsza z mutacją w locus Snrnp
odziedziczonym po ojcu (PWScrm
+/p−
) wykazuje
opóźnienie wzrastania, druga z mutacją w allelu
odziedziczonym po matce (PWScrm
−/p+
) rozwija
się prawidłowo.
Przeprowadzono szereg badań na myszach, które pozwoliły
zaproponować mechanizmy patogenezy schorzenia u ludzi. W jednym
z nich mysz z mutacją wewnątrzgenową genu Snrpn była fenotypowo
normalna, co sugerowało, że mutacje ludzkiego genu SNRPN nie są
wystarczające do wywołania fenotypu PWS. Myszy z większymi
delecjami obejmującymi zarówno Snrpn jak i domniemane centrum
imprintingu (PWS-IC) nie wykazywały ekspresji piętnowanych genów
Zfp127 (mysi ortolog ZNF127), Ndn i lpw, a ich fenotyp wykazywał
podobieństwo do obrazu klinicznego PWS u ludzkich noworodków
[25]
. W jednym z najnowszych badań poddano delecji region krytyczny
zawężony do klastra genów MBII-85 kodujących małe jąderkowe RNA
(snoRNA). Myszy dziedziczące allel po matce (PWScrm
−/p+
) są
nieodróżnialne od myszy dzikiego typu; wszystkie myszy z
odziedziczonym allelem po ojcu (PWScrm
+/p−
) wykazywały

Zespół Pradera-Williego
4
opóźnienie wzrostu i 15% śmiertelność w okresie pourodzeniowym
[26]
.
Czynniki ryzyka
Niemal wszystkie przypadki PWS są sporadyczne, co oznacza, że zespół nie jest dziedziczony. Opisywano
przypadki rodzinnego występowania, przyjmuje się jednak, że ryzyko wystąpienia PWS u kolejnego dziecka jest
rzędu 0,1% (patrz: Poradnictwo genetyczne).
ryzyka wystąpienia PWS u dziecka
[27]
. W jednym retrospektywnym badaniu stwierdzono, że około połowa ojców
dzieci z PWS była narażona na te mutageny w związku z wykonywaną pracą
[28]
.
Objawy kliniczne i przebieg
15-letni chłopiec z zespołem Pradera-Williego o
słabo wyrażonym fenotypie (brak cech
dysmorficznych twarzy), z otyłością typu
centralnego, małymi dłońmi i stopami
(odpowiednio 20 i 5 percentyl dla płci i
wieku)[29]
Charakterystyczne objawy w zespole Pradera-Williego to:
• hipotonia mięśniowa (obniżenie napięcia mięśni) w okresie
prenatalnym objawia się słabymi ruchami płodu, brakiem postępu
akcji porodowej; utrzymuje się u niemowląt i małych dzieci,
napięcie mięśniowe poprawia się około 2–4 roku życia; z hipotonią
wiąże się słaby odruch ssania i trudności w karmieniu. PWS jest
jednym z zespołów predysponujących do ułożenia pośladkowego
płodu.
• opóźniony rozwój psychoruchowy;
• hipogonadyzm hipogonadotropowy
, manifestujący się jako
hipogenitalizm (niedorozwój narządów płciowych), małe prącie,
mała, hipoplastyczna, nie pomarszczona moszna o słabej
pigmentacji (zabarwieniu skóry), jednostronne lub obustronne
[31]
;
• nadmierne łaknienie (hiperfagia) i wynikające z niej nadwaga i otyłość. Szczegółowe badanie zachowania
pacjentów z PWS pozwoliło zasugerować, że istotą hiperfagii nie jest nadmierny głód, tylko zahamowanie
uczucia sytości
[32]
.
• zmieniona budowa (cechy dysmorficzne) twarzy: węższe czoło, antymongoloidalne ustawienie szpar
powiekowych, "migdałowaty" kształt szpar powiekowych, skierowane do dołu kąciki ust, małe i trójkątne usta
("rybie"), wąska czerwień wargowa górnej wargi. Dysmorfia twarzy może być bardzo delikatna lub nieobecna u
noworodka i ewoluować z wiekiem
[33]
;
• małe dłonie i stopy (akromikria), zwężające się ku końcom palce;
[34]
;
• niezwykle gęsta ślina, zwiększająca podatność na próchnicę
[35]
i stwarzająca problemy przy anestezjologicznej
kontroli pracy dróg oddechowych podczas zabiegów operacyjnych;
• wysoki próg odczuwania bólu;
• skłonność do hipertermii
[36]
;
• opóźnienie rozwoju mowy; później mogą wystąpić zaburzenia artykulacji;
• zmiany zachowania – dzieci z PWS mogą być kapryśne, uparte, czasem agresywne, skłonne do wybuchów złości,
zachowań obsesyjno-kompulsyjnych i psychoz; typowym zachowaniem jest skubanie skóry (ang. skin picking),
nieraz poważnie utrudniające gojenie się ran pooperacyjnych
[37]
;
• niezwykłe zdolności poznawcze u pacjentów wykazujących jednocześnie opóźnienie umysłowe: opisywano
zamiłowanie do układania puzzli
i szczególną umiejętność zapamiętywania czytanego tekstu
[39]
;

Zespół Pradera-Williego
5
• hipopigmentacja skóry, tęczówek i włosów, wynikająca z tyrozynazo-dodatniego albinizmu (około 30%
[40]
);
• niski wzrost (niskorosłość), spowodowany niedoborem hormonu wzrostu
[41]
;
• wady ośrodkowego układu nerwowego (szczególnie mózgu): poszerzenie układu komorowego
(wentrikulomegalia, do 100%), zmniejszona objętość płatów ciemieniowych i potylicznych (50%), spłycenie
bruzdy Sylwiusza (60%), niecałkowite zamknięcie wyspy (65%)
[42]
.
Obraz kliniczny PWS w zależności od wieku
Ciąża i poród
Okres noworodkowy i
niemowlęcy
Dzieciństwo
Dojrzewanie i
dorosłość
Zmniejszona aktywność
ruchowa płodu
Ułożenie pośladkowe
Poród przedwczesny lub po
czasie
Wąskie czoło
Niezstąpione jądra
Małe narządy płciowe
zewnętrzne i jądra
Słabe napięcie mięśni
Problemy z karmieniem
Słaby odruch ssania
Lepka ślina
Cichy płacz
Zmienna temperatura ciała
Opóźnienie w rozwoju
Niskorosłość
Małe dłonie i stopy
Jasna skóra i tęczówki
Migdałowate szpary powiekowe
Zez
Krótkowzroczność
Skubanie skóry
Próchnica zębów
Nadmierny apetyt/ szukanie jedzenia
Otyłość
Deficyt intelektualny
Zaburzenia zachowania (napady złości, upór, zachowania
obsesyjno-kompulsywne)
Niskorosłość
Małe dłonie i stopy
Skolioza
Osteoporoza
Opóźnione dojrzewanie
płciowe
Cukrzyca
Depresja
Nadmierna senność
Powikłania
Powikłania otyłości są główną przyczyną zgonu pacjentów z PWS. Należą tu:
• niewydolność krążeniowo-oddechowa
• cukrzyca
• zakrzepowe zapalenie naczyń
• obrzęki nóg
• ostre rozdęcie żołądka, mogące skutkować jego martwicą
[43]
[44]
.
Rozpoznanie
Opracowano kryteria diagnostyczne dla PWS. Dla dzieci w wieku do 3 lat do rozpoznania wystarcza 5 punktów, z
czego co najmniej 4 za kryteria duże; dla dzieci w wieku powyżej 3 lat rozpoznanie wymaga 8 punktów, z czego co
najmniej 5 za kryteria duże. Kryteria dodatkowe jedynie zwiększają lub zmniejszają pewność postawionej
diagnozy
[45]
[46]
:
Kryteria duże (1 punkt każde)
1. Hipotonia mięśniowa w okresie noworodkowym i wczesnodziecięcym, ze słabo wykształconym odruchem ssania
i ustępująca w miarę dorastania
2. Problemy z karmieniem i (lub) niechęć do ssania w niemowlęctwie, wymuszające konieczność specjalnych
technik karmienia
3. Szybki wzrost masy ciała między 1. a 6. rokiem życia powodujący otyłość centralną
4. Hiperfagia, obsesja na punkcie jedzenia, szukanie jedzenia
5. Charakterystyczne cechy dysmorficzne twarzy (dolichocefalia w niemowlęctwie, wąski wymiar czołowy,
migdałowate szpary powiekowe, skierowane w dół kąciki ust – wymagane przynajmniej 3)

Zespół Pradera-Williego
6
6. Hipogonadyzm (hipoplazja narządów płciowych, małe wargi sromowe mniejsze u dziewcząt, hipoplastyczna
moszna i wnętrostwo u chłopców; niecałkowite lub opóźnione dojrzewanie płciowe; niepłodność)
7. Opóźnienie rozwoju psychoruchowego, łagodne do średniego opóźnienie umysłowe, trudności w uczeniu się u
dziecka <6 roku życia.
8. Delecja 15q11-13 stwierdzona w badaniu cytogenetycznym wysokiej rozdzielczości (>650 prążków), lub inna
nieprawidłowość chromosomu w PWSCR, w tym matczyna disomia
Kryteria małe (0,5 punktu każde)
1. Obniżone ruchy płodu, letarg płodu, poprawa z wiekiem
2. Zaburzenia zachowania u dziecka (zmiany nastroju, zachowania obsesyjno-kompulsywne, upór, skłonność do
kradzieży i kłamstwa)
3. Zaburzenia snu/ okresy bezdechu
4. Niskorosłość w wieku 15 lat (przy niewdrożeniu terapii GH; należy wziąć pod uwagę wzrost rodziców)
5. Hipopigmentacja skóry i włosów w porównaniu do reszty rodziny
6. Małe i wąskie dłonie (<25 percentyla), małe stopy (<10 percentyla) – sprawdzane na siatkach centylowych
7. Zez zbieżny, krótkowzroczność
8. Wydzielanie gęstej i lepkiej śliny, zbierającej się w kącikach ust
9. Zaburzenia artykulacji
10. Nawykowe skubanie skóry
Kryteria dodatkowe
1. Wysoki próg bólu
2. Osłabiony odruch wymiotny
3. Skolioza (w zależności od wieku występuje u 30–80% osób z zespołem
[47]
) i (lub) kifoza
5. Osteoporoza
6. Niezwykłe zamiłowanie do układania puzzli
7. Prawidłowe wyniki badań nerwowo-mięśniowych (biopsji mięśnia, EMG, NCV).
Metody cytogenetyczne
Rozpoznanie kliniczne PWS powinno być potwierdzone cytogenetycznie. Dostępne metody to:
• analiza metylacji DNA
• metoda FISH
• techniki oparte na markerach mikrosatelitarnych
Testy stosowane w diagnostyce PWS
[40]
Metoda
Rozpoznawane mutacje
Odsetek
pacjentów
Analiza metylacji
Nieprawidłowa metylacja DNA 99%
FISH/Ilościowe metody
Delecja PWCR
70%
Badania w kierunku UPD
UPD regionu PWCR
25%
Defekt centrum imprintingu
<1%

Zespół Pradera-Williego
7
Różnicowanie
uszkodzenie podwzgórza przez guz, zabieg neurochirurgiczny albo radioterapię, zwłaszcza w młodym wieku,
wywołuje niskorosłość i zaburzenia łaknienia. Dawniej zespół objawów wynikających z uszkodzenia podwzgórza
rozpoznawano jako dystrofię tłuszczowo-płciową albo podwzgórzycę.
• Hiperfagiczna niskorosłość (hyperphagic short stature) jest nabytym stanem związanym z psychospołecznym
stresem skutkującym niedoborem GH, hiperfagią i zaburzeniami poznawczymi.
Hipotonia w okresie noworodkowym wymaga uwzględnienia w diagnostyce różnicowej następujących chorób:
• depresji ośrodkowego układu nerwowego;
• wrodzonej dystrofii miotonicznej typu 1;
• rzadkich zespołów spowodowanych aberracjami chromosomalnymi (dup Xq27.2-ter, del q16.2, del 1p36, del
10q26);
Opóźnienie rozwoju psychoruchowego, otyłość i hipogonadyzm występują w takich zespołach jak:
• jednorodzicielska disomia chromosomu 14;
• wrodzona osteodystrofia Albrighta – występuje niskorosłość, ale nie stwierdza się hipotonii, a dysmorfia twarzy
jest odmienna (okrągła twarz);
• zespół Cohena – charakterystyczna dysmorfia twarzy (krótka rynienka podnosowa, mikrocefalia, ciężkie
opóźnienie umysłowe);
• zespół Börjesona-Forssmana-Lehmana – chorują mężczyźni, znaczenie różnicujace mają mikrocefalia, ciężkie
opóźnienie umysłowe, charakterystyczne grube rysy twarzy, głęboko osadzone oczy, ptoza.
Zespoły podobne do PWS z przykurczami stawowymi w obrazie klinicznym:
• zespół Urbana-Rogersa-Meyera;
• zespół Camery-Marugi-Cohena;
• zespół Vasqueza.
Leczenie
Nie ma metody leczenia przyczynowego PWS. Dowiedziono, że korzystne efekty daje terapia hormonem wzrostu,
poprawiająca stosunek tkanki mięśniowej do tłuszczowej i pozwalająca na normalizację wzrostu. Obecnie, PWS jest
obok zespołu Turnera i somatotropinowej niedoczynności przysadki, wskazaniem do standardowego leczenia GH.
W Polsce program terapii GH objął dzieci z zespołem Pradera-Williego w 2006 rokuWikipedia:Weryfikowalność.
Terapia wymaga jednoczesnej diety, ograniczania dostępu do jedzenia i intensywnej rehabilitacji. Leczenie zostaje
przerwane gdy wystąpią przeciwwskazania (np. cukrzyca), w przypadku zakończenia wzrastania, braku rehabilitacji.
Terapie eksperymentalne
Podejmowano próby chirurgicznego leczenia pacjentów z PWS, przez wytworzenie zespolenia omijającego żołądek
sposobem Roux-en-Y
[48]
. Chirurgiczne możliwości obok gastroplastyki obejmują jeszcze wagotomię, są jednak
stosunkowo mało skuteczne
[49]
.
Leki teoretycznie mogące poprawić gospodarkę lipidową pacjentów z PWS: orlistat i tiazolidynediony, nie mają do
tej pory sprawdzonej skuteczności
[50]
.
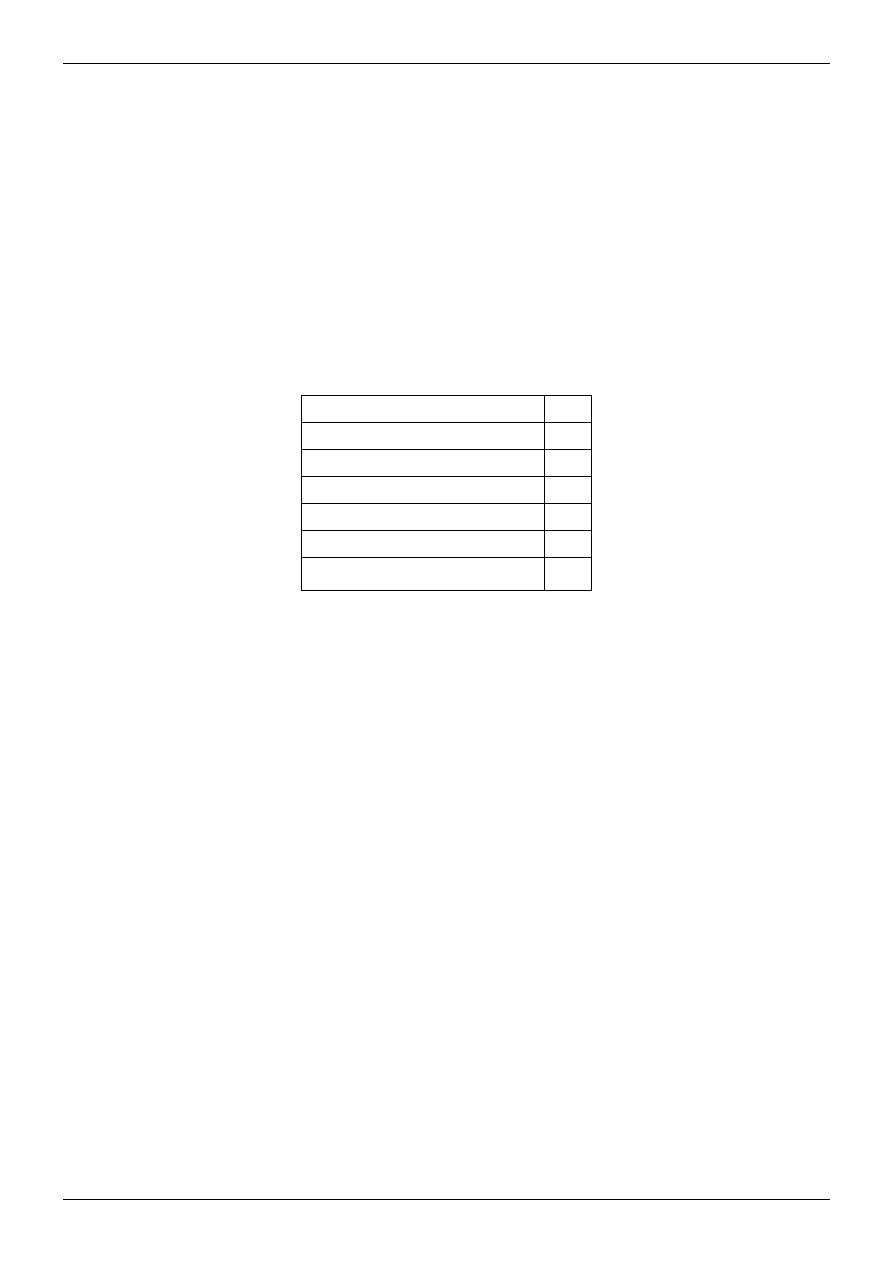
Zespół Pradera-Williego
8
Rokowanie
Jakość życia pacjentów z PWS jest znacznie obniżona i zazwyczaj osiągana długość życia jest krótsza niż średnia w
populacji. Jednak postępy w prowadzeniu chorych z PWS przyczyniły się w ostatnich latach do wydłużenia
oczekiwanej długości życia; opisano przypadek zgonu pacjenta z PWS w wieku 71
[51]
i 68 lat
[52]
.
Poradnictwo genetyczne
Większość chorych z PWS jest niepłodnych i nie wymaga konsultacji genetycznej pod kątem ryzyka urodzenia
chorego potomstwa. W większości przypadków ryzyko wystąpienia PWS u kolejnych dzieci rodziców dziecka z
PWS jest małe, zależy jednak od typu choroby i może sięgać nawet 50%. Informacje podsumowane są w tabeli:
Ryzyko wystąpienia PWS u rodzeństwa probanda w zależności od mechanizmu
genetycznego, który wywołał chorobę
[40]
Mechanizm genetyczny
Ryzyko
Delecja PWCR
<1%
UPD
<1%
Defekt imprintingu i mutacja
≤50%
Defekt imprintingu bez mutacji
<1%
Translokacja zrównoważona powstała de novo
<1%
Odziedziczona translokacja zrównoważona[53] .
25%
Zespół Pradera-Williego w kulturze
Za najstarsze przedstawienie zespołu Pradera-Williego uważane są dwa obrazy hiszpańskiego malarza barokowego
Juana Carreño de Mirandy (1616–1685), La Monstrua vestida i La Monstrua desnuda, przedstawiające dziewczynkę
o imieniu Eugenia Martinez Vallejo, przebywającą wówczas na dworze króla Karola II i przezywaną "La Monstrua"
z powodu monstrualnej otyłości. Podobno dr Andrea Prader po zobaczeniu obrazów w madryckim Museo del Prado
od razu rozpoznał charakterystyczne cechy PWS
[54]
[55]
.
Zauważono też, że Joe sportretowany w Klubie Pickwicka Dickensa ma wiele cech zespołu Pradera i Williego
[50]
.
Bibliografia
• Couper RT, Couper JJ. Prader-Willi syndrome. „Lancet”. 356. 9230, ss. 673-5 (2000). PMID 10968453.
DOI:10.1016/S0140-6736(00)02617-9
• Wattendorf DJ, Muenke M. Prader-Willi syndrome
. „Am Fam Physician”. 72. 5, ss. 827-30 (2005). PMID
16156341.
• Douglas C. Bittel, Merlin G. Butler. Prader–Willi syndrome: clinical genetics, cytogenetics and molecular
biology. „Expert Rev Mol Med”. 7. 14 (2005). DOI:10.1017/S1462399405009531
• Gunay-Aygun M, Schwartz S, Heeger S, O'Riordan MA, Cassidy SB. The changing purpose of Prader-Willi
syndrome clinical diagnostic criteria and proposed revised criteria
. „Pediatrics”. 108. 5, ss. E92 (2001).
PMID 11694676.
• Schüle B, Albalwi M, Northrop E, Francis DI, Rowell M, Slater HR, Gardner RJ, Francke U. Molecular
breakpoint cloning and gene expression studies of a novel translocation t(4;15)(q27;q11.2) associated with
Prader-Willi syndrome. „BMC Medical Genetics”. 6. 18 (2005). PMID 15877813. DOI:10.1186/1471-2350-6-18

Zespół Pradera-Williego
9
• Joyce E Whittington, Jill V Butler, Anthony J Holland. Changing rates of genetic subtypes of Prader–Willi
syndrome in the UK. „European Journal of Human Genetics”. 15, ss. 127–130 (2007).
Linki zewnętrzne
w bazie Online Mendelian Inheritance in Man
• Suzanne B Cassidy, Stuart Schwartz: Prader-Willi Syndrome
• Polskie Stowarzyszenie Pomocy Osobom z Zespołem Pradera-Williego
)
• Ann Scheimann: Prader-Willi Syndrome
. eMedicine Genetics. [dostęp 5 stycznia 2008].
• Prader-Labhardt-Willy syndrome
w bazie Who Named It
)
Przeczytaj zastrzeżenia dotyczące pojęć medycznych i pokrewnych w Wikipedii!
Przypisy
[1] Butler MG. Prader-Willi syndrome: current understanding of cause and diagnosis. „Am J Med Genet”. 35, ss. 319-332 (1990). PMID
2309779.
[2] Historical introduction. W: Brain RT: Mongolism. Wolstenholme GEW, Porter R. Boston: Little, Brown and Co, 1967, ss. 1-5.
[3] O Conor Ward. Prader-Willi syndrome. „Lancet”. 356, ss. 1586 (2000). DOI: 10.1016/S0140-6736(05)73324-9 (http:/
[4] Ward OC. Down's 1864 case of Prader Willi syndrome: a follow-up report. „J R Soc Med”. 90, ss. 694-696 (1997). PMID 9496304.
[5] Langdon Down J. Polysarcia and its treatment. „London Hasp Rep”. 1, ss. 97-103 (1864).
[6] Langdon Down J: Mental Affections of Childhood and Youth. London: Churchill, 1887.
[7] Hopkins ID. A case of polysarcia. „Buffalo Med Surg J”. 1, ss. 114-115 (1861).
[8] Prader A, Labhart A, Willi H. Ein Syndrom von Adipositas, Kleinwuchs, Kryptorchismus und Oligophrenie nach Myatonieartigem Zustand im
Neugeborenenalter. „Schweiz Med Wschr”. 86, ss. 1260-1261 (1956).
[9] Prader A, Willi H: Das Syndrom von Imbezilität, Adipositas, Muskelhypotonie, Hypogenitalismus, Hypogonadismus und Diabetes mellitus
mit "Anamnese". T. Part 1. Basel & New York: Karger, 1961, s. 353.
[10] Schneider HJ, Zellweger H. Forme fruste of the Prader-Willi syndrome (HHHO) and balanced D-E translocation. „Helv Paediatr Acta”. 23.
2, ss. 128-35 (1968). PMID 4387003.
[11] Endokrynologia kliniczna, tom I. Walenty Hartwig (red.). Warszawa: Państwowy Zakład Wydawnictw Lekarskich, 1978, s. 1047.
[12] Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD. Deletions of chromosome 15 as a cause of the Prader-Willi
syndrome. „New England Journal of Medicine”. 304. 6, ss. 325-9 (1981). PMID 7442771.
[13] Butler MG, Palmer CG. Parental origin of chromosome 15 deletion in Prader-Willi syndrome. „Lancet”, ss. 1285-1286 (1983). PMID
6134086.
[14] Nicholls RD et al. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. „Nature”. 342, ss. 281-285
(1989). PMID 2812027.
[15] Hudgins L, Geer JS, Cassidy SB. Phenotypic differerencss in African Americans with Prader-Willi syndrome. „Genet Med”. 1. 1, ss. 49-51
(1998).
[16] Ann Scheimann: Prader-Willi Syndrome (http:/
htm). eMedicine Genetics. [dostęp 5 stycznia
2008].
[17] Akefeldt A, Gillberg C, Larsson C. Prader-Willi syndrome in a Swedish rural county: epidemiological aspects. „Dev Med Child Neurol”. 33.
8, ss. 715-21 (1991).
[18] Ehara H, Ohno K, Takeshita K. Frequency of the Prader-Willi syndrome in the San-in district, Japan. „Brain Dev”. 17. 5, ss. 324-6 (1995).
[19] Oiglane-Shlik E, Talvik T, Zordania R, Põder H, Kahre T, Raukas E, Ilus T, Tasa G, Bartsch O, Väisänen ML, Ounap K. Prevalence of
Angelman syndrome and Prader-Willi syndrome in Estonian children: sister syndromes not equally represented. „Am J Med Genet A”. 140.
18, ss. 1936-43 (2006). PMID 16906556.
[20] Vogels A, Van Den Ende J, Keymolen K, Mortier G, Devriendt K, Legius E, Fryns JP. Minimum prevalence, birth incidence and cause of
death for Prader-Willi syndrome in Flanders. „Eur J Hum Genet”. 12. 3, ss. 238-40 (2004). PMID 14679397.
[21] Smith A, Egan J, Ridley G, Haan E, Montgomery P, Williams K, Elliott E. Birth prevalence of Prader-Willi syndrome in Australia. „Arch
Dis Child”. 88. 3, ss. 263-4 (2003). PMID 12598399.
[22] Whittington JE, Holland AJ, Webb T, Butler J, Clarke D, Boer H. Population prevalence and estimated birth incidence and mortality rate
for people with Prader-Willi syndrome in one UK Health Region. „J Med Genet”. 38. 11, ss. 792-8 (2001). PMID 11732491.

Zespół Pradera-Williego
10
[23] Douglas C. Bittel, Merlin G. Butler. Prader–Willi syndrome: clinical genetics, cytogenetics and molecular biology. „Expert Rev Mol Med”.
7. 14 (2005). DOI: 10.1017/S1462399405009531 (http:/
[24] Rodriguez-Jato S, Nicholls RD, Driscoll DJ, Yang TP. Characterization of cis- and trans-acting elements in the imprinted human
SNURF-SNRPN locus. „Nucleic Acids Res”. 33, ss. 4740-4753 (2005). PMID 16116039.
[25] Yang T, Adamson TE, Resnick JL, Leff S, Wevrick R, Francke U, Jenkins NA, Copeland NG, Brannan CI. A mouse model for Prader-Willi
syndrome imprinting-centre mutations. „Nature Genet”. 19, ss. 25-31 (1998). PMID 9590284.
[26] Skryabin BV, Gubar LV, Seeger B, Pfeiffer J, Handel S, et al. Deletion of the MBII-85 snoRNA Gene Cluster in Mice Results in Postnatal
Growth Retardation. „PLoS Genet”. 3. 12, ss. e235 (2007). DOI: 10.1371/journal.pgen.0030235 (http:/
[27] Strakowski SM, Butler MG. Paternal hydrocarbon exposure in Prader-Willi syndrome (Letter). „Lancet”, ss. 1458 (1987).
[28] Cassidy SB, Gainey AJ, Butler MG. Occupational hydrocarbon exposure among fathers of Prader-Willi syndrome patients with and without
deletions of 15q. „Am J Hum Genet”. 44, ss. 806-810 (1989). PMID 729276.
[29] Schüle B, Albalwi M, Northrop E, Francis DI, Rowell M, Slater HR, Gardner RJ, Francke U. Molecular breakpoint cloning and gene
expression studies of a novel translocation t(4;15)(q27;q11.2) associated with Prader-Willi syndrome. „BMC Medical Genetics”. 6. 18 (2005).
PMID 15877813. DOI: 10.1186/1471-2350-6-18 (http:/
[30] Hamilton CR, Jr, Scully RE, Kliman B. Hypogonadotropism in Prader-Willi syndrome: induction of puberty and spermatogenesis by
clomiphene citrate. „Am J Med”. 52, ss. 322-329 (1972). PMID 5011391.
[31] Crinò A, Schiaffini R, Ciampalini P, Spera S, Beccaria L, Benzi F, Bosio L, Corrias A, Gargantini L, Salvatoni A, Tonini G, Trifirò G,
Livieri C; Genetic Obesity Study Group of Italian Society of Pediatric endocrinology and diabetology (SIEDP). Hypogonadism and pubertal
development in Prader-Willi syndrome. „Eur J Pediatr”. 162. 5, ss. 327-33 (2003). PMID 12692714. DOI: 10.1007/s00431-002-1132-4 (http:/
[32] Lindgren AC, Barkeling B, Hagg A, Ritzen EM, Marcus C, Rossner S. Eating behavior in Prader-Willi syndrome, normal weight, and obese
control groups. „J Pediat”. 137, ss. 50-55 (2000). PMID 10891821.
[33] Aughton DJ, Cassidy SB. Physical features of Prader-Willi syndrome in neonates. „Am J Dis Child”. 144. 11, ss. 1251-4 (1990). PMID
2239867.
[34] Hered RW, Rogers S, Zang YF, Biglan AW. Ophthalmologic features of Prader-Willi syndrome. „J Pediatr Ophthalmol Strabismus”. 25. 3,
ss. 145-50 (1988). PMID 3397859.
[35] Scardina GA, Fucà G, Messina P. Oral diseases in a patient affected with Prader-Willi syndrome. „Eur J Paediatr Dent”. 8. 2, ss. 96-9
(2007). PMID 1757193.
[36] Wise MS, Zoghbi H, Edwards M, Byrd LK, Guttmacher AE, Greenberg F. Hyperthermia in infants with Prader-Willi syndrome (Abstract).
„Am J Med Genet”. 41, ss. 528 (1991).
[37] Bhargava SA, Putnam PE, Kocoshis SA, Rowe M, Hanchett JM. Rectal bleeding in Prader-Willi syndrome. „Pediatrics”. 97, ss. 265-267
(1996). PMID 8584392.
[38] Dykens EM. Are jigsaw puzzle skills 'spared' in persons with Prader-Willi syndrome?. „J Child Psychol Psychiatry”. 43. 3, ss. 343-52
(2002). PMID 11944876.
[39] Pacjent opisany przez Couperów (2000) potrafił cytować z pamięci wybrane wersy Bleak House Dickensa.
[40] Ann Scheimann: Prader-Willi Syndrome (http:/
htm). eMedicine Genetics. [dostęp 5 stycznia
2008].
[41] Burman P, Ritzén EM, Lindgren AC. Endocrine dysfunction in Prader-Willi syndrome: a review with special reference to GH (http:/
787). „Endocr Rev”. 22. 6, ss. 787-99 (2001). PMID 11739333.
[42] Miller JL, Couch JA, Schmalfuss I, He G, Liu Y, Driscoll DJ. Intracranial abnormalities detected by three-dimensional magnetic resonance
imaging in Prader-Willi syndrome. „Am J Med Genet”. 143A, ss. 476-483 (2007).
[43] Wharton RH, Wang T, Graeme-Cook F, Briggs S, Cole RE. Acute idiopathic gastric dilation with gastric necrosis in individuals with
Prader-Willi syndrome. „Am J Med Genet”. 73, ss. 437-41 (1997). PMID 9415471.
[44] Stevenson DA, Heinemann J, Angulo M, Butler MG, Loker J, Rupe N, Kendell P, Cassidy SB, Scheimann A. Gastric rupture and necrosis
in Prader-Willi syndrome. „J Pediatr Gastroenterol Nutr”. 45(. 2, ss. 272-4 (2007). PMID 17667731.
[45] Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greenswag LR, Whitman BY, Greenberg F. Prader-Willi syndrome: consensus diagnostic
criteria. „Pediatrics”. 91. 2, ss. 398-402 (1993). PMID 8424017.
[46] Gunay-Aygun M, Schwartz S, Heeger S, O'Riordan MA, Cassidy SB. The changing purpose of Prader-Willi syndrome clinical diagnostic
criteria and proposed revised criteria (http:/
e92). „Pediatrics”. 108. 5, ss. E92
(2001). PMID 11694676.
[47] de Lind van Wijngaarden RF., de Klerk LW., Festen DA., Duivenvoorden HJ., Otten BJ., Hokken-Koelega AC. Randomized controlled trial
to investigate the effects of growth hormone treatment on scoliosis in children with Prader-Willi syndrome.. „The Journal of clinical
endocrinology and metabolism”. 4 (94), ss. 1274–80 (kwiecień 2009). doi:10.1210/jc.2008-1844 (http:/
2008-1844). PMID 19158197.
[48] Kobayashi J, Kodama M, Yamazaki K, Morikawa O, Murano S, Kawamata N, Kawamura I. Gastric Bypass in a Japanese Man with
Prader-Willi Syndrome and Morbid Obesity. „Obesity Surgery”. 13. 5 (2003). PMID 14627483. DOI: 10.1381/096089203322509453 (http:/

Zespół Pradera-Williego
11
[49] Scheimann AO, Butler MG, Gourash L, Cuffari C, Klish W. Critical analysis of bariatric procedures in Prader-Willi syndrome. „J Pediatr
Gastroenterol Nutr”. 46. 1, ss. 80-3 (2008). PMID 18162838.
[50] Couper RT, Couper JJ. Prader-Willi syndrome. „Lancet”. 356. 9230, ss. 673-5 (2000). PMID 10968453. DOI:
10.1016/S0140-6736(00)02617-9 (http:/
[51] Carpenter PK. Prader-Willi syndrome in old age. „J Intellect Disabil Res”. 38. Pt 5, ss. 529-531 (1994). PMID 7841690.
[52] Butler MG. A 68-year-old white female with Prader-Willi syndrome. „Clin Dysmorphol”. 9, ss. 65-67 (2000). PMID 10649803.
[53] Sytuacja teoretyczna
[54] Management of Prader-Willi Syndrome. Merlin G. Butler, Phillip D.K. Lee, Barbara Y. Whitman. Springer, 2006. ISBN 0-387-25397-1.
[55] Pozzilli P, Khazrai YM. "La Monstrua Vestida", a case of Prader-Willi syndrome. „J Endocrinol Invest”. 28. 2, ss. 199 (2005). PMID
15887871.
[56] http:/
[57] http:/
[58] http:/
[59] http:/
[60] http:/
[61] http:/
[62] http:/
[63] http:/
[64] http:/
[65] http:/
[66] http:/

Źródła i autorzy artykułu
12
Źródła i autorzy artykułu
Zespół Pradera-
Williego Źródło: http://pl.wikipedia.org/w/index.php?oldid=20991866 Autorzy: Admirał Bum, DrPZ, Enejsi, Filip em, Gładka, Kauczuk, Kenraiz, Kpjas, Leesec75, Louve,
Masur, MesserWoland, Milen, Mroman, Mrug, Nevermore, Psyche, Remedios44, Sati, SkyMaja, Szoltys, Tescobar, ToSter, Vuvar1, Żbiczek, 5 anonimowych edycji
Źródła, licencje i autorzy grafik
Plik:Caduceus.svg Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Caduceus.svg Licencja: Public Domain Autorzy: User:Fadookie, User:Rama
Plik:Eugenia Martinez Vallejo. Carrena.jpg Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Eugenia_Martinez_Vallejo._Carrena.jpg Licencja: nieznany Autorzy: Ecummenic, Enrique
Cordero, Gryffindor, Nevit, Shakko, 3 anonimowych edycji
Plik:Pws.svg Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Pws.svg Licencja: Creative Commons Attribution 2.0 Autorzy: User:Filip em
Plik:PWS mice.jpg Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:PWS_mice.jpg Licencja: Creative Commons Attribution 2.0 Autorzy: Skryabin BV, Gubar LV, Seeger B, Pfeiffer J,
Handel S, et al.
Plik:Pws.jpg Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Pws.jpg Licencja: Creative Commons Attribution 2.0 Autorzy: Schüle B et al
Plik:Star of life2.svg Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Star_of_life2.svg Licencja: Attribution Autorzy: User:Verdy p
Licencja
Creative Commons Attribution-Share Alike 3.0 Unported
http:/
Document Outline
Wyszukiwarka
Podobne podstrony:
Zespół Pradera Williego
Zespół Pradera Williego
zespół pradera Williego
PRZEMÓWIENIE SĄDOWE OSKARŻAJĄCE WILLIEGO SONNENBRUCHA
Zespół Pradera Willego i Angelmana
LIST W IMIENIU MARIKI DO WILLIEGO LEKTURA NIEMCY
zespol Angelmana i zespol Pradera - Willego, VI rok, Genetyka, Genetyka, Egzamin
03 0000 037 02 Leczenie dzieci z zespolem Prader Willi hormonem wzrostu
willie riley
Blind Willie
Zespół Pradera
The Space Willies Eric Frank Russell
Konspekt lekcji z zespoowych gier sportowyc1 doc
WILLIE BOY TU BYĹ 90 MIN
Wee Willie Winkie
Willie Nelson On My Mind
więcej podobnych podstron