
METABOLIZM GLIKOGENU
Glikogen- zapasowa forma glukozy, łatwa do wykorzystania. Jest to rozgałęziony polimer, złożony
z reszt glukozy, połączone wiązaniami alfa-1,4-glikozydowymi, przeciętnie co dziesięć reszt
glukozy występuje wiązanie alfa-1,6-glikozydowe. Glikogen pełni funkcję buforu, utrzymującego
odpowiednie stężęnie glukozy we krwi. Glukoza pochodząca z glikogenu, może być szybko
uwalniana w razie intensywnego głodu czy zapotrzebowania. Stężenie glikogenu jest większe w
wątrobie niż w mięśniach, natomiast wagowo jest go więcej w mięśniach. Podczas syntezy
glikogenu potrzebna jest aktywowana forma glukozy, glukozodifosforan urydyny (UDP-glukoza),
utworzona w reakcji, której substratami są UTP i glukozo-1-fosforan.
Rozkład glikogenu – przebiega w 3 etapach:
1. Uwolnienie glukozo-1-fosforanu z glikogenu
2. Przekształcenie glikogenu do dalszej degeneracji
3. Przekształcenie glukozo-1-fosforanu w glukozo-6-fosforan.
Natomiast glukozo-6-fosforan może być dalej metabolizowany trzema sposobami:
1. użyty jako pierwszy substrat w glikolizie
2. przekształcony w szlaku pentozofosforanowym z wytworzeniem NADPH i pochodnych
rybozy
3. przetwarzany na wolną glukozę do krwioobiegu
Przekształcenia te odbywają się w wątrobie, w mniejszym stopniu z jelitach i nerkach.
Fosforylaza glikogenowa- jest istotnym enzymem rozkładu glikogenu, zostaje uwolniony glukozo-
1-fosforan, poprzez odłączanie pojedynczych reszt glukozy poprzez dodawanie Pi. Miejsce
katalityczne znajduje się w głębokiej szczelinie utworzonej przez reszty aminokwasowe domeny
aminowej i karboksylowej. Rozszczepienie wiązania poprzez dodanie Pi nazywa się fosforolizą.
Reszty są usuwane od strony nieredukującego końca cząsteczki glikogenu (końca z wolną grupą
OH przy węglu C-4). Rozrywane są wiązania alfa-1,4-glikozydowe. Reakcja ta jest łatwo
odwracalna in vitro. Fosforolityczne rozszczepienie glikogenu jest energetycznie korzystne, gdyż
uwolbiony cukier jest już ufosforylowany. Fosforylaza przestaje rozszczepiać wiązanie alfa-1,4-
glikozydowe jeśli osiągnie resztę glukozy odległą o cztery reszty od rozgałęzienia. Do
katalitycznego działania fosforylazy potrzebny jest fosforan pirydoksalu. Dwa dodatkowe enzymy
transferaza i alfa-1,6-glukozydaza przekształcają glikogen, by mógł być dalej fosforylowany.
Transferaza przenosi grupę 3 reszt glukozowych z jednego zewnętrznego odgałęzienia na drugie.
Przeniesienie to odsłania pojedynczą resztę glukozy połączoną wiązaniem alfa-1,6-glikozydowym.
Alfa-1,6-glukozydaza, hydrolizuje wiązanie alfa-1,6-glikozydowe, uwalniając cząsteczkę wolnej
glukozy.
Fosfoglukomutaza- przenosi grupę fosforanową z glukozo-1-fosforanu i powstaje glukozo-6-
fosforan. Miejsce katalityczne zawiera fosforylowaną resztę seryny. Przechodzi z glukozo-1-
fosforanu na glukozo-1,6-bisfosforan a następnie powstaje glukozo-6-fosforan i regeneruje się
fosfoenzym.
Glukozo-6-fosfataza – znajduje się w wątrobie, w błonie gładkiego retikulum
endoplazmatycznego, od strony światła pęcherzyków. Odłącza grupę fosforanową i powstaje
glukoza i ortofosforan.
Regulacja fosforylazy – w mięśniach: fosforylaza istnieje w dwóch formach: aktywna fosforylaza
a i nieaktywna fosforylaza b. Są ze sobą w stanie równowagi. Fosforylaza a przekształca się w
fosforylazę b przez fosforylację pojedynczej reszty seryny w każdej z podjednostek. Tę
kowalencyjną modyfikację katalizuje enzym kinaza fosforylazowa. Forma T jest nieaktywna,
ponieważ jej miejsce katalityczne jest częściowo zablokowane, przez pętle. Stan równowagi zależy
od warunków występujących w komórce. W mięśniach fosforylaza b jest aktywna tylko w
obecności dużych stężeń AMP, ATP przesuwa równowagę w stronę tworzenia formy R
(rozluźnionej). Przemiany fosforylazy b między stanami T i R są kontrolowane przez ładunek
energetycznych komórki mięśniowej. Glukozo-6-fosforan faworyzuje formę T. Fosforylaza a jest w
pełni aktywna, niezależnie od stężenia ATP,AMP i glukozo-6-fosforanu. Wysiłek fizyczny

powoduje przejście fosforylazy b ze stanu R do stanu T i powstawanie formy a.
Regulacja fosforylazy- w wątrobie: w wątrobie forma a jest wrażliwsza na przechodzenie z formy
T do R. Związanie glukozy przesuwa równowagę z formy R do T, co powoduje inaktywację
enzymu. Fosforylaza w wątrobie jest niewrażliwa na stężenie AMP, ponieważ nie występują tam
gwałtowne zmiany ładunku energetycznego.
Regulacja kinazy fosforylazowej: aktywność katalityczna kinazy jest zlokalizowana w
podjednostce gamma, a inne jej podjednostki pełnią funkcję regulacyjną. Kinaza przekształca się z
formy mało aktywnej w bardzo aktywną przez fosforylację podjednostki beta. Enzymem
katalizującym aktywację kinazy jest kinaza białkowa A (PKA). Aktywacja zachodzi również dzięki
zmianom stężenia jonów Ca2+. Kalmodulina jest czynnikiem wrażliwym na wapń, jest w
podjednostce gamma. Kinaza osiąga maksymalną aktywność tylko wtedy, gdy zachodzi zarówno
fosforylacja podjednostki beta, jak i aktywacja podjednostki gamma przez związanie Ca2+.
Rozkład glikogenu: Glukagon i adrealina wywołują rozkład glikogenu. Adrealina stymuluje
bardziej rozkład glikogenu w mięśniach, niż w wątrobie, glukagon rozkłada bardziej w wątrobie.
Kaskada przekaztwania sygnału odpowiedzialnego za rozkład glikogenu:
1. Adrealina i glukagon wiążą się do receptorów 7TM. Adrealina wiążę się do receptora beta-
adrenergicznego, glukagon- z receptorem glukagonu. Procesy te aktywują podjednostkę alfa
białka G.
2. GTP związany z podjednostką alfa białka G aktywuje cyklazę adenylanową, tworzącą
cAMP.
3. Zwiększone stężenie cAMP w cytozolu aktywuje PKA. Powoduje to odłączenie jednostki
regulatorowej od katalitycznej, która jako wolna staje się aktywna.
4. PKA fosforyluje podjednostkę beta kinazy fosforylazowej, która aktywuje fosforylazę
glikogenową. Kaskada ta silnie wzmacnia działanie hormonów.
W wątrobie adrealina wiązę się nie tylko z receptorem beta-adrenergicznym ale też z receptorem
7TM alfa-adrenergicznym, który aktywuje fosfolipazę C, co zapoczątkowuje kaskadę
fosfoinozytolową. Wzrost poziomu 1,4,5-trisfosforanu inozytolu indukuje uwolnienie Ca2+ z
retikulum endoplazmatycznego.
Zahamowanie rozkładu glikogenu: Białko G, przekształca związany ATP w ADP co hamuje
przekazywanie sygnału. Komórki zawierają aktywność fosfodiestrazy, która przekształca cAMP w
AMP. PKA uczestniczy w wyłaczaniu procesu degradacji glikogenu przez dodawanie grupy
fosforanowej do podjednostki alfa kinazy fosforylazowej, tuż po ufosforylowaniu podjednostki
beta. Enzym ulega inaktywacji przez fosfatazę 1 białek PP1.
Szlaki syntezy i degradacji glikogenu: UDP-glukoza, donor glukozy w syntezie glikogenu, jest
aktywną formą glukozy. Jest syntetyzowana z glukozo-1-fosforanu i trifosforanu urydyny w reakcji
katalizowanej przez pirofosforylazę UDP-glukozy. Uwolniony w tej reakcji pirofosforan pochodzi z
dwóch zewnętrznych reszt fosforanowych UTP. Reakcja ta jest łatwo odwracalna. Jednak in vivo
pirofosforan szybko ulega hydrolizie do ortofosforanu przez enzym pirofosforan szybko ulega
hydrolizie do ortofosforanu przez enzym pirofosfatazę nieorganiczną. Ta nieodwracalna reakcja
napędza syntezę UDP-glukozy.
Aktywowana jednostka UDP-glukozy przenoszona jest do grupy hydroksylowej węgla C-4 na
końcu glikogenu- powstaje wiązanie alfa-1,4-glikozydowe. Reakcję tę katalizuje syntaza
glikogenowa. Syntaza może dodawać reszty glukozowe tylko wtedy, gdy łańcuch zawiera już 4
albo więcej reszt glukozy. Syntaza wymaga więc startera. Funkcję tę pełni glikogenina. Każda
podjednostka glikogeniny katalizuje przyłączenie ośmiu jednostek glukozy do drugiej podjednoski
w dimerze glikogeniny. Donorem reszt glukozowych jest UDP-glukoza. Po przyłączeniu 8 reszt
glukozy, włącza się syntaza glikogenowa, która wydłuża cząsteczkę glikogenu. Odgałęzienie
tworzone jest przez rozerwanie wiązania alfa-1,4-glikozydowego i utworzenie połączenia alfa-1,6.
Enzym rozgałęziający jest dość wymagający. Grupa ok. 7 reszt glukozy musi zawierać koniec
nieredukujący i pochodzić z łańcucha zbudowanego z co najmniej 11 reszt. Nowy punkt
rozgałęziający musi być oddalony o co najmniej 4 reszty od rozgałęzienia już istniejącego.
Rozgałęzienia w glikogenie zwiększają jego rozpuszczalność.

Regulacja syntazy glikogenowej- jest regulowana przez modyfikację kowalencyjną. Syntaza
glikogenowa ulega w wielu miejscach fosforylacji przez kinazę białkową A i kilka innych kinaz.
Fosforylacja przekształca aktywną formę a w nieaktywną formę b.
Podczas włączania glukozo-6-fosforanu do glikogenu ulega hydrolizie 1 cząsteczka ATP. 90% reszt
jest rozczepiane przez fosforolizę do glukozo-1-fosforanu, który bez nakładów energetycznych jest
przekształcany do glukozo-6-fosforanu. 10% stanowią reszty rozgałęzione, które są rozszczepiane
hydrolitycznie. Całkowite utlenienie glukozo-6-fosforanu dostarcza ok. 31 cząsteczek ATP, a proces
zmagazynowania zużywa trochę więcej niż 1 cząsteczkę ATP.
Fosfataza białkowa 1 – enzym, odgrywa kluczową rolę w metaboliźmie glikogenu. PP1
inaktywuje kinazę fosforylazową i fosforylazę a przez defosforylazę. PP1 zmniejsza szybkość
rozkładanego glikogenu, ponieważ odwraca efekt kaskady. PP1 usuwa grupę fosforanową z syntazy
glikogenowej b i przekształca ją w aktywną formę a. PP1 przyspiesza syntezę glikogenu. PP1
składa się z 3 podjednostek: Rgi – decyduje o powinowactwie do glikogenu, samej PP1 i z
inhibiotra 1, który w stanie ufosforylowania hamuje PP1. Regulacja aktywności fosfatazy białkowej
1- gdy przeważa degradacja glikogenu- fosforylacja podjednostki Rgi przez PKA zapobiega jej
łączeniu z podjednostka katalityczna PP1. Wskutek tego aktywacja kaskady cAMP prowadzi do
inaktywacji PP1, ponieważ nie może ona dłużej wiązać substratu. Gdy działa cAMP, włączana jest
degradacja glikogenu.
Stymulacja syntezy glikogenu: Gdy stężęnie glukozy we krwi jest duże, insulina stymuluje
syntezę glikogenu uruchamiając szlak, który aktywuje fosfatazę białkowa 1. Pierwszy etap
działania insuliny polega na jej związaniu z receptorową kinazą tyrozynową, która znajduje się w
błonie plazmatycznej. Wielokrotne fosforylacje wywołują falę defosforylacji. Związanie insuliny z
receptorem powoduje aktywację kinazy białkowej wrażliwej na insulinę, która fosforyluje
podjednostkę Rgi, ale w innym miejscu niż w tym, w którym fosforyluje ją PKA. Fosforylacja ta
prowadzi do połączenia podjednostki Rgi z PP1 i z cząsteczkę glikogenu. Defosforylacja syntezy
glikogenowej, kinazy fosforylazowej i fosforylazy powoduje syntezę glikogenu.
Poziom glukozy we krwi: Ilość fosforylazy a w wątrobie zmniejsza się szybko, gdy pojawia się
glukoza. Po fazie opóźnienia zwiększa si e ilosc syntazy glikogenowej a, co powoduje rozpoczęcia
syntezy glukogenu. Fosforylaza a jest czynnikiem reagującym na poziom glukozy w wątrobie.
Wiązanie glukozy do fosforylazy a przesuwa równowagę z aktywnej formy R do nieaktywnej T. Te
zmiany powodują, że grupa fosforanowa seryny staje się substratem dla fosfatazy białkowej 1.
Fosforylaza b nie wiążę się z fosfatazą. W konsekwencji, przekształceniu fosforylazy a w
fosforylazę b towarzyszy uwolnienie fosfatazy PP1, która może aktywować syntazę glikogenową.
Aktywność syntazy glikogenowej wzrasta wtedy, gdy dominuje forma b fosforylazy.
METABOLIZM KWASÓW TŁUSZCZOWYCH
Kwasy tłuszczowe to długie łańcuchy węglowodorowe, zakończone grupą karboksylową. Są
składnikami lipidów i fosfolipidów (składniki błon biologicznych), magazynują energię (w formie
triacylogliceroli), niektóre hormony i wewnątrzkomórkowe przekaźniki sygnałów są pochodnymi
kwasów tłuszczowych.
Triacyloglicerole – są to związki zredukowane i bezwodne, mają duży zasób energii metabolicznej.
Główne miejsce akumulacjii triacylogliceroli u ssaków jest cytoplazma komórek tłuszczowych
(adypocytów), wyspecjalizowanych do przechowywania i uruchamiania ich zapasów. W jelicie
triacyloglicerole są włączane do miceli za pomocą soli źółciowych. W komórkach błony śluzowej
jelit triacyloglicerole są ponownie syntetyzowane z kwasów tłuszczowych i monoacylogliceroli, a
nastepnie upakowane do chylomikronów. Uczestniczą one w transporcie cholesterolu i witamin
rozpuszczalnych w tłuszczach. Są uwalniane do układu limfatycznego, a następnie do krwi.
Wykorzystanie energii z triacylogliceroli:
1) Hydroliza triacylogliceroli przez lipazy: ten etap nazywa się lipolizą. Lipazy tkanki
tłuszczowej są aktywowane przez traktowanie jej komórek hormonami- adrealiną,
noradrealiną, glukagonem lub adrenokortykotropiną. W adypocytacg te hormony za

pośrednictwem 7TM aktywują cyklazę adenylanowa. Zwiększone stężęnie AMP stymuluje
kinazę białkową A, która aktywuje lipazy przez fosforylację. Wyżej wymienione hormony
indukują lipolizę, natomiast insulina ją hamuje. Uwalniane kwasy wiążą się z albuminą,
przez co stają się dostępnym źródłem energii dla innych tkanek. Powstający glicerol jest
wchłaniany w wątrobie, ulega fosforylacji i utlenieniu do fosfodihydroksyacetonu, a
następnie izomeryzacji dla aldehydu-3-fosfoglicerynowego.
2) Przyłączenie do koenzymu A: kwasy tłuszczowe są utleniane w mitochondrium, są
aktywowane zanim tam dotrą. Powstawanie wiązania tioestrowego miedzy grupą
karboksylową kwasu tłuszczowego a grupą hydrosulfidową coA napędza ATP. Aktywacja
przebiega na zewnętrznej błonie mitochondrialnej dzięki syntetazie acylo-coA. Najpierw
powstaje acyloadenulan (w reakcji z ATP) potem acylo-coA i AMP. Te reakcje są łatwo
odwracalne.
3) Przeniesienie aktywowanych kwasów tłuszczowych do matriks: przenoszone są przez
karnitynę. Grupa acylowa zostaje przeniesiona z atomu siarki coA na grupę hydroksylową
karnityny i powstaje acylokarnityna. Reakcję katalizuje acylotransferaza karnitynowa I.
Acylokarnitynę przez błonę przemieszcza odpowiednia translokaza. W błonie wewnętrznej
po stronie matriks reszta acylowa jest ponownie przenoszona na CoA. Reakcję tę katalizuje
acylotransferaza karnitynowa II. Na koniec translokaza przenosi karnitynę z powrotem na
stronę cytozolu, wymieniając ją na acylokarnitynę wprowadzaną do mitochondrium.
Szlak beta-oksydacji: nasycony łańcuch tłuszczowy acylo-coA jest degradowany w powtarzającej
się sekwencji 4 reakcji: utlenienia z udziałem FAD, uwodnienia, utlenienia z udziałem NAD+ i
tiolizy przez coA. W wyniku tej reakcji powstaje FADH2, NADH i acetylo-coA, a łąńcuch kwasu
tłuszczowego skraca się o 2 atomy węgla. Jest to szlak beta-oksydacji. Dotyczy on kwasów
tłuszczowych o parzystej liczbie węgli.
1) Utlenienie – utlenienie acylo-coA z udziałem dehydrogenazy acylo-coA, a produktem jest
enoilo-coA z podwójnym wiązaniem. Elektrony z FADH2 (gr. Prostetyczna) są przenoszone
na flawoproteinę przenoszącą elektrony (ETF). ETF przekazuje elektrony na białko
żelazowo-siarkowe: reduktazę ETF-ubichonon. Ubichinon ulega redukcji do ubichinoli. Na
tym etapie z 1 cząsteczki FADH2 powstaje 1,5 ATP.
2) Uwodnienie- uwodnione zostaje podwójne wiązanie między C-2 i C-3 przez hydratazę
enoilo-coA.
3) Utlenianie – powoduje przekształcenie grupy hydroksylowej przy atomie C-3 w ketonową, z
wytworzeniem NADH. Reakcję katalizuje dehydrogenaza L-3-hydroksyacylo-coA.
4) Tioliza – rozszczepienie 3-ketoacylo-coA przez grupę tiolową innej cząsteczki koenzymu A,
w wyniku czego powstaje acetylo-coA oraz acylo-coA. To cięcie katalizuje beta-ketotiolaza.
Skrócony acylo-coA wchodzi w 2 cykl utleniania, który rozpoczyna się od reakcji katalizowanej
przez dehydrogenazę acylo-coA.
Utlenianie nienasyconych kwasów tłuszczowych:
Utlenianie palmitooleinianu: kwas tłuszczowy C16, z 1 podwójnym wiązaniem między C-9 a C-10,
ulega aktywacji i jest transpotowany przez błonę tak samo jak nasycone kwasy tłuszczowe.
Palmitooleilo-coA podlega 3 cyklom degradacji, przez te same enzymy. W 3 cyklu powstaje cis-
(delta^3)-enoilo-coA. Odpowiednia izomeraza przekształca wiązanie podwójne między C-3 i C-4 w
wiązanie trans-(delta^2). Kolejne reakcje są takie same jak w szlaku utleniania kwasów nasyconych
Linolan- wielonienasycony kwas tłuszczowy C18 z wiązaniami pdwójnymi cis-delta^9 i cis-
delta^12. Reakcje wspomaga reduktaza-2,4-dienoilo-coA. Formy są przekształcane w trans-delta^2.
Z nieparzystymi wiązaniami podwójnymi radzi sobie izomeraza, a z parzystymi- izomeraza i
reduktaza. Produktem ostatecznej tiolizy łańcuchów kwasów tłuszczowych o nieparzystej liczbie
węgli jest propionylo-coA, który włącza się do cyklu Krebsa po przekształceniu w bursztynylo-
coA. Potrzebna jest do tego witamina B12. Pewna część kwasów tłuszczowych ulega również
utlenieniu w peroksysomach, które skracają długie łańcuchy, czyniąc je podatniejszymi na beta-
oksydację w mitochondriach.
Ciała ketonowe: U osób będących na czczo i u cukrzyków szczawiooctan jest wykorzystywany do

syntezy glukozy w szlaku glukoneogenezy. W takich warunkach następuje wykorzystanie acetylo-
coA do syntezy acetooctanu i D-3-hydroksymaślanu, które są nazywane ciałkami ketonowymi.
Synteza acetooctan z acetylo-coA przebiega w 3 etapach: Dwie cząsteczki acetylo-coA kondensują
do acetoacetylo-coA. Acetoacetylo-coA reaguje z acetylo-coA i wodą, dając 3-hydroksy-
3metyloglutarylo-coA i coA. 3-hydroksy-3metyloglutarylo-coA ulega rozszczepieniu co acetylo-
coA iacetooctanu. Głównym miejscem produkcji acetooctanu i 3-hydroksymaślanu jest wątroba.
Substancje te dyfundują z mitochondriów wątroby do krwi i są transportowane do tkanek
docelowych. Są one paliwem komórkowym. Mięsień serca i kora nerek wykorzystują acetooctan.
W wyniku utleniania 3-hydroksymaślanu powstaje acetooctan oraz NADH, wykorzystywane w
fosforylacji oksydacyjnej. Acetooctan może ulec aktywacji przez przeniesienie coA z bursztynylo-
coA. Reakcję tę katalizuje odpowiednia transferaza coA. Acetoacetylo-coA jest rozcinany do 2
cząsteczek acetylo-coA, wchodzących do cyklu Krebsa.
Różnice między szlakami degradacji i syntezy kwasów tłuszczowych:
1. Synteza przebiega w cytozolu, degradacja w matriks mitochondriów
2. Produkty pośrednie syntezy kwasów tłuszczowych są kowalencyjnie związane z ACP, a
intermediaty rozkładu są kowalencyjnie związane z grupą hydrosulfidową koenzymu A.
3. Enzymy z syntezy są połączone w pojedynczy łańcuch polipeptydowy (syntaza kwasów
tłuszczowych), a enzymy degradacji nie są połączone.
4. Rosnący łańcuch kwasu tłuszczowego ulega wydłużaniu przez kolejne dobudywowanie
dwuwęglowych jednostek, pochodzących z acetylo-coA. Aktywowanym donorem tych
jednostek jest malonylo-ACP. Reakcja elongacji napędza uwalnianie CO2
5. Reduktorem w syntezie jest NADPH, utleniaczami w redukcji NAD+ i FAD
6. Wydłużanie ulega zahamowaniu po zsyntetyzowaniu palmitynianu.
Synteza malonylo-coA – synteza kwasów tłuszczowych rozpoczyna się od karboksylacji acetylo-
coA do malonylo-coA. Jest to reakcja nieodwracalna. Syntezę malonylo-coA, katalizuje
karboksylaza acetylo-coA. Produktem pośredniem jest CO2-biotyna-enzym. Aktywowana grupa
CO2 tego produktu pośredniego jest przenoszona na acetylo-coA, tworząc malonylo-coA.
Wydłużanie kwasów tłuszczowych: faza wydłużania rozpoczyna się od powstania acetylo-ACP i
malonylo-ACP. Reakcje te są katalizowane przez acylotransferazy: transacylazę acetylową i
transacylazę malonulową. Kwasy tłuszczowe o nieparzystej liczbie atomów węgla są
syntetyzowane począwszy od piopionylo-ACP, który powstaje z propionylo-coA. Acetylo-ACP i
malonylo-ACP reagują, tworząc acetoacetylo-ACP. Reakcję katalizuje anzym kondensujący
acylomalonylo-ACP. Nie mogą kondensować dwie jednostki acetylo-ACP, gdyż jest to niekorzystna
energetycznie reakcja. Wszystkie kwasy z parzystą liczbą węgli pochodzą od acetylo-coA.
Czynnikiem redukującym jest NADPH. Cykle wydłużania trwają aż do powstania C16-actylo-ACP,
dając palmitynian i ACP. Z palmitynianu uzyskujemy 106 cząsteczek ATP.
Całkowita stechiometria syntezy palmitynianu:
8 acetylo-coA + 7ATP + 14NADPH + 6H+ -> palmitynian + 14NADP+ + 8coA + 6H2O+ 7ADP + 7Pi
Cytrynian przenosi reszty octanu przez wewnętrzną błonę mitochondriów, czyli acetylo-coA. Jeśli
cytrynian osiągnie duże steżęnie, ulega w cytozolu rozpszczepieniu przez liazę ATP-cytrynianową.
W ten sposób acetylo-coA i szczawiooctan są przenoszone z mitochondriów do cytozolu kosztem
hydrolizy cząsteczki ATP.
NADPH jest potrzebne do syntezy kwasów. Podczas transportu jednej cząsteczki acetylo-coA do
cytozolu powstaje 1 NADPH. Kiedy 8 cząsteczek acetylo-coA potrzebnych do syntezy
palmitynianu wędruje do cytozolu, powstaje 8 NADPH. 6 dodatkowych cząsteczek NADPH
powstaje ze szlaku pentozofosforanowego.
Hormony ikozanoidowe: arachidonian, kwas tłuszczowy 20:4, jest głównym prekursorem wielu
klas cząsteczek sygnałowych: prostaglandyn, prostacyklin itp. Prostaglandyna jest wytwarzana
przez reduktazy i izomerazy.Stymulują stan zapalnu, regulują przepływ krwi, kontrolują przepływ
jonów przez błony itp. Hormony ikozanoidowe mają 20 atomów węgla.

PRZEMIANA BIAŁEK I KATABOLIZM AMINOKWASÓW:
Aminokwasy: podstawową funkcją aminokwasów jest wykorzystywanie ich jako elementów
budulcowych w syntezie białek i peptydów, oraz jako źródło azotu. Aminokwasy występują w
nadmiarze i nie mogą być przechowywane. Nadmiar jest wykorzystywany jako paliwo komórkowe,
przekształcana w mocznik w cyklu kwasu mocznikowego, ich szkielety węglowe w acetylo-coA,
acetoacetylo-coA, pirogronian itp. Mogą powstawać kwasy tłuszczowe, ciała ketonowe i glukoza.
Rozkład białek: rozpoczyna się w żołądku pod wpływem kwaśnego środowiska, które denaturuje
białko. Ulegają wtedy łatwiej proteolizie. Dalszy rozkład zachodzi w jelicie dzięki wytwarzanym
przez trzustkę enzymom. Aminokwasu są transportowane ze światła jelita do komórek ściany jelita i
uwalniane do krwioobiegu. Białka ulegają degradacji z różną szybkością.
Ubikwityna: małe białko, występuje u wszystkich eukariotów, pełni funkcję znacznika białek
przeznaczonych do degradacji. W przyłączaniu ubikwityny do białek biorą udział 3 enzymy: enzym
aktywujący ubikwitynęE1, enzym koniugujący E2 i ligaza ubikwitynowo-białkowa E3.
1. etap: utworzenie wiązania tioestrowego między grupą karboksylową ubikwityny a grupą
hydrosulfidową enzymu E1. Reakcja ta wymaga ATP. Adenylan przyłącza się do grupy
karboksylowej na końcu C ubikwityny, a towarzyszy temu uwolnienie pirofosforanu.
Następnie ubikwityna zostaje przeniesiona na grupę hydrosulfidową kluczowej reszty
cysteiny w białku E1.
2. Przeniesienie aktywowanej ubikwityny na gr. Hydrosulfidową enzymu E2
3. Katalizowany etap przez E3: przeniesienie ubikwityny z E2 na grupę E-aminową białka
docelowego.
Enzymy: E1- enzym aktywujący ubikwitynę E2- enzym koniugujący E3- ligaza ubikwityno-
białkowa.
O okresie półtrwania białek cytoplazmatycznych w dużej mierze decydują reszty aminokwasowe
występujące na końcu aminowym białka. Zależność ta nazywa się regułą końca N. Zależy to
również od tego czy an końcu występują reszty degradujące lub sekwencja PEST.
Proteasom: proteasom 26S jest kompleksem białkowym złożonym z dwóch części- proteasomu
20S o aktywności katalitycznej i dwóch podjednostek regulatorowych 19S. Rozkłada on
nieporządane białka, oznaczone przez ubikwitynę. Uwalniana ubikwityna jest ponownie
wykorzystana, a zdegradowane białka są rozkładane do pojedynczych aminokwasów.
Degradacja aminokwasów: Grupa alfa-aminowa wielu aminokwasów jest przenoszona na alfa-
ketoglutaran i powstaje glutaminian, którego deaminacja prowadzi do odszczepienia jonu
amonowego NH4+. Te reakcje katalizują aminotransferazy. Aminotransferaza asparaginianowa
katalizuje przeniesienie grupy aminowej z asparaginianu na alfa-ketoglutaran. Aminotransferaza
alaninowa katalizuje przeniesienie na alfa-ketoglutaran z alaniny. Reakcje te są odwracalne.
Dehydrogenaza glutaminianowa przenosi atom azotu i uwalnia go jako jon amonowy. Enzym ten
wykorzystuje zarówno NAD+ i NADP+.
Fosforan pirydoksalu: zawierają go wszystkie aminotransferazy. Ma on grupę aldehydową, która
pozwala na tworzenie produktu pośredniego o charakterze zasady Sniffa pomiędzy fosforanem
pirydoksalu a substratami aminokwasowymi. Jest wykorzystywany jako koenzym w reakcjach
transaminacji przez aminotransferazę asparaginianową. Oprócz transaminacji katalizuje również
dekarboksylacje, deaminację, racemizacje i rozszczepienie aldolowe. Usuwaja i wymieniają
podstawniki przy węglach beta i gamma.
Transport azotu z mięśni do wątroby: jest transportowany w 2 formach zasadniczych. W wyniku
reakcji transaminacji tworzy się glutaminian, a następnie azot jest przenoszony na pirogronian, w
wyniku czego powstaje alanina, która zostaje uwolniona do krwi. Alanina jest wychwytywana przez
wątrobę i tam przekształcana z powrotem w pirogronian przez transaminację. Pirogronian może być
wykorzystywany w glukoneogenezie a grupa aminowa ulega przekształceniu w mocznik. Transport
ten określa się jako cykl alaninowy. Azot może być również przekształcany jako glutamina.
Cykl mocznikowy: prowadzi do syntezy mocznika
Reakcje
Enzymy
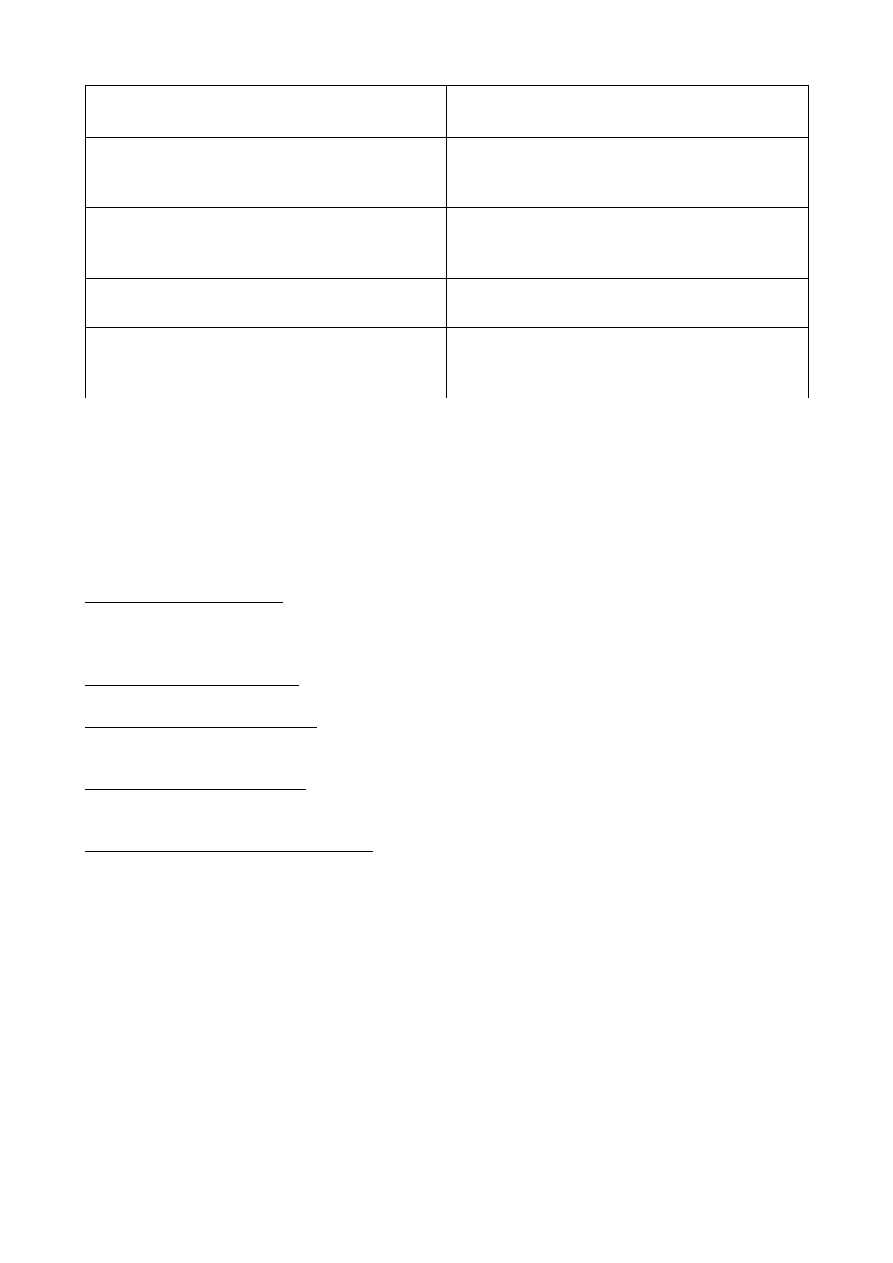
Powstanie karbamoilofosforanu z jonów NH4+ i
HCO3-
Syntetaza karbamoilofosforanu
Grupa karbamiolowa z karbamoilofosforanu
zostaje przeniesiona na ornitynę, co prowadzi do
powstania cytruliny (w mitochondrium)
Karbamoilotransferaza ornitynowa
Cytrulina jest transportowana do cytoplazmy,
gdzie ulega kondensacji z asparaginianem i
powstaje argininobursztynian
Synteraza argininobursztynianu + ATP
Argininobursztynian jest rozszczepiany do
argininy i fumaranu
Liaza argininobursztynianowa
Argininę zostaje zhydrolizowana do mocznika i
ornityny. Ornityna jest transportowana do
mitochondrium, a mocznik jest wydalany
Arginaza
Stechiometria cyklu mocznikowego:
CO2 + NH4+ + 3ATP + asparaginian + 2H2O -> mocznik + 2ADP + 2Pi + AMP + Ppi + fumaran
Synteza fumaranu zachodząca w cyklu mocznikowym łaczy go z cyklem Krebsa.
Proces degradacji aminokwasów prowadzi do powstawania intermediatów: łańcuchy 20
węglowe są przekształcane do: pirogronianu, acetylo-coA, acetoacetylo-coA, alfa-ketoglutaranu,
bursztynylo-coA, fumaranu i szczawiooctanu. Aminokwasy nazwane ketogennymi degradują się do
ciał ketonowych lub kwasów tłuszczowych. Aminokwasy glukogenne degradują się do
intermediatów które wchodzą w skład cyklu Krebsa, glikolizy i glukoneogenezy ( do glukozy).
Mogą być również aminokwasy, które są zarówno glukogenne i ketogenne.
Powstawanie pirogronianu:
alanina + alfa-ketoglutaran <=> pirogronian + glutaminian
seryna -> pirogronian + NH4+
Również pirogronian powstaje z cysteiny, lub z aminoacetonu, który powstaje z treoniny.
Powstawanie szczawiooctanu:
Asparaginian + alfa-ketoglutaran <=> szczawiooctan + glutaminian
Powstawanie alfa-ketoglutaranu:
Z glutaminy, proliny, argininy lub histydyny powstaje glutaminian, który za pomocą dehydrogenazy
glutaminianowej zostaje przekształcony w alfa-ketoglutaran.
Powstawanie bursztynylo-coA:
Metionina, walina, izoleucyna – powstaje propionylo-coA, następnie metylomalonylo-coA i w
końcu bursztynylo-coA. Powstaje również z metioniny w 9 różnych etapach.
Rozkład aminokwasów rozgałęzionych:
Prowadzi do powstawania acetylo-coA, acetooctanu i propionylo-coA. Tymi aminokwasami
rozgałęzionymi są leucyna i walina.
SYSTEMY CZUCIA
Odbiór zapachu – kształt cząsteczki jest bardziej odpowiedzialny za jej zapach, a nie pozostałe
właściwości fizyczne. Cząsteczki zapachowe są wykrywalne w określonej części nosa zwanej
nabłonkiem węchowym, która znajduje się w szczycie jamy nosowej. Rzęski czuciowe, zawierające
białkowe receptory wiążące cząsteczki zapachowe, wystają z tych neuronów do śluzówki
wyściełającej jamę nosową.W procesie przekazywania sygnału zapachowego biorą udział białka G
u receptory 7TM. Jednostka białka G, zwana G(olf) ulega ekspresji wyłącznie w rzęskach
węchowych. Białka receptorów węchowych RW wykazują w 20% identyczne sekwencje z
receptorem beta-adrenergicznym. Środkowy rejon białek jest wysoce zmienny, co sugeruje, ze jest
on miejscem wiązania substancji zapachowej. Wśród setek istniejących genów RW w każdym
neuronie węchowym ekspresji ulega tylko jeden z nich. Wiązanie substancji zapachowej z RW na
powierzchni neuronu inicjuje kaskadę przewodzenia sygnału i powstanie potencjału
czynnościowego. RW związany z ligandem aktywuje G(olf), początkowo związany z GDP. Po

aktywacji wiąże się z GTP i zostają uwolnione związane dotychczas podjednoski beta-gamma.
Podjednostka alfa aktywuje specyficzną cyklazę adenylanową, zwiększając stężenie cAMP. Wzrost
stężenia cAMP aktywuje niespecyficzny kanał kationowy, przez który przedostaje się wapń i inne
kationy do komórki. Przepływ kationów pozwala na zainicjowanie powstawania potencjału
czynnościowego, co wywołuje odczucie specyficznego zapachu. Prawie wszystkie substancje
zapachowe aktywują pewną liczbę receptorów i prawie każdy receptor jest aktywowany przez
więcej niż jedną substancję zapachową. Neurony wykazujące ekspresję specyficznych RW są
połączone ze ściśle określonymi miejscami w mózgu. Czynnościowy rezonans magnetyczny
pozwala na ujawnienie regionów przetwarzających informacje czuciowe. Gdy specyficzna część
mózgu jest aktywna, naczynia krwionośne rozszerzają się, bardziej aktywna część mózgu będzie
zawierała więcej oksyhemoglobiny w danym momencie.
Odbiór smaku- odbieramy tylko 5 podstawowych smaków- gorzki, słodki, słony, kwaśny i umami
(smak glutaminianu). Cząsteczki smakowe są wykrywane przez wyspecjalizowane struktury zwane
kubkami smakowymi. Na jednym końcu neuronu czuciowego, w kierunku powierzchni języka,
wystają mikrokosmki. Włókna nerwowe na przeciwległym końcu każdego neuronu przenoszą
implusy elektryczne w odpowiedzi na stymulację spowodowaną przez substancje smakowe. Białko
G również uczestniczy w procesie odbierania smaku, a szczególnie podjednostka zwana
gustducyną- smak gorzki. Analiza locus wykazała, że region odpowiedzialny za wykrywanie
gorzkiego smaku może kodować receptor 7TM, reagujący na PROP. Wykryto jeden gen
wyizolowany z receptora 7TM- T2R-1. Za wykrywanie smaku słodkiego odpowiedzialna jest
rodzina receptorów 7TM a za wykrywanie smaku słonego przepływ jonów sodowcyh przez kanały
wrażliwe na amiloryd. Smak kwaśny powstaje w wyniku oddziaływania jonów wodorowych na
kanały. Przepływające jony indukują znaczny prąd w poprzek błony. Glutaminian ma smak zwany
umami, rozpoznawany przez metabotropowe receptory glutaminianu.
Proces widzenia – mamy 2 rodzaje komórek fotoreceptorowych, zwane pręcikami i czopkami,
które są wrażliwe na fale o długości 300 do 850 nm. Czopki odbierają kolory i funkcjonują w
jasnym świetle, pręciki funkcjonują w świetle przyćmionym. Mamy 3 miliony czopków i 100
milionów pręcików. Budowa pręcików- są smukłymi komórkami, mają segment zewnętrzny
zawierający dyski. Cząsteczka fotoreceptora w pręciku stanowi rodopsyna, która składa się z białka
opsyny, połączoną z gr. Prostetyczną 11-cis-retinalem.
W wyniku wzbudzenia rodopsyny przez światło zachodzą następujące zdarzenia:
1. przekształcenie z 11-cis-retinalu do całkowicie trans-retinalu podczas absorpcji światła,
następuje izomeryzacja zasad Sniffa
2. przemiana w batorodopsynę
3. przemiana w metarodopsynę II, zasada Sniffa ulega deprotonacji, a białko opsyna
przechodzi reorganizację
4. aktywacja transducyny białek G, metarodopsyna powoduje wymianę GDP na GTP w
podjednostce alfa transducyny. Po związanie GTP podjednostka aktywuje fosfodiesterazę
cGMP, hydrolizuje cGMP do GMP. Zmniejszenie stężenia cGMP powoduje zamknięcie
kanałów jonowych bramkowanych cGMP, polaryzację błony i wytworzenie sygnału
nerwowego
5. uruchomienie kaskady enzymatycznej ( spadek stężenia cGMP -> kanały jonowe zamknięte
-> spadek stężenia jonów Ca2+ -> aktywność cyklazy guanylanowej wzrasta -> wzrost
stężenia cGMP)
6. przekształcenie z trans-retinalu do 11-cis-retinalu. Poziom wapnia reguluje szybkość z jaką
układ wraca do stanu wyjściowego.
W ludzkich czopkach 11-cis retinal jest wykorzystywany jako chromofor. Czopki mją 3 różne
białka fotoreceptorowe o różnych maksimach absorpcji odpowiadające kolorom czerwonemu,
niebieskiem i zielonemu.

BIOSYNTEZA AMINOKWASÓW
Reakcja wiązania azotu netto i źródła elektronów oraz ATP:
N2 + 8e- + 16ATP + 16H2O -> 2NH3 + 16ADP + 16Pi + 8H+ + H2
Elektrony – pochodzą z procesu utleniającego, pochodzi od organizmów wiążących azot i przez
światło energii słonecznej
ATP – zazwyczaj pochodzi z oksydacji lub mechanizmów fotosyntezy komórek.
Procesy biosyntezy rozpoczynają się od redukcji N2 do NH3,w procesie zwanym wiązaniem azotu.
Aby mogła zajść reakcja wiązania azotu potrzebny jest kompleks nitrogenazy, zawierający dwa
białka: reduktazę, która dostarcza elektrony o wysokim potencjale redukcyjnym i nitrogenazę, która
wykorzystuje je od redukcji. Transfer elektronów wymaga hydrolizy ATP prowadzonej przez
reduktazę. Kompleks ten jest inaktywowany przez tlen.
Aminokwasy egzogenne (nie mogą być syntetyzowane): fenyloalanina, izoleucyna, leucyna,
lizyna, metionina, tryptofan, treonina, walina.
Aminokwasy endogenne ( sami syntetyzujemy): alanina, asparagina, asparaginian, cysteina,
glutamina, glutaminian, glicyna, prolina, seryna.
Aminokwasy wywodzące się z pirogronianu: alanina, walina, leucyna
Aminokwasy wywodzące się z alfa-ketoglutaranu: glutaminian, prolina, glutamina, arginina,
ornityna.
Wbudowanie jonu amonowego: następnym etapem asymilacji azotu jest wbudowanie NH4+ do
aminokwasów. Zasadniczą rolę w tym procesie pełni glutaminian i glutamina.
Dehydrogenaza glutaminianowa:
NH4+ + alfa-ketoglutaran + NADPH + H+ -> glutaminian + NADP+ + H2O
Działanie tego enzymu dąży do tego, aby powstał glutaminian. Enzym ten nie rozróżnia NADH i
NADPH.
Syntetaza glutaminianowa: prowadzi do powstania glutaminy
Glutaminian + ATP-> glutaminian-P + NH3 -> Glutamina + Pi
Syntaza glutaminianowa: katalizuje redukcyjną aminację alfa-ketoglutaranu.
Alfa-ketoglutaran + glutamina+ NADPH -> 2 Glutaminian + NADP+
Gdy ilość NH4+ jest ograniczona, większość glutaminianu powstaje w wyniku działania kolejno
syntetazy glutaminowej i syntazy glutaminianowej. Suma tej reakcji:
NH4+ alfa-ketoglutaran + NADPH+ ATP -> Glutaminian + NADP+ + ADP + Pi
Cząsteczki które dostarczają szkieletów węglowych sześciu biosyntetycznych rodzin
aminokwasów:
Ze szlaku glikolizy- pirogronian, 3-fosfoglicerynian i fosfoenolopirogronian
Z cyklu Krebsa- szczawiooctan (również z glukoneogenezy) i alfa-ketoglutaran
Ze szlaku pentozofosforanowego- rybozo-5-fosforan i erytrozo-4-fosforan
Regulacja biosyntezy aminokwasów: pierwsza nieodwracalna reakcja szlaku, zwana etapem
kluczowym, ma wysokie znaczenie. Produkt ostateczny szlaku jest inhibitorem enzymu
odpowiedzialnego za reakcję etapu A-> B. Ten rodzaj regulacji pozwala na optymalne
wykorzystanie zarówno elementów budulcowych jak i energii zużywanej podczas procesów
metabolicznych.
Hamowanie i aktywacja na drodze sprzężenia zwrotnego: przykładem jest biosynteza waliny,
leucyny i izoleucyny. Reakcja hydroksyetylo-TPP z alfa-ketomaślanem rozpoczyna szlak
prowadzący do powstania izoleucyny. Względne stężenie alfa-ketomaślanu i pirogronianu decyduje
o tym, ile izoleucyny może powstać w stosunku do waliny i leucyny. Aktywność deaminazy
treoninowej, enzymu PLP, podlega allosterycznemu hamowaniu przez izoleucynę. Enzym podlega
allosterycznej aktywacji przez walinę. Mechanizm ten polega na zachowaniu właściwych proporcji
pomiędzy powstającymi aminokwasami.
Skumulowane sprzężęnie zwrotne: enzym reguluje przepływ azotu, a tym samym odgrywa
zasadnicza rolę w kontroli metabolizmu bakterii. Skumulowane sprzężenie zwrotne polega na tym,
że każdy z czynników hamujących może redukować aktywność enzymu w określonym zakresie.
Oznacza to, że aktywność enzymatyczna praktycznie zanika, kiedy enzym wiążę się ze wszystkimi

regulatorowymi produktami końcowymi.
Regulacja aktywności syntetazy glutaminowej: polega na tworzeniu odwracalnych modyfikacji
kowalencyjnych w cząsteczce tego białka. Modyfikacja polega na przyłączeniu AMP do grupy
hydroksylowej określonej reszty tyrozyny w każdej z podjednostek tego enzymu. Adenylacja
powoduje, że enzym jest mniej aktywny i bardziej podatny na skumulowane sprzężenie zwrotne niż
w postaci nieadenylowanej. Przyłączenie AMP jest ostatnim etapem w kaskadzie enzymatycznej,
która rozpoczyna się kilka etapów wcześniej z udziałem różnych związków, w tym bezpośrednich
produktów powstających w trakcie syntezy glutaminy. Reakcję adenylacji i fosforolizy
katalizowane są przez ten sam enzym, transferazę adenylową. Jest on zbudowany z dwóch
podjednostek, jedna odpowiada za adenylację, druga za usunięcie grupy adenylowej. Przyłaczanie
AMP jest kontrolowane przez białko regulatorowe. Kaskada pozwala na wzmocnienie sygnału.
Wyszukiwarka
Podobne podstrony:
Finanse pubiczne II cz 2
Mathcad Projekt wytrzymałość II cz 3
biochemia 2-2 z odp plus, biochemia II, biochemia 2 kolokwia
Oleksyszyn, biochemia II, biosynteza nukleotydów
Biochemia II
Odpowiedzi cwiczymy czytanie kl II cz II
dr hab RG I II II cz swoboda przeplywu pracownikow
RECEPTY Z MAŚCI dla semestru II? CZ III
BIOCHEMIA II termin egzaminu 06 i 07 LEK i STOMA by KaMilka
Księga 1. Proces, ART 502 KPC, II CZ 93/09 - postanowienie z dnia 17 listopada 2009 r
Oleksyszyn, Biochemia II, zagadnienia do glikoliza i glukoneogeneza
pytania II cz
biochemia II 1 plus id 86425 Nieznany (2)
Rozdział II, cz 1
Analiza matematyczna II cz I
biochemia II 1 cw
stentor Odpowiedzi cwiczymy czytanie kl II cz I
Biochemia II odpowiedzi koło
więcej podobnych podstron