CZĘŚĆ I.
WYBRANE ZAGADNIENIA BIOCHEMII
Biochemia jest nauką zajmującą się poznaniem budowy chemicznej składników organizmów
żywych oraz przemian, jakie w nich zachodzą. Tradycyjnie biochemię dzieli się na dwa działy, a
mianowicie:
1) biochemię statyczną
2) biochemię dynamiczną.
Pierwszy z tych działów skupia uwagę na strukturze i właściwościach związków chemicznych
znajdowanych w przyrodzie ożywionej i stąd coraz częściej nazywany bywa chemią bioorganiczną.
Z kolei biochemia dynamiczna za pomocą metod chemicznych wyjaśnia istotę procesów
przemiany materii i stąd identyfikuje się ją z metabolizmem komórkowym.
Ponieważ każdy organizm ma pewne odrębne właściwości, biochemię w zależności od badanego
obiektu dzieli się na:
1) biochemię drobnoustrojów,
2) biochemię roślin
3) biochemię zwierząt.
Z tych trzech podstawowych działów wyodrębnia się dalsze bardziej wąskie specjalistyczne
kierunki (np. biochemia człowieka, biochemia bakterii itd.).
W rozdziale tym, w formie zwięzłego kompendium, omówiono jedynie wybrane zagadnienia
biochemii związane bezpośrednio z problematyką poruszaną w kolejnych rozdziałach tej książki. Tak,
więc, przede wszystkim, ma on ułatwić ich studiowanie, a nie zastąpić podręczników niezbędnych do
dokładnego i systematycznego przyswajania wiedzy biochemicznej.
ROZDZIAŁ 1.
CHEMIA BIOORGANICZNA
Jak już wspomniano, biochemia zajmuje się m.in. ustaleniem budowy chemicznej składników
żywych organizmów. Ustalono, że do najbardziej rozpowszechnionych w organizmach żywych
związków należą: sacharydy, białka (w tym enzymy) i tłuszczowce. Wśród innych na uwagę
zasługują: kwasy nukleinowe, witaminy, hormony, barwniki (m.in. pirolowe i karotenoidy),
glikozydy, alkaloidy i kwasy organiczne. Ich budowa zostanie omówiona w kolejnych rozdziałach.
Przy jej rozpatrywaniu, w większości przypadków, konieczne jest uwzględnienie zjawiska izomerii.
Dlatego też pierwszy rozdział poświęcony zostanie omówieniu tego niezwykle ważnego zagadnienia.
1. Izomeria
Izomeria polega na występowaniu związków o identycznym składzie atomowym, a różniących się
jedynie sposobem, kolejnością lub miejscem powiązania atomów albo ich rozmieszczeniem (lub ich
grup) w przestrzeni. Cząsteczki takich związków nazywane są i zo me r a mi . Znanych jest kilka
różnych odmian izomerii. Tu zostaną omówione jedynie te odmiany, które mają znaczenie z
biochemicznego punktu widzenia, a przede wszystkim będą przydatne przy studiowaniu dalszych
rozdziałów podręcznika (m.in. przy nazewnictwie enzymów).
Regioizomery (izomery pozycyjne) to izomery różniące się jedynie rozmieszczeniem tych samych
grup atomów (np. grup funkcyjnych, podstawników) w
podstawowym szkielecie cząsteczki. Mogą one różnić się
właściwościami fizyko-chemicznymi, ale przede wszystkim
najczęściej wykazują diametralnie różne właściwości
biologiczne.
Przykładem
regioizomerów
może
być
mieszanina dwu diestrów etylowych glicerolu.
C H
2
- CO OC
2
H
5
C H
2
- CO OC
2
H
5
C H- CO OH
C H
2
- CO OC
2
H
5
C H
2
- CO OH
C H- CO OC
2
H
5
I
II
regioizomery
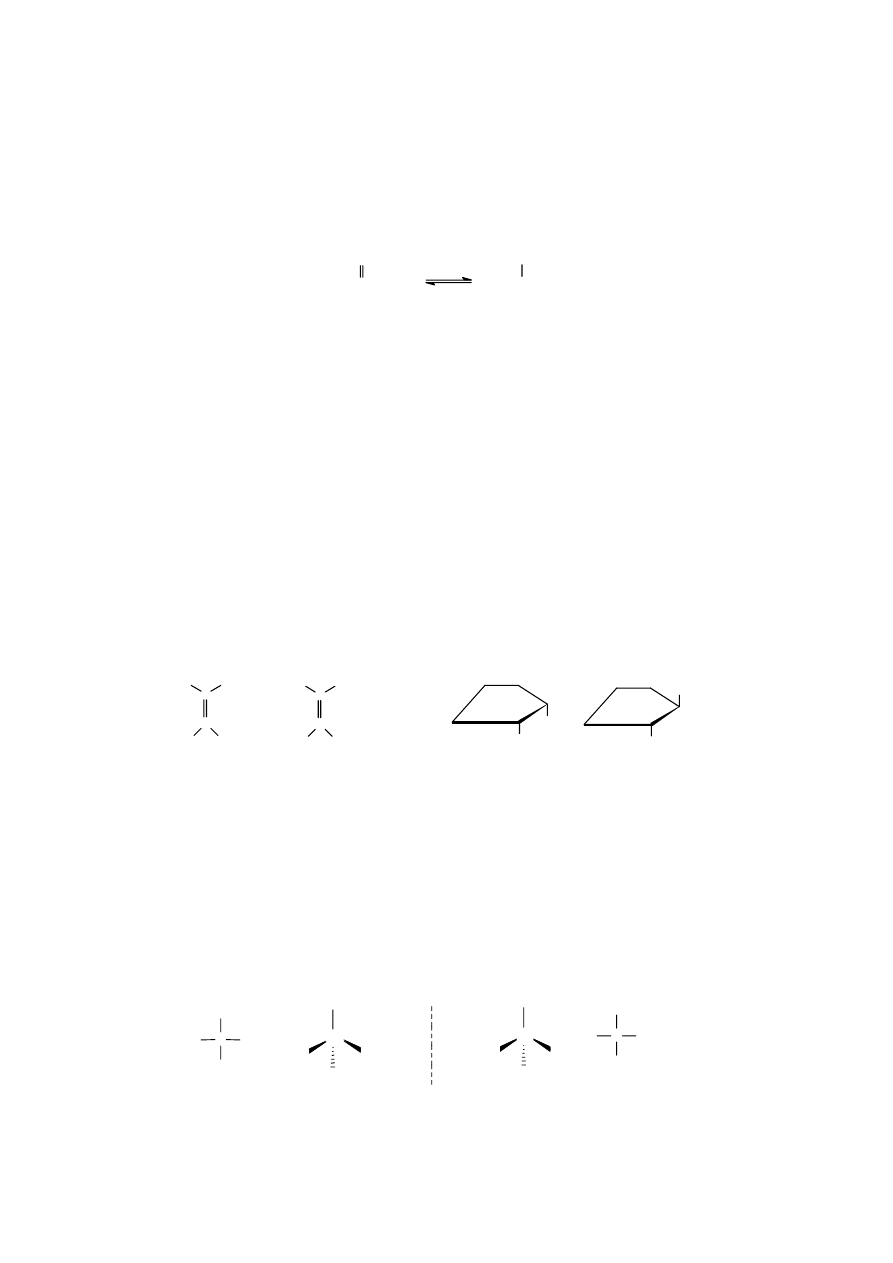
Tautomery (izomery funkcyjne - różnią się obecnością w cząsteczce odmiennych grup
funkcyjnych) to dwa izomery, które przechodzą nawzajem w siebie w wyniku przesunięcia atomu
wodoru z jednoczesnym przemieszczeniem układu wiązań w cząsteczce. Zjawisko występowania
obok siebie takich izomerów nazywamy t a u t o me r i ą. Stan równowagi pomiędzy tautomerami kwasu
pirogronowego można zapisać następująco:
Odmiana ketonowa większości tautomerów jest znacznie trwalsza i stąd stan równowagi reakcji
przesunięty jest na jej korzyść.
-izomery to dwa izomery różniące się położeniem
podwójnego wiązania (lub kilku takich wiązań) w
cząsteczce, np.:
Stereoizomeria
Stereoizomeria, czyli izomeria przestrzenna jest związana z różnym przestrzennym
rozmieszczeniem atomów (lub grup atomów) i układem wiązań w cząsteczce. Według klasycznego
podziału obejmuje ona izomerię geometryczną i izomery optyczne.
Izomery cis-trans -izo me r y ge o me t r yc zn e , zaliczane do stereoizomerów - różnią się
konfiguracją, czyli przestrzennym położeniem wyróżnionych atomów względem wybranej
płaszczyzny, przy czym przyjmuje się, że atomy te nie mają możliwości swobodnego obrotu wokół
podwójnego wiązania (np. w alkenach) lub – w związkach cyklicznych – wokół wiązania C-C.
Izomery, w których takie same lub wyróżnione atomy (albo grupy atomów) znajdują się po tej samej
stronie są nazywane odmianami cis, po przeciwnych stronach – odmianami trans. Izomery cis-trans
mogą różnić się nieco właściwościami fizyko-chemicznymi, natomiast ich właściwości biologiczne są
z reguły całkowicie różne.
Izomery optyczne lub ogólnie związki optycznie czynne, tj. skręcające w różny sposób
płaszczyznę światła spolaryzowanego Ta właściwość jest konsekwencją obecności w ich cząsteczkach
centrum chiralnego. Chiralność, to właściwość obiektu polegająca na tym, iż nie pokrywa się on ze
swoim odbiciem w zwierciadle płaskim. Z reguły centrum chiralne izomerów optycznych stanowi
asymetryczny (chiralny) atom lub atomy węgla (oznaczone na rysunkach gwiazdką), wokół których
może występować różna konfiguracja, czyli przestrzenne rozmieszczenie innych atomów lub grup
atomów.
CH
2
=C-COOH
OH
CH
3
-C-COOH
O
odm iana ketonowa
odm iana enolowa
tautomery
CH
3
C
C
CH
3
H
H
H
C
C
CH
3
H
3
C
H
cis
-buten-2
trans
-buten-2
OH
OH
cis
-cyklopentano-
-1,2-diol
trans
-cyklopentano-
-1,2-diol
OH
OH
cis-trans
izomery
CHO
C*
H
CH
2
OH
CHO
C*
CH
2
OH
H
OH
OH
aldehyd L(-)-glicerynowy
aldehyd D(+)-glicerynowy
CHO
C*
CH
2
OH
HO
H
płaszczyzna
zwierciadła
CHO
C*
HO
H
CH
2
OH
-izomery
- buten
1
CH
2
=CH-CH
2
-CH
3
- buten
2
CH
3
-CH=CH-CH
3
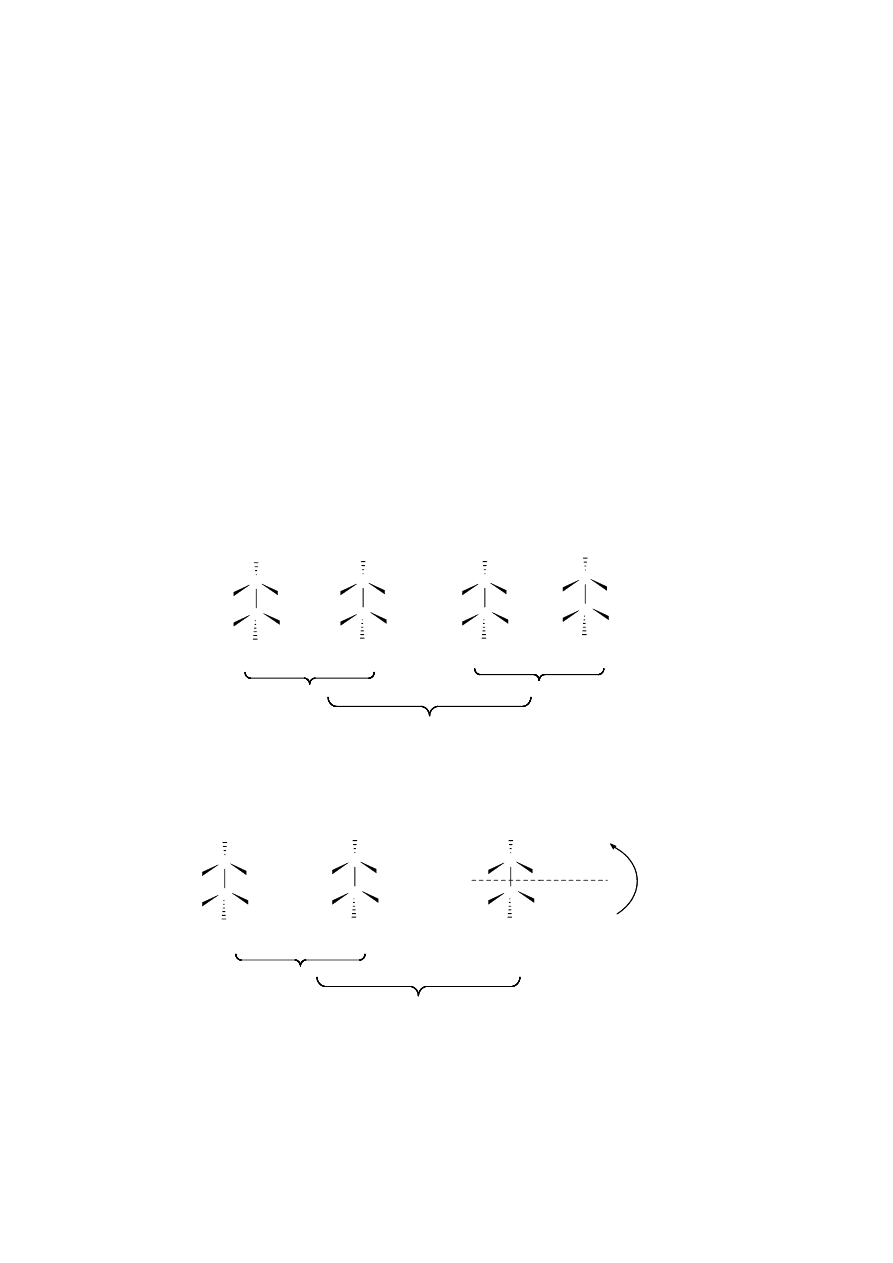
Izomery optyczne dzieli się na prawoskrętne (+) lub lewoskrętne (-), w zależności od kierunku, w
którym skręcają płaszczyznę światła spolaryzowanego. W celu ujednolicenia przedstawiania ich
konfiguracji za wzorce przyjęto prawo- i lewoskrętne aldehydy glicerynowe. Grupa aldehydowa jest
lokowana na „górze” wzoru, natomiast grupę hydroksylową umieszcza się po lewej stronie (z punktu
widzenia patrzącego), otrzymując w ten sposób wzór aldehydu
L(-)-glicerynowego, lub po prawej,
otrzymując wzór aldehydu D-(+)-glicerynowego.
Enancjomery, to pary prawo- i lewoskrętnych izomerów optycznych, stanowiących wzajemne
odbicie w lustrze. Ich właściwości fizyko-chemiczne są identyczne, z wyjątkiem zdolności do
kierunku skręcania płaszczyzny światła spolaryzowanego. Natomiast różnią się właściwościami
biologicznymi. Np. pleśnie Penicillium glaucum z roztworu racemicznego winianu amonowego
zużywają wyłącznie odmianę prawoskrętną.
Racemat (postać racemiczna) to mieszanina równych ilości cząsteczek enancjomerów, nie
wykazująca aktywności optycznej (z uwagi na kompensację) oznaczana znakiem (±) lub literami
D
,
L
-
umieszczanymi przed nazwą lub wzorem związku chemicznego.
Racemizacja, to zjawisko polegające na wzajemnym przekształcaniu się jednego enancjomeru w
drugi, aż do ustalenia się równowagi ilościowej pomiędzy obu odmianami.
Diastereoizomery to izomery optyczne nie będące wzajemnymi odbiciami w lustrze. Różnią się
właściwościami fizyko-chemicznymi i biologicznymi. Poniżej przedstawione są konfiguracje czterech
stereoizomerów kwasu 2,3-dihydroksymasłowego. Wzory (I) i (II) oraz (III) i (IV) przedstawiają pary
enancjomerów, które w stosunku do siebie nawzajem są d i a s t e r e oi zo me r a mi .
Mezo-izomery. Niektóre związki, pomimo obecności asymetrycznych atomów węgla, są
nieczynne optycznie na skutek wewnętrznej kompensacji; nazywamy je związkami mezo. Przykładem
takiego związku jest kwas mezowinowy, który jest diastereoizomerem w stosunku do swoich odmian
prawo- i lewoskrętnej.
* * *
Niektóre inne aspekty stereoizomerii poruszone zostaną w następnym rozdziale, przy omawianiu
sacharydów.
C
C
COOH
CH
3
H
*
*
H
OH
OH
C
C
COOH
CH
3
*
*
HO
HO
H
H
OH
C
C
COOH
CH
3
*
*
HO
H
OH
HO
H
C
C
COOH
CH
3
*
*
(I)
(II)
(III)
(IV)
enancjomery
enancjomery
diastereoizomery
diastereoizomery
enancjomery
OH
HO
H
*
*
C
C
COOH
COOH
OH
HO
H
*
*
C
C
COOH
COOH
kwas
L(+)-winowy
kwas
D(-)-winowy
C
C
COOH
H
*
*
H
OH
OH
COOH
zwierciadlo
180o
kwas mezowinowy
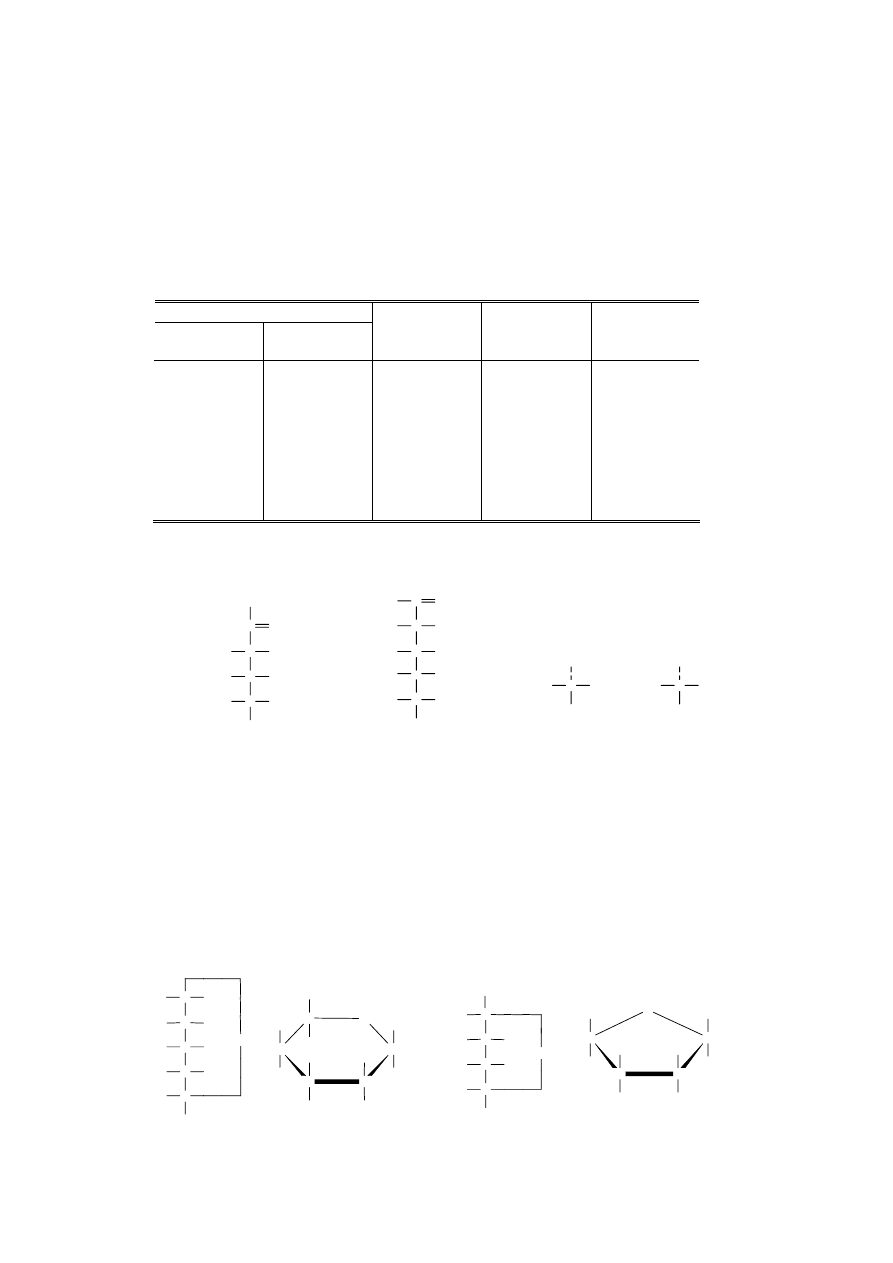
2. Węglowodany (sacharydy)
Duża grupa związków zaliczanych do węglowodanów, zwana jest również sacharydami lub
cukrami. Termin „węglowodany” obejmuje monosacharydy (cukry proste), oligosacharydy,
polisacharydy oraz związki pokrewne (aminocukry, kwasy uronowe, itp.). Natomiast do innych klas
związków chemicznych zalicza się glikozydy aglikonowe (sacharyd połączony wiązaniem
glikozydowym z aglikonem – związkiem niecukrowym), np. nukleozydy, glikozydy roślinne, itp. W
tabeli wymieniono sacharydy najczęściej spotykane w przyrodzie.
Tabela 1.1. Klasyfikacja sacharydów najczęściej spotykanych w przyrodzie.
Monosacharydy
Pentozy
Heksozy
Oligosacharyd
y
Polisacharydy
Pochodne
polisacharydó
w
aldo
Arabinoza
Ksyloza
Ryboza
keto
Rybuloza
Ksyluloza
aldo
Glukoza
Mannoza
Galaktoza
keto
Fruktoza
Sorboza
disacharydy
Sacharoza
Laktoza
Maltoza
Celobioza
trisacharydy
Rafinoza
pentozany
Araban
Ksylan
heksozany
Skrobia
Glikogen
Celuloza
Hemicelulozy
Pektyny
Aminocukry
W skład węglowodanów wchodzą: węgiel, wodór i tlen. Z chemicznego punktu widzenia są to
alkohole wielowodorotlenowe, zawierające w cząsteczce grupę aldehydową (aldozy) lub grupę
ketonową (ketozy).
Związki zbudowane z jednej cząsteczki aldozy lub ketozy nazywa się monosacharydami. Ich
charakterystyczną cechą jest obecność w cząsteczkach asymetrycznych atomów węgla (oznaczonych
na rysunku gwiazdką), co warunkuje istnienie izomerów przestrzennych, czynnych optycznie tzn.
skręcających płaszczyznę światła spolaryzowanego w lewo (-) lub prawo (+). Ich konfigurację
przestrzenną (wywodzącą się od aldehydu glicerynowego) rozróżnienia się na podstawie układu na
przedostatnim węglu, dodając przed nazwą monosacharydu literę
D
lub
L
. Izomery
D
i
L
różnią się
między sobą właściwościami chemicznymi i fizycznymi.
W środowisku alkalicznym monosacharydy występują w formie łańcuchowej, natomiast w
kwaśnym lub obojętnym w formie pierścieniowej (półacetalowej): piranozowej (sześcioczłonowy
pierścień) lub furanozowej (pięcioczłonowy pierścień).
C*
C
O
1
C
*
2
C
CH
2
-OH
H
H
H
OH
OH
HO
3
4
5
6
*
D(-)-fruktoza
ketoheksoza
CH
2
-OH
C*
C
O
1
C
*
2
C
CH
2
-OH
H
H
H
OH
OH
HO
3
4
5
6
*
H
C
OH
H
D(+)-glukoza
aldoheksoza
*
szereg
D
C*
CH
2
OH
H
OH
C*
CH
2
OH
HO
H
szereg
L
-
D
-fruktofuranoza
6
5
4
3
HO
OH
H
H
H
CH
2
-OH
C
O
2
C
1
C
C
HO
CH
2
-OH
wzór pionowy
C
O
C
C
C
HOH
2
C
H
H
H
OH
OH
OH
1
2
3
4
5
6
CH
2
OH
wzór Hawortha
-
D
-glukopiranoza
H
OH
C
HO
OH
H
H
H
CH
2
-OH
C
O
C
C
1
2
3
4
5
6
H
C
OH
wzór pionowy
6
5
4
3
2
1
C
C
O
C
C
C
CH
2
OH
HO
H
H
OH
H
H
H
OH
OH
wzór Hawortha
Pierścień furanozowy jest w zasadzie płaski,
natomiast pierścień piranozowy nie leży na płaszczyźnie,
lecz na powierzchni pofałdowanej, przypominającej
formę krzesełkową cykloheksanu.
Monosacharydy
w
postaci
cyklicznej
mają
dodatkowo jeden asymetryczny atom węgla więcej, co
powoduje powstanie nowych izomerów (tzw. anomerów
- i -). Jeśli grupa hydroksylowa przy pierwszym
atomie węgla cyklicznej postaci cukru, czyli grypa
półacetalowa –OH, jest skierowana przestrzennie tak
samo jak grupa wodorotlenowa przy atomie drugim, to izomer (anomer) ten oznaczamy
, jeśli jest
skierowana przeciwnie, izomer nosi nazwę
.
Glukoza wykazuje zjawisko mutarotacji: w odpowiednich warunkach izomery
- i - mogą
przechodzić jeden w drugi. Związki, w skład których wchodzą izomery
- lub - różnią się znacznie
między sobą wieloma właściwościami (np. amyloza zbudowana jest z
-glukozy, a celuloza w sposób
analogiczny z
-glukozy).
Grupa hydroksylowa półacetalowa, czyli leżąca przy pierwszym atomie węgla piranozowej postaci
cukru lub przy drugim furanozowej postaci, ze względu na swoją reaktywność chemiczną, jest
odpowiedzialna za tworzenie wiązań O-glikozydowych, N-glikozydowych i S-glikozydowych.
Dwie aldozy (np.
D
-glukoza i
D
-mannoza) różniące się jedynie konformacyjnym położeniem
grupy –OH przy drugim atomie węgla (sąsiadującym z grupą aldehydową) nazywa się epimerami.
Epimeryzacja to przekształcanie jednej takiej aldozy w drugą.
Uwaga. Przy nazewnictwie enzymów, dla wygody, pojęcie epimeryzacji rozszerzono i dwie aldozy różniące
się jedynie przestrzennym położeniem jednej grupy –OH przy którymś z asymetrycznych atomów węgla traktuje
się jako epimery. Np. nazwa „4-epimeraza UDP-glukozowa” wskazuje, że enzym może przy czwartym atomie
węgla dokonać epimeryzacji (w tym konkretnym przypadku przekształca UDP-1-glukozę w UDP-1-galaktozę).
2.1. Monosacharydy
Związki zbudowane z jednej cząsteczki aldozy lub ketozy nazywa się ogólnie monosacharydami
(monozami) lub cukrami prostymi. W zależności od ilości atomów węgla w cząsteczce dzieli się je na
triozy, tetrozy, pentozy, heksozy, itd.
Triozy.
Spośród trioz w przyrodzie występuje dihydroksyaceton i oba izomery
D
- i
L
- aldehydu
glicerynowego.
C
C
O
C
C
C
H
H
HO
H
OH
H
H
CH
2
OH
HO
OH
wzor konformacyjny Reeves`a
/
-
D
-glukopiranoza
OH
OH
H
H
H
OH
H
H
HO
CH
2
OH
C
C
O
C
C
C
1
2
3
4
5
6
OH
OH
H
H
H
OH
H
H
HO
CH
2
OH
C
C
O
C
C
C
1
2
3
4
5
6
*
*
-
D
-glukopiranoza
-
D
-glukopiranoza
dihydroksyaceton
aldehyd
L-glicerynowy
CH
2
-OH
C
O
CH
2
-OH
CHO
C
CH
2
OH
HO
H
CHO
C
CH
2
OH
H
OH
aldehyd
D-glicerynowy
Związki te w postaci estrów fosforanowych tworzą się w zielonych częściach roślin podczas
fotosyntezy, w organizmach zwierzęcych między innymi podczas glikolizy oraz podczas fermentacji
alkoholowej.
Tetrozy
Spośród tetroz na uwagę zasługują
D
-erytroza, która pojawia się w cyklu pentozowym, a także
podczas fotosyntezy w zielonych częściach roślin w postaci estru fosforanowego oraz izomery
D
- i
L
-
treozy tworzące się podczas degradacji izomerów
D
- i
L
- kwasu ksylonowego.
Pentozy
Pentozy występują obficie w przyrodzie. Tworzą się one w organizmach zwierzęcych i roślinnych
w znacznych ilościach i to zarówno w stanie związanym, jako O-glikozydy, N-glikozydy lub
wielocukry zwane pentozanami, jak i w stanie wolnym.
D
-ryboza i
D
-dezoksyryboza są składnikami kwasów nukleinowych.
D
-rybuloza, m.in. jest związkiem, który przyłącza CO
2
podczas fotosyntezy cukrów, w pierwszej
fazie procesu.
L
-arabinoza jest jednym z nielicznych
L
- cukrów występujących w przyrodzie. W postaci
spolimeryzowanej jest składową częścią m.in. śluzów, gum roślinnych, pektyn i hemiceluloz.
D
-ksyloza, zwana dawniej cukrem drzewnym, występuje w drewnie w postaci polisacharydu
ksylanu, będącego składnikiem hemiceluloz.
D
-ksyluloza pojawia się w postaci fosforanu podczas wzajemnych przemian pentoz w cyklu
pentozowym.
Na uwagę zasługuje również
L
-ramnoza (uważana za metylopentozę), składnik wielu glikozydów i
antocyjanów.
Heksozy
Obok wymienionych wcześniej glukozy i fruktozy do najszerzej rozpowszechnionych w świecie
ożywionym monosacharydów z tej grupy należą:
D
-mannoza i
D
-galaktoza.
CHO
CH
2
OH
C
H
OH
C
H
OH
C
HO
H
C
HO
H
CHO
C
H
OH
CH
2
OH
C
H
OH
C
HO
H
C
HO
H
C
H
OH
C
H
C
H
OH
C
HO
H
C
HO
H
COOH
O
CH
2
OH
C
HO
H
HO
OH
H
H
C
O
C
C
CH
2
OH
D
-mannoza
D
-galaktoza
L
-sorboza
kwas
galakturonowy
C
OH
H
CHO
CHO
C
HO
H
CH
2
OH
C
H
OH
C
HO
H
CH
2
OH
C
H
OH
CHO
C
HO
H
CH
2
OH
D-treoza
D-erytroza
L-erytroza
CH
2
OH
C
H
OH
C
OH
H
CHO
C
HO
H
D-ksyloza
CH
2
OH
C
O
CH
2
OH
C
H
OH
C
HO
H
D-ksyluloza
C
H
OH
CH
3
C
HO
H
C
OH
H
CHO
C
HO
H
L-ramnoza
C
HO
H
CH
2
OH
C
OH
H
CHO
C
HO
H
L-arabinoza
CHO
CH
2
OH
C
H
OH
C
H
OH
CH
2
D-dezokyryboza
C
OH
H
CHO
CH
2
OH
C
H
OH
C
H
OH
D-ryboza
D-rybuloza
CH
2
OH
C
O
CH
2
OH
C
H
OH
C
H
OH
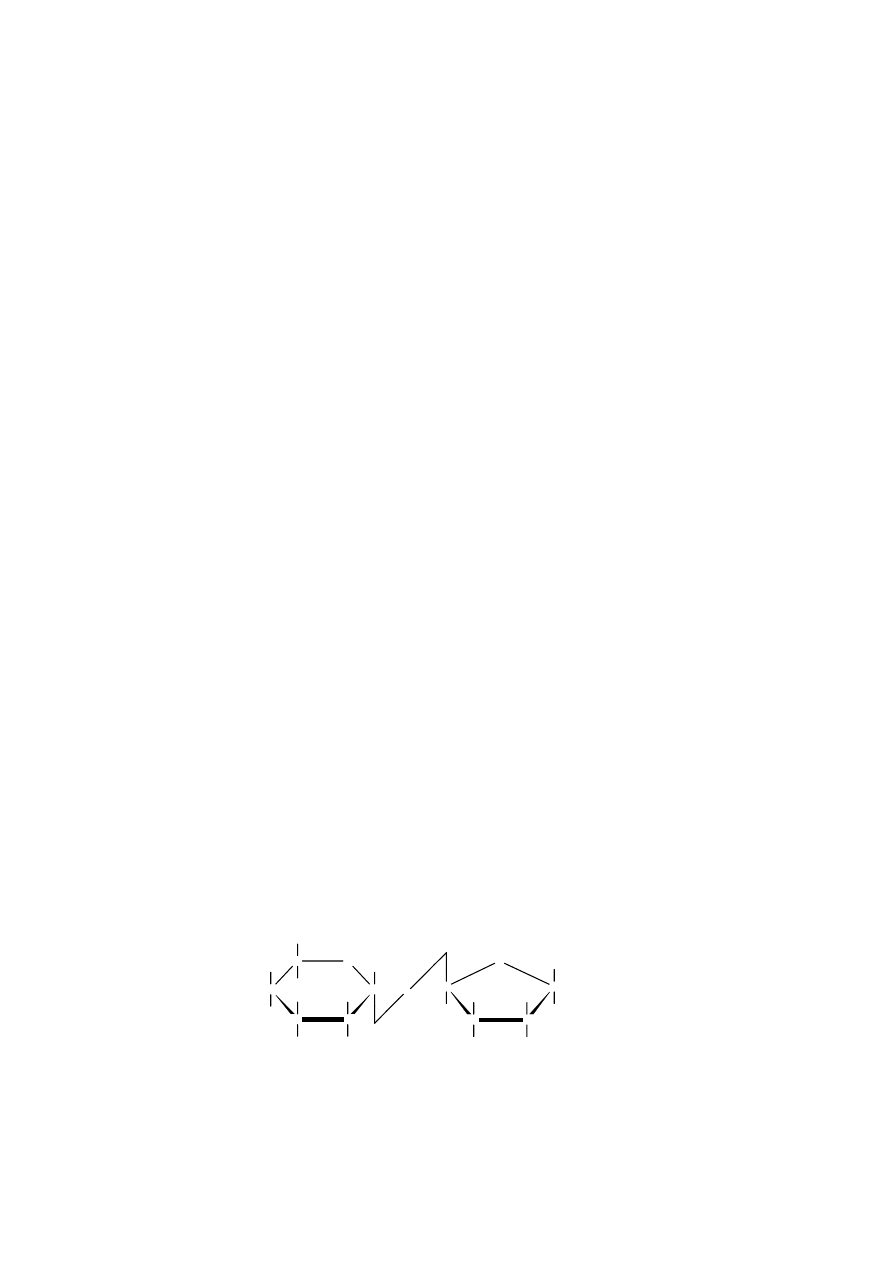
D
- gl u ko za jest najpospolitszą aldozą. Występuje w stanie wolnym w owocach, kwiatach,
miodzie, krwi. Jest częścią składową wielu oligosacharydów (sacharozy, maltozy, laktozy). W
olbrzymich ilościach występuje jako składnik polisacharydów, przede wszystkim skrobi, glikogenu,
celulozy.
D
-f r u kt o za , najważniejsza z ketoz, w stanie wolnym występuje w owocach, zielonych częściach
roślin, miodzie. Jest częścią składową sacharozy, rafinozy oraz polisacharydów – inuliny, lewanu.
D -
ga l a kt o za w stanie wolnym spotykana bywa rzadko (m.in. owoce bluszczu). Jest częścią
składową disacharydu zwierzęcego – laktozy oraz dwóch innych oligosacharydów – melibiozy i
rafinozy. Szczególnie szeroko są rozpowszechnione pochodne zawierające kwas galakturonowy takie,
jak: agar, gumy, pektyny oraz polisacharydy otoczek bakteryjnych.
D -
ma n n o za w stanie wolnym występuje raczej wyjątkowo, np. w skórkach pomarańczy.
Spotykana bywa natomiast w postaci polisacharydów o budowie glikozydowej, zwanych
mannozydami. Do pospolitszych mannozydów należy mannan, występujący w łupinach orzechów, w
drożdżach. Polisacharyd zbudowany z kwasu mannurowego (pochodnej mannanu) występuje dość
powszechnie w wodorostach morskich.
D -
s o r b o za , również jedna z heksoz zasługująca na uwagę (z uwagi na jej wykorzystanie jako
podstawowego surowca do produkcji witaminy C), występuje jako produkt utlenienia sorbitolu
obecnego w soku nasion jarzębiny.
2.2. Oligosacharydy
Monosacharydy mogą kondensować tworząc disacharydy (zbudowane z dwóch cząsteczek cukru
prostego), trisacharydy, tetrasacharydy, itd. o ogólnej nazwie oligosacharydów (połączenie od 2 do 10
cząsteczek monosacharydów). Charakter i właściwości powstałego cukru zależą od ilości połączonych
monosacharydów, samych monosacharydów, z jakich zbudowany jest cukier złożony, rodzaju
wiązania glikozydowego z punktu widzenia konfiguracji
- lub - i wreszcie od miejsca ich
połączenia, tzn. miejsca grupy wodorotlenowej jednego z monosacharydów, która utworzyła wiązanie
glikozydowe (połączone mostkiem tlenowym) z grupą hydroksylową półacetalową drugiego
monosacharydu.
Disacharydy
Disacharyd może być zbudowany z dwóch jednakowych lub różnych monosacharydów. Jeżeli w
wiązaniu glikozydowym wzięły udział obie grupy hydroksylowe półacetalowe połączonych
monosacharydów (jak ma to miejsce np. w sacharozie), to taki disacharyd nie wykazuje właściwości
redukujących. Jeżeli w utworzonym disacharydzie jedna z grup hydroksylowych półacetalowych jest
nadal wolna (np. w maltozie), to taki disacharyd wykazuje właściwości redukujące.
Do najczęściej spotykanych w przyrodzie disacharydów należą: sacharoza, maltoza, laktoza,
celobioza, trehaloza.
Sacharoza, zwana również cukrem trzcinowym lub buraczanym, zasługuje na szczególne
wyróżnienie wśród disacharydów, z uwagi na szerokie rozpowszechnienie w przyrodzie. Występuje w
liściach, łodygach, nasionach, owocach, korzeniach i bulwach różnych roślin. Jej zawartość w
łodygach trzciny cukrowej i bulwach buraków cukrowych może dochodzić do 24%. Znaczne jej ilości
zawiera miód naturalny.
Zbudowana jest z jednej cząsteczki
-D-glukozy i jednej cząsteczki -D-fruktozy połączonych
wiązaniem 1-2 i stąd jej nazwa
-1-
D
-glukopiranozylo-
-2-
D
-fruktofuranozyd. Jest cukrem nie
redukującym.
6
5
4
3
2
1
C
C
O
C
C
C
O
CH
2
OH
HO
H
H
OH
H
H
H
OH
CH
2
OH
6
5
4
3
2
1
OH
OH
H
H
H
HOH
2
C
C
O
C
C
C
sacharoza
rafinoza
6
sacharoza
CH
2
OH
6
5
4
3
2
1
OH
OH
H
H
H
HOH
2
C
C
O
C
C
C
5
4
3
2
1
C
C
O
C
C
C
CH
2
O
HO
H
H
OH
H
H
H
OH
v
melibioza
6
5
4
3
2
1
CH
2
O H
HO
H
H
OH
H
H
H
OH
O
C
C
C
O
C
C
^
6
5
4
3
2
1
C
C
O
C
C
C
CH
2
OH
HO
H
H
OH
H
H
H
OH
OH
OH
H
H
H
OH
H
H
CH
2
OH
C
C
O
C
C
C
1
2
3
4
5
6
O
,
maltoza
Maltoza cukier słodowy, tworzy się podczas enzymatycznego rozpadu skrobi. Jest
-glikozydem
zbudowanym z dwóch cząsteczek glukozy połączonych wiązaniem
-1,4 i stąd jej nazwa -1,4-
D
-
glukopiranozylo-glukopiranozyd. Jest cukrem redukującym.
Celobioza w stanie wolnym nie znaleziona w przyrodzie. Powstaje podczas enzymatycznej
hydrolizy celulozy. Jest
-glikozydem zbudowanym z dwóch cząsteczek glukozy połączonych
wiązaniem
-1,4 i stąd jej nazwa -1,4-
D
-glukopiranozylo-glukopiranozyd. Jest cukrem redukującym.
Laktoza, czyli cukier mlekowy, jest związkiem dobrze znanym, występującym w mleku ssaków.
Zbudowanym jest z galaktozy i glukozy połączonych wiązaniem
-1 od strony galaktozy i stąd jej
nazwa
-1,4-
D
-galaktopiranozylo-glukopiranozyd. Jest cukrem redukującym.
Spośród innych disacharydów wypada wspomnieć o melibiozie, zbudowanej z galaktozy i
glukozy, połączonych wiązaniem
-1 od strony galaktozy i stąd jej nazwa -1,6-
D
-galaktopiranozylo-
glukopiranozyd. Jest cukrem redukującym.
Rafinoza, trisacharyd występujący w burakach cukrowych, który podczas ich przeróbki gromadzi
się w melasie. Zbudowana jest z galaktozy, połączonej wiązaniem
-1,6 z glukozą, a ta wiązaniem -
1-
-2 z fruktozą. Jest cukrem nie redukującym.
6
5
4
3
2
1
CH
2
OH
HO
H
H
OH
H
H
H
OH
celobioza
OH
OH
H
H
H
OH
H
H
CH
2
OH
C
C
O
C
C
C
O
C
C
C
O
C
C
1
2
3
4
5
6
6
5
4
3
2
1
CH
2
OH
HO
H
H
OH
H
H
H
OH
OH
OH
H
H
H
OH
H
H
CH
2
OH
C
C
O
C
C
C
O
C
C
C
O
C
C
1
2
3
4
5
6
laktoza
2.3. Polisacharydy
Polisacharydy zbudowane są z setek a nawet tysięcy połączonych ze sobą cząsteczek
monosacharydów, tworząc jedną olbrzymią cząsteczkę. Ich skład z reguły przedstawia się wzorem
sumarycznym: (C
6
H
10
O
5
)
n
. Są szeroko rozpowszechnione w przyrodzie. Niektóre, jak skrobia i
celuloza, spotykane są powszechnie u roślin. Do związków pokrewnych polisacharydom zalicza się
także hemicelulozy, aminocukry i pektyny.
Skrobia jest szeroko rozpowszechniona w świecie roślinnym; jest zawarta w ziarnach zbóż i
bulwach roślin. Skrobia jest mieszaniną dwóch polisacharydów: rozpuszczalnej w wodzie amylozy
(około 20%) oraz nierozpuszczalnej w wodzie amylopektyny (około 80%).
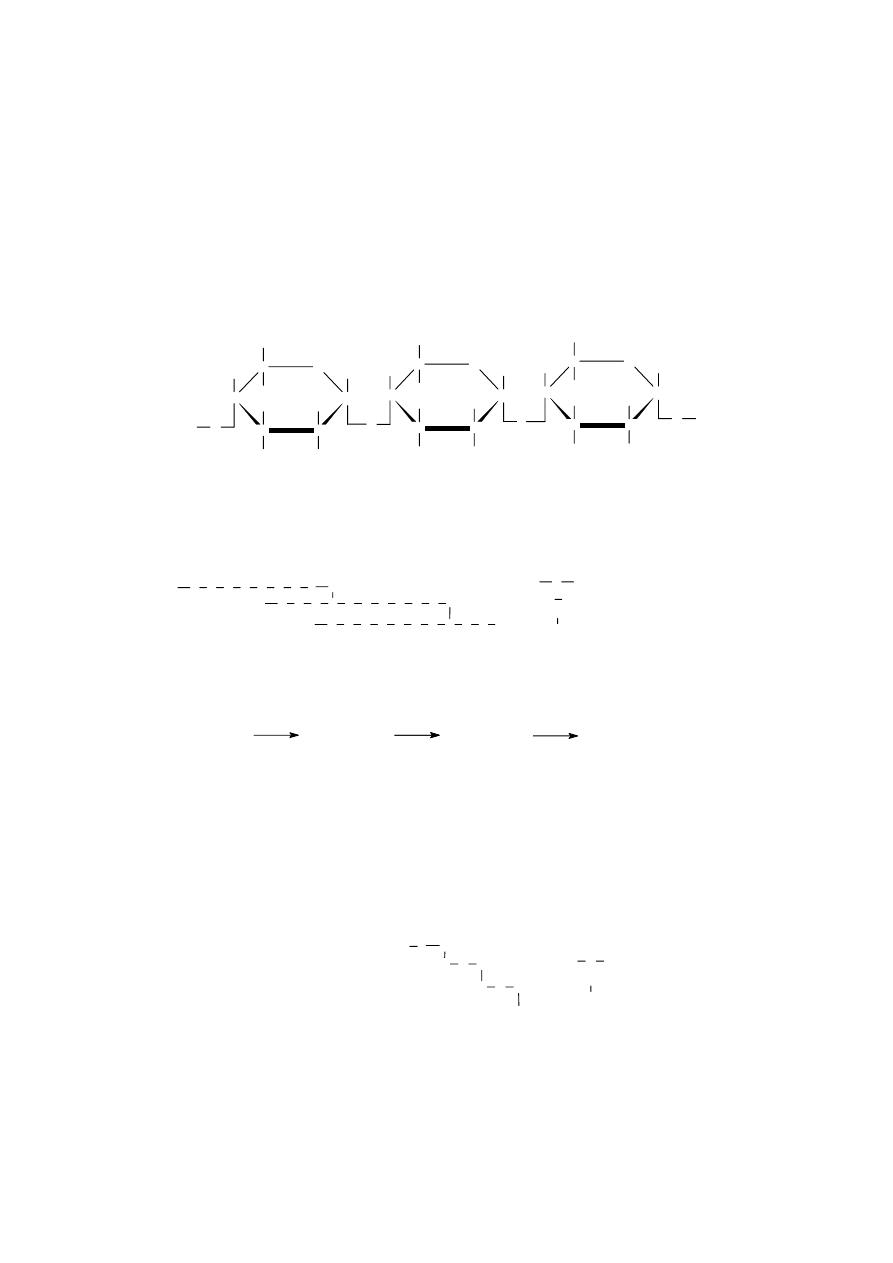
A m y l o z a zbudowana jest z reszt α-D-glukopiranoz połączonych wiązaniem α-1,4-
glikozydowym. Jej łańcuch jest nie rozgałęziony.
A m y l o p e k t y n a , podobnie jak amyloza, zbudowana jest z reszt glukopiranozy połączonych
wiązaniami α-1,4-glikozydowymi. W przeciwieństwie do amylozy jest jednak rozgałęziona; od jej
głównego łańcucha, co 24-30 reszt glukozowych, odchodzą łańcuchy boczne utworzone przez
wiązania α-1,6-glikozydowe. Jej budowę można schematycznie przedstawić następująco:
Dekstryny są produktem częściowej hydrolizy skrobi. Biorąc pod uwagę ciężar cząsteczkowy
można je uszeregować następująco:
Glikogen, zapasowy węglowodan zwierząt, o bardziej rozgałęzionych łańcuchach aniżeli
amylopektyna; co 8-12 reszt glukozowych pojawia się rozgałęzienie utworzone przez wiązanie
-1,6-
glikozydowe.
Dekstran to wielocukier o masie cząsteczkowej od 40 do 200 kDa, którego budowa przypomina
amylopektynę. Większość cząsteczek glukozy połączona jest jednak wiązaniem
-1,6-glikozydowym,
a tylko nielicznie występują wiązania
-1,4 (około 10%).
Inulina, zbudowana podobnie jak skrobia z tym, że jest polifruktozofruktozydem. Reszty
fruktofuranozydowe połączone są za sobą wiązaniem
-1,2-glikozydowym.
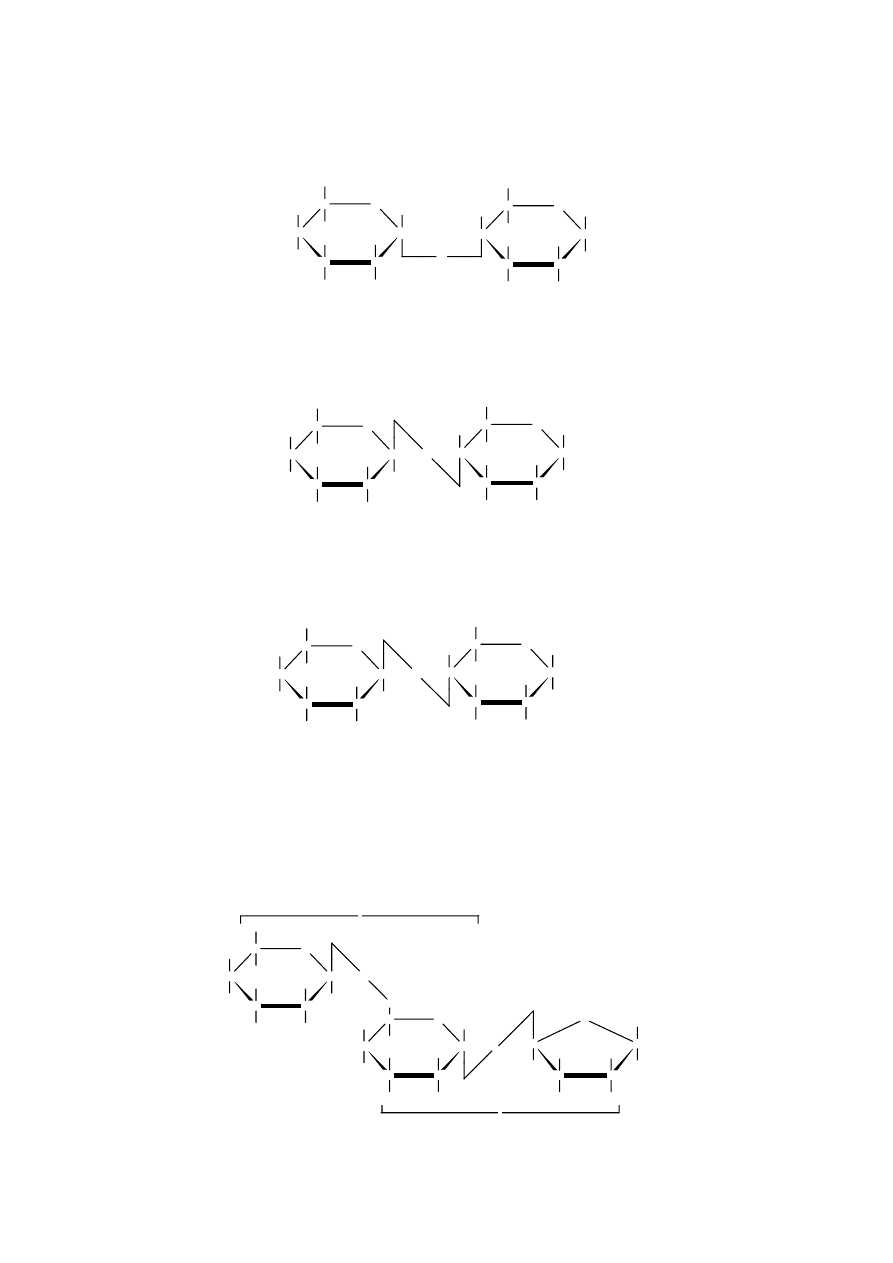
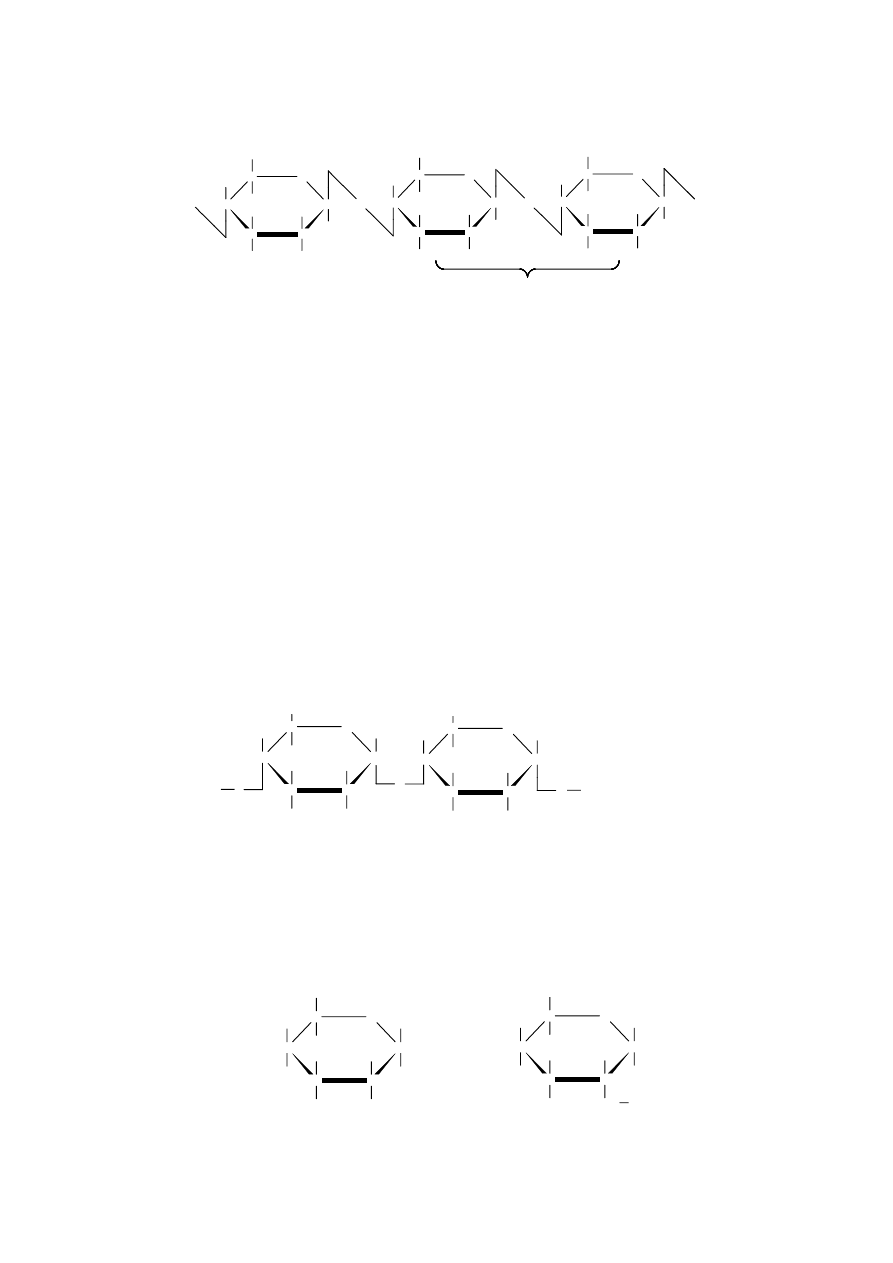
Pulullan, polisacharyd zbudowany z
cząsteczek maltotriozy połączonych
wiązaniem
-1,6-glikozydowym.
Schematycznie jego budowę można
przedstawić następująco:
Celuloza, szeroko rozpowszechnio-
na w przyrodzie, jako główny składnik ścianek komórkowych roślin. Pod względem chemicznym nie
jest jednorodnym związkiem, lecz można ją zaklasyfikować do przynajmniej dwóch głównych klas,
alfa i beta.
-Celuloza rozpuszcza się w 17% roztworze wodorotlenku sodowego, podczas gdy -
celuloza w tych warunkach jest nierozpuszczalna.
-Celulozę używa się do produkcji sztucznego
włókna.
erytrodekstryna
achrodekstryna
amylodekstryna
skrobia
pulullan
O O
O
O
O
O
O
O O
O
O
- wiązanie -1,6
O O
O
- maltotrioza (wiązanie -1,4)
O O O O O O O O
O
O
O
O O O O O O O
O
O
O
O
O
O O O O O O O O
amylopektyna
O
O
O
- wiązanie -1,6
- końcowa redukująca grupa
O O - wiązanie -1,4
O
- reszta glukozowa
6
5
4
3
2
1
C
C
O
C
C
C
CH
2
OH
H
H
OH
H
H
H
OH
O
6
5
4
3
2
1
C
C
O
C
C
C
O
CH
2
OH
H
H
OH
H
H
H
OH
O
6
5
4
3
2
1
C
C
O
C
C
C
O
CH
2
OH
H
H
OH
H
H
H
OH
amyloza
6
5
4
3
2
1
C
C
O
C
C
C
CH
2
OH
HO
H
H
OH
H
H
H
OH
NH COCH
3
N-acetylo-2-dezoksy-
2-aminoglukoza
NH
2
6
5
4
3
2
1
C
C
O
C
C
C
CH
2
OH
HO
H
H
OH
H
H
H
OH
2-dezoksy-2-aminoglukoza
Budową przypomina amylozę z tym, że reszty glukozowe połączone są wiązaniem
-1,4-
glikozydowym.
Hemicelulozy, niejednorodna grupa polisacharydów występujących, obok celulozy i ligniny, w
zdrewniałych tkankach roślin. Wśród hemiceluloz rozróżnia się ksylany (z D-ksylozą), galaktany (z
D
-galaktozą), arabany (L-arabinozą) oraz mannany (z D-mannozą). W skład hemiceluloz wchodzą
również kwasy uronowe.
Ksylan, polisacharyd zaliczany do pentozanów, pochodzący z drewna, otrąb i łupin orzeszka
ziemnego. Po hydrolizie daje D-ksylozę Niektóre ksylany, podobnie jak celuloza, mają strukturę
liniową, inne – rozgałęzioną i dodatkowo zawierają cząsteczki L-arabinozy lub kwasu D-
glukuronowego. Główny łańcuch zbudowany jest z reszt D-ksylopiranozy połączonych wiązaniami
-
1,4-glikozydowymi, od którego odchodzą liczne rozgałęzienia - głównie przy węglach 2 i 3.
Pektyny zazwyczaj występuje w postaci złożonego kompleksu zwanego protopektyną, którego
skład schematycznie można przedstawić następująco:
araban
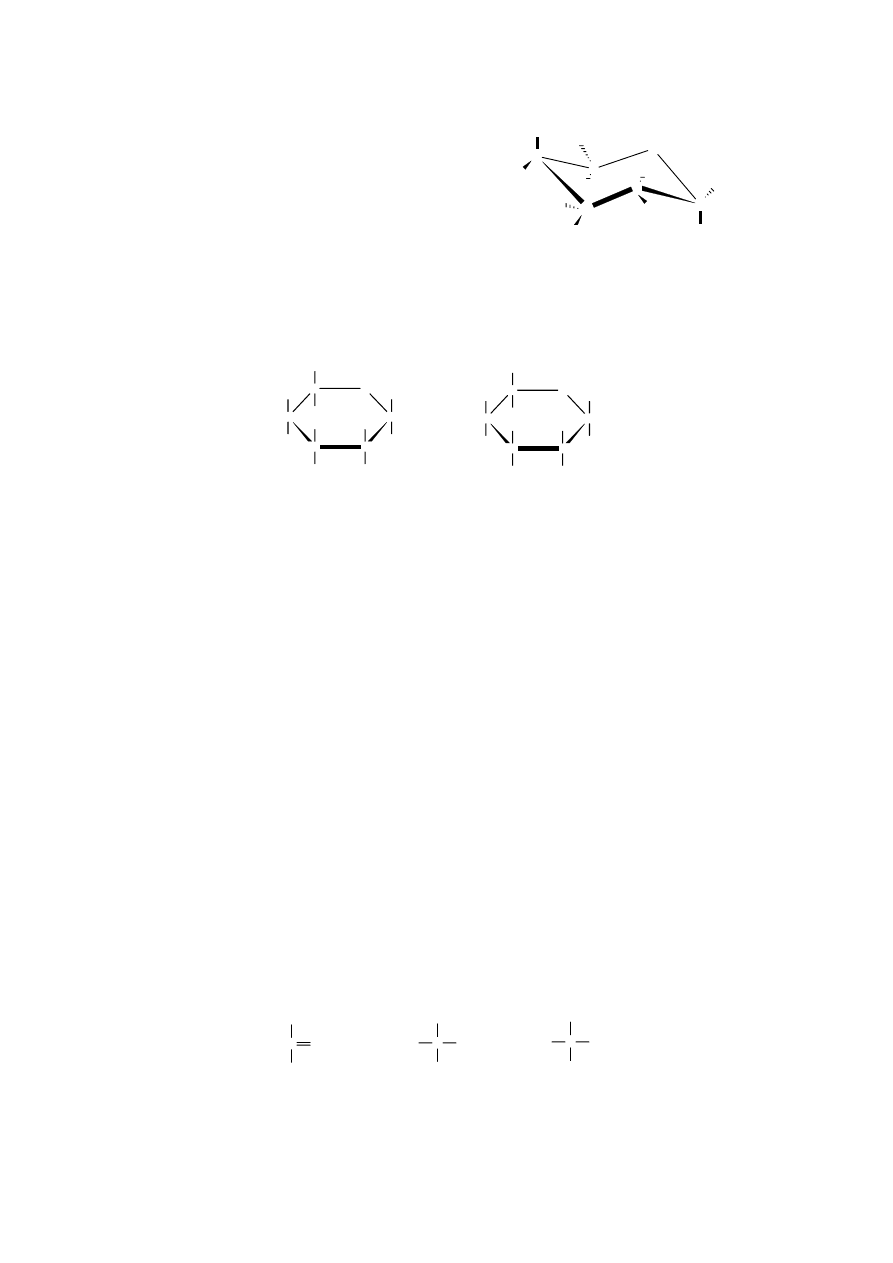
zmetoksylowany kwas poligalakturonowygalaktan
Główną strukturę stanowi łańcuch zbudowany z reszt kwasu D-galakturonowego, połączonych
wiązaniami
-1,4-glikozydowymi. Grupy karboksylowe reszt kwasu galakturonowego są w
mniejszym lub większym stopniu zestryfikowane alkoholem metylowym. Od węgli 2 i 3 głównego
łańcucha odgałęziają się łańcuchy boczne.
Aminocukry. W przyrodzie znaleziono dwie pochodne aminowe cukrów: 2-dezoksy-2-
aminoglukozę i 2-dezoksy-2-aminogalaktozę. Reszty N-acetylo-2-aminoglukozy połączone są
wiązaniem
-1,4-glikozydowym są podstawowym składnikiem chityny. Stąd chitynę uważa się za
pochodną aminową celulozy acetylowaną przy azocie. D-Glukozoamina wchodzi również w skład
kwasu hialuronowego, podstawowej substancji tkanki łącznej.
celuloza
6
5
4
3
2
1
CH
2
OH
H
H
OH
H
H
H
OH
OH
H
H
H
OH
H
H
CH
2
OH
C
C
O
C
C
C
O
C
C
C
O
C
C
O
1
2
3
4
5
6
OH
H
H
H
OH
H
H
CH
2
OH
1
2
3
4
5
6
C
C
O
C
C
C
O
O
reszta celobiozy
6
5
4
3
2
1
C
C
O
C
C
C
COOH
H
H
OH
H
H
H
OH
O
OH
H
H
H
OH
H
H
C
C
O
C
C
C
COOCH
3
1
2
3
4
5
6
O
O
fragment zmetoksylowanego kwasu poligalakturonowego
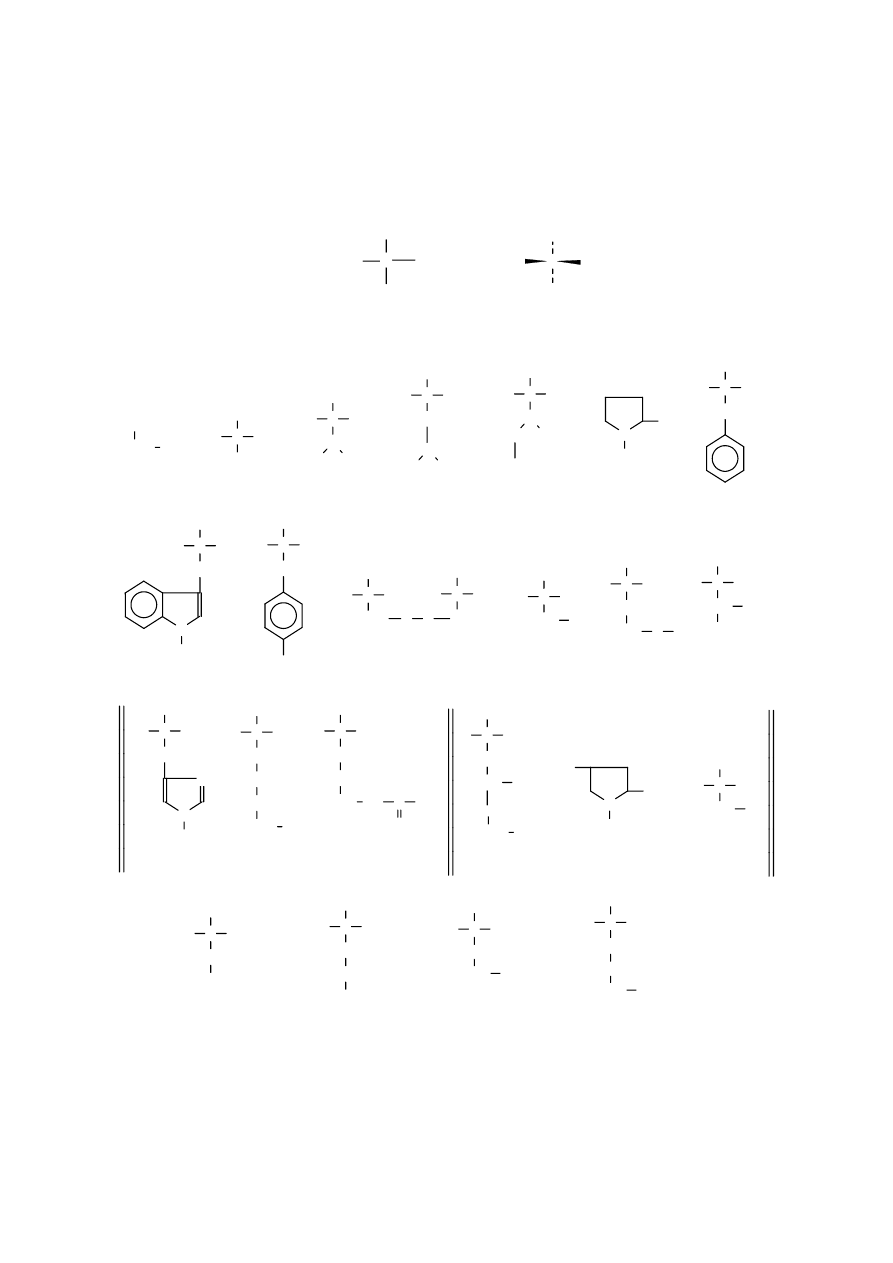
11
3. Aminokwasy, peptydy i białka
3.1. Aminokwasy
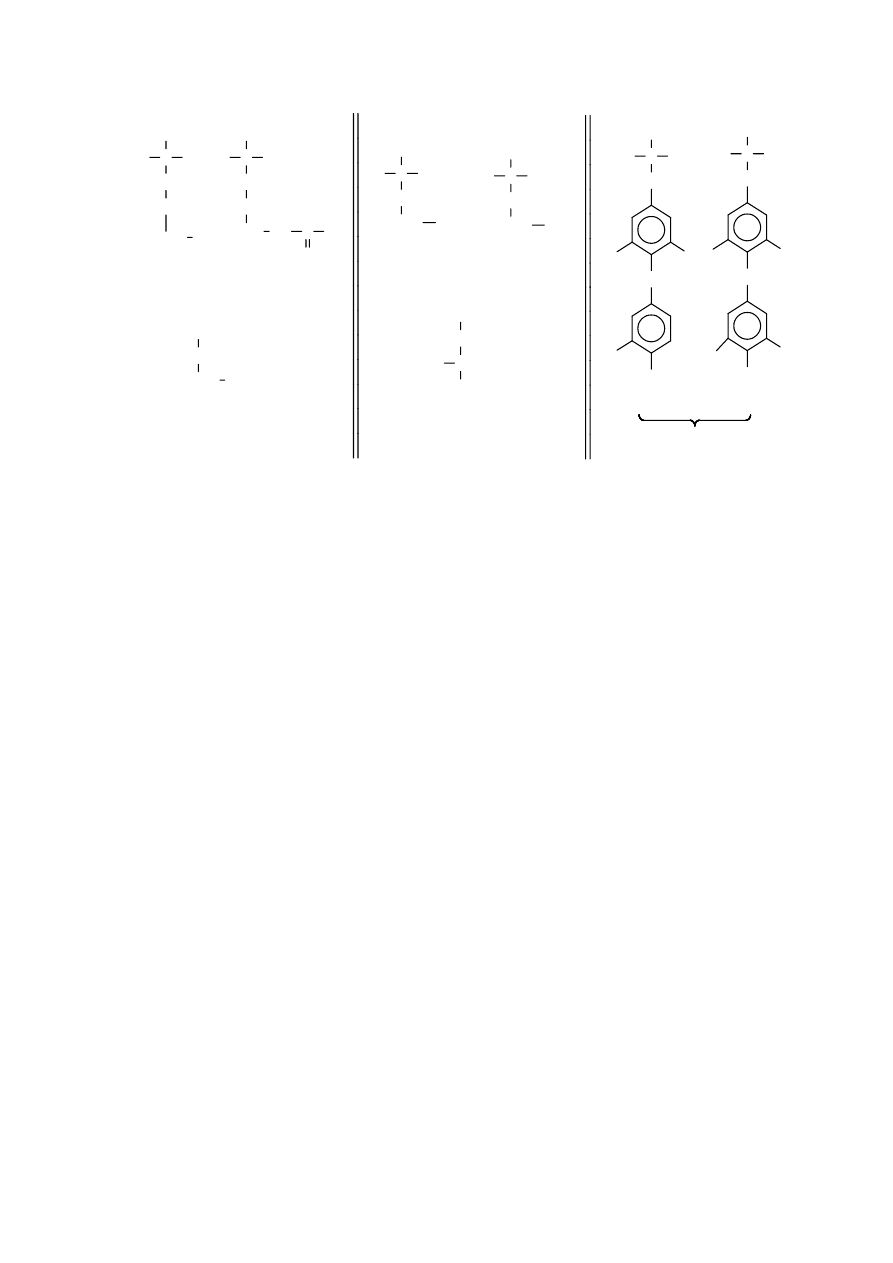
Wszystkie aminokwasy wchodzące w skład białek mają konfigurację
L
i należą do grupy
aminokwasów, to znaczy mają grupę aminową przy węglu sąsiadującym z grupą karboksylową.
Ogólny ich wzór jest następujący:
W organizmach żywych występują również aminokwasy nie będącymi składnikami białek.
Poniżej podano wzory najwazniejszych aminokwasów spotykanych w przyrodzie ożywionej.
Aminokwasy spotykane w białkach
C
COO
-
R
+
H
3
N
H
C
H
R
COO
-
+
H
3
N
lub
hydroksylizyna
C NH
2
COOH
CH
3
H
C NH
2
COOH
CH
H
H
3
C
CH
3
C NH
2
COOH
CH
H
CH
3
OH
CH
2
COOH
NH
2
glicyna
C NH
2
COOH
CH
2
H
CH
H
3
C
CH
3
C NH
2
COOH
CH
H
CH
3
H
2
C
H
3
C
alanina
walina
leucyna
izoleucyna
N
H
COOH
prolina
C NH
2
COOH
CH
2
H
OH
C NH
2
COOH
CH
2
H
S S
C NH
2
COOH
CH
2
H
cystyna
C NH
2
COOH
CH
2
H
CH
2
CH
2
CH
2
NH
2
C NH
2
COOH
CH
2
H
OH
seryna
treonina
tyrozyna
N
H
COOH
HO
hydroksyprolina
C NH
2
COOH
CH
2
H
SH
cysteina
C NH
2
COOH
CH
2
H
CH
2
CH
2
NH C NH
2
NH
lizyna
arginina
C NH
2
COOH
CH
2
H
N
N
H
histydyna
CH
2
CH
2
NH
2
C NH
2
COOH
CH
2
H
CH OH
C NH
2
COOH
CH
2
H
fenyloalanina
C NH
2
COOH
CH
2
H
N
H
tryptofan
C NH
2
COOH
CH
2
H
CH
2
S CH
3
metionina
C NH
2
COOH
CH
2
H
COOH
kwas
asparaginowy
C NH
2
COOH
CH
2
H
CH
2
COOH
kwas
glutaminowy
C NH
2
COOH
CH
2
H
CO NH
2
asparagina
C NH
2
COOH
CH
2
H
CH
2
CO NH
2
glutamina
12
Aminokwasy niebiałkowe
Tyroksyna i trijodotyronina pochodne nie występującej w przyrodzie tyroniny, występują jedynie
w tyreoglobulinie, hormonalnym białku tarczycy. β-alanina, karnityna oraz N-metylohistydyna są
aminokwasami biologicznie czynnymi lub też są składnikami biologicznie czynnych substancji
niskocząsteczkowych. Homocysteina i homoseryna są metabolitami pośrednimi przemian metioniny i
cysteiny. Kwas β-aminomasłowy jest produktem końcowym rozkładu nukleotydów pirymidynowych.
Z kolei ornityna i cytrulina biorą udział w syntezie mocznika.
Aminokwasy o konfiguracji D występują w przyrodzie rzadko i to wyłącznie w związkach
względnie prostych, nie będącymi białkami (np. w niektórych antybiotykach pochodzenia
bakteryjnego o budowie peptydowej).
W laboratoriach sztucznie otrzymuje się całą gamę aminokwasów nie spotykanych w przyrodzie
ożywionej. Z ich wykorzystaniem np. do produkcji leków wiąże się duże nadzieje.
Podział α-aminokwasów
Poszczególne α-aminokwasy różnią się strukturą rodnika. Stosuje się różne kryteria ich podziału
Rozróżnia się aminokwasy alifatyczne i aromatyczne, a w zależności od ilości grup aminowych i
karboksylowych aminokwasy mono- lub di- aminowe lub karboksylowe. Rodniki aminokwasów
cysteiny i metioniny w swojej budowie zawierają atom siarki (są to aminokwasy siarkowe). Rodniki
waliny, leucyny i izoleucyny posiadają rozgałęziony łańcuch alifatyczny (aminokwasy rozgałęzione).
Prolina jest z kolei iminokwasem (kwasem pirolidyno-karboksylowym).
Często aminokwasy dzieli się biorąc pod uwagę hydrofobowość lub hydrofilowość łańcucha
bocznego. Aminokwasy z niepolarnymi rodnikami węglowodorowymi (glicyna, alanina, walina,
leucyna i izoleucyna) lub zawierające grupę funkcyjną o znikomej polarności (prolina, fenyloalanina,
tryptofan, metionina i cystyna) zalicza się do aminokwasów hydrofobowych.
Z punktu widzenia wartości pokarmowych (dietetycznych) aminokwasy dzieli się na egzogenne
(niezbędne) i endogenne (nie niezbędne). Aminokwasy egzogenne muszą być dostarczone
organizmowi zwierzęcemu z zewnątrz (np. z pokarmem), z uwagi na zanik zdolności do ich
syntetyzowania. Dla człowieka niezbędnymi aminokwasami są w al i n a , l e ucyn a , i zol eu c yn a ,
f e n yl o al an i n a, t r yp t of a n, t r e o ni na , me t i oni na i l i zyn a .
C NH
2
COOH
CH
2
H
CH
2
CH
2
NH C NH
2
O
C NH
2
COOH
CH
2
H
CH
2
CH
2
NH
2
ornityna
cytrulina
CH
2
NH
2
CH
2
COOH
-alanina
C NH
2
COOH
CH
2
H
O
OH
J
J
J
C NH
2
COOH
CH
2
H
O
OH
J
J
J
J
trijodotyronina
tyroksyna
tyreoglobulina
C NH
2
COOH
CH
2
H
CH
2
SH
C NH
2
COOH
CH
2
H
CH
2
OH
homocysteina
homoseryna
CH
2
COOH
CH
CH
3
H
2
N
*
kwas
-aminomaslowy
13
3.2. Peptydy
Grupa aminowa jednego α-aminokwasu może reagować z grupą karboksylową innego
α-aminokwasu tworząc w i ą za n i e p ep t yd o we
–CO–NH–
, które z chemicznego punktu widzenia
jest szczególnym przypadkiem wiązania amidowego (co ma pewne znaczenie, z uwagi na
specyficzność działania niektórych enzymów). Związki powstałe z takiego połączenia nazwano
p e pt yd a mi .
Podane wyżej równanie przedstawia jedynie schemat syntezy peptydu i jego uproszczony wzór, nie
uwzględniający budowy przestrzennej.
3.2.1. Budowa peptydów - wiązanie peptydowe
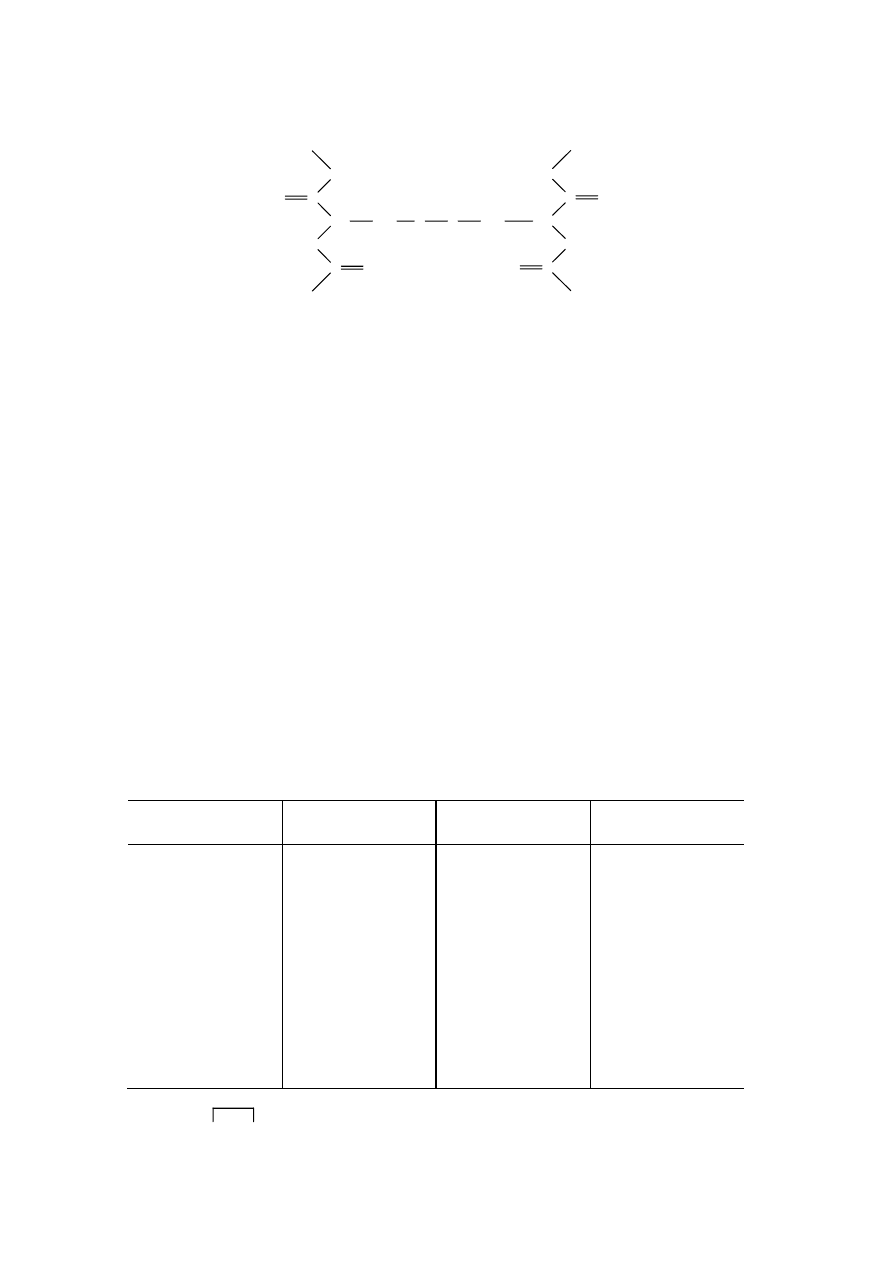
Badania krystalograficzne wykazały, że wiązanie C–N w wiązaniu peptydowym jest znacznie
krótsze niż zwyczajne pojedyncze wiązanie tego typu. Jest to spowodowane, mezomerycznym
charakterem wiązania peptydowego (hybrydyzazja typu sp
2
). Wynikiem tej mezomerii jest brak
swobodnego obrotu wokół wiązania C – N i stąd istnienie tylko dwóch stabilnych konformacji cis i
trans. Ogólnie w natywnych peptydach i białkach występuje głównie forma trans.
Zgodnie z tym właściwsze jest przedstawianie wiązania peptydowego za pomocą dwu wzorów
granicznych lub wzorem, na którym symbolicznie zaznaczono delokalizację elektronów.
Z badań rentgenograficznych wynika, że konfiguracja wiązania peptydowego nie zależy od rodzaju
i właściwości łańcuchów bocznych aminokwasów tworzących wiązanie. Wyjątek pod tym względem
stanowi prolina i hydroksyprolina, których grupy iminowe związane są z łańcuchami bocznymi.
Delokalizacja elektronów w wiązaniu peptydowym sprawia, że wszystkie cztery jego atomy:
CONH a również i oba atomy C
α
łańcuchów bocznych, leżą w jednej płaszczyźnie (mówi się o
planarności wiązania peptydowego). W konsekwencji wiązanie łączące atom węgla i azotu wykazuje
znaczną sztywność właściwą wiązaniu podwójnemu, a swobodny obrót wokół niego wymaga
przezwyciężenia oporu około 88 kJ/mol. Na poniższym rysunku przedstawiono perspektywicznie
strukturę wiązania peptydowego w hipotetycznym dipeptydzie.
Drugim ważnym wiązaniem kowalencyjnym, spotykanym w cząsteczkach peptydów, jest wiązanie
disulfidowe (disiarczkowe), tworzone przez sąsiadujące ze sobą grupy tiolowe – SH pochodzące z reszt
cysteiny. Rozróżnia się wewnątrzłańcuchowe wiązania disulfidowe, które występują w tym samym
CH
H
2
N
R
1
COOH +
CH
H
2
N
COOH
R
2
H
2
O
CH
H
2
N
R
1
CO NH
CH COOH
R
2
aminokwas 1
aminokwas 2
dipeptyd
C
N
O
C
H
C
C
N
C
H
C
O
+
-
..
-
+
delokalizacja elektronow w wiazaniu peptydowym
H
H
H
R
2
H
N
O
C
C
H
N
O
C
C
C
H
2
N
R
1
COOH
R
2
14
łańcuchu peptydowym, oraz międzyłańcuchowe wiązania disulfidowe występujące między dwoma
różnymi łańcuchami peptydowymi:
Wewnątrzcząsteczkowe wiązania S – S występują np. w oksytocynie, wazopresynie, w łańcuchu A
insuliny i w rybonukleazie. Wiązania te mogą spowodować, że części cząsteczki o zupełnie różnych
sekwencjach znajdują się w bezpośrednim zbliżeniu, jak np. w rybonukleazie. W utlenionej formie
glutationu dwa identyczne łańcuchy peptydowe połączone są przez kowalencyjne wiązanie disulfidowe.
W insulinie w ten sam sposób połączone są dwa różne łańcuchy.
Sąsiadujące ze sobą wiązania peptydowe, np. w dwu równoległych peptydach, mogą tworzyć
wiązania wodorowe >CO...HN<, w których odległość atomu tlenu od atomu azotu wynosi nieco mniej
niż 0,3 nm.
Oprócz tych wiązań, czynnikami mającymi wpływ na przyjęcie przez cząsteczkę peptydu
określonej konformacji są oddziaływania kwasowych, zasadowych, aromatycznych i alifatycznych
łańcuchów bocznych różnych aminokwasów ze sobą lub z łańcuchem głównym peptydu. Te
oddziaływania, zwykle stabilizujące, staja się w pełni efektywne dopiero w białkach, niemniej mają one
także znaczenie dla utrzymania odpowiednich efektywnych konformacji wielu polipeptydów
biologicznie aktywnych.
3.2.2. Nazwy i wzory peptydów
Nazwy peptydów są tworzone w zależności od liczby rodników aminoacylowych tworzących
cząsteczkę peptydu. Peptyd zbudowany z dwu aminokwasów nazywa się dipeptydem, z trzech —
tripeptydem, z dziesięciu — dekapeptydem, itp. Peptydy zawierające w cząsteczce do 10 rodników
aminoacylowych nazywa się o l i go pe pt yd a mi , zawierające zaś większą liczbę —
p o l i pe pt yd a mi . Te ostatnie stanowią przejście do białek (zaliczanych do makropeptydów), których
cząsteczki są zbudowane ze 100 i więcej rodników aminoacylowych.
Tabela 1.2. Nazwy i symbole najpospolitszych
-aminokwasów
Rodnik
aminoacylowy
Symbol
Rodnik
aminoacylowy
Symbol
Alanyl
Arginyl
Aspargil
Asparginyl
Cysteil
Cysteinyl
Fenyloalanyl
Glicyl
Glutamil
Glutaminyl
Histydyl
Hydroksylizyl
Ala
Arg
Asp
Asn lub Asp(NH
2
)
Cys
Cys
Cys
1
Fen (Phe)
Gli (Gly)
Glu
Gln lub Glu(NH
2
)
His
Hyl lub Liz(OH)
Hydroksyprolil
Izoleucyl
Leucyl
Lizyl
Metionyl
Ornityl
Prolina
Seryl
Treonyl
Tryptofanyl
Tyrozyl
Walil
Hyp lub Pro(OH)
Ileu (Ile)
Leu
Liz (Lys)
Met
Orn
Pro
Ser
Tre (Thr)
Try (Trp)
Tyr
Wal (Val)
W nawiasach podano symbole zalecane przez Unię Biochemiczną.
1
również Cys Cys
C
HN
CH
C
O
O
NH
CH
2
S
S
H
2
C
C
HN
O
HC
NH
C
O
mostek disiarczkowy
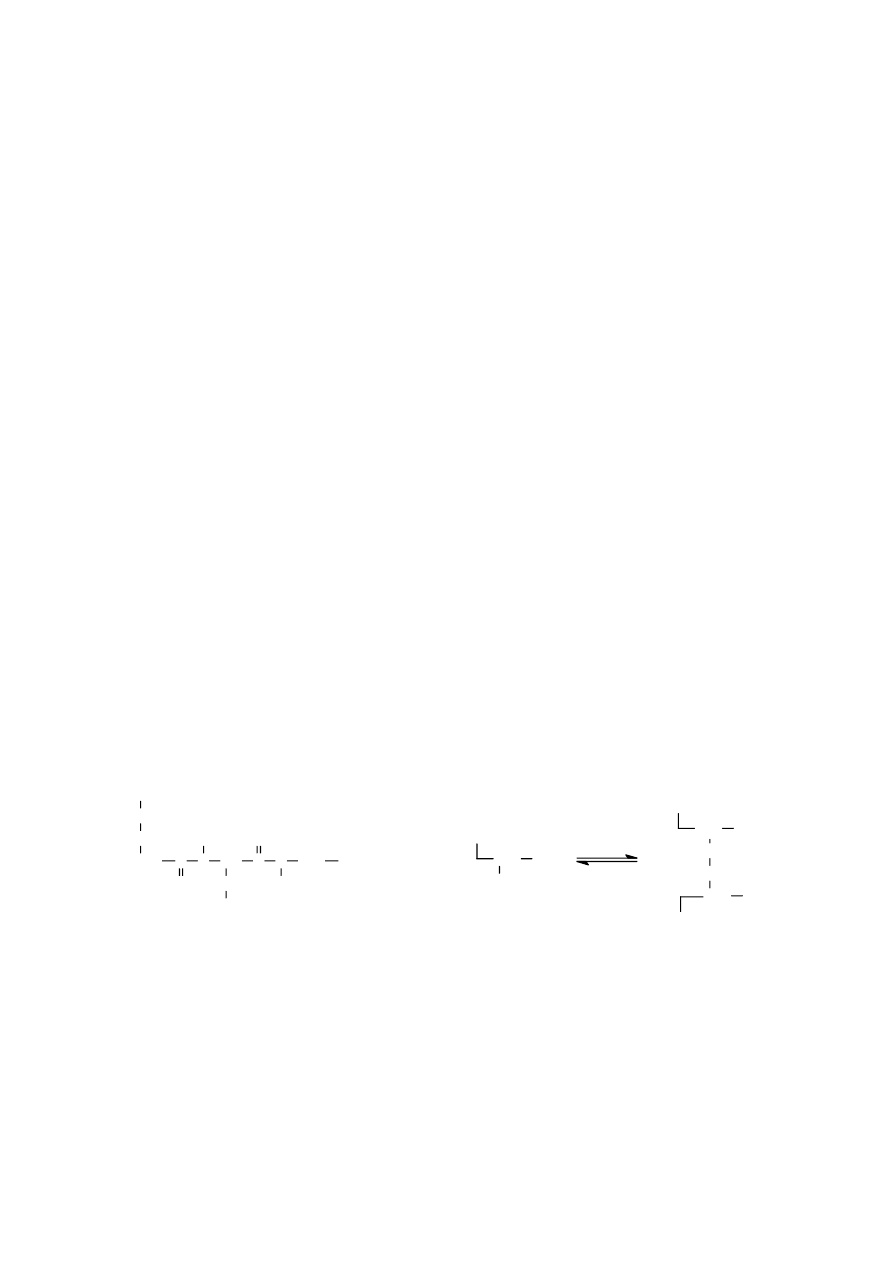
15
Szczegółowe nazwy peptydów są albo zwyczajowe, albo mają charakter systematycznych nazw
chemicznych określających je jako aminoacyloaminokwasy. Na przykład tripeptyd zbudowany z
glicyny, alaniny i cysteiny nosi nazwę glicyloalanylocysteiny. Nazwa ta podaje skład aminokwasowy
peptydu i jednocześnie kolejność łączenia się aminokwasów. I tak tripeptyd wspomniany wyżej jest
zupełnie różnym od alanyloglicylocysteiny, peptydu zbudowanego z identycznych aminokwasów, ale
połączonych w innej kolejności.
W celu uproszczenia zapisywania wzorów peptydów i uniknięcia długich nazw, ustalono, by
rodniki aminoacylowe oznaczać pierwszymi trzema literami nazwy aminokwasu. W tabeli 1.2 podano
nazwy i symbole rodników najpospolitszych
-aminokwasów.
Przy pisaniu wzorów peptydów symbole łączy się krótką kreską, przy czym lewa strona symbolu
odpowiada grupie aminowej, a jego prawa strona - grupie karboksylowej. Wzory przytoczonych wyżej
tripeptydów można więc zapisać symbolicznie: Gli-Ala-Cys oraz Ala-Gli-Cys. Jeżeli zachodzi
specjalna potrzeba wyraźnego wskazania kierunku, w jakim połączone są ze sobą cząsteczki
aminokwasów w peptydzie (np. przy przedstawianiu struktury peptydów pierścieniowych) wówczas
zaznacza się strzałką
kierunek powstawania wiązań od grupy karboksylowej do aminowej:
Gli
AlaCys. Jeżeli kolejność łączenia się aminokwasów nie jest znana dla jakiegoś fragmentu
łańcucha polipeptydowego, to fragment ten ujmuje się w nawiasy, wewnątrz których symbole oddziela
się przecinkami, np. Gli-Ala-Cys-(Arg, Tyr, Leu)-Pro-Ala-Fen. Końcowy aminokwas z wolną grupą
aminową, tzw. N-terminalny aminokwas, symbolizuje się dopisaniem litery H- lub H
2
N-, zaś końcowy
aminokwas z wolną grupą karboksylową, czyli C-terminalny aminokwas, dodaniem liter -OH lub -
COOH: H-Gli-Ala-Cys-OH lub H
2
N-Gli-Ala-Cys-COOH.
3.2.3. Ważniejsze peptydy naturalne
W ciągu ostatnich 20 lat liczba peptydów znalezionych w materiale zwierzęcym, roślinnym i
bakteryjnym ogromnie wzrosła. Większość znajdowanych w przyrodzie peptydów składa się
wyłącznie z aminokwasów, które zwykle spotykamy w białkach. Jednak w naturalnych peptydach
często występują inne proste aminokwasy, takie jak β-alanina i kwas γ-aminomasłowy.
Najprostszym i jednocześnie najwcześniej poznanym dipeptydem wyodrębnionym z tkanek była
ka r n o zyn a , czyli
-alanylohistydyna. Obok niej w mięśniach kręgowców spotyka się także
anserynę:
-alanylo–N-metylohistydynę. Rola obydwu dipeptydów jest do dzisiaj nie poznana.
Glutation (GSH) jest tripeptydem, który szeroko rozpowszechniony jest w zarówno w tkankach
zwierzęcych jak i roślinnych. Zawiera on kwas glutaminowy, cysteinę i glicynę. Był pierwszym
znanym peptydem zawierającym wiązanie inne niż α-peptydowe.
Związek ten łatwo ulega odwracalnym przemianom oksydacyjno-redukcyjnym. Reakcja
utlenienia jest katalizowana solami Cu i Fe. Redukcja disulfidu i przekształcenie do GSH
katalizowane jest przez reduktazę glutationową.
Funkcje biochemiczne glutationu są bardzo różne. Jest to koenzym hydrolazy
hydroksyacyloglutationowej, dehydrogenazy formaldehydowej, tautomerazy indolilopirogronianowej,
izomerazy maleiloacetooctanowej i innych enzymów. Stanowi on również grupę prostetyczną
dehydrogenazy fosfogliceroaldehydowej oraz odgrywa pewną rolę w działaniu dehydrochlorynazy
DDT, którą odkryto w odpornych na DDT muchach domowych. Glutation jest także bardzo ważnym
związkiem w wielu biologicznych reakcjach redox, ponieważ może stabilizować wolne grupy – SH.
COOH
CH-NH
2
CH
2
CH
2
C N
O
H
CH
CH
2
SH
C
O
N
H
CH
2
COOH
glutation
Glu
Cys
SH
Gli
2
-2H
+
Glu
Cys Gli
S
S
Cys Gli
Glu
utlenianie i redukcja glutationu

16
Biosynteza glutationu zachodzi następująco:
Glu + Cys +ATP → Glu(Cys) + ADP + H
3
PO
4
I
Gly + Glu(Cys) + ATP → Glu(Cys-Gly) + ADP + H
3
PO
4
II
Reakcja I jest katalizowana przez syntetazę γ-glutamylocysteinową, a reakcja II przez syntetazę
glutationową.
Hormony peptydowe
H o r mo n y są substancjami chemicznymi, produkowanymi w specjalnych organach lub tkankach
i przynoszonymi do miejsca działania poprzez układ krwionośny. Ta chemicznie zróżnicowana grupa
substancji – regulatorów, między innymi, obejmuje pewną liczbę peptydów i białek. Znaczna liczba
hormonów peptydowych i białkowych produkowana jest w gruczołach wydzielania wewnętrznego
(podwzgórze, przysadka, tarczyca, przytarczyce, komórki Langerhansa w trzustce, itp.). Pozostała
część jest produkowana w specjalnych tkankach. Na tej podstawie rozróżniamy hormony gruczołowe i
tkankowe.
Wiele ze znanych hormonów peptydowych ma prekursory białkowe, z których w skutek
działania enzymów następuje uwalnianie aktywnych związków. Typowymi przykładami są
angiotensyna, bradykinina, kalidyna i insulina. Przypuszcza się, że podobne metaboliczne prekursory
istnieją dla wazopresyny, gastryny, glukagonu, hormonu paratyreotropowego i ACTH. Najpierw, na
rybosomach zachodzi synteza biologicznie nieaktywnych prekursorów o dużej masie cząsteczkowej.
Następnie jeden lub więcej enzymów uwalnia hormony lub prohormony. Okres półtrwania wolnych
hormonów nie przekracza zwykle 30 min. Są one dezaktywowane przez różne egzo i endopeptydazy.
A n gi o t e ns yn a I I zaliczana do oligopeptydów jest oktopeptydem wytwarzanym w nerkach o
wzorze: H-Asp-Arg-Wal-Tyr-Ileu-His-Pro-Fen-OH. Hormon ten powoduje wzrost ciśnienia krwi i
pobudza wytwarzanie hormonów kory nadnercza. Jest to jeden z najefektywniejszych regulatorów
ciśnienia krwi. Związek ten wywołuje silny skurcz naczyń krwionośnych, przez co automatycznie
zwiększa się ciśnienie krwi.
Peptydy regulacyjne
Od czasów odkrycia pierwszych hormonów rośnie stale liczba „chemicznych posłańców”, czyli
związków niosących określoną informację biologiczną od komórek wydzielniczych do komórek
docelowych. Ostatnie 20 lat przyniosło wiele doniesień na temat nieznanych dotąd peptydów o
aktywności: hormonalnej, neuromodulacyjnej, immunoregulacyjnej i mitogennej. Wszystkie te
peptydy określane są często wspólną nazwą p ep t yd ów r e gul ac yj n yc h .
Przyjmuje się obecnie trzy podstawowe sposoby przekazywania informacji międzykomórkowej z
udziałem peptydów regulacyjnych: endokrynny, parakrynny i autokrynny.
(1)
Transport endokrynny, w którym peptyd syntetyzowany przez wyspecjalizowane
komórki wydzielany jest do krwioobiegu i przenoszony z krwią do komórek docelowych. Ten
sposób transportu jest typowy dla hormonów dokrewnych.
(2)
Hipoteza transportu parakrynnego zakłada, że peptydy regulacyjne przenoszone są w
drodze dyfuzji międzykomórkowej. Przypuszczalnie wiele peptydów działa lokalnie tą drogą.
Podobny sposób transportu (neuroparakrynny) przyjmuje się w przypadku przenoszenia sygnału
przez synapsy chemiczne w obrębie układu nerwowego.
(3)
Hipoteza regulacji autokrynnej przyjmuje, że komórkami docelowymi są te same
komórki, które syntetyzują peptyd regulacyjny. Regulacja autokrynna ma przypuszczalnie istotne
znaczenie w kontroli wzrostu komórek neoplastycznych, aczkolwiek autoregulacja jest zjawiskiem
stwierdzonym wielokrotnie w hodowlach komórek nietransformowanych.
Antybiotyki o budowie peptydowej peptydowe
Liczne antybiotyki wytwarzana przez mikroorganizmy mają budowę peptydową. Niektóre z nich
produkowane są na skalę przemysłowa w oparciu o wgłębną hodowlę bakterii Bacillus. Jako przykład
można wymienić gr a mi c yd yn ę S, pierścieniowy dziesięciopeptyd wytwarzany przez Bacillus
brevis. Gramicydynę S stosuje się jako składnik maści i roztworów używanych zewnętrznie do
leczenia owrzodzeń oraz zainfekowanych ran i oparzeń.
17
Bakterie Bacillus brevis wytwarzają jeszcze inne antybiotyki, m.in. t yr o c yd yn y A, B i C,
będące również cyklicznymi oligopeptydami, zawierającymi
-
D
-fenyloalaninę, podobnie jak
gramicydyna S. Znanych jest obecnie ponad 25 antybiotyków wytwarzanych przez różne szczepy tych
bakterii.
Innym przykładem antybiotyku o budowie peptydowej jest b ac yt r a c yn a , wytwarzana na skalę
przemysłową (m.in. w PZF „POLFA” w Pabianicach) w oparciu o hodowlę bakterii Bacillus subtilis.
Poszczególne szczepy bakterii Bacillus subtilis produkują ponad 65 antybiotyków o budowie
polipeptydowej. Z kolei Bacillus polymyxa wytwarza p o l i my ks yn y. Wszystkie te antybiotyki
działają na bakterie gramujemne, gramdodatnie i grzyby. Ich działanie jest różne. Niektóre blokują
syntezę ściany komórkowej lub zakłócają funkcje błon biologicznych, inne, mniej liczne, zakłócają
replikację, transkrypcję lub translację.
A kt yn o m yc yn y (nazywane chromopeptydami gdyż zawierają chromofor) są bardzo
toksycznymi antybiotykami produkowanymi przez różne gatunki Streptomyces. Wydzielono i
wykrystalizowano więcej niż 20 różnych związków należących do tej grupy. Różnią się one jedynie
sekwencja aminokwasową układu cyklicznego zbudowanego z dwóch pierścieni laktonowych
przyłączonych do aktynocyny wiązaniami amidowymi. W tej grupie antybiotyków, a kt yn o myc yn a
C
1
wykazuje najsilniejsze działanie przeciwbakteryjne.
P e ni c yl i na jest najstarszym i najlepiej poznanym antybiotykiem - mówi się o niej, że jest
prototypem antybiotyków. Jej budowa ma jednak mało wspólnego z peptydami, tym niemniej
rozpatruje się ją przy okazji omawiania antybiotyków o budowie peptydowej. W niektórych
publikacjach mówi się o pseudopeptydowej budowie penicyliny:
D-Fen
L-Wal
L-Orn
L-Pro
L-Leu
L-Leu
L-Wal
L-Orn
D-Fen
L-Pro
gramicydyna S
aktynocyna
Aktynomycyna C1
O
N
CO
CO
H
3
C
H
3
C
NH
2
O
NH
NH
Thr
Thr
Sar
Pro
Me-Val
D-Val
D-Val
Me-Val
Pro Sar
Sar = sarkozyna (N-metyloglicyna)
Me-Val = metylowalina
cysteina
C
HC
N
CH
S
C
CH
O
H
3
C
H
3
C
HOOC
NH CO R
walina
R =
CH
2
benzylopenicylina
penicylina
N
S
CH
NH
2
CH
CH
2
-CH
3
CH
3
C
O
D-Glu
L-Leu
L-His
D-Fen
L-Asp
L-Ile
D-Orn
D-Asn
L-Liz
L-Ile
bac ytracy na
18
Wytwarzana jest przez pleśń Penicillium notatum. Biogeneza jej wynika z połączenia waliny
oraz cysteiny. W jej cząsteczce występuje silnie napięty czteroczłonowy pierścień
-laktamowy oraz
drugi pierścień zawierający siarkę. Grupa aminowa cysteiny jest N-acylowana, przy czy rodnik R
kwasu acylującego może mieć różną naturę. Np. benzylopenicylina zawiera rodnik benzylowy.
Depsipeptydy
Nazwa d e p s i p ept yd , jest kombinacją terminów depsyd (ester hydroksykwasu) i peptyd.
Nazwę tę zaproponował Szemiakin w 1953 roku w celu określenia związków, które zawierają
zarówno wiązania estrowe jak i peptydowe.
R
1
R
2
R
1
R
2
R
1
│ │ │ │ │
- NH – CH – CO – O – CH – NH – CH - CO – O – CH – CO – NH – CH – CO –
R
1
- reszty aminokwasu
R
2
– reszta hydroksykwasu
Związki cykliczne tego rodzaju wykryto w dużych ilościach w mieszaninie produktów
metabolizmu bakterii. Wykazują one zadziwiająco duża aktywność antybiotyczną. Szersze
zastosowanie tych związków w medycynie jest ograniczone ze względu na to, że wiązania estrowe
bardzo łatwo ulęgają rozszczepieniu w warunkach sprzyjających hydrolizie.
Do depsipeptydów, zawierających wyłącznie aminokwasy (często także ich pochodne
N-metylowe i D–aminokwasy) i hydroksyaminokwasy, zaliczamy: eniatyny, amidomycynę,
walinomycynę, sporydesmolidy, serratomolid i esperynę.
W innych depsipeptydach mogą istnieć wiązania estrowe utworzone z udziałem grup
hydroksylowych hydroksyaminokwasów, takich jak seryna i treonina. Depsipeptydy mogą także
zawierać skomplikowane jednostki strukturalne, których nigdy nie wykryto w białkach. Do tej grupy
należą:
pektynomycyny,
etamycyna
i
echinomycyna. Depsipeptydy w zależności od ich
struktury dzieli się na O–peptydy, peptolidy i
laktony peptydowe.
Najprostszym, występującym w przyrodzie
depsipeptydem jest fitopatogenetyczna toksyna z
Pseudomonas tabaci. Cząsteczka tego związku
składa się z cząsteczek kwasu mlekowego i
2,5-diaminoadypinowego, związanych ze sobą w
taki
sposób,
że
powstaje
pierścień
dioksomorfolinowy:
Struktura innego depsipeptydu, a mi d o myc yn y jest do tej pory nie wyjaśniona. Wiadomo
tylko, że depsipeptyd ten składa się z kwasu
D
–hydrosywalerianowego i
D
–waliny. Jego działanie
antybiotyczne polega na hamowaniu wzrostu niektórych mikroorganizmów wywołujących choroby
roślin oraz niektórych typów drożdży.
W al i n o myc yn a , nazywana tak ze względu na dużą zawartość waliny, jest cyklicznym
dwunastopeptydem. Działa ona na M.tuberculosis
Se r r at o mo l i d , cykliczny tetradepsipeptyd, oraz e s p e r yna zawierają β–hydroksykwasy. W
wyniku całkowitej hydrolizy serratomolidu stwierdzono, że zawiera on
L
–serynę i kwas
D-
β-hydroksydekanowy.
Protaminy
Do peptydów zalicza się również p r ot a mi n y , polipeptydy o masie cząsteczkowej rzędu 1
6 kDa
wyizolowane z jąder plemników ryb. Zawierają one znaczne ilości argininy, przekraczające połowę
wszystkich aminokwasów w cząsteczce, stąd wybitnie zasadowe właściwości.
CH
HN
C
CH
O
C
CH
3
O
O
CH
2
CH
2
CH
NH
2
HOOC
depsipeptyd z Pseudomonas tabaci
19
Toksyny peptydowe
Śmiercionośne toksyny zawarte w kapeluszu muchomora Amanita phalloides dzieli się na
fallatoksyny (falloidyna, falloina i fallacydyna) i amatoksyny (α-, β-, γ– amanidyny), które są bardziej
toksyczne, ale działają wolniej. Związki te są cyklicznymi siedmiopeptydami.
Fallacydyna zamiast jednostki Ala–
D-
Thr, wchodzącej w skład falloidyny, zawiera kwas
Val-
D-
erytro–β-hydroksyasparaginianowy.
Amatoksyny, które składają się wyłącznie z
L
–aminokwasów są także siedmiopeptydami
cyklicznymi. Zamiast grupy tioeterowej występuje grupa sulfotlenkowa. O toksyczności stanowi
obecność grupy γ – hydroksylowej w łańcuchu bocznym izoleucyny. Amanulina nie zawiera tej grupy
hydroksylowej i mimo to, że pod względem struktury jest bardzo podobna do γ–amatyliny, jest
nietoksyczna.
Można się całkowicie zabezpieczyć przed działaniem tych toksyn jedynie wtedy, gdy w tym
samym czasie, co toksynę spożyje się odpowiednią dawkę antamanidyny. Zabezpieczające działanie
antamanidyny i niektórych jej analogów związane jest z ich działaniem na błony. Związki te tworzą
kompleksy z jonami K
+
i Na
+
, analogicznie do tworzenia eniatyny i walinomycyny.
Peptydy strepogeninowe
S t r e po ge n i n y pobudzają wzrost bakterii, głównie bakterii kwasu mlekowego. Ich obecność
stwierdzono np. w ekstraktach z wątroby, soku pomidorowym i częściowych hydrolizatach
enzymatycznych, uzyskanych z insuliny, kazeiny, rybonukleazy, itp.
Wzorcową jednostka aktywności strepogeninowej jest przyrost tempa wzrostu Lactobacillus
casei, spowodowany przez działanie 1 mg wzorcowego ekstraktu z wątroby. Aktywność
strepogeninowa produktów syntetycznych związana jest z obecnością cysteiny. Najwyższą aktywność
stwierdzono w przypadku, gdy cysteina jest albo powiązana z N–końcową leucyną, albo umieszczona
między dwoma resztami leucynowymi.
Peptydy endorfinowe
E n d or f i n y to peptydy będące ligandami receptorów opiatowych, tzn. działających podobnie
jak morfina. Najważniejsze z nich – e n kef al i n y –są pentapeptydami. Obok naturalnie
znajdowanych enkefalin (np. Tyr-Gly-Gly-Phe-Leu), w ostatnim czasie wytwarzane są syntetyczne ich
analogi:
syntetyczny analog enkefaliny
Tyr - D-Ala - Gly - Phe - Hse-lakton
Hse-lakton =
C
CH
O
NH
O
H
2
lakton homoseryny
N
CH
2
S
CH CO
NH
H
2
C
CH
NH
CO
HO
CH
3
CH
CH
H
OH
N CO
CO
H
3
C
NH
CH CO
H
NH CH CH
2
CO
R
NH
NH
CO
CH
3
HC
Ala
Trp
Ala
D-Thr
Cys
Hyp
falloidyna: R=
C
CH
2
OH
CH
3
OH
C
CH
3
CH
3
OH
falloina: R=
fallotoksyny

11
3.3. Białka (proteiny)
Białka, w zasadzie, są olbrzymimi polipeptydami (makropeptydami). Jako kryterium podziału
pomiędzy polipeptydami a białkami umownie przyjmuje się ilość aminokwasów wchodzących w ich
skład. Związki zbudowane z ponad 100 aminokwasów połączonych wiązaniami peptydowymi zalicza
się do białek. Poprawniejsze jest jednak założenie, że masa cząsteczkowa białek jest większa od
10 kDa. Jednakże, w kilku przypadkach, o przynależności do klasy białek decydują właściwości
polipeptydu i jego biologiczna funkcja. Ta koncepcja podziału ma coraz więcej zwolenników.
Rozwiązuje wiele sprzecznych i problematycznych przypadków. Na przykład, wspomniane w
poprzednim rozdziale protaminy, pomimo masy cząsteczkowej od 1000Da do 6000Da, wygodnie i
rozsądniej jest rozpatrywać w kategorii białek.
Składnikami większości białek zwykle jest 21 różnych aminokwasów w rozmaitych kombinacjach
(a ściślej permutacjach), plus Pro(OH) oraz Liz(OH) w kolagenie. Charakterystyczne jest, że
wszystkie aminokwasy występujące w białkach mają konfigurację L. Warto zauważyć, że już z 4
różnych aminokwasów można otrzymać 24 różne peptydy. Różną kolejnością wiązania się
poszczególnych aminokwasów (tzw. sekwencją aminokwasów), tłumaczy się zjawisko ogromnej
różnorodności białek, zarówno pod względem właściwości fizycznych, chemicznych i biologicznych.
Struktura przestrzenna białek zdeterminowana jest właśnie sekwencją aminokwasów (a tak na
marginesie, ta z kolei zdeterminowana jest sekwencją nukleotydów w DNA).
Białka można podzielić na dwie zasadnicze grupy: pr o t e i n y i pr ot e i d y.
P r ot ei n y to białka właściwe (inaczej, białka proste), wśród których można wyróżnić:
albumi ny – białka rozpuszczalne w wodzie i w rozcieńczonych roztworach soli; występują w
białku jaj ptasich oraz w nasionach roślin strączkowych,
gl obul iny - występują w surowicy krwi, w nasionach roślinnych oraz tkance mięśni,
pr ol ami ny i gl ut eli ny – to są białka wyłącznie roślinne,
skl er opr ot ei ny –z kolei białka wyłącznie zwierzęce; wśród których dalej można wyróżnić:
kol agen – zasadniczy składnik białkowy tkanki kostnej,
ker at yna – główny składnik substancji rogowatych (włosy, paznokcie, skóra),
elast yna – główny składnik komórek w tkankach elastycznych.
P r ot ei d y to białka złożone, zawierające oprócz aminokwasów składniki chemiczne niebiałkowe
(tzw. grupy prostetyczne). Do proteidów zalicza się m.in.:
nukl eoprotei dy – główny składnik białkowy jąder żywych komórek,
gl i koprotei dy – związki białek właściwych z węglowodorami,
l ipopr oteidy – połączenia białek z tłuszczami,
chromopr ot ei dy – połączenia białek z barwnymi związkami (np. flawoproteidy).
met aloprot ei dy – połączenie białka z jonami metali.
Wiele białek będących enzymami należy do proteidów. W skład ich centrum katalitycznego
wchodzą witaminy lub atom metalu. Enzymy, z uwagi na ich rolę i specyficzne właściwości zostaną
omówione oddzielnie w dalszej części podręcznika.
W zależności od b ud o wy p r ze s t r zen n ej cząsteczki białka dzieli się na dwie podstawowe
grupy:
1) b i a ł ka f i br yl ar n e (włókienkowe),
2) b i a ł ka gl o b ul ar ne (kuliste).
Umownym kryterium tego podziału jest stosunek liczbowy osi dłuższej (l) cząsteczki białka do osi
krótszej (d). Białka, dla których ten stosunek osiowy ma wartość nie większą niż 20 (l/d<20), zalicza
się do b i ał e k gl o bu l a r n yc h . Ich rozmiary są rzędu kilku do kilkunastu tysięcy nm. Ich kształt
przypomina elipsoidy obrotowe. Kuliste cząsteczki spotyka się rzadko, głównie w przypadku
lipoproteidów.
Białka o stosunku osiowym większym od 20 (l/d>20) zalicza się do f i br yl ar nyc h . Długość ich
cząsteczek przekracza nierzadko sto tysięcy nm, przy średnicy włókna nie większej niż kilkaset nm.

12
3.3.1. Zasady organizacji strukturalnej cząsteczek białek
Cząsteczka białka stanowi trójwymiarowe ułożenie jednego lub kilku łańcuchów
polipeptydowych. Jej przestrzenna struktura jest hierarchicznie uporządkowana i można w niej
wyróżnić cztery poziomy uporządkowania, z których każdy wykazuje odmienność i zależność od
różnych typów wiązań między atomami. W latach pięćdziesiątych Lindestrom Lang badając budowę
białek wprowadził pojęcie ich struktury pierwszo-, drugo- i trzeciorzędowej później uzupełnione o
strukturę czwartorzędową.
Poziom I (czyli struktura pierwszorzędowa) w hierarchicznym skonstruowaniu cząsteczki
białkowej determinuje poziomy następne (tzw. strukturę wtórną), czyli strukturę przestrzenną danego
białka zwaną też konformacją łańcuchową, która z kolei decyduje o jego funkcji biologicznej.
Struktura I-rzędową mówi o kolejności, czyli s ekw e n cj i a mi n o kw a s ów w łańcuchu
polipeptydowym. Ta struktura uwarunkowana jest wiązaniami peptydowymi. W strukturze
pierwszorzędowej uwzględnia się również położenie wszystkich innych wiązań kowalencyjnych. Są to
głównie wiązania disiarczkowe między resztami cysteiny, sąsiadującymi ze sobą w przestrzeni.
Struktura II-rzędowa białka określa p r zes t r zen ne w zaj e mn e uł o że ni e p ł a s zc zyzn
w i ą za ń p ep t yd o w ych (ściśle biorąc, atomów tworzących te wiązania).
Uwarunkowana jest przede wszystkim wiązaniami wodorowymi głównie między grupami
karbonylowymi i aminowymi łańcucha polipeptydowego oraz planarnością wiązania peptydowego.
Można rozpatrywać tę strukturę jako lokalne uporządkowanie w przestrzeni fragmentów łańcucha
peptydowego o identycznej konformacji. Elementami tej konfiguracji w białkach globularnych mogą
być: α - h e l i s , s t r u kt ur a β -f a ł d o w a oraz za kr ę t y. W kolagenie indywidualnie występuje
s t r u kt u r a ko l a ge n u .
Struktury α-helis, β-fałdowa i kolagenu zostaną szczegółowo przedstawione w dalszej części
podręcznika przy okazji omawiania białek fibrylarnych. Za kr ę t bet a ( β-zakręt) jest to element
struktury drugorzędowej łańcucha polipeptydowego zapewniający zmianę - odwrócenie - kierunku w
jego przebiegu. Ma postać ciasnej pętli, w której tlen z grupy karbonylowej jednego aminokwasu jest
połączony wiązaniem wodorowym z wodorem grupy aminowej czwartego z kolei aminokwasu.
Regiony łańcucha polipeptydowego pozbawione regularnej struktury drugorzędowej chętnie
przyjmują postać zakrętu beta. Taką postać może przyjąć nawet połowa łańcucha polipeptydowego
typowego białka.
Struktura III-rzędowa cząsteczki białka lub jej podjednostki określa u ł o żen i e w
p r ze s t r ze ni ł a ńc uc h a p o l i pe pt yd o w e go (a ściślej, rozlokowanie w przestrzeni wszystkich
jego atomów). To przestrzenne upakowanie łańcucha polipeptydowego wywołane jest wewnątrz
cząsteczkowymi wzajemnymi oddziaływaniami łańcuchów bocznych aminokwasów oraz miedzy tymi
łańcuchami a cząsteczkami wody. Czynnikami warunkującymi powstawanie i utrzymywanie w
białkach struktury III–rzędowej są wiązania chemiczne wszystkich typów, w tym wspomniane
wcześniej wiązanie disiarczkowe i wodorowe, a ponadto oddziaływania jonowe, hydrofobowe i siły
van der Waalsa.
Struktura drugorzędowa tripeptydu
Kąt ψ odpowiada rotacji wokół wiązania C
α
-C,
Kąt φ odpowiada rotacji wokół wiązania C
α
-N

13
Struktura IV-rzędowa jest to s p o s ó b u ł o że ni a w pr ze s t r zen i ki l ku ł ań cu c hó w
p o l i pe pt yd o w yc h stanowiących podjednostki cząsteczki białka oraz zespół oddziaływań między
nimi
Czynniki strukturotwórcze białek
Rola wiązań peptydowych w budowie białek jest oczywista. Omówione teraz zostaną krótko
wszystkie pozostałe wiązania i oddziaływania chemiczne ogrywające rolę w powstawaniu i
utrzymaniu właściwej konformacji łańcucha polipeptydowego cząsteczki białka. Kolejność ich
omawiania wynika z roli i znaczenia, jakie pełnią.
Oddziaływania hydrofobowe (e f e kt h ydr of o bow y). Cząsteczki niepolarne nie zawierają
spolaryzowanych wiązań ani atomów - powoduje to, że są one nierozpuszczalne w wodzie.
Właściwość tą nazywa się h yd r of o bo w o ś ci ą . W środowisku wodnym oddziaływanie z polarnymi
cząsteczkami H
2
O doprowadzi do takiego układu, w którym fragmenty hydrofobowe będą "unikały"
kontaktu z wodą grupując się razem, natomiast części hydrofilowe "chętnie" będą z nią oddziaływać.
Źródłem energii oddziaływań hydrofobowych jest niechęć cząsteczek wody zwiększenia stopnia
organizacji.
Efekt tych oddziaływań sprawia, że syntetyzowany wewnątrz komórki w środowisku wodnym
łańcuch polipeptydowy fałduje się spontanicznie w ten sposób, że większość jego hydrofobowych
rodników bocznych (przede wszystkim w a l i n y, l e uc yn y, i zo l e uc yn y, a l a ni n y i
f e n yl o al an i n y) zostaje skierowana do wnętrza powstającej struktury, a większość jego polarnych,
obdarzonych ładunkiem bocznych łańcuchów znajduje się na powierzchni.
Wiązania disiarczkowe (m o s t k i d i s i a r c z k o w e ). Najsilniejszym wiązaniem (pomijając
peptydowe) jest kowalencyjne w i ą z a n i e d i s i a r c z k o w e ( S S ) powstające pomiędzy
dwoma grupami —SH sąsiadujących ze sobą w przestrzeni dwóch reszt cysteiny w tym samym
łańcuchu lub łącząc dwa różne łańcuchy.
Energia wiązania disiarczkowego wynosi 300 kJ/mol, a odległość między atomami siarki S—S
jest nie większa niż 0,15nm.
Wiązanie to odgrywa znaczną rolę jako czynnik strukturotwórczy ke r a t yn , łącząc między sobą
sąsiadujące łańcuchy polipeptydowe, jednocześnie uodparniając te białka na działanie czynników
chemicznych. W cząsteczce β-keratyny ilość tych wiązań może sięgać nawet kilkudziesięciu.
Natomiast w białkach globularnych mostek disiarczkowy odgrywa jedynie rolę strukturotwórczą
w odniesieniu do pewnych fragmentów łańcucha polipeptydowego. Bez wątpienia usztywnia jednak
kształt tych białek i zwiększa ich termostabilność. W wielu enzymach reszty cysteiny tworzące mostki
disiarczkowe pełnią rolę aminokwasów pomocniczych (patrz: budowa centrum aktywnego enzymów).
Ilość wiązań disiarczkowych w cząsteczce białka globularnego jest rzędu kilku. Znanych jest ponadto
wiele białek (zwłaszcza pozakomórkowych enzymów), które nie posiadają mostków disiarczkowych.
Ta cecha czyni cząsteczki tych białek bardziej plastycznymi, co ma niewątpliwie znaczenie przy ich
sekrecji (wydzielaniu poza komórkę). Do takich enzymów należą rybonukleaza czy subtilizyna
Wiązania jonowe. Wiązania te są wywołane elektrostatycznym przyciąganiem się jonów o
przeciwnych znakach. W białkach wiązania jonowe tworzone są pomiędzy dodatnio naładowanymi
grupami amoniowymi łańcuchów bocznych lizyny i argininy oraz naładowanymi ujemnie grupami
karboksylanowymi kwasu glutaminowego i asparaginowego (—NH
3
+
....-
OOC—). Ponadto udział w
powstaniu tego wiązania w pewnym stopniu może mieć dodatnio naładowany rodnik histydyny.
Energia tych wiązań waha się w granicach 14-29 kJ/mol, ich zasięg wynosi ponad 1 nm, a siła
oddziaływania jest odwrotnie proporcjonalne do r
2
. Tak więc, wiązania te są względnie silne. Ich ilość

14
w cząsteczce białka może dochodzić nawet do kilkudziesięciu. Na przykład we wspomnianej
subtilizynie (pozakomórkowej proteinazie serynowej z Bacillus subtilis) na powierzchni cząstki
enzymu występuje 16 krzyżowych wiązań jonowych stabilizujących skutecznie trzeciorzędową
strukturę białka. U enzymów tych brak jest mostków disiarczkowych, a mimo to są one odporne na
działanie wielu czynników denaturujących.
Wiązania wodorowe. Tworzą się one pomiędzy atomem wodoru związanym kowalencyjnie z
atomem o dużej elektroujemności a atomem z wolnymi parami elektronowymi (np. w białkach
najczęściej: >C=O.....H—N< lub —O—H...O=C<). Wiązanie to symbolizowane we wzorach jest
kropkowaną linią, jak to widać w prezentowanych układach. Wiązanie wodorowe wykazuje wyraźne
ukierunkowanie. Jest ono tym silniejsze, im bardziej linearnie są ułożone wszystkie trzy tworzące je
atomy. Dla struktury białek, istotne wiązania wodorowe mają zasięg oddziaływania rzędu 0,26-0,31
nm, a energia pojedynczego wiązania waha się w granicach od 12 do 29 kJ/mol – a więc są to słabe
oddziaływania. Wiązania wodorowe mogą powstawać zarówno między atomami tej samej cząsteczki
jak i atomami różnych cząsteczek.
Pomimo, że wiązania wodorowego należą do słabych oddziaływań, są one chyba
najważniejszymi siłami, które utrzymują przestrzenną konformację cząsteczki białka. Wynika to z
faktu, iż w cząsteczce białka występują tysiące wiązań wodorowych, w sumie mają więc one dużą
wartość energetyczną.
Ogólnie należy stwierdzić, że wiązania wodorowe są niezmiernie ważnym elementem
strukturalnym układów biologicznych. Biorą one udział w tworzeniu struktur cząsteczek białek,
kwasów nukleinowych i polisacharydów. Powstawanie wiązań wodorowych między cząsteczkami
wody i cząsteczkami substancji rozpuszczonej jest warunkiem rozpuszczania w wodzie wielu
związków organicznych. Nadto tworzenie się i rozpad wiązań wodorowych warunkuje szereg tak
ważnych biologicznie funkcji, jak np. działanie enzymów.
Siły van der Waalsa są wynikiem wzajemnego oddziaływania elektronów i jąder w cząsteczkach,
są więc przykładem oddziaływań elektrostatycznych. Ogólnie biorąc energia wiązań powstałych na
bazie sił van der Waalsa jest bardzo mała, rzędu 4-8 kJ/mol. Mechanizm powstawania tych
oddziaływań może być różny. Rozróżnia się m.in. efekt dyspersyjny, efekt indukcyjny czy efekt
kierunkowy.
E f e kt d ys p er s yj n y (siły dyspersyjne) pojawia się jedynie wtedy, gdy cząsteczki znajdują się
bardzo blisko siebie, tak że prawie się stykają. W wyniku ruchu elektronów walencyjnych gęstość
ładunku ujemnego na zewnętrznej powłoce atomów ulega szybkim fluktuacjom wzbudzając podobną
fluktuację w powłoce walencyjnej sąsiednich atomów. Powstają szybkozmienne dipole, które
wzajemnie przyciągają się zwiększając, w miarę zbliżania się, wzajemną polaryzację elektronową.
Siły te występują przez bardzo krótki czas (rzędu 10
-9
s) i są bardzo słabe (ok. 4 kJ/mol).
E f e kt i nd u kc yj n y polega na oddziaływaniu indukowanych dipoli (jego znaczenie jest
niewielkie).
E f e kt ki e r un ko w y związany jest z asymetrią elektryczną cząsteczek (dipole) powodującą
oddziaływanie orientacyjne i przyciąganie odpowiednio utrwalających się dipoli.
S i ł y va n d er W aal s a maleją odwrotnie proporcjonalnie z siódmą potęgą odległości (r
7
) i są
one bardzo małe w porównaniu do innych omówionych wiązań. Tym niemniej, sumując się wywołują
efekty o dużym znaczeniu dla stabilizacji niektórych struktur białka.
Do czynników strukturotwórczych, w e d ł u g a ut o r a , można też zaliczyć białka zwane
molekularnymi opiekunkami.

15
Molekularne opiekunki (chaperones). Nawet cząsteczki białka, którym udało się uzyskać prawidłowy kształt
w fazie posttranslacyjnej (po zakończeniu biosyntezy), mogą w każdej chwili ulec deformacji wskutek działania
różnych czynników denaturujących. Od lat zadawano sobie pytania:
Jak ludzkie komórki radzą sobie z rozpoznawaniem białek, które mają złą strukturę przestrzenną?
Czy takie źle zwinięte białka mogą powrócić do prawidłowego kształtu?
Co dzieje się, kiedy cząsteczka białka jest tak beznadziejnie zniekształcona, że już w żaden sposób
nie może przybrać prawidłowej konformacji?
W ostatnich latach częściowo znaleziono odpowiedź na te pytania. Okazuje się, że w tych przypadkach
wkracza do akcji specjalna grupa białek: białka opiekuńcze, zwane też przyzwoitkami.
Białka opiekuńcze łączą się z odkształconymi białkami i pomagają im w uzyskaniu odpowiedniej struktury
trójwymiarowej. Często wymaga to zużycia pewnej ilości energii zmagazynowanej w
ATP
. Następnie komórkowe
opiekunki odłączają się od białek, które uzyskały odpowiedni kształt, i zajmują się innymi cząsteczkami białek o
nieprawidłowej konformacji. Same białka opiekuńcze są bardzo odporne na denaturację.
Białka opiekuńcze są bardzo wszędobylskie. Można je znaleźć w cytoplazmie (tam biorą udział w
nadawaniu kształtu białkom wytwarzanych w procesie translacji oraz w naprawianiu zdeformowanych białek), w
jądrze komórkowym, w szorstkiej siateczce śródplazmatycznej (gdzie uczestniczą w kształtowaniu białek
przeznaczonych do wydzielenia przez komórkę) oraz w pobliżu błon białkowo-lipidowych (białka opiekuńcze są
potrzebne w procesie transportu innych białek przez błony biologiczne: z jednej strony błony jedna przyzwoitka
rozwija białko i wpuszcza je do kanału w błonie, a po przeciwnej stronie błony białkowo-lipidowej inna przyzwoitka
odbiera transportowany łańcuch polipeptydowy i nadaje mu odpowiednią konformację). Bez udziału białek
opiekuńczych wiele procesów metabolicznych nie mogłoby przebiegać prawidłowo, a naprawienie popsutej
cząsteczki białka przekraczałoby możliwości komórki.

16
H
H
H
H
N
O
C
C
N
O
C
C
C
H
2
N
COOH
R
2
H
2
C
CH
2
CH
2
H
108o
110o
3.3.2. Białka fibrylarne
Białka fibrylarne (skleroproteiny) to białka o budowie włókienkowej, nierozpuszczalne w wodzie,
rozcieńczonych kwasach, alkoholu; odporne na działanie enzymów proteolitycznych i czynników
chemicznych. Są głównym składnikiem substancji podporowych i strukturalnych organizmu, np.
skóry i kości (kolagen), ścięgien i wiązadeł (elastyna), paznokci, piór, włosów (keratyna), jedwabiu
naturalnego (fibroina), rzęsek bakterii (flagellina), pancerzy owadów (sklerotyna). Białka te nie mają
wartości odżywczych.
Białka fibrylarne wykazują wysoki stopień regularności i uporządkowania struktury. Obok
wiązań peptydowych podstawowym czynnikiem strukturotwórczym białek fibrylarnych są
wiązania wodorowe. Długie łańcuchy polipeptydowe tych białek przyjmują najczęściej regularną
strukturę drugorzędową, w której szkielet skręcony jest wokół własnej osi, tak że powstaje
przestrzenna możliwość utworzenia stabilizujących układ wiązań wodorowych między atomami
oddalonych od siebie ugrupowań peptydowych.
Struktura kolagenu
.
Kolagen jest głównym składnikiem białek skóry, tkanki łącznej (m.in.
wiązadła i ścięgna) oraz białek wchodzących w skład chrząstek i kości. Kolagen odznacza się dużą
wytrzymałością na rozciąganie. Jego skład aminokwasowy jest
następujący: glicyna 33%, prolina 12%, hydroksyprolina 9%,
ponadto 9,5% Ala, 3% Wal, l,5% Liz(OH); przy absolutnym
braku Cys i Try. Ustalono, że we włókienkowych cząsteczkach
kolagenu trzy podstawowe aminokwasy tworzą łańcuch
peptydowy układający się wzdłuż linii śrubowej o bardzo dużym
skoku (3,3 reszty aminokwasowe na 1 skręt), a trzy takie
łańcuchy splatają się ze sobą na kształt liny (rysunek obok).
Poszczególne łańcuchy łączą się między sobą wiązaniami
wodorowymi tworzonymi przez atomy azotu i tlenu wiązań
peptydowych.
O przestrzennej strukturze kolagenu decyduje przede wszystkim jego niezwykły skład
aminokwasowy. Periodyczna struktura pierwszorzędowa wygląda następująco: -Gly-X-Y-Gly-X-Y-.
X często jest proliną (nigdy hydroksyproliną), Y jest często hydroksyproliną. Należy sobie
uświadomić, że wiązania peptydowe utworzone z udziałem proliny i hydroksyproliny są
„zdeformowane” (patrz rysunek powyżej) na skutek pierścieniowej budowy tych aminokwasów.
Stąd kąt rozwarcia płaszczyzn wiązań peptydowych za proliną wynosi 110
, podczas gdy normalnie
wynosi on około 100
.
Hydroksyprolina (Hyp) i hydroksylizyna (Lys-OH) nie są wbudowywane w łańcuch podczas
syntezy. Oba te aminokwasy powstają w gotowych łańcuchach prokolagenu z proliny i lizyny pod
działaniem dioksygenaz - enzymów wbudowujących atomy tlenu z wytworzeniem grup
hydroksylowych.
Trzy enzymy modyfikują prokolagen:
4-dioksygenaza prokolageno-prolinowa (EC 1.14.11.2),
3-dioksygenaza prokolageno-prolinowa (EC 1.14.11.7) i 5-dioksygenaza prokolageno-lizynowa (EC`1.14.11.4).
- glicyna
- prolina
- hydroksyprolina
Struktura II-rzędowa kolagenu
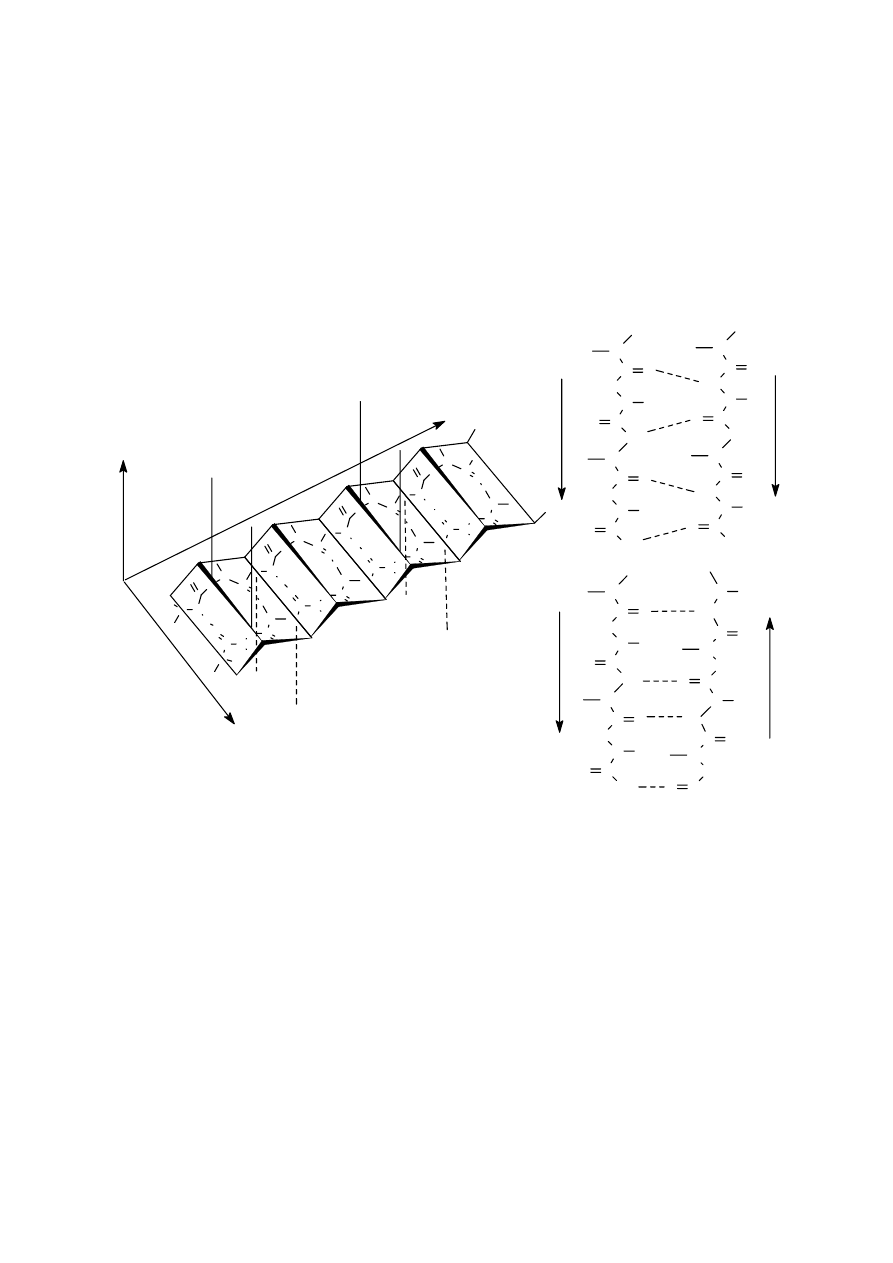
17
C
O
H
H
O
N
C
R
O
C
N
R
H
C
N
C N
C
H
O
C
C
H
H
C
O
H
H
O
N
C
O
C
N
H
C
N
C N
C
H
O
C
C
R
R
C
O
H
H
O
N
C
R
O
C
N
R
H
C
N
C N
C
H
O
C
C
C
O
H
H
O
N
C
C
O
C
N
H
C
N
C N
C
H
O
C
C
R
R
C
Struktura fałdowa. Białka zwane β-ke r a t yn a mi stanowią składniki naskórka, włosów,
paznokci, kopyt. Podobnie jak kolagen są nierozpuszczalne wodzie i cechuje je odporność na
czynniki chemiczne. Ich cząsteczki są zbudowane głównie z aminokwasów o hydrofobowych
łańcuchach bocznych i aminokwasów zasadowych oraz znacznych ilości cysteiny.
Keratyna rozciągniętego włosa (β-keratyna) charakteryzuje się regularną strukturą. Struktura ta
zwana jest fałdową, inaczej: strukturą pofałdowanej kartki, harmonijkową lub beta fałdową. Między
różnymi łańcuchami polipeptydowych lub różnymi częściami tego samego łańcucha powstają
wiązanie wodorowe wytworzone głównie w obrębie wiązań peptydowych. Płaskie wiązanie
peptydowe sprawia, że łańcuch przyjmuje postać pofałdowanej kartki z łańcuchami bocznymi
znajdującymi się poniżej lub powyżej tej płaszczyzny. Łańcuchy boczne sterczą teraz niemal pionowo
w górę, względnie w dół, jak widać na modelu przedstawionym poniżej. Łańcuchy sąsiadujące ze sobą
mogą być równoległe lub antyrównoległe zależnie czy przebiegają one odpowiednio: w tym samym
kierunku lub kierunku przeciwnym (rysunek poniżej).
A - Struktura fałdowa, B – układ peptydów równoległy, C – układ peptydów antyrównoległy
Struktura α-helisy. Helisa (z grec. heliks, helikos = skręcony) to linia śrubowa, linia leżąca na
powierzchni walca (poprawniej: cylindra). Strukturę α-helisy w 1951 roku zaproponowali Pauling i
Corey. α-He l i s a w przypadku struktury białek to łańcuch polipeptydowy płaszczyznowo nawijający
się na hipotetyczny walec (rysunek poniżej). Sposób jego ułożenia umożliwia utworzenie się
wewnątrz łańcuchowych wiązań wodorowych pomiędzy atomami wiązań peptydowych w kolejno po
sobie następujących zwojach helisy. Grupa >C=O każdego aminokwasu wiąże się wiązaniem
wodorowym z grupą HN< aminokwasu, zajmującego w sekwencji liniowej pozycję wysuniętą do
przodu o cztery reszty aminokwasowe. Ostatecznie wszystkie grupy >NH i >C=O łańcucha głównego
łączą się wiązaniami wodorowymi. Każda reszta aminokwasowa jest przesunięta w stosunku do
sąsiedniej o 0,15 nm wzdłuż osi helisy i obrócona o kąt 100° wokół osi. Na jeden obrót helisy
przypada więc 3,6 reszt aminokwasowych. W ten sposób, co czwarte reszty aminokwasowe w
α-helisie znajdują się przestrzennie blisko siebie. Łańcuchy boczne aminokwasów wystają na zewnątrz
cylindra w ułożeniu śrubowym. Prolina „deformuje” α-helisę. Odmiana helisy prawoskrętna jest
bardziej stabilna dla łańcuchów polipeptydowych.
R
H
CH
C O
HN
C R
C
O
NH
NH
O C
R
C
HN
O
C
CH
R
H
CH
C O
HN
C R
C
O
NH
NH
O C
R
C
HN
O
C
CH
R
H
R
H
C
N
C
N
C
N
R
H
R
H
H
R
H
R
C
C
NH
O
C R
HN
CH
C
O
CH
C O
HN
C R
C
O
NH
R
C
NH
O
HN
CH
C
O
NH
O C
R
C
HN
O
C
CH
C
N
A
B
C
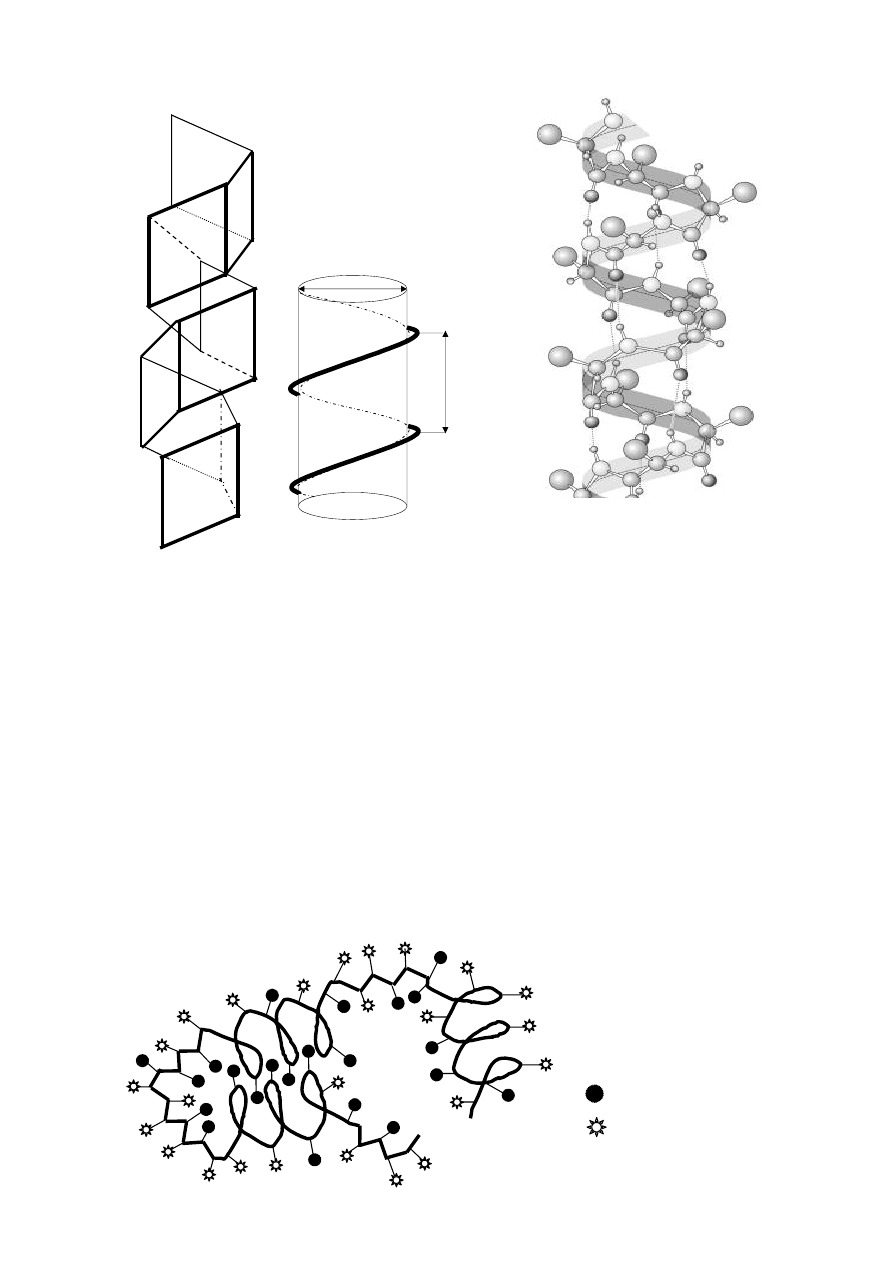
18
- Rodniki hydrofobowe
- Rodniki hydrofilowe
3,6Å
5,4Å
Struktura α-helisy charakterystyczna jest dla niezbyt długich łańcuchów polipeptydowych takich,
jakie występują w α-ke r at yn i e (białko fibrylarne) lub mi o gl o bi ni e (białko globularne).
Włókienka α - ke r at yn y zbudowane są z dwóch typów łańcuchów polipeptydowych. Oba mają
budowę α-helisy, ale oś jednego (I) jest prosta, drugiego (II) zaś tworzy linię śrubową o znacznie
większym skoku aniżeli w α-helisie. Łańcuch typu I stanowi trzon, wokół którego oplata się 6
śrubowych łańcuchów typu II.
3.3.3. Białka globularne
Omawiane poprzednio białka fibrylarne stanowią agregaty wielocząsteczkowe o uporządkowanej
budowie. W przeciwieństwie do nich białka globularne w stanie naturalnym to pojedyncze, niezależne
od siebie cząsteczki, przypominające „kłębuszki poplątanej wełny”. Łańcuch polipeptydowy białek
globularnych jest z reguły nieregularne poskręcany i pofałdowany. Brak w strukturze trzeciorzędowej
tych białek wyraźnej regularności i uporządkowania. Na rysunku przedstawiono schematyczny model
cząsteczki białka globularnego. Wewnątrz „kłębuszka” znajduje się strefa hydrofobowa, a na zewnątrz
strefa hydrofilowa, co decydująco wpływa na właściwości tych białek.

19
Schematyczny model cząsteczki białka globularnego.
W białkach globularnych występują jedynie dwa typy struktur drugorzędowych: α-helisa i
β-fałdowa. W wielu białkach globularnych fragmenty łańcucha polipeptydowego o strukturze α-helisy
są poprzedzielane strukturą β-fałdową. Ponadto obie te struktury mogą oddziaływać pomiędzy sobą
tworząc bardziej złożone struktury „ponad” drugorzędowe zwane motywami. Motywy strukturalne,
powstają wskutek asocjacji α-helisy lub struktury β-fałdowej. Powszechnie występującym
motywem β, czyli powstałym jedynie na bazie struktury β-fałdowej jest motyw „szpilki do włosów”.
Ten motyw zbudowany jest z jednego łańcucha polipeptydowego, przyjmującego antyrównoległą
strukturę typu harmonijki. Innym przykładem bardziej złożonej struktury opierającej się na strukturze
β-fałdowej jest motyw „klucza greckiego”. Jest to bardziej rozbudowany motyw „szpilki do włosów”,
gdzie jeden łańcuch polipeptydowy tworzy ze sobą cztery struktury β-fałdowe ułożone względem
siebie antyrównoległe. Struktura α-helisy podobnie jak harmonijkowa tworzy własne motywy
strukturalne. Najczęściej jest to motyw „helisa-pętla-helisa". Innym przykładem motywów α, są
struktury tzw. „suwaków leucynowych”, zbudowanych z dwóch oplecionych ze sobą α-helis,
bogatych w leucynę. Oprócz motywów β lub α, występują struktury mieszane typu α / β. Za przykład
może posłużyć motyw βαβ, w którym pomiędzy dwoma ułożonymi równolegle łańcuchami struktury
β-fałdowej znajduje się α-helisa. Hydrofobowa strona łańcuchów β jest ciasno upakowana i kontaktuje
się z hydrofobową stroną α-helisy.
Wszystkie omówione motywy dotyczą białek globularnych zbudowanych jedynie z jednego
łańcucha polipeptydowego. Jednakże wiele białek zbudowanych jest z kilku łańcuchów
polipeptydowych, fałdujących się niezależnie od siebie i pomiędzy którymi również formują się
swego rodzaju motywy, zwane domenami. Domeny można zdefiniować, jako precyzyjnie splecione ze
sobą fragmenty łańcuchów polipeptydowych, tworzących w przestrzeni szczególnie regularne rejony.
Zbudowane są one ze 100 do 400 reszt aminokwasowych. Można za przykład podać domenę
składającą się z czterech α-helis tworzących złożony motyw „helisa-pętla-helisa", ułożonych
wzajemnie równolegle, w ten sposób, że reszty aminokwasowe poszczególnych łańcuchów zazębiają
się między sobą tworząc przestrzeń hydrofobową w centralnej części domeny. Inną, bardzo podobną
strukturą, charakterystyczną dla białek globularnych jest domena „globinowa”. Powstaje ona w
wyniku dopasowania grzbietów jednej α-helisy w grzbiet drugiej. Domeny często są składnikami
części funkcjonalnych białek.

20
Niektóre białka mają niezwykle skomplikowaną strukturę trzeciorzędową, nawet z kilkunastoma
motywami i kilkoma domenami, jak np. dystrofina.
Dystrofina jest białkiem kodowanym przez gen leżący na chromosomie X. Mutacje tego genu wywołują
choroby zwane dystrofiami mięśniowymi Duchenne’a i Becker’a. D y s t r o f i n a zbudowana jest z 3685
aminokwasów (M
cz
= 427 kDa). Podzielona jest na cztery domeny - pierwsza domena na N-końcu o masie
cząsteczkowej 30 kDa jest homologiem α-aktyniny, domena centralna zbudowana z 25 powtórzonych motywów
potrójnych helis podobnych do występujących w spektrynie, domena bogata w cysteiny (280 reszt) oraz C-
końcowa domena. C-końcowa domena zawiera kilka charakterystycznych motywów – domenę WW (posiadają
dwie konserwatywne reszty tryptofanu), motyw „EF hands”, domenę ZZ (motyw wiążący cynk) oraz dwie α-helisy.
Allosteria (z grec. allos = obcy, inny; stereos = stanowiący bryłę) to innokształtność
przestrzenna, czyli zmiana konformacji łańcuchowej. Cząsteczki większości białek globularnych
odznaczają się względnie dużą elastycznością. Trzeciorzędowa struktura wielu z nich może ulegać
odwracalnym „odkształceniom” na skutek niekowalencyjnego przyłączenia jakiejś innej cząsteczki
(tzw. l i ga nd u ). Takie białka nazywa się bi ał ka mi a l l o s t e r yc zn ymi . E f ekt a l l o s t e r yc zn y
polega na odwracalnej zmianie konformacji białka w pewnym ściśle określonym obszarze jego
cząsteczki na skutek przyłączenia ligandu (efektora allosterycznego) w innym miejscu. Ta zmiana
konformacji pociąga za sobą zaburzenia aktywności biologicznej białka. Najlepiej poznanym białkiem
allosterycznym jest hemoglobina. Wiele enzymów również jest białkami allosterycznymi. Na zasadzie
allosterii działają aktywatory i inhibitory niektórych enzymów.
Hemoglobina zawarta w erytrocytach działa jako przenośnik tlenu we krwi oraz odgrywa decydującą rolę w
transporcie dwutlenku węgla i jonów wodorowych (pokrewnym białkiem jest mioglobina, odpowiedzialna za
transport tlenu w mięśniach). Kształt cząsteczki hemoglobiny jest zbliżony do kuli o średnicy 5,5 nm.
Hemoglobina jest białkiem złożonym. Składa się z dwóch łańcuchów α (po 141 aminokwasów) i dwóch łańcuchów
β (po 146 aminokwasów). Łańcuchy te są upakowane w formę czworościanu. Oddziałują ze sobą za
pośrednictwem wiązań niekowalencyjnych tworząc charakterystyczny tetramer oznaczany jako α-β-α-β lub α
2
β
2
.
Oba typy łańcuchów mają bardzo zbliżoną strukturę trzeciorzędowa i zawierają taką samą niebiałkową grupę
prostetyczną — cząsteczkę hemu. Grupy hemowe są umiejscowione, pojedynczo w każdej podjednostce, w
zagłębieniach na ich powierzchni. Hem jest związkiem kompleksowym, w którym atom żelaza dwuwartościowego
(Fe
2+
) znajdujący się w środku układu porfirynowego tworzy wiązania koordynacyjne z czterema atomami azotu
porfiryny. Atom żelaza może przyłączyć cząsteczkę O
2
bez zmiany swojej wartościowości, tworząc tzw.
utlenowaną formę hemoglobiny (zwaną też oksyhemoglobiną) o jasnoczerwonym kolorze. Cztery miejsca
wiązania tlenu są znacznie od siebie oddalone; odległość między dwoma najbliżej położonymi atomami żelaza
wynosi 2,5 nm. Przyłączanie tlenu jest odwracalne i zależy od ciśnienia cząstkowego tlenu w narządach.
Przyłączenie tlenu (będącego w stosunku do hemoglobiny ligandem) do jednego z protomerów zwiększa
zdolność przyłączania O
2
przez pozostałe protomery. W miarę postępującego utlenowania hemoglobiny jej
protomery łączą się coraz łatwiej z tlenem na skutek zmian konformacyjnych wywołanych efektem
allosterycznym. Utlenowana cząsteczka hemoglobiny jest bardziej upakowana. Na przykład, w wyniku
utlenowania odległość między atomami żelaza zmniejsza się z 4,0 do 3,3 nm. Reszty aminokwasów C-terminalne
wszystkich czterech łańcuchów mają prawie całkowitą swobodę rotacji. Terminalne grupy karboksylowe i

21
łańcuchy boczne aminokwasów na końcach C tworzą wiązania jonowe, które krępują tetramer. Ze względu na
obecność ośmiu wiązań jonowych cząsteczka nieutlenowanej hemoglobiny jest bardziej napięta i skrępowana niż
hemoglobiny utlenowanej.
Właściwości białek globularnych
Białka, które posiadają określone właściwości biologiczne (tzn. wypełniają określone funkcje w
komórce), i te właściwości nadal zachowują niezależnie od tego, czy są wewnątrz komórki czy też
zostały z niej wyizolowane określa się nazwą bi a ł ka n at yw n e (rodzime). Białka natywne wykazują
kilka charakterystycznych właściwości.
Punkt izoelektryczny. Cechą charakterystyczną każdego białka jest jego p u n kt
i zo el e kt r yc zn y . Jest to takie pH, w którym ilość ładunków dodatnich i ujemnych cząsteczki białka
jest jednakowa. Taka cząsteczka, oczywiście, nie wędruje w polu elektrycznym.
Denaturacja. Część białek natywnych należy do grupy związków bardzo delikatnych. Pod
wpływem wielu czynników zarówno fizycznych, jak i chemicznych, np. wysokiej temperatury,
znacznych zmian pH, promieni ultrafioletowych, rozpuszczalników organicznych itp., struktura białek
ulega trudno odwracalnym lub całkowicie nieodwracalnym zmianom połączonym z utratą natywnych
właściwości biologicznych (m.in. białka będące enzymami tracą swoje właściwości katalityczne).
Proces ten nazywa się de na t u r a cj ą .
D e n at ur acj ę bi ał ka można więc zdefiniować jako utratę natywnych właściwości
biologicznych lub innej indywidualnej cechy charakterystycznej tego białka spowodowaną
zniszczeniem jego struktury wtórnej (przy zachowaniu struktury pierwszorzędowej). W wyniku
denaturacji białka mogą wystąpić zmiany rozpuszczalności i przesunięcie punktu izoelektrycznego.
Rozwinięcie łańcucha peptydowego może prowadzić do wzrostu lepkości, a także zmian absorpcji w
nadfiolecie. Obserwuje się również często procesy agregacji i wytrącania, co jest związane ze
zmianami stopnia hydratacji i rozpuszczalności zdenaturowanych białek.
Ogólnie biorąc, mechanizm denaturacji związany jest ze zniesieniem oddziaływań czynników
strukturotwórczych, które odpowiedzialne są za stabilizację struktury przestrzennej cząsteczki białka.
Podczas denaturacji rozrywane mogą być wiązania wodorowe, jonowe, disulfidowe oraz niszczone
oddziaływania elektrostatyczne van der Waalsa. I tak:
działanie p od w yżs zo n ej t e mp e r a t u r y doprowadza do zerwania wiązań wodorowych oraz
zniesienia oddziaływań van der Waalsa, co w konsekwencji skutkuje zniszczeniem struktur II-,
III- i IV-rzędowej białka,
pod wpływem roztworu m o c zn i ka (6-8 mol/dm
3
) lub c h l or ku gu a ni d yn y (4 mol/dm
3
) i
innych związków chemicznych silnie polarnych zerwaniu ulegają wiązania wodorowe,
j o n y me t a l i ci ę żki c h (Hg
2+
, Pb
2+
, Cu
2+
, itp.) powodują zerwanie mostków
disiarczkowych, a ponadto częściowo wiązań jonowych, co w niektórych przypadkach może
prowadzić do wbudowania się tych jonów w cząsteczkę białka,
na skutek działania kw a s ó w l ub za s ad (wartość pH poniżej 3 lub powyżej 9) zerwaniu
ulegają wiązania jonowe i w pewnym stopniu wodorowe, co przede wszystkim skutkuje
zniszczeniem III-rzędowej struktury białka,
p r o mi en i o w a ni e ( nadfioletowe, rentgenowskie i radioaktywne) w skrajnych przypadkach
może nawet prowadzić do rozrywania wiązań kowalencyjnych,
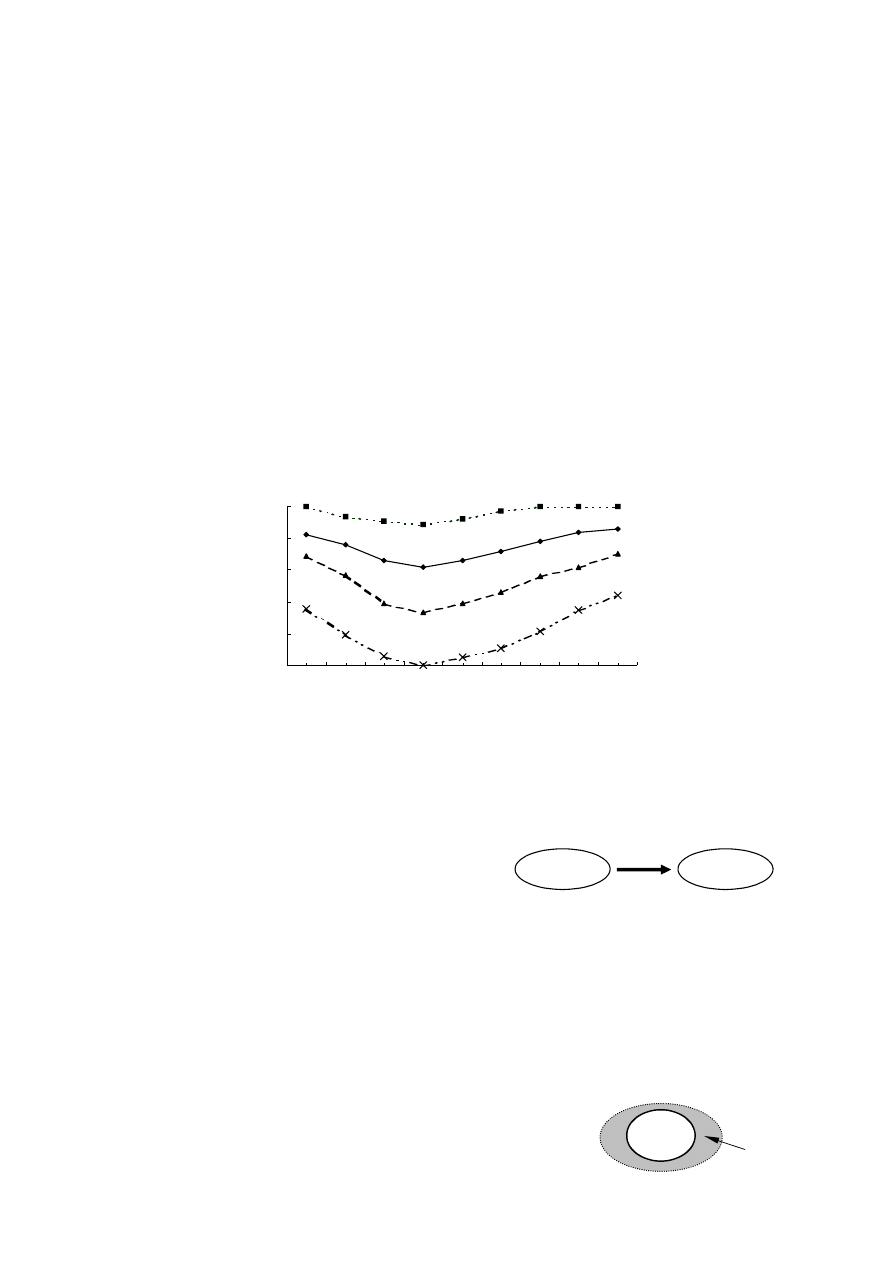
22
+
+
+
+
-
-
-
-
+
-
+
+
innymi czynnikami mogącymi wywoływać denaturację białek są: intensywne mi e s za n i e ,
w yt r zą s a ni e l ub d zi ał a ni e u l t r a d źwi ę ka mi .
Wpływ pH i stężenia elektrolitu na rozpuszczalność białek globularnych. Ze względu na
r o zp us z c za l n o ś ć w wodzie białka globularne dzieli się na dwie grupy:
1) a l b u mi n y - rozpuszczalne w wodzie,
2) gl o b ul i n y - rozpuszczalne w rozcieńczonych elektrolitach (np. w 0,1-1%-owych roztworach
NaCl).
Białka najmniejszą rozpuszczalność posiadają w punkcie izoelektrycznym. Im pH roztworu jest
bardziej oddalone od pI białka, tym lepsza jest jego rozpuszczalność. Zjawisko to można wyjaśnić
następująco. W roztworach o pH różnym od pI cząsteczki białka posiadają ładunek (dodatni lub
ujemny). Dwa ładunki jednoimienne odpychają się (m.in. tym silniej im większy jest ten ładunek). Nic
więc dziwnego, że naładowane cząsteczki białka też się odpychają, co ułatwia ich hydratację i w
efekcie zwiększa rozpuszczalność. Podobnie podczas wysalania (o czym będzie mowa poniżej)
jednoimiennie naładowane cząsteczki białka znacznie trudniej ulegają agregacji i trudniej wytrącić je
w postaci osadu.
0
20
40
60
80
100
3
4
5
6
7
8
9
10
11
pH
R
o
zp
u
sz
cz
a
ln
o
ść
[
%
]
Wpływ pH i stężenia elektrolitu na rozpuszczalność modelowego białek globularnych o pI = 6.
W wielu przypadkach niewielkie stężenie elektrolitu - np. 0,8% wodny roztwór NaCl, czyli sól
fizjologiczna - znacznie poprawia rozpuszczalność wielu białek (tzw. pr o ce s w s al a ni a bi ał e k ).
Niektóre białka w ogóle nie rozpuszczają się w wodzie i
wprowadzenie ich do roztworu wymaga obecności jonów
elektrolitu. Mechanizm zjawiska polega na tym, że
niezależnie od ogólnego (sumarycznego) ładunku
molekuły, cząstkowe ujemne i dodatnie ładunki nie są
równomiernie rozmieszczone na powierzchni cząsteczki białka. W efekcie cząsteczki białka są
dipolami. W punkcie izoelektrycznym dipol taki posiada znaczny moment, kilkaset razy większy od
mementu dipolowego wody. Zjawisko to wpływa na silne, wzajemne przyciąganie pomiędzy
molekułami białka. Dodatek NaCl sprawia, że w roztworze pojawiają się jony Na
+
i Cl
-
, które
neutralizując te cząstkowe ładunki na powierzchni białka znoszą efekt przyciągania się cząsteczek,
tym samym ułatwiają ich rozpuszczanie się.
Przekroczenie pewnego progowego stężenia elektrolitu powoduje wytrącanie się białka z roztworu
w postaci osadu (tzw. p r o c es w ys al a ni a b i ał ek ), co po oddzieleniu od cieczy i wysuszeniu
pozwala otrzymać białko w stanie stałym. Dobrym odczynnikiem
wysalającym białka jest siarczan amonu. Okazuje się, że zarówno jony
H
2
O
1%NaCl
40%(NH
4
)
2
SO
4
20%(NH
4
)
2
SO
4
wsalanie
wysalanie
pI
H
2
O
Molekuła
białka
-
+
-
-

23
NH
4
+
, jak i SO
4
2-
mają bardzo duże powinowactwo do cząsteczek wody i odrywają je od otoczki
hydratacyjnej białka.
Frakcjonowane wysalanie białek. Dobierając odpowiednio pH i stężenie elektrolitu można na
drodze wysalania z grubsza rozdzielić mieszaninę białek na pojedyncze frakcje, zawierające znacznie
podczyszczone indywidualne białka.

24
Wytrącanie białek globularnych rozpuszczalnikami organicznymi. Niektóre rozpuszczalniki
organiczne o silnych właściwościach hydrofilowych, takie jak np. etanol, aceton mają zdolność
wytrącania białka z roztworu. Związki te mają bardzo silne powinowactwo do wody i wyrywają jej
cząsteczki z otoczki hydratacyjnej białka. Niestety, część molekuł białka ulega denaturacji.
Skuteczność procesu wytrącania aktywnego biologicznie białka, czyli niezdenaturowanego, można
poprawić poprzez obniżenie temperatury wytrącania do 4
C, dbania o względnie niskie stężenie
rozpuszczalnika wobec cząsteczek białka, (co, m.in. zapewnia dozowanie rozpuszczalnika do
roztworu białka!) i maksymalnie możliwe skrócenia czasu kontaktu rozpuszczalnika z cząsteczkami
białka. W ten sposób można z dużą wydajnością (sięgającą nawet 86%) skutecznie wytrącać natywne
białko z roztworu. Za takim sposobem otrzymywania białka w stanie stałym, w porównaniu z
wysalaniem, przemawia łatwość regeneracji odczynnika wytrącającego. [Od autora: siarczan
amonowy stosowany do wysalania białek jest trudny do regeneracji, a jego obecność w ściekach
powoduje bardzo groźną korozję wszelkich elementów betonowych (umocnień wałów
przeciwpowodziowych, filarów mostów, itp.)].
Suszenie rozpyłowe. W ostatnich latach, dzięki zastosowaniu ultrafiltracji, białko w stanie stałym (m.in.
enzymatyczne) korzystnie jest otrzymywać z zastosowaniem suszenia rozpyłowego. Wydajność tego procesu
sięga nawet 94%, a otrzymane białko w stanie stałym może być jednocześnie znacznie podczyszczone.
Odbiałczanie roztworów. W niektórych sytuacjach nieodzowne jest pozbycie się białka z
roztworu wodnego. Można efekt taki osiągnąć stosując odczynniki odbiałczające, m.in. 5% roztwór
kwasu trichlorooctowego lub 10% roztwór kwasu sulfosalicylowego. Oba te związki prawie ze 100%
skutecznością wytrącają zdenaturowane białko z wodnych roztworów.
Roztwory koloidalne białek. Roztwory wodne białek globularnych mają charakter ko l o i d ó w
h yd r of i l o w yc h . I jako takie, posiadają charakterystyczne cechy koloidów hydrofilowych, o których
naucza chemia fizyczna. M.in. obniżają napięcie powierzchniowe, wykazują znaczną lepkość, są silnie
uwodnione, żelatynują, wykazują słabą opalescencję i mały efekt Tyndala, itp.
Dializa. Białka są olbrzymimi molekułami i jako takie nie d i al i zuj ą , tzn. nie wykazują zdolności
do dyfundowania przez błony półprzepuszczalne. Zjawisko to wykorzystuje się m.in. do odsalania
roztworów białek. Wszystkie związki niskocząsteczkowe (np. jony, sole, mono- i disacharydy,
aminokwasy, peptydy, itp.) ulegają dializie, tzn. dyfundują przez błonę półprzepuszczalną woreczka
dializacyjnego do otaczającego go roztworu, natomiast białko pozostaje wewnątrz woreczka.
Elektroforeza. Cząsteczki białka zawieszone w roztworze wodnym o pH różnym od ich pI
(punktu izoelektrycznego) posiadają ładunek elektryczny. Oczywiście, ładunek ten jest tym większy,
im większa różnica pomiędzy pH roztworu a pI. Takie białko umieszczone w polu elektrycznym
zacznie wędrować w kierunku elektrody o przeciwnym znaku, z prędkością tym większą, im większy
posiada ładunek.
Punkty izoelektryczne białek są dość zróżnicowane. Istnieje duże prawdopodobieństwo, że w
mieszaninie białek zawieszonych w buforze o jakimś dobranym pH, część cząsteczek naładowana
zostanie dodatnio, część ujemnie, a nadto znajdą się i takie, które nie będą miały ładunku. Jeżeli taką
mieszaninę umieści się w polu elektrycznym, to cząsteczki białek zaczną wędrować w kierunku
elektrod o przeciwnych znakach (oczywiście, te bez ładunku pozostaną w punkcie startowym).
Umożliwia to na skuteczne rozdzielenie mieszaniny białek. Ogólnie rozpowszechnionymi metodami
elektroforezy są elektroforeza bibułowa lub płytkowa cienkowarstwowa.
Oczyszczanie białek. W obecnej dobie istnieje wiele metod pozwalających szybko i skutecznie wyizolować
białko z mieszaniny różnych związków (w tym, innych białek) i oczyścić je do stanu homogenności (jednolitości).
Metody te opisywane są w fachowej literaturze dotyczącej omawianego problemu.

20
CH
2
CH
CH
2
O
O
O
CO-R
1
CO-R
2
CO-R
3
4. Tłuszczowce (lipidy)
Do grupy związków objętych nazwą tłuszczowce lub lipidy zalicza się estry wyższych kwasów
tłuszczowych z mono- lub wielowodorotlenowymi alkoholami. Są one nierozpuszczalne w wodzie.
Dzieli się je na dwie podstawowe grupy:
lipidy proste, w skład których wchodzą wyłącznie alkohole i kwasy,
lipidy złożone, zawierające oprócz tych podstawowych składników inne związki.
Lipidy proste dzieli się na:
1) tłuszcze właściwe - estry kwasów tłuszczowych z glicerolem.
Tłuszcze właściwe, zwane w skrócie tłuszczami, stanowią substancje zapasowe żywych
organizmów. Są szeroko rozpowszechnione w świecie roślinnym, jak i zwierzęcym. Pod
względem budowy chemicznej stanowią mieszaninę estrów glicerolu z kwasami tłuszczowymi
gdzie: R-CO- reszta acylowa wyższego
kwasu tłuszczowego
ogólny wzór tłuszczów właściwych
Wszystkie kwasy tłuszczowe, wchodzące w skład tłuszczów występujących w przyrodzie,
poza nielicznymi wyjątkami, mają parzystą ilość atomów węgla
(co jest zrozumiałe, biorąc pod
uwagę sposób ich syntezy).
W zależności od budowy chemicznej kwasy tłuszczowe dzieli się na
2 grupy:
kwasy tłuszczowe nasycone - nie zawierające wiązań podwójnych,
kwasy tłuszczowe nienasycone - zawierające wiązania podwójne.
W przyrodzie tłuszcze występują przeważnie jako glicerydy mieszane, o różnym składzie
kwasowym, głównie zawierające trzy różne kwasy tłuszczowe.
Przykłady wyższych kwasów tłuszczowych:
C
15
H
31
COOH kwas palmitynowy,
C
17
H
35
COOH kwas stearynowy,
C
17
H
33
COOH kwas olejowy (kwas nienasycony),
rycynolowy (hydroksylowa pochodna kwasu olejowego), a także inne wielonienasycone linolowy,
linolenowy, arachidonowy.
Kwasy wielonienasycone występują szczególnie obficie w olejach roślinnych. Niektóre ssaki nie posiadają
zdolności syntetyzowania tych związków i w takim przypadku stanowią one nieodzowny składnik ich
pożywienia (tzw. związki egzogenne) i stąd noszą nazwę witaminy F.
2) woski - estry kwasów tłuszczowych z wyższymi alkoholami jednowodorotlenowymi, głównie
alifatycznymi, np.
C
30
H
61
-CO-O C
16
H
33
melisynian cetylowy
Lipidy złożone również dzielą się na kilka grup w zależności od dodatkowych składników
nietłuszczowych, np. fosfolipidy, sfingomieliny, glikolipidy, itp. Do tłuszczowców zalicza się, ze
względu na niektóre podobne cechy, również sterydy, które są estrami kwasów tłuszczowych i
jednowodorotlenowych wielkocząsteczkowych alkoholi pierścieniowych.
Wśród tłuszczowców złożonych na uwagę zasługują fosfolipidy (zwane też fosfatydami).
Głównym reprezentantem tej grupy jest lecytyna (fosfatydylocholina). Zbudowana jest ona z glicerolu,
którego dwie grupy wodorotlenowe zestryfikowane są kwasami tłuszczowymi, zaś trzecia - kwasem
fosforowym połączonym z choliną.
21
CH
2
CH
CH
2
O
O
O
CO
CO
R
1
R
2
P
O CH
2
CH
2
N(CH
3
)
3
+
lecytyna
R
1
- reszta kwasu tluszczowego nasyconego
R
2
- reszta kwasu tluszczowego nienasyconego
HO CH
2
CH
2
N(CH
3
)
3
+
cholina
Z kolei wśród sterydów na uwagę zasługuje cholesterol, występujący niemal we wszystkich
komórkach ludzkiego organizmu. Jego stężenie we krwi wynosi od 140 do 250 mg%.
* * *
Bardziej szczegółowe informacje dotyczące dotychczas omówionych związków można znaleźć w
wielu popularnych, łatwo dostępnych podręcznikach (np. z zakresu chemii organicznej, biochemii,
itp.).
HO
cholesterol
P
umownie
oznacza fosforan
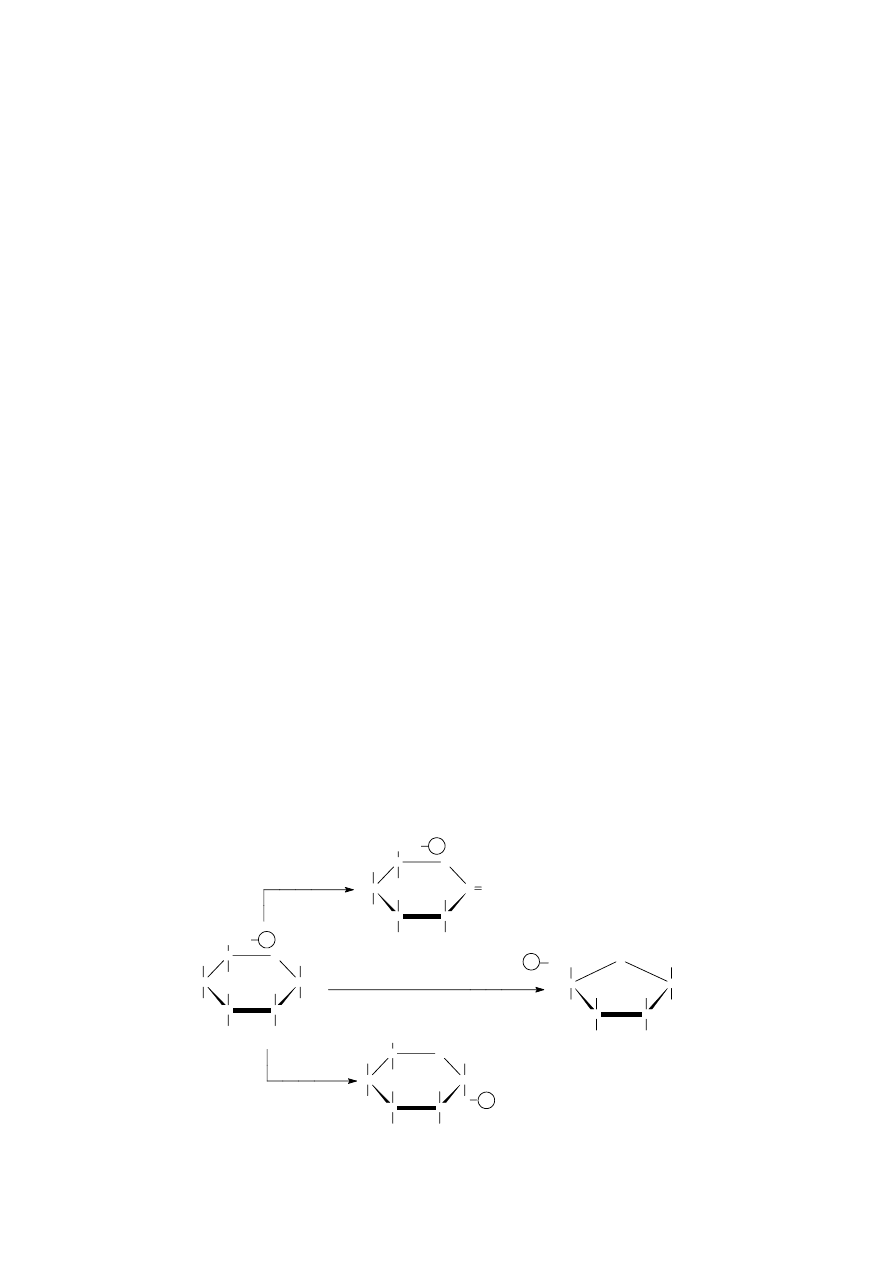
33
dehydrogenaza
glukozo-6-fosforanowa
OH
H
H
OH
H
H
HO
C
C
O
C
C
C
CH
2
O
O
P
lakton kwasu 6-fosfoglukonowego
glukozo-1-fosforan
OH
H
H
H
OH
H
H
HO
C
C
O
C
C
C
CH
2
OH
P
O
fruktozo-6-fosforan
C
O
C
C
C
H
H
H
OH
OH
OH
CH
2
OH
OH
2
C
P
izomeraza
heksozofosforanowa
fosfoglukomutaza
glukozo-6-fosforan
OH
OH
H
H
H
OH
H
H
HO
C
C
O
C
C
C
CH
2
O
P
ROZDZIAŁ 2. ELEMENTY ENZYMOLOGII
Enzymy są swoistymi białkami, katalizującymi zachodzące w przyrodzie ożywionej reakcje
chemiczne. Pod pojęciem katalizy enzymatycznej rozumie się zwiększenie szybkości reakcji termody-
namicznie możliwych (nawet milionkrotne), jedynie poprzez zmniejszenie energii aktywacji cząste-
czek substratu bez naruszania stanu końcowej równowagi. Enzym nie wchodzi w skład końcowych
produktów reakcji i zasadniczo pozostaje w stanie nie zmienionym po zakończeniu reakcji.
Zdecydowana większość dotychczas poznanych enzymów ma budowę białkową. Niektóre z nich
są białkami prostymi, zbudowanymi wyłącznie z aminokwasów (np. większość hydrolaz). Jednak w
przeważającej liczbie przypadków składają się one z części białkowej (a p oe n zy mu ) i niebiałkowej
(witamin i ich pochodnych, jonów metali oraz małocząsteczkowych związków organicznych) tzw.
gr u p p r o s t et yc zn yc h i ko e n zy mó w . Pod nazwą gr u p a p r os t et yc zn a rozumie się
różnorodne niebiałkowe związki chemiczne (sacharydy, żelazoporfiryny) i jony metali luźno związane
z apoenzymem oraz silnie związane z częścią białkową koenzymy. K o e n zym y są witaminami lub
ich pochodnymi. Połączenie koenzymu z apoenzymem określa się mianem h o l o e n zymu . Trwałość
tego połączenia jest różna; bardzo często koenzym łatwo oddysocjowuje od apoenzymu.
A p o e n zym jest czynny wyłącznie w połączeniu z koenzymem lub grupą prostetyczną i
decyduje o swoistości substratowej enzymu, a niekiedy również kierunkowej, np. dekarboksylację i
transaminację aminokwasów katalizują enzymy o różnych apoenzymach i tych samych koenzymach.
Dla porządku należy wspomnieć, iż aktywność biokatalityczną mogą również wykazywać cząsteczki kwasów
nukleinowych (Nagroda Nobla w 1989 roku dla G. Altmana i T. Czecha).
2.1. Ogólne właściwości katalityczne enzymów
Szczególną cechą enzymów jest ich duża specyficzność (określana także jako wybiórczość czy
swoistość) zarówno pod względem rodzaju katalizowanej reakcji chemicznej - tzw. specyficzność
kierunkowa, jak i wobec związków (substratów) biorących w niej udział - tzw. specyficzność
substratowa. Swoistość idzie często tak daleko, że enzym rozróżnia odmianę stereoizomeryczną
substratu, działając wyłącznie na jedną z nich – tzw. stereospecyficzność. Nic więc dziwnego, iż
enzymy mogą katalizować w sposób wybiórczy takie przekształcenia, których trudno lub wręcz nie
można dokonać w inny znany sposób.
Specyficzność kierunkowa. Określony enzym katalizuje na ogół pojedynczą reakcję lub grupę
ściśle spokrewnionych przemian chemicznych. Dokonuje on niejako selekcji wśród możliwych
reakcji, wybierając jedną z nich. Na przykład cząsteczka glukozo-6-fosforanu może w komórce ulec
wielu różnym przemianom, w tym utlenieniu (do laktonu kwasu 6-fosfoglukonowego), izomeryzacji
(do fruktozo-6-fosforanu) czy też mutarotacji (do glukozo-1-fosforanu).
Każda z tych reakcji jest katalizowana przez inny enzym: pierwszą katalizuje dehydrogenaza
glukozo-6-fosforanu, drugą – izomeraza heksozofosforanowej, wreszcie trzecią - fosfoglukomutaza.
34
Specyficzność substratowa. Od specyficzności działania enzymu należy odróżnić jego
specyficzność substratową, polegającą na katalizowaniu przez dany enzym przemiany chemicznej
tylko jednego określonego substratu lub ograniczonej ich liczby. Znane są enzymy, wykazujące
absolutną wybiórczość wobec substratu: np. ureaza działa tylko i wyłącznie na mocznik powodując
jego rozpad, dehydrogenaza metanolowa utlenia jedynie metanol. Jednakże nie wszystkie enzymy
wykazują tak daleko posuniętą specyficzność substratową, np. proteinazy, lipazy czy amylazy
hydrolizują większość swoich substratów tj. odpowiednio białek, lipidów oraz pochodnych α-glukanu
niezależnie od specyficznych cech budowy tych związków. O tych enzymach mówi się, że wykazują
szeroką specyficzność (są mało specyficzne). Ich przeciwieństwem są enzymy o wąskiej
specyficzności (wysoko specyficzne).
Stereospecyficzność. W Rozdziale 1 tego podręcznika omówiono zjawisko izomerii związków
chemicznych. Wiele enzymów rozróżnia izomery i wykazuje aktywność katalityczną wyłącznie w
stosunku do jednej z odmian izomerycznych związku lub też tworzy jedynie jedną z nich, np. cis lub
trans, α- lub β-,
D
- lub
L
-, itp. Klasycznym
przykładem jest dehydrogenaza bursztynianowa,
która przekształca bursztynian do fumaranu
(odmiana trans kwasu etenodikarboksylowego-
1,2). Kwas maleinowy (odmiana cis kwasu
etenodikarboksylowego-1,2) nie tylko, że nie
powstaje w tej reakcji, lecz ją hamuje.
Innym przykładem, który dobrze obrazuje zagadnienie stereospecyficzności enzymów, jest
α-glukozydaza. Ta hydrolaza działa na wszystkie α-
D
-glukopiranozydy, nie biorąc jednak udziału w
reakcjach hydrolizy innych glikozydów (np. β-
D
-glukopiranozydów, fruktofuranozydów, itp.). Z kolei
subtilizyna
(serynowa
proteinaza
z
B.subtilis)
z
mieszaniny
racemicznej
estrów
N-acetyloaminokwasów hydrolizuje jedynie formę
L
-aminokwasu.
Znane są enzymy wykazujące regioselektywność. Rozpoznają one miejsce w cząsteczce związku
chemicznego (np. atom węgla, grupę funkcyjną, itp.), w którym pod
działaniem enzymu następuje przekształcenie właściwe dla jego
specyficzności kierunkowej. Takie właściwości przejawiają m.in.
monooksygenazy, które mogą selektywnie wbudowywać atom tlenu w
jedno, ściśle określone miejsce cząsteczki, hydroksylując ją. Niektóre z
nich, hydroksylujące np. progesteron, mogą przejawiać jednocześnie
stereospecyficzność. To, czy hydroksylowanie nastąpi w pozycji
lub
oraz w którym pierścieniu i przy którym atomie węgla zależy, z
jakiego drobnoustroju pochodzi enzym. Monooksygenazy bakterii
B.megaterium ATCC, jako jedne z nielicznych, hydroksylują
progesteron przy 15 atomie węgla w pozycji
i Monooksygenazy
szczepu B.megaterium KM hydroksylują progesteron w kilku
pozycjach: 15
i 6 oraz 11.
Natomiast tautomerazy, epimerazy lub Δ-izomerazy przekształcają jeden izomer w drugi.
Przykładem może być epimeraza UDP-glukozowa przekształcająca UDP–1-glukozę w UDP-1-
galaktozę.
2.2. Istota katalizy enzymatycznej
Według aktualnej wiedzy, katalityczne działanie enzymów związane jest ze zjawiskiem
międzycząsteczkowych oddziaływań. Enzym (E) i substrat (S) - ewentualnie substraty - tworzą
przejściowo połączenie ES, w którym na skutek specyficznych właściwości centrum katalitycznego
enzymu następuje aktywacja cząsteczki substratu z jednoczesnym osłabieniem niektórych jego wiązań
kowalencyjnych. Uaktywniony kompleks ES przekształca się w połączenie EP, gdzie P oznacza
produkt reakcji. W końcowym etapie połączenie EP rozpada
się na wolny enzym i produkt reakcji. Schematycznie reakcję
taką można zapisać równaniem:
Dokładne poznanie natury oddziaływań odpowiedzialnych za katalizę enzymatyczną jest
przedmiotem badań i rozważań w literaturze naukowej od wielu lat. Pionierami w tej dziedzinie byli
H
2
C COOH
H
2
C COOH
HC
CH
COOH
HOOC
bursztynian
fumaran
FAD
FADH2
dehydrogenaza
bursztynianowa
O
C H
3
Progesteron
C
O
15
11
6
E + S
ES
E + P
EP
35
E. Fischer, pierwszy sugerujący komplementarność centrów aktywnych enzymów do substratów, oraz
L.Pauling, autor koncepcji silnej stabilizacji stanu przejściowego przez centrum aktywne. Od tego
czasu, pojawiało się wiele alternatywnych propozycji jak teoria naprężeń sterycznych, optymalnego
nałożenia orbitali, elektrostatycznego klucza i zamka, itp.
Jedno z bardziej nowoczesnych opracowań na temat czynników odgrywających rolę w katalizie
enzymatycznej zawarł w swojej książce Warshel*. Na drodze rozważań teoretycznych (półempirycznych)
dochodzi on do wniosku o dominującej roli oddziaływań o naturze elektrostatycznej w katalizie enzymatycznej.
Warshel rozpatruje następujące czynniki, które w różnych modelach teoretycznych proponowano jako źródło
aktywności katalitycznej enzymów:
naprężenia steryczne;
desolwatację reagentów;
optymalne nakładanie orbitali (OS);
wiązania wodorowe o niskiej barierze przeniesienia protonu (LBHB);
efekty dynamiczne;
zmiany entropii w wyniku wiązania z enzymem,
oddziaływania elektrostatyczne w preorientowanym, polarnym centrum aktywnym.
Zmiany energii swobodnej podczas reakcji niekatalizowanej i enzymatycznej można przedstawić
schematycznie jak na rysunku obok. Wielkość energii swobodnej tworzenia stanu przejściowego
wyznacza barierę energetyczną dla całej
reakcji.
Ta
bariera
dla
reakcji
enzymatycznej jest wyraźnie niższa (o ∆G
E
)
niż bariera energetyczna dla reakcji
niekatalizowanej. Enzymy: zmniejszając
energię
swobodną
tworzenia
stanu
przejściowego
znacznie
przyspieszają
przekształcenie substratów w produkty.
2.2.1. Mechanizm działania enzymów
W czasie reakcji enzymatycznej
cząsteczka substratu jest wiązana w
określonym obszarze do cząsteczki enzymu
w tzw. centrum aktywnym, przez co tworzy się kompleks enzym–substrat. Dzięki specyficznemu
rozkładowi grup chemicznych w centrum, enzym oddziałuje na ugrupowania chemiczne substratu
rozluźniając konkretne wiązanie chemiczne. W wyniku rozerwania tych wiązań substrat przekształca
się w produkt, który uwalniany jest z kompleksu z enzymem. Enzym po powrocie do formy
pierwotnej (w enzymach złożonych po przyłączeniu przenoszonych grup do innego związku) tworzy
nowy kompleks z następną cząsteczką substratu itd.
Mechanizm łączenia enzymu z substratem tłumaczy się obecnie indukowanym dopasowaniem,
polegającym na dopasowaniu kształtu enzymu do substratu i przekształceniu go w produkt. Przy tym
enzym może zniekształcić substrat wymuszając
w nim konformację podobną do stanu
przejściowego. Przykładem może być wiązanie
glukozy z heksokinazą.
Centrum aktywne enzymów. Centrum aktywne to określony obszar cząsteczki białka
enzymatycznego, w którym podczas katalizy następuje wiązanie substratu i następnie jego
przekształcenie w produkt względnie produkty reakcji. Specyficzność kierunkowa i substratowa
enzymów, a także wiele ich właściwości, zdeterminowanych jest budową centrum aktywnego.
Przyjmuje się, że centrum aktywne enzymów zlokalizowane jest w głębi globularnej cząsteczki białka
w kieszonce kształtem przypominającej szczelinę lub rowek. Obszar centrum aktywnego w
przeważającej ilości tworzą reszty aminokwasów o właściwościach hydrofobowych, a co za tym idzie,
panują w tym obszarze warunki hydrofobowe. Tak więc, jest to mikrośrodowisko o małej stałej
*
[Warshel, A.: Computer Modelling Of Chemical Reactions In Enzymes And Solutions", John Wiley & Sohns,
Inc., New York, Chichester, Brisbane, Toronto and Singapore, 1991 ]
+
E
S
S
E
Postęp reakcji
E
ne
rg
ia
s
w
ob
od
na
stan przejściowy
∆G
E
produkty
substraty
reakcja enzymatyczna
36
dielektrycznej, a stąd o zwielokrotnionych oddziaływaniach elektrostatycznych. Dzięki temu możliwe
jest rozluźnienie określonych wiązań kowalencyjnych w cząsteczce substratu i ich przekształcenie w
inne układy wiązań.
Aminokwasy wchodzące w skład centrum aktywnego - z uwagi na funkcję, jaką pełnią – dzieli się
na cztery kategorie:
1. aminokwas lub a mi n o kw a s y b e zp o ś r e d ni o dzi a ł aj ą ce ; w enzymach złożonych ich
rolę pełnią koenzymy lub grupy prostetyczne,
2. a mi n o kw a s y w s po ma gaj ą ce ,
3. a mi n o kw a s y ko n t a kt ow e , zwane również w i ą żą c ymi ,
4. a mi n o kw a s y p o mo c ni c ze .
A mi n o kw a s y p o mo c ni c ze nie biorą bezpośredniego udziału w katalizie, lecz niezbędne są
do utrzymanie właściwej konformacji przestrzennej pozostałych aminokwasów centrum aktywnego
(można je traktować, jako czynniki strukturotwórcze centrum aktywnego).
A mi n o kw a s y ko n t a kt o w e (wi ą żąc e ) odpowiedzialne są za rozpoznanie substratu i
„wciągniecie” go do kieszonki centrum aktywnego. Z reguły są zlokalizowane na obrzeżach
kieszonki. Odpowiedzialne są za specyficzność substratową enzymów.
Rola a mi no kw a s u b e zp o ś r ed ni o d zi ał aj ącego i a mi n o kw a s ó w w s p o ma ga j ą c ych
zostanie wyjaśniona na przykładzie centrum katalitycznego endopeptydaz serynowych. Enzymy te
hydrolitycznie rozszczepiają wiązania peptydowe w białkach. Omawiane aminokwasy zlokalizowane
są w głębi kieszonki; w przestrzeni znajdują w bliskiej od siebie odległości przyjmując z góry ściśle
określoną konfigurację (tak, jak to pokazano na rysunku).
Mechanizm działania centrum katalitycznego proteinaz serynowych (opis w tekście).
W centrum katalitycznym wszystkich proteinaz serynowych występuje triada aminokwasów.
Aminokwasem bezpośrednio działającym jest seryna, która w subtilizynach zajmuje pozycję 221 w
łańcuchu polipeptydowym, a np. w chymotrypsynie 195. Aminokwasami wspomagającymi są
histydyna i kwas asparaginowy. W środowisku alkalicznym zdysocjowana, silnie ujemna grupa
karboksylanowa rodnika aspartylowego wywołuje efekt indukcyjny, który w wyniku rezonansu
przenosi się poprzez rodnik imidazolowy histydyny i na azocie 3 pojawia się silny ładunek ujemny; w
konsekwencji wodór grupy hydroksylowej seryny zostaje przeniesiony na azot i jednocześnie
powstaje ujemny jon alkoholanowy seryny. Jeżeli w jego pobliżu znajdzie się odpowiednio ustawiony
przestrzennie węgiel wiązania peptydowego – naładowany dodatnio – to oba przeciwnie naładowane
atomy zaczną się zbliżać do siebie. Na skutek silnych oddziaływań elektrostatycznych panujących w
tym obszarze wiązanie peptydowe ulegnie rozerwaniu. Proton z cząsteczki wody przyłączy się do
atomu azotu grupy aminowej i ten fragmentu łańcucha peptydowego odłączy się od centrum
Asp
CH
2
C
O
O
-
32
His
N
N
H
CH
2
64
Ser
221
CH
2
O
H
Asp
CH
2
C
O
OH
32
His
N
N
CH
2
H
64
Ser
221
CH
2
O-
C
NH
R
2
R
1
O
-
+
-
Ser
221
CH
2
O
Ser
221
CH
2
O
C
R
2
O
H
2
O
OH
-
R
2
COOH
R
1
NH
2
+
-
Ser
221
CH
2
O

37
Lip
S
S
aktywnego. Drugi fragment łańcucha peptydowego przejściowo utworzy wiązanie estrowe z resztą
seryny centrum aktywnego, by po chwili ulec hydrolizie pod działaniem wolnej grupy OH
-
i też
oddzieli się od centrum aktywnego proteinazy.
2.2.2. Koenzymy i grupy prostetyczne.
W enzymach złożonych rolę aminokwasu bezpośrednio działającego pełnią koenzymy lub grupy
prostetyczne. Spełniają one rolę przenośników elektronów, atomów lub niewielkich grup
chemicznych. Biorą udział w 2 kolejnych reakcjach enzymatycznych: w pierwszej pobierają z jednego
substratu grupę chemiczną, w drugiej oddają ją drugiemu substratowi, odtwarzając się w pierwotnej
postaci, po czym proces się powtarza. Przenoszenie takie może również odbywać się w obrębie tej
samej cząsteczki i mamy wtedy do czynienia z izomeryzacją substratu. Trwałość połączenia
apoenzymu z koenzymami jest różna; jeśli koenzym łatwo dysocjuje, reakcje przenoszenia grup
chemicznych na koenzymy i z koenzymów katalizuje układ złożony z 2 enzymów o wspólnym
koenzymie; jeśli koenzym jest związany z enzymem trwale (pełniąc rolę grupy prostetycznej), enzym
ten katalizuje kolejno obie reakcje. Uogólniając, niebiałkowe składniki enzymów można traktować,
jako kosubstraty reakcji enzymatycznej. W tabeli 2.1. zestawiono ważniejsze koenzymy i grupy
prostetyczne, których budowa chemiczna i mechanizm działania zostaną omówione dalej.
Tabela 2.1. Ważniejsze koenzymy i grupy prostetyczne
Nazwa koenzymu lub grupy prostetycznej
Skrót
Przenoszone grupy lub
charakterystyczna cecha
Koenzymy oksydoreduktaz
Dinukleotyd nikotynamidoadeninowy
Fosforan dinukleotydu
nikotynamidoadeninowego
Dinukleotyd flawinoadeninowy
Mononukleotyd flawinowy
Metaloflawoproteidy
Flawoproteidy transportujące elektrony
Ubichinon
Liponian
Porfiryny
Koenzymy transferaz
Adenozynometionina
Adenozynotrifosforan
Biotyna
Difosfotiamina
Fosforan pirydoksalu
Koenzym A
Koenzym B
12
Kwas foliowy
Siarczan fosfoadenilowy
Urydynodifosforan
NAD
NADP
FAD
FMN
Me-Fp
ETF
Q
-
-
ATP
-
DPT
TPP
CoASH
B
12
FH
4
PAPS
UDP
Atomy wodoru i epimeryzacja
Atomy wodoru
Atomy wodoru i koenzym oksydaz
Atomy wodoru
Transport elektronów
Transport elektronów
Transport elektronów
Atomy wodoru i grupy acylowe
Cytochromy, oksydaza cytochromowa
koenzym katalazy i peroksydaz
Grupy metylowe
Grupy fosforanowe, pirofosforanowe i
AMP
CO
2
(koenzym karboksylaz)
Grupy aldehydowe lub dekarboksylacja
Przemiany aminokwasów (w tym
deaminacja)
Grupy acylowe
Wewnątrzcząsteczkowe przeniesienie
grupy -COOH
Grupy formylowe, hydroksymetylowe,
formimidowe
Reszta kwasu siarkowego
Grupy glikozylowe
38
Koenzymy i grupy prostetyczne oksydoreduktaz
Nukleotydy nikotynamidowe. Koenzymem wielu dehydrogenaz, czyli enzymów odrywających
lub przyłączających atomy wodoru do substratów, jest dinukleotyd nikotynamidoadeninowy (NAD
+
)
lub jego fosforan - NADP
+
. Jak wskazuje nazwa, koenzymy te są dinukleotydami
zbudowanymi z nukleotydu adeninowego i nukleotydu nikotynamidowego lub jego
fosforanu. Podstawowym składnikiem nukleotydu nikotynamidowego jest amid
kwasu nikotynowego. Kwas nikotynowy, (znany także jako niacyna), jest pochodną
pirydyny i jest jedną z witamin z grupy B o nazwie witamina PP.
Niedobór witaminy PP w organizmie człowieka powoduje zaburzenia czynności przewodu pokarmowego
oraz ośrodkowego układu nerwowego a także powoduje zmiany skórne znane jako pelagra. Kwas nikotynowy w
wystarczających dla człowieka ilościach występuje w mięsie, rybach oraz nasionach zbóż. Dzienne
zapotrzebowanie człowieka na tę witaminę wynosi około 30mg.
Postać utleniona dinukleotydu nikotynamidoadeninowego przedstawiana jest skrótem NAD
+
, a
jego forma zredukowana NADH + H
+
. Umownie, dla wygody, dopuszcza się również symboliczne
zapisy NAD (zamiast NAD
+
) i NADH
2
(zamiast NADH+H
+
).
Poznano przeszło 200 dehydrogenaz sprzężonych z NAD lub NADP. Właściwości i działanie
obu koenzymów jest analogiczne. Stężenie NAD w komórkach jest znacznie większe niż NADP i stąd
dehydrogenazy sprzężone z NAD są liczniejsze. Ogólnie można przyjąć, że NAD pełni w żywej
komórce rolę akceptora wodoru, natomiast jego fosforan jest donorem wodoru (pełni rolę reduktora).
W wielu przemianach enzymatycznych oba koenzymy mogą być nawzajem wymieniane. Ale znane są
przemiany, w których różnice pomiędzy NAD i NADP są bardzo wyraźne. Np. dehydrogenaza
alkoholowa z NAD (EC 1.1.1.1) jest 10000 razy aktywniejsza niż z NADP (EC 1.1.1.2); stąd
rozróżnienie w klasyfikacji enzymów. Istnieje możliwość wymiany H pomiędzy obu koenzymami:
Zredukowane formy NADH
2
– wyjątkowo również NADPH
2
- w żywych komórkach
regenerowane (utleniane) są w łańcuchu oddechowym. Ale NADH
2
może też regenerować się w
reakcjach, w których staje się donorem wodoru
(w tym, jako donor wodoru dla oksygenaz z
pod-podklas EC 1.14.12. i EC 1.14.13.).
Większość
reakcji
katalizowana
przez
dehydrogenazy
sprzężone
z
NAD
jest
odwracalna. Jeżeli reakcja redukcji ma przewagę
nad reakcją utlenienia, enzym nazywa się reduktazą, np. przyłączenie atomów wodoru do podwójnego
wiązania w kwasie fumarowym katalizuje reduktaza fumaranowa (sprzężona z NAD - EC 1.3.1.6).
Identyczną reakcję tyle, że biegnącą głownie w przeciwnym kierunku katalizuje dehydrogenaza
bursztynianowa (sprzężona z FAD - EC 1.3.99.1).
Swoistość substratowa dehydrogenaz sprzężonych z NAD lub NADP zależy od rodzaju
apoenzymu. Nie jest ona jednak absolutna. W wielu przypadkach wyraża się bardzo dużymi różnicami
N
C
O
NH
2
amid kwasu
nikotynowego
dinukleotyd nikotynamidoadeninowy (NAD)
i jego fosforan (NADP)
P
P
+
N
CONH
2
HO
OH
CH
2
O
O
O
OH
OH
OH
2
C
(
P
)
N
N
N
N
NH
2
N
Ryb
CONH
2
H
H
P
P
Aden
N
CONH
2
Ryb
P
P
Aden
+
+
H
+
XH
2
X
mechanizm odrywania wodoru od substratu XH
2
przez NAD
NAD
NADH + H+
NADPH
2
+ NAD
NADP + NADH
2
transdehydrogenaza
EC 1.6.1.1
NADH+H
+
NAD
reduktaza fumaranowa
H
2
C COOH
H
2
C COOH
kwas bursztnowy
HC
CH
COOH
HOOC
kwas fumarowy
(EC 1.3.1.6)
39
H
ryboflawina
N
N
N
NH
H
3
C
H
3
C
CH
2
O
O
CHOH
CHOH
CHOH
CH
2
O
Lip
S
S
szybkości katalizowanych reakcji, np. dehydrogenaza aldehydu fosfoglicerynowego utlenia również
aldehyd glicerynowy, ale około 1000 razy wolniej.
Apoenzymy licznych dehydrogenaz sprzężonych z NAD lub NADP zawierają związane jony
metali Mg
2+
, Zn
2+
, Mn
2+
. Jony te są aktywatorami działania tych dehydrogenaz. Np. jony Mg
2+
lub
Zn
2+
odgrywają ważną rolę podczas utleniania alkoholi, ułatwiając powstawanie jonu
alkoholanowego.
Niektóre dehydrogenazy sprzężone z NAD lub NADP podczas swojego „właściwego” działania, czyli
utleniania substratu mogą równocześnie prowadzić do jego d e k a r b o k s y l a c j i . Takiej dekarboksylacji ulegają
tylko α-ketokwasy lub α-hydroksykwasy. Utlenianie α-ketokwasów połączone z ich dekarboksylacją nazywa się
α - o k s y d a c j ą i katalizowane jest przez u k ł a d y w i e l o e n z y m a t y c z n e (sprzężone z ,DPT, FAD,
CoASH i NAD). Przykładem jest przemiana pirogronianu do acetylo~SCoA katalizowana przez
d e h y d r o g e n a z ę p i r o g r o n i a n o w ą ( d e k a r b o k s y l u j ą c ą ) , o czym będzie mowa dalej. Natomiast
przykładem dekarboksylacji α-hydroksykwasów podczas ich utlenienia jest działanie dehydrogenaz
jabłczanowych (dekarboksylujących). Ogólnie znane są aż cztery dehydrogenazy jabłczanowe sprzężone z NAD
lub NADP, w tym trzy o właściwościach dekarboksylujących:
EC 1.1.1.37 – d e h y d r o g e n a z a j a b ł c z a n o w a - znana z cyklu Krebsa, sprzężona z NAD, utlenia jabłczan do
szczawiooctanu,
EC 1.1.1.38 – d e h y d r o g e n a z a j a b ł c z a n o w a ( s z c z a w i o o c t a n - d e k a r b o k s y l u j ą c a ) - sprzężona z
NAD, podczas utleniania jabłczanu, jednocześnie dekarboksyluje go; posiada zdolność
dekarboksylacji szczawiooctanu,
EC 1.1.1.39 – d e h y d r o g e n a z a j a b ł c z a n o w a ( d e k a r b o k s y l u j ą c a ) - sprzężona z NAD, podczas
utleniania jabłczanu, jednocześnie dekarboksyluje go; nie posiada zdolności dekarboksylacji
szczawiooctanu,
EC 1.1.1.40 – d e h y d r o g e n a z a j a b ł c z a n o w a ( s z c z a w i o o c t a n - d e k a r b o k s y l u j ą c a ) - sprzężona z
NADP, podczas utleniania jabłczanu, jednocześnie dekarboksyluje go; posiada zdolność
dekarboksylacji szczawiooctanu.
Nukleotydy flawinowe. W skład tych nukleotydów wchodzi r yb of l a wi na . Związek ten
zbudowany jest z heterocyklicznej izoalloksazyny i przyłączonego do niej rybitolu,
pięciowodorotlenowego alkoholu, będącego pochodną rybozy. Ryboflawina jest witaminą B
2
.
Ryboflawina posiada charakterystyczne żółte zabarwienie. Jej niedobór w organizmie człowieka powoduje
zmiany chorobowe w obrębie jamy ustnej; m.in. pękanie kącików ust i śluzówki, obrzęki warg i ich łuszczenie się,
zmiany zapalne języka a także zaburzenia ze strony narządu wzroku. Duże ilości tej witaminy występują w
drożdżach, w ziarnach zbóż i jajach. Zapotrzebowanie człowieka wynosi 2mg na dobę.
Fosforan ryboflawiny – m o n o nu kl e ot yd f l a wi no w y – zapisuje się symbolicznie FMN, a
jego połączenie z nukleotydem adeninowym – d i nukl e ot yd f l a w i n o ad en i n o w y – oznacza się
skrótem FAD. Oba nukleotydy flawinowe są nierozerwalnie związane z odpowiednimi białkami
enzymatycznymi, tworząc f l a w o pr ot e i d y , będąc jednocześnie przykładem grup prostetycznych.
FMN i FAD są grupami prostetycznymi dehydrogenaz, przenoszących głównie atomy wodoru.
Ich cechą charakterystyczną jest zdolność odrywania atomów wodoru od dwóch sąsiadujących ze sobą
atomów węgla, tworząc podwójne wiązanie: -CH
2
-CH
2
-→ -CH=CH-. Przykładem może być
wspomniana już dehydrogenaza bursztynianowa (EC 1.3.99.1). Zredukowaną formę FAD
dinukleotyd flawinoadeninowy (FAD)
O
OH
OH
N
N
N
N
NH
2
OH
2
C
N
N
N
NH
H
3
C
H
3
C
CH
2
O
O
CHOH
CHOH
CHOH
CH
2
O
P
P
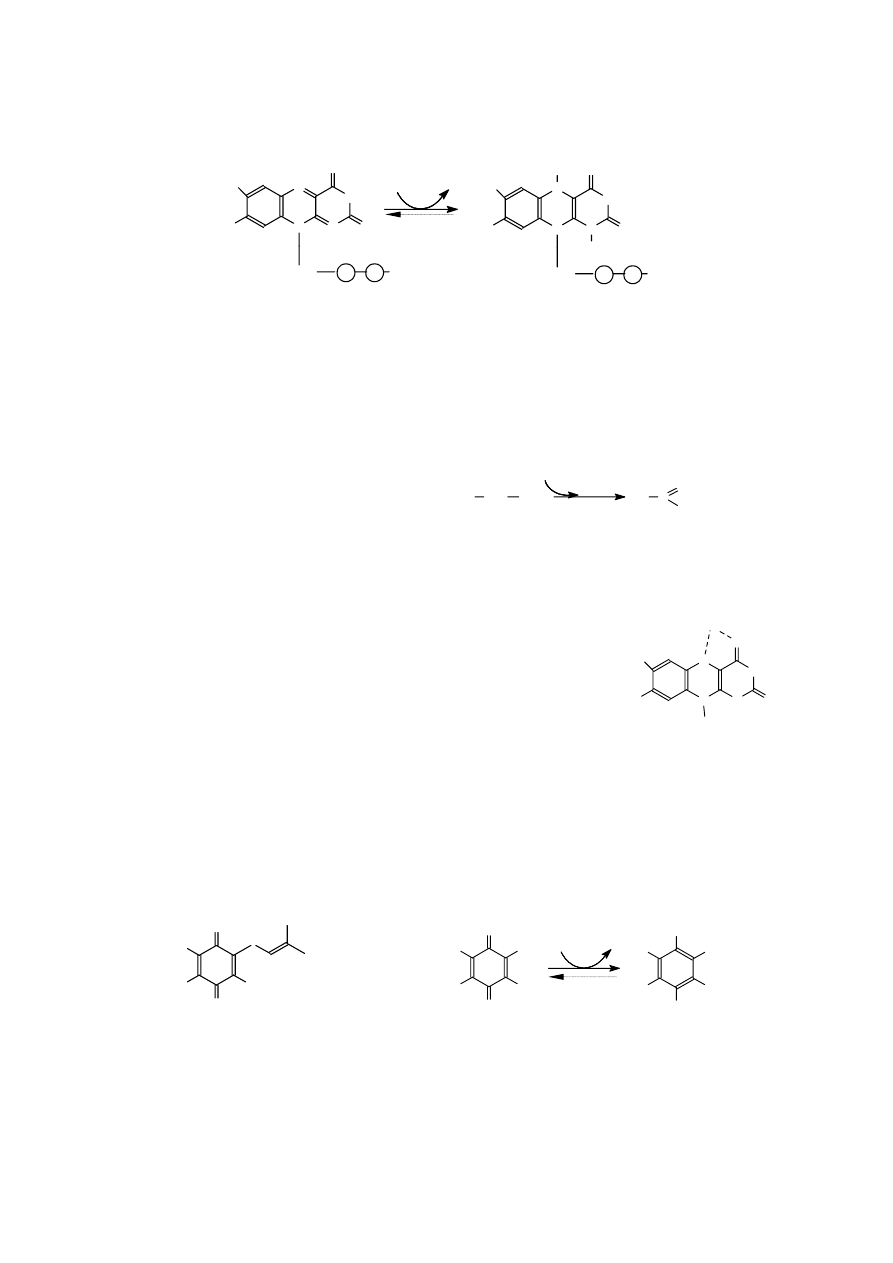
40
ubichinon (Q)
O
O
H
3
C
H
3
C
CH
3
)
(
n H
symbolicznie zapisuje się FADH
2
. Podobnie jak zredukowany NADH
2
, zredukowany FADH
2
w
komórce regeneruje się (utlenia) w łańcuchu oddechowym.
Ale FMN i FAD mogą być również koenzymami oksydaz: enzymów posiadających zdolność
bezpośredniego przenoszenia oderwanych od substratu atomów wodoru na tlen – uaktywnianie
cząsteczki tlenu prowadzą in situ, tj. w miejscu zetknięcia się enzymu z cząsteczką tlenu. Reakcja ta
przebiega bez zmiany formy grup prostetycznych na ich formy zredukowane. Przykładem takiego
enzymu może być o ks yd a za a mi n o w a
za w i er aj ąc a F A D (EC 1.4.3.4). Reakcję taką
symbolicznie zapisuje się z FAD ujętym w nawias
dla podkreślenia, że nie zmienia się jego forma po
zakończeniu reakcji. Dla podkreślenia roli FAD
lub FMN w tego typu reakcjach, nazywa się je kofaktorami reakcji utlenienia.
FAD jest również elementem składowym systemu enzymatycznego c yt o chrom u
P 450. Zredukowany FADH
2
może z kolei być donorem wodoru dla oks ygenaz z
podpodklasy EC 1.14.14.
Dość często apoenzym oksydoreduktaz, sprzężonych z FMN lub
FAD, zawiera w swojej budowie jony metali Fe
2+
, Zn
2+
lub Mo
2+
, które
współdziałają podczas reakcji utleniania i redukcji; enzymy tego typu
nazywa się metaloflawoproteidami i oznacza symbolem Me-Fp.
Transportują one przede wszystkim elektrony.
Ubichinon (koenzym Q). Przenośnikiem atomów wodoru może być również ubichinon, zywany
też koenzymem Q. Związek ten pod względem budowy przypomina witaminy E i K, choć sam
witaminą nie jest, gdyż w komórkach zwierzęcych jest syntetyzowany (z tyrozyny). Łańcych boczny
zbudowany jest z jednostek izoprenoidowych. Ich liczba bywa rózna w zależności od źródła
pochodzenia ubichinonu. Np. ubichinon z serca świni ma ich 50, a z serca wołu 10, co zapisuje się
symbolicznie Q-50 i odpowiednio Q-10. Ilośc jednostek izoprenoidowych w ubichinonach
drobnoustrojowych waha się od 5 do 13.
Mechanizm przenoszenia atomów wodoru przez ubichinon polega na utlenianiu i redukcji
pierścienia chinonowego.
N
N
N
NH
H
3
C
H
3
C
O
O
R
Fe
2+
metaloflawoproteid
H
2
O
oksydaza
O
2
(FAD)
+
+ H
2
O
2
CH
2
NH
2
R
C
O
H
R
NH
3
aminowa
przez FAD
XH
2
X
mechanizm odrywania wodoru od substratu XH
2
FAD
FADH2
N
N
N
NH
H
3
C
H
3
C
O
O
rybitol
P
P
Aden
N
N
N
NH
H
3
C
H
3
C
O
O
rybitol
H
H
P
P
Aden
Q
O
O
H
3
C
H
3
C
R
CH
3
QH2
H
3
C
H
3
C
R
CH
3
OH
OH
XH
2
X
mechanizm odrywania wodoru od substratu XH
2
przez ubichinon
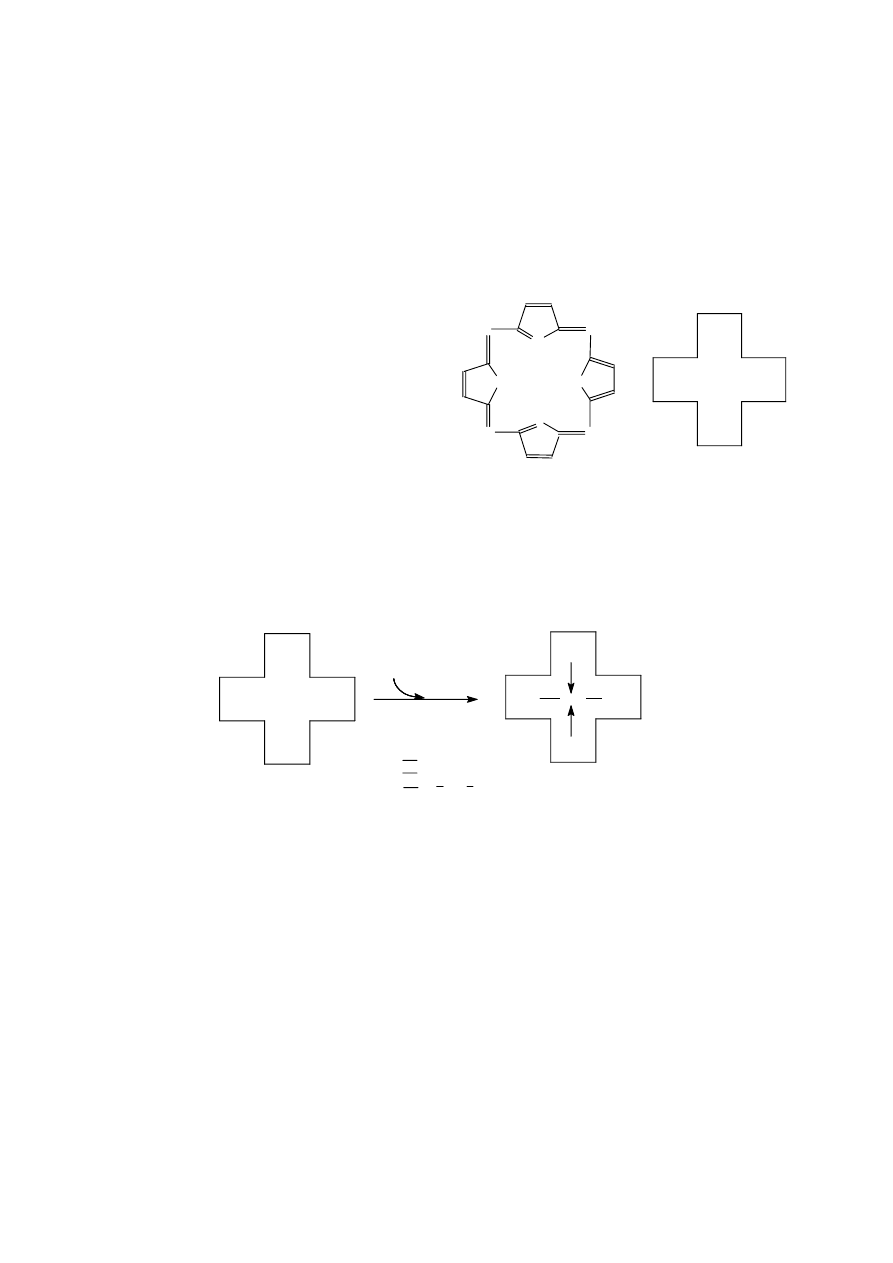
41
Ubichinon jako koenzym dehydrogenaz bądź reduktaz spotykany jest rzadko. Jednym z
nielicznych przykładów może być d e h yd r o ge n a za N A D H ( u bi c hi no n o wa ) –EC 1.6.5.3, która
katalizuje reakcję utlenienia NADH
2
do NAD z jednoczesną redukcją ubichinonu do ubichinolu.
Ciekawym enzymem jest d e h yd r o ge n a za me t ano l o wa (EC 1.1.99.8) znaleziona u niektórych
metylotrofów, której grupą prostetyczną jest ubichinonoproteina (kofaktor PQQ).
Natomiast ubichinon w komórkach pełni bardzo ważną rolę, będąc elementem składowym
ł ań c uc ha o dd ec h o w e go .
Związki porfirynowe. Porfiryny to związki, w których 4 pierścienie pirolowe połączone są ze
sobą mostkami metinowymi (=CH–). Przy atomach węgla β (3 i 4) pierścieni pirolowych zamiast
atomów wodoru występują różne grupy
chemiczne (metylowe, winylowe, octanowe i
inne). W celu ujednolicenia zapisu związków
tego typu Fisher zaproponował symboliczny,
uproszczony ich wzór. Pominięto w nim mostki
metinowe a pierścienie pirolowe zaznaczano
kodem kreskowym. Pierścienie ponumerowano
cyframi rzymskimi od I do IV, atomy węgla w
pierścieniach od 1 do 8, a mostki metinowe
literami alfabetu łacińskiego od α do δ, jak to
przedstawiono na rysunku obok.
Z uwagi na możliwość podstawiania atomów wodoru 1-8 w pierścieniach pirolowych różnymi
podstawnikami, istnieje teoretycznie olbrzymia ilość izomerów pochodnych porfiryny. Jednakże w
naturalnych systemach biologicznych głównie występuje izomer typu III, w którym podstawniki przy
IV pierścieniu pirolowym są ułożone asymetrycznie w stosunku do innych pierścieni. Oznaczony i
nazwany jest on przez Fishera pr ot op or f i r yn ą IX . Izomer ten jest prekursorem me t a l o po r f i r yn
wchodzących w skład he m o p r o t e i n i c hl or of i l i .
M e t a l o p o r f i r y n y to pochodne protoporfiryny IX z atomem metalu (żelaza, magnezu, miedzi)
centralnie wbudowanym w układ porfirynowy za pośrednictwem atomów azotu pierścieni pirolowych.
Metaloporfiryny z wbudowanym żelazem (hem i heminy) występują jako grupy prostetyczne w
h e mo p r ot ei na ch . Metaloporfiryny z wbudowanym Mg
2+
są składnikami c h l or of i l u .
H e m o p r o t e i n y to białka zawierające jako grupę prostetyczną hem lub heminę.
H e m to połączenie żelaza występującego na II stopniu utlenienia (Fe
2+
) z protoporfiryną IX.
Hem jest barwną grupą prostetyczną hemoglobiny i mioglobiny. W organizmach zwierząt,
hemoglobina – czerwony barwnik krwi - uczestniczy w transporcie tlenu, CO
2
a także jonów
wodorowych. Mioglobina uczestniczy w transporcie tlenu w mięśniach i w jego magazynowaniu.
H e m i n y to połączenia protoporfiryny IX z jonem żelaza występującym na III stopniu utlenienia
(Fe
3+
). Heminy we wszystkich organizmach pełnią funkcję przenośników elektronów w łańcuchu
oddechowym (układ cytochromów), a także są grupami prostetycznymi katalaz i peroksydaz,
katalizując procesy utleniania. Powstają z hemoglobiny pod wpływem kwasu solnego, a reakcja ta
służy w medycynie sądowej do wykrywania śladów krwi.
HC
N
CH
N
N
HC
CH
N
porfiryna
I
II
III
IV
1
2
5
7
6
8
4
H
H
3
3
I
II
IV
III
1
2
4
5
6
7
8
symboliczny, uproszczony
zapis wzoru porfiryny
M =
P =
V =
CH
3
CH=CH
2
CH
2
CH
2
COOH
I
II
IV
III
V
M
M
M
M
V
P
P
I
II
IV
III
V
M
M
M
M
V
P
P
Fe
2+
Fe
2+
protoporfiryna IX
hem
ferrochelataza
EC 4.99.1.1
enzymatyczna transformacja protoporfiryny IX w hem
42
fragment łańc
ń
ucha polipeptydowego
cytochromu c
His
Liz
S
S
Cys Ser
Cys
Glu(NH
2
)
miejsce przyłączenia heminy
N
CH
3
HC
CH
N
CH
3
N
CH
2
CH
2
HOOC
H
3
C
Fe
+3
CH
3
HOOC CH
2
CH
2
HC
CH
N
CH
2
H
2
C
CH
2
CH
2
hemina
cytochrom c
C y t o c h r o m y to hemoproteiny, które dzięki odwracalnej zmianie stopnia utlenienia żelaza
grupy heminowej (z Fe
2+
na Fe
3+
) stanowią układ przenośników elektronów w łańcuchu oddechowym
w żywych organizmach. Na podstawie budowy chemicznej, widma absorpcyjnego i przede wszystkim
miejsca w łańcuchu oddechowym, cytochromy dzieli się na grupy: a, b i c (np. cytochromy b) oraz
podgrupy (np. cytochromy a
3
). Kompleks hemoprotein a + a
3
jest enzymem znanym jako o ks yd a za
c yt o ch r o mo w a .
O k s y d a z a c y t o c h r o m u c (EC 1.9.3.1) to enzym będący kompleksem hemoprotein a i a
3
,
składający się z kilku łańcuchów polipeptydowych o różnej masie cząsteczkowej, 2 grup heminowych
oraz 2 atomów miedzi. Oksydaza cytochromowa jest końcowym enzymem łańcucha oddechowego;
aktywuje tlen atomowy do przyłączenia atomów wodoru, przenosząc na niego elektrony.
C h l o r o f i l e , pochodne protoporfiryny IX, zielone barwniki występujące u fotosyntetyzujących
roślin, glonów i bakterii (bakteriochlorofil). Chlorofile pełnią rolę grupy prostetycznej chromoproteidu
zwanego chloroplastyną. Są to metaloporfiryny, zawierające wbudowany w centrum jon magnezu
(Mg
2+
). Połączone są wiązaniem estrowym z
fitolem. Rozróżnia się 4 rodzaje chlorofilu. U
wszystkich roślin wyższych i glonów
występuje chlorofil a (niebieskozielony, co
najmniej w 3 formach: P700, Ca680, Ca670 -
liczby oznaczają długość fali świetlnej, przy
której występuje maksimum absorpcji światła).
Chlorofil b - żółtozielony, występujący u
roślin wyższych i niektórych glonów. Chlorofil
c — występuje w małych ilościach w
czerwono zabarwionych roślinach wodnych
(m.in. z rodzaju Alternanthera), brunatnicach,
niektórych
okrzemkach
i
wiciowcach.
Chlorofil d znajdowany jest u krasnorostów.
Chlorofil a absorbuje głównie światło
fioletowe i czerwone, oraz żółtozielone, chlorofil b absorbuje głównie światło niebieskie
i pomarańczowe. Chlorofile z karotenoidami wchodzą w skład fotosystemów PS-I i PS-II.
Kwas liponowy (tiooktanowy). Związek ten uczestniczy w transporcie elektronów, a także w
przenoszeniu grup acylowych w reakcjach α-oksydacji (oksydacyjnej dekarboksylacji α-ketokwasów).
Jest elementem składowym układów wieloenzymatycznych wraz z DPT, CoASH, FAD i NAD, np. w
dehydrogenazie pirogronianowej (dekarboksylującej). Jest to kwas 6,8-ditiolooktanowym. Łatwo
ulega odwracalnemu utleenieniu i redukcji.
C
20
H
39
OOC
CH
3
fityl
chlorofil a
N
CH
3
H
C
CH
N
CH
3
N
HC
CH
N
CH
3
CH
CH
3
CH
2
CH
2
C
2
H
5
CH
2
CH
OOC
C
O
H
Mg
2+
43
C
Enzym-NH
CH
2
CH
2
CH
2
CH
2
CH
CH
2
S
CH
2
S
O
liponian
Lip
S
S
Lip
S
S H
~
C
O
CH
3
DPT
enzym
DPT CH
CH
3
OH
enzym
Lip
S
S
H
H
FAD
FADH2
CoASH
CH
3
C
O
~
SCoA
O
OH
OH
N
N
N
N
NH
2
CH
2
S
CH
2
CH
2
CHNH
2
COOH
CH
3
+
adenozynometionina
H
H
kwas tetrawodorofoliowy (H
4
folian)
1
2
3
4
5 6
7
8
9
10
COOH
CH
2
CH
2
COOH
CH
O
NH
NH
C
CH
2
H
2
N
OH
N
N
N
N
Jak już wspomniano, liponian jest składnikiem układów wieloenzymatycznych. W układach tych,
jako kosubstrat, transportuje rodniki acylowe.
Koenzymy i grupy prostetyczne transferaz
Adenozynometionina. Jest to koenzym transferaz przenoszących jednowęglową grupę –CH
3
.
Grupa metylowa w tym układzie jest wybitnie reaktywna (tzw. „aktywny metyl”). I dlatego, w formie
jonu CH
3
+
, może być przenoszona na elektroujemnie naładowane inne grupy chemiczne
(najkorzystniej z wolną parą elektronową). Koenzym ten bierze udział w wielu szlakach
anabolicznych, metylując m.in. grupę aminową (np. w biosyntezie choliny).
Kwas foliowy. Koenzym ten jest pochodną pterydyny, heterocyklicznego dipierścieniowego
związku, do którego bocznej gupy metylowej przy C6 przyłączona jest cząsteczka kwasu
p-aminobenzoesowego i dalej poprzez wiązanie amidowe jedna lub więcej cząstwczek kwasu
glutaminowego. Kwas foliowy jest witaminą. Jej niedobór w ustroju człowieka może prowadzić do
pojawienia się trombocytopenii i pokrewnych odmian niedokrwistości. Do organizmu człowieka
dostarczany jest przez bakterie jelitowe.
Kwas foliowy - a właściwie produkt jego redukcji, kwas 5,6,7,8-tetrawodorofoliowy (H
4
folian) -
jest grupą prostetyczną enzymów przenoszących jednowęglowe rodniki typu:
CH
3
–
HOCH
2
–
–CH
2
–
–CH=
HOC–
HN=CH–
Miejscem przyłączenia grup C
1
są atomy N-5 lub N-10 tetrawodorofolianu. Natomiast źródłem
ich pochodzenia są przemiany aminokwasów: histydyny, tryptofanu, seryny i metioniny.
Lip
S
S
XH
2
X
Lip
S
S
H
H
liponian
uteniony
liponian
zredukowany
44
Biotyna. Biotyna jest grupą prostetyczną karboksylaz i dekarboksylaz. Przyłączenie grupy
-COO
-
do substratu wymaga dostarczenia energi chemicznej; jej dawcą jest ATP, który transformuje
do aż AMP.
Biotyna jest witaminą H. Jej brak w organizmie człowieka wywołuje zaburzenia skórne oraz zmiany
psychomotoryczne: depresję, brak łaknienia i snu, itp. Witamina ta jest wytwarzana przez bakterie jelitowe.
Koenzym A. Na pierwszy rzut oka przypomina on opisane wcześniej dinukleotydy. Jednakże
koenzym ten nie jest zaliczany do dinukleotydów. Zbudowany jest z adenozyno-3’-fosforanu–5’-
pirofosforanu połączonego wiązaniem estrowym z kwasem pantotenowym, a ten z kolei wiązaniem
peptydowym z cystoaminą. Cysteoamina jest produktem dekarboksylacji cysteiny. Kwas pantotenowy
jest amidem kwasu 2,4-dihydroksy-3,3-dimetylomasłowego i β-alaniny. Symbolicznie koenzym A
zapisuje się jako CoASH.
Kwas pantotenowy jest witaminą B
5
. Niezbędna jest ona do prawidłowego metabolizmu białek, cukrów i
tłuszczów oraz do syntezy niektórych hormonów, uczestniczy w regeneracji tkanek i przyspiesza gojenie ran,
zapobiega przemęczeniu i usprawnia układ sercowo-naczyniowy, nerwowy i pokarmowy, bierze udział w
wytwarzaniu tłuszczów, cholesterolu, poprawia pigmentację i stan włosów. Kwas pantotenowy wytwarzany jest w
organizmach roślin, drobnoustrojów, a także niektórych pleśni. Bogatym źródłem tego związku są drożdże,
grzyby, groch, wątróbka, otręby pszenne, ryby (np. śledzie, makrele, pstrągi), mleko pełne, mięso kurczaka,
mleczko pszczele, pestki słonecznika, sery, orzechy, jajka, owoce awokado, pomarańcze, ziemniaki, brokuły,
ciemny ryż, melony, pełnoziarnisty chleb, soja, masło orzechowe, banany.
Koenzym A odgrywa podstawową rolę podczas aktywacji i
przenoszenia rodników acylowych. Reakcje przyłączenia CoASH
do kwasu karboksylowego katalizują syntetazy, które do swego
działania wymagają dostarczenia energii, której źródłem jest
ATP.
Powstałe
wiązanie
tioestrowe
jest
wiązaniem
wysokoenergetycznym (zaznaczane we wzorach chemicznych
tyldą
„~”
). Przykładem może być synteza acetylo~SCoA z kwasu octowego, katalizowana przez
syntetazę acetylo-CoA (EC 6.2.1.1).
Difosfotiamina. Tiamina jest związkiem zbudowanym z pierścienia pirymidynowego
połączonego z heterocyklicznym pierścieniem tiazolowym grupą metylenową. W difosfotiaminie
grupa hydroksylowa łańcucha bocznego pierścienia tiazolowego jest zestryfikowana pirofosforanem.
Z apoenzymem wiąże się właśnie przez ugrupowanie pirofosforanowe. Symbolicznie diosfotiaminę
zapisuje się jako DPT.
HN
NH
O
S
C
O
NH-Enzym
biotyna
HN
NH
O
S
R
biotyna
NH
O
N
R
S
HOOC
ATP
AMP+PP
CO
2
karboksylaza
karboksybiotyna
adenozyno-3'-fosforan-5`-pirofosforan
koenzym A (CoA-SH)
P
O CH
2
C
CH
3
CH
3
CH
OH
C
O
NH CH
2
CH
2
C
O
NH CH
2
CH
2
SH
P
O
OH
CH
2
O
N
N
N
N
NH
2
P
cysteoamina
kwas pantotenowy
ATP
AMP+PP
syntetaza
X-CoA
CoASH
X COOH
~
SCoA
X C
O
45
N
N
H
3
C
NH
2
CH
2
N
S
CH
2
CH
3
CH
2
O
P
P
+
difosfotiamina (DPT)
Tiamina jest w i t a m i n ą B
1
. Skutkami jej niedoboru są zaburzenia czynności centralnego układu
nerwowego (uczucie osłabienia i zmęczenie, możliwy oczopląs, zaburzenia pamięci i koncentracji a nawet
depresja), niewydolność krążenia (przyspieszona akcja serca, powiększenie wymiarów serca, obrzęki kończyn),
zaburzenia ze strony przewodu pokarmowego (utrata łaknienia, nudności, wymioty, biegunki, bóle brzucha, brak
apetytu, spadek wagi). W przypadku silnej awitaminozy B
1
może wystąpić choroba Beri-beri, objawiająca się
bólami kończyn, osłabieniem mięśni i ich drżeniem oraz wyraźną niewydolnością układu krążenia. Etanol
powoduje rozkład tego związku, o czym powinni pamiętać ludzie nadużywający alkoholu i dbać o dostarczanie jej
do organizmu. Ź r ó d ł e m t e j w i t a m i n y są produkty zbożowe, mięso, groch, fasola, drożdże, orzechy, ryby,
owoce i warzywa. Zapotrzebowanie człowieka na tę witaminę wynosi ok. 1,5 mg na dobę.
Difosfotiamina jest grupą prostetyczną enzymów przenoszących aldehydy: glikolowy, octowy i
semialdehyd bursztynowy.
Jako grupa prostetyczna k e t o l a z , DPT przenosi aldehyd glikolowy, który odrywa od ksylulozo-
5-fosforanu i transportuje go na erytrozo-4-fosforan.
Jako e l e m e n t u k ł a d ó w w i e l o e n z y m a t y c z n y c h - podczas α-oksydacji - np.
pirogronianu lub α-ketoglutaranu zachowuje się jak transferaza, a równocześnie jak liaza. Jako
dehydrogenaza pirogronianowa (acetyl-transportująca) – EC 1.2.4.1, prowadzi dekarboksylację
pirogronianu do aldehydu octowego, a jako dehydrogenaza ketoglutaranowa (bursztynian-
transporująca) – EC 1.2.4.2, dekarboksyluje α-ketoglutaran do semialdehydu bursztynianowego, które
dalej przenosi na kwas liponowy.
Pirydoksyna. Mieszanina trzech naturalnych związków: pirydoksyny, pirydoksalu i
pirydoksaminy. Są one pochodnymi pirydyny.
DPT
N
S
CH
2
CH
3
R
H
CH
2
O
+
PP
Enzym
aldehyd
3-fosfoglicerynowy
P
CH OH
CH
2
O
C
O
H
transketolaza
ksylulozo-5-fosforan
CH
2
OH
C O
CH
HO
CH OH
CH
2
O
P
N
S
CH
2
CH
3
R
CH
2
O
+
PP
Enzym
~
C
H
HO
CH
2
OH
aktywny aldehyd glikolowy
~
DPT
N
H
3
C
HO
CH
2
OH
CH
2
OH
N
H
3
C
HO
CH
2
OH
CH
2
NH
2
N
H
3
C
HO
CH
2
OH
C
O
H
pirydoksyna
pirydoksamina
pirydoksal
DPT
N
S
CH
2
CH
3
R
H
CH
2
O
+
PP
Enzym
~
DPT
CH
3
C O
COOH
pirogronian
hydroksyetylo
CO
2
fragment dehydrogenazy
pirogronianowej
(dekarboksylującej)
N
S
CH
2
CH
3
R
CH
2
O
+
PP
Enzym
~
C
OH
H
CH
3
46
5 - F o s f o r a n p i r y d o ks a l u (PLP) jest koenzymem lub grupą prostetyczną przeszło 50
enzymów. Bierze udział w przemianach aminokwasów. Przede wszystkim współdziała z
aminotransferazami przenoszącymi grupę aminową z aminokwasów na α-ketokwasy i odwrotnie. Jest
koenzymem dekarboksylaz aminokwasów. Uczestniczy ponadto w przemianie tryptofanu w amid
kwasu nikotynowego, transformacji glicyny w serynę i w biosyntezie cysteiny. Szczegóły dotyczące
reakcji transaminacji i dekarboksylacji aminokwasów zostaną opisane w rozdziale omawiającym
metabolizm aminokwasów.
Mieszanina pirydoksyny, pirydoksalu i pirydoksaminy nazywana jest witaminą B
6
. Wszystkie trzy związki
ulegają w organizmie wzajemnym przekształceniom i wykazują podobne działanie biologiczne. Niedobór witaminy
B6 występuje tylko wyjątkowo, np. u małych dzieci karmionych sztucznymi odżywkami lub karmionych przez
matki, które długo przyjmowały doustne środki antykoncepcyjne oraz u alkoholików i ich potomstwa. Do
najczęściej spotykanych objawów należą stany zapalne skóry (łojotokowe zmiany na twarzy), podrażnienie języka
i błon śluzowych jamy ustnej (języka, kącików warg) zmiany w ośrodkowym układzie nerwowym (apatia,
bezsenność, nadwrażliwość, napady drgawek), zwiększona podatność na infekcje. Witamina B
6
wytwarzana jest
przez florę bakteryjną przewodu pokarmowego, w dużych występuje w wątrobie, jajach, jarzynach i mięsie.
Cyjanokobalamina. Jej nazwa wywodzi się od jonu cyjankowego połączonego koordynacyjnie
z atomem kobaltu, wbudowanym w rdzeń porfirynowy. Rdzeń porfirynowy wraz z jonem kobaltu
noszą nazwę ko b al a mi ny. Zamiast jonu CN
-
, z kobalaminą może być połączony inny anion, np.
hydoksylowy, chlorkowy czy azotynowy. W tym ostatnim przypadku związek nosi nazwę
nitrokobalaminy, a znajdowany jest w podłożach hodowlanych Streptomyces griseus.
Cyjanokobalamina jest koenzymem niektórych dehydrataz, amoniako-liaz i mutaz.
Witamina B
12
, znana jako czynnik przeciwdziałający niedokrwistości złośliwej lub jako czynnik
przeciwanemiczny. Brak tej witaminy powoduje charakterystyczne zmiany w obrazie krwi, związane z anemią
złośliwą. Obserwuje się również zmiany zapalne języka oraz brak kwasu solnego w żołądku. Organizmy roślin i
zwierząt nie produkują cyjanokobalaminy. Zdolność tą mają niektóre gatunki bakterii (choć u niektórych
mikroorganizmów jest ona także czynnikiem wzrostowym). Zwierzęta mięsożerne zapotrzebowanie na tę
witaminę pokrywają, spożywając mięso innych zwierząt. Szczególnie obfita w cyjanokobalaminę jest wątroba.
Zwierzęta roślinożerne wykorzystują cyjanokobalaminę wytwarzaną przez florę bakteryjną ich przewodu
pokarmowego. Źródło to jest czasem niewystarczające i u niektórych gatunków zwierząt dochodzi niekiedy do
zjadania własnych odchodów, co prowadzi do zwiększenia ilości tej witaminy w organizmie. Organizm dorosłego
człowieka potrzebuje ok. 3 mg cyjanokobalaminy dziennie. Zapotrzebowanie to jest w zupełności pokrywane
przez normalną dietę.
Nukleozydofosforany. Nukleotydy, które do swojej reszty fosforanowej dodatkowo mają
przyłączoną jedną lub dwie cząsteczki kwasu fosforowego, czyli nukleozydodi- i trifosforany są
koenzymami, a właściwie poprawniej definiując kofaktorami czy też kosubstratami reakcji
katalizowanych przez liczne enzymy. Zaliczane są one do związków bogatych w energię chemiczną;
tzw. związków makroergicznych lub inaczej związków wysokoenergetycznych. Wśród nich
najczęściej kosubstratem w reakcjach enzymatycznych bywa a de n o zyn o t r i f os f or a n – ATP. Jest
on kosubstratem większości ligaz (enzymów z klasy EC 6.), kinaz (transferaz z podklasy EC 2.7.,
H
+
CN
P
N
N
CH
3
CH
3
O
OH
O
HOH
2
C
CH
3
CH
CH
2
NH
CO
CH
2
Co
H
3
C
H
2
NOH
2
C
H
2
NOH
2
CH
2
C
H
2
NOH
2
C
CH
2
CH
3
CH
2
CH
2
CONH
2
CH
3
CH
3
CH
3
CH
2
CH
2
CONH
2
CH
2
CONH
2
H
3
C
CH
3
CH
3
N
CH
N
C
N
C
N
cyjanokobalamina
47
O
OH
CH
2
O
O
P O
OH
N
N
N
N
NH
2
H
adenozyna
AMP
ADP
ATP
O
P O
OH
P
OH
OH
O
O
przenoszących grupę fosforanową na różne substraty) czy wreszcie niektórych karboksykinaz
(enzymów z pod-podklasy EC 4.1.1.).
T e r m o d y n a m i k a r e a kc j i e n z y m a t y c z n y c h . Zgodnie z prawami termodynamiki reakcje
endoergiczne nie mogą przebiegać spontanicznie, nawet jeśli są katalizowane przez enzymy. Tego
typu reakcje muszą być sprzężone z inną reakcją silnie egzoergiczną, tak by suma ich energii
swobodnej ΔG była równa zero lub ujemna.
Przykładowo reakcja glukozy z kwasem
fosforowym jest reakcją endoergiczną; zmiana
energii swobodnej ΔG = 12 kJ/mol. Stąd
równowaga tej reakcji jest silnie przesunięta
na
lewą
stronę;
glukozo-6-fosforan
praktycznie nie powstaje. Reakcja hydrolizy
ATP do ADP + P
i
* jest z kolei silnie egzoergiczna; ΔG = −29 kJ/mol. Przy tak dużej ujemnej wartości
ΔG jest to reakcja praktycznie nieodwracalna. W przypadku enzymatycznego sprzęgnięcia obu tych
reakcji suma energii swobodnej ΔG = −17 kJ/mol. W takim układzie równowaga reakcji przesunięta
jest zdecydowanie na prawą stronę i praktycznie cała pula glukozy jest przemieniona w glukozo-6-P.
W ATP pierwsza cząsteczka kwasu fosforowego przyłączona jest do grupy –OH rybozy
zwykłym wiązaniem estrowym. Energia swobodna jego hydrolizy wynosi ΔG = −14 kJ/mol.
Natomiast kolejne cząsteczki kwasu fosforowego wiążą się ze sobą wiązaniem bezwodnikowym.
Energia swobodna ich hydrolizy odpowiednio wynosi:
Stąd ATP i ADP (adenozynodifosforan) należą do związków bogatych w energię chemiczną.
Natomiast AMP (adenozynomonofosforan) jest związkiem niskoenergetycznym. Przyjmuje się, że
dolną wartością energii swobodnej hydrolizy, która decyduje o zaliczeniu związku do
makroergicznych jest ΔG zbliżone do −29 kJ/mol.
W tabeli 2.2. przykładowo podano przybliżone średnie wartości energii swobodnej hydrolizy
kilku związków zawierających grupę fosforanową. Energia swobodna hydrolizy ATP do ADP+P
i
wynosi ΔG = −29 kJ/mol, co sprawia, że ten nukleozydotrifosforan wraz z ADP i pirofosforanem
należą do grupy fosforanów znajdujących się pośrodku listy cytowanych związków
wysokoenergetycznych i niskoenergetycznych. Dlatego ATP może być donorem fosforanów dla
związków niskoenergetycznych znajdujących na liście poniżej, o ΔG >−29 kJ/mol. Z tego samego
powodu ADP może być akceptorem wysokoenergetycznego fosforanu od związków znajdujących się
na tej liście powyżej, o ΔG <−29 kJ/mol.
* umownie P
i
oznacza fosforan nieorganiczny
ATP
G= -29 kJ/mol
ADP + P
i
,
G= -28 kJ/mol
ADP
AMP + P
i
glukoza
G= -17 kJ/mol
ATP
ADP + P
i
glukozo-6- P
G= -29 kJ/mol
ATP
ADP
glukoza + H
3
PO
4
glukozo-6- P
-H
2
O
+H
2
O
G= 12 kJ/mol

48
Należy jeszcze dodać, że wiązania bezwodnikowe w ATP łatwo ulegają rozerwaniu. Oderwana
reszta fosforanowa lub pirofosforanowa może być przenoszona na różne substraty, tym łatwiej, iż
rozkład ATP jednocześnie dostarcza energii swobodnej niezbędnej dla takich reakcji.
Tabela 2.2. Energia swobodna hydrolizy niektórych fosforanów
Nazwa związku
ΔG [kJ/mol]
1
Fosfoenolopirogronian
Karbamoilofosforan
1,3-difosfoglicerynian
Fosfokreatyna
Acetylo~SCoA
−62
−51
−49
−43
−34
ATP
AMP+PP
ATP
ADP+P
ADP
AMP+P
PP (pirofosforan)
−31
−29
−28
−27
Glukozo-1-fosforan
AMP
Glukozo-6-fosforan
−21
−14
−14
1
cytowane dane są przybliżonymi wartościami ΔG
ATP zawiera dwa wysokoenergetyczne wiązania bezwodnikowe. Oderwanie fosforanu, prowa-
dzące do powstania ADP, powoduje utratę jednego z nich (ΔG =−29 kJ/mol). Oderwanie fosforanu od
ADP prowadzi do powstania AMP (ΔG =−28 kJ/mol). Teoretycznie wyliczona, sumaryczna energia
swobodna takiej dwuetapowej przemiany ATP do AMP obniża się więc o ΔG =−57 kJ/mol*. Ale od
ATP może być oderwany również pirofosforan, jednoetapowo prowadząc do powstania AMP. Zmiana
energii swobodnej ΔG takiej przemiany równa jest −31 kJ/mol. Dla pełnego jej bilansu należy jednak
uwzględnić dodatkowo energię swobodną zawartą w pirofosforanie
(ΔG =−27 kJ/mol), co po podsumowaniu daje ΔG =−58 kJ/mol*. Ta
swobodna energia zawarta w PP
i
jest uwalniana podczas jego
enzymatycznej hydrolizy pod działaniem pirofosfatazy nieorganicznej (EC 3.6.1.1).
W komórkach żywych organizmów pomiędzy ATP, ADP i AMP istnieje pewnego typu
specyficzna równowaga. Kinaza adenylanowa (EC 2.7.4.3)
przenosi jeden z wysokoenergetycznych fosforanów z ATP na
AMP, co prowadzi do powstania dwóch cząsteczek ADP, tak jak
to przedstawiono na rysunku obok.
Dla pełnego obrazu należy jeszcze wspomnieć, że w komórkach ATP uczestniczy w reakcjach
enzymatycznych w postaci kompleksu z jonem Mg
2+
. Stąd jony Mg
2+
są aktywatorami enzymów
współdziałających z ATP.
Reasumując, r o l a A T P j a k o k o s u b s t r a t u w reakcjach enzymatycznych jest następująca:
1. ATP jako d o n o r e n e r g i i s w o b o d n e j w reakcjach syntez katalizowanych przez ligazy. W
większości takich przypadków ATP ulega rozkładowi do AMP + PP
i
. Jak już wspomniano,
ilość energii swobodnej dostarczona do takiego układu jest równoważna ΔG =−58 kJ/mol, co
wystarcza do przeprowadzenia rozmaitych syntez, niekiedy nawet związków
wysokoenergetycznych. Za przykład niech posłuży synteza acetylo~SCoA katalizowana przez
syntetazę acetylo-CoA (EC 6.2.1.1).
*
Sumaryczna energia swobodna wyzwolona w jednoetapowym i dwuetapowym przekształceniu ATP do AMP
oczywiście musi być taka sama. Różnica 1 kJ/mol pomiędzy wyliczeniami wynika ze stosowania przybliżonych wartości ΔG.
PP
i
2P
i
pirofosfataza
nieorganiczna
ATP + AMP
2 ADP
kinaza
adenylanowa
49
NADH
ADP
ATP
NADPH
kinaza NADH
metionina + ATP
adenozynotransferaza
metioninowa
EC 2.5.1.6
PP + P
adenozynometionina
ATP dużo rzadziej, jako koenzym ligaz, ulega podczas reakcji rozkładowi do ADP. Tym
niemniej znanych jest sporo i takich przypadków. Dla podkreślenia, że ATP rozkłada się
jedynie do ADP w nazwie ligazy w nawiasie umieszcza się (ADP-tworząca). Jako przykład
podana zostanie syntetaza acetylo-CoA (ADP-tworząca) – EC 6.2.1.13. Wybrano ją celowo,
gdyż tylko wyjątkowo niektóre bakterie posiadają ten enzym, a z energetycznego „punktu
widzenia” komórki jest on korzystniejszy od wcześniej opisanej syntetazy acetylo-CoA.
2. ATP jako d o n o r o r t o f o s f o r a n u . Odpowiednie transferazy (kinazy) przenoszą rodnik
fosforanowy na rozmaite substraty, np. na monosacharydy,
glicerol, nukleozydy, itp. Za przykład posłuży synteza NADPH z
NADH katalizowana przez kinazę NADH (EC 2.7.1.86).
Na marginesie omawianego zagadnienia warto zwrócić uwagę na fakt, że w wyjątkowych sytuacjach
źródłem ortofosforanu może być pirofosforan. Nie powinno to budzić większego zdziwienia, bowiem
energia swobodna jego hydrolizy ΔG =−27 kJ/mol. Przykładem może być kinaza octanowa
(pirofosforanowa) – EC 2.7.2.12, katalizująca przeniesienie fosforanu z PP
i
na kwas octowy z
wytworzeniem acetylofosforanu.
3. ATP jako d o n o r p i r o f o s f o r a n u . W niektórych przypadkach konieczne jest przyłączenie
do substratu rodnika pirofosforanowego. Jego donorem jest ATP, a reakcję katalizują
pirofosfokinazy. Jako przykład może posłużyć synteza
difosfotiaminy
z
tiaminy.
Reakcję
katalizuje
pirofosfokinaza tiaminowa (EC 2.7.6.2).
4. ATP jako d o n o r r o d n i ka a d e n yl i l o w e g o . Adenylacja to reakcja przeniesienia rodnika
adenylilowego z ATP na substrat z jednoczesnym odłączeniem PP
i
. Reakcja ta ma
podstawowe znaczenie dla syntezy dinukleotydów adenilowych i związków o podobnej
budowie (choćby wspomnianego wcześniej CoASH). Reakcję katalizują adenylilotransferazy
(zwane zwyczajowo pirofosforylazami). Jako przykład posłuży synteza FAD z FMN
katalizowana przez adenylilotransferazę FMN (EC 2.7.7.2).
Ale enzymy z pod-podklasy EC 2.7.7. mogą również transportować inne nukleotydilowe
rodniki z nukleotydotrifosforanów. Jedną z takich reakcji o podstawowym znaczeniu dla
procesów biochemicznych jest synteza UDPG (urydyliloglukozy). Reakcja katalizowana jest
przez urydylilotransferazę glukozo-1-fosforanową (EC 2.7.7.9).
5. ATP jako d o n o r a d e n o z y n y . W pewnych szczególnych przypadkach ATP staje się
donorem adenozyny. Przykładem może być synteza adenozynometioniny. Koenzym ten
powstaje w reakcji metioniny z ATP, katalizowanej przez adenozynotransferazę metioninową
(EC 2.5.1.6).
Niekiedy rolę ATP w omówionych wyżej reakcjach może pełnić inny z
nukleozydotrifosforanów. O urydynotrifosforanie (UTP) i jego roli już wspomniano.
Guanozynotrifosforan (GTP) lub inozynotrifosforan (ITP) są koenzymami syntetazy bursztynylo-CoA
ATP
AMP+PP
syntetaza
acetylo-CoA
CoASH
~
SCoA
C
O
CH
3
CH
3
COOH
ATP
ADP+P
CoASH
~
SCoA
C
O
CH
3
CH
3
COOH
syntetaza
acetylo-CoA
(ADP-tworzaca)
FMN
ATP
PP
i
FAD
adenylilotransferaza FMN
tiamina
ATP
AMP
pirofosfokinaza
tiaminowa
difosfotiamina
glukozo-1- P
UTP
PP
i
urydylilotransferaza
glukozo-1-fosforanowa
UDPG

50
(patrz cykl Krebsa). Cytydynotrifosforan (CTP) bierze udział w syntezie substancji lipidowych pod
postacią cytydylodifosfocholiny.
Pomiędzy ATP a pozostałymi nukleotydotrifosforanami występuje zależność:
2.2.3. Kinetyka reakcji enzymatycznych
S z y b k o ś ć r e a k c j i e n z y m a t y c z n e j , podobnie jak każdej reakcji chemicznej, wyraża się
ubytkiem stężenia jednego z substratów lub też przyrostem stężenia któregoś z produktów:
dt
P
d
dt
S
d
v
]
[
]
[
(2.1)
R ó w n o w a g a r e a k c j i e n z y m a t y c z n y c h . Teoretycznie wszystkie reakcje chemiczne
(również katalizowane enzymatycznie) są odwracalne. Po pewnym czasie ustala się równowaga
pomiędzy substratami reakcji a jej produktami, objawiająca się tym, że szybkość powstawania
produktów jest równa szybkości ich rewersji do substratów. Szybkość powstawania produktów v
1
jest
proporcjonalna do stężeń reagujących substratów. Szybkość reakcji rewersji produktów v-
1
jest
proporcjonalna do stężeń produktów. W stanie równowagi:
2
1
v
v
]
[
]
[
1
1
B
A
k
v
]
[
1
1
AB
k
v
(2.2)
gdzie: A,B – substraty; AB – produkt; k
+1
, k
-1
– stałe szybkości reakcji
Dla przypomnienia: stałe szybkości reakcji k są charakterystyczne dla konkretnych reagentów. Rosną wraz
ze wzrostem temperatury reakcji.
Zgodnie z prawem działania mas Guldberga i Waagego w stanie równowagi, stężenia reagentów
spełniają zależność:
K
B
A
AB
k
k
]
[
]
[
]
[
1
1
gdzie: K jest stałą równowagi konkretnej reakcji.
(2.3)
Im większa jest stała równowagi K, tym większe stężenie produktu AB, czyli równowaga reakcji
bardziej przesunięta na prawo. Reakcja pomiędzy A i B zachodzi tym energiczniej, im większa jest
stała równowagi reakcji K.
Jeszcze raz należy wyraźnie podkreślić: obecność enzymu w układzie nie zmienia wartości
stałej K. Enzym jedynie przyspiesza moment osiągnięcia równowagi przez reagenty. Jeżeli K>1
to reakcja jest spontaniczna.
Stałą K można wyrazić liczbowo, o ile znane są stężenia substratów i produktów reakcji w stanie
równowagi. Wartości K można również wyliczyć na drodze termodynamicznej. Pomiędzy zmianami
energii swobodnej ∆G a stałą równowagi reakcji K istnieje zależność:
K
ln
-RT
G
gdzie: R - stała gazowa, T – temperatura bezwzględna
(2.4)
Z zależności tej wynika prosta formuła potwierdzająca wcześniejsze stwierdzenia: im większa
stała równowagi reakcji K, tym większa różnica pomiędzy wartościami energii swobodnej substratów
i produktów. I odwrotnie: im większe ∆G, tym równowaga reakcja jest bardziej przesunięta na korzyść
produktów. W krańcowych przypadkach, gdy ujemna wartość ∆G jest bardzo duża cały substrat ulega
przekształceniu w produkt, a reakcja praktycznie staje się nieodwracalna.
Z kolei pomiędzy zmianami energii swobodnej ∆G (w chemii fizycznej znanej pod słuszną
nazwą potencjału termodynamicznego), entalpią układu i zmianami entropii występuje zależność:
S
T
H
G
(2.5)
S z y b k o ś ć p o c zą t k o w a r e a k c j i e n z y m a t y c z n e j .
Szybkość początkowa reakcji v jest to szybkość wyznaczona przed
powstaniem dostatecznie dużej ilości produktu, który by umożliwiał
zachodzenie reakcji odwrotnej. Szybkość początkowa reakcji
enzymatycznej jest zawsze proporcjonalna do stężenia enzymu.
nukleozydotrifosforan + ADP
kinaza
nukleozydodifosforanowa
nukleozydodifosforan + ATP
EC 2.7.4.6
S
P
enzym
A + B
AB
v
+1
, k
+1
v
-1
, k
-1
V
[E]
Wpływ stężenia enzymu [E] na
szybkość reakcji enzymatycznej V
[S]>>[E]
51
m
K
[S]
2
max
V
max
V
V
Stąd oznaczenie ilości enzymu (jego aktywności) powinno być oparte, o ile jest to możliwe, na
pomiarach początkowej szybkości reakcji przy znacznym nadmiarze substratu S. W takim
układzie szybkość reakcji nie zależy od stężenia substratu i reakcja jest rzędu zerowego.
A k t y w n o ś ć e n z y m a t y c z n a . Oznaczenie bezwzględnej ilości enzymu (np. w gramach lub
molach) jest trudne do wykonania, gdyż wiąże się z oczyszczeniem białka enzymatycznego do stanu
homogenności. Stąd umówiono się, że za miarę ilości enzymu w biopreparatach przyjmuje się jego
aktywność katalityczną (enzymatyczną), czyli szybkość, z jaką w założonych warunkach preparat
enzymatyczny przekształca określoną ilość wybranego arbitralnie substratu. Miarą tej szybkości jest
ubytek substratu lub przyrost produktów reakcji enzymatycznej w przeliczeniu na jednostkę czasu.
Zdefiniowano również oficjalnie obowiązujące standardowe jednostki enzymatyczne.
Pierwszą z nich, obowiązującą w układzie SI, nazwano ka t al e m (Kat). 1 Kat to taka ilość
enzymu, która katalizuje przemianę 1 mola substratu w czasie 1 sekundy w temperaturze 30
C.
Druga z tych oficjalnie obowiązujących jednostek, rekomendowana przez Międzynarodową Unię
Biochemiczną, nazwana została m i ę d z y n a r o d o w ą j e d n o s t ką e n z y m a t y c z n ą (w skrócie
m.j.e.). 1 m.j.e. to taka ilość enzymu, która katalizuje przemianę 1 μmola substratu w ciągu 1 minuty
w temperaturze 30
C.
Jednakże dla wielu enzymów obie te oficjalne obowiązujące jednostki nie znalazły praktycznego
zastosowania. Natomiast powszechnie przyjęły się jednostki enzymatyczne skojarzone z
odpowiednimi metodami oznaczania aktywności opracowanymi dla całej gamy konkretnych enzymów
a nawet grup enzymów.
Teoria Michaelisa-Menten. W 1913 roku L.Michaelis i M.L.Menten przedstawili swoją
koncepcję katalizy enzymatycznej. Model Michaelisa–Menten opiera się na założeniu powstawania
przejściowego kompleksu enzymu-substrat:
(2.6)
Zgodnie z tym równaniem, przy niewielkich stężeniach substratu - równoważnym stężeniom
enzymu - reakcja enzymatyczna staje się reakcją I rzędu i jej szybkość staje się proporcjonalna do
stężenia zarówno substratu [S], jak i enzymu [E]. Przy stałym stężeniu enzymu [E] szybkość reakcji
będzie zależała jedynie od stężenia substratu. Wpływ stężenia substratu [S] na początkową szybkość
reakcji enzymatycznej przy stałym stężeniu enzymu przedstawia poniższy wykres.
Dla pewnych stężeń substratu - równoważnym ilościom dostępnych centrów aktywnych
enzymu - reakcja osiągnie szybkość maksymalną V
max
. Dalsze zwiększanie stężenia substratu nie
przyspiesza jego przemiany. Jak już wspomniano wcześniej szybkość reakcji enzymatycznej zależy
od łatwości tworzenia kompleksu enzymu z substratem. Z równania (2.6) wynika, że w stanie
równowagi szybkość tworzenia kompleksu ES jest równa szybkości jego rozpadu:
1
2
2
1
v
v
v
v
(2.7)
]
[
]
[
1
1
S
E
k
v
]
[
1
1
ES
k
v
]
[
2
2
ES
k
v
]
[
]
[
2
1
P
E
k
v
(2.8)
Po podstawieniu zależności (2.8) do równania (2.7) i po prostych matematycznych
przekształceniach otrzymuje się:
E + S
ES
E + P
k
-1
k
1
k
2
k
-2
52
[S]
v
V
max
K
m2
K
m1
S
1
S
2
V
max
1
v
-1
K
m
1
[S]
tgα = K
m
/V
max
2
1
2
2
1
1
]
[
]
[
]
[
]
[
k
k
P
k
k
k
S
k
E
ES
(2.9)
Uwzględniając fakt, że na początku reakcji [P]
0 oraz po dalszych prostych przekształceniach
równania (2.9), otrzymuje się:
m
K
1
2
1
]
[
]
[
]
[
k
k
k
ES
S
E
(2.10)
Wielkość K
m
nosi nazwę s t a ł e j M i c h a e l i s a . J est ona charakterystyczna dla danego układu
enzym-substrat. Łatwo zauważyć, że im mniejsza wartość K
m
, tym większe stężenie kompleksu
przejściowego [ES].
Analizując równania (2.6)
(2.10) należy zauważyć, że
1. stężenie enzymu [E] w równaniu (2.10), w rzeczywistości jest stężeniem enzymu wolnego w
danym momencie reakcji, czyli niezwiązanego w kompleks przejściowy ES. Na każdym
etapie reakcji część enzymu wyjściowego [E
0
] obecnego w układzie reakcyjnym jest związana
w ES. Stąd [E]= [E
0
]-[ES].
2. Szybkość reakcji enzymatycznej v, czyli szybkość powstawania produktu P, zależy tak
naprawdę od szybkości rozpadu kompleksu ES, czyli od v
+2
. Stąd: v = v
+2
=k
+2
[ES].
3. Gdy cały enzym jest w kompleksie z substratem, czyli [E
0
]=[ES] szybkość reakcji
enzymatycznej osiąga maksymalną szybkość V
max
.
Uwzględniając powyższe założenia w równaniu (2.10) otrzymuje się r ó w n a n i e
m a t e m a t y c z n e M i c h a e l i s a - M e n t e n :
]
[
]
[
max
S
K
S
V
v
m
(2.11)
Równanie (2.11) pozwala w prosty sposób zdefiniować stałą Michaelisa K
m
:
gdy: v= 1/2
V
max
to
K
m
= [S]
Stała Michaelisa K
m
jest to takie stężenie substratu [S], przy którym szybkość reakcji
enzymatycznej równa się połowie szybkości maksymalnej V
max
.
Łatwo wykazać, że:
1. Jeśli [S]<<K
m
(czyli przy małych stężeniach substratu), to reakcja jest I rzędu: v= f([S]),
2. Jeśli [S]=K
m
, to v=1/2
V
max
3. Jeśli [S]>>K
m
, to reakcja jest 0 rzędu i v=V
max
.
Odwrotność stałej Michaelisa 1/K
m
nazywana jest powinowactwem danego enzymy do substratu.
Im mniejsza stała Michaelisa K
m
, tym większe powinowactwo enzymu do substratu, co pokazuje
poniższy rysunek. Celowo dla obu substratów - S
1
i S
2
– przyjęto identyczne szybkości maksymalne
reakcji V
max
. K
m2
>K
m1
, co oznacza, że ten enzym wykazuje większe powinowactwo do substratu S
1
aniżeli do S
2
. Oznacza to, że szybciej przekształca on substrat S
1
aniżeli S
2
.
Wpływ rodzaju substratu na szybkość reakcji
Wykres Lineweavera-Burka
enzymatycznej
Stałe Michaelisa K
m
dla różnych układów enzym-substrat mogą mieć różne wartości; nawet od
10
–2
mola/dm
3
do 10
–7
mola/ dm
3
.
Stałą K
m
można wyznaczyć graficznie korzystając ze wzoru Lineweavera-Burka..

53
max
max
1
]
[
1
1
V
S
V
K
v
m
Szczegółowo kinetyką reakcji enzymatycznych zajmuje się enzymologia i tam można znaleźć więcej danych
odnośnie tematu.
2.2.4. Klasyfikacja i nomenklatura enzymów
Pierwotne nazewnictwo enzymów było nieuporządkowane i dopuszczało stosowanie dowolnych
nazw takich, jak pepsyna, papaina, subtilizyna. W późniejszym okresie nazwę enzymu wywodzono od
typu reakcji lub substratu, na który działał, przy czym za charakterystyczną dla enzymów uznano
końcówkę -aza (np. reduktaza, lipaza, itp.).
Od 1964 roku Komisja Enzymowa Międzynarodowej Unii Biochemicznej zaleciła stosowanie
s y s t e m a t y c z n y c h n a zw e n z y m ó w , składających się przeważnie z dwu części. Pierwsza jest
utworzona od nazwy katalizowanej reakcji i określa jej rodzaj (np. oksydoreduktaza, glukohydrolaza,
itp.). Druga część nazwy enzymu składa się z nazwy substratu lub substratów biorących udział w
reakcji. Np.:
maltohydrolaza 1,4-α-D-glukanu (zwana oficjalnie i zwyczajowo β-amylazą) - EC 3.2.1.2,
oksydoreduktaza alkohol:NAD (oficjalnie zwana dehydrogenazą alkoholową) - EC 1.1.1.1.
Nazwy systematyczne enzymów są długie i niekiedy bardzo skomplikowane. Z tego powodu ich
stosowanie napotyka na spory opór ze strony biochemików. Ponadto do wielu enzymów przylgnęły
nazwy stosowane pierwotnie. Dlatego zdecydowano, że obok nazwy systematycznej, dopuszczalne
jest stosowanie nazw zwyczajowych. Ostatnio (po 2000 roku) pojawiła się koncepcja wprowadzenia
tzw. nazw oficjalnych enzymów wywodzących się od nazw zwyczajowych. I tak np. nazwa α-amylaza
jest w świetle tej koncepcji nazwą oficjalną enzymu sklasyfikowanego pod numerem EC 3.2.1.1.
Należy jeszcze uzupełnić, że pierwotne, zwyczajowe nazwy niektórych enzymów (np. sacharaza,
maltaza) okazały się w świetle aktualnej wiedzy bez pokrycia i dla nich nie zaleca się ich stosowania.
Ponadto zdarza się, że pod jedną i tą samą nazwą zwyczajową są znane nawet trzy różne enzymy, np.
tyrozynaza. Nazwa „tyrozynaza” dla dwóch z nich oczywiście jest nazwą błędną.
W numerze 1 czasopisma "Postępy Biochemii" z 1985 roku zamieszczono słownik zalecanych i
zwyczajowych nazw enzymów w języku polskim.
Wszystkie poznane enzymy zostały ujęte przez Komisję Enzymową w jednolity system
klasyfikacji (EC). Każdy enzym w ramach tego systemu posiada odpowiedni numer złożony z
czterech członów liczbowych oddzielonych kropkami (np. dehydrogenaza alkoholowa ma numer EC
1.1.1.1). Podanie tego numeru jednoznacznie określa konkretny enzym, co pozwala uniknąć
nieporozumień i pomyłek. Zasady klasyfikacji i numeracji zbadanych enzymów oparte zostały o ich
specyficzność kierunkową (rodzaj katalizowanej reakcji) i substratową. Wszystkie enzymy podzielono
na sześć głównych klas:
EC.1. Oksydoreduktazy - enzymy katalizujące reakcje odłączania lub przyłączania atomów
wodoru, przyłączenia atomu tlenu lub przenoszenia elektronów.
EC.2. Transferazy - enzymy przenoszące grupy funkcyjne, rodniki, fragmenty cząsteczek itp.
EC 3. Hydrolazy - enzymy katalizujące reakcje hydrolizy.
EC 4. Liazy - enzymy niehydrolitycznie rozrywające wiązania.
EC 5. Izomerazy - enzymy katalizujące przemiany wewnątrzcząsteczkowe (racemizacja,
izomeryzacja, przenoszenie grup itp.).
EC 6. Ligazy - enzymy syntetyzujące, katalizujące tworzenie wiązań.
Każda klasa główna dzieli się na szereg podklas, których kolejne numery stanowią drugi człon
numeru klasyfikacyjnego i wynikają, ogólnie biorąc, z uściślonej specyficzności kierunkowej. Na
przykład EC.3.4. jest numerem podklasy hydrolaz peptydowych, czyli enzymów hydrolizujących
wiązanie peptydowe. W obrębie podklas rozróżnia się pod-podklasy - trzeci człon numeru
klasyfikacyjnego. Określa on dokładniej specyfikę katalizowanej reakcji. Dla oksydoreduktaz określa
grupę funkcyjną będącą akceptorem; dla transferaz uściśla rodzaj przenoszonej grupy; dla hydrolaz -
typ hydrolizowanego wiązania; dla liaz - rodzaj odszczepianej grupy; dla izomeraz - charakter
przekształcenia i wreszcie dla ligaz - rodzaj powstałego związku.

54
Dostęp do internetowej bazy danych dotyczącej nomenklatury enzymów (Enzyme Nomenclature
Database) można znaleźć pod adresem
http://www.expasy.org/enzyme/
. Na następnej stronie
przykładowo pokazano stronę internetową ExPASy dla dehydrogenazy alkoholowej – EC 1.1.1.1.

55
Wzór strony E xP A S y dla EC 1.1.1.1 – dehydrogenaza alkoholowa.
Official Name
Alcohol dehydrogenase.
Alternative Name(s)
Aldehyde reductase.
Reaction catalysed
An alcohol
+
NAD(+)
<=>
an aldehyde or ketone
+
NADH
Cofactor(s)
Zinc or Iron.
Comment(s)
Acts on primary or secondary alcohols or hemiacetals.
The animal, but not the yeast, enzyme acts also on cyclic secondary alcohols.
Cross-references
Biochemical
Pathways; map
number(s)
D8
;
E6
;
J10
PROSITE
PDOC00058
;
PDOC00059
;
PDOC00060
BRENDA
1.1.1.1
PUMA2
1.1.1.1
PRIAM enzyme-
specific profiles
1.1.1.1
Kyoto University
LIGAND chemical
database
1.1.1.1
IUBMB Enzyme
Nomenclature
1.1.1.1
IntEnz
1.1.1.1
MEDLINE
Find literature relating to 1.1.1.1
Swiss-Prot
P80222
, ADH1_ALLMI;
P49645
, ADH1_APTAU;
P06525
, ADH1_ARATH;
P41747
, ADH1_ASPFL;
P12311
, ADH1_BACST;
Q17334
, ADH1_CAEEL;
P43067
, ADH1_CANAL;
P48814
, ADH1_CERCA;
P23991
, ADH1_CHICK;
P23236
, ADH1_DROHY;
P48586
, ADH1_DROMN;
P09370
, ADH1_DROMO;
P22246
, ADH1_DROMT;
P07161
, ADH1_DROMU;
P12854
, ADH1_DRONA;
P08843
, ADH1_EMENI;
P05336
, ADH1_HORVU;
P20369
, ADH1_KLULA;
Q07288
, ADH1_KLUMA;
P00333
, ADH1_MAIZE;
P80512
, ADH1_NAJNA;
Q9P6C8
, ADH1_NEUCR;
P20306
, ADH1_ORYSA;
P12886
, ADH1_PEA;
P14219
, ADH1_PENAM;
P25141
, ADH1_PETHY;
O00097
, ADH1_PICST;
Q03505
, ADH1_RABIT;
P22797
, ADH1_RANPE;
P14673
, ADH1_SOLTU;
P80338
, ADH1_STRCA;
P13603
, ADH1_TRIRP;
P00330
, ADH1_YEAST;
Q07264
, ADH1_ZEALU;
P20368
, ADH1_ZYMMO;
P42327
, ADH2_BACST;
O45687
, ADH2_CAEEL;
O94038
, ADH2_CANAL;
P48815
, ADH2_CERCA;
P27581
, ADH2_DROAR;
P25720
, ADH2_DROBU;
P23237
, ADH2_DROHY;
P48587
, ADH2_DROMN;
P09369
, ADH2_DROMO;
P07160
, ADH2_DROMU;
P24267
, ADH2_DROWH;
P37686
, ADH2_ECOLI;
P54202
, ADH2_EMENI;
Q24803
, ADH2_ENTHI;
P10847
, ADH2_HORVU;
P49383
, ADH2_KLULA;
Zasady klasyfikacji enzymów w podklasach i pod-podklasach.
Oksydoreduktazy. W tej klasie enzymów drugi człon numeru - czyli podklasa - wskazuje, na
jaką grupę funkcyjną lub grupę związków działa dana oksydoreduktaza. Pewnego typu odstępstwo
występuje w podklasach EC 1.13. i EC 1.14. Te dwie podklasy skupiają oksygenazy – enzymy
56
wbudowujące atom lub dwa atomy tlenu do substratów, choć oficjalnie mówi się o
„oksydoreduktazach działających na pojedyncze lub podwójne donory z jednoczesnym
wybudowaniem atomów tlenu”.
Poniżej przedstawiono przykłady podklas (w EC 1.13. i EC 1.14. również pod-podklas) w tej
klasie enzymów.
EC 1. Oksydoreduktazy
EC 1.1. działające na grupę –CH
2
-OH jako donor
EC 1.2. działające na grupę –CHO lub >C=O jako donor
EC 1.3. odrywające atomy wodoru od –CH
2
-CH
2
, prowadząc do –CH=CH
EC 1.4. działające na grupę –CH
2
-NH
2
jako donor
EC 1.5. działające na grupę -CH-NH- jako donor
EC 1.6. działające na NADH lub NADPH
EC 1.7. działające na inne związki azotowe jako donory
EC 1.8. działające na związki siarki jako donory
EC 1.9. działające na grupę hemową jako donor
EC 1.10. działające na związki difenolowe i pokrewne jako donory
EC 1.11. działające na H
2
O
2
jako akceptor
peroksydazy
Są to p e r o k s y d a z y . W tej podklasie brak jest pod-podklas, stąd ich numer zaczyna się od
EC 1.11.1. Na przykład numer EC 1.11.1.6 odnosi się do k a t a l a z y .
EC 1.12. działające na wodór jako donor
EC 1.13. oksygenazy, enzymy wbudowujące atom lub dwa atomy tlenu do substratu, nie wymagają
dodatkowego donoru wodoru
EC 1.13.11. dioksygenazy- wbudowują dwa atomy tlenu
EC 1.13.12. monooksygenazy – wbudowują jeden atom tlenu
EC 1.14. oksygenazy, enzymy wbudowujące atom lub dwa atomy tlenu do substratu, wymagają
dodatkowego donoru wodoru
Schematycznie, reakcje katalizowane przez te enzymy można przedstawić następująco:
EC 1.14.11. dioksygenazy, DH
2
= 2-ketoglutran
EC 1.14.12. dioksygenazy, DH
2
= NADH
EC 1.14.13. monooksygenazy, DH
2
= NADH
EC 1.14.14. monooksygenazy, DH
2
= FADH
2
lub zredukowany FMN
EC 1.14.15. monooksygenazy, DH
2
= zredukowane żelazowo-siarkowe białka
EC 1.14.16. – EC 1.14.20. monooksygenazy, DH
2
= inne zredukowane związki
EC 1.15. działające na ponadtlenki jako akceptory
EC 1.16. utleniające jony metali
EC 1.17. działające na grupy =CH
lub –CH
2
jako donory
EC 1.18. działające na siarkowo-żelazowe białka jako donory
EC 1.19. – EC 1.21. działające na inne związki jako donory
EC 1.97. inne oksydoreduktazy
W klasie oksydoreduktaz trzeci człon numeru - czyli pod-podklasa - wskazuje, co jest donorem lub
akceptorem atomów wodoru względnie elektronów. Poniżej podano kilka przykładów pod-podklas z
klasy oksydoreduktaz.
EC 1.x.1. akceptorem wodoru jest NAD lub NADP,
dehydrogenazy i reduktazy
EC 1.x.2. akceptorem elektronów są cytochromy
EC 1.x.3. akceptorem wodoru jest tlen cząsteczkowy,
oksydazy
Oksydazy są flawoproteidami (np. z FAD), metaloflawoproteidami bądź hemoproteidami. Znane są
dwa typy oksydaz: oksydazy typu I wytwarzają podczas działania wodę, oksydazy typu II wytwarzają
nadtlenek wodoru.
XH
X-OH
O
2
DH
2
D
H
2
O
mechanizm działania
monooksygenazy z podklasy EC 1.14.
XH
2
O
2
DH
2
D
mechanizm działania
dioksygenazy z podklasy EC 1.14.
X
OH
OH
XH
2
O
2
H
2
O
(koenzym)
X
1/2
oksydazy typu I
XH
2
O
2
(koenzym)
X
oksydazy typu II
H
2
O
2

57
Przykładem oksydazy typu I jest oksydaza cytochromu-c (EC 1.9.3.1), a przykładem oksydazy typu II
oksydaza glukozowa (EC 1.1.3.4).
EC 1.x.4. akceptorem są związki disulfidowe (min. liponian) dehydrogenazy (dekarboksylujące)
EC 1.x.5. akceptorem wodoru jest ubichinon
dehydrogenazy
EC 1.x.7. akceptorem są żelazo-siarkowe białka
EC1.x.99.z innym akceptorem (najczęściej z FAD)
dehydrogenazy
Transferazy. W tej klasie enzymów drugi człon numeru - czyli podklasa - wskazuje, jaką grupę
funkcyjną transportuje dana transferaza. Trzeci człon numeru – pod-podklasa – uściśla o jaki rodnik
chodzi lub wskazuje akceptor przenoszonej grupy.
EC 2. Transferazy
EC 2.1. transportujące grupy jednowęglowe (C
1
)
EC 2.1.1. metylotransferazy (
CH
3
)
EC 2.1.2. hydroksymetylo- i formylotransferazy (
CH
2
OH, CHO)
EC 2.1.3. karboksy
i karbamoilotransferazy
EC 2.1.4. amidynotransferza (
CONH
2
)
EC 2.2. transportujące grupy aldehydowe lub ketonowe
EC 2.3. Acylotransferazy
EC 2.3.1. transportujące grupy inne niż acetylo-aminowe
EC 2.3.2. aminoacylotransferazy
EC 2.3.3. acylowe grupy przekształcane na alkilowe rodniki podczas transportu
EC 2.4. glikozylotransferazy
EC 2.4.1. heksozylotransferazy
EC 2.4.2. pentozylotransferazy
EC 2.4.99 transportujące inne glikozylowe grupy
EC 2.5. transportujące alkilowe lub arylowe grupy, inne niż metylowe grupy
EC 2.6. transportujące grupy z azotem
EC 2.6.1. Transaminazy
aminotransferazy
EC 2.6.2. Oksymotransferazy
EC 2.6.99. transportujące inne grupy z azotem
EC 2.7. transportujące grupy zawierające fosfor
EC 2.7.1. Fosfotransferazy z grupą alkoholową jako akceptorem
kinazy
EC 2.7.2. Fosfotransferazy z grupą karboksylową jako akceptorem
kinazy
EC 2.7.3. Fosfotransferazy z grupą z azotem jako akceptorem
kinazy
EC 2.7.4. Fosfotransferazy z grupą fosforową jako akceptorem
kinazy
EC 2.7.6. Difosfotransferazy
difosfokinazy
EC 2.7.7. Nukleotydilotransferazy
EC 2.7.8. Transferazy dla innych pochodnych grup fosforowych
EC 2.7.9. Fosfotransferazy z parą akceptorów
dikinazy
EC 2.8. transportujące grupy zawierające siarkę
EC 2.8.1. Siarkotransferazy
EC 2.8.2. Siarczanotransferazy
EC 2.8.3. CoA-transferazy
EC 2.8.4. transportujące grupy tioalkylowe
EC 2.8. transportujące grupy zawierające selen
Hydrolazy W tej klasie enzymów drugi człon numeru (podklasa) wskazuje, jakiego rodzaju
wiązanie ulega hydrolizie. Trzeci człon numeru – pod-podklasa – uściśla typ hydrolizowanego
wiązania.
EC 3.Hydrolazy
EC 3.1. działające na wiązanie estrowe
EC 3.1.1. Hydrolazy estrów karboksylowych
esterazy, lipazy i laktonazy
EC 3.1.2. Hydrolazy tioestrów
EC 3.1.3. Hydrolazy monoestrów fosforowych
fosfatazy i nukleotydazy
EC 3.1.4. Hydrolazy diestrów fosforowych
fosfolipazy i fosfodiesterazy
EC 3.1.5. Hydrolazy monoestrów trifosforowych
EC 3.1.6. Hydrolazy estrów kwasu siarkowego
EC 3.1.7. Hydrolazy monoestrów difosforowych
difosfatazy
EC 3.1.8. Hydrolazy triestrów fosforowych
EC 3.1.11.
3.1.31. Egzo- i endorybonukleazy
EC 3.2. Glikozylazy

58
EC 3.2.1. Glikozydazy, i inne enzymy hydrolizujące O- i S-glikozyle
EC 3.2.2. hydrolizujące związki N-glikozylowe
EC 3.3. działające na wiązanie eterowe
EC 3.3.1. Hydrolazy tioeterowe i trialkilosulfonianowe
EC 3.3.2. Hydrolazy eterowe
EC 3.4. działające na wiązanie peptydowe
hydrolazy peptydowe
EC 3.4.11. Aminopeptydazy
EC 3.4.13. Dipeptydazy
EC 3.4.14. Dipeptydylo- i tripeptydylo-peptydazy
EC 3.4.15. Peptydylo-dipeptydazy
EC 3.4.16 Karboksypeptydazy serynowego typu
EC 3.4.17 Metalokarboksypeptydazy
EC 3.4.18 Karboksypeptydazy cysteinowego typu
EC 3.4.19 Omega peptydazy
EC 3.4.21. Endopeptydazy serynowe
EC 3.4.22. Endopeptydazy cysteinowe
EC 3.4.23. Endopeptydazy aspartylowe
EC 3.4.24. Metaloendopeptydazy
EC 3.4.25. Endopeptydazy treoninowe
EC 3.4.99. Endopeptydazy o nierozpoznanym mechanizmie działania
EC 3.5. działające na wiązanie C
N, inne niż peptydowe
EC 3.5.1. w linearnych amidach
m.in. amidazy i deacylazy
EC 3.5.2. w cyklicznych amidach
EC 3.5.3. w linearnych amidynach
EC 3.5.4. w cyklicznych amidynach
EC 3.6. działające na bezwodniki kwasowe
EC 3.6.1. w związkach zawierających fosforanowe bezwodniki
m.in. difosfatazy
EC 3.6.2. w związkach zawierających sulfonowe bezwodniki
EC 3.6.3. działające na bezwodniki kwasowe; katalizujące przenoszenie transmembranowe
EC 3.6.5. działające na GTP; związane z komórkowym transportem
EC 3.7. działające na wiązanie C
C
EC 3.7.1. w ketonowych substancjach
EC 3.8. działające na halogenkowe wiązania
EC 3.8.1. w związkach C
halogen
EC 3.9. działające na wiązanie fosforo-azotowe
EC 3.10. działające na wiązanie siarkowo-azotowe
EC 3.11. działające na wiązanie węglowo-fosforowe
EC 3.11. działające na wiązanie siarkowo-siarkowe
EC 3.11. działające na wiązanie węglowo-siarkowe
Liazy W tej klasie enzymów drugi człon numeru (podklasa) wskazuje, jakiego rodzaju wiązanie
ulega rozerwaniu. Trzeci człon numeru – pod-podklasa – uściśla typ rozrywanego wiązania.
EC 4. Liazy
EC 4.1. Liazy węgiel-węgiel
EC 4.1.1. karboksy-liazy
dekarboksylazy
EC 4.1.2. aldehydo-liazy
EC 4.1.3. liazy ketokwasów
EC 4.1.99. inne węgiel-węgiel liazy
EC 4.2. Liazy węgiel-tlen
EC 4.2.1. hydro-liazy
dehydratazy
EC 4.2.2. działające na polisacharydy
EC 4.2.3. działające na fosforany
syntazy
EC 4.2.99 inne węgiel-tlen liazy
EC 4.3. Liazy węgiel-azot
EC 4.3.1. amoniako-liazy
EC 4.3.2. liazy działające na amidy, amidyny, itp.
EC 4.3.3. amino-liazy
EC 4.3.99. inne węgiel-azot liazy
EC 4.4. Liazy węgiel-siarka
EC 4.5. Liazy węgiel-halogen
EC 4.6. Liazy fosfor-tlen
Izomerazy W tej klasie enzymów drugi człon numeru (podklasa) uściśla typ katalizowanej
reakcji. Trzeci człon numeru – pod-podklasa – wskazuje, jakie związki lub ugrupowania ulegają
izomeryzacji.
EC 5. Izomerazy

59
EC 5.1. Racemazy i epimerazy
EC 5.1.1. działające na aminokwasy i ich pochodne
EC 5.1.2. działające na hydroksykwasy i ich pochodne
EC 5.1.3. działające na węglowodory i ich pochodne
EC 5.1.99. działające na inne związki
EC 5.2. cic-trans izomerazy
EC 5.3. wewnątrzcząsteczkowe oksydoreduktazy
EC 5.3.1. wzajemnie przekształcające się aldozy i ketozy
EC 5.3.2. wzajemnie przekształcające się ugrupowania enolowe i ketonowe
tautomerazy
EC 5.3.3. przenoszące wiązania C=C
Δ-izomerazy
EC 5.3.4. przenoszące wiązania S
S
EC 5.3.99. inne wewnątrzcząsteczkowe oksydoreduktazy
EC 5.4.wewnątrzcząsteczkowe transferazy
mutazy
EC 5.4.1. transportujące grupy acylowe
EC 5.4.2. fosfotransferazy
fosfomutazy
EC 5.4.3. transportujące grupy aminowe
EC 5.4.99. transportujące inne grupy
EC 5.5. wewnątrzcząsteczkowe liazy
EC 5.99. inne izomerazy
Ligazy W tej klasie enzymów drugi człon numeru (podklasa) wskazuje, jakiego typu wiązanie jest
tworzone. Trzeci człon numeru – pod-podklasa – uściśla, jakiego typu związki lub ugrupowania
powstają w wyniku syntezy.
EC 6. Ligazy
EC 6.1. tworzące wiązanie węgiel-tlen
EC 6.1.1. ligazy tworzące aminoacylo-tRNA i spokrewnione związki
EC 6.2. tworzące wiązanie węgiel-siarka
EC 6.2.1. ligazy kwas-tiol
syntetazy acylo-CoA
EC 6.3. tworzące wiązanie węgiel-azot
EC 6.3.1. ligazy kwas-amoniak
syntetazy amidowe
EC 6.3.2. ligazy kwas- D-aminokwas
syntetazy peptydowe
EC 6.3.3. cyklo-ligazy
EC 6.3.4. inne ligazy tworzące wiązanie węgiel-azot
EC 6.3.5. ligazy wiązania węgiel-azot z glutaminą, jako amido-N donorem
EC 6.4. tworzące wiązanie węgiel-węgiel
EC 6.5. tworzące wiązanie fosforowoestrowe
EC 6.6. tworzące wiązanie azot-metal
chelatazy
Izoenzymy
Izoenzymy, to odmiany tego samego enzymu o identycznym centrum aktywnym i mechanizmie
działania, a różniące się w niewielkim stopniu sekwencją aminokwasów w łańcuchu polipeptydowym.
Konsekwencją tego mogą być pewne różnice właściwości fizyko-chemicznych i biologicznych takich
odmian enzymu, np.: różne pI, optymalne pH i temperatura działania, K
M
i odczyn immunologiczny.
Izoenzymy są umieszczane w Klasyfikacji i Nomenklaturze Enzymów pod tym samym numerem.
Zazwyczaj w komentarzu do danego enzymu wspomina się o jego odmianach i podaje odpowiednie
odnośniki literaturowe.
2.2.5. Czynniki wpływające na efektywność działania enzymów
Zaletą enzymów są łagodne pod względem temperatury, pH i ciśnienia warunki działania, co
obniża koszt procesów, w których uczestniczą oraz pozwala na zachowanie w otrzymanych
produktach cennych, a nieodpornych na drastyczne warunki obróbki, składników. Ponadto przebieg
procesów katalizowanych przez enzymy można łatwo regulować, zmieniając stężenie enzymu bądź
temperaturę lub pH środowiska reakcyjnego.
Do podstawowych czynników określających przebieg (kinetykę) reakcji enzymatycznych należą:
pH środowiska, temperatura, stężenie jonów, potencjał oksydoredukcyjny, obecność inhibitorów,
aktywatorów oraz stabilizatorów (substancji hamujących lub podwyższających efektywność działania
enzymu) i oczywiście tak istotne parametry procesu, jak stężenie enzymu i substratu, o czym mowa
była już wcześniej. Optymalne pH

60
0
20
40
60
80
100
0
2
4
6
8
10
12
pH
A
kt
yw
no
ść
w
zg
lę
dn
a
[%
]
pepsyna
metaloproteinaza
B.subtilis
subtilizyna
pH środowiska. Szybkość reakcji enzymatycznych w znacznym stopniu zależy od pH
środowiska. Każdy enzym wykazuje charakterystyczne dla siebie optimum pH. Na poniższym
rysunku przedstawiono wpływ pH na aktywność trzech endopeptydaz: pepsyny, metaloproteinazy z
Bacillus subtilis oraz subtilizyny.
Wpływ pH na aktywność trzech różnych endopeptydaz
Jak widać, każda z tych endopeptydaz działa w zupełnie innym zakresie pH: pepsyna w
środowisku kwaśnym (optymalne pH 1,8), metaloproteinaza w obojętnym (optymalne pH 7,3), a
subtilizyna w alkalicznym (optymalne pH 10,2). Związane jest to z wpływem stężenie jonów
hydronowych na konformację centrum aktywnego każdej z nich. Pepsyna, której aminokwasem
bezpośrednio działającym w centrum aktywnym jest kwas asparaginowy, wymaga jego formy
niezdysocjowanej (czyli grupy -COOH). Grupa taka w przewadze występuje w środowisku kwaśnym.
Endopeptydazy serynowe, których centrum aktywne opisano wyżej, wymagają do aktywnego
działania zdysocjowanej formy kwasu asparaginowego (grupy karboksylanowej –COO
-
), która
występuje w przewadze w środowisku alkalicznym.
Należy ponadto zwrócić uwagę na to, że o ile subtilizyna jest aktywna w szerokim zakresie pH od
6 do 11, to pozostałe dwie proteinazy działają w bardzo wąskim przedziale pH i niewielkie jego
zmiany wykazują znaczny wpływ na wzrost lub obniżenie ich aktywności. Optymalne pH niektórych
enzymów (np. aktywność subtilizyny) zmieniać się może w zależności od substratu na który działają,
choć są to wahania nie większe niż 0.5 jednostki (np. subtilizyna wobec hemoglobiny optimum pH
wykazuje przy 10,2, a wobec kazeiny 10,5).
Optymalna temperatura. Temperatura z jednej strony przyspiesza samą reakcję enzymatyczną, z
drugiej jednak przyspiesza denaturację enzymu,
co wiąże się z jego inaktywacją. Na poniższym
rysunku przedstawiono wpływ temperatury na
aktywność subtilizyny. Do osiągnięcia pewnej
granicznej wielkości (w tym przypadku
temperatury
60
C),
szybkość
reakcji
katalizowanej przez ten enzym wzrasta, podobnie
jak to się dzieje przy wszystkich reakcjach
chemicznych, a następnie dość gwałtownie spada
do wartości zerowych. Większość enzymów
drobnoustrojowych najefektywniej działa w
granicach 60
C. Enzymy roślinne i zwierzęce z
reguły już powyżej 40-50
C ulegają denaturacji. Znane są enzymy z bakterii termofilnych,
wykazujące optimum działania nawet w temperaturach sięgających 90
C.
Stabilność enzymów w roztworach wodnych. Jak już wspomniano pH środowiska i temperatura
bezpośrednio wpływają na szybkość reakcji enzymatycznej. Jednakże mogą również niekorzystnie
oddziaływać na sam enzym, prowadząc do jego inaktywacji. Dlatego niezbędna jest znajomość
wpływu tych dwóch czynników na aktywność enzymu w subtilizyny stabilność subtilizyny warunkach
rzeczywistych lub do nich zbliżonych, panujących podczas procesu prowadzonego z jego udziałem.
0
20
40
60
80
100
-20
0
20
40
60
80
Temperatura [
o
C]
A
kt
yw
no
ść
w
zg
lę
dn
a
[%
]
Wpływ temperatury na aktywność subtilizyny
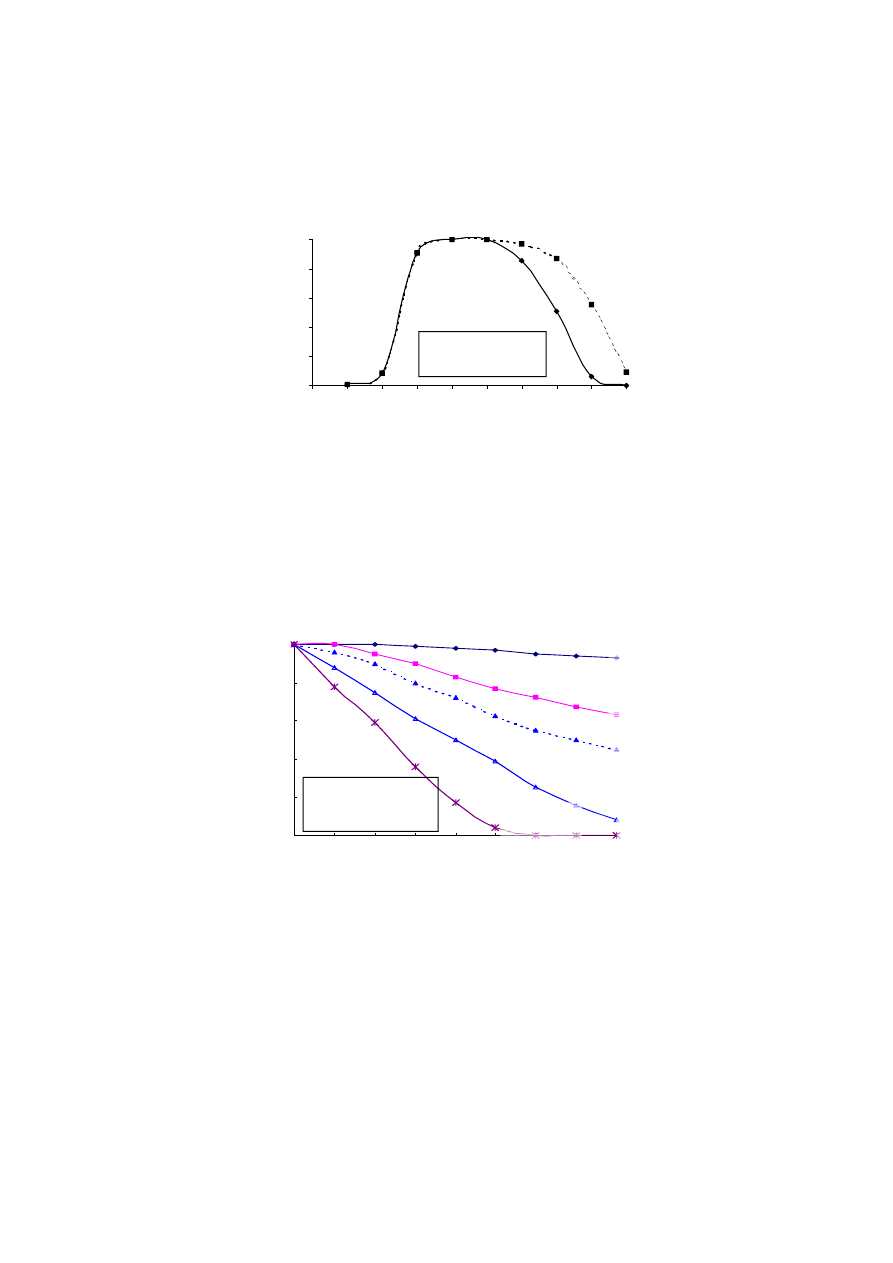
61
0
20
40
60
80
100
3
4
5
6
7
8
9
10
11
12
pH
A
kt
yw
no
ść
w
zg
lę
dn
a
[%
]
w buforze
Z dodatkiem
1% CaCl
2
Preinkubacja:
45
C, 60min
Większość enzymów jest względnie stabilna w obszarze pH zbliżonym dla ich optymalnego działania,
jakkolwiek nie jest to regułą. Na rysunku 3 przedstawiono wpływ pH na stabilność subtilizyny. Jej
optymalne pH (porównaj rys.1) przekracza 10, jednakże obszar stabilności tego enzymu w warunkach
przykładowej reakcji (1% kazeiny, 45 C, 60 min.) zawarty jest w zakresie 6.5-9.5, a w pH zbliżonym
do optymalnego straty aktywności (w wyniku postępującej autolizy) sięgają 50% wyjściowej.
Wpływ pH na stabilność subtilizyny w roztworze buforowym i z dodatkiem 1% CaCl
2
Dużo poważniejsze straty enzymu spowodowane mogą być wpływem podwyższonej temperatury
reakcji. Znaczna część enzymów jest bardzo wrażliwa na ten czynnik (wyjątkiem są niektóre enzymy
termostabilne otrzymywane ze szczepów drobnoustrojów termofilnych). Na rys.4 pokazano wpływ
temperatury na zachowanie się subtilizyny podczas przykładowego procesu hydrolizy kazeiny (1%
roztwór substratu w buforze o pH 9).
Wpływ temperatury na stabilność subtilizyny w roztworze buforowym i z dodatkiem 1%CaCl
2
Należy wyraźnie zaznaczyć, że subtilizyna (konsekwentnie w tekście niniejszego opracowania
rozpatrywana jako enzym modelowy) jest wyjątkowo odporna na działanie wielu czynników fizyko-
chemicznych, w tym temperatury. Tym niemniej w warunkach testu, po 40 minutach reakcji
prowadzonej w 60 C straty jej aktywności sięgają 70%. W tym samym czasie w temperaturze 70 C
enzym ulega już pełnej inaktywacji. Natomiast w 50
C po 60 minutach straty enzymu są stosunkowo
niewielkie i wynoszą 36%. Oczywiście, ostatecznego wyboru temperatury dokonuje się po
szczegółowej analizie wyników wszystkich badań, uwzględniającej aspekty technologiczne i
ekonomiczne procesu. Przy tego typu analizach przydatne jest wyznaczenie produktywności reakcji
enzymatycznej w kilku wytypowanych temperaturach (rys.5). Wybór optymalnej temperatury
skojarzony jest w tym przypadku z czasem procesu, który może być determinowany innym
czynnikiem (np. kosztem energii).
0
20
40
60
80
100
0
15
30
45
60
75
90
105
120
c z as [minuty ]
A
kt
yw
n
o
ść
w
zg
lę
d
n
a
[%
]
20
C
40
C
60
C
60
C
70
C
Preinkubacja:
w buforze o pH 9
Z dodatkiem
1%CaCl
2
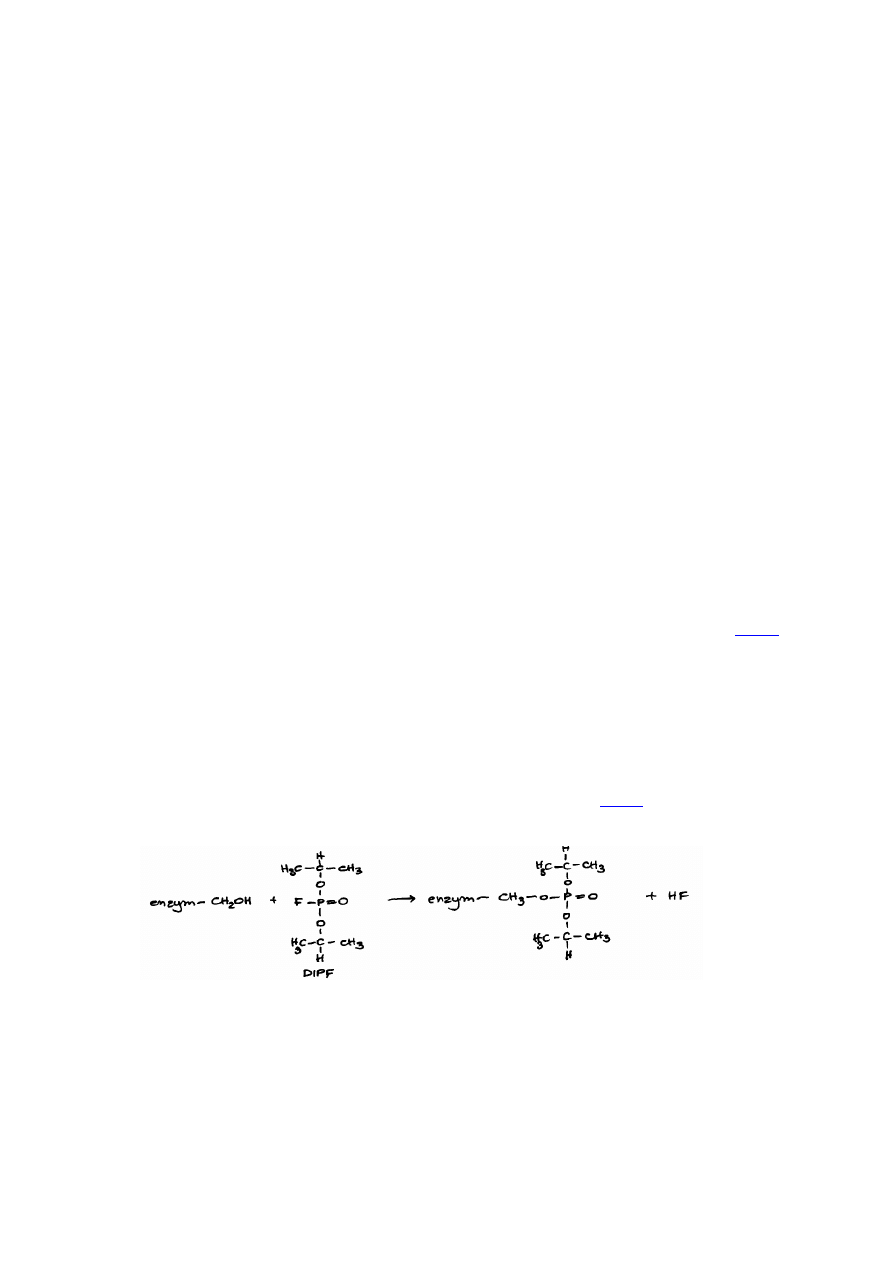
62
Rys. 5 Produktywność subtilizyny w hydrolizie kazeiny
Inhibitory, aktywatory, stabilizatory.
Oprócz wymienionych wyżej czynników inaktywujących działanie enzymów, istnieje grupa
substancji zdolnych do wybiórczego hamowania ich aktywności katalitycznej (są to tzw. inhibitory
specyficzne). W opracowaniu pominięto szczegóły dotyczące tego działu enzymologii. Należy jednak
wiedzieć, że tego typu naturalne inhibitory występują w przyrodzie, co może mieć istotne znaczenie
dla aplikacji enzymów w niektórych przypadkach. Przykładem mogą być specyficzne inhibitory
proteinaz serynowych (w tym subtilizyny) znalezione m.in. w ziemniakach czy soi, które skutecznie
blokują aktywność tej grupy enzymów.
Istnieje wiele typów cząsteczek , które są zdolne do zakłócania aktywności danego enzymu.
Każda cząsteczka działająca bezpośrednio na enzym w kierunku zmniejszenia jego aktywności
katalitycznej jest określana jako inhibitor. Pewne inhibitory enzymów są normalnymi metabolitami
komórkowymi, które hamują dany enzym w ramach naturalnej metabolicznej kontroli odpowiedniego
szlaku. Inne inhibitory mogą być substancjami obcymi dla organizmu, takimi jak toksyny i leki, i w
tym przypadku hamowanie enzymu może mieć działanie terapeutyczne , ale również
letalne
.
Rozróżnia się dwa główne typu inhibicji : - nieodwracalną - odwracalną.
Inhibicję odwracalną można podzielić na:
1) kompetycyjną
2) niekompetycyjną
Inhibicja nieodwrcalna polega na wiązaniu się inhibitora i enzymu w sposób trwały ,
nieodwracalny, często tworząc wiązania kowalencyjne z resztami aminokwasów, znajdującymi się w
miejscu aktywnym lub jego pobliżu i w ten sposób inaktywują enzym na stałe. W wiązaniu tym biorą
udział reszty Ser i Cys mające , odpowiednio, reaktywne grupy -OH i -SH . Przykładem może być
związek diizopropylofluorofosforan (DFP) , składnik gazów bojowych (
soman
) działający na układ
nerwowy, reaguje z resztą Ser w miejscu aktywnym enzymu esterazy acetylocholinowej,
nieodwracalnie hamując enzym i uniemożliwiając przekazywanie impulsów nerwowych.
Inhibitor kompetycyjny jest zazwyczaj strukturalnie podobny do normalnego substratu danego
enzymu. Dzięki temu współzawodniczy z cząsteczkami o wiązanie się z miejscem aktywnym. Enzym
może się wiązać albo z cząsteczkę substratu, albo cząsteczkę inhibitora, ale nie z obiema
jednocześnie. Inhibitor kompetycyjny wiąże się z miejscem aktywnym odwracalnie. Przy dużych
stężeniach substratu działanie inhibitora kompetycyjnego zostaje przezwyciężone , ponieważ duże
stężenie substratu będzie z powodzeniem współzawodniczyć z cząsteczką inhibitora o wiązanie się w
miejscu aktywnym. Nie nastąpi więc żadna zmiana w wartości V
max
enzymu , ale w obecności
inhibitora kompetycyjnego zmniejsza się powinowactwo enzymu do jego substratu i dlatego wratośc
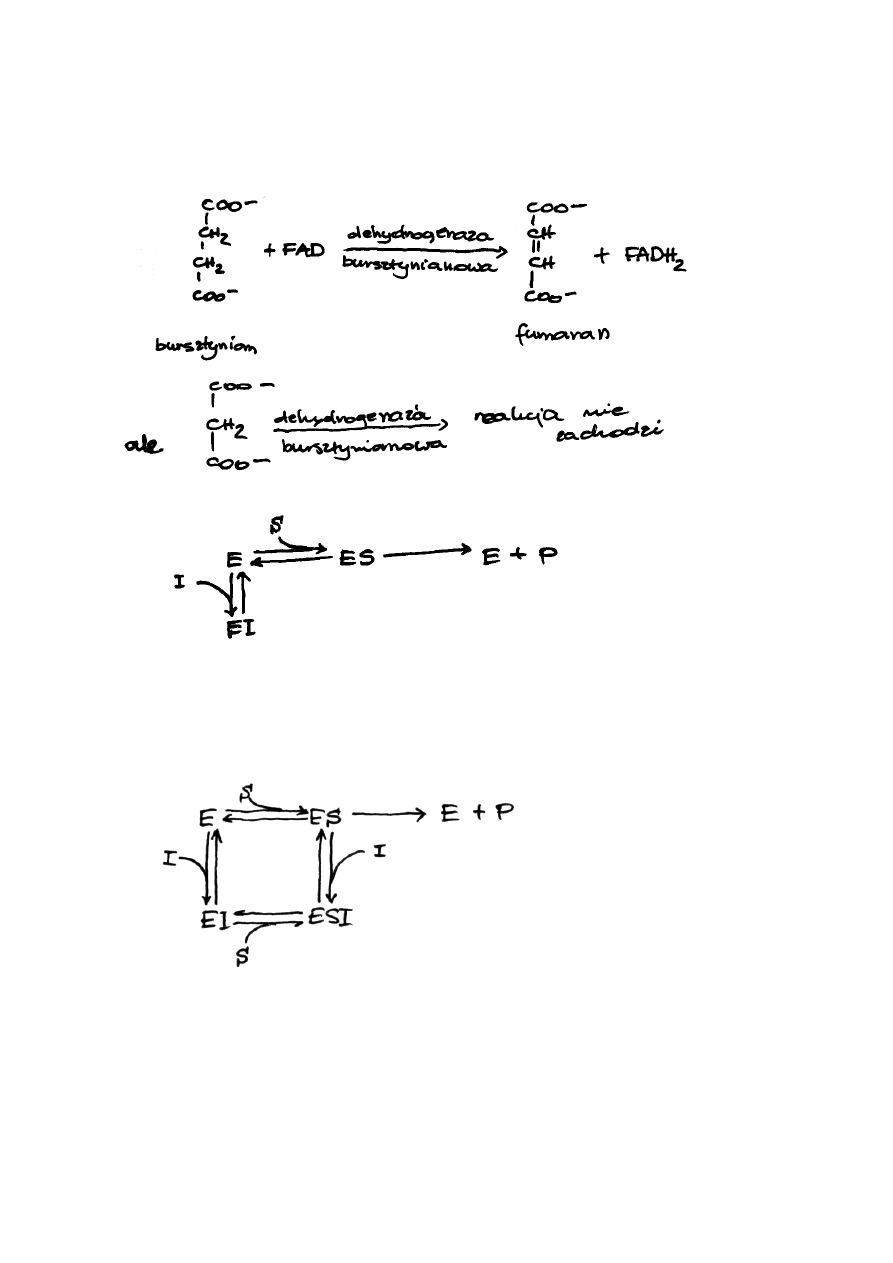
63
K
m
wzrasta. Przykładem hamowania kompetycyjnego może być dehydrogenaza bursztynianowa.
Enzum ten używa bursztyniany jako substratu i jest hamowany kompetycyjnie przez malonian, który
różni się od bursztynianu posiadniem tylko jednej, a nie dwóch grup metylenowych.
Inhibitor niekompetycyjny wiąże się odwracalnie w innym miejscu enzymu niż jego miejsce
aktywne i powoduje zmianę przestrzennego kształtu enzymu, co prowadzi do zmniejszenia
aktywności katalitycznej. Ponieważ inhibitor wiąże się w innym miejscu niż substrat, enzym może
wiązać albo inhibitor, albo substrat równocześnie. Efektu inhibitora niekompetycyjnego nie można
przezwyciężyć przez zwiększanie stężenia substratu i dlatego zmniejsza się wartość V
max
. W inhibicji
niekompetycyjnej powinowactwo enzymu do substratu pozostaje nie zmienione, a więc wartość K
m
nie zmienia się . Przykładem inhibicji niekompetycyjnej jest działanie pepstatyny na enzym reninę .
Na działanie enzymów wpływ mogą wywierać jony obecne w środowisku reakcyjnym. Jony
dwuwartościowych metali, jak magnez, wapń, cynk, mangan lub kobalt w specyficzny sposób
aktywują wielu enzymów (t.zn. zwiększa- ją szybkość reakcji przez nie katalizowanej). Aktywatorami
mogą być też aniony np. amylaza ślinowa jest aktywowana jonami Cl . Jony metali mogą wykazywać
również wpływ stabilizujący na enzym (tzn. podwyższać jego stabilność wobec niekorzystnego
wpływu m.in. pH i temperatury). Przy- kładem może być wpływ jonów Ca na subtilizynę. Na rys. 3 i
4 pokazano, że jony te podwyższają termostabilność subtilizyny (z 50 do 60 C) i jej trwałość w
alkalicznym środowisku (z pH 9.5 do 10.5). Natomiast jony wielu metali ciężkich (np. Hg, Cu, Pb) są
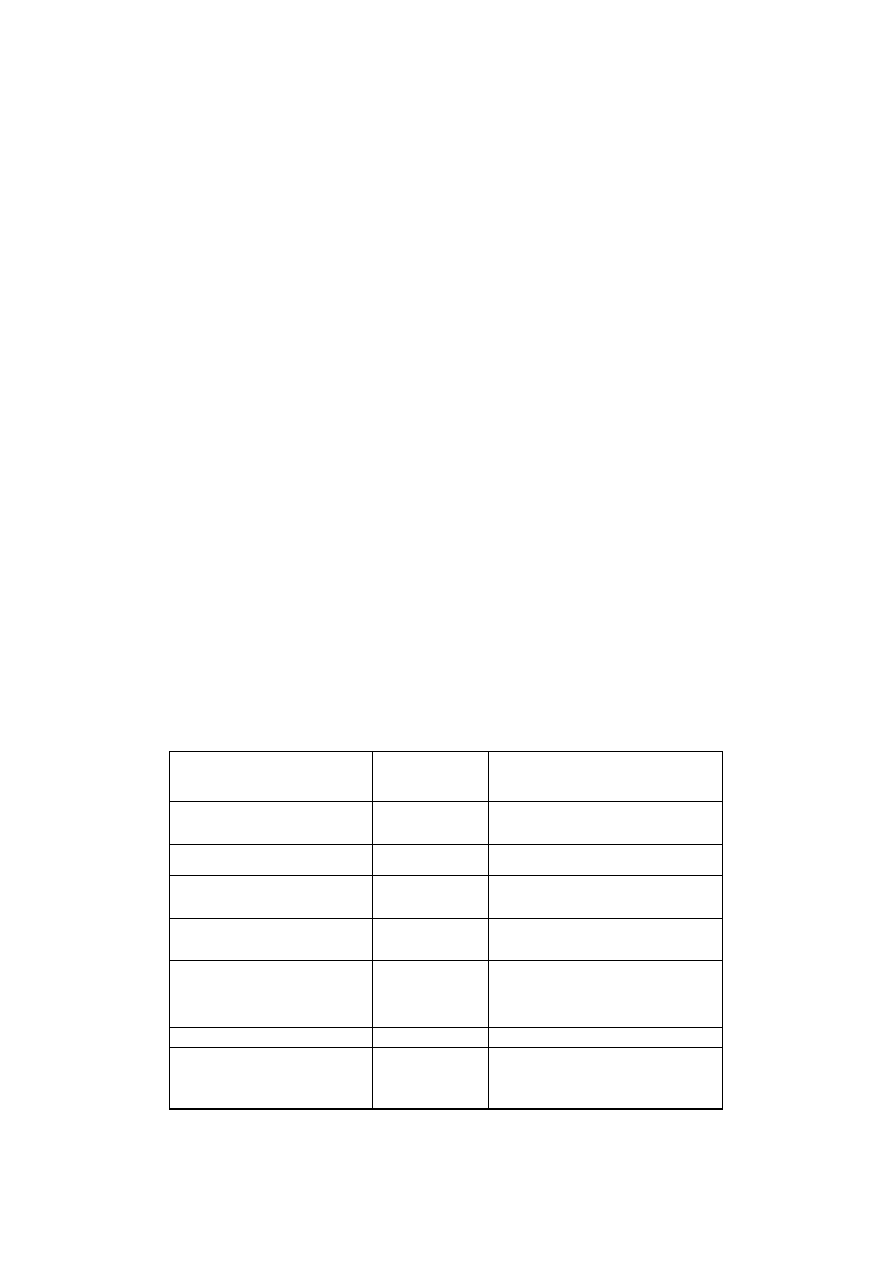
64
silnymi inhibitorami enzymów (tzn. inaktywują one enzym, tym samym hamując reakcję
przebiegającą przy jego udziale). Jest to inhibicja niespecyficzna, której podlegają wszystkie znane
enzymy. Istnieje ponadto wiele innych substancji mających zdolność niespecyficznej inaktywacji
enzymów (związanej ze zniszczeniem naturalnej struktury białka). Do nich zalicza się mocznik,
aldehyd mrówkowy czy glutarowy, silne utleniacze (np. nadmanganian lub chlor).
Koncentracja wody.
W praktyce nie wszystkie reakcje katalizowane przez enzym przebiegają do końca. Należy
bowiem pamiętać, że są one odwracalne i po pewnym czasie ustala się równowaga pomiędzy reakcją
przebiegającą do "przodu" i odwrotną. Jednym z czynników wpływających na ich stałą równowagi
może być stężenie wody w środowisku reakcyjnym. Dotyczy to zwłaszcza prze- mian, w których
woda jest jednym z reagentów (np. w reakcjach enzymatycznej hydrolizy). Wykazano, że stężenie
wody decyduje o kierunku ich przebiegu. I tak, subtilizyna w środowisku wodnym katalizuje
hydrolizę wiązań peptydowych w białkach lub wiązań estrowych w licznych estrach, natomiast w
środowisku bezwodnym lub o obniżonej koncentracji wody katalizuje rewersję tych reakcji. Dla
pożądanego przesunięcia stałej równowagi takich reakcji stosuje się rozmaite zabiegi. M.in. rewersję
enzymatycznej hydrolizy (czyli syntezę) osiąga się, stosując w środowisku reakcyjnym
rozpuszczalniki organiczne, prowadząc proces w układzie dwufazowym (z rozpuszczalnikiem
apolarnym nasyconym wodą), korzystnie z immobilizowaną formą enzymu itp. Obecnie jest to jeden z
najbardziej preferowanych kierunków badawczych, m.in. z uwagi na potencjalnie możliwe do
uzyskania efekty (również o charakterze aplikacyjnym w wielu procesach przemysłowych).
Charakterystyka biokatalizatorów przemysłowych
Termin "biokatalizatory przemysłowe" (używany obecnie w bardzo szerokim znaczeniu) oznacza
katalizatory otrzymywane w skali przemysłowej na drodze biologicznej. Pod tym pojęciem rozumie
się zarówno enzymy: natywne, spreparowane rozmaitymi technikami (np. ich immobilizowane
formy), ich mikstury, rozmaite proszki zawierające ich dodatek, jak i komórki: całe lub ich fragmenty,
żywe i martwe.
Enzymy wyodrębniane są z tkanek zwierzęcych lub roślinnych oraz wytwarzane przez
drobnoustroje. Te pochodzące z dwóch pierwszych wymienionych źródeł występują na rynku
stosunkowo w małych ilościach i są względnie drogie. Niektóre z nich: renina, pepsyna, trypsyna,
papaina i bromelaina są ekstrahowane ze spożywczych surowców.
Tabela 1 Enzymy produkowane przez mikrobiologiczną fermentację (adaptowane z Trampera, 1994).
Enzym
Substrat
Mikroorganizm
-amylaza
Skrobia
Bacillus amyloliquefaciens,
Bacillus licheniformis
-amylaza
Skrobia
Bacillus polymyxa
Amyloglukozydaza
(glukoamylaza)
Dekstryny
Aspergillus niger
Rhizopus niveus
Celulazy
Celuloza
Phanerochaete chrysosporium
Trichoderma reesei
Izomeraza glukozowa
Glukoza
Actinoplanes missouriensis
Streptomyces murinus
Streptomyces olivochromogenus
Oksydaza glukozowa
Glukoza
Aspergillus niger
-glukozydaza
Maltoza
Aspergillus niger
Bacillus amyloliquefaciens
Saccharomyces cerevisiae
65
Lipazy
Lipidy
Aspergillus niger
Candida rugosa
Geotrichum candidum
Rhizopus arrhizus
Pektynoesteraza
Pektyna
Aspergillus niger
Aspergillus oryzae
Proteinaza alkaliczna
(serynowa)
Białka
Aspergillus oryzae
Bacillus licheniformis
Bacillus subtilis
Proteinaza kwaśna
(pepsyno-podobna)
Białka
Aspergillus oryzae
Aspergillus saitoi
Proteinaza kwaśna (renino-
podobna)
białka
Endothia parasitica
Mucor michei
Mucor pusillus
Proteinaza obojętna białka
Bacillus stearothermophilus
Bacillus subtilis
Pullulanaza
Amylopektyna
Aerobacter aerogenes
Bacillus cereus var.mycoides
W wyniku rozwoju biotechnologii, mikroorganizmy stanowią podstawowe źródło enzymów.
Zastosowanie mutacji i specjalnych technik selekcji drobnoustrojów pozwoliło osiągnąć wysoki
poziom produkowanych przez nie enzymów, w niektórych przypadkach sięgający 10% wszystkich
wytwarzanych białek . Ponadto użycie dużych fermentorów (do 300 m ) sprawia, że obecnie w
krótkim czasie i przy niskich kosztach produkuje się na skalę przemysłową olbrzymie ilości
rozmaitych enzymów (tabela 1).
Niezależnie od źródła pochodzenia enzymy mogą katalizować te same reakcje chemiczne, ale nie
muszą posiadać identycznej budowy chemicznej, a tym samym mogą znacznie różnić się optymalnymi
warunkami działania. I tak na przykład α -amylaza katalizuje hydrolizę wiązań ŕ-1,4- między
cząsteczkami D-glukozy w polisacharydach, takich jak amyloza. α-amylaza trzustkowa posiada
optymalne pH około 7 i optymalną temperaturę 37 C, podczas gdy enzym ten wyodrębniony z
Aspergillus oryzae - optymalne pH 4,7 i optymalną temperaturę działania 50 C, a z Bacillus subtilis
odpowiednio pH 6,5 i 75 C. W zasadzie te dwa parametry (pH i temperatura) decydują o możliwości
zastosowania enzymów w procesach produkcji środków spożywczych przebiegających w różnych
warunkach (np. w różnych gałęziach przemysłu spożywczego). Fakt ten tłumaczy występowanie na
rynku rozmaitych preparatów homologicznych enzymów. Należy jednak mieć na uwadze, że tylko
część drobnoustrojów i wytwarzanych przez nie bioproduktów (w tym enzymów) traktowana jest jako
nieszkodliwa dla stosowania w środkach spożywczych (tabela 2).
W ogólnym zarysie proces produkcji enzymów drobnoustrojowych polega na hodowli
wyselekcjonowanych kultur mikroorganizmów na specjalnie opracowanych dla każdej z nich
pożywkach, z zachowaniem specyficznych warunków fizycznych środowiska hodowlanego, a w
następnym etapie wyodrębnieniu wytworzonego enzymu. Jeśli enzym jest nagromadzany
pozakomórkowo, to można go wyodrębnić z podłoża pohodowlanego na drodze wirowania lub
filtracji. Filtrat lub supernatant poddaje się ogólnie stosowanym proce- durom wydzielania i
oczyszczania białek. Jeśli zanieczyszczenia zawarte w preparacie nie działają ujemnie na własności
katalityczne enzymu oraz nie stanowią toksykologicznego zagrożenia dla konsumenta nie zachodzi
konieczność poddawania ich skomplikowanym i drogim procesom oczyszcza- nia. W dużej mierze
przeznaczenie enzymu decyduje o stopniu czystości jego handlowych preparatów. Ogólnie biorąc,
preparaty enzymów wykorzystywane dla celów analitycznych charakteryzują się najwyższą czystością
(niekiedy są one homogennym białkiem enzymatycznym). Również enzymy stosowane w farmacji
posiadają wysoką czystość. Natomiast te same enzymy przeznaczone dla przemysłu spożywczego nie
wymagają już tak wysokiego stopnia czystości. Dla niektórych technologii nie muszą być nawet
wyjaławiane. Coraz częściej dla konkretnego procesu wytwarza się specjalnie przeznaczony preparat
enzymu, dobierając jego aktywność tak, aby dodana ilość wynosiła 0.05-0.5 % na kg substratu.

66
Tabela 1 Mikroorganizmy używane dla produkcji enzymów (adaptowane z Gacesa i Hubble, 1987).
Grupa A
Mikroorganizmy tradycyjnie używane w produkcji żywności (nie wymagające
testowania)
Bacillus subtilis (włączając szczepy znane jako mesentericus, natto i
amyloliquefaciens),
Aspergillus niger (włączając awamori, foetidus, phoenicis, saitoi i usamii),
Asp. oryzae (włączając sojae i effesus)
Mucor javanicus
Rhizopus arrhizus, oligosporus, oryzae
Saccharomyces cerevisiae
Kluyveromyces fragilis, lactis
Leuconostoc oenos
Grupa B
Mikroorganizmy uważane za nieszkodliwe w żywności (enzymy wytwarzane
przez nie, testowane są jedynie pod kątem ewentualnej toksyczności)
Bacillus stearothermophilus, licheniformis, coagulans, megaterium, circulans,
Klebsiella aerogenes
Grupa C
Mikroorganizmy nie włączone do grup A i B (ewentualne stosowanie enzymów
produkowanych przez nie w produkcji żywności, wymaga specjalistycznych
testów)
Mucor miehei, pusillus
Endothia parasitica
Actinoplanes missouriensis
Streptomyces albus, olivaceus
Bacillus cereus
Trichoderma reesei (T. viride)
Penicillium dimorphosporum, simplicissium, funiculosum
Zwykle handlowe preparaty zawierają od 2 do 10% suchej masy czystego enzymu. Reszta to
zanieczyszczenia pochodzące z podłoża hodowlanego (np. inne białka wytworzone przez rosnącą
kulturę, niektóre z nich będące również enzymami) oraz celowo dodane stabilizatory, wypełniacze itp.
Z aplikacyjnego punktu widzenia istotna jest zwłaszcza wiedza na temat enzymów stanowiących
zanieczyszczenie handlowych preparatów. Niektóre z nich mogą bowiem katalizować niekorzystne,
uboczne reakcje chemiczne. Niekiedy jednak gotowe preparaty celowo są mieszaniną kilku enzymów
(np. preparaty typu "Protogal" przeznaczone dla detergentów, a otrzymywane w oparciu o technologię
opracowaną w Instytucie Biochemii Technicznej PŁ zawierają w swoim składzie proteinazę, ŕ-
amylazę i lipazę- wszystkie trzy przydatne w środkach piorących).
Przeznaczenie preparatu enzymatycznego określa jego postać końcową. Mogą być one
otrzymywane w postaci ciekłego koncentratu lub ciała stałe- go, o różnym stopniu oczyszczenia,
ewentualnie z dodatkiem stabilizatorów. Preparaty enzymatyczne w stanie stałym charakteryzują się
znacznie wyższą stabilnością od preparatów ciekłych. Te drugie podatne są na działanie
drobnoustrojów, a ponadto w środowisku wodnym enzym ulega łat- wiej denaturacji i dodatkowo
narażony jest na działanie enzymów proteolitycznych, których obecność w gotowych preparatach
(choćby w nieznacznych ilościach) jest dość powszechna. Preparaty stałe enzymu nie powinny być
pyliste (z uwagi na możliwość wywołania reakcji alergicznych u pracowników), stąd najczęściej
wytwarzane są w postaci granulowanej.
Zasadniczym przełomem w biotechnologii było zastosowanie enzymów immobilizowanych. W
uogólnieniu, immobilizacją enzymu można nazwać za- biegi umożliwiające wielokrotne jego użycie.
Jeden ze sposobów immobilizacji polega na unieruchomieniu enzymu na nośniku. Takie formy
enzymów w wielu przypadkach wykazują wyższą stabilność od natywnych enzymów, zarówno w
67
trakcie przechowywania jak i w warunkach samej reakcji bio- chemicznej. Jednakże największa
korzyść związana jest z możliwością ich wielokrotnego stosowania, w tym w procesach o charakterze
ciągłym (np. w reaktorze o działaniu ciągłym z immobilizowaną izomerazą glukozową produkuje się
wysoko fruktozowy syrop). Metody stosowane w immobilizacji enzymów mogą być również
wykorzystane w unieruchamianiu komórek zarówno martwych jak i żyjących. Unieruchomione
biokatalizatory (enzymy i komórki) są szczególnie obiecujące w zastosowaniu do procesów
przetwórstwa żywności. Chemiczne katalizatory nie mogą konkurować z enzymami, chociażby ze
względu na przepisy sanitarne obowiązujące w produkcji żywności. Najnowszym kierunkiem badań w
tym zakresie jest ko-immobilizacja enzymów, czyli jednoczesne unieruchomienie na wspólnym
nośniku dwu lub więcej enzymów przeznaczonych do katalizowania pojedynczego procesu.
Cena preparatów enzymatycznych jest bardzo zróżnicowana (od 3 $/kg w przypadku preparatu
glukoamylazy przeznaczonego do scukrzania dekstryn, aż do 1 miliona $/kg w przypadku czynnika
VIII enzymów trombolitycznych stosowanych w medycynie). Zależy ona od wiele czynników, w tym
tak oczywistych jak koszty produkcji (a głównie stopień oczyszczenia enzymu), ale także w dużej
mierze zależy od rynkowego zbytu, konkurencyjności i skuteczności działania (ich produktywności w
aplikacyjnych warunkach). Ogólnie biorąc, najdroższe są enzymy przeznaczone dla analityki, a naj-
tańsze stosowane masowo w różnych gałęziach przemysłu (tabela 3). Przyjmuje się ponadto, że koszt
aplikacyjny enzymów przemysłowych powinien stanowić 2-4 % kosztów całego procesu, co w pewien
sposób determinuje ich cenę.
Tabela 2 Wielkość produkcji enzymów i ich cena (adaptowane z Trampera, 1994)
Typ aplikacji
Wielkość produkcji (tony)
Cena ($/kg)
Analityka
10
-3
- 1
10
6
– 10
9
Farmacja
1 – 10
2
10
2
– 10
5
Specjalna
1 – 10
2
10
2
– 10
5
Przemysł masowy
10
2
– 10
4
3 – 50
Potencjał aplikacyjny enzymów tkwi w specyficzności i ich katalitycznej wydajności, przy czym
działają one w umiarkowanych warunkach pH, temperatury i ciśnienia zapewniając wysoką
wydajność nawet w skomplikowanych reakcjach lub ich ciągach. Jednakże z aplikacyjnego punktu
widzenia dla każdego konkretnego procesu i enzymu w nim biorącego udział konieczna jest
skojarzona optymalizacja temperatury, pH środowiska reakcyjnego, stężenia reagentów oraz innych
parametrów technologicznych. Niepowodzenia napotykane niekiedy w stosowaniu enzymów w
krajowych technologiach, o których się nieraz słyszy, wynikać mogą z niepełnej wiedzy o
właściwościach samych enzymów, ich preparatów lub procesach, w których biorą udział.
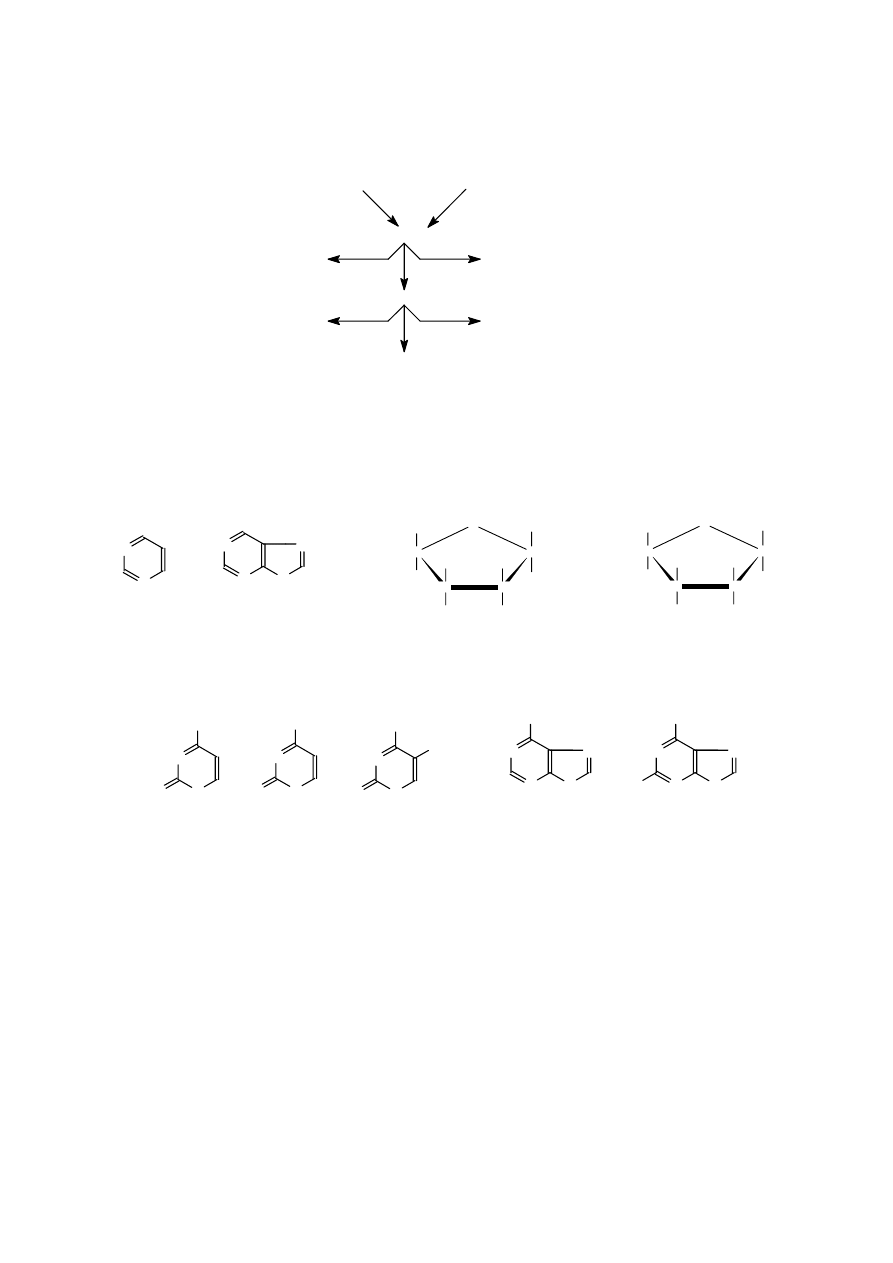
68
ROZDZIAŁ 3. BIOCHEMIA GENÓW
RNA
Nukleotyd
DNA
Nukleozyd
H
3
PO
4
H
3
PO
4
ryboza
deoksyryboza
zasady purynowe
i pirymidynowe
C
O
C
C
C
OH
H
OH
H
HOH
2
C
H
H
OH
3'
4'
5'
2'
1'
C
O
C
C
C
OH
H
H
OH
H
HOH
2
C
H
H
3'
4'
5'
2'
1'
D-ryboza
D-deoksyryboza
N
N
pirymidyna
N
N
N
N
1
1
2
2
3
3
4
4
5
6
7
8
9
H
5
6
puryna
N
N
NH
2
O
H
cytozyna
N
N
O
OH
N
N
O
OH
CH
3
H
H
uracyl
tymina
N
N
N
N
NH
2
H
N
N
N
N
OH
H
2
N
H
adenina
guanina

69
Biochemia ma olbrzymie znaczenie praktyczne dla wielu dziedzin życia codziennego, w tym
również w przemyśle spożywczym. Powstały nowe gałęzie przemysłu spożywczego oparte na
osiągnięciach biochemii (m.in. biotechnologia). Wielka rola przypada biochemii w unowocześnianiu
procesów technologicznych przemysłu spożywczego oraz w opracowaniu nowych metod przeróbki
surowców (a zwłaszcza wykorzystania enzymów). Dzięki temu udaje się zredukować do minimum
różnorodne straty zarówno ilościowe, jak i jakościowe, wynikające z procesów niepożądanych dla
technologii.

70

71
PISMIENNICTWO
Stryer L.: Biochemia, PWN, W-wa 1986
Gacesa P., Hubble J.: Enzyme technology. Open University Press, 1987.
Chaplin M.F., Bucke C.: Enzyme technology. Cambridge University Press, 1990.
Chmiel A., Biotechnologia, PWN, W-wa 1991
Murray R.K., Granner D.K., Mayes P.A., Rodwell V.W.: Biochemia Harpera, Wyd. Lekarskie
PZWL, W-wa 1994
Tramper J. W: Applied Biocatalysis. ed.by Cabral J.M.,Best D.,Boross L., and Tramper J.,
Harwood Ac. Publish., 1994, s.1-45
Galas E.: Wiadomości chemiczne (1984), 11, 11-30
Galas E., Trzmiel T.: Kosmos (1989), 38, 15-24
72
dinukleotyd nikotynamidoadeninowy (NAD)
i jego fosforan (NADP)
P
P
+
N
CONH
2
HO
OH
CH
2
O
O
O
OH
OH
OH
2
C
(
P
)
N
N
N
N
NH
2
dinukleotyd flawinoadeninowy (FAD)
N
N
N
NH
H
3
C
H
3
C
CH
2
O
O
CHOH
CHOH
CHOH
CH
2
O
P
adenozyna
P
C
Enzym-NH
CH
2
CH
2
CH
2
CH
2
CH
CH
2
S
CH
2
S
O
liponian
ubichinon (Q)
O
O
H
3
C
H
3
C
CH
3
)
(
n H
N
CH
2
CH
3
CH
HC
CH
N
CH
3
CH
CH
2
N
CH
2
CH
2
HOOC
H
3
C
Fe
+3
CH
3
HOOC CH
2
CH
2
HC
CH
N
hemina (porfiryna)
O
OH
OH
N
N
N
N
NH
2
CH
2
S
CH
2
CH
3
CH
2
CHNH
2
COOH
+
adenozynometionina
N
N
N
N
OH
H
2
N
CH
2
C
NH
NH
O
CH
COOH
CH
2
CH
2
COOH
H
H
kwas tetrawodorofoliowy (H
4
folian)
N
N
H
3
C
NH
2
CH
2
N
S
CH
2
CH
3
CH
2
O
P
P
+
difosfotiamina (DPT)
N
H
3
C
CH
2
OH
HO
C
O
H
pirydoksal
HN
NH
O
S
C
O
NH-Enzym
biotyna
adenozyno-5`-fosforan
koenzym A (CoA-SH)
P
P
O CH
2
C
CH
3
CH
3
CH
OH
C
O
NH CH
2
CH
2
SH
Dodatek
S
Lip
<
S
Wyszukiwarka
Podobne podstrony:
# Skrypt Biochemia czesc2(1)
# Skrypt Biochemia czesc4 id 30 Nieznany
# Skrypt Biochemia czesc1
# Skrypt Biochemia czesc3(1)
# Skrypt Biochemia spis tresci
# Skrypt Biochemia spis tresci
# Skrypt Biochemia czesc1
Biochemia skrypt AGH
BIOCHEMIA SKRYPT
BIOCHEMIA skrypt 2010 id 86508 Nieznany
chemia zywności wykłady, Zachomikowane, Naukowe, Medycyna, Biochemia, Skrypty
Biochemia zwierząt skrypt UR
biochemia skrypt aminokwasy i bia id 86612
Skrypt - Rozdzial 2 Izolowanie i amplifikacja kwasów nukleinowych CALOSC, materiały medycyna SUM, bi
Kwasy nukleinowe nowy skrypt, Studia, UR OŚ, semestr III, biochemia
więcej podobnych podstron