
1
W
PROWADZENIE DO SPEKTROSKOPII W PODCZERWIENI
Opracownie: dr Katarzyn
a Ostrowska, dr Maria Burgieł
Konsultacja: dr Jarosław Wilamowski, dr Bartłomiej Kozik
1. WIĄZANIA W ZWIĄZKACH ORGANICZNYCH
Związki organiczne zawierają głównie atomy C, H, N, O, S, P, które połączone są ze sobą
wiązaniami atomowymi (kowalencyjnymi), powstałymi w wyniku uwspólnienia elektronów
walencyjnych. Różnica elektroujemności pomiędzy poszczególnymi pierwiastkami powoduje
przesunięcie uwspólnionej pary elektronów w stronę bardziej elektroujemnego atomu pierwiastka.
Wiązanie z niesymetrycznym rozkładem elektronów nazywa się wiązaniem atomowym
spolaryzowanym. Każde spolaryzowane wiązanie (C-H, C-N, C-O, C-S, C-P, N-H, O-H) posiada
moment dipolowy wiązania μ [C m D (debaj)], który równa się iloczynowi wartości ładunku e
[C] i długości wiązania d [m]. Moment dipolowy jest wielkością wektorową, a suma wektorów
momentów dipolowych wszystkich wiązań w cząsteczce daje wypadkowy moment dipolowy
cząsteczki.
Elektroujemność wybranych pierwiastków w skali
Paulinga
Wartości momentów dipolowych wybranych,
izolowanych, wiązań występujących w związkach
organicznych
Wiązanie
Moment dipolowy μ [D]
H
Li
Be
2,1
1,0
1,5
B
C
N
O
F
Cl
Br
I
Na
Mg
Al
Si
P
S
K
Ca
2,0
2,5
3,0
3,5
4,0
1,0
1,2
0,9
1,0
1,5
1,8
2,1
2,5
3,0
2,8
2,5
C-H
N-H
O-H
C-C
C-N
C-O
C-F
C-Cl
C-Br
C-I
0,4
1,3
1,5
0
0,2
0,7
1,4
1,5
1,4
1,2
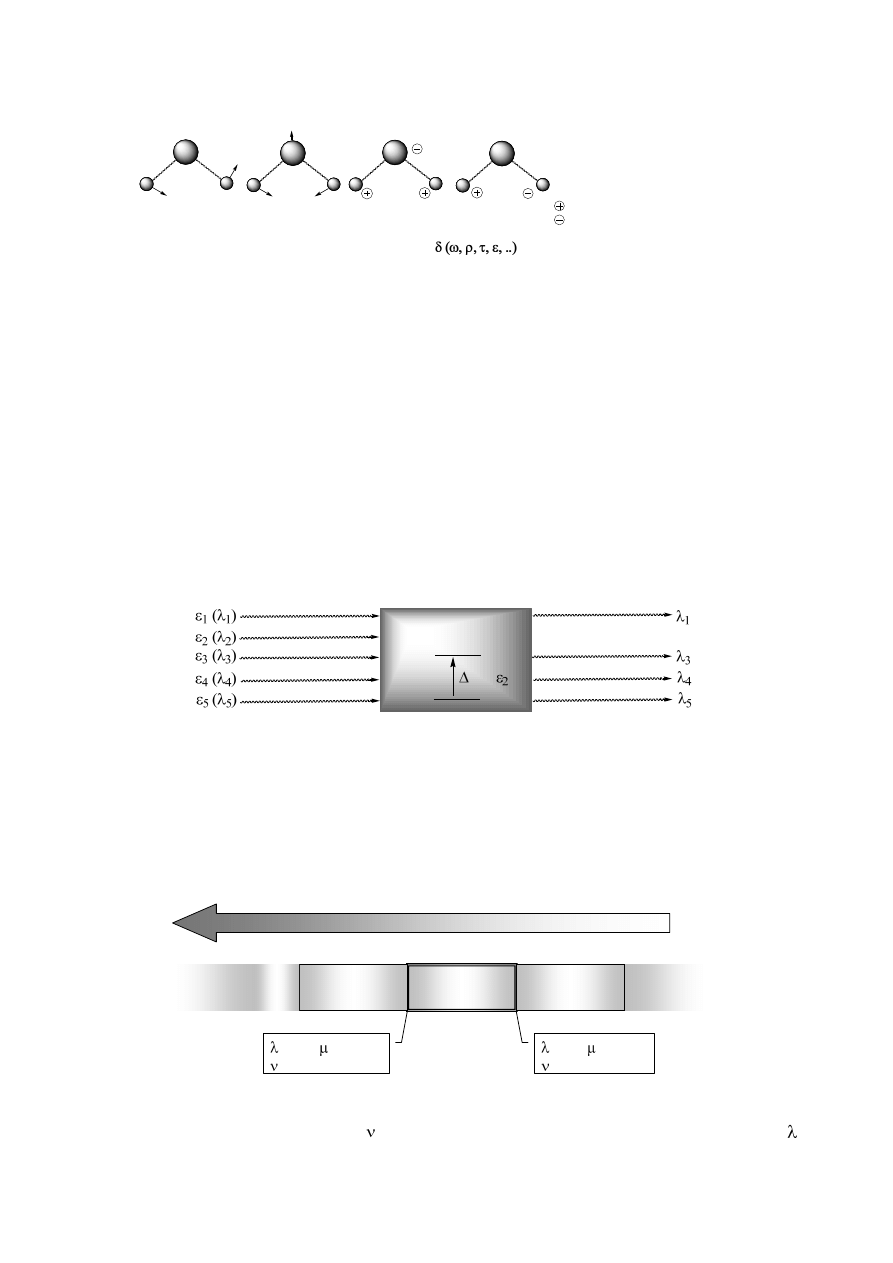
2. RODZAJE DRGAŃ WIĄZAŃ
W temperaturze pokojowej cząsteczka wykonuje trzy rodzaje ruchu: translacje, rotacje
i oscylacje. Z punktu widzenia spektroskopii w podczerwieni, najważniejsze są tutaj oscylacje,
polegające na cyklicznym skracaniu i wydłużaniu się wiązań w cząsteczce, następującymi
z określoną częstotliwością i amplitudą. Oprócz tego wiązania mogą również ulegać deformacjom,
w wyniku których następują zmiany kątów między nimi. Tego typu ruchy są możliwe, ponieważ
wiązania atomowe w cząsteczkach zachowują się jak sprężyny, które mogą:
● rozciągać się symetrycznie
(ν
s
)
lub niesymetrycznie
(ν
as
)
; są to tzw. drgania walencyjne;
w wyniku których następuje głównie zmiana długości wiązań;
DRGANIA ROZCIĄGAJĄCE (WALENCYJNE); oznaczane:
symetryczne
asymetryczne
(następuje zmiana głównie długości wiązań)
2
● zginać się w płaszczyźnie
(δ
s
,
ρ)
lub poza płaszczyzną
(ω, τ)
; są to tzw. drgania
deformacyjne (zginające), które powodują przede wszystkim zmiany kątów między wiązaniami:
DRGANIA (ZGINAJĄCE) DEFORMACYJNE; oznaczane:
wachlarzowe
nożycowe
(dominuje zmiana kątów między wiązaniami)
wahadłowe
wachlarzowe
(poza płaszczyzną)
(w płaszczyźnie)
- ruch
nad płaszczyznę
- ruch pod
płaszczyznę
3. ABSORPCJA ENERGII A WZBUDZENIE DRGAŃ
Absorpcja dostarczonej z zewnątrz energii może spowodować zwiększenie amplitudy drgań
wiązań. Energia cząsteczki nie zmienia się jednak w sposób ciągły, ale jest skwantowana. Przejście
cząsteczki do wyższego stanu oscylacyjnego może nastąpić jedynie w wyniku absorpcji ściśle
określonej ilości energii dopasowanej do różnicy między poziomami energetycznymi. W
temperaturze pokojowej około 99% cząsteczek znajduje się w stanie podstawowym (gdy
oscylacyjna liczba kwantowa v = 0), dlatego najbardziej prawdopodobne jest przejście E
0
→ E
1
(E
0
- stan podstawowy, E
1
– pierwszy stan wzbudzony).
Każdej oscylacji odpowiada jedno pasmo podstawowe, czyli przejście E
0
→ E
1
. Przejścia na
wyższe stany energetyczne
E
0
→ E
2
oraz E
0
→ E
3
widoczne są w widmie w postaci pasm zwanych
nadtonami.
Schemat absorpcji:
PRÓBKA
v=0
v=1
E
0
E
1
E =
Energia oscylacji jest rzędu 1,2 - 120 kJ/mol, co odpowiada energii promieniowania elektro-
magnetycznego w zakresie podczerwieni. Zakres w podczerwieni mieści się pomiędzy
promieniowaniem mikrofalowym a promieniowaniem ultrafioletowym i widzialnym. W zakresie 1
– 100 μm (1 μm = 10
-4
cm = 10
-6
m). W wyniku pochłonięcia promieniowania ε
2
o długości fali λ
2
,
odpowiadającej różnicy energii ΔE między stanem podstawowym E
0
i wzbudzonym E
1
, cząsteczki
związku ulegają wzbudzeniu do wyższego stanu oscylacyjnego (o oscylacyjnej liczbie kwantowej v
= 1).
ENERGIA
= 2,5 m
= 4000 cm
-1
= 25 m
= 400 cm
-1
przejścia elektronowe
podczerwień
IR
bliska
podczerwień
UV
mikrofale
daleka
podczerwień
VIS
przejścia oscylacyjne
przejścia rotacyjne
Zgodnie ze znanymi zależnościami energia absorbowana promieniowania jest wprost
proporcjonalna do częstości fali (
)
, a odwrotnie proporcjonalna do długości fali ( ).
W spektroskopii w podczerwieni tradycyjnie przyjęło się, że określenie absorbowane

3
promieniowanie wyrażane jest tzw. liczbą falową ( ν , czyt. „ni z kreską”), której jednostką jest
1/cm czyli cm
-1
, zwany „centymetrem odwrotnym”. Jest to wielkość wprost proporcjonalna do
częstości (i energii) promieniowania, ale wyrażana w innych jednostkach (
[Hz] = 100 c
ν )!
Poniżej podano sposób przeliczania liczby falowej na długość fali wyrażoną w μm (alternatywny
sposób określania promieniowania absorbowanego podczas pomiaru widm IR).
hc
hν
E
]
m
[
[m/s]
c
[Hz]
ν
[nm, m, cm, m] – długość fali
]
m
[
10000
]
cm
[
1
]
[cm
ν
1
-
μ
]
[cm
ν
10000
]
m
[
1
-
μ
h = 6,62 10
-34
[J s] - stała Plancka
c = 3 10
8
[m/s]- prędkość światła,
[Hz = 1/s] – częstość
ν [cm
-1
] - liczba falowa
W celu ustalania struktury związków organicznych najczęściej wykorzystuje się pomiary
promieniowania pochłanianego przez związek w przedziale długości fal 2,5 - 25 μm (1 μm = 10
-4
cm = 10
-6
m), co odpowiada liczbie falowej 4000 - 400 cm
-1
. Widma w zakresie tzw. dalekiej
podczerwieni (poniżej 400 cm
-1
) oraz bliskiej podczerwieni (powyżej 4000 cm
-1
) nie
będą
omawiane podczas tego kursu.
4. DRGANIA AKTYWNE W PODCZERWIENI – REGUŁY WYBORU
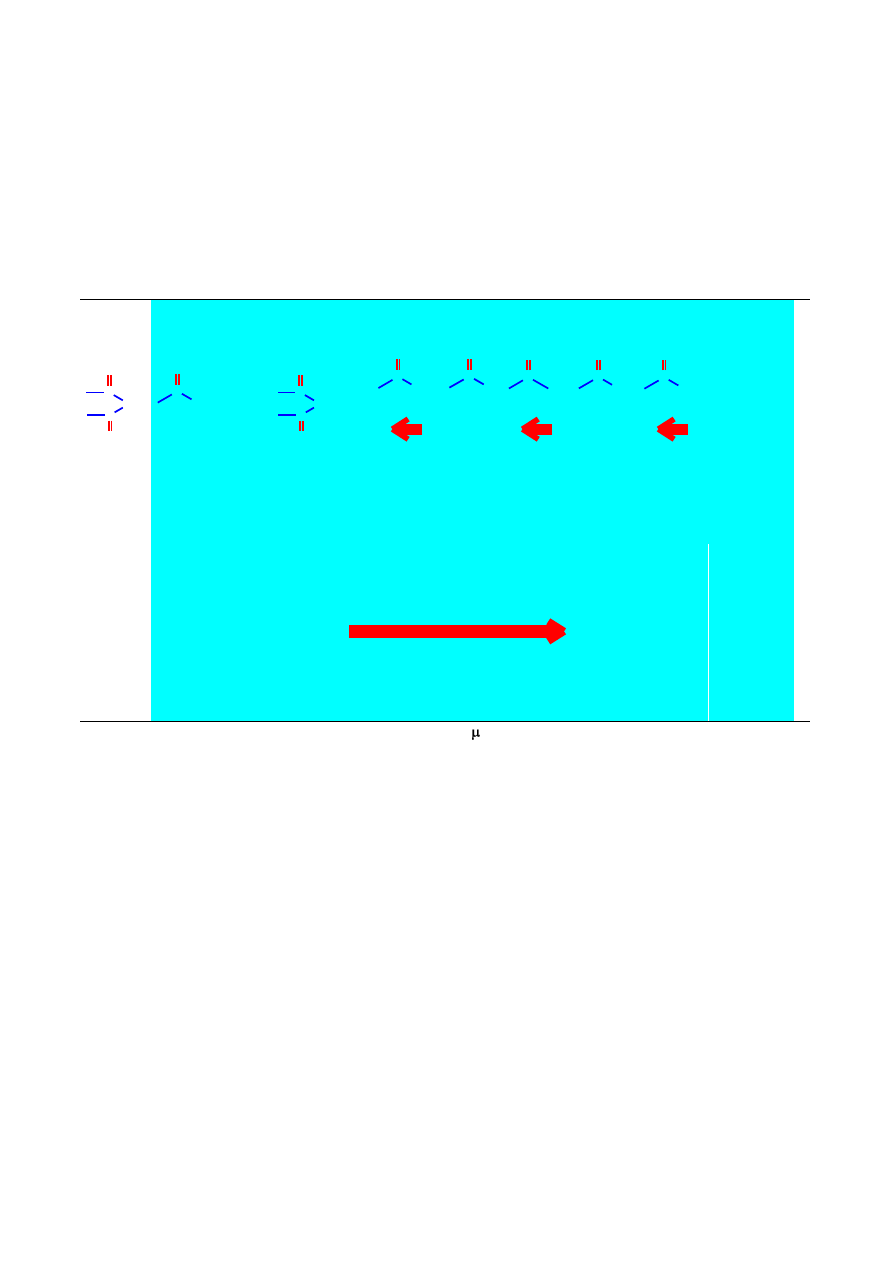
Poza odpowiednim dopasowaniem kwantu energii, warunkiem absorpcji promieniowania,
czyli przejścia cząsteczki do wyższego stanu oscylacyjnego, jest oddziaływanie promieniowania
elektromagnetycznego z dipolem wiązania spolaryzowanego. Absorpcja kwantu promieniowania
z zakresu podczerwieni, o odpowiedniej energii, zajdzie tylko wtedy, kiedy wzbudzanemu w ten
sposób drganiu będzie towarzyszyła zmiana momentu dipolowego cząsteczki (Δμ 0). Mówi się
wtedy, że drganie takie jest aktywne w podczerwieni. Symetryczne wiązania, obecne w
cząsteczkach np. H
2
, Br
2
lub wiązanie podwójne w etenie są nieaktywne i nie absorbują
promieniowania w zakresie IR, ponieważ wiązania nie są spolaryzowane i nie posiadają momentu
dipolowego (brak różnicy elektroujemności). Przykładowo, wiązanie podwójne w alkenach lub
wiązanie potrójne w alkinach będą aktywne i będą absorbować promieniowanie tylko wtedy, gdy
atomy połączone tymi wiązaniami będą niesymetrycznie podstawione.
Widmo absorpcyjne przedstawione jest zazwyczaj w postaci zależności transmitancji T [%]
od liczby falowej
ν [cm
-1
]
.
Transmitancja określona jest przez stosunek natężenia wiązki światła po
przejściu przez próbkę (I) do natężenia wiązki światła monochromatycznego (I
0
) padającego na
próbkę:
T [%] = I / I
0
Transmitancja może przyjmować wartości od 0 do 100 %. Wartość T wynosząca 100%
odpowiada przypadkowi, gdy substancja nie absorbuje światła o określonej długości fal, a T = 0%
oznacza całkowitą absorpcję promieniowania.
Transmitancja nie jest liniową funkcją stężenia substancji pochłaniającej. Taką cechę
posiada natomiast absorbancja (A), definowana jako logarytm odwrotności transmitancji:
A = log (1/T) = log (I
0
/I) ~ l c
gdzie:
c – stężenie roztworu; l – grubość warstwy roztworu absorbującego
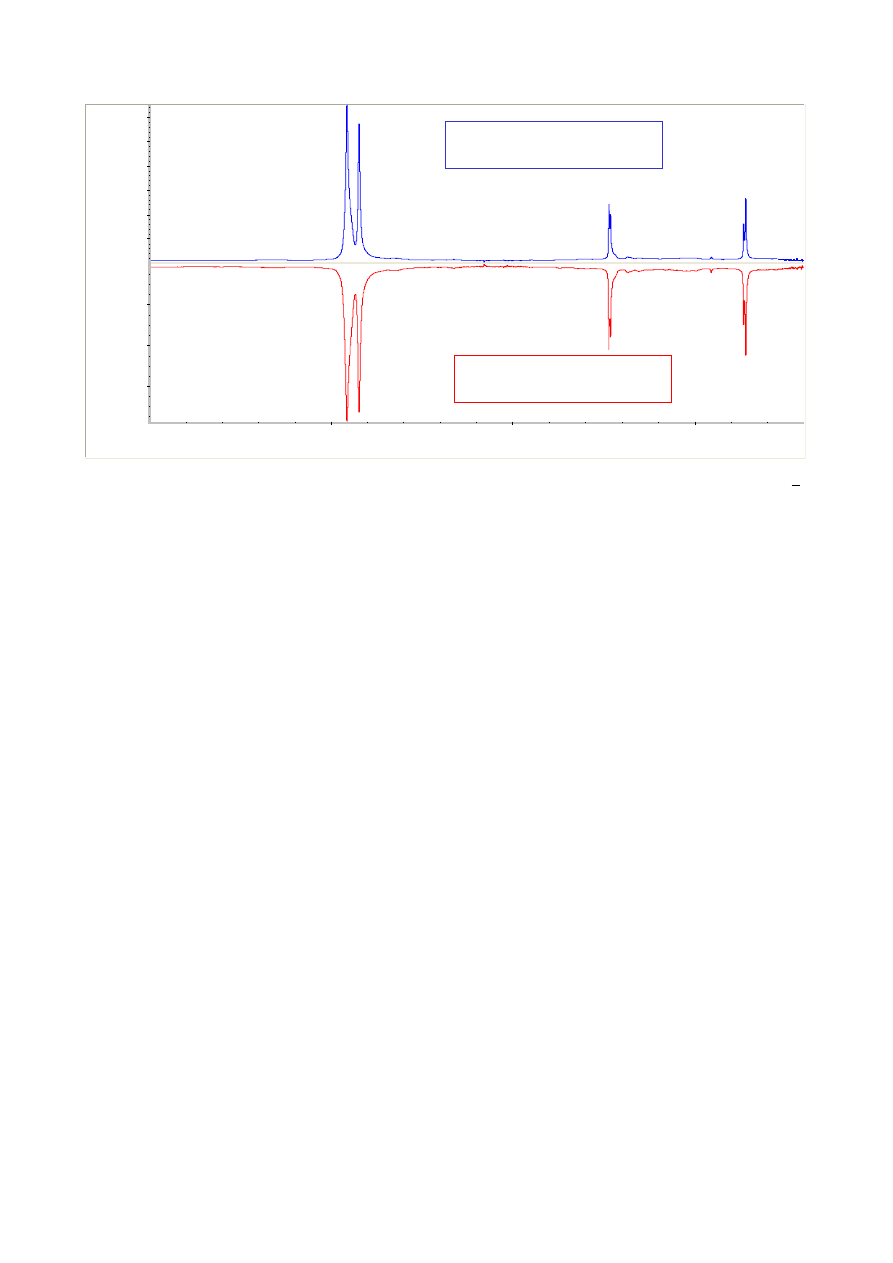
4
Porównanie widma IR zarejestrowanego w skali absorbancji i transmitancji
0,1
0,2
0,3
0,4
0,5
0,6
Ab
s
40
60
80
%T
1000
2000
3000
Liczby falowe (cm-1)
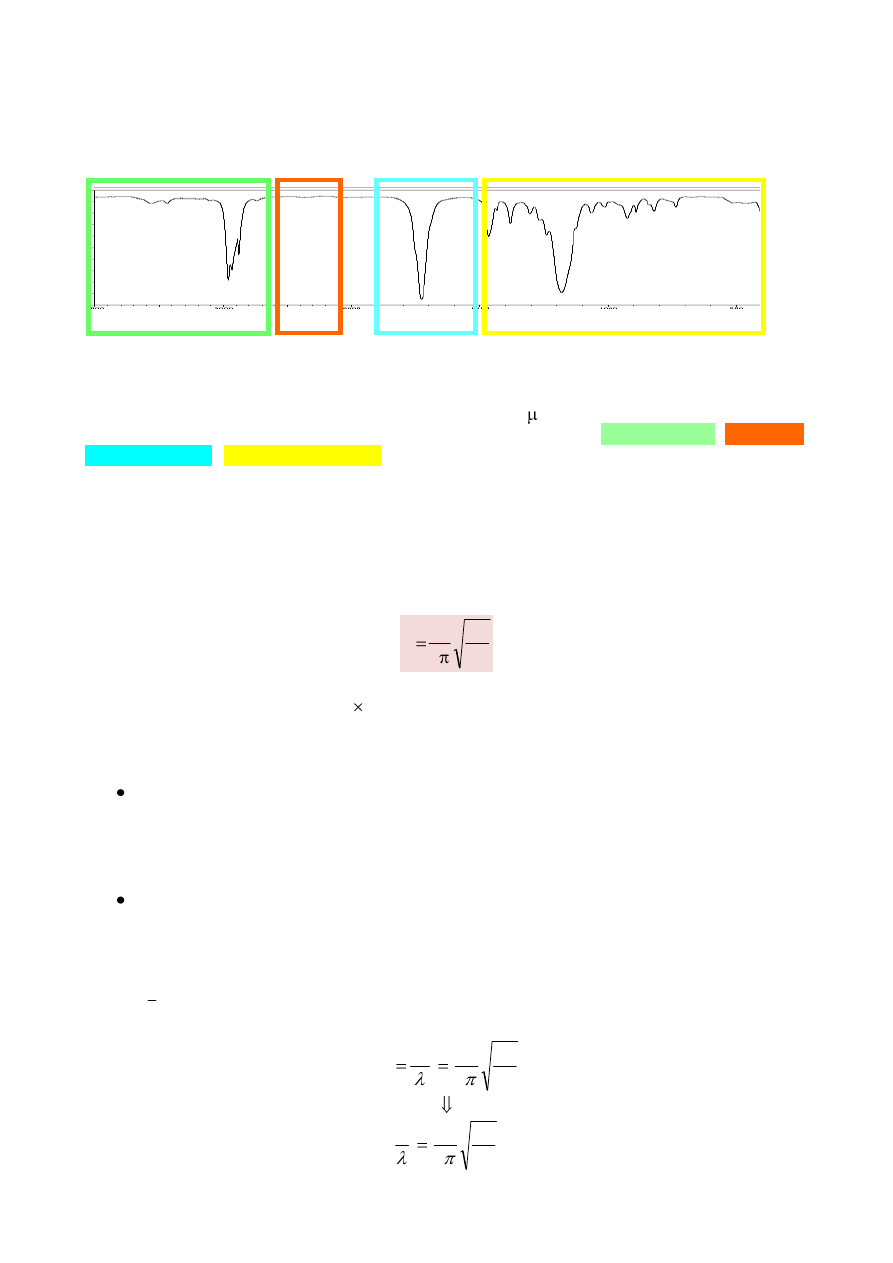
Ważną cechą każdego obserwowanego pasma, oprócz wartości liczby falowej ν ,
odpowiadającej określonej długości absorbowanej fali λ, jest również jego intensywność, której
przybliżoną miarą może być transmitancja T. Wartość momentu dipolowego zależy od różnicy
elektroujemności pomiędzy atomami tworzącymi wiązanie oraz od jego długości. Ponieważ długość
wiązania zmienia się w trakcie drgań walencyjnych, to zmianie ulega również wartość momentu
dipolowego wiązania. Wielkość tej zmiany (Δμ) decyduje o intensywności pasma absorpcji
proporcjonalnie do wartości momentu dipolowego. Dlatego z pewnym uproszczeniem można
przyjąć zależność, że wraz ze wzrostem wartości momentu dipolowego μ spolaryzowanego
wiązania atomowego wzrasta intensywność odpowiadającego mu pasma w widmie w podczerwieni.
Widmo polietylenu
w skali absorbancji (Abs)
Widmo polietylenu
w skali transmitancji (T [%])
5
5. PRZEWIDYWANIE POŁOŻENIA PASM ABSORPCJI
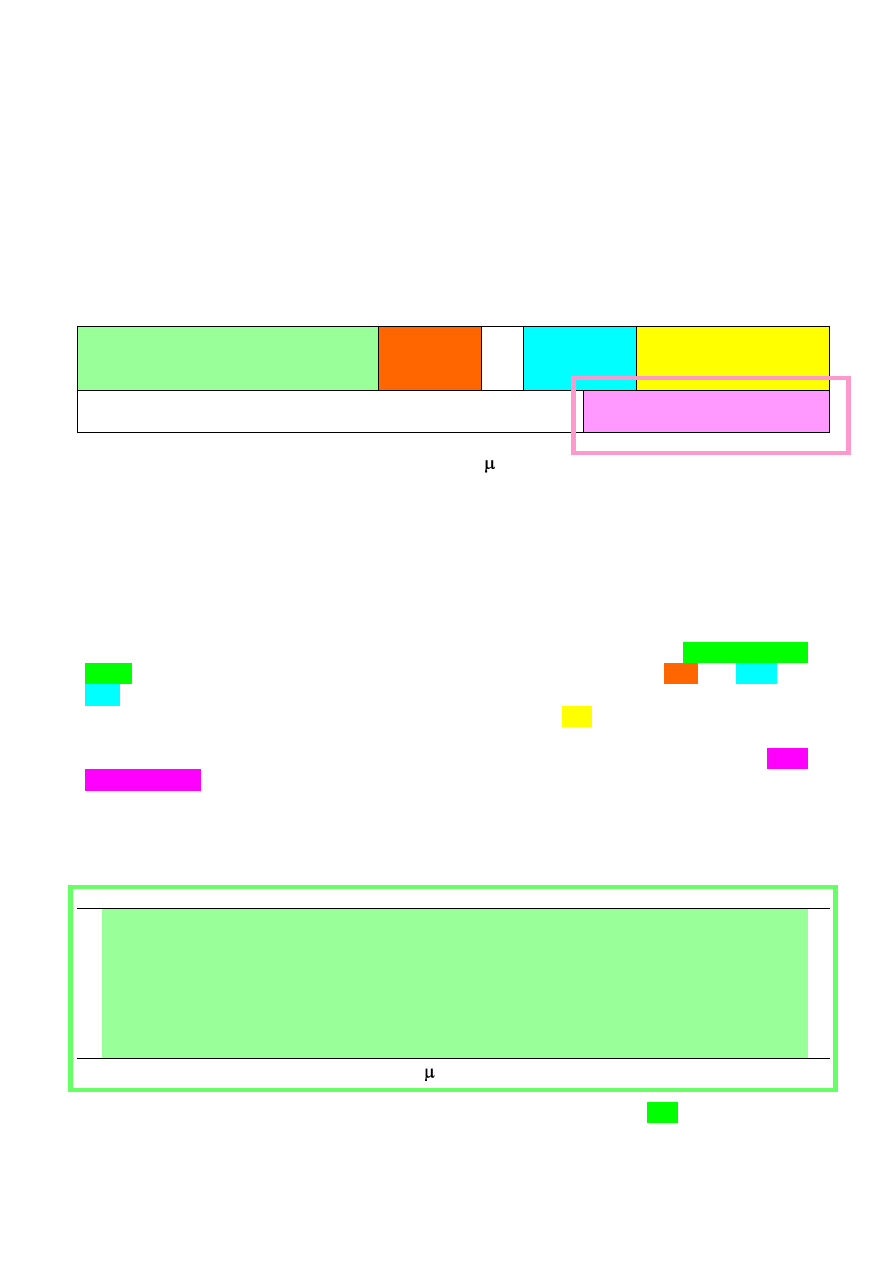
Typowe zakresy absorpcji dla różnych typów wiązań oznaczone różnymi kolorami
Liczba falowa [cm
-1
]
Każde pasmo absorpcji w widmie odpowiada wzbudzeniu innego rodzaju drgań atomów, a
położenie tych pasm określane w cm
-1
(znacznie rzadziej w m), dostarcza informacji o strukturze
cząsteczki. Absorpcja promieniowania przez spolaryzowane wiązanie (O-H, N-H, C-H, C≡C, C≡N,
C=O, C=N, C=C, C-C, C-N, C-O, C-X) daje pasmo absorpcji w określonym fragmencie widma.
Część widma w zakresie od 1500 do 400 cm
-1
nazywana jest często obszarem daktyloskopowym
(tzw. fingerprint). Określenie to wynika z faktu, iż układ pasm w tym zakresie jest bardzo
charakterystyczny dla danego związku, jednak równocześnie jest on też zazwyczaj bardzo złożony i
przez to trudny do interpretacji. Największą ilość jednoznacznych informacji o grupach funkcyjnych
znajdujących się w badanym związku dostarczają pasma położone w obszarze 4000-1500 cm
-1
.
Jeżeli taki układ dwuatomowy o masach m
1
i m
2
potraktujemy jako oscylator harmoniczny (co
stanowi duże uproszczenie!), to jego energię można wyrazić równaniem:
r
m
k
2
h
E
gdzie: k - stała siłowa,
m
r
- masa zredukowana = (m
1
m
2
)/(m
1
+
m
2
).
Stała siłowa k jest miarą „sztywności wiązania”. Wartość stałej k wzrasta między innymi
wraz:
ze wzrostem rzędu (krotności) wiązania, czy też zmniejszeniem jego długości; długość
wiązania zależy bowiem nie tylko od krotności wiązania i rodzaju atomów, ale również od
rodzaju hybrydyzacji, jakiej ulega atom tworząc dane wiązanie – im większy jest w niej
udział orbitalu s (np. 50% dla hybrydyzacji sp, a tylko 25% dla hybrydyzacji sp
3
), tym
wiązanie tworzone przez dany atom jest krótsze.
ze wzrostem polarności (momentu dipolowego) wiązania.
Energia promieniowania elektromagnetycznego zaabsorbowana przez wiązanie musi
odpowiadać różnicy między kolejnymi poziomami oscylacji. Na podstawie poniższych równań
opisujących energię promieniowania elektromagnetycznego można określić zależność liczby
falowej
ν od stałej siłowej wiązania k oraz od mas atomów m
1
i m
2
, czyli przewidywać wartość
długości fali λ promieniowania zaabsorbowanego.
r
m
k
2
h
hc
hν
r
m
k
2
1
c
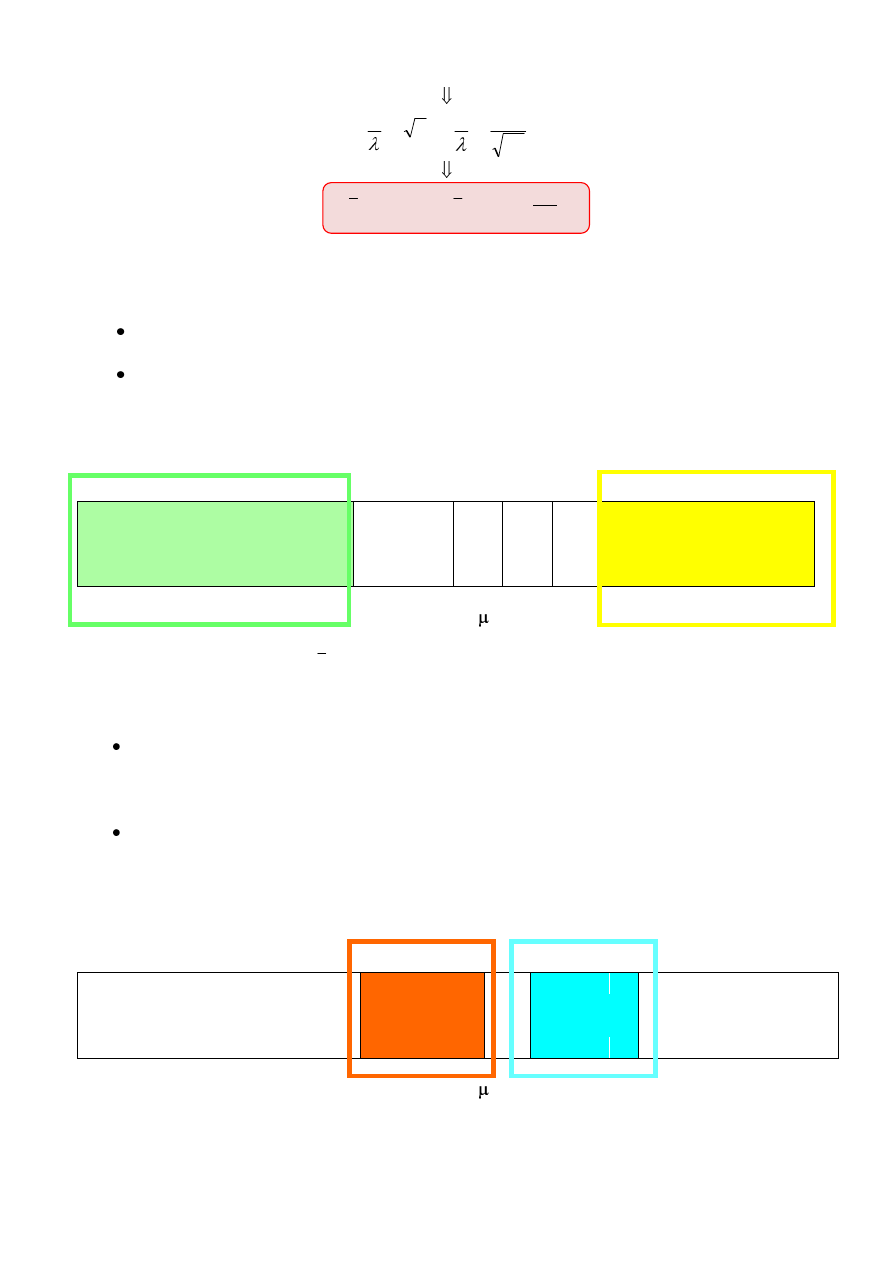
6
k
~
1
r
m
1
~
1
k
~
]
[cm
ν
-1
~
]
[cm
ν
-1
r
m
1
Zgodnie z zależnością, wraz ze wzrostem masy atomów połączonych wiązaniem, które
zostaje wzbudzone, maleje liczba falowa wzbudzającego promieniowania. Na tej podstawie
można określić dwa zakresy występowania pasm absorpcji pochodzących od drgań walencyjnych
wiązań pojedynczych:
4000 - 2500 cm
-1
– typowy wyłącznie dla wiązań tworzonych przez atomy wodoru,
charakteryzujące się małą masą atomową (np. ok. 3000 cm
-1
dla wiązań C-H);
1550 - 400 cm
-1
– charakterystyczny dla wiązań między atomami o większych masach
atomowych (np. ok. 1200 cm
-1
dla C-C i C-N; ok 1100 cm
-1
dla C-O; ok. 750 cm
-1
dla
C-Cl; ok. 600 cm
-1
dla C-Br oraz poniżej 500 cm
-1
dla C-I).
Zakresy występowania pasm absorpcji pochodzących od drgań walencyjnych wiązań pojedynczych.
Liczba falowa [cm
-1
]
4000
3000 2500
2000 1800 1650
1550
400
O
-
H
≡C
–
H
C
-
C
N
-
H
=C
–
H
C
-
N
–C
–
H
C
-
O
C
-
X
2,5
4,0
5,0
5,5
6,1
6,5
25
Długość fali [
m]
Zgodnie z zależnością ν ~ k, gdy stała siłowa wiązania wzrasta, to rośnie również
wartość liczby falowej. Z tego powodu wiązania wielokrotne (krótsze i mocniejsze niż wiązania
pojedyncze, cechujące się więc wyższą stałą siłową) absorbują promieniowanie o wyższej energii
(liczbie falowej) niż wiązania pojedyncze:
wiązania podwójne (tworzone przez atomy o hybrydyzacji sp
2
) w zakresie 1800 - 1550 cm
-1
(C=O: 1650-1800 cm
-1
; C=N: 1650 -1640 cm
-1
; C=C 1640 - 1550 cm
-1
); należy zwrócić
uwagę, że im większa wartość momentu dipolowego wiązania, tym wiązanie absorbuje
promieniowanie o większej energii (liczbie falowej)
wiązania potrójne i skumulowane wiązania podwójne (czyli ugrupowania zawierające atomy
o hybrydyzacji sp) w zakresie 2500 – 2000 cm
-1
(niesymetrycznie podstawione C≡C oraz
C≡N: ok. 2200 cm
-1
; X=C=X ok. 2100 cm
-1
).
Zakresy występowania pasm absorpcji pochodzących od drgań walencyjnych wiązań wielokrotnych.
Liczba falowa [cm
-1
]
4000 3000
2500
2000
1800 1650 1550
400
C
≡
C
C
=
O
C
≡
N
C
=
N
X
=
C
=
Y
C
=
C
X=C,O,N,S
2,5
4,0
5,0
5,5
6,1
6,5
25
Długość fali [
m]
Na pasma absorpcyjne drgań walencyjnych
(ν)
nakładają się pasma absorpcyjne drgań
deformacyjnych
(δ)
. Ze względu na to, że do zginania potrzebna jest niższa energia niż do
7
rozciągania i ściskania wiązania, zakres drgań deformacyjnych występuje od 1650 do 400 cm
-1
.
Zakres drgań deformacyjnych pokrywa zakres drgań walencyjnych wiązań pojedynczych C-C, C-N,
C-O, C-X oraz część zakresu drgań wiązań podwójnych C=N i C=C. Przewidywanie zakresów
absorpcji charakterystycznych dla poszczególnych typów drgań deformacyjnych jest dość złożone i
wykracza poza ramy tego kursu. Aby nie popełniać błędów przy interpretacji widm IR, należy
zdawać sobie jednak sprawę, że np. zakresie 1650 - 1550 cm
-1
, charakterystycznym dla wiązań
podwójnych, obserwuje się drgania zginające wiązań N-H amin pierwszorzędowych.
Porównanie zakresów występowania pasm absorpcji pochodzących od drgań walencyjnych (górny
diagram) i deformacyjnych (dolny diagram).
Liczba falowa [cm
-1
]
4000 3000 2500 2000 1800
1650 1550
400
O
-
H
C
-
H
C
≡
C
C
=
O
C
=
N
C
-
C
C
-
O
C
≡
N
C
=
C
C
-
N
C
-
X
N
-
H
Zakres drgań deformacyjnych
2,5
4,0
5,0
5,5
6,1 6,5
25
Długość fali [
m]
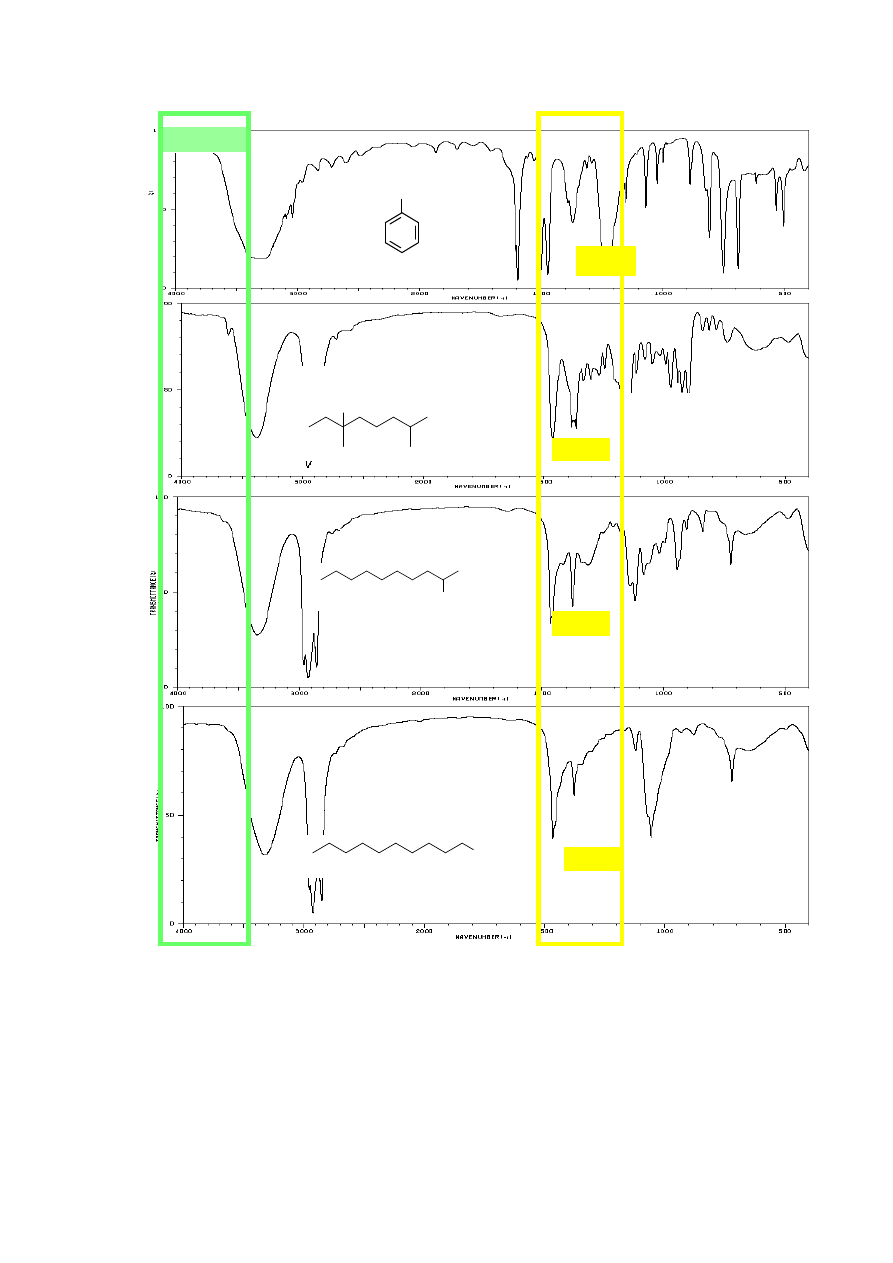
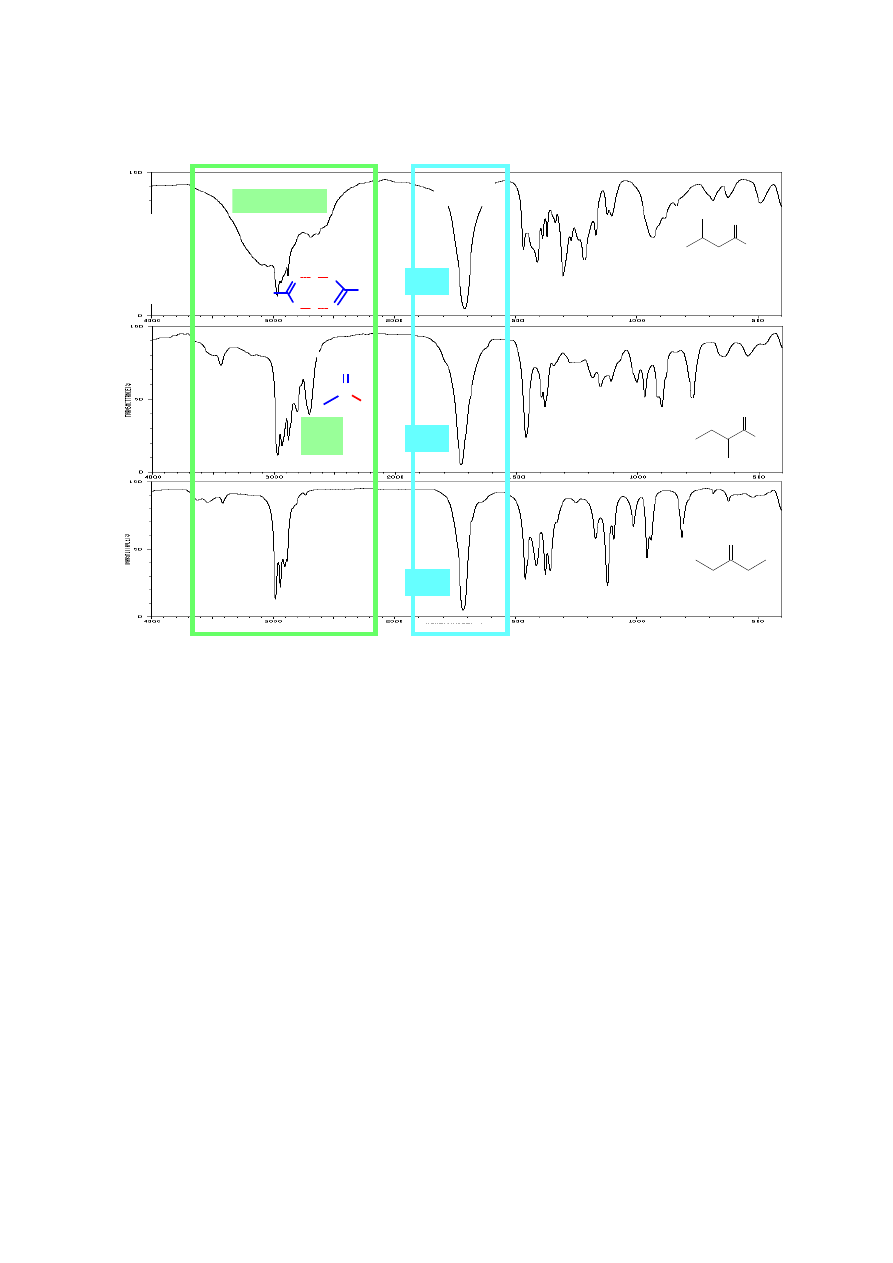
6. ANALIZA ZAKRESU PASM ABSORPCJI DRGAŃ WALENCYJNYCH
WIĄZAŃ
C
-
H
(alkany, alkeny, alkiny, związki aromatyczne, aldehydy).
Zróżnicowany udział orbitalu s w hybrydyzacji atomów węgla ma również wpływ na
długość i wartość stałej siłowej k wiązań C-H. Najkrótsze jest wiązanie atomu wodoru z atomem
węgla o hybrydyzacji sp (3300 cm
-1
), następnie o hybrydyzacji sp
2
(3100-3010 cm
-1
) i najdłuższe w
związkach nasyconych (3000-2840 cm
-1
). Obecność pasm absorpcji drgań wiązań Csp
3
-H, Csp
2
-H i
Csp-H oraz występowanie (lub brak) pasm absorpcji wiązań wielokrotnych (C≡C lub C=C oraz
C=O) umożliwia rozróżnienie między alkanem, alkenem, alkinem i aldehydem na podstawie analizy
widm IR. Pasma drgań walencyjnych wiązań pojedynczych C-C nie podlegają interpretacji,
ponieważ nie są one intensywne ze względu na niewielką wartość momentu dipolowego wiązań.
Dodatkowych informacji o szkielecie węglowym może dostarczyć obecność pasm drgań
deformacyjnych grup CH
3
występujących zazwyczaj przy 1375 cm
-1
i CH
2
przy około 1465 cm
-1
,
ale dla grupy CH
2
podstawionej fluorowcem pasma drgań występują odpowiednio przy 1300-1230
cm
-1
(CH
2
Cl), 1250-1190 cm
-1
(CH
2
Br ) i 1200-1150 cm
-1
(CH
2
I).
Zakresy wy
stępowania pasm absorpcji pochodzących od drgań walencyjnych wiązań C-H.
4000
3500
Liczba falowa [cm
-1
]
3000
2500
3300
3100
3000
2850
2750
≡C
–
H
=C
–
H
–C
–
H
–C(=O)
–
H
sp
– 1s
sp
2
– 1s
sp
3
– 1s
sp
2
– 1s
winylowy
alifatyczny
aldehydy
aromatyczny
O
–C
–
H
N
–C
–
H
sp
3
– 1s
cyklopropyl
np. etery, alkohole, aminy
2,5
Długość fali [
m]
4,0
Jak wspomniano wcześniej, na położenie pasm wiązań walencyjnych C-H ma wpływ udział
orbitalu s w hybrydyzacji atomów węgla, stąd w widmach alkanów widoczne są pasma przy 3000-
2840 cm
-1
, alkeny posiadające wiązania Csp
2
-H absorbują promieniowanie o liczbie falowej ok.
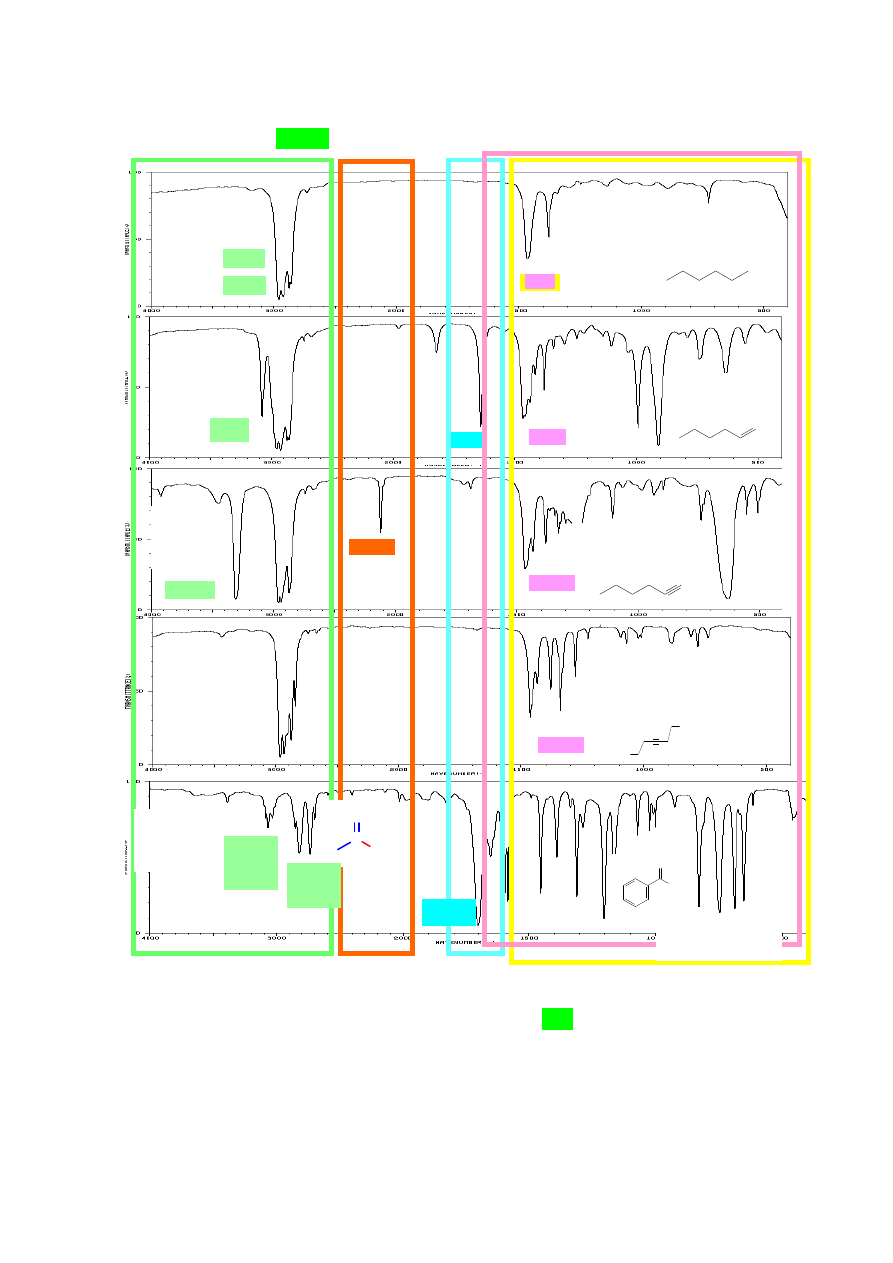
8
3100-3010 cm
-1
, a terminalne alkiny dają charakterystyczne, ostre pasma przy ok. 3300 cm
-1
.
Również widma związków zawierających pierścień aromatyczny charakteryzują się obecnością
pasm drgań walencyjnych Csp
2
-H ok. 3100 – 3000 cm
-1
, czyli w tym samym zakresie co alkeny.
Należy zwrócić uwagę, że obecność atomu o wysokiej elektroujemności (np. tlenu, azotu)
przy atomie węgla powoduje zmianę stałej siłowej wiązania C-H i przesunięcie pasma absorpcji
w stronę niższych wartości liczb falowych. Dlatego pasmo drgań walencyjnych C-H w grupie CHO
aldehydu, występuje zazwyczaj w postaci dwóch średnio intensywnych pasm przy ok. 2820 i ok.
2700 cm-
1
. Pojawienie się dwóch pasm spowodowane jest przez rezonans Fermiego (sprzężenie
drgań rozciągających C-H z nadtonem drgań deformacyjnych wiązań O=C-H). Pierwsze z pasm
może być zakryte przez pasma drgań walencyjnych grup alifatycznych C-H, ale to drugie jest na
ogół dość intensywne.
2928
2959
1470
3079
1650
3316
2120
ALKENY:
heks-1-en
ALKANY:
n-heksan
ALKINY:
heks-1-yn
(terminalny)
okt-4-yn
–C
–
H
sp
3
-1s
=C
–
H
sp
2
-1s
≡C
–
H
sp-1s
–C
–
H
sp
3
-1s
1465
1464
1462
C
≡
C
C
O
H
ALDEHYDY:
benzaldehyd
H
O
O
H
1703
=C
–
H
sp
2
-1s
3086
3065
3061
2820
2756
9
8. ANALIZA ZAKRESU PASM ABSORPCJI DRGAŃ WALENCYJNYCH
WIĄZAŃ
O
-
H
(alkohole, fenole, kwasy karboksylowe).
W podobnym zakresie co pasma absorpcyjne drgań walencyjnych C-H pojawiają się również
pasma drgań walencyjnych O-H i N-H (2500- 4000 cm
-1
). Intensywność oraz szerokość pasm grupy
O-H zależy od obecności wiązań wodorowych. Wolne, nie związane grupy OH alkoholi dają ostre
mało intensywne pasmo przy 3600 cm
-1
, a związane wiązaniem wodorowym grupy dają szerokie,
charakterystyczne pasmo o dużo większej intensywności, przesunięte w stronę niższych wartości
liczby falowej (dla alkoholi i fenoli przy 3400 - 3000 cm
-1
oraz dla kwasów karboksylowych przy
3400 -2500 cm
-1
).
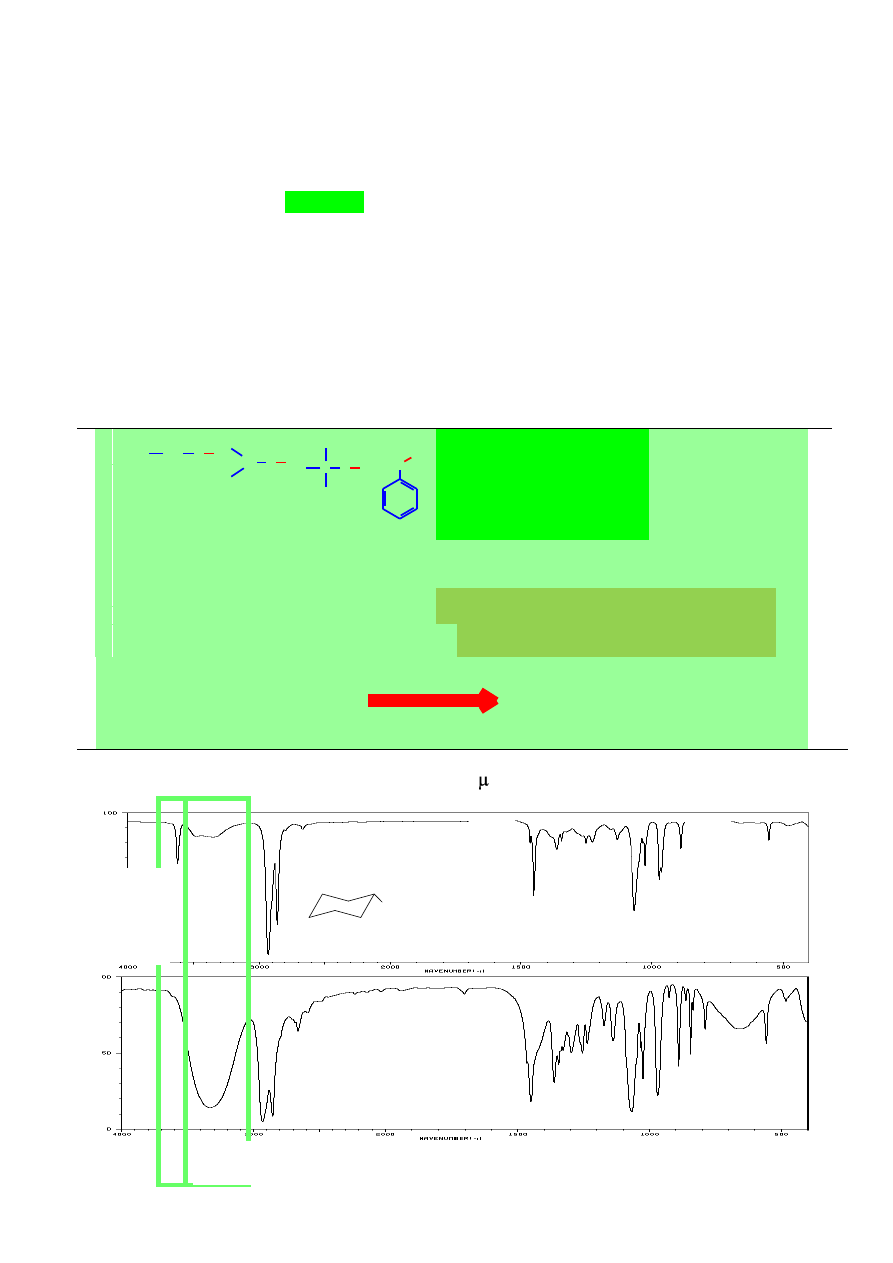
Zakresy wys
tępowania pasm absorpcji pochodzących od drgań walencyjnych wiązań O-H
niezwiązanych i związanych wiązaniem wodorowym.
Liczba falowa [cm
-1
]
4000
2400
3640
3630
3620
3610
3400
-
3000
O
-
H
alkoholi
szerokie
pasmo (OH związane
wiązaniem wodorowym)
O
-
H
alkoholi
ostre pasmo nie związane
wiązaniem wodorowym
3400
-
2500
O
-
H
kwasów
szerokie
pasmo dimerów związanych wiązaniem
wodorowym
kierunek przesunięcia pasma
(wiązanie wodorowe osłabia stałą siłową wiązania O-H)
2,5
Długość fali [
m]
O
H
CH
2
O
H
CH
O
H
C
O
H
cykloheksanol (CCl
4
)
cykloheksanol (film)
Pasmo drgań
O
-
H
związanych wiązaniem
wodorowym
Pasmo drgań
O
-
H
nie
związanych
wiązaniem
wodorowym
OH
10
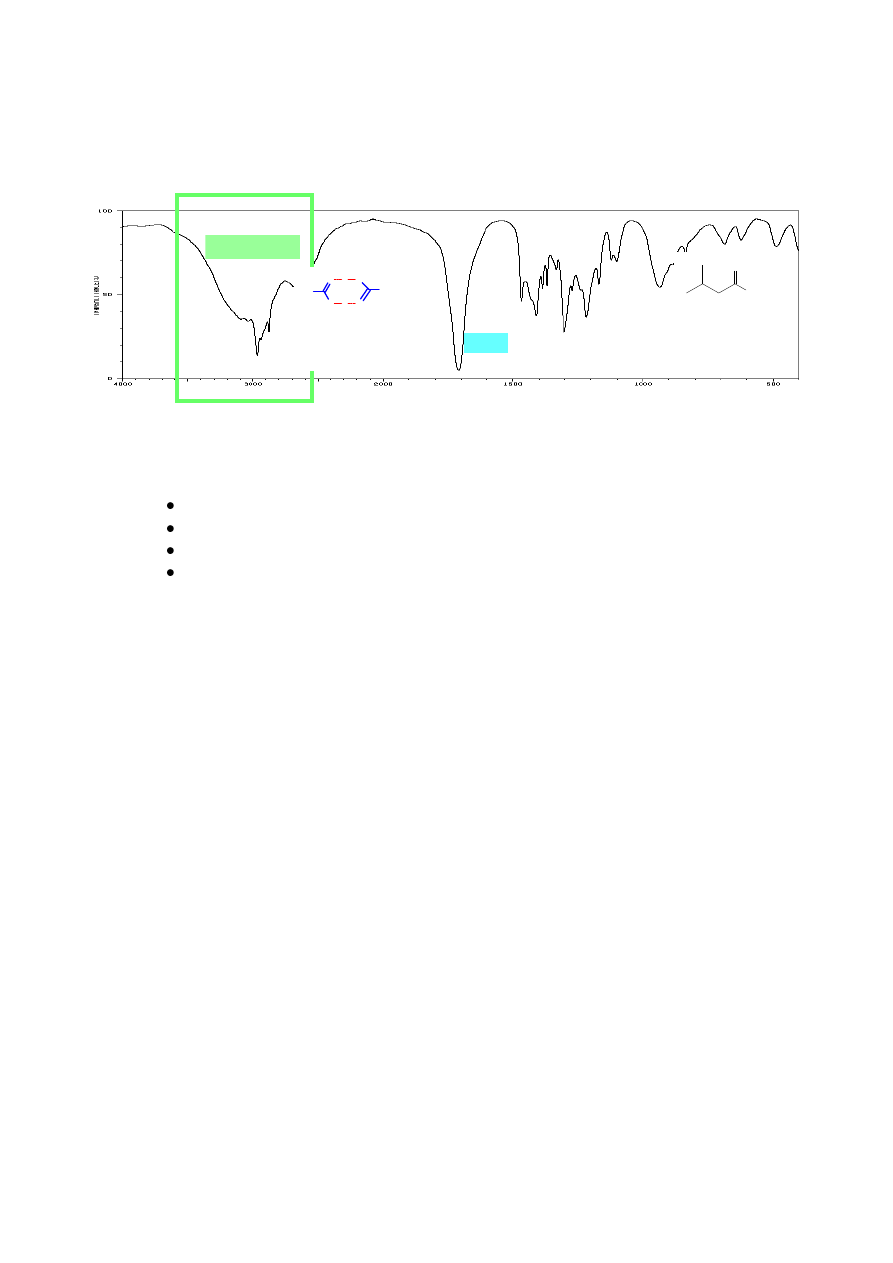
W widmie cykloheksanolu wykonanym dla czystej cieczy występuje szerokie intensywne
pasmo związanej wiązaniem wodorowym grupy O-H. Ostre pasmo drgań wolnej grupy O-H jest
obserwowane gdy widmo alkoholu jest wykonane w rozpuszczalniku (zazwyczaj w CCl
4
).
Rozcieńczenie, w dużym stopniu zmniejsza liczbę międzycząsteczkowych wiązań wodorowych.
W zakresie drgań walencyjnych alkoholi obecne jest również pasmo C-O przy około 1100 cm
-1
. Na
podstawie położenia tych pasm można określić rzędowość alkoholi i odróżnić od nich fenol.
Położenie pasm drgań walencyjnych C-O występujących przy około:
1220 cm
-1
wskazuje na obecności fenolu,
1150 cm
-1
wskazuje na obecność alkoholu 3˚
1100 cm
-1
wskazuje na obecność alkoholu 2˚
1060 cm
-1
wskazuje na obecność alkoholu 1˚
3500 - 2500
1710
KWASY KARBOKSYLOWE:
kw. 3-metylobutanowy
(izowalerianowy)
O
OH
O
H
O
H
O
R
R
O
Szerokie pasmo
dimerów związanych
wiązaniem
wodorowym
11
3400-3000
fenol
OH
3,7-dimetylooktan-3-
ol (3°)
OH
Pasmo drgań
walencyjnych
C
-
O
1236
1154
Pasmo drgań
O
-
H
związanych
wiązaniem
wodorowym
1116
dekan-2-
ol (2°)
OH
1058
dekan-1-
ol (1°)
OH
12
9. ANALIZA ZAKRESU PASM ABSORPCJI DRGAŃ WALENCYJNYCH
WIĄZAŃ
N
-
H
(aminy i amidy).
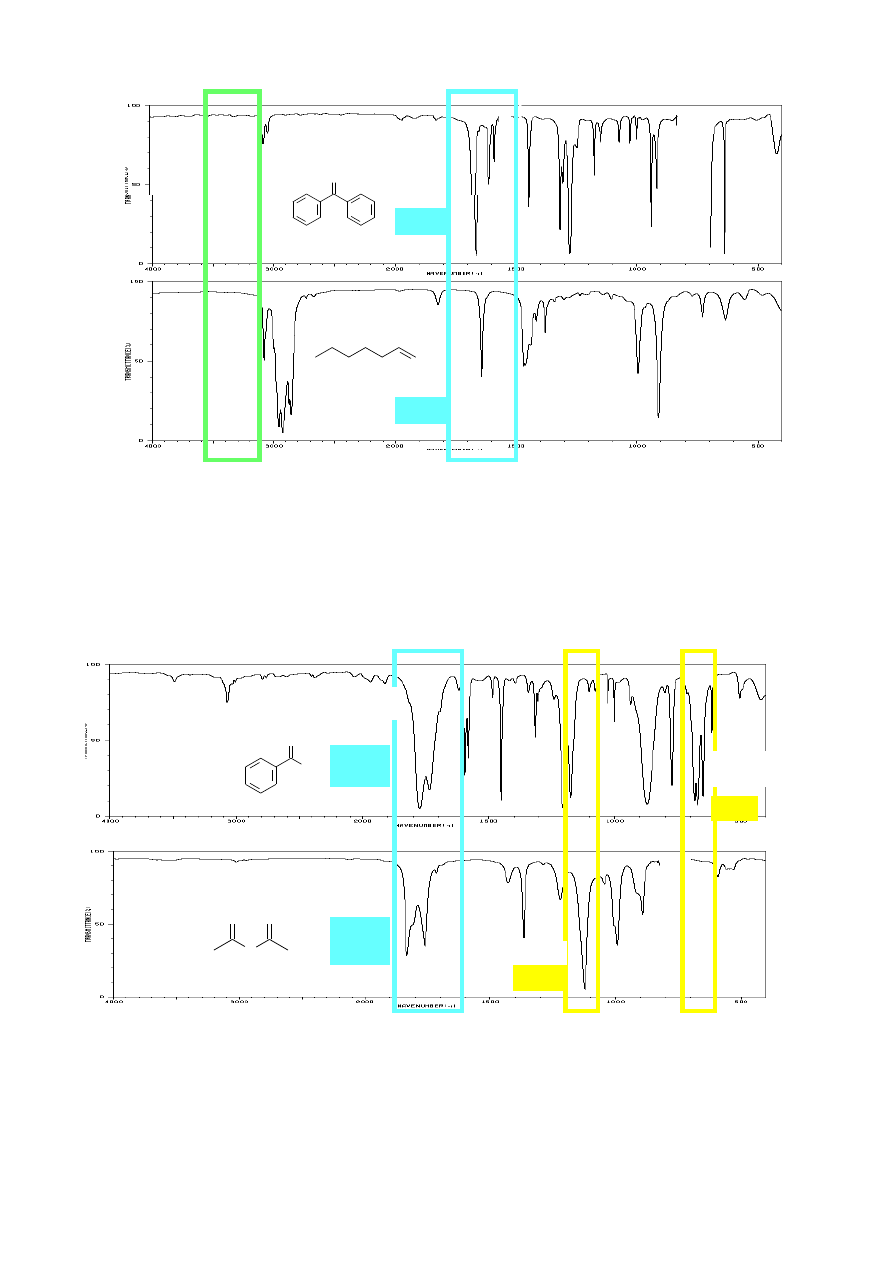
Pierwszorzędowe aminy dają dwa (a czasami trzy) pasma absorpcji dla grupy NH
2
, dla drgań
symetrycznych (ν
s
) i asymetrycznych (ν
as
) w charakterystycznym zakresie drgań walencyjnych 3500
- 3300 cm
-1
oraz pasmo drgań deformacyjnych 1640 - 1550 cm
-1
. Pojawienie się trzeciego pasma w
zakresie 3500 - 3300 cm
-1
jest związane z obecnością nadtonu drgań deformacyjnych N-H.
Drugorzędowe aminy można zidentyfikować na podstawie jednego pasma N-H w zakresie 3500 -
3300 cm
-1
, podczas gdy aminy trzeciorzędowe nie dają żadnego pasma w tym zakresie.
Amidy pierwszorzędowe i drugorzędowe można rozróżnić na podstawie liczby pasm N-H
w zakresie w zakresie 3400 – 3100 cm
-1
. Amidy pierwszorzędowe dają dwa pasma przy około 3180
i 3350 cm
-1
, Amidy drugorzędowe dają jedno pasmo przy około 3300 cm
-1
. Na pasmo drgań
deformacyjnych (δ) grupy N-H może nałożyć się pasmo grupy karbonylowej. Amidy
trzeciorzędowe ze względu na brak pasm N-H trudno jest zidentyfikować
10. ANALIZA ZAKRESU PASM ABSORPCJI DRGAŃ WALENCYJNYCH
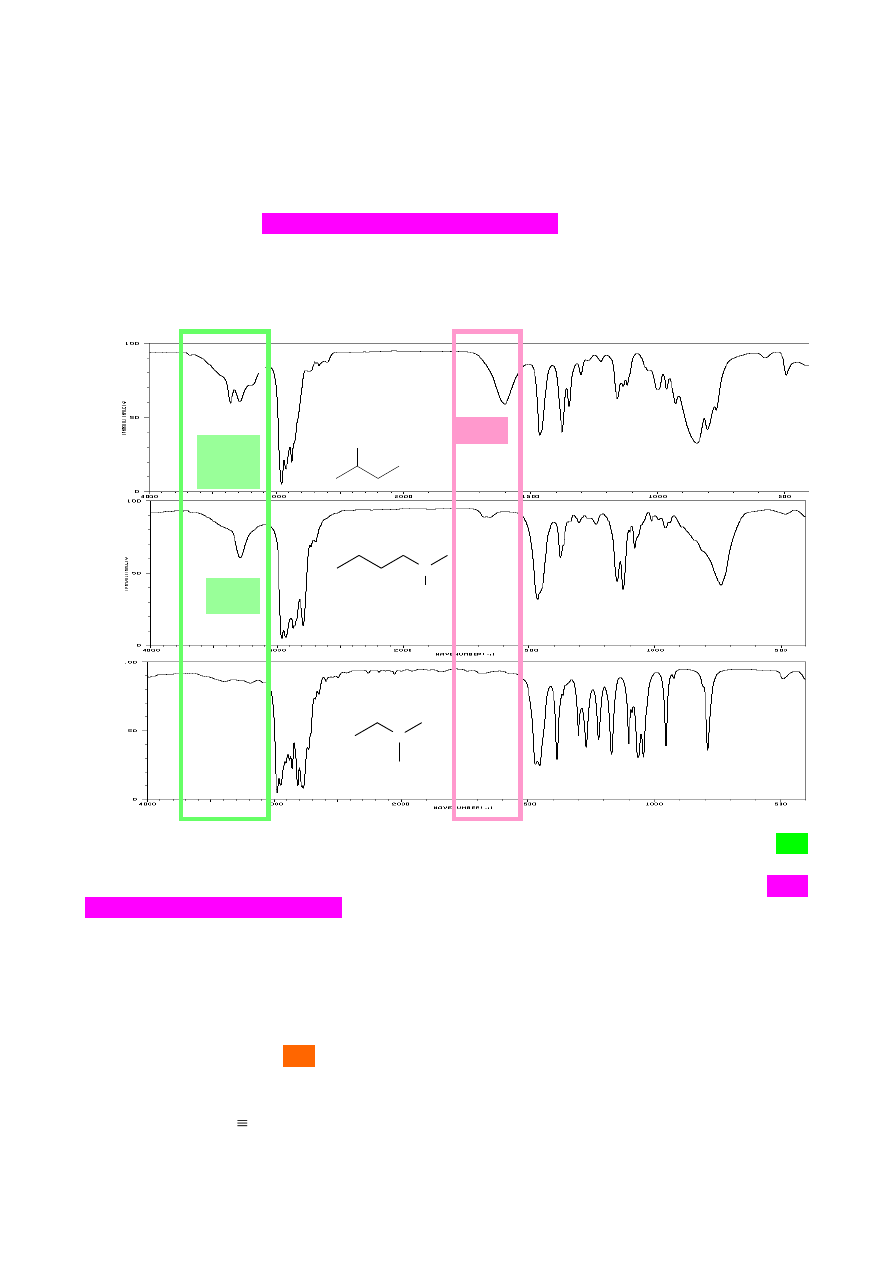
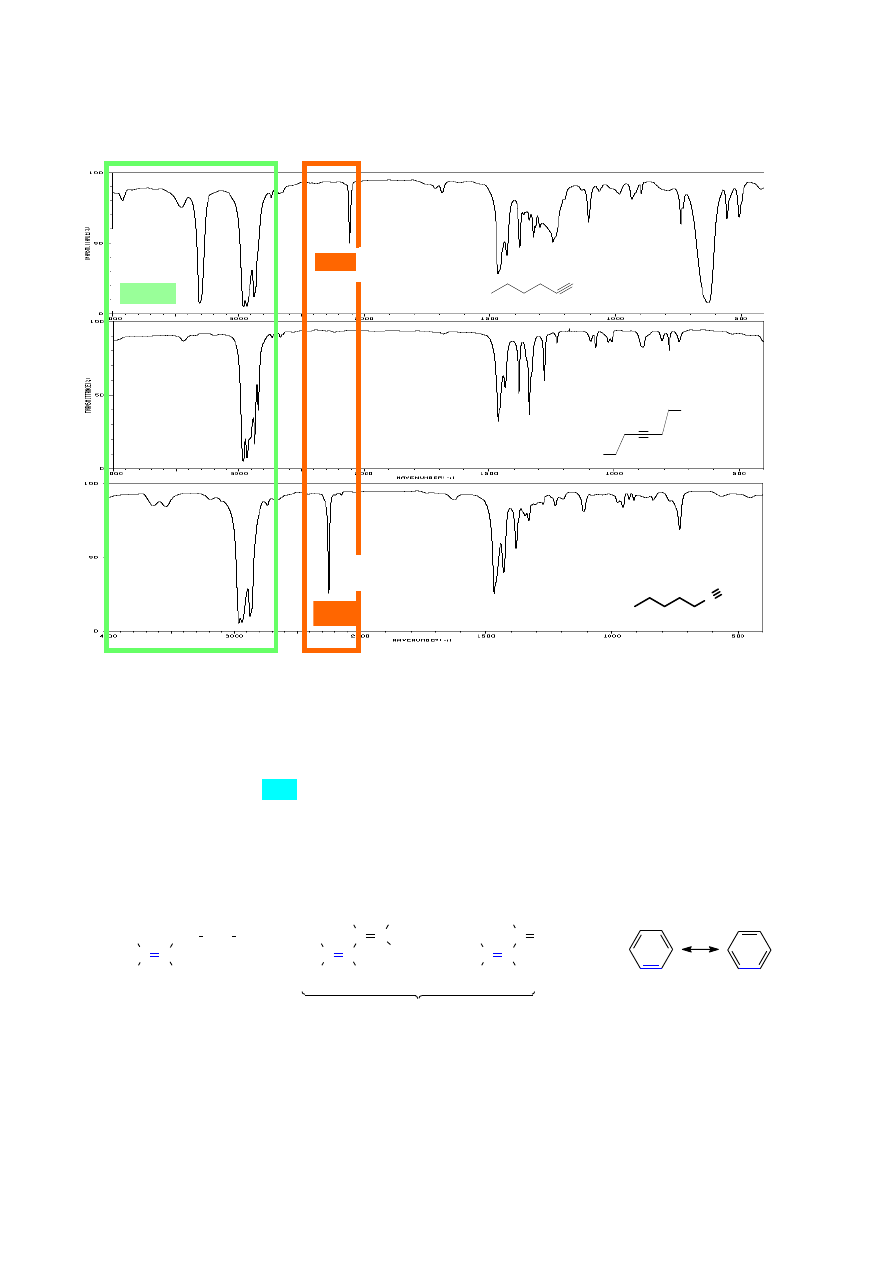
WIĄZAŃ POTRÓJNYCH:
C
≡
C
I
C
≡
N
(alkiny, nitryle).
Wiązania potrójne C≡C absorbują promieniowanie w charakterystycznym zakresie 2100-
2200 cm
-1
. Należy jednak pamiętać, że symetrycznie podstawione alkiny wewnętrzne takie jak okt-
4-yn dają jedynie pasma absorpcji charakterystyczne dla drgań walencyjnych alkanów (brak
wiązania Csp–1s dla C-H, oraz brak momentu dipolowego wiązania potrójnego).
Pasmo absorpcji alkinów i grupy nitrylowej leżą w tym samym zakresie, tj. około 2200 cm
-1
.
Rozróżnienia obu grup można dokonać na podstawie intensywności pasm oraz położenia. Większy
N
-
H
N
H
AMINY:
sec-
butyloamina (1˚)
NH
2
N,N-dimetyloetyloamina (3
o
)
3362,
3286
3292
N-
metylobutyloamina (2˚)
1603
pasma drgań
deformacyjnych
wiązań
N
-
H
N
13
moment dipolowy ma wiązanie grupy nitrylowej, dlatego pasmo to jest bardziej intensywne
i przesunięte do wartości liczb falowych powyżej 2200 cm
-1
. Pasmo drgań wiązania potrójnego
w alkinach leży poniżej wartości 2200 cm
-1
.
11. ANALIZA ZAKRESU PASM ABSORPCJI DRGAŃ WALENCYJNYCH
WIĄZAŃ PODWÓJNYCH
C
=
C
(alkeny, związki aromatyczne).
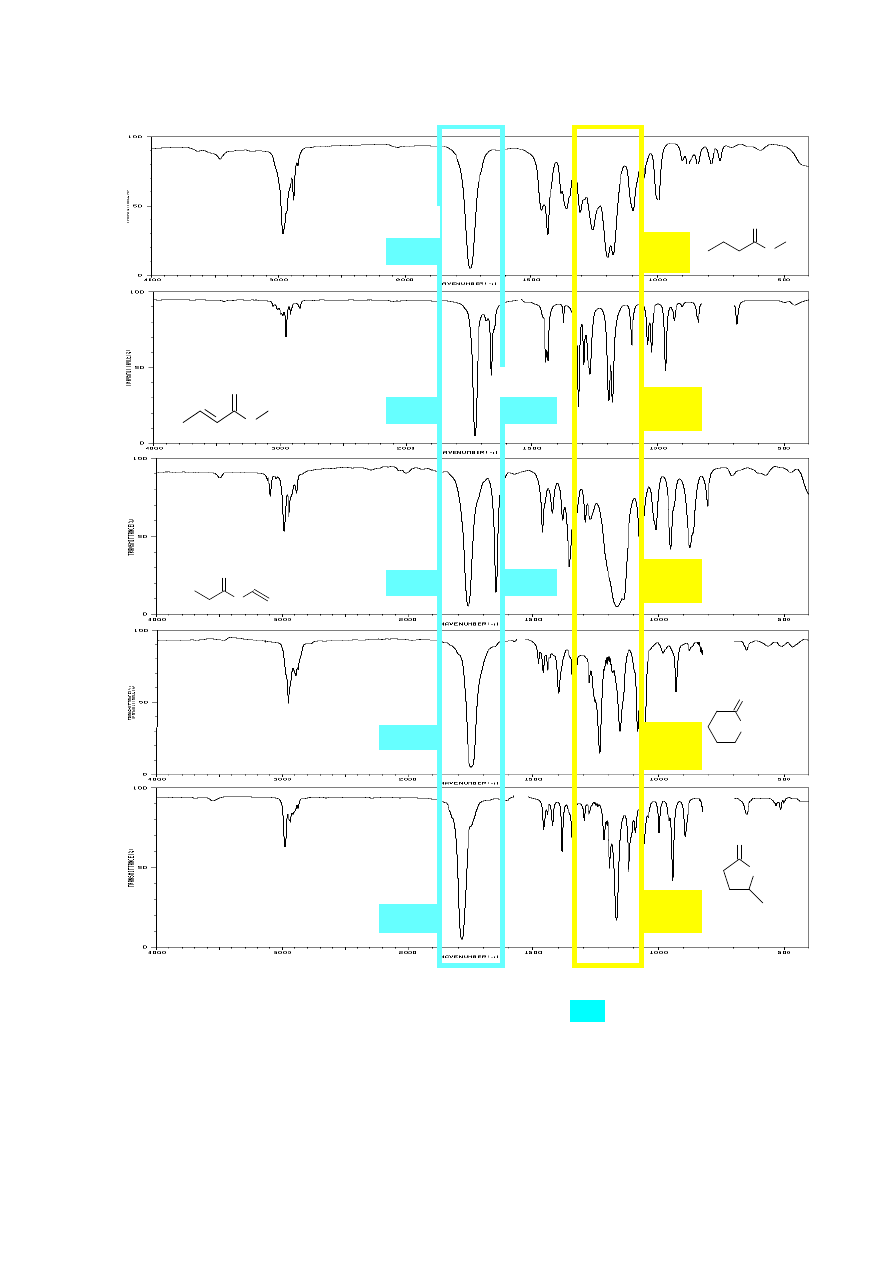
Drgania wiązań C=C są źródłem pasm absorpcyjnych przy ok. 1650 cm
-1
, jednak często
pasma te mają dość niską intensywność, a w przypadku symetrycznie podstawionych alkenów mogą
być praktycznie niewidoczne.
Osłabienie wiązania podwójnego poprzez sprzężenie z innym wiązaniem podwójnym lub
heteroatomem posiadającym wolne pary elektronowe, powoduje obniżenie wartości liczby falowej.
Im więcej atomów obejmuje takie sprzężenie, tym stała siłowa wiązań k jest mniejsza:
C C
H
H
H
CH
2
CH
2
R
C C
H
H
H
C C
H
R
H
C C
H
H
H
C O
R
izolowane C=C
1640-1670 cm
-1
sprzężone C=C
1620-1640 cm
-1
aromatyczne C=C
ok. 1600 cm
-1
2247
NITRYLE:
heksanonitryl
C
≡
N
C
N
≡C
–
H
sp-1s
2120
C
≡
C
3316
ALKINY:
heks-1-yn
(terminalny)
okt-4-yn
–C
–
H
sp
3
-1s
–C
–
H
sp
3
-1s
–C
–
H
sp
3
-1s
14
O C
R
CH
2
CH
2
R'
O C
R
C C
H
R'
H
izolowane C=O
1720 cm
-1
sprzężone C=O
1670-1700 cm
-1
O C
R
O C
R
N R'
R''
O C
R
N R'
R''
amidowe C=O
1640-1680 cm
-1
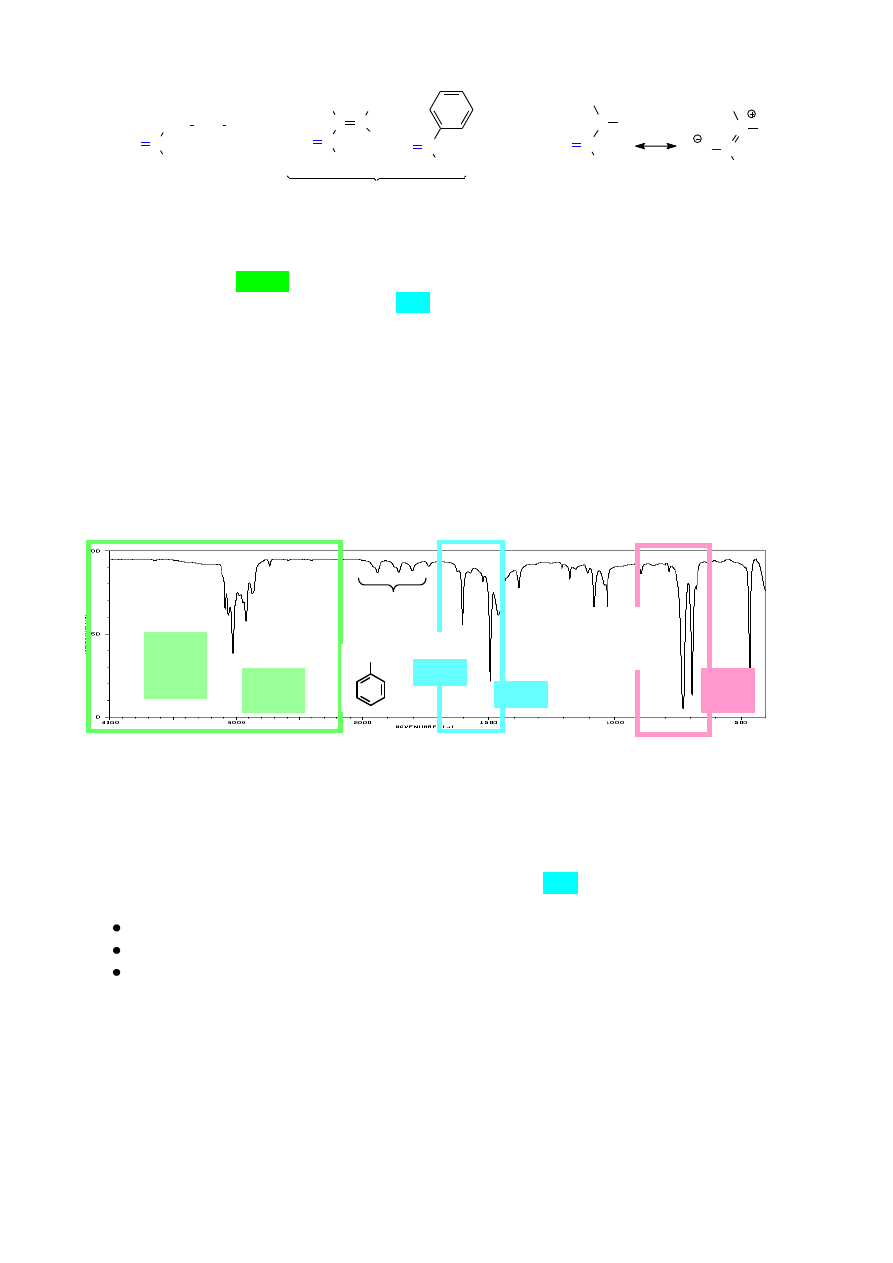
Widma związków zawierających pierścień aromatyczny charakteryzują się obecnością pasm
drgań walencyjnych Csp
2
-H ok. 3100 – 3000 cm
-1
, czyli w tym samym zakresie co alkeny. Jednak
położenie pasm drgań rozciągających C=C (będących zarazem drganiami deformacyjnymi
pierścienia) jest, ze względu na sprzężenie, przesunięte w stronę niższych wartości liczb falowych
i występuje zazwyczaj w postaci dwóch pasm przy 1600 cm
-1
oraz przy ok. 1500 - 1475 cm
-1
.
Diagnostyczne dla różnie podstawionych pierścieni aromatycznych są również pasma drgań
deformacyjnych poza płaszczyznę pierścienia (oop – out of plane) wiązań Ar-H. Pasma te występują
w zakresie 900 – 690 cm
-1
. Na podstawie liczby i intensywności tych pasm można określić np.
układ podstawników w mono-, di- i tripodstawionych pochodnych pierścienia benzenowego.
Sposób podstawienia pierścienia aromatycznego można wywnioskować również z układu nadtonów
w zakresie 1670 – 2000 cm
1
(przykłady można znaleźć w szczegółowych tabelach - lit. [1]), jednak
widma IR często nie dają jednoznacznych rozstrzygnięć i należy posłużyć się innymi technikami
spektroskopowymi.
12. ANALIZA ZAKRESU PASM ABSORPCJI DRGAŃ WALENCYJNYCH
WIĄZAŃ PODWÓJNYCH
C
=
O
(aldehydy, ketony, kwasy karboksylowe i ich
pochodne)
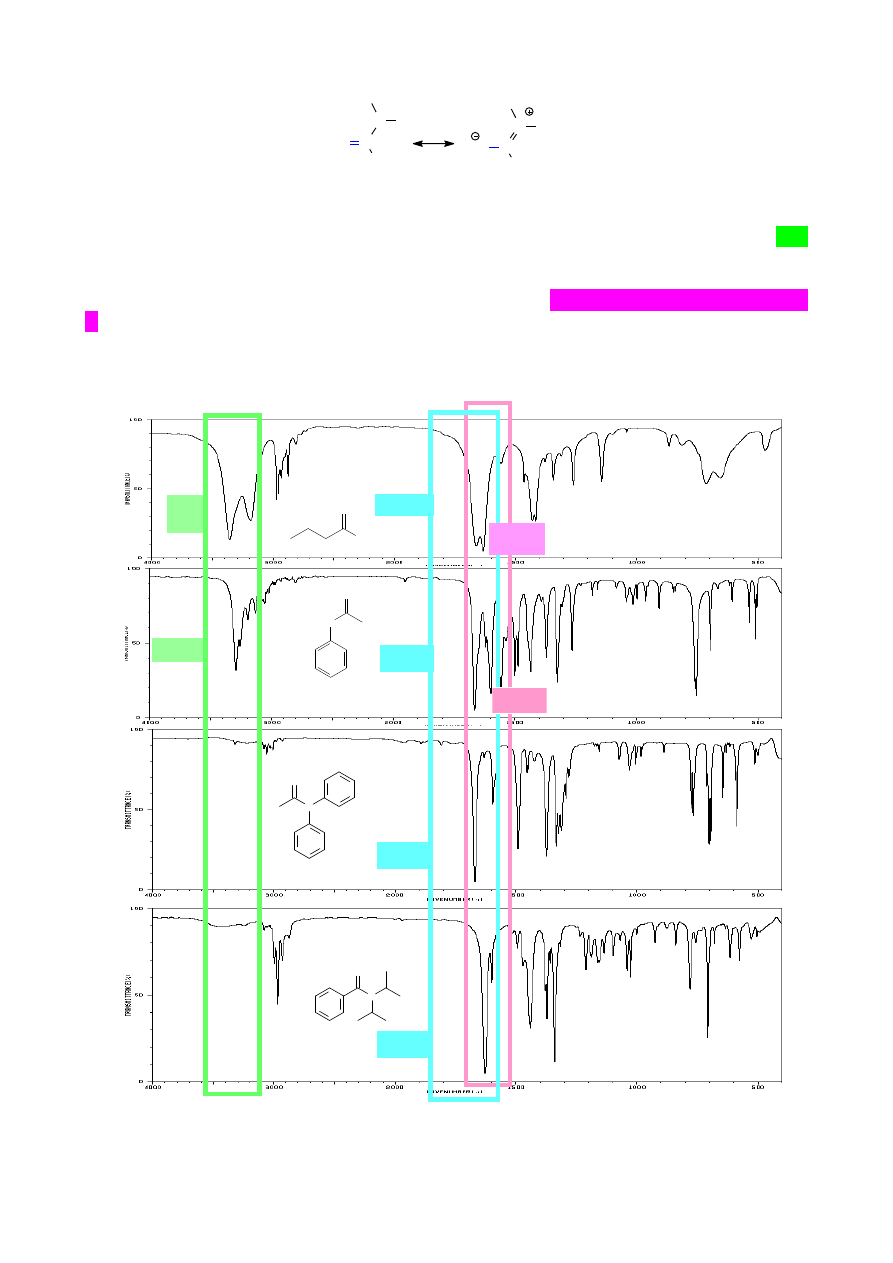
Związki karbonylowe można zidentyfikować na podstawie charakterystycznych
intensywnych pasm drgań walencyjnych grupy karbonylowej C=O w zakresie 1800 – 1650 cm
-1
.
Wartość stałej siłowej wiązania podwójnego w grupie karbonylowej zależy od trzech czynników:
możliwości utworzenia wiązania wodorowego przez atom tlenu grupy karbonylowej,
efektu rezonansowego (sprzężenia z innym układem π-elektronowym),
efektu indukcyjnego.
Obecność wiązania wodorowego oraz efekt rezonansowy zmniejszają stałą siłową wiązania
C=O, natomiast efekt indukcyjny (wynikający z różnicy elektroujemności między atomem węgla
i atomami odpowiednio wodoru, azotu, tlenu i chloru) wpływa na skrócenie lub wydłużenie
wiązania czyli zwiększenie (wpływ grup elektonoakceptorowych) lub zmniejszenie (wpływ grup
elektronodonorowych) wartości stałej siłowej wiązania. Kwasy i amidy, jako jedyne z pochodnych
karbonylowych, tworzą międzycząsteczkowe wiązania wodorowe. Atom tlenu jest jednak bardziej
elektroujemny niż atom azotu dlatego atom azotu w amidach łatwiej oddaje swoją wolną parę
elektronów w efekcie rezonansowym niż atom tlenu w kwasach. Grupy alkilowe w ketonach są
grupami elektronodonorowymi w przeciwieństwie do atomu wodoru w aldehydach, dlatego stała
nadtony
toluen
3087
3062,
3026
=C
–
H
sp
2
-1s
2948,
2920
–C
–
H
sp
3
-1s
C
=
C
1605
1496
729,
696
=C
–
H
deformacyjne
(oop)
CH
3
15
siłowa wiązania C=O w aldehydach (obecna jest tylko jedna grupa alkilowa) jest większa. Efekt
rezonansowy w bezwodnikach jest mniejszy niż w estrach (dwie grupy acylowe), natomiast efekty
indukcyjne są podobne. Efekty indukcyjne wywierane przez atomy chloru i tlenu są podobne (oba
atomy mają podobną elektroujemność), jednak oddziaływanie rezonansowe atomu chloru jest dużo
słabsze (ze względu na wielkość atomu), dlatego stała siłowa wiązania C=O w chlorkach
kwasowych jest dużo większa niż w estrach i bezwodnikach kwasowych.
Zakresy występowania pasm absorpcji pochodzących od drgań walencyjnych wiązań C=O
Liczba falowa [cm
-1
]
1800
1750
1700
1650
1810
1800
1760
1735
1725
1715
1710
1680
Bezwodniki
Chlorki
Bezwodniki
Estry
Aldehydy Ketony
Kwasy
Amidy
pasmo 1
kwasowe
pasmo 2
Pasmo dla cyklicznych
estrów (laktonów)
przesuwa się do wyższych
wartości wraz ze
zmniejszaniem się
pierścienia
Pasmo dla cyklicznych
ketonów przesuwa się do
większych wartości wraz
ze zmniejszaniem się
pierścienia
Pasmo dla cyklicznych
amidów (laktamów)
przesuwa się do
wyższych wartości wraz
ze zmniejszaniem się
pierścienia
Sprzężenie od strony
grupy alkoksylowej
przesuwa pasmo w stronę
wyższych wartości
Sprzężenie od strony węgla grupy karbonylowej przesuwa pasmo
o około 20-25 cm
-1
w stronę niższych wartości liczby falowej (sprzężenie
zmniejsza wartość stałej siłowej wiązania)
5,5
Długość fali [ m]
6,1
C
NH
2
O
C
OH
O
C
O
C
H
O
C
OR
O
C
O
O
C
O
C
Cl
O
C
O
O
C
O
16
Kwasy karboksylowe można łatwo odróżnić od innych związków karbonylowych dzięki
szerokiemu pasmu dimerów związanych wiązaniem wodorowym w zakresie 3400 – 2500 cm
-1
.
Aldehydy można łatwo odróżnić od ketonów na podstawie drgań walencyjnych C-H
w grupie CHO aldehydu, dających zazwyczaj dwa średnio intensywne pasma przy ok. 2820 i ok.
2700 cm-
1
. Pojawienie się dwóch pasm spowodowane jest przez rezonans Fermiego (sprzężenie
drgań rozciągających C-H z nadtonem drgań deformacyjnych wiązań O=C-H). Pierwsze z pasm
może być zakryte przez pasma drgań walencyjnych grup alifatycznych C-H, ale to drugie jest na
ogół dość intensywne.
Estry od ketonów można odróżnić na podstawie drgań walencyjnych C-O dających dwa
pasma w zakresie 1300 – 1000 cm
-1
. Pasmo drgań walencyjnych grupy C=O estru występuje przy
wyższych częstościach, w stosunku do analogicznych pasm ketonu (dla estrów o grupie
karbonylowej niesprzężonej z innym układem π-elektronowym to ok. 1735 cm
-1
, a dla ketonów ok.
1715 cm
-1
).
2711
2811
ALDEHYDY:
2-metylobutanal
O
H
KETONY:
pentan-3-on
O
2500-3500
C
O
H
KWASY KARBOKSYLOWE:
kw. 3-metylobutanowy
(izowalerianowy)
O
OH
Szerokie pasmo
dimerów
związanych
wiązaniem
wodorowym
C
=
O
O
H
O
H
O
R
R
O
1728
1710
1716
17
Intensywne pasmo pochodzące od drgań rozciągających C=O w amidach jest położone
najniżej ze wszystkich pochodnych zawierających grupę karbonylową przy 1680-1630 cm
-1
, ze
względu na bardzo silne oddziaływanie rezonansowe grupy aminowej i karbonylowej i wynikające
stąd zmniejszenie stałej siłowej wiązania C=O:
ESTRY:
butanian metylu
1198,
1097
(E)-but-2-enian metylu
1742
1726
1652
propanian winylu
1760
1649
1167
1078
1196,
1180
C
-
O
O
O
C
=
O
C
=
C
O
O
O
O
1747
1786
O
O
pentano-5-lakton
lakton kwasu
δ-hydroksywalerianoego
pentano-4-lakton
lakton kwasu
γ-hydroksywalerianowego
O
O
1236,
1155
1168,
1119
18
O C
R
N R'
R''
O C
R
N R'
R''
amidowe C=O
1630-1680 cm
-1
Amidy pierwszorzędowe i drugorzędowe można rozróżnić na podstawie liczby pasm N-H
w zakresie w zakresie 3400 – 3100 cm
-1
. Amidy pierwszorzędowe dają dwa pasma przy około 3180
i 3350 cm
-1
. Amidy drugorzędowe dają jedno pasmo przy około 3300 cm
-1
.
Na pasmo grupy karbonylowej może nałożyć się pasmo drgań deformacyjnych (δ) grupy N-
H. Amidy trzeciorzędowe ze względu na brak pasm N-H trudno jest zidentyfikować. Tym bardziej,
że niesprzężone amidy dają pasmo drgań grupy C=O w zakresie typowym dla sprzężonych
ketonów, natomiast sprzężone amidy - w zakresie drgań wiązań podwójnych C=C. Intensywność
tych pasm jest jednak mniejsza ze względu na mniejszą wartość momentu dipolowego.
3294
acetanilid (2
o
)
1665
HN
O
AMIDY:
butanoamid (1
o
)
O
NH
2
3366
3184
1662
ν
C
=
O
δ
N
-
H
1663
N,N-
difenyloacetamid (3˚)
1671
N,N-
diizopropylobenzamid (3˚)
1630
O
N
O
N
ν
N
-
H
1557
19
Chlorki kwasowe i bezwodniki można odróżnić na podstawie obecności intensywnych
pasm rozciągających C-Cl występujących w zakresie 730-550 cm
-1
. Chlorki kwasowe dają często
dwa pasma absorpcji grupy karbonylowej przy około 1800 – 1775 cm
-1
. Niżej położone pasmo jest
zazwyczaj mniej intensywne i powstaje w wyniku rezonansu Fermiego. Bezwodniki wykazują dwa
intensywne pasma przy odpowiednio 1775 - 1740 cm
-1
oraz 1830 - 1800 cm
-1
. Dwa pasma absorpcji
obecne dla bezwodników pochodzi od odpowiednio asymetrycznych (as, 1830 -800 cm
-1
) i
symetrycznych (s, 1775 - 1740 cm
-1
) drgań walencyjnych.
13. ANALIZA
ZAKRESU
PASM
ABSORPCJI
DRGAŃ
WIĄZAŃ
POJEDYNCZYCH
C
-
O
,
CH
2
-
X
,
C
-
X
(etery, alkohole, halogenki alkilowe).
Rozróżnienie alkoholu od eteru wydaje się proste. Brak pasm OH w zakresie 3400 - 3300
cm
-1
i obecność dość intensywnego pasma przy 1300 - 1000 cm
-1
może świadczyć o obecności
eteru. Etery arylowo – alkilowe i winylowo – alkilowe dają dwa pasma absorpcji w tym zakresie
chlorek benzoilu
bezwodnik octowy
Cl
O
O
O
O
C
=
O
C
-
O
C
-
Cl
1775,
1733
1832,
1761
1124
671
benzofenon
hept-1-en
1665
1642
O
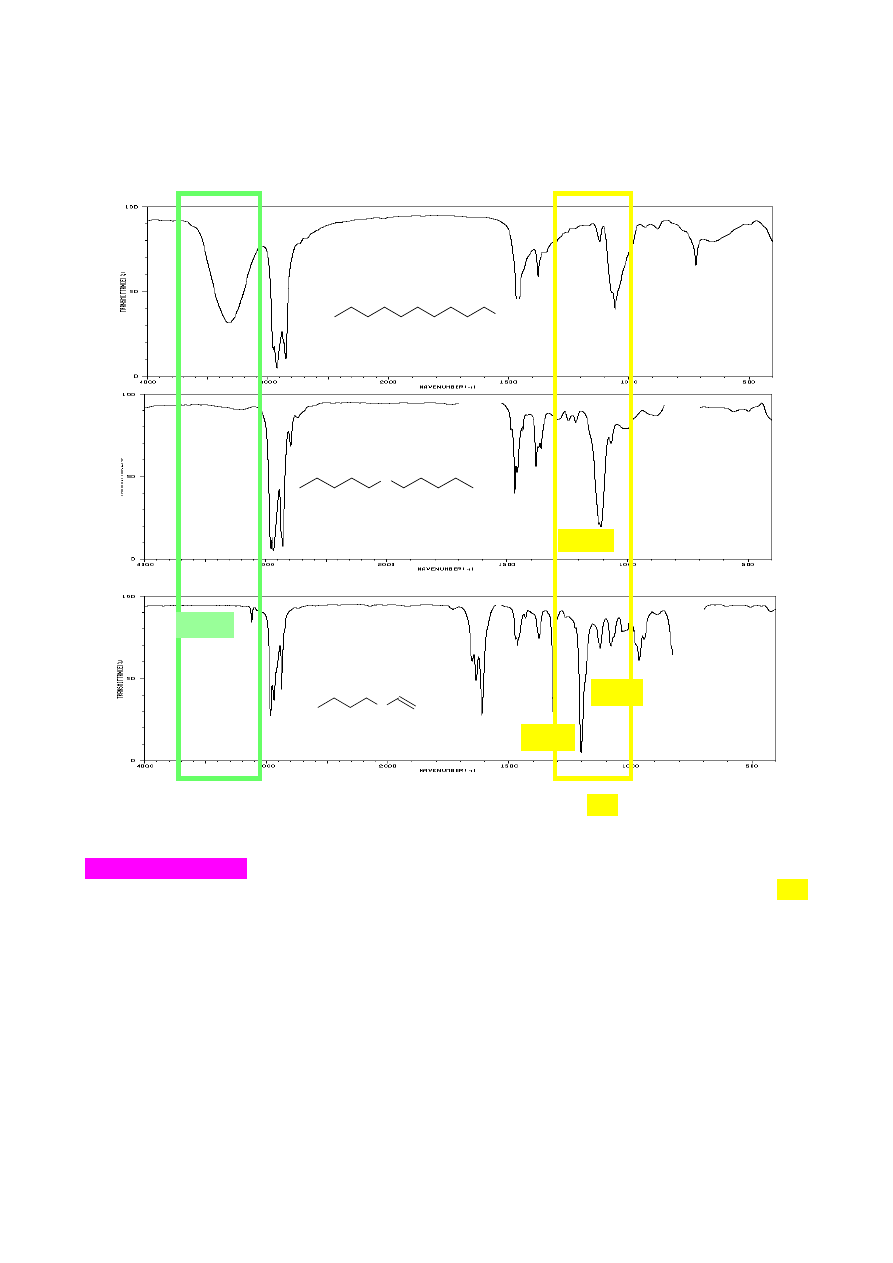
20
1250 i 1050 cm
-1
, ponieważ związek zawiera dwa nierównocenne wiązania C-O i jedno z nich, ze
względu na sprzężenie wiązań wielokrotnych z atomem tlenu, posiada większą wartość stałej
siłowej wiązania.
W tym samym zakresie co pasma drgań rozciągających grupy C-O występują również pasma
drgań deformacyjnych CH
2
X. Dlatego należy rozróżnić etery od związków zawierających grupę
CH
2
X na podstawie obecności (lub braku) pasm C-X, co często bywa dość trudne. Obecność pasm
drgań deformacyjnych CH
2
podstawionej fluorowcem występują odpowiednio przy 1300-1230 cm
-1
(CH
2
Cl), 1250-1190 cm
-1
(CH
2
Br ) i 1200-1150 cm
-1
(CH
2
I). Pasma drgań normalnych C-X
występują przy ok. 750 cm
-1
dla C-Cl; ok. 600 cm
-1
dla C-Br oraz poniżej 500 cm
-1
dla C-I).
14. ANALIZA ZAKRESU PASM ABSORPCJI DRGAŃ WALENCYJNYCH
WIĄZAŃ
N
=
O
,
S
=
O
C
-
S
,
C
=
S
(związki nitrowe, pochodne kwasów sulfonowych,
związki siarki).
W widmach IR związków organicznych, które mają grupę nitrową obserwuje się dwa silne
pasma w zakresach 1560-1500 cm
-1
i 1350-1300 cm
-1
pochodzące od asymetrycznych (as) i
symetrycznych (sym) drgań rozciągających silnie polarnych wiązań między atomami azotu i tlenu.
Nitrozwiązki aromatyczne dają te pasma przy nieco mniejszych częstościach niż nitrozwiązki
alifatyczne
dekan-1-
ol (1°)
eter dipentylowy
OH
O
1109
eter butylowo-
winylowy
1204
O
=C
–
H
sp
2
-1s
3121
–HC
=
O
+
–
–H
2
C
–
O
–
Pasma drgań
walencyjnych
C
-
O:
1037
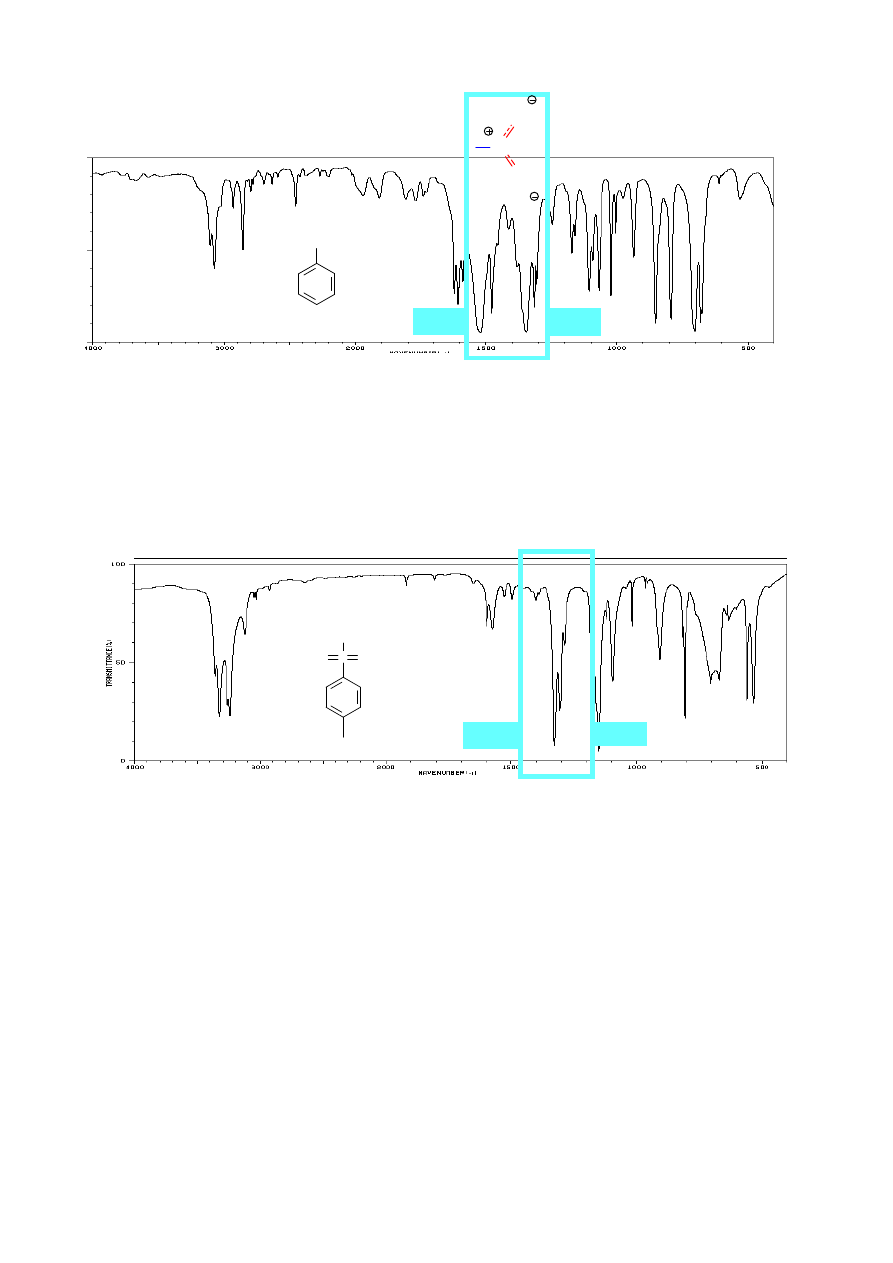
21
Widma organicznych związków siarki (tioli, tioketonów, ditioestrów i sulfotlenków) są
bardzo trudne do interpretacji. Pasma pochodzące od tych grup funkcyjnych są zazwyczaj mało
intensywne, a niektóre z nich znajdują się w zakresie daktyloskopowym (fingerprint).
Jedynie związki które mają grupę -SO
2
- (sulfony, sulfonamidy, chlorki sulfonylu, kwasy
sulfonowe, sulfoniany i siarczany organiczne) można łatwo zidentyfikować na podstawie dwóch
silnych pasm występujących w zakresach 1415 –1300 cm
-1
i 1200-1120 cm
-1
, które pochodzą od
asymetrycznych i symetrycznych drgań
rozciągających.
15. TECHNIKI PRZYGOTOWANIA PRÓBEK I REJESTRACJI WIDM
W podczerwieni można badać widma gazów, cieczy i substancji stałych. Dokonując pomiaru
widma cieczy lub ciała stałego należy pamiętać, że większość substancji chemicznych na tyle silnie
absorbuje promieniowanie podczerwone, że już warstwa grubości 0,1 mm staje się całkowicie
nieprzeźroczysta. Widmo transmisyjne musi być więc rejestrowane dla bardzo cienkiej warstwy
próbki względnie dla jej rozcieńczonego roztworu lub zawiesiny w substancji, która nie pochłania
promieniowania podczerwonego. Jednocześnie materiały, które znajdują się na drodze optycznej
(np. kuwety) nie mogą pochłaniać promieniowania w analizowanym zakresie. Najczęściej stosuje
się płytki i kuwety wykonane bromku potasu (nie absorbuje w zakresie 400 – 4000 cm
-1
) lub
chlorku sodu (nie absorbuje w zakresie 500 – 4000 cm
-1
) - są względnie tanie, jednak cechują się
niską odpornością mechaniczną i chemiczną. Przykładowo, płytki i kuwety wykonane z tych soli
ulegają zmatowieniu już pod wpływem śladów wilgoci, więc analizowane próbki lub
rozpuszczalniki stosowane do sporządzania roztworów muszą być całkowicie bezwodne. Inne
materiały, o wyższej odporności chemicznej i mechanicznej (np. fluorek wapnia – od 1000 cm
-1
,
p-toluenosulfonoamid
1328
1163
nitrobenzen
1521
1347
O
=
S
=
O
S
O
O
NH
2
NO
2
N
O
O
1/2
1/2
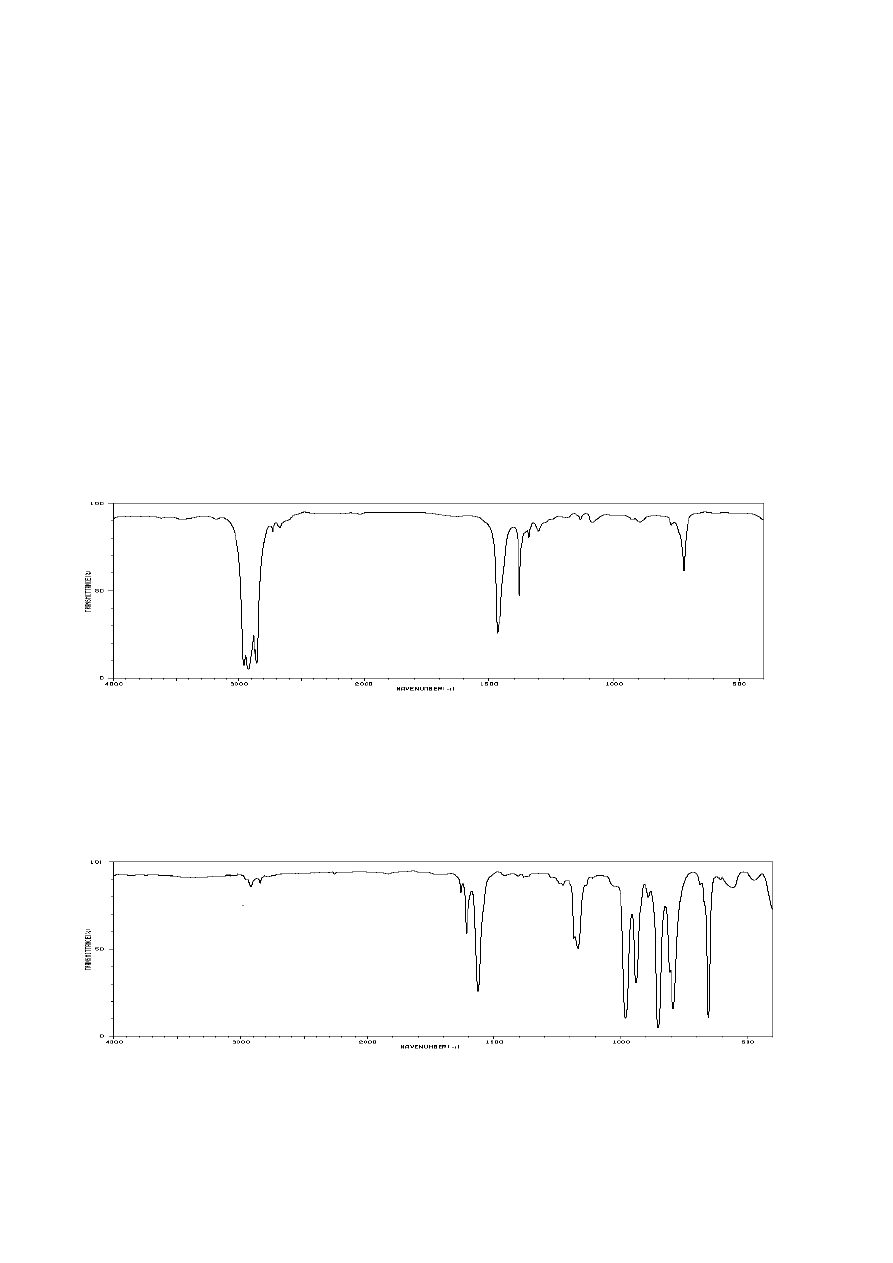
22
KRS-5, czyli bromek jodek talu(II) lub selenek cynku – w całym zakresie tzw. średniej
podczerwieni) są znacznie droższe i rzadziej stosowane.
Widma czystych cieczy można rejestrować w postaci filmu. Małą kroplę ciekłej substancji
(kilka mg) umieszcza się między dwoma płytkami z chlorku sodu lub bromku potasu i całość
umieszcza w oprawie dociskającej do siebie płytki, uzyskując warstewkę cieczy grubości kilku
setnych milimetra. Płytki z substancją umieszcza się w komorze pomiarowej i rejestruje widmo.
Wszystkie pasma obserwowane na widmie tak przygotowanej próbki pochodzą od badanej
substancji.
Najprostszą techniką rejestrowania widm substancji stałych polega na przygotowaniu
zawiesiny próbki w Nujolu (nazwa handlowa specjalnie oczyszczonej mieszaniny wysokowrzących
węglowodorów alifatycznych, rafinowany olej parafinowy). W tym celu 2 - 6 mg substancji rozciera
się w moździerzu agatowym, następnie proszek miesza się z 1 kroplą Nujolu i tak przygotowaną
zawiesinę umieszcza się między płytkami z chlorku sodu lub bromku potasu, dociska i umieszcza w
komorze pomiarowej aparatu. Nujol jest medium obojętnym chemicznie, tanim, a ponadto jego
widmo w podczerwieni ma tylko cztery intensywne pasma pochodzące od drgań walencyjnych i
deformacyjnych grup -CH
2
i -CH
3
, które są tak położone, że pozwalają na obserwację pasm
zdecydowanej większości innych grup funkcyjnych obecnych w analizowanej próbce.
Widmo nujolu
Oprócz Nujolu do wykonania zawiesiny można stosować heksachlorobutadien (HCB). Ma
on jednak ograniczone zastosowanie, ponieważ w zakresie 2000 – 400 cm
-1
posiada wiele pasm
własnych, natomiast znakomicie nadaje się do wykonania widma w zakresie 4000– 2000 cm
-1
, czyli
jest medium komplementarnym wobec Nujolu.
Widmo HCB
Optymalnym sposobem wykonywania widma w podczerwieni substancji stałej jest
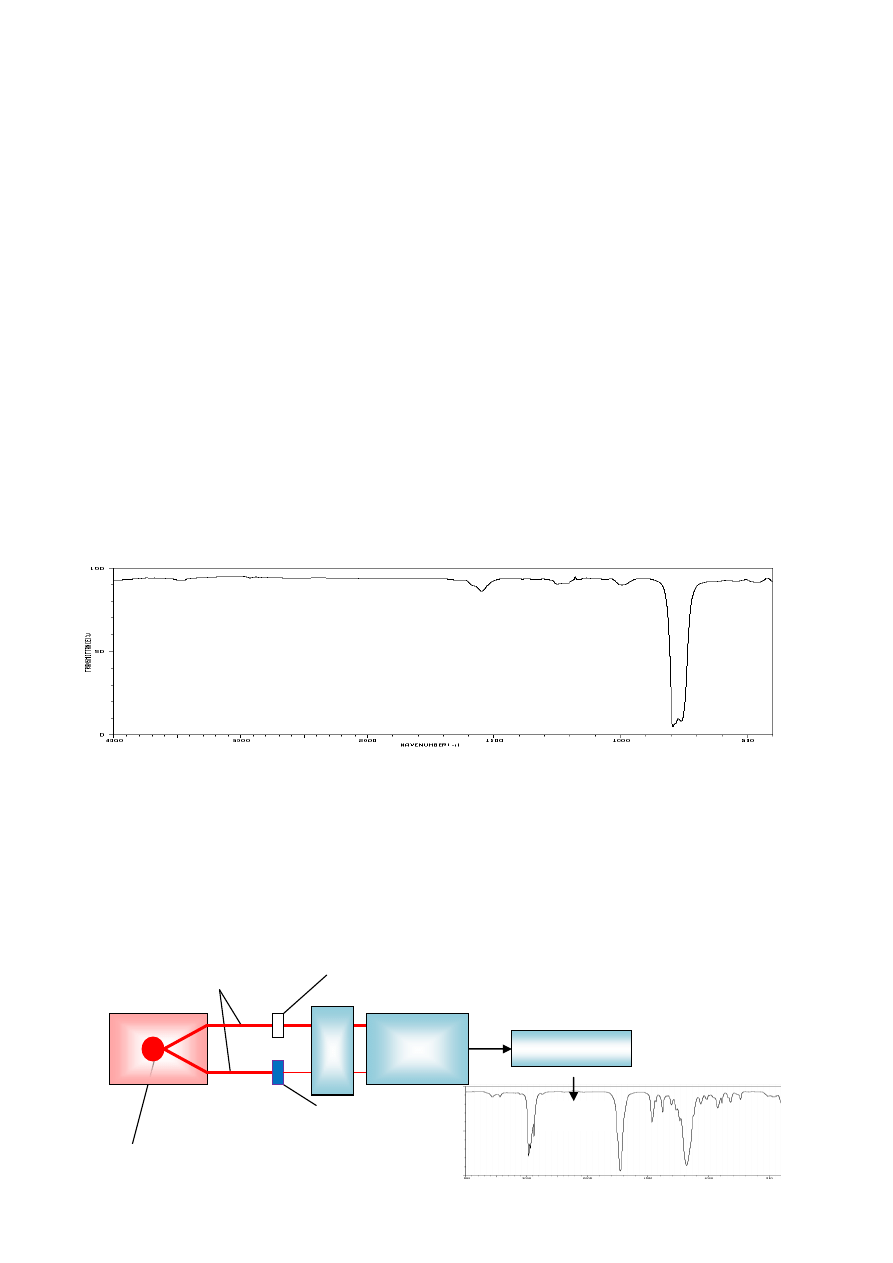
sporządzenie jej zawiesiny w sprasowanym bromku potasu, czyli tak zwanej pastylki KBr. W tym
celu odważa się 500 mg wstępnie zmielonego, bezwodnego bromku potasu i miesza się go
dokładnie z ok. 5 mg próbki (ilość użytego bromku potasu i substancji może być proporcjonalnie
23
mniejsza, zależnie od wielkości przygotowywanej pastylki). Mieszaninę można dokładniej zmielić
w młynku wibracyjnym. Próbkę umieszcza się w matrycy i prasuje przy użyciu prasy hydraulicznej
stosując ciśnienie ok. 120-140 MPa. Otrzymany przeźroczysty krążek umieszcza się następnie w
odpowiednim uchwycie w przedziale pomiarowym aparatu. Bromek potasu przepuszcza
promieniowanie podczerwone od 2 – 25 µm, czyli wszystkie obserwowane pasma pochodzą od
związku.
Jeśli zachodzi konieczność zarejestrowania widma IR substancji stałej nie dającej się
rozdrobnić, lecz rozpuszczalnej w lotnych rozpuszczalnikach organicznych, stosuje się technikę
folii. Próbkę rozpuszcza się w odpowiednim rozpuszczalniku, kroplę roztworu rozprowadza się na
powierzchni płytki z chlorku sodu lub bromku potasu i odparowuje rozpuszczalnik. Na płytce
pozostaje cienka błonka badanej substancji. Płytkę pokrytą błonką wstawia się do aparatu i
rejestruje widmo.
Widma IR można rejestrować również dla roztworów, szczególnie w przypadku pomiarów
ilościowych. Jednak bardzo trudnym problemem jest dobór odpowiedniego rozpuszczalnika. Żaden
rozpuszczalnik nie jest przeźroczysty w całym zakresie 400 – 4000 cm
-1
. Im mniej złożone są
cząsteczki rozpuszczalnika, tym mniejsza jest liczba jego pasm absorpcji. Jednak każdy
rozpuszczalnik w pewnych zakresach zakłóca widmo substancji swoimi silnymi pasmami
własnymi. Do najlepszych, łatwo dostępnych rozpuszczalników należą disiarczek węgla i
tetrachlorometan. Użyteczne zakresy rozpuszczalników stosowanych w podczerwieni można
znaleźć w cytowanej literaturze [1] (str. 193) oraz [2] (str. 119).
Widmo CCl
4
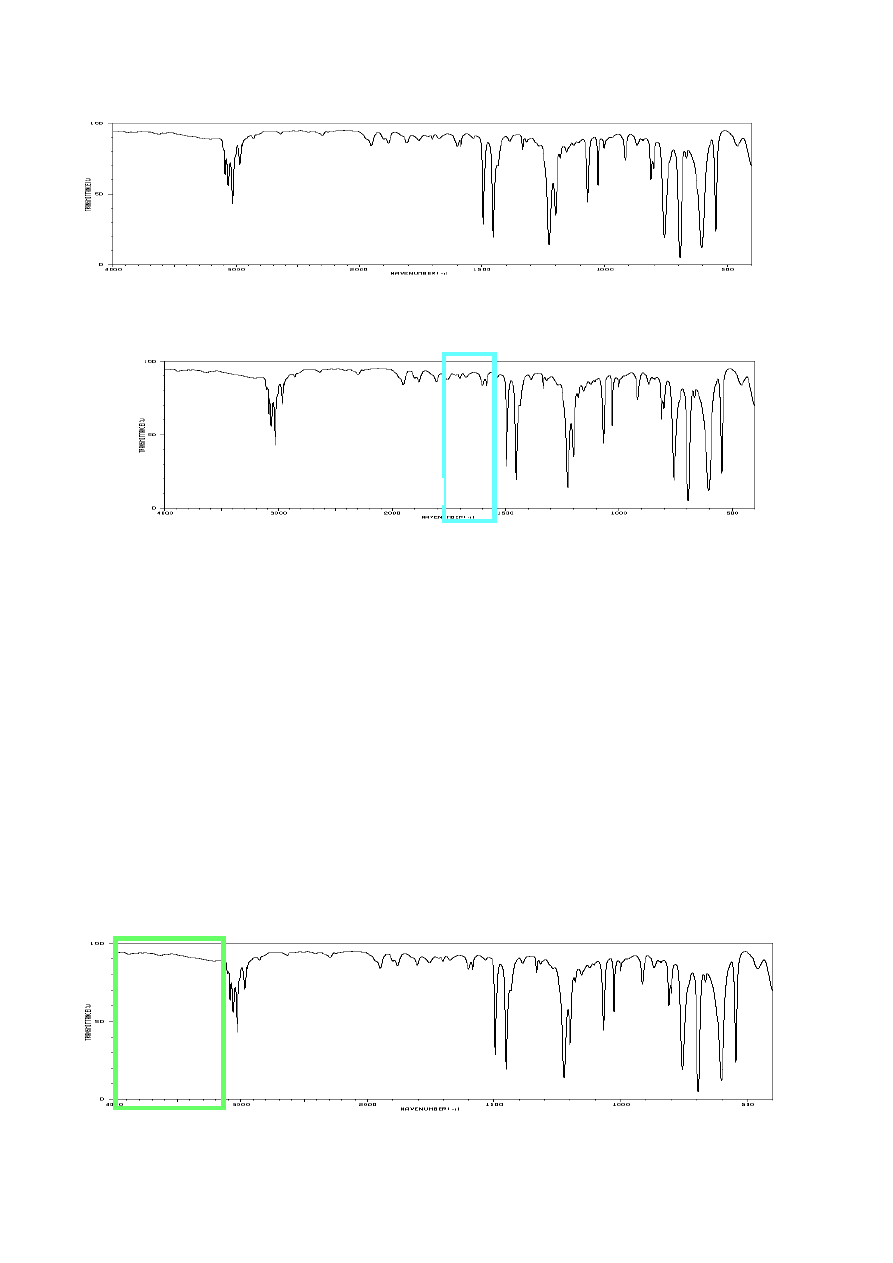
Poniższy rysunek przedstawia ogólny schemat tradycyjnego dwuwiązkowego dyspersyjnego
spektrofotometru IR. Promieniowanie podczerwone wytwarzane przez rozgrzany pręt Nernsta
(mieszanina tlenków cyrkonu, itru, toru i cezu) lub tzw. Globar (pręt z węgliku krzemu) jest
rozdzielane w układzie wklęsłych zwierciadeł na dwie wiązki. Jedna z nich przechodzi przez
komorę pomiarową z próbką, a na drodze drugiej wiązki umieszcza się kuwetę wypełnioną czystym
rozpuszczalnikiem (pastylkę z czystego bromku potasu lub czyste płytki), aby skompensować
pasma rozpuszczalnika, powietrza itp. Obie wiązki kieruje się następnie naprzemiennie do
monochromatora (pryzmat lub siatka dyfrakcyjna) i detektora, gdzie zostają porównane ich energie
dla poszczególnych długości fal.
widmo
próbka
kuweta odnośnikowa
źródło promieni IR
(np. pręt Nernsta)
wiązka
pomiarowa
i
porównawc
za
detektor,
analizator
i wzmacniacz
rejestrator
monochro
-
ma
to
r

24
Wzmocniony sygnał jest kierowany do rejestratora, którym dawniej był pisak poruszający
się po przesuwającej się wstędze lub arkuszu papieru, a w nowszych rozwiązaniach –
skomputeryzowany system rejestrujący zależność.
Wadą opisanych powyżej rozwiązań był względnie długi czas rejestracji widma (kilka
minut), niski stosunek sygnał:szum, wysoki koszt produkcji i podatność na rozregulowanie, ze
względu na dużą liczbę ruchomych części. Tych wad nie posiadają obecnie najczęściej stosowane
jednowiązkowe spektrometry wykorzystujące transformatę Fouriera (FT-IR). Dokładne omówienie
konstrukcji i zasady działania tzw. aparatów fourierowskich można znaleźć w cytowanej literaturze:
[5], str. 314. Jedną z głównych zalet przyjętego w nich rozwiązania jest niezwykle krótki czas
pomiaru pełnego widma, rzędu ułamka sekundy. Pozwala to na wielokrotną rejestrację i sumowanie
widm tej samej próbki (tzw. skanowanie), co znacznie poprawia stosunek sygnał:szum.
W przypadku rejestracji widm w aparatach jednowiązkowych należy pamiętać o konieczności
zapisu zarówno widma próbki, jak również, podczas niezależnego pomiaru, tzw. widma tła (np.
pastylki czystego bromku potasu, kuwety z czystym rozpuszczalnikiem). Widmo analizowanej
substancji otrzymuje się po matematycznym odjęciu widma tła od widma próbki.
W ostatnich latach coraz szersze zastosowanie znajdują aparaty FT-IR wyposażone w tzw.
przystawkę ATR. Przystawki ATR wykorzystują zjawisko osłabionego wewnętrznego odbicia,
w wyniku którego promieniowanie odbijające się od granicy faz zostaje zubożone o fale
zaabsorbowane przez materiał, od którego nastąpiło odbicie. Konsekwencją tego efektu jest
możliwość rejestracji widm IR próbki, która zostanie dociśnięta do odpowiednio przygotowanego i
wyszlifowanego kryształu, przez który przepuszczana jest wiązka promieniowania podczerwonego.
Standardowymi kryształami wykorzystywanymi w spektrometrach FT-IR/ATR są:
A. diament – najwyższa odporność i chemiczna i twardość, praktycznie nie ulega zużyciu
podczas użytkowania; jedyną jego wadą jest ograniczona dokładność pomiaru w zakresie
1800 – 2000 cm
-1
B. german – wysoka odporność chemiczna i twardość; zakres spektralny 550 – 5000 cm
-1
;
komplementarny dla diamentu
C. selenek cynku – tani, lecz charakteryzujący się niską odpornością chemiczną i mechaniczną;
może być stosowany do konstruowania przystawek wieloodbiciowych dedykowanych m. In.
do ilościowych pomiarów analitycznych (np. roztworów)
Widma uzyskiwane techniką ATR, po odpowiedniej matematycznej transformacji, są
zbieżne z klasycznymi widmami transmisyjnymi. Do licznych zalet tej techniki można zaliczyć:
niski koszt eksploatacji;
brak konieczności wstępnego przygotowania próbek, jak ma to miejsce przy pomiarach
transmisyjnych;
możliwość pomiaru widm substancji zawierających wodę;
bezstratnej rejestracji widm praktycznie każdego materiału, który może być dociśnięty do
monokryształu lub nań naniesiony (np. substancje chemiczne i ich roztwory - w tym
roztwory wodne, tworzywa sztuczne, folie, tekstylia, tkanki organizmów żywych).
16. UPROSZCZONY SCHEMAT POSTĘPOWANIA PRZY INTERPRETACJI
WIDM IR ZWIĄZKÓW ORGANICZNYCH
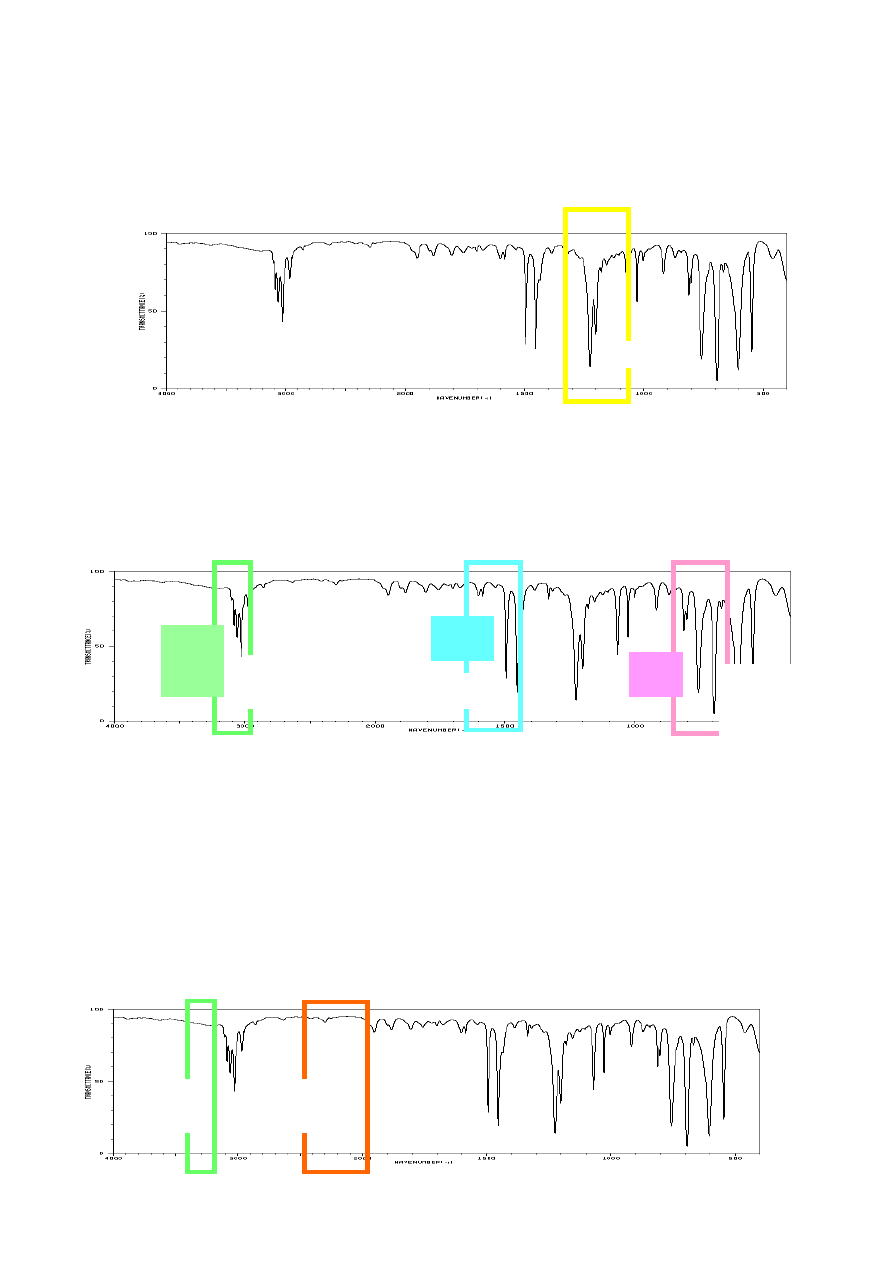
Analizując widmo IR nieznanego związku organicznego należy w pierwszej kolejności
zwrócić uwagę na warunki w jakich było wykonane widmo. Jeżeli widmo wykonane było w takich
rozpuszczalnikach jak HCB, nujol, czy też CCl
4
, to należy wyeliminować pasma pochodzące od
tych związków. Następnie należy zwrócić uwagę na obecność lub brak pasm podstawowych grup
funkcyjnych: C=O, OH, NH
2
(lub -NHR), C-O, C=C, C≡C, C≡N, i NO
2
, które dają natychmiastową
informację o budowie związku.
25
Widmo nieznanego związku wykonane jako film.
Analizę obecności grup funkcyjnych należy zacząć od sprawdzenia czy są obecne :
1. Intensywne pasma grupy karbonylowej występujące w zakresie 1820-1630 cm
-1
2. Jeżeli grupa C=O jest obecna należy określić czy jest to:
a)
kwas karboksylowy: obecność szerokiego pasma grup O-H 3500-2500 cm
-1
;
b)
amid: dwa pasma N-H dla amidu 1° oraz jedno pasmo dla amidu 2 °w zakresie
3500-3300 cm
-1
; jeżeli brak jest pasm N-H a pasmo drgań C=O leży w zakresie
1680-1630 cm
-1
to może to być amid 3°;
c)
ester: obecne są intensywne pasma drgań rozciągających C-O w zakresie 1300-1100
cm
-1
. Ponadto pasmo drgań C=O dla niesprzężonych estrów leży przy wyższych
wartościach liczby falowej (około 1740 cm
-
);
d)
bezwodnik kwasu karboksylowego: dwa intensywne pasma od drgań C=O
występujące przy 1800 cm
-1
i 1760 cm
-1
;
e)
chlorek kwasowy: możliwe dwa pasma C=O przy 1800-1770 cm
-1
oraz obecne
intensywne pasmo C-Cl leżące w zakresie 730-550 cm
-1
;
f)
aldehyd: obecne są zazwyczaj dwa mało intensywne pasma drgań C-H (w -CHO)
przy 2820 cm
-1
i 2700 cm
-1
;
g)
keton: jeżeli wyeliminowano wszystkie poprzednie przypadki.
3. Jeżeli brak jest pasm grupy karbonylowej C=O należy sprawdzić obecność:
a) alkoholu lub fenolu: szerokie pasmo O-H w zakresie 3600 - 3000 cm
-1
, położenie
pasma C-O umożliwi identyfikację fenolu, alkoholu 3°, 2° i 1°;
C
=
O
O
-
H
N
-
H
26
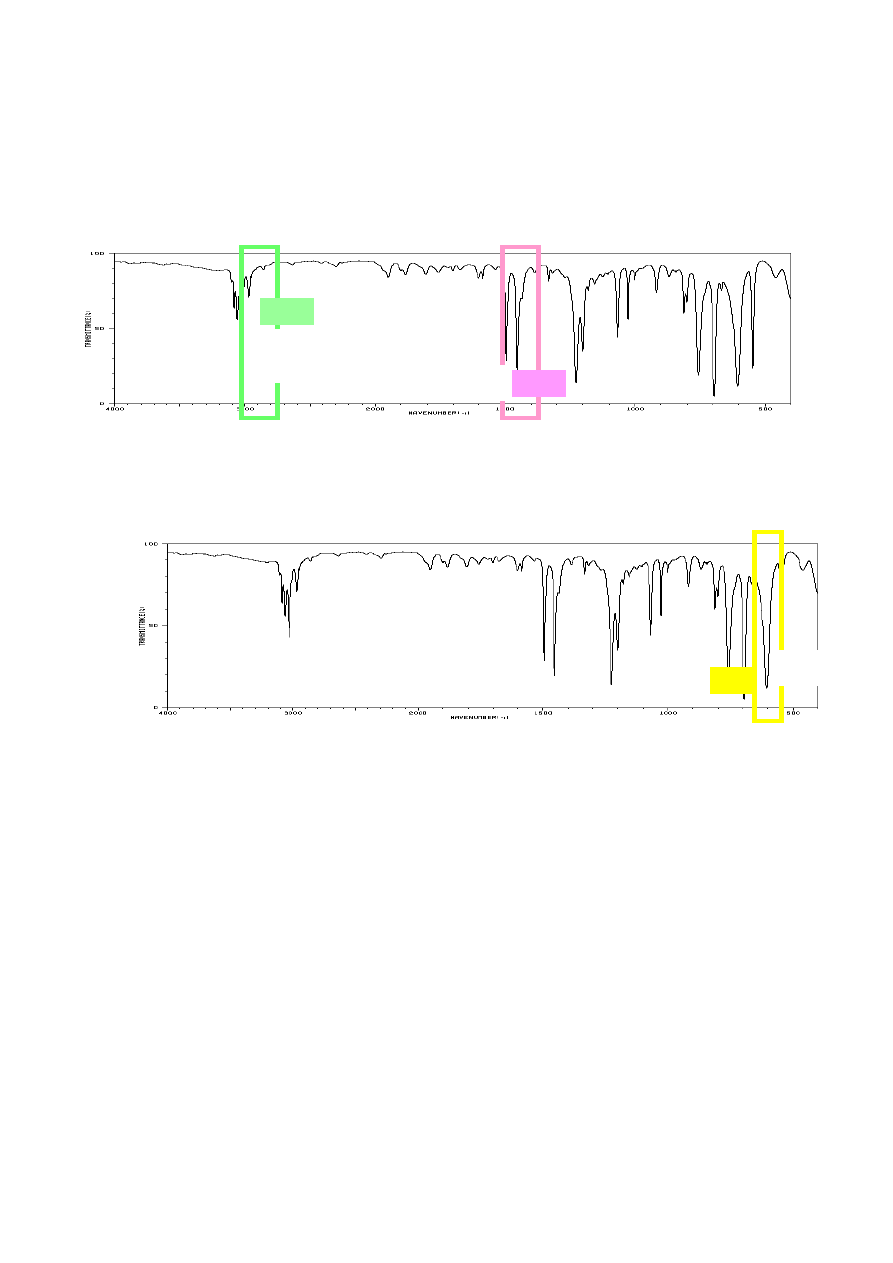
b) aminy: dwa pasma dla NH
2
i jedno dla grupy N-H leżące w zakresie 3400 cm
-1
. Dla
amin 1° jest również obecne pasmo drgań deformacyjnych N-H w zakresie 1650-1550
cm
-1
; amina trzeciorzędowa nie daje pasm w tym zakresie;
c) eteru: intensywne pasmo C-O występujące w zakresie 1300-1100 cm
-1
(przy braku O-
H). Należy jednak pamiętać, że w tym samym zakresie występuje pasmo drgań
deformacyjnych CH
2
X.
4. Analiza sugeruje, że związek nie posiada żadnych pasm charakterystycznych dla grup
funkcyjnych wymienionych powyżej. Następnym krokiem jest sprawdzenie obecności pasm
pochodzących od drgań rozciągających wiązań C=C leżących w zakresie około 1650 cm
-1
dla alkenów oraz 1600 – 1500 cm
-1
dla wiązań C=C pierścienia aromatycznego. Obecność
tych grup należy również potwierdzić w zakresie 3100 – 3000 cm
-1
, w którym pojawiają się
pasma =C-H.
W widmie analizowanego związku są obecne pasma pochodzące od drgań =C-H pierścienia
aromatycznego: 3087, 3064, 3031 cm
-1
oraz dwa charakterystyczne pasma 1602 cm
-1
i 1496
cm
-1
pochodzące od drgań deformacyjnych pierścienia. Obecne są również dwa pasma drgań
deformacyjnych =C-H (oop) 758, 695 cm
-1
charakterystyczne dla monopodstawionego
pierścienia benzenowego. Porównując układ sygnałów w zakresie nadtonów (1670 – 2000
cm
-1
) z wzorcowymi przykładami zawartymi w tablicach [1], można uzyskać potwierdzenie
przypuszczenia, że związek zawiera monopodstawiony pierścień benzenowy.
5. Należy również sprawdzić obecność wiązań potrójnych C≡N (wąskie pasmo powyżej 2200
cm
-1
), C≡C (poniżej 2200 cm
-1
) oraz ≡C-H (około 3300 cm
-1
). W analizowanym widmie
pasma te nie występują.
3106,
3087,
3064’
3031
1586,
1496
758,
695
C
-
O lub CH
2
X
=C
–
H
sp
2
-1s
=C
–
H
deformacyjne
(oop)
C
ar
=
C
ar
C
≡
C
C
≡
N
≡C
–
H
sp-1s
1227
27
6. Następnie należy określić czy w związku znajdują się charakterystyczne pasma drgań
rozciągających C-H poniżej 3000 cm
-1
należące do grup alifatycznych oraz pasma drgań
deformacyjnych grup CH
2
i CH
3
około 1410 i 1375 cm
-1
. W widmie są pasma 2968 cm
-1
i
1454 cm
-1
świadczące o obecności grupy CH
2
.
7. Ostatnim etapem analizy widma jest sprawdzenie obecności intensywnych pasm C-X,
leżących w zakresie C-Cl 750 cm
-1
, C-Br 600 cm
-1
oraz C-I 500 cm
-1
. Obecność
fluorowców musi być wtedy potwierdzone widmem masowym.
Podsumowanie analizy:
Analizowana cząsteczka jest związkiem aromatycznym, zawierającym podstawnik alkilowy
oraz atom fluorowca (prawdopodobnie bromu). Jeżeli założyć, że pierścień zawiera tylko jeden
podstawnik, to atom halogenu musi znajdować się przy łańcuchu alkilowym
17. LITERATURA
1.
Określenie struktury związków organicznych metodami spektroskopowymi- tablice i
ćwiczenia; Praca zbiorowa, red. M. Szafran, Z. Dega-Szafran, PWN, Warszawa 1988 .
2.
Spektroskopowe metody identyfikacji związków organicznych R. M. Silverstein, F. X.
Webster, D. J. Kiemle, PWN, Warszawa 2007.
3.
Preparatyka organiczna A. I. Vogel, B. S Furniss, A. J. Hannaford, P. W. G. Smith, A.
R. Tatchell, Wydawnictwo Naukowo-Techniczne, Warszawa 2006, (rozdział 3. str. 255).
4.
Introduiction to spectroscopy D. L. Pavia, G. M. Lampman, G. S. Kriz, Harcourt, Inc.
2001.
5.
Metody spektroskopowe i ich zastosowanie do identyfikacji związków organicznych;
Praca zbiorowa, red. W. Zieliński, A. Rajca, Wydawnictwo Naukowo-Techniczne,
Warszawa 1995.
1454
2968
606
C
-
Br
–C
–
H
sp
3
-1s
–CH
2
Wyszukiwarka
Podobne podstrony:
IR zadania gr D1 II rok 2011 12Z
2011 02 Pojazd z radarem IR
Odległości kolejowe IR Giewont 2011 2012r
IR materialy dodatkowe gr D1 II rok 2011 12Z
Odległości kolejowe IR Piast 2011 2012r
Odległości kolejowe IR Piast 2011 2012r
Odległości kolejowe IR Giewont 2011 2012r
IR Lecture1
2011 2 KOSZE
higiena dla studentów 2011 dr I Kosinska
Plan pracy na 2011 pps
W 8 Hormony 2010 2011
wm 2011 zad 2
Zawal serca 20 11 2011
PRK 23 10 2011 org
więcej podobnych podstron