
Metody badań minerałów i skał – wykłady
Całość została napisana w darmowym pakiecie biurowym
Każda pomoc i uwagi będą mile widziane, wszelkie skargi natomiast ignorowane.
Wykład 1 Ilnicki 1 4.10.2007
„Elektronowe mikroskopy – transmisyjne TEM i skaningowe SEM”
Literatura:
1) „Metody Badań minerałów i skał” Praca zbiorowa. Wydawnictwa geologiczne.
2) „Phisicochemical methods of mineral analysis” Plenum Press
3) „Minerals and reactions at the atomic scale: transmission electron microscopy
Reviews in Mineralogy vol. 27
około setnego roku były znane już prototypy szkieł powiększających
ok. 1284 roku pierwsze okulary
1590 rok – Zacharias Jannsen – prototyp mikroskopu i teleskopu.
Robert Hooke w 1665 roku opracował pierwszy mikroskop.
Zdolność rodzielcza B (wzór Abbego)=
1,22
n∗sinV
Zdolność rozdzielcza B jest to najmniejsza odległość dwóch punktów przedmiotu dająca się za
pomocą danego urządzenia optycznego jednoznacznie rozróżnić.
Jest funkcją długości fali (im większa tym lepiej) użytego promieniowania,
oraz warunkowana aperturą n*sinV (im mniejszy kąt V tym lepiej).
Zdolność rozdzielcza zwiększa się wraz ze wzrostem napięcia
przyspieszającego i długości fali.
Dla św. widzialnego o dł. 0,40,7 mikrometra jest to 1/3lambda 0,2285pm
→
ok. 1000 – kamień do czytania, uzyskiwano powiększenie na szkle.
ok. 1284 – pierwsze okluary do czytania – Włochy, Salvio d'Armate
1590 – prototyp mikroskopu i teleskopu, nakładanie iluś soczewek – Holandia
1897 Thompson odkrywa elektron.
1924 – de Broglie przypisał fali poruszające się elektrony. lambda = h l m V
1926 – pole magnetyczne i elektryczne działąją na elektrony jak soczewki
1931 – pierwszy mikroskop elektronowy. Ernest Ruska.
1938 – pierwszy TEM (Siemens) – rozdzielczość 10nm i pierwszy SEM
1965 – pierwszy komercyjny SEM
Techniki badawcze dostępne w polskich instytutach geologicznych.
–
Mikroskopia elektronowa SEM (skaningowy) , TEM (transmisyjny , HRTEM (High
→
resolution), STM (
skaningowy mikroskop tunelowy)
–
EMPA
–
XRF, WOS, EDS (Energy Dispersive XRay Spectroscopy)
–
AES, ICP – AES, ICP – MS
–
AAS, FAAS, ETA – AAS, CV – AAS, Hg – AAS
–
FI (badanie inkluzji)
–
FT – JR
–
CL
–
Kolumetria ?
–
XRD (High resolution Dyfraction)
–
MS
–
Chromatografia GL – MSD, GL – FID, ECD
→
–
DTA ( Analiza termiczna)
Charakterystyka elektronu
–
energia = 1,6 * 10
19
Coulomba
–
masa = 9,1 * 10
28
grama
–
prędkość ruchu bliska prędkości c światła.
–
masa się zmienia w strumieniu efekt relatywistyczny
Efekt wzrastającej masy w zależności od napięcia:
1 kV v = 1,873* 10
→
9
cm/s m
→
o
zmienia się o 0,2 %
50 kV v = 1,238 * 10
→
10
cm/s m
→
o
zmienia się o 9,8 %
–
długość fali elektronu liczymy np. w Angsztremach → = h
p
=
h
m
o
⋅
V
=
1− V
2
C
2
≈
150
Va
h stała Plancka, p pęd, m
o
– masa spoczynkowa el., V prędkość el, C – prędkość światła,
Va (ze wzoru) – napięcie przyspieszające [A] ma także wpływ na długość fali w strumieniu. Wraz z
jego wzrostem zmniejsza się długość fali
Ruch elektronu w polu elektrycznym F :
∂
m V
∂
t
=−
e [E V B]=−e E−e V B
Pole elektryczne i magnetyczne zmieniają tor strumienia elektronów, ale można je modulować.
Zwiększenie napięcia pozwala na zwiększenie pędu elektronów, co zmniejsza długości fali.
Składowa elektryczna zależy od natężenia pola i jego ładunku.
Składowa magnetyczna zależy od natężenia pola elektrycznego i kąta alfa 0
→
o
=0, 90
o
=max
Elektrony nie będą poruszać się po linii prostej, ale po spirali.
także dotyczy elektronów w polu magnetycznym:
n 2
n 1
=
V 2
V 1
=
p 2
p 1
Przy przejściu przez różne ośrodki następuje zmiana załamania światła.
Zakres zmienności załamania światła jest większy o optyce elektronowej.
Mamy tu brak powierzchni łamiących, tj. oddzielających obszary o różnych n.
Nie ma tu straty energii na soczewkach mikroskopu optycznego były.
→
Mamy płynne zmiany w zależności od zmian potencjału elektrycznego powiększenie zmienia się
→
jak zoom w aparacie fotograficznym ( na zwykłych mikr. mieliśmy zmiany skokowe).
Zasada Cartera z 1937 roku: Na zakrzywnienie toru w polu elektrycznym ma wpływ zwiększenie
potencjału eletrycznego. Na ruch eletronów ma wpływ skłądowa elektryczna.
Wykład 2 Ilnicki 2 08.10.2007 Budowa mikroskopu i funkcje jego części.
Mikroskopy optyczny i elektronowy mają wiele współnych elementów. Różnią
sie przede wszystkim innym ustawieniech soczewek.
Polaryzaja wiązki jest w mikroskopach elektronowych niemożliwa.
W mikroskopie optycznym mamy za to niestety bardzo ograniczone i skokowe
powiększenie.
Plusem EM jest możliwość badania dyfrakcji i składu chemicznego.
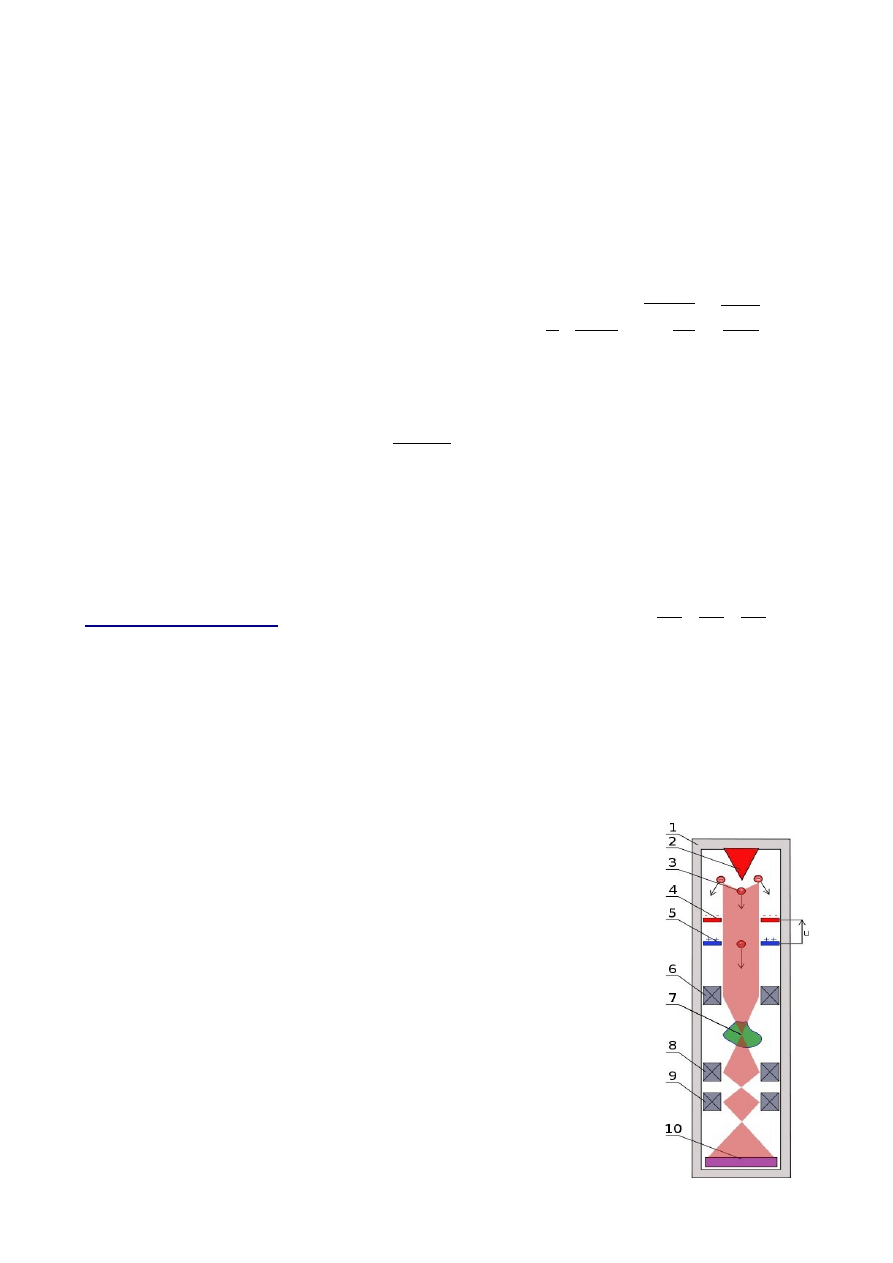
Elektronowy mikroskop transmisyjny
TEM:
Wiązka elektronowa po przejściu przez preparat może być kształtowana
podobnie jak promienie świetlne poprzez estaw soczewek elektronowych.
W przypadku elektronów zamiast szklanych elementów optycznych
wykorzystywane są cewki zmieniające bieg naładowanych cząstek.
Mikroskop może pracować w trybie obrazu wówczas wiązka tworzy obraz
preparatu na detektorze (10). Każda z soczewek pośrednich ma odpowiednie stymulatory, potrafiące
ogniskować wiązkę elektronu w odpowiedni sposób.
Działo elektronowe element urządzeń wytwarzający odpowiednio skierowany strumień
elektronów o odpowiedniej energii. Składa się z trzech elementów:
•
żarzona pośrednio za pomocą spirali, emitująca w wyniku termoemisji elektrony.
•
niewielki cylinder z otworkiem, otaczający katodę, cylinder ma
ujemny względem katody, zmiana potencjału zmienia
wiązki el.
Zmieniając potencjał możemy rozproszyć lub skupić wiązkę elektronów.
•
składająca się z jednej lub kilku cylindrycznych elektrod o różnych średnicach,
stanowią układ przyspieszający i ogniskujący (odpowiednik soczewek w układach
optycznych).
Energia prądu jest zależna od temperatury włókna W. Przepływający przez włókno prąd powoduje
jego rozgrzanie. Emisja termiczna powoduje wydostanie się elektronów przez opornościowe grzanie
włókna. Jest ono robione najczęsciej z wolframu przez jego wysoką temperaturę topnienia (3410 K)
przy temperaturze żarzenia 2700
Niektóre źródła elektronów są wytwarzane z LaB
6
. Jest to pojedynczy pręcik o temp. żarzenia 1700
2100
. Daje ono wysoką jasność. Źródło może skupić do niewielkiej plamki wysoka
→
rozdzielczość. Jest to jednakże kruchy element i bardzo drogi. W trakcie żarzenia jest dość
reaktywny reaguje z otoczeniem i preparatem, który może sam ulec rozpadowi pod wpływem
→
elektronów. Otoczenie może zatem dojść do katody i ją pokryć zaniżając jego wartości.
Najnowsze źródła katody z emisją polową
→
„ma zimno”, wysoka bariera potencjału
– bariera potencjału. którą musi pokonać elektron aby wyjść poza
powierzchnię metalu. Przy innych metodach potrzebne są duże temperatury.
Potrzebny jest wysoki gradient pola elektrycznego.
Kryształ wolframu jest tu używany w kształcie żyły o bardzo małej końcówce. W polu o dużym
gradiencie potencjału elektrony będą się wydobywać z tejże końcówki. Uzyskuje się w ten sposób
bardzo wysoką jasność.
Niestety jest wrażliwy na zanieczyszczenia stojące na drodze wydostających się elektronów →
także katody z emisją polową są nowszą technologią, ale która wymaga efektywniejszych systemów
próżniowych i opróżniających z gazów do pokonania barier.
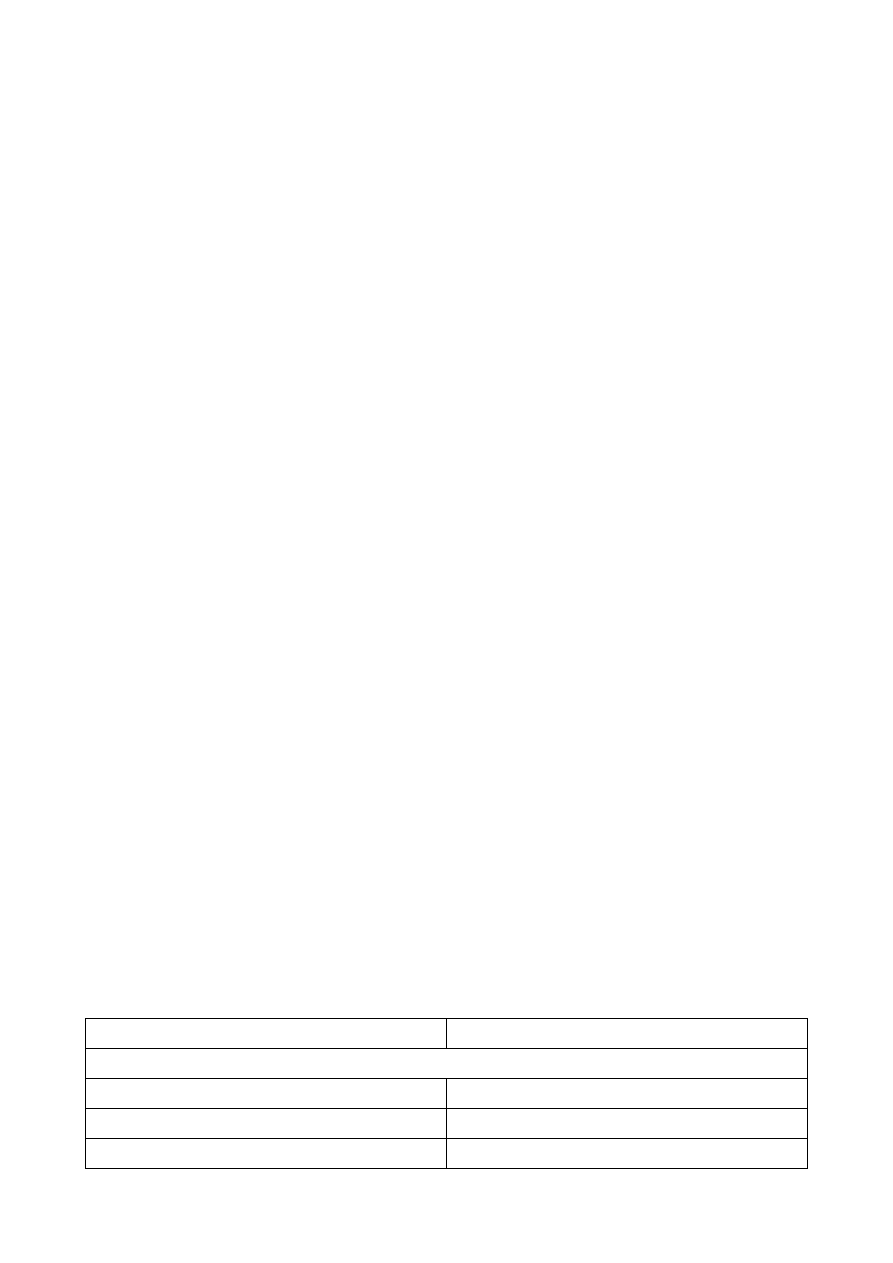
? Odmiana Schottky'go – termiczna, anoda ma podwójny układ porządkujący elektrony, pierwsze
przecięcie elektronów następuje za anodą.
Rozmiar źródła – średnica wiązki elektronów w pierwszym przecięciu za albo przed anodą.
wolframowe
LaB6
Schottky
emisja polowa
rozmiar źródła
100
5
<100A
<100A
jasność
1 A/cm2
2050 A/cm2
100500 A/cm2
1001000 A/cm2
wymagana próżnia 10
5
Tora
10
6
Tora
10
8
Tora
10
9
Tora
Dwa typy soczewek elektronowych:

–
soczewki elektrostatyczne – są w niewielkim stopniu używane. Są to np. cylindry, lub
pierścienie do których przyłożony jest potencjał. Przykładając pole elektryczne będziemy
generować pole elektryczne o różnym potencjale.
–
elektromagnetyczne – nie jest możliwe generowanie wiązki rozbieżnej. Częściej używane.
Dzięki nim wiązka zmienia swój kierunek, przyspiesza, a później spowalnia.
Soczewki są czułe na zanieczyszczenia i wykazują np. zjawisko
Można w sposób płynny zmieniać ogniskowanie poprzez zmiany płynącego przez uzwojenia prądu.
Defekty obrazu wynikające ze zjawiska aberracji:
1)
– zjawisko różnego ogniskowania dla różnych widm
Nie każdy elektron ma taką samą energię, więc różnie będą oddziaływać z polem
magnetycznym różnie ogniskować.
→
Następuje rozmycie obrazu obraz gorszej jakości.
→
Stabilizatory – mają za zadanie odchylać elektrony do prawidłowego położenia.
2)
wada układu optycznego polegająca na tym, że promienie padające w dwóch
prostopadłych płaszczyznach są
w różnych punktach. Wywołuje obraz nieostry
i zniekształcony.
Wiązka powinna w przekroju okrągła a w wyniku tego ma elipsoidalny kształt.
W jakimś miejscu wiązka jest rozciągana w którąś stronę, bo pole magnetyczne jest
niejednorodne. Zależy to np. od rodzaju uzwojenia.
Korekcja – należ wiązkę upakować znów w kulisty kształt.
Stygmatory – magnesiki i elektromagnesiki służące do regulacji wiązki. Jest ich kilka dla
każdej soczewki.
3)
„beczkowanie obrazu” wada optyczna układu optycznego polegająca na różnym
powiększeniu obrazu w zależności od jego odległości od
instrumentu.
Obraz może ulec tzw beczkowaniu, czyli wykrzywieniu się jego ścian w krzywe, bądź też
ulec np. skręceniu. Obraz jest najbardziej prawdiłowy w środku.
Jest to wada jakościowa soczewek
Wykład 3 Ilnicki 3
Soczewki kondensatorowe – 2 soczewki ze stygmatora
Układ pojedynczego kondensatora – sprawia że obszar oświetlony jest w obrębie preparatu na 3050
mikronów. Taka jest wielkość plamki na preparacie.
Układ podwójnego kondensatora – obecnie stosowany
–
jedna soczewka – zmniejsza obszar efektywnego źródła elektronów ogniskuje wiązkę (z 30
→
50 mikronów do jednego)
–
druga soczewka – tworzy powiększenie do 23 mikronów
Zadania podwójnego kondensatora:
–
dobrze oświetlona próbka – odpowiednio działając natężeniem można sterować zakresem
intensywności.
–
formowanie wiązki
–
zwiększanie wiązki

–
zmniejszanie uszkodzeń radiacyjnych (zwłaszcza poza badanym obszarem), reszta próbki nie
jest niszczona wiązką
–
ciepło wyzwalane w próbce na skutek promieniowania łatwiej będdzie można odprowadzić
(obszar niewiele większego obszaru niż obszar aktualnie prowadzonej obserwacji – ok 1 mikro).
–
poprawa kontrastu wskutek spójności zmniejszonego efektywnego źródła elektronów.
–
ograniczenie obszaru zanieczyszczeń preparatu wiązka może również nieść zanieczyszczenia.
→
–
większa wydajność kondensatora – zwiększa żywotność włókna poprzez redukcję jego jasności.
Rodzaje soczewek
1) kondensatorowe – podwójny układ kodensatorów. Znajdują się pomiędzy działem
elektronowym, a soczewkami elektronowymi.
a) krótkoogniskująca – charakteryzuje się dużą siłą pola
b) długoogniskująca obszar dyfrakcyjny z niewielkiego obszaru (mała siła pola)
2) Soczewki elektronowe stanowią układ powiększający.
→
a) obiektywowa – ogniskowanie obrazu i wiązki elektronów po przejściu przez próbkę
b) pośrednie – powiększenie obrazu, także dyfrakcyjnego
c) projektorowa – dalsze powiększenie obrazu, obrócenie do właściwej orientacji
Liczbę soczewek powiększających obraz dostosowujemy do przewidywanej zdolności rozdzielczej.
Korona preparatu – musi być przygotowana do różnych funkcji.
Umiejscawiamy w uchwycie, który doprowadzany jest do stolika.
Bardzo precyzyjny przesuw następuje we wszystkich trzech płąszczyznach x, y, z.
Korona preparatu musi być odporna na odkształcenia i zjawiska fizchem w obszarze próbki.
Możliwe ustawienie wg orientacji krystalograficznej dzięki goniometrom.
Urządzenia grzewcze max do 2500 stopni Celsjusza.
Urządzenia chłodnicze do 186 stopni Celsjusza.
Spektrometr (analiza składu chemicznego) – stosowane różne rodzaje widma.
Zapis obrazu:
1) metody fotograficzne – płyty szklane z błonami.
2) kamery wideo – zapis cyfrowy dający możliwość komputeroej obróbki obrazu
3) matryca CCD – liniowa rozdz. nawet do 12mln pixeli, powyżej 10
4
poziomów szarości,
stosuje się do źródeł o małej intensywności (materiały wrażliwe na reakcje np. min. ilaste).
Typy mikroskopów elektronowych:
1) SEM
Elektronowy mikroskop skaningowy
2) TEM – Transmisyjny mikroskop elektronowy
3) analityczne
4) STEM – skaningowy transmisyjny
5) TEM + spektrometr elektronów (dla Z<Si)
6) SEM + spektrometr RTG (dla Z>B)
7) mikroskopy emisyjne
8) mikroskop odbiciowy ?
9) HRTEM – wysokorozdzielczy TEM (wysokie napięcie przyspieszające)
10) STM
Próbka na kilka sposobów będzi oddziaływać z preparatem:
–
absorbcja – funcja gęstości i składu
–
rozpraszanie niskolatowe
–
–
wsteczne rozproszenie elektronów
–
emisja elektronów wtórnych – wybite danej próbki przez strumień elektronów

–
emisja fotonów – katodoluminescencja – zakres widzialny światła
–
oddziaływanie nieelastyczne z materią próbki, elektrony przekazuą część energii próbce podczas
przechodzenia. Wiąze się to z spektroskopią uuuaty energii przez elektrony EELS
–
elastyczne elektrony obrazy dyfrakcyjne SAED i CBED.
→
Próbka i prepatyka:
Próbka musi być trwała próżni, oraz w czasie odziaływania wiązki elektronów, musi być cienka to
tego stopnia, by wiązka elektronów mogła się przebić.
Wymiary – kładziemy na siateczce miedzianeo średnicy 3mm
Grubość preparatu musi być poniżej 0,5 mikrometra,
dla napięcia przyspieszającego 220kV preparat poniżej 0,1 mikrometra=100nm
→
TEM – zasosowanie – metale cemika, stopy, minerały
–
badania sukturalne,
–
składu chemicznego i
–
mikrostruktur (orientacja faz, struktura krzyształów, poprzez dyfrakcję elektronów, prom. X,
spetroskopię energii elektronów)
dobre do badań geotektoniki o czytania procesów ale próbki muszą być zorientowane
→
–
morfologii,
–
ułożenia atomów i badania strkturalne defektów (błędy ułożenia, wakanse, dyslokacje)
–
techni Burgensa – badania minerałów i ułożenia atomów w sieci krystalicznej
–
Powiększenie do 1.000.000 razy
–
Rozdzielczość od 1mm do 2A.
Wady TEM:
1) Metoda droga i destrukcyjna dla próbki
2) Bardzo trudna preparatyka (obrabianie do odpowiedniej wielkości)
3) Część materiałów nie nadaje się do badań (min. uwodnione, te które będą się rozkładać pod
wpływem ciepłabo zniszczą się przed uzyskaniem danych)
TEM – techniki specjalne
1) tworzenie obrazu –
a) tworzenie jasnego i ciemnego pola
obrazy mikrostruktulne, konieczne jest tutaj zastosowanie specjalnej aparatury
pierścieniowej o różnej średnicy w zależności od chcianego efektu.
BF – obraz wytworzony przez promieniowanie pierwotne (kontrast przez różnicę w
rozpraszaniu).
DF – (dark field) – promienie ugięte na płaszczyznach hkl, zmiana ustawień aparatury
obiektywowej, zmiana układu oświetlającego (odchylenie wiązki), badanie defektów
pojedynczych faz krystalicznych.
b) HRTEM – można badać pojedyncze kolumny atomów, odpowiednio zorientowane
krystalicznie.
2)
elektronów – obraz dyfrakcyjny mamy przy wykorzystaniu soczewki
długoogniskowej.
a) linie kikuchiego – pozwalają uzyskać orientację kryształów, domeny
SAED
gruby fragment cienkiego preparatu.
b) CBED – analiza krystaliczna małych obiektów jak
domen, mały obszar badań,
potrzebna zbieżna wiązka elektronów.
c) obraz pierścieniowy – pozwala na określenie sposobu ułożenia kryształów
d) obraz puktowy – przełożenie płaszczyzn krystalograficznych w punkty. Służy do

identyfikacji pojedynczych kryształów i ich orientacji krystalograficznej.
3) spektroskopia energii elektronów (utrata ich energii) EECS
Elektronowy mikroskop skaningowy
–
próbkę umiejscawiamy na dole mikroskopu
–
badamy powierzchnię próbki, a nie całą objętość (jak w TEM) ważna orientacja próbki.
→
–
może być różnej wielkość i nierównej powierzchni
Co się dzieje przy powierzchni oddziaływania:
–
elektrony wtórne SE
→
–
elektrony Angera
–
widmo promieniowania rentgenowskiego
–
elektrony wstecznie rozproszone BSE
→
–
promieniowanie ciągłe
–
promieniowanie charakterystyczne
Budowa SEM ma podobną strukturę, ale inne ułożenia.
Wiązka jest zogniskowana do plamki na 40
(im mniejsza plamka tym większa rozdzielczość).
Przesuwamy ją po badanym polu aż pokryjemy transwersami całą powierzchnię próbki, stąd
średnica wiązki jest istotna. Przesuwanie następuje bardzo dynamicznie, także mamy wrażenie
stabilnego trwałego obrazu. Częstotliwość przechodzenia wiązki po powierzchni preparatu jest taka
sama jak w tubie kineskopu ( w starych telewizorach).
Źródła elektronów i soczewki są takie same w SEM i TEM.
Tworzenie obrazu w SEM:
a) elektrony wtórne wybijane z próbki.
b) elektrony wstecznie rozproszone o energi na ogół wynoszącej 50 eV
c) skanowanie pewnego prostokątnego rastra obszaru
d) zmiany amplitudy sygnałów elektronów wtórnych topografia preparatu
→
e) zdolność rozdzielcza 510
(z reguły 34x gorzej niż TEM).
Obraz jest dynamiczny, a nie statyczny w tym mikroskopie.
f) powielanie – długość linii skanu na preparacie długość linii skanu na ekranie CCD.
→
Detektory i tryby pracy SEM:
–
topografia powierzchni
–
emisja elektronów
–
katodoluminescencja
–
absorbcja
–
refleksja ??
Zastosowanie:
–
badanie morfologii i topografii kryształów. SE wtórne elektrony
→
–
badania ilościowe (składu chemicznego badanie względnej koncentracji BSE e. wstecznie
→
→
rozproszone)
–
badania krystaliczne pojedynczych kryształów.
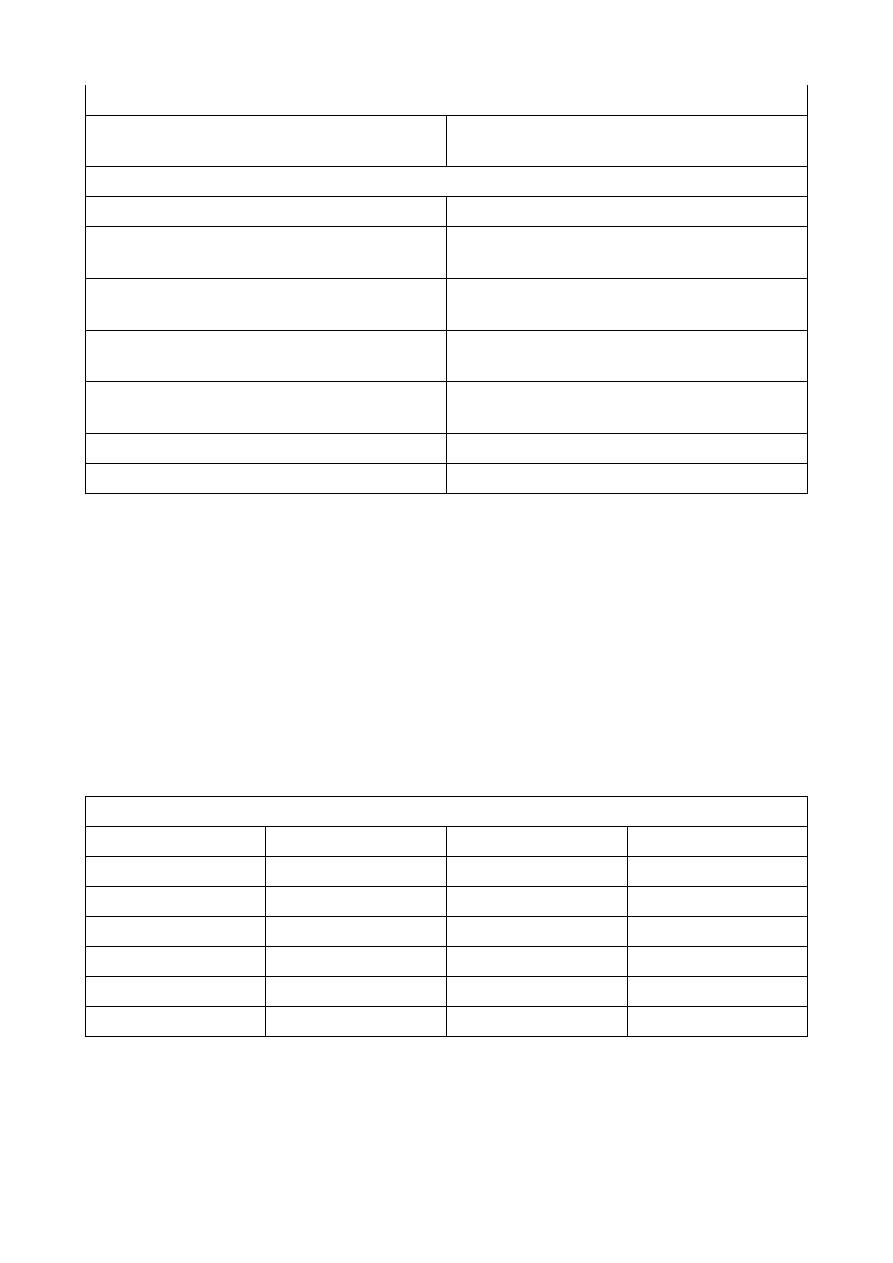
TEM
SEM
wiązka statyczna
wiązka dynamiczna
napięcie przyspieszające do 300400kV
czasem nawet powyżej 1250 kV
lepsza rozdzielczość – 12
gorsza rozdzielczość 5
badamy zmiany w wiązce rozproszonych, bądź obraz z wykorzystania elektronów wzbudzonych
ugiętych elektronów przebijających próbkę.
wtórnie i elastycznie odbitych od powierzchni
badamy wnętrze próbki: objętość, struktury,
mikrostruktury, ułożenie kolumn atomowych,
obrazy dyfrakcyjne (geometria sieci odwrotnej
małych monokryształów), skład chemiczny
badamy powierzchnię próbki: obrazy w 3D,
możliwe zdjęcia stereoskopowe dzięki
obrotowemu stolikowi, katodoluminescencja
utrata próbki
można odzyskać próbkę
próbka umiejscawiana w środku aparatury
próbka umiejscawiana na dole mikroskopu
Trudna preparatyka (Próbka musi być cienką
płytką o grubości mniejszej od 0,1 mikrometra).
łatwiejsza preparatyka (możliwe badanie
większych próbek, ich świeżych przełamów).
Wymagana próżnia w kolumnie mikroskopu do
badania na 10
7
– 10
9
Tora – pompy próżniowe
ograniczenie dla próbek biologicznych.
→
Mniejsze pociśnienie wymagane.
Wykład 4 Kozłowski 1
Trzy wytyczne do określenia warunków tworzenia minerałów
→
1) T Temperatura 2) P Ciśnienie 3) x Skład środowiska
Są trzy metody odczytywania:
1) T określamy zakres temperatur danych minerałów
2) P – pola twardości – siatki petrogenetyczne. Zawartość pierwiastka w jakimś minerale, np.
Al zastępującego Si.
3) określony skład w danym minerale wskazuje na pierwiastki jakie były w danym środowisku.
Bywają sobie wakuole wypełnione roztworem wodnym L
H2O
czy również z ciekłym L
CO2
np. w
pegmatytach, czy kwarcach. Zjawisko obecności różnofazowych inklucji odkrył Brewster.
W drugiej połowie XIX w. skonstruowano mikroskop petrograficzny.
Kwarc biały jest biały, bo ma mnóstwo
wielkości kilku do kilkudziestu mikrometrów.
Przyrosty spiralne, czy warstwy dochodzące to przykładowe metody wzrostu kryształów. Kiedyś
uważano, że kryształy rosną poprzez dodawanie kolejnych „warstw”.
Dwie niemieszające się ciecze mogą także powodować inkluzje ze względu na powstający przez nie
defekt sieci krystalicznej.
Roztwór zamknięty w inkluzji reprezentuje warunki i roztwór, w jakich wykrystalizował kryształ.
Inkluzja powstała, a temperatura w końcu zaczęła maleć. Ciała podczas chłodzenia zmniejszają
swoją objętość, tak więc kryształ zaczyna maleć wraz z inkluzją. Jednak ciecze szybciej tracą na
objętości niż ciała stałe, także objętość cieczy zmniejszyła się bardziej niż objętość samej wakuoli.
Skutkiem tego jest powstanie pustej przestrzeni uzupełnionej nasyconą parą roztworu. Im niższa
temperatura tym pęcherzyk gazowy się bardziej powiększa względem cieczy. Inkluzja zamienia się
w ciekłogazową.
Podgrzanie preparatu powoduje zmniejszenie pęcherzyka, aż do jego całkowitego zamknięcia na
koniec. Jest to oczywiście proces odwracalny. Możemy zmierzyć temperaturę kiedy nastąpiło
przejście fazowe gazu w ciecz. Jest to temperatura powstania kryształu.
Inkluzja mówi nam o ciśnieniu i temperaturze jakie towarzyszyły tworzeniu kryształu.
1) Zakładamy, że roztwór zamknięty w krysztale jest homogeniczny, a kryształ tworzył się w
temperaturze Tkr i ciśnieniu Pkr w ciekłym roztworze.
Ponieważ ciecz (w wakuoli) podczas chłodzenia kurczy się szybciej niż otaczający kryształ, to
powstaje ciekłogazowa inkluzja.
kontrakcja = kurczliwość spowodowana stygnięciem pęcherzyk kontrakcyjny.
→
Istnieje metoda podgrzewania kryształów i w ten sposób zmniejszenia pęcherzyków w celu
obliczenia temperatury tworzenia się kryształu. Teraz legenda do wykresu:
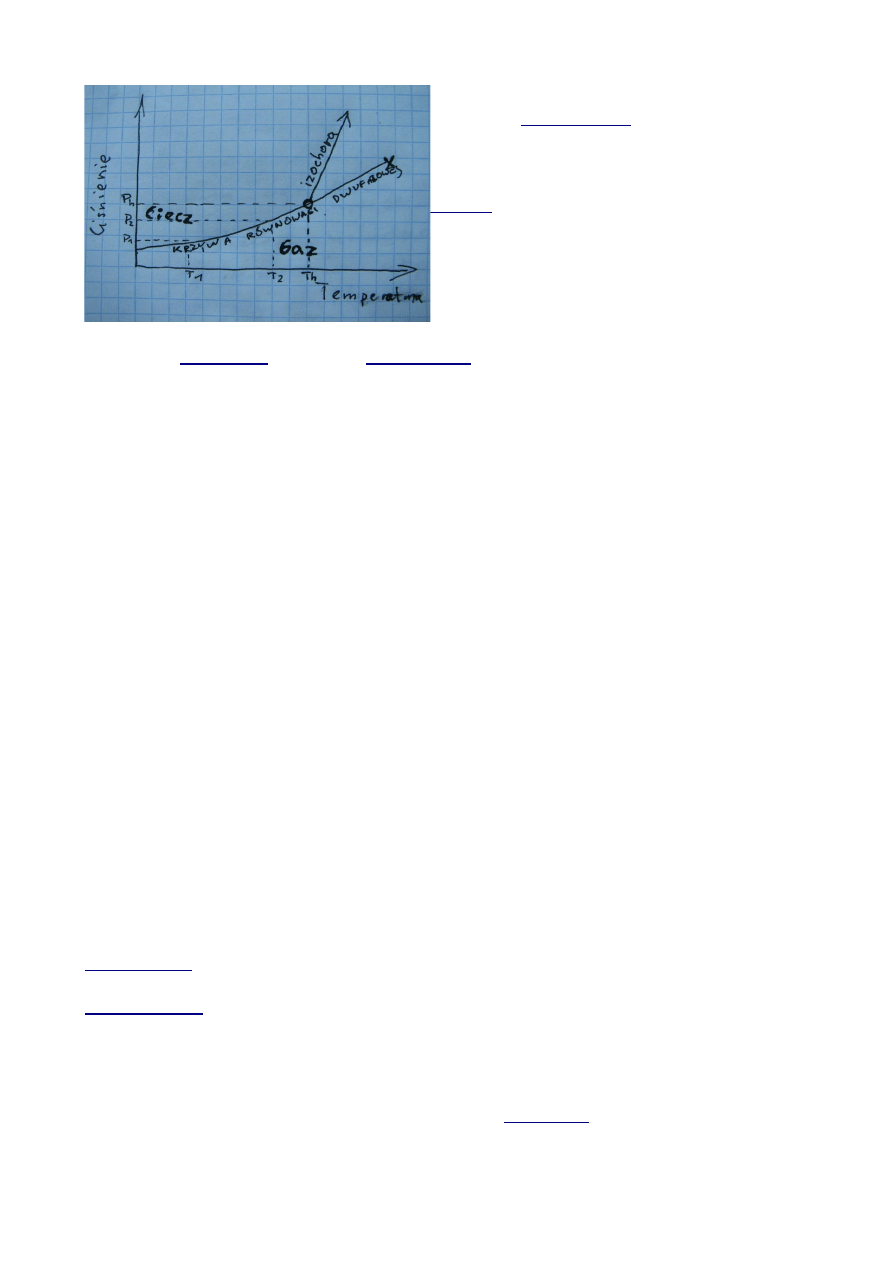
Gdy pęcherzyki się zamkną to mamy Th →
temperaturę
(na wykresie jako „o”).
punkt x przejście w stan krytyczny zanik
→
menisku (granicy między stanem ciekłym i
gazowym).
linia o stałej objętości. Zaczyna się w
miejcu Th. Inkluzja mogła powstać w każdej
temperaturze na izochorze. Jest to stroma krzywa,
co oznacza, że tym samym wzrostom temparatury
odpowiadają większe wzrpsty ciśnienia, niż to
miało miejsce przy krzywej równowagi 2fazowej.
punkt o – zanik pęcherzyka gazowego (na krzywej równowagi dwufazowej)
Temperatura
= delta T (zawsze dodatnia lub zero).
→
Krystalizacja w warunkach wrzenia roztworu to Th= Tkr delta T = 0. Wrzenie oznacza się w
→
fazie ciekłej. Są pęcherzyki pary nasyconej. Spadek ciśnienia także powoduje wrzenie.
Temperatura homogenizacji Th jest dolną granicą, gdzie mogła powstać inkluzja.
Delta T jest zatem zakresem temperatur, powyżej Th, gdzie mogła powstawać inkluzja.
Po podgrzaniu do Tk ścianka inkluzji pęka, ponieważ nie ma ciśnienia otaczającego macierzystego
wokół kryształu.
Fazę gazową można wydzielić podwyższając temperaturę, lub obniżając ciśnienie.
Inkluzja może być gazowa (G), gazowa z cieczą(L/G), lub z samą cieczą(L).
W przypadku przylgnięcia do minerału nowego małego ziarna (L/G?) może wskutek
niekompatybilności jonów nie być szczelnie otoczony i utworzyć wakuolę z roztworem. Taki
pęcherzyk nie powstaje z kurczenia. Ciecz w nim jest przypadkowa niezależnie od pęcherzyka.
Intruzja L/G nie powie nam o temperaturze homogenizacji, Pkr i Tkr.
Wszystkie inkluzje tworzone w tych samych warunkach mają tą samą gęstość.
Jednofazowe są zazwyczaj roztwory CO
2
, ze względu na niską temp. jego homogenizacji <31
o
C
Metoda skrzyżowanych izochor CO2 ma więszą ściśliwość niż woda. Znajdujemy gęstość
→
inkluzji LCO2, która ma swoją izochorę i to samo dla wody. Moment przecięcia ich izochor
wskazuje na P i T krystalizacji. Temperaturę nawet w zakresie kilku stopni, a ciśnienie dokładnością
12 kB metoda dotyyczy także innych mediów jak np. azot, ciekły H2S i różne węglowodory.
To, że mamy inkluzję homogeniczną nie musi wskazywać na homogeniczne środowisko
krystalizacji.
Duża inkluzja zawiera uśrednione warunki przy rozroście na tych etapach rozrostu minerału, który
obejmuje także im mniejsza tym bardziej dokładna „średnia kontrolowana”. Podgrzewając
→
→
dużą inkluzję doprowadzamy do jej eksplozji (gdy inkluzja była generowana, na zewnątrz było dużo
większe ciśnienie teraz pogrzewając obniżamy ciśnienie).
→
Inkluzja mówi nam o T i P kiedy powstawała dana strefa wzrostu (w środku krzyształu była
największa temperatura). Jeśli mam odpowiednie innkluzje możemy zaanalizować kolejne strefy
wzrostu.
– W odpowiednim ciśnieniu i temperaturze dana substancja może istnieć w trech
stanach skupienia jednocześnie i w równowadze.
– przejście w stan krytyczny. Ujednolicenie wypełnienia inkluzji przez zanik
menisku (366 stopni dla wody). Od punktu krytycznego zaczyna się krzywa krytyczna.
Spotyka się inkluzje, które mają gęstość krytyczną.
Metoda zamrażania inkluzji i kryształu do 100
→
o
C 150
o
C np. ciekłym azotem.
następnie pogrzewamy i widzimy główne składniki
→
obniżające temp.
np. NaCl, KCl, CaCl
2
, MgCl
2
Wykład 5 Kozłowski 2 inkluzji ciąg dalszy

–
Środowiska homogeniczne:
a) inkluzje gazowe – reakcje w środowisku pneumatolitycznym.
b) inkluzje ciekłe – powstają pęcherzyki kontrakcyjne. 1*) stop, 2) roztwory wodne CO2,
→
metan, azot, H2S
1*) stop należy niby do cieczy, ale znacznie inne właściwości fizchem. jak gęstość i lepkość
→
Rodzaje stopów
a) krzemianowe
b) węglanowe, np. aktywny wulkan w E Afryce Olduyinyo Oldvai
c) fosforanowe – często współwystępujące z krzemianami, np. prekambryjskie nelsonity
d) tlenkowe – brak ezpośrednich dowodół na ich istnienie – magnetytowe i tytanowe z
domieszkami obniżającymi temperaturę topnienia.
e) solne – z podgrzania serii ewaporatowych przez magmę w warunkach subwulkanicznych, np.
Masyw Elbrus.
W przybadkach stopów a), b), c) i e) znaleziono inkluzje.
–
Heterogeniczne środowisko:
a) dwie ciecze niemieszające się
b) ciecz + gaz
c) ciecz + kryształ
d) gaz + kryształ
e) wszytkie pośrednie warianty
a) dwie ciecze niemieszające się
Im większa temperatura tym mniejsza luka mieszalności.
Podgrzewanie powinno nam pokazać tą samą Th z dokładnością do 12 stopnia.
Wszystkie inkluzje powstałe w takim środowisku mają różną zawartość CO2, ale taką samą
temperaturę homogenizacji.
Obliczając proprcje objętościowe składników gazu L
H2O
i L
CO2
i znając temperaturę możemy z
wykresu obliczyć ciśnienie homogenizacji.
Przy obniżaniu się temperatury powstaje także obok niemieszjących się cieczy pęcherzyk z gazem.
W przypadku solnych inkluzji podczas schładzania następnuje krystalizacja spowodowana
zwiększającym się wskutek malejącej temperatury przesyceniem ciekłego roztworu. W
temperaturze pokojowej mamy zatem pęcherzyk kontrakcyjny i kryształ. Żeby mieć pewność przy
tego rodzaju badania analizujemy czy Th i temperatura powstawania kryształu się zgadzają dla
dwóch inkluzji z tej samej strefy wzrostu.
d) ze względu na duże różnice współczynników załamania światła gazów do kryształów, dokładne
pomierzenie Th jest praktycznie niemożliwe.
Dla wszytkich przykładów musimy wyeliminować możliwość istnienia gradientu temp. w inkluzji.
Inkluzje stopu dzielimy ponadto na inkluzje te z szkliwa i z kryształów.
Inkluzje stopu (krzemianowego) – ich Th jest do wyliczenia pewnie tylko w kwarcu
inkluzje szkliwa (podobne wizualnie do wodnych)
1) jest dużo mniejsza różnica wsp. załamania światła między ... a wsp. inkluzji.
2) dużo większa będzie różnica między wypełnieniem, a pęcherzykami kontrakcyjnymi.
3) w inkluzjach ruchy pęcherzyka kontrakcyjnego praktycznie zanikają w porównaniu do
inkluzji wodnych (w małych inkluzjach może nawet nie być pęcherzyka)
4) Th są rzędu 800900 stopni dla skał kwaśnych i 10001300 stopni dla maficznych – dużo

wyższe niż dla inkluzji wodnych.
5) często zaznaczają się w inkluzji jakieś objawy rekrystalizacji (igiełki są charakterystyczne
dla inkluzji stopu)
inkluzje kryształów – te częściej zawierają pęcherzyki kontrakcyjne
Warunek równowagi dwufazowej:
przy pogrzewaniu pęcherzyk w inkluzji się zmniejsza i przemieszcza (ruchy Browna, prądy
gęstościowe i termiczne.
Heterogeniczne środowisko powstawania, ale homogeniczne lub heterogeniczne środowisko
występujące w inkluzji.
Dla homogenicznego środowiska powstawania mamy zawsze homogeniczne środowisko inkluzji.
Wykład 6 Kozłowski 3 – nie odbył się jeszcze
Wykład 7 Parafiniuk 1 5.11.2007 Analiza termiczna
Literatura:
„Termiczna analiza różnicowa” Dietrich Schultze
„Termogramy minerałół ilastych” Anna LangierKuźniarowa
„Analiza derywatograficzna skał ilastych” Ryszard Wyrwicki
Badanie zachowania danej sunstancji pod wpływem zmian temperatury.
Zaletą metod jest dobra analiza składu fazowego (ale nie zawsze, bo niektóre próbki są obojętne
termicznie dla tego zakresu badań).
Dobre narzędzie do badań ilościowych (analiza derywatograficzna, termograwimetria).
Służy także do badań ilościowych zawartości wody.
Metoda derywatograficzna istnieje już od 50 lat.
1) Metody statyczne – w warunkach izotermicznych (próbkę doprowadza się do odpowiedniej
temperatury aż przestaną zachodzić procesy).
Wymagają dłuższego czasu przeprowadzania, ale są to metody dokładne np. do badania
kinetyki procesów. Tą metodą wykreśla się diagramy fazowe.
Metody dynamiczne – nie czekamy do uzyskania równowagi, podczas liniowego podnoszenia
temperatury. Jest to metoda szybka (ok. 100min). Przebieg procesów jest skomplikowany i
interpretacja jest skomplikowana. Badania mają charakter porównawczy.
Przykłady badań metodą dynamiczną:
1) Termograwimetria – TG – mieszana jest masa badanej substancji w funkcji temperatury.
Próbka umieszczana jest na wadze. Podgrzewa się ją i patrzy się ile zyskuje lub traci na
wadze. Wynikiem jest krzywa na wadze.
2) Termiczna analiza różnicowa – DTA rejestruje się tempereturę wewnątrz badanej próbki
→
ogrzanej wg określonego programu temperaturowego.
Reakcja endotermiczna – temperatura próbki robi się niższa niż substancji inertnej.
Reakcja egzotermiczna – temperatura próbki jest wyższa niż punktu odniesienia.
Mierzymy temperaturę substancji obojętnej termicznie i próbki. Musimy analizować
różnicę, która na wykresie zapisze się w postaci piku. Dla reakcji endotermicznej będzie to
pik skierowany w dół, a dla reakcji egzotermicznej w górę.
3) Skaningowa kalorymetria różnicowa DSC – podobna do DTA
W tej metodzie mierzymy różnicę energii dostarczanej do badanej próbki i substancji
odniesienia w funkcji temperatury. Zwykle na DTA jest nałożony system, który porównuje
temperatury między dwoma próbkami i mierzy ciepło. Wynikiem jest krzywa.

Powierzchnia piku jest wprostproporcjonalna do pochłoniętego lub wydzielonego ciepła.
4) Termodylatometria (TD, TDA) – pomiar wymierów liniowych, lub objętości próbki podczas
ogrzewania. Stosuje się np. wśród minerałów ilastych, np. do określenia jakości porcelany.
Do próbki są podłączone czujniki, wynikiem badań jest także krzywa.
Termosonimetria – badanie efektów akustycznych emitowanych przez próbkę podczas
ogrzewania (trzaski). Otrzymuje się krzywą termosonimetryczną.
5) Termooptometria – zmiany parametrów optycznych takich jak promieniowanie, zmiany
współczynnika załamania światła.
6) Termofluproscencyjna metoda – minerały wykazują świecenie przez naświetlanie fotonami,
np. do oznaczania wieku bezwzględnego minerału (ile pochłonął minerał promieniowania),
ale jest to metoda niedokładna.
7) Termoelektrometria – badanie elektrycznej charakterystyki próbki w funkcji temperatury,
np. oporności/przewodności, stałej dialektrycznej. Można badać zmiany na krzywej.
8) Termomagnetometria – pomiar podatności magnetycznej substancji w funkcji temperatury,
np. na termoszalce jedna z szalek znajduje się w polu magnetycznym.
→
Badanie reakcji utlenienia i redukcji, określenie
dla substancji, czyli
kiedy ciało przestaje być ferromagnetykiem.
9) Analiza wydzielanego gazu (EGD) – detekcja objętości wydzielanego gazu.
Stosuje się gaz nośny (inertny) np. hel, którym przepłukuje się próbkę.
Potem ten gaz analizuje się. Jeżeli coś się zmieniło, to bada się zmiany. Pomiar objętości w
biurecie gazowej.
10) Analiza wydzielanego gazu (EGA) określenie ilościowe składu chemicznego za pomocą
→
chromatografu gazowego. Może być zastąpiony przez zestaw biuret do miareczkowania.
Można określić parę wodną, siarkowodów, dwutlenek węgla itd.
Gazowe produkty reakcji wynoszone są za pomocą gazu nośnego.
Przenoszą one gazy ze środowiska reakcji do biurety. Są tam elektrody pHmetru. Jeżeli
nastąpi zmiana pH urządzenie dodaje tyle substancji, aby utrzymać pierwotny skład.
11) Analiza termocząsteczkowa – wykrywanie elementów o wielkich masach cząsteczkowych.
Gaz nośny wynosi elementyo dużych masach cząsteczkowych. Wprowadza się do komory
jonizującej. Na cząsteczkach osadza się para wodna i można pomierzyć ilość cząsteczek.
Można badać także polimery.
Efekty termiczne w DTA i TG – można rejestrować procesy fizyczne (endo) i chemiczne (egzo)
1) Topienie (endo) – połączenie DTA (w dół) i TG (nic się nie zaznaczy)
2) Sublimacja – ciało stałe przechodzi w gaz – DTA w dół, utrata masy przy TG, np. Salmiak
rodzimy, arsenid
3) Wrzenie – rozkład próbki – np. wody krystalizacyjnej. DTA w dół, TG ubytek masy
Trudno odróżnić wrzenie od sublimacji.
4) Przemiany fazowe (polimorficzneendo) pod wpływem ogrzewania, np. kwarc z
trygonalnego w heksagonalny. DTA w dół, TG – nic się nie zaznacza.
chemiczne dysocjacja chemiczna (zależna od ciśnieniaPrzy wysokim ciśnieniu węglan jest
→
bardziej trwały rozkład na czynniki prostsze, zwykle z wydzieleniem fazy gazowej, np. kalcyt,
magnezyt, syderyt, dolomit, cerusyt (węglan ołowiu) rozkład w trzech etapach.
→
Oddawanie wody
a) woda krystalizacyjna np. gips CaSO4*2H20 podgrzewając woda zostaje oddana (w pewnej
→
określonej temperaturze zazwyczaj niewysokiej, do 300 stopni).
b) woda związana w postaci grup OH, np. minerały ilaste, ałunit jest to woda konstytucyjna,
→
trzeba więcej ciepła, aby wyrwać tą wodę.
Hemimorfit ma grupy OH i wodę krystalizacyjną, każdą stratę z nich będzie oddawał w postaci
innych efektów (i w innych temperaturach).
c) woda zeolityczna – luźno związana w strukturze minerału w lukamch siłami Van der Waalsa, np
w rzeczonych zeolitach.
Nie ma jednej określonej temperatury oddawania wody. Zeolit po oddaniu wody nie traci swoich
własności.
Wykład 8 Parafiniuk 2 12.11.2007 Podstawy analizy derywatograficznej
Egzotermiczne procesy fizycznych procesów przejść odmian niskotemperaturowych w
wysokotemperaturowe są związane z wydzielaniem ciepła na zewnątrz układu.
Azotany rozkładają się z wydzieleniem NO
2
, siarczany metali ciężkich (jak Cu, Zn, Pb) z
wydzieleniem dwultlenku lub tritlenku siarki (desulfuryzacja), oba widoczne jako endopiki.
Rozpad struktury jest widoczny jako endopik (nie wiem czy skrót myślowy „endopik” jest
poprawny) i w przeciwnieństwie do procesów chemicznych jest niewidoczny na krzywej TG.
przy podgrzewaniu tracą grupy hydroksylowe, oddają ciepło i przechodzą w forsteryt.
Utlenianie jest reakcją chemiczną egzotemiczną ! np. siarczki siarczany, tlenki siarki; 100400
→
o
C.
Procesy utleniania wiążą się ze wzrostem masy np. przy syderycie, gdy mamy (już) FeO Fe
→
2
O
3
.
Krzemiany rozkładające się w temperaturze do 1000
o
C (zakres analizy) są bardzo dobre do analizy.
Węglany i minerały strefy hipergenicznej również.
Siarczki, arsenki, siarkosole także jednak już w mniejszym stopniu.
Do badań używamy próbkę inertną (nieczułą chemicznie na zmiany temperatur w badaniu).
Obecnie wystarcza kilka mg próbki, kiedyś było to 200300 mg.
Przy badaniu liczymy także wskaźnik symetri S jako iloraz stycznych do wykresu prowadzonych od
maksimum piku termicznego (miejsca gdzie najwięcej w tym momencie przereagowuje, lub inaczej
miejsce ugięcia piku termicznego).
→
Wzrost temperatury, by badanie miało sens, musi następować liniowo.
Jeśli zaistnieje np. reakcja endoenergetyczna, to w próbce mamy temperaturę niższą, ponieważ
ciepło zostało zużyte do reakcji. Jej intensywność ukazuje skierowany w dół pik na krzywej DTA.
Warto obliczyś wskaźnik symetrii S reakcji termicznej. Jednym ze sposobów jest skorzystanie ze
wzoru S=
a
b
, gdzie a i b są odcinkami łączących poziomo na wykresie na wysokości
temperatury próbki inernej, dwóch półprostych powstałych z kolejnych stycznych do krzywej,
prowadzonych od punktu przegięcia piku (jego maksimum) do półprostej pionowej na wykresie
prowadzonej również od punktu przegięcia. Ale zamotałem.
Metoda Stocka dla S – różniczkowa. Wykres piku termicznego wskutek tego zamienia się na dwa
odwrotnie skierowane piki po obu stronach maksimum piku termicznego. Mierzymy wtedy
stosunek pionowego wychylenia tychże pików.
Szybkość ogrzewania warunkuje wielkość pików.
Amplituda i wielkość pików świadczy o ilości pochłoniętego/oddanego ciepła (energii termicznej).
Szerokość piku może poświadczyć o gwałtowności.
Im szybciej ogrzewamy, tym bardziej rozmyte są granice pomiędzy poszczególnymi pikami.
Przy szybkim ogrzewaniu zmienia się także w kierunku wyższych temperatur położenie piku.
Za słabo grzać też nie można, bo piki też się przesuną. Najczęsciej grzanie do 100
o
C trwa 100minut.
Wielkość próbki – Nie może być za duża bo działało by to negatywnie na gradient temperaturowy
(w środku by była mniejsza temperatura niż ogrzana szybciej powierzchnia. Przesuwa zatem znów
piki na wyższe temparatury. Zbyt mała masa daje też z kolei mało wiarygodne wyniki.
Wielkość ziaren i stopień zdefektowania struktury także ma wpływ pik leci w dół, a przy
→
defektach może powstać nawet nowy.
Atmosfera pieca – niektóre substancje gazowe mogą wejść w reakcję np. utleniania (by temu
zapobiec używa się azotu i argonu, lub innego gazu szlachetnego.

Analiza derywatograficzna:
1) Próbkę (zazw. 200300mg) sypiemy do tygielka przymocowanego do wagi analitycznej.
Należy uważać z ubijaniem jej zbyt ubite przesuwają reakcje w kierunku wyższych temp.
→
2) Do drugiego tygielka sypiemy substancję inertną np.Al
2
O
3
, MgO, wyprażony kaolinit.
3) Piec elektryczny grzeje z prędkością dziesięciu stopni na minutę.
4) Galwanometr zwierciadlany zapisuje w postaci różnicy temperatur na tygielkach, zmierzoną
z różnicy potencjałów wynikającej z pracy elektromotorycznej. gdy nie ma różnicy
temperatur, to zwierciadło się nie wychyla.
5) Derywatograf – umożliwia jednoczesne zapisywanie krzywych TG, DTA, oraz DTG.
6) Temperatura mierzymy różnicę temperatur porównując tą z próbki inertnej do próbki
badanej z różnicy napięcia. Materiał jest najczęściej z drutów Pt, lub Pt z radem.
7) Różnica temperatur jest zapisywana na papierze zwierciadlanym w postaci krzywej DTA
8) krzywa TG podczas reakcji masa próbki ulega redukcji na wadze i się zmienia. Waga się
porusza a jej ruch jest także zapisywany w postaci krzywej (TG) na papierze śwczułym..
9) Krzywa DTG – cewka EM której ruch zależny jest od masy próbki ruchu wagi
→
analitycznej co też jest odzwierciedlaniej reakcji zachodzących w kolejnych
→
temperaturach. gdy cewka się wychyla jest tworzy się pole elektryczne, którego wielkość jest
rejestrowana w postaci krzywej DTG
Innym sposobem pomiaru temperaturowego jest termopara. Wykorzystuje ona fakt różnego
przewodnictwa sybstancji w funkcji temperatury. Jako termopary używa się dwóćh drutu, jeden z
platyny, drugi z platyny z radem. Układ ma mierzyć różnicę temperatur pomiędzy próbką
wzorcową, a badaną.
Wykład 9 Parafiniuk 3 Derywatogramy minerałów 19.11.2007
Badamy minerały czułe na zmiany temperatury do 1000 stopni Celsjusza.
Węglany się nadają oprócz węglanów metali alkalicznych. Wszytkie oprócz (na szczęście rzadkich
w przyrodzie) Na i Ca mają rozkład endotermiczny w zakresie 5001000 stopni. Jest jeden wielki
pik termiczny związany z dekarboksylacją, podczas którego wydziela się CO
2
. Efekt ten jest
asymetryczny. temperatura w jakiej zachodzi jest już indywidualną cechą każdego minerału. Kalcyt
ma np. przy wysokich temperaturach, magnezyt przy niższych, smitsonit prz najniższych. Wiąże się
ztym spora strata masy. W przypadku węglanów lekkich metali mamy większe straty masy?
Kalcyt ma pik związany z rozkładem przy 950970 stopniach. Następuje wtedy wysoka strata masy
bo aż 40%. Aragonit ma podobnie, ale ma jeszcze jeden słabszy pik endotermiczny przy 420,
związany z przejściem polimorficznym z aragonitowej w kalcytową strukturę krystaliczną. Kryształ
aby to się stało musi zostać spękany podzielony na fragmenty, by mogło dojść do przebudowy.
→
Oczywiście w tym czasie wszelkie zrosty i inkluzje ulegają w tym czasie również zmianom. W
skrajnych przypadkach może się zdarzyć nawet, że ziarna zaczną wręcz wyskakiwać z tygielka z
próbką.
Syderyt – Dwa nakładające się niemal konkurencyjne efekty termiczne – proces dwufazowy jeden
pik przy 450 i zaraz potem jeden (egzo, w górę) związany z powstaniem nietrwałego FeO i zaraz
potem procesem utleniania do Fe
2
O
3
. Ponieważ jest tu też utlenianie następuje przyrost masy, lecz
niewielki w stosunku do zauważalnego podczas dekarboksylacji. Jednakże przyrost masy nie równy
teoretycznemu związanemu z utlenianiem, ponieważ część została zrółnoważona z jej stratami
związanymi z rozpadem.
Oczywiście należy pamiętać, ze nie wszytko jest efektem termicznym związanym z jakąś reakcją.
Często jest to po prostu zmiana pozycji zerowej związana ze zmianą np. ciepła właściwego próbki,
dlatego też linia nigdy nie będzie idealną linią prostą.
– podobny jak u kalcytu, alecz temperatura reakcji niższa bo ok. 560 stopni i większa
strata masy bo prawie 50% (Mg jest lżejszy niż wapń).
3
– Efekt podobny do kalcytu, jeden pik w temp. 300500 stopni. Strata masy
mniejsza bo cynk jest cięższym metalem.
Dolomit – (Mg,Ca)CO
3
ma dwa piki endotermiczne związane najpierw z rozkładem części
magnezowej do MgO przy temp. 720870, potem wapniowej przy 900 stopniach. Kształt piku przy
rozpadzie częsci magnezowej jest asymetryczny. Związane jest to z równoczesnym rozpadem
struktury dolomitu.
– PbCO
3
3stopniowy rozkład z wydzieleniem Pb kolejno w temperaturach 420500
stopni.procesy zlewają się ze sobą w tym przypadku na wykresie.
Mimo, że mają bardziej złożone wzory, widać, że zarówno OH jak CO2 rozpada
→
się w jednym momencie i pozostaje nam tylko tlenek miedzi.
i witeryt się nie nadają. Potrzebują wyższych temp. do rozpadu i jeyne co w nich
zachodzi to procesy polimorficzne.
Siarczany – jest ich ok. 300. Są dobre. Powstają na drodze wietrzenia siarczków i ewaporacji, więc
w niskich temperaturach i w takich też często się rozpadają w związku z tym?
Przy odwodnieniu siarczany ukazują (niejednokrotnie wieloetapowo złożone) endotermiczne efekty.
Grupy hydroksylowe mają efekty endotrmiczne w wyższych temperaturach (najczęsciej przy ok.
300 stopni) niż przy utracie wody krystalizacyjnej np. przy kopiapicie. Czasem razem z wodą.
Przy wielometalowych różne piki też wskaują na rozpady kolejnych wodorotlenków metali.
– ma dwa duże piki endotermiczne w bassenit CaSO
→
4
*1/2 H
2
0 CaSO
→
4
(ale higroskopijny
w przeciwieństwie do anhydrytu) w temp. kolejno 170 i 200 stopni. Proces odwadniania prowadzi
się na dużą skalę by uzyskać tzw. gips wiążący, czyli szybko chłonący wodę (zawierający gips
bezwodny i półwodny).
, rozenit one wszystkie i inne złożene siarczany mają złożone wykresy DTA z
→
wieloma pikami endotermicznymi. Jest to przede wszytkim związane z obecnością H
2
O i OH.
Tlenki i wodorotlenki
FeOOH – ma jeden pik kiedy przechodzi w Fe
2
O
3
w temp 30420 stopni. Stopień
krystaliczności ma tutaj duży wpływ na kształty krzywej DTA. Mała strata masy 89%.
– wykres podobny do getytu na tyle, że trudno oba odróżnić.
Boehmit i
(składniki boksytów) mają też podobne efekty (jeden) w temp 500600 stopni.
Krzemiany
Bardzo dobre są minerały ilaste dzięki swym grupom hydroksylowym np. zeolity jak desmin
(dawniej stylbit), oraz inne minerały posiadające w swej strukturze wodę krystaliczną. Nie nadają
się skalenie, bo nnie wykazują istotnych reakcji termicznych.
– (min. ilasty 1:1) oddanie wody cząteczkowej 90 stopni jeśli jest to kaolinit powstały z
→
wietrzenia; potem endopik przy 580 stopniach związany z oddaniem grup OH i ostatni przy
przejściu w mullit egzopik na 980 stopniach.
Smektyty (2:1) – najczęsciej jako dwa wyraźne efekty endo– 1) oddanie wody międzypakietowej 2)
oddanie wody hydroksylowej. W zależności z jakim podstawionym katonem mamy do czynienia,
pik na krzywej temperaturowej DTA się przesuwa. Można w ten sposób wyznaczyć, ile jakiego
pierwiastka mamy.
Smektyty od kaolinitu różnią się przede wszystkim pierwszym efektem, który w tym przypadku jest
silniejszy.
Beidelit – ma trochę więcej wody międzypakietowej niż kaolinit.
Chaloizyt – podobny do kaolinitu, ale też wody ma więcej wody międzypakietowej.
– Skała składająca się ze smektytów (sporo montmoryllonitu) ma dwudzielny efekt
związany z dehydroksylacją.
– kiepsko się bada, bo ma słabo uporządkowaną strukturę. Lekko zauważalne
odwodnienie.

Łyszczyki z grupy glaukonitu – trzy efekty kolejno – odwodnienie, dehydratacja i utlenianie Fe.
Chloryty – trudno rozróżnić dokładniej. Mają 23 efekty związane ze stratami H2O i OH,
zachodzące w kilku etapach. Prawie zawsze na koniec jeszcze jeden egzopik z tego co zostało.
Szamozyt – kiepski do badania, bo mało charakterystyczne właściwości termiczne. Widać egzopik
związany z utlenianiem żelaza (i jak zwykle wzrost masy w związku z tym).
Węgiel i kerogen – ciężkie w interpretacji. Zależne od atmosfery w jakiej się wykonuje badanie.
tj. kruszce, siarczki – widoczne piki z utleniania.
Antygoryt i chryzotyl (serpentyny) – mają słąby efekt z odwodnienia, a i to nie zawsze widać.
Nie nadają się amfibole, bo mają za wysokie temperatury topnienia (powyżej 1200 stopni)
Kwarc – pik endotermiczny na 573 stopniach związany z przejściem w odmianę
wysokotemperaturową (i dlatego na krzywej TG się nie zaznacza).
Wykład 10 Bagiński 1 26.11.1007 Termometria geologiczna
Ciśnienie i temperatura – podstawowe parametry brane pod uwagę podczas badań skał magmowych,
metamorficznych, jak i osadowych.
Badanie składu chemicznego w mikroobszarze.
Aby otrzymać analizy musimy dokładnie zapoznać się z materiałem. Metoda ta oparta jest na
wzorcach. Badana próbka musi mieć wyskalowane parametry gładkość i równoległość do stolika.
→
Analiza zależy od jakości zachowania się materiału w próbce.
Procesy przemian wtórnych, często zakłócają badania. Dopiero badania mikrosondą mogą określić,
czy dany minerał się nadaje do badań.
Większość minerałów ma wyliczoną dokładnie stechiometrię.
Nie da się polegać tylko na jednej analizie należy zrobić kilka.
→
Odwzorowanie min. odwzorowanie poszczególnych pierwiastków.
→
Na analizę mają wpływ: jakość próbki, niedokłądność struktury mineralnej, własności sprzętu.
Dlatego właśnie robimy kilka badań. Średnią wyznaczamy na podstawie najbardziej
reprezentatywnych strefach, nawet w jednym minerale. Natomiast jeśli minerał jest homogeniczny,
to rozkład reprezentatywnych wartości jest podobny dla całego jego obszaru, a nie rozłożony na
poszczególne strefy.
geotermometria – przykład granatbiotyt oba krystalizując wybierają te same kationy a
→
mianowicie Mg i Fe. Badania eksperymantalne wykazały, że w zależności od temperatury i
ciśnienia będziemy mieli inne zależności składu tychże kationów w tych minerałach. Śledząc
zmiany zawartości Mg i Fe można wykalibrować odpowiednio system dla naszych potrzeb.
Minerały te ze względu na szeroki zakres trwałości bo 40700 stopni Celsjusza, są powszechnie
stosowane. Mamy kilkanaście odmian tego termometru i każda stosowana dla innych warunków.
Jednakże różne odmiany tego termometru mają różny zakres błędu, który może wynosić nawet 200
stopni Celsjusza. Trzeba zweryfikować wtedy ów termometr innym termometrem. Czasem nie jest
to możliwe, ale zazwyczaj możemy na różne sposoby da się rozpoznać historię skały. Oczywiście
zdarzają się i skały, które w ogóle nie da się badać.
Ustalenie piku metamorfizmu jest możliwe przy wykorzystaniu dwóch różnych termometrów, choć
nie musi to oznaczać, że oba wykażą tą samą temperaturę.Przykładami takich par mogą być np.
hornblendabiotyt i hornblendaplagioklaz.
Ścieżka rozwoju skały jest otrzymywana poprzez składanie wyników z różnych pomiarów
temperatury i ciśnienia dla danych minerałów; np. niektóre minerały dążą do ustalenia równowagi
chemicznej, ale minerały zamknięte w innych minerałach nie wymieniają pierwiastków, stąd inne
warunki. zatem zapisany w „zamkniętych” minerałąch skał nie może się zmienić, bo był
odizolowany, czyli zarówno inne ciśnienie i temperatura będą zapisane przez te same minerały,
pomimo obecności metamorfizmu.
Granaty inaczej się zachowują niż pospolite minerały wykazują pasowość podczas zmiany
→

warunków.
Biotyty są homogeniczne (jednorodne) jak się zmienia ich skład to po całości minerału.
→
W każdym momencie metamorfimu mogą powstawać różne fazy, jak choćby proces chlorytyzacji.
Dla biotytu takie zmiany mogą okazywać ścieżkę rozwoju skały, zdradzając tzy temperatury
różnych procesów: 1) odizolowany biotyt, 2) zmieniony biotyt pod wpływem metamorfizmu 3)
przeobrażony biotyt w wyniku np. procesu cholrytyzacji (traci potas).
Uniwersalne wytrychy (lecz nieprecycyjne niestety):
–
zawartość glinu w hornblendzie – mówi o ciśnieniu barometr geologiczny
→
–
Zawartość tytanu w biotycie – mówi o temperaturze termometr geologiczny
→
Ciśnienie szacujemy w innych układach niż przy termometrach geologicznych
Wykład 11 Bagiński 2 Datowanie skał krystalicznych
Wiek izotopowy wiek bezwzględny
→
Badając wiek skały krystalicznej musimy brać pod uwagę, iż jest ona efektem
wieloetapowego rozwoju. Zatem różne były warunku zarówno temperaturowe jak i ciśnienia w
danych etapach. Klasycznym minerałem używanym do badań wieku skał krystalicznych jest cyrkon.
Jeśli powstaje on podczas jednego epizodu, to jest najlepiej, ale może ich być kilkadziesiąt (niektóre
mogą nawet pochodzić z innej skały magmowej, mając ten sam skład powstaje zonalność).
→
Jak odróżnić jedne populacje od drugich separacją frakcji.
→
Katodoluminescencja badanie wyglądu i zarysu kryształu cyrkonu? Kiedyś można było datować
→
tylko jeden etap powstawania. Często najstarsze datowania cyrkonu pochodzą z wyników jego
środka.
EPMA Mikrosonda jonowa pozwala badać stosunki izotopowe w mikroobszarze. Nie daje ona
→
→
wyników stosunków izotopowych, a jedynie ich sumaryczną zawartość pierwiastków, które
posiadają izotopy).
Mikrosonda jonowa i EMPA – rozgrzana katoda emituje elektrony, kontrolowane przez zestaw
soczewek skupiające się na małym obszarze. Możemy analizować obiekty mające mniej niż 5
mikrometrów. W zależności od ustawionego napięcia przyspieszającego modulujemy wiązkę
elektronów, a co za tym idzie sterować wielkością plamki od 1mm do kilkunastu mikrometrów.
Kryształy minerałów o homogenicznej naturze można badać przy mniejszym powiększeniu i przy
mniejszej ilości analiz.
Istnieje wskazanie, by korzystać do badań lekkich pierwiastków jak Li, Na, czy boru, aby. Stosuje
się rozmytą wiązkę aby detektor mógł dokładniej wybrać je, ponieważ ma problemy z analizą
większych ich ilości.
3 spektrometry trzy różne liczniki zbierają informacje z próbki na temat trzech różnych
→
sondowanych pierwiatków. Za pomocą tychże badań dokonuje się dwóch analiz:
1) EDS – analiza dyspersji
2) WD – analiza długości fali i dyspersji. Jest to dokładna analiza, ale wymaga, by każdy
pierwiastek był oddzielnie skanowany.
Może być maksymalnie 5 spektrometrów 5 analizowanych pierwiastków.
→
Jest to ułatwienie,
jednakże minerały mają raczej więcej pierwiastków. Wynikiem jest zawartość pierwiastków i
tlenków. Proste minerały bada się 34. Bardziej skomplikowane pierwiastki takie jak monacyt czy
apatyt, mają pierwiastki ziem rzadkich które mają barzdiej skomplikowane parametry, spektra
energetyczne często tym samym zakresie widma.
Zwykle analizuje się od lantanu po europ, jak jest więcej, analiza może zawierać również te z
cięższych ziem rzadkich. Analiza monacytu może trwać ponad 20 minut, aby uzyskać wszyskie
rozsądne analizy. Trend analityki zresztą idzie w kierunku badań pierwiastków, których zawartości
są niewielkie. Próg dokłądności zawartości poszczegółnych pierwiastków wynosi obecnie ok.
10ppm. Należy wykonać kilka analiz tego samego obszaru, gdyż tło może zaburzyć analizy
poszczególnych pierwiastków. Zawartość ich wyznacza wielkość piku.
Nie zawsze inny odczyt musi oznaczać inny epizod danego procesu, może to być po prostu inny.
Wzorce wykorzystywane w badaniach są zależne od analizowanych próbek.
SHRIMP II potrafi badać pierwiastki promieniotwórcze, ale bada dużo większą powierzchnię
→
mikroobszaru na raz (wielkość plamkitam 12 mikrometra, tutaj ok. 20). Tutaj mamy badany każdy
izotop oddzielnie, wykorzystując fakt różnej masy każdego z nich.
Monacyt bogaty w ziemie rzadkie jak uran i tor po wykrystalizowaniu mamy tzw. zegar
→
→
izotopowy. Zegar zaczyna „tykać”, gdy temperatura zjedzie poniżej 700 stopni. Okazuje się, że
mamy w nim rzadko do czynienia z pierwotnym ołowiem. Cała ilość ołowiu w próbce ma
radiogeniczne pochodzenie. Można obliczając sumaryczną ilość ołowiu wyznaczyć w ten sposób
wiek minerału. Występuje razem z cyrkonem, ma podobną wielkość kryształów. Preferują skały
magmowe kwaśne i obojętne. Monacyt powstaje chętnie także w warunkach metamorficznych, oraz
w diagenezie. Widywany jest nawet w okruchowych dobry wskaźnik protolitu. Powstaje w
→
szerokich warunkach temperaturowych, dzięki czemu możemy go stosować w szerokim zakresie
badań. W warunkach facji zieleńcowej ulega metamorfizmowi i rozpada się na allanit (epidot ziem
rzadkich), czy apatyt.
Inne minerały chętnie przyjmujące ziemie rzadkie to np. tytanit, granat, monacyt, allanit. Zajmują
one mała objętość w skale, ale decydują o zmianach krzywych zawartości ziem rzadkich.
Przy wykorzystaniu SHRIMPa można określić wiek z dokładnością + 1 mln lat.
Monacyt wychwytuje wszelkie zmiany w historii skały, na tej zasadzie, że jeśli raz wykrystalizował,
to w kolejnym etapie jego część ulega rozpuszczeniu i ponownej krystalizacji zachowując nowy
skład. Nalepiej widać to na obrazie BSE (wstecznie rozproszone elektrony), gdzie widać strefy o
takich samych odcieniach, charakteryzujących ten sam skład. Inne będą się różnić. Potrzeba około
20 analiz by podać wartość.
Skład monacytu jest niestabilny. Widoczna zmienność Th+U przy w miarę stałej proporcji Th/U.
W różnych partiach monacytu mamy inny skład inny wiek
→
CHIME – metoda wykorzystująca wiedzę z zakresu statystyki. Należy wykonać jak najwięcej analiz
i w ten sposób wyliczyć.
Uwagi:
Należy uważać przy interpretacji zewnętrznych stref monacytu mogą to być strefy powstałe
→
wskutek rozpadu przy obecności fluidów zwłaszcza silnie alkalicznych, gdyż jest to minerał bardzo
na nie czuły i łatwo z nimi reaguje (przy obojętnych i zasadowych na szczęście niechętnie).
W temperaturach 400500 stopni monacyt jest nietrwały. powstaje z niego wtedy allanit.
Monacyty lepiej zapisują zmiany od cyrkonów.
PiXi jest to inna metoda wykorzytująca mikrosondę protonową, mamy tu jednakże problemy ze
→
sterowaniem wiązką. Potrzeba tu także stabilnych i homogenicznych minerałów. Nie ma możliwości
sterowania wiązką nastawiamy kryształ. Mniejsza procyzja pomiaru. Plusem natomiast jest brak
→
potzrebnego we wcześniejszych metodach wzorca. Wielkość wiązki pomiędzy EMPA i SHRIMP.
Wykład 12 Dubińska 1 Geodynamika chemiczna
Geodynamika – zajmuje się próbami określenia w jakiej pozycji geodynamicznej powstała skała.
Ryft oceaniczny
Ryft kontynentalny
geometria
ryft spowodowany rozsuwaniem się dwóch płyt rozszczepienie jednej płyty
linia ryftu, długość i kształt są zmienne
stały kształt. Powiększanie zw. z ekspansją term.
stale tworzona nowa skorupa w osi i ramionach oś i ramiona ryftu stale w skorupie kontynent.
przyczyna magmatyzmu
rozsuwanie się płyt powoduje termiczne
wynoszenie astenosfery
w strefach osłabienia litosfery następuje
uwalnianie składników lotnych
charakterystyka termiczna i magmowa
chemizm stopu zgodny z perydotowym
wytapianie zmetasomatyzowanej litosfery
generalnie magmatyzm ala dekompresyjne
topienie podnoszonej astenosfery
topienie litosfery pod wpływem metasomatozy
i przepływu składników lotnych
magmatyzm i duży przepływ ciepła związany
głównie z przepływem mas płaszcza
magmatyzm i duży przepływ ciepła
sporadyczny, bez związku z płaszczem
duże objętości bazaltów grzbietów
śródoceanicznych o stosunkowo stałym składzie
bardzo zmienny magmatyzm, zwykle bogaty w
alkalia i gazy
położenie miejsc erupcji zmienia się w
zależności od ruchu płyt i migracji ryftu
powtarzają się erupcje o tym samym składzie o
prekambru
źródła magmowe zubożone w LILE
wzbogacenie w LILE
czasem występują ksenolity płaszczowe
ksenolity płaszczowe są częstsze i pozmieniane
LILE – pierwiatki litofilne o dużym promieniu atomu, np. K, Rb, Ba
MORB – bazalty grzbietów śródoceanicznych
OIB – bazalty wysp oceanicznych
IAT – toleity łuków wysp powstające nad strefą subdukcji
Boninity – skały występujące pomiędzy łukiem wysp, a strefą subdukcji
Pierwiastek śladowy
–
zawartość jego w układzie jest tak niska, że zmiana jego stężenia nie wpłynie na stabilność
żadnej fazy układu
–
zachowuje się zgodnie z prawem Henry'ego: stężenie składnieka w roztworze jest
wprostproporcjonalne do jego prężności cząśtkowej nad roztworem
–
można obliczyć podział pierwiasków pomiędzy dwie fazy układu stosując współczynnik
podziału minerałstop lub minerałminerał nie rozważając występowania pierwiastka w żadnej
innej fazie układudla pełnego zakresu analizowanych stężeń
wahania składu toleitów w różnych sytuacjach geotektonicznych
łuki wysp
grzbiety śródocean.
wyspy oceaniczne
FeO/MgO
1,7
0,82,1
0,0052,5
SiO2 %
4676
4751
4565
FeO %
616
614
816
Na2O %
1,13,6
1,73,3
0,74,5
K2O %
0,12,0
0,070,04
0,062,0
TiO2 %
0,32,0
0,72,3
0,25,0
Oparcie badań o główne składniki skał nie daje jednoznacznych odpowiedzi (czy magmatyzm był
toleitowy czy alkaliczny). Najbardziej wskaźnikowym z powyższych są TiO2 oraz K2O.
Rodzaje wykresów zawartościdiagramów: Sherrera, dwuwymiarowe, trójkątne, pajęcze itd
Diagramy dyskryminujące – służą do oznaczenia rodzaju skał magmowych i określenia pozycji
geotektonicznych skał.
Na podstawie zawartości pierwiastków śladowych można określic skład źródła i stopu.

Model wytapiania równowagowego: C
s
=
C
M
F 1−F D
C
S1
/
C
S2
C
M1
/
C
M2
=
C
s
=
F1−F D
1
D
1
F1−F D
2
D
2
, gdzie Cs stęż pierw. w źródle (uwzględniamy 2 źródła).
→
Cm stężenie pierwiastka w źródle, Ffrakcja stopu (udział w stosunku do źródła)
→
D współczynnik podziału pierwiastków (pomiędzy stop a residuum) D1 i D2
→
Różne fazy są dobrymi wskaźnikami dla różnych miejsc np.głębokie źródło płaszcowe perowskit.
→
1) pierwiatki silnie kompatybilne
np. Ni
D>5
2) Pierwiatki umiarkowanie kompatybilne
np. Cr, Co Mg
5>D>1,5
3) pierwiastki słabo kompatybilne/słabo inkompatybilne
np. Fe, Mn
1,5>D>0,5
4) pierwiastki umiarkowanie inkompatybilne
np. Sc, Y, Ga, Ca Al 0,5>D>0,05
5) pierwiastki silnie inkompatybilne
np. Yb, Y, Ti
0,15>D>0,05
6) pierwiastki bardzo silnie inkompatybilne
np. Zr, Nb
D<0,05
Wykład 13 Dubińska 2 geodynamika chemiczna
W zależności od stopnia wytopienia skład pierwiastkowy w stopie ulega zmianie (np. dla
klinopiroksenów anomalia ujemna zawartości europu maleje w trakcie wytapiania).
1) Ntype MORB – normal ridge sediments, synonim toleity ubogie w potas
HREE (ciężkie, rzadkie od GD do Lu) – 1315x więcej niż w chondrycie
LREE (lekkie rzadkie od La do Eu) – conajmniej 910 razy mniej niż w chondrycie
LIL i HFS (high fields strength, np. Nb, Zr, Ta)– obniżone zawartości
2) Etype MORB – anomalne grziety śródoceaniczne, synonim – bazalty typu P (plume)
HREE – podobnie jak w chondrycie
LREE 1525x więcej niż w chondrycie
LIL i HFS – podwyższone zawartości
3) Ttype MORB „transitional”ridge sediments
Przemieszczaniu się pierwiastków, pomaga metamorfizm dna oceanicznego, poprzez gorące fluidy
infilrujące skały i osady. Powstaje np. epidozyt pod powierzchnią. Pierwiastki w związkach są
ługowane i mogą tworzyc później złoża, gdy wskutek spadku temperatury są strącane.
Zawartość Sr jest zakłóconaw oceanach w zależności od intensywności procesów przemiany skał.
Lantan i Cez często tworzą anomalie dodatnie na diagramach pajęczych a Sm i Eu ujemne.
1) Pierwiastki inertne
→
Zr vs Zr/Y;
Ti/Zr; ZrTiY; ZrYNb, Ni vs Ti/Cr Zr/P
2
O
5
vs TiO
2
2) pierwiastki dość często ruchliwe np. Th, Ce, V
→
HfThTa; HfThNb; Th/Nb vs Ce/Nb
3) pierwiastki ruchliwe w procesach zmian →
K/Nb vs Ce/Nb; Ba/Nb vs Ce/Nb ZrTiSr
Pierwiastki inkompatybilne w procesach maficznych D<0,1 K, Cs, Rb, U, Th, Ba, Ta, Nb, LREE
→

bardzo inkompatybilne natomiast D<<0,01 HREE, Ti, Y, Zr, Hf, P
→
Współczynniki ilustrujące zmiany (frakcjonowanie) i skład płaszcza
→
Tb/Y, Zr/Hf,
→
Ta/Nb Sm/Nd
Yb/Lu Ti/Zr
La/Ce
Ce/Yb
Wstępny bilans geochemiczny wymaga rozeznania się w różnych aspektach sytuacji geologicznej:
1) ruchliwość pierwiastka w analizowanym środowisku
2) jednorodność skału macierzytej
3) dystrybucja danego pierwiastka pomiędzy fazy mineralne
Stosunek B/Be jest nowym narzędziem w ocenie wpływu subdukowanej płyty na zapis
geochemiczny łuków wysp (dla świerzych skał).
lawy wapniowoalkaliczne i toleitowe łuków wysp B/Be – 10200
osady oceaniczne ~100
zmienione częście skorupy oceanicznej – 90170
MORB i OIB – 35
Bor w porółnaniu z berylem jest ekstrahowany w sposób uprzywilejowany z subdukowanego
materiału przez uwodniony fluid.
Ba/La zależy od stopnia wytopienia i pośrednio głębokości żródła płaszczowego.
Rola fluidów hydrotermalnychw powstawaniu magm strefy subdukcji bazuje na podstawie składu i
zależności pierwiastków syderofilnych, chalkofilnych i boru.
Zasady:
–
zakładamy że mamy układ zamknięty (nic nie ucieka ani nie przybywa)
–
stosunek pierwiastków syderofilnych czy chalkofilnych do litofilnych o tym samym składzie
można wykorzystać do oceny procesów pomagmowych
–
stosunki Pb/Ce; As/Ce; Mo/Ce; Sn/Sm; W/Th i B/La są niezależne od stopnia ewolucji
magmowej i można je wykorzystać do oceny wzbogacenia lub zubożenia pierwiastków
syderofilnych czy chalkofilnych w stosunku do innych pierwiastków inkompatybilnych, czy
małoruchliwych.
Fluidy hydrotermalne, a powstawanie magm SSZ ( na podstawie pierw. sydero czy chalkofilnych):
–
Procesy magmowe nie wpływają na stosunki Pb/Ce, czy B/La natomiast domieszka fluidu
wpływa na ich wzrost. Zawartość tychże pierwiastków jest więc pośrednio funkcją odległości od
frontu wulkanicznego.
Epsilon neodymu:
Nd
T =
10
4
∗[
Nd
143
/
Nd
próbka
144
T − Nd
143
/
Nd
CHUR
144
T ]
Nd
143
/
Nd
CHUR
144
T
143
Nd – produkt rozpadu alfa
147
Sm (T½ = 106mld lat);
144
Nd – izotop trwały
CHUR – chondric uniform reservoir (jednorodny zbiornik chondrytowy).
Wykład 14 Dubińska 3 14.01.2007 Skały ultramaficzne
Strefa Dupal anomalia izot. tworząca pas od równika do 60
→
o
S przejaw zmienności płaszcza.
→
Składniki płaszcza o odmiennej charakterystyce izotopowej.
–
zubożony zapis izotopowy NMORB
wysokie wartości
→
143
Nd/
144
Nd;
niskie wartości
87
Sr/
86
Sr i
206
Pb/
204
Pb →
istnienie zubożonego członu płaszcza MORB (DMM depleted end member MORB mantle).
→
→
–
bardzo wysokie wartości
87
Sr/
86
Sr i
206
Pb/
204
Pb oraz średnie wartości
143
Nd/
144
Nd płaszcz
→

wzbogacony w U i Th w stosunku do Pb (MIMU), któremu nie towarzyszy wzrost stos Rb/Sr.
–
na grzbietach Samoa obserwuje się trendy zmian
143
Nd/
144
Nd i
87
Sr/
86
Sr, przekraczające BSE
(skład krzemionkowego płaszcza Ziemi)
–
w skałach wulkanicznych PREMA (skład płaszcza o stosunkowo stałej charakterystyce
izotopowej)
→
87
Sr/
86
Sr ~ 0,7033 i
143
Nd/
144
Nd ~0,5130
–
bazalty oceaniczne wysoki stosunek izotopowy
→
206
Pb/
204
Pb i niski
87
Sr/
86
Sr
Pierwiastki śladowe
Z metasomatozą związana jest np. zmiana zawartości pierwiastków alkalicznych.
Pierwiastki ruchliwe K, Cs, Rb, Ba, U
→
Pierwiastki w miarę inertne P, LREE
→
Pierwiastki inertne Ta, Nb, Ti, Y, Zr, Hf
→
Bazalty świerze mają ujemną anomalię europu, zmienione (f. zieleńcowa) dodadnia anomalia Eu.
→
Metamrorfizm morskiego dna ma wysoki gradient geotermiczny, dużo wody bierze udział. Świerze
skały zaw. np. plg, cpx, szkliwo mają jako zmienione np. chloryt, kwarc widoczne. Co się dzieje:
–
duży dopływ Mg powstają chloryty
→
–
następuje nie mal całkowite odprowadzenie Ca, Na, Sr, Cu
→
–
Odprowadzenie częściowe (>50%) K, Mn, Zn, CO
→
2
–
zachowanie inertne P, Ti, V, Cr, Co, Ni Y, Zr
→
–
zachowanie zmienne (często lokalna redystrybucja) Fe
→
Spility i zieleńce:
–
pierw. inertne Th, Nb, Ta, Y, Ti, Zr, Hf, REE
→
–
pierw. ruchliwe K, Rb, Cs, U, Ba, Sr
→
–
Współczynniki geochemiczne ilustrujące skład płaszcza:
Tb/Y, Zr/Hf, Ta/Nb, Sm/Nd, Sm/Nd, Yb/Lu, Zr/Nb, La/Yb, Ti/Zr, Zr/Tb, Zr/Ti vsTi, Zr/YbvsYb
Strefy wpływu roztworów hydrotermalnych (kwaśnych):
–
pierw. inertne Nb
→
–
niekoherentne zachowanie REE, względna ruchliwość LREE (zwłaszcza LA i Ce)
–
wzbogacenie REE we fluidzie LREE 250x, HREE 30x, Ce 1500x w stosunku do pierw. składu
Sprawa
87
Sr/
86
Sr:
–
współczesna woda morska ma
87
Sr/
86
Sr na poziomie 0,70916 (zależnie od wieku jest różny)
–
czynniki kształtujące wartość
87
Sr/
86
Sr w wodzie morskiej:
dopływ wód z kontynentów, aktywność hydrotermalna, diageneza osadów węglanowych
–
Sr we fluidach hydrotermanych pochodzących ze sk. oceanicznej:
wymywanie Sr z bazaltów, Sr z wody morskiej (który nie uległ wytr.), duża zmienność
87
Sr/
86
Sr.
–
zakres zmienności Sr w wodzie morskiej sięga ok 2 km.
K, Ce, Ba nie nadają się do badań skał powstałych z maficznego protolitu, spilitu, zieleńców,
→
amfibolitów, granulitów i eklogitów.
Wyszukiwarka
Podobne podstrony:
pytania z metod, Geologia, UNIWERSYTET WARSZAWSKI, SEMESTR I, METODY BADAŃ MINERAŁÓW I SKAŁ, Metody
metody sciaga, Geologia, UNIWERSYTET WARSZAWSKI, SEMESTR I, METODY BADAŃ MINERAŁÓW I SKAŁ, Metody ba
ściaga metodyszczaki 1, Geologia, UNIWERSYTET WARSZAWSKI, SEMESTR I, METODY BADAŃ MINERAŁÓW I SKAŁ,
metody bada˝ 3, Geologia, UNIWERSYTET WARSZAWSKI, SEMESTR I, METODY BADAŃ MINERAŁÓW I SKAŁ, Metody b
Sciaga Metody, Geologia, UNIWERSYTET WARSZAWSKI, SEMESTR I, METODY BADAŃ MINERAŁÓW I SKAŁ, Metody ba
metody, Geologia, UNIWERSYTET WARSZAWSKI, SEMESTR I, METODY BADAŃ MINERAŁÓW I SKAŁ, Metody badan min
Od Ilnickiego, Geologia, UNIWERSYTET WARSZAWSKI, SEMESTR I, METODY BADAŃ MINERAŁÓW I SKAŁ, Metody ba
chemia analityczna emisyjna spektrometria atomowa, Geologia, UNIWERSYTET WARSZAWSKI, SEMESTR I, METO
Aktywator, Geologia, UNIWERSYTET WARSZAWSKI, SEMESTR I, METODY BADAŃ MINERAŁÓW I SKAŁ
Budowa TEM, Geologia, UNIWERSYTET WARSZAWSKI, SEMESTR I, METODY BADAŃ MINERAŁÓW I SKAŁ, Metody badan
Spektroskopia l R, Geologia, UNIWERSYTET WARSZAWSKI, SEMESTR I, METODY BADAŃ MINERAŁÓW I SKAŁ, Metod
pytania z metod semestr letni, Geologia, UNIWERSYTET WARSZAWSKI, SEMESTR I, METODY BADAŃ MINERAŁÓW I
SPEKTROSKOPIA IR, Geologia, UNIWERSYTET WARSZAWSKI, SEMESTR I, METODY BADAŃ MINERAŁÓW I SKAŁ, Metody
Metody badań wieku skał
metody badan spolecznych msm wyklad 3
metody badan spolecznych msm wyklad 2
porownywanie populacji, Pedagogika, Metody Badan pedagogicznych, KOZUH, WYKŁADY
więcej podobnych podstron